

Abstract
The cerebral cortex is a cellularly-complex structure comprised of a rich diversity of neuronal and glial cell types. Cortical neurons can be broadly categorized into two classes—glutamatergic excitatory neurons and GABAergic inhibitory interneurons. Previous developmental studies in rodents have led to the prevailing model that while excitatory neurons are born from progenitors located in the cortex, cortical interneurons are born from a separate population of progenitors located outside of the developing cortex in the ganglionic eminences1–5. However, the developmental potential of human cortical progenitors has not been thoroughly explored. Here we show that in addition to excitatory neurons and glia, human cortical progenitors are also capable of producing GABAergic neurons with the transcriptional characteristics and morphologies of cortical interneurons. By developing a cellular barcoding tool called “ScRNAseq-compatible Tracer for Identifying Clonal Relationships” (STICR), we were able to perform clonal lineage tracing of 1912 primary human cortical progenitors from six specimens and capture both the transcriptional identities and clonal relationships of their resulting progeny. A subpopulation of cortically-born GABAergic neurons were transcriptionally similar to cortical interneurons born from the caudal ganglionic eminence and these cells were frequently related to excitatory neurons and glia. Thus, our results demonstrate that individual human cortical progenitors can generate both excitatory neurons and cortical interneurons, providing a new framework for understanding the origins of neuronal diversity in the human cortex.
The neocortex is responsible for performing many higher order cognitive functions such as decision making, language comprehension, and sensory perception. During cortical development, progenitors in the dorsal forebrain and ganglionic eminences produce the diverse array of neurons and glia that comprise the neocortex. Extensive developmental studies in rodents have demonstrated that the two principal types of cortical neurons—glutamatergic excitatory neurons and GABAergic inhibitory interneurons—are produced by two distinct groups of progenitors.1–5 Excitatory neurons are generated by progenitors in the dorsal forebrain and migrate radially to occupy their terminal positions in the cortical plate. In contrast, cortical interneurons are generated by progenitors located in the ganglionic eminences of the ventral telencephalon - and then migrate dorsally into the developing neocortex.
While there have been a small number of limited studies that have suggested the possibility that cortical neural progenitors might give rise to cortical interneurons, these studies have been conflicting6–10. Short-term clonal labeling of progenitors in the human cortex revealed the local generation of newborn GABAergic neurons7, but did not determine whether they were cortical interneurons or another type of interneuron such as olfactory bulb interneurons, which have previously been shown to derive from cortical progenitors11–13. Similarly, in vitro cultures derived from human cortical progenitors have been shown to generate GABAergic inhibitory neurons10. Conversely, a later study that labeled dividing cells in short-term cultures of human organotypic tissue slice did not observe a substantial fraction of newborn inhibitory neurons in the cortex8. The overwhelming consensus remains that human cortical progenitors give rise to excitatory neurons but not cortical inhibitory neurons, however this has not been thoroughly examined and the developmental potential of individual human cortical progenitors remains largely unknown.
Design and Validation of Lineage Tracer
In order to perform high-throughput clonal lineage tracing of primary human neural progenitors, we developed STICR, an ultra-high complexity barcoding strategy that allowed us to permanently label cells and their progeny using a lentivirus encoding a heritable, transcribed molecular barcode within the 3’ untranslated region of the enhanced green fluorescent protein (EGFP) reporter gene (Fig. 1a). The combinatorial, single molecule barcode design of STICR allows for a pre-defined, error-correctable barcode library with a maximum diversity of 125 million sequences (Extended Data Fig. 1a, see methods). Deep sequencing of STICR plasmid and lentiviral libraries confirmed ultra-high barcode diversity (~50–65 million unique barcodes per library) without any overrepresented barcodes (Extended Data Fig. 1b). Using the observed barcode diversity and frequency from each STICR library, we modeled the rate of barcode “collision”—the event in which two different cells are independently labeled with the same barcode—and found that STICR could be used to label more than ~250,000 cells before reaching an estimated barcode collision rate of ~0.5% (Extended Data Fig. 1c). To confirm the accuracy of STICR barcode recovery from single cells, we performed a cell mixing, or “barnyard”, experiment where we labeled mouse and human cells with different STICR libraries that were readily distinguishable by a constant “viral index” sequence unique to each library (Extended Data Fig. 1d). Following scRNA-seq, recovered STICR barcodes were 100% concordant with the STICR library used to infect each population (Extended Data Fig. 1e,f), indicating that our method is accurate and can be robustly applied to perform high-throughput clonal lineage tracing.
Figure 1. STICR-labeled progenitors generate all three principal cortical cell types.
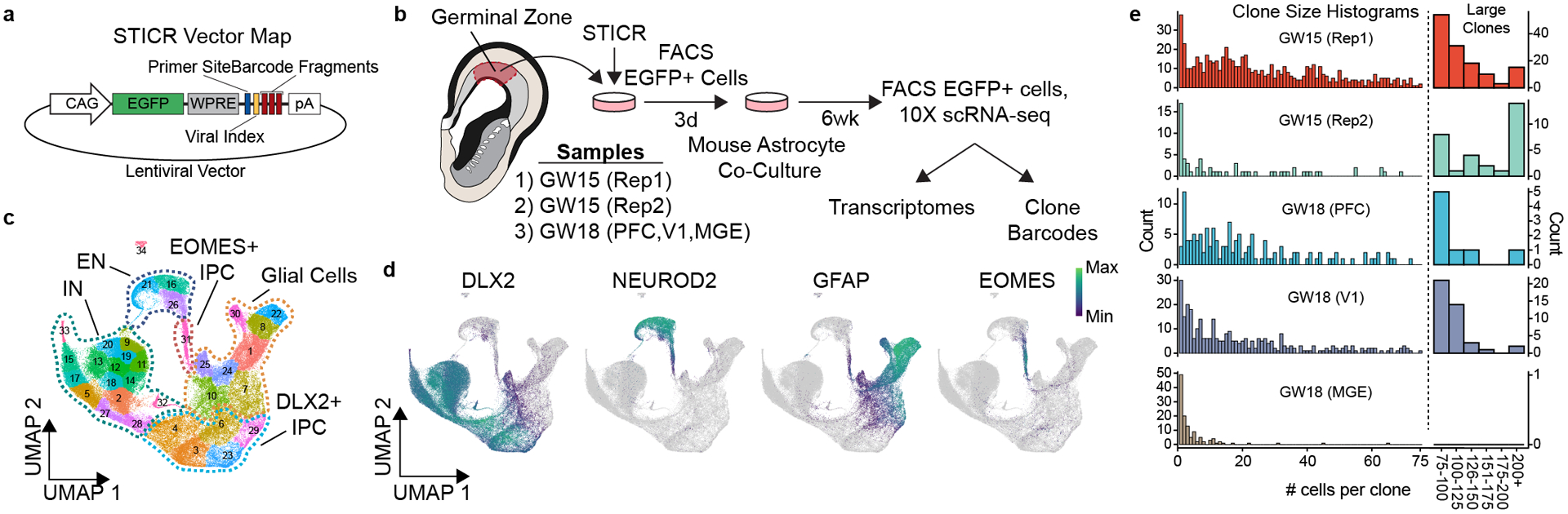
a) Lentiviral vector design of STICR b) Schematic of experimental design used to label and capture samples. c) UMAP embedding and Louvain clustering of STICR-labeled cells following scRNA-seq. d) Feature plot of principal cell trajectory marker genes DLX2 (GABAergic inhibitory neurons and DLX2+ IPCs), NEUROD2 (excitatory neurons), GFAP (glia), and EOMES (EOMES+ IPCs). e) Histogram of clone sizes within each sample. Left, clone sizes from 1–75 cells. Right, clone sizes of >75 cells in 25 cell bins.
Human Neural Progenitor Lineage Tracing
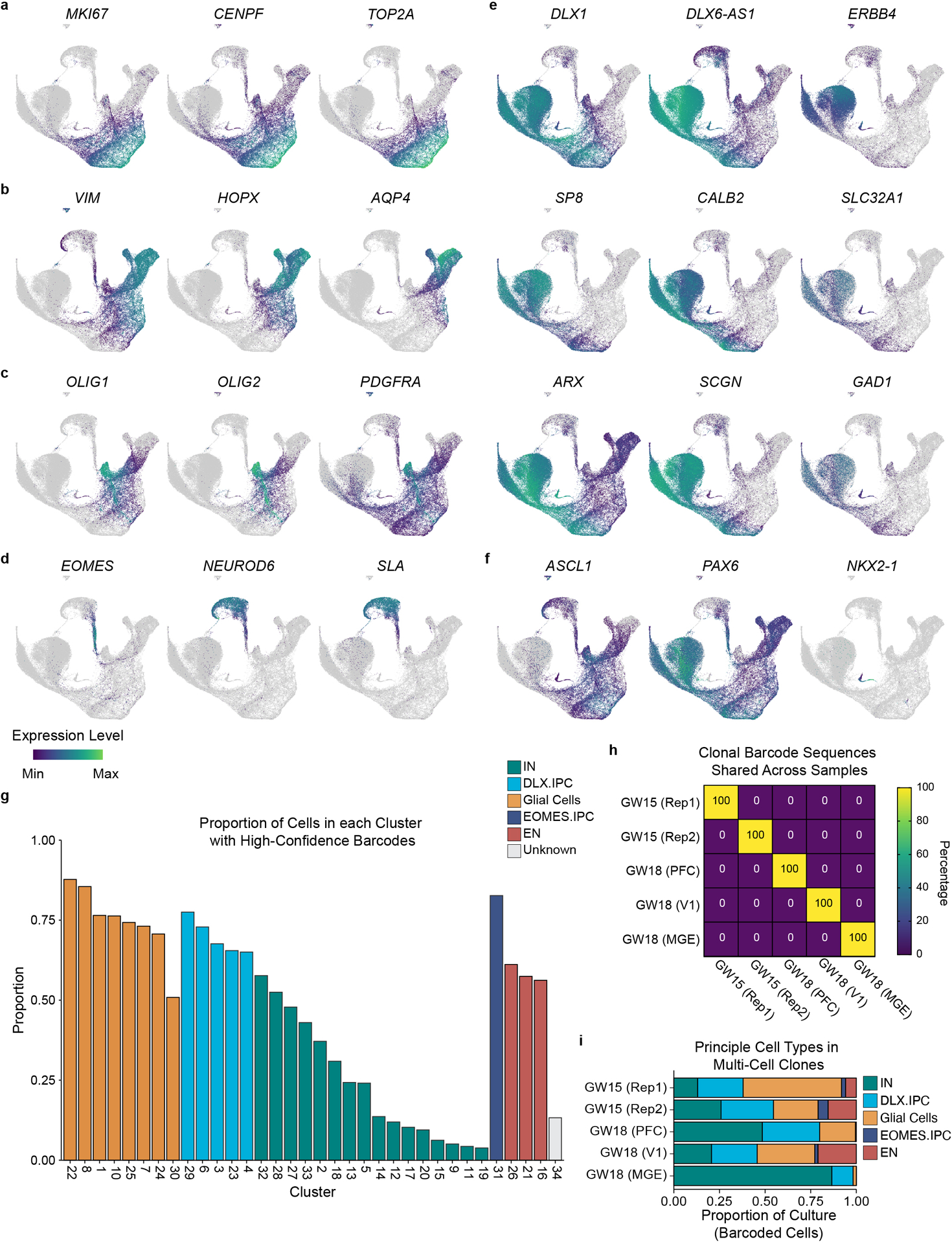
To determine the developmental potential of individual human cortical progenitors, we derived primary human cell cultures from the cortical germinal zone of three different specimens at stages of peak neurogenesis (gestational weeks 15 and 18—GW15 and GW18) (Fig. 1b). Prior to clonal labeling, one of the specimens (GW18) was further dissected based on known anatomical landmarks, allowing us to generate region-specific cultures from the germinal zones of the prefrontal cortex (PFC), primary visual cortex (V1), and medial ganglionic eminence (MGE). We then labeled cells with STICR and cultured them for six weeks in vitro before performing scRNA-seq. Transcriptome analysis from 121,290 cells identified three principal cortical cell type trajectories— excitatory neurons, GABAergic inhibitory neurons, and glia—based on differential gene expression including that of marker genes NEUROD2, DLX2, and GFAP, respectively (Fig. 1c,d, Extended Data Figs. 2,3). We identified intermediate progenitor cells (IPCs) within both the inhibitory and excitatory neuron trajectories, which we refer to as DLX2+ IPCs (inhibitory trajectory) and EOMES+ IPCs (excitatory trajectory) (Extended Data Fig 3). Cluster correlation analysis of STICR datasets with an scRNA-seq atlas of the developing primary human cortex at comparable developmental time points and regions14 further supported these cell-type designations (Extended Data Fig 2d).
We recovered STICR barcodes in 63±23% (mean±SD, n=5 libraries) of cells per culture including cells of each principal cell type (Supplemental Table 1, Extended Data Fig. 3g). In total, we identified 1461 unique clonal barcodes, 1324 of which belonged to multicellular clones with a median size of 23 cells per clone (Fig. 1e). While there is very little known about the output of human cortical progenitors over this time scale, we observe a maximum clone size of 1209 cells which is congruent with a prior study that measured the output from three individual human outer radial glia.15 Importantly, no STICR barcodes were shared between experimental groups (Extended Data Fig 3h), further indicating that cells identified as members of multicellular clones in this study are not the result of barcode collision. While all three principal cortical cell types (excitatory glutamatergic neurons, GABAergic interneurons, and glia) were found in multi-cell clones in both GW15 samples and the GW18 V1 sample, GW18 PFC clones contained relatively more GABAergic inhibitory neurons than excitatory neurons or glia (Extended Data Fig 3i). Consistent with previous studies, the GW18 MGE culture completely lacked excitatory neurons and instead was comprised almost entirely of interneurons (Extended Data Fig 3i). Thus, STICR reveals clonal lineage relationships of all principal cell types derived from human forebrain progenitors.
Clonal Relationship of Human ENs and INs
In order to determine the lineage relationships of cells born from cortical progenitors, we first analyzed the relative proportions of principal cell types in multicellular clones. The majority (66%, 829 of 1,252) of multicellular clones from cortical cultures contained at least 1 excitatory neuron, with these clones containing a median of 3 excitatory neurons per clone (Extended Data Fig. 4a–b, Supplemental Table 1). While our study of regional cortical progenitors is limited to a single GW18 sample, we found that PFC clones contained proportionally fewer excitatory neurons than clones in the V1 sample (Extended Data Fig. 4b,c). This is consistent with the neurodevelopmental gradient in the cortex where developmental milestones are reached in the PFC several weeks prior to the V1 region16. Across all samples, only 1.5% (19 of 1252) of cortical clones were comprised entirely of excitatory neurons, and 50.7% (635 of 1252) contained a combination of excitatory neurons, inhibitory neurons, and glia (Fig. 2a–c, Extended Data Fig 4c, and Supplementary Table 1). Due to extensive aggregation of excitatory neurons within in vitro cultures and the gentle dissociation used (Extended Data Fig. 4d,e), excitatory neuron production may be underestimated in the dataset derived from cell culture experiments. Nevertheless, across GW15 samples, the coincidence of excitatory and inhibitory neurons within the same clone occurred across the range of clone sizes (Fig. 2b). Together, these results suggest that individual human cortical progenitors reproducibly generate both excitatory and inhibitory neurons.
Figure 2. Individual human cortical progenitors can generate both excitatory and inhibitory cortical neurons in vitro.
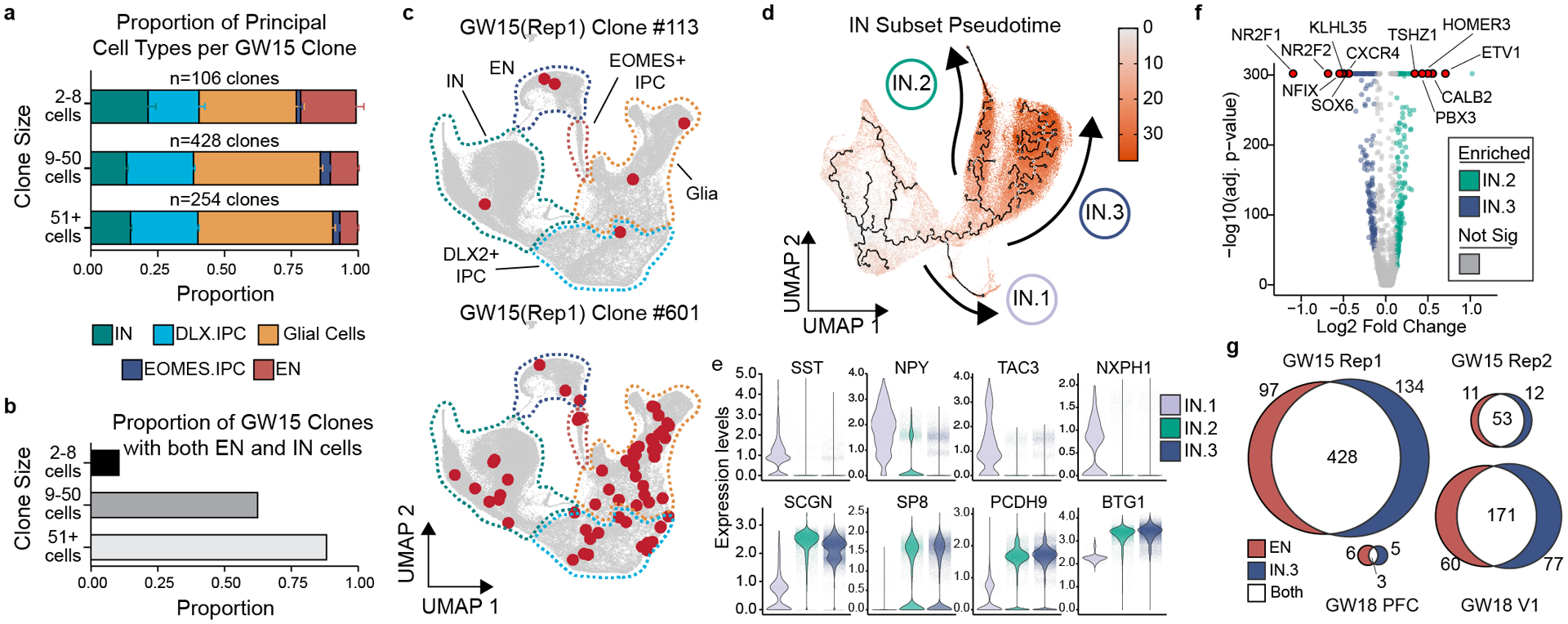
a) Stacked boxplot depicting the average (mean±SD) proportion of GABAergic inhibitory neurons, DLX2+ IPCs, EOMES+ IPCs, excitatory neurons, and glia in different sized clones. b) Bar graph depicting the proportions of clones of different sizes that contain both EN and IN cells. c) Representative 6 cell (GW15, Rep1, #113) and 61 cell (GW15, Rep1, #601) clones depicted in UMAP space. Cells within each clone colored in red. Dashed lines depict borders of principal cell types from Fig. 1c. d) Pseudotime transcriptional trajectories of subclustered inhibitory neurons. Three different interneuron transcriptional trajectories (IN.1, IN.2 and IN.3) indicated with arrows. e) Violin plots depicting gene expression of IN.1 marker genes (SST, NPY, TAC3, and NXPH1) and general CGE marker genes (SCGN, SP8, PCDH9, and BTG1) in IN.1, IN.2, and IN.3 trajectories. f) Volcano plot comparing gene expression differences between IN.2 and IN.3 trajectory cells. g) Venn diagram depicting the number of multicellular cortical clones that contain IN.3 or EN cells.
EN Clones Contain OB and Cortical INs
We next sought to determine the transcriptional identities of cortically-born GABAergic neurons. In addition to DLX2 (Fig. 1d), IN trajectory cells were broadly enriched for general markers of interneurons including GAD1, ARX, SLC32A1, and DLX6AS-1 (Extended Data Fig. 3e). Iterative subclustering and transcriptional trajectory analysis of the IN cells along with the DLX2+ IPCs revealed three distinct subgroups of GABAergic inhibitory neurons that we termed IN.1, IN.2, and IN.3 (Fig. 2d, Extended Data Fig. 5a–c). IN.1 cells were enriched for markers of SST+ cortical interneurons including SST, NPY, TAC3, and NXPH1 (Fig. 2e). Consistent with developmental studies in mice which demonstrated that SST+ cortical interneurons derive primarily from the MGE17, 73% (492 of 671 cells) of IN.1 cells are produced by MGE progenitors (Extended Data Fig. 5d). In contrast, IN.1 cells comprised only 0.3% (179 of 56,244 cells) of progeny born from cortical progenitors (Extended Data Fig. 5e). Furthermore, MGE-derived IN.1 cells expressed canonical MGE-born interneuron genes including LHX6, NKX2–1, ACKR3 (CXCR7), PDE1A, and MAF, while cortically-born IN.1 cells did not (Extended Data Fig. 5f). Together, these data suggest that IN.1 cells are transcriptionally similar to SST+ cortical interneurons and that the majority of IN.1 cells derive from the MGE. In contrast to IN.1 trajectory cells, IN.2 and IN.3 trajectory cells were transcriptionally similar to cells born from the caudal ganglionic eminence (CGE) and expressed marker genes such as SCGN, SP8, PCDH9, and BTG1 (Fig. 2e)18. Furthermore, IN.2 and IN.3 cells differed from IN.1 cells in that they were derived entirely from cortical progenitors, with no contribution from MGE progenitors (Extended Data Fig. 5d). Top IN.2 markers included TSHZ1, PBX3, MEIS2, CALB2, CDCA7L, SYNPR and ETV1 which are enriched in mouse olfactory bulb interneurons (Fig 2f)19,20. In contrast, IN.3 cells were enriched for NR2F1, NFIX, PROX1, and NR2F2, which are enriched within the CGE, as well as SOX6 and CXCR4, which are enriched in cortical interneurons (Fig. 2i), suggesting that these cells are transcriptionally similar to CGE-derived cortical interneurons.14,21–25 Comparison of IN.2 and IN.3 marker genes to orthogonal datasets including those from the Allen Brain Atlas26 similarly support this distinction (Extended Data Fig. 5g,h). Thus, while there are currently no marker genes that can unequivocally distinguish cortical interneurons from olfactory bulb interneurons, our transcriptome-wide data suggest that IN.2 cells resemble olfactory bulb interneurons while IN.3 cells are similar to CGE-born cortical interneurons.
Previous fate-tracing studies in mice have demonstrated that cortical progenitors can produce a subset of embryonically-born OB interneruons11. In agreement with these findings, we find that multicellular cortical clones frequently contain OB-like (IN.2 branch) GABAergic inhibitory neurons (Extended Data Fig. 5d,e). Interestingly, many clones containing OB-like GABAergic inhibitory neurons also contained excitatory neurons (61%, 321 of 530 clones) (Extended Data Fig. 5j). Such clones were found in all cortical samples but were especially common in both GW15 samples (Supplementary Table 1). GW18 PFC clones contained the highest proportion of OB-like GABAergic neurons (41%, 1637 of 3949 cells) out of any sample analyzed, and although relatively few of the GW18 PFC clones contained excitatory neurons (9 of 141 clones), 8 of these also contained at least one OB-like interneuron (Extended Data Fig. 5d, Supplementary Table 1). These results suggest that similar to mice13, human cortical progenitors can generate OB interneurons during embryogenesis and that some excitatory neurons and embryonically-born OB interneurons are clonally related.
Surprisingly, we found that the majority (79%, 655 of 829) of multicellular clones that contained excitatory neurons also included putative cortical interneurons (IN.3 cells) (Fig. 2g), indicating that some human cortical progenitors were capable of generating both excitatory neurons and cortical interneurons. Many of these clones contained multiple cells of both types, and mixed clones were especially abundant in GW15 and GW18 V1 samples. Further subclustering analysis of excitatory neurons revealed both deep-like and upper-like excitatory neuron subgroups, both of which were frequently clonally related to IN.3 cells (Extended Data Fig. 6). Notably, every cortical sample we analyzed contained clones with both excitatory and inhibitory cortical neurons (Fig. 2g). These mixed clones also frequently contained olfactory bulb interneurons as well as glial-trajectory cells (Extended Data Fig. 5i,j). Taken together, our results suggest that human cortical progenitors cultured in vitro are unexpectedly multipotent in their ability to generate a wide variety of principal neural cell types including both excitatory neurons and putative cortical interneurons, two cell types previously thought to be produced by different pools of spatially restricted progenitors in the developing forebrain.
Mixed Cortical EN/IN Clones in Xenografts
To confirm that the observed lineage relationship between excitatory neurons and cortical interneurons was not an in vitro culture artifact, we STICR-labeled cortical germinal zone cells from three additional GW15 specimens as above and transplanted them into the cortex of early postnatal immunodeficient mice, where we allowed them to develop for six weeks before analysis (Fig. 3a). Immunohistochemistry revealed xenografted human cells at the injection site itself as well as distributed throughout the adjacent tissue (Figs. 3b, Extended Data Fig. 7a–c). To quantify the proportion of the principal cell types in the xenografts, we performed IHC for excitatory neuron marker NEUROD2, interneuron marker GABA, and glia markers SOX9 and OLIG2. Excitatory neurons were the most common cell type, accounting for 75.7±8.4% of transplanted cells, followed by glia at 16.8±6.3%, and interneurons at 7.9±3.2% (mean±SD, n=7 recipient mice, Extended Data Figs. 7a–c). In parallel, we FACS-isolated EGFP+ xenograft cells and performed scRNA-seq to determine their transcriptional identities and clonal relationships. Similar to our in vitro cultures, xenograft cells formed distinct transcriptional clusters of GABAergic neurons, excitatory neurons, and glia (Fig. 3c, Extended Data Fig. 7d–f). The proportion of excitatory and inhibitory neurons within multicellular clones from xenograft experiments differed from we observed in our in vitro cultures (Fig 2g). This was likely due to the specific loss of excitatory neurons from in vitro culture during dissociation as these cells frequently associated in tightly formed masses that were difficult to dissociate (Extended Data Figs. 4d,e). Quantification of principal cell types by scRNA-seq closely matched cell proportions observed by immunohistochemistry, and was reproducible across biological replicates (Extended Data Fig. 7g). These data suggest that xenograft-derived STICR-labeled cells analyzed by scRNA-seq accurately reflected the cell proportions produced in vivo.
Figure 3. Xenografted human cortical progenitors generate both excitatory and inhibitory cortical neurons in the same clone.
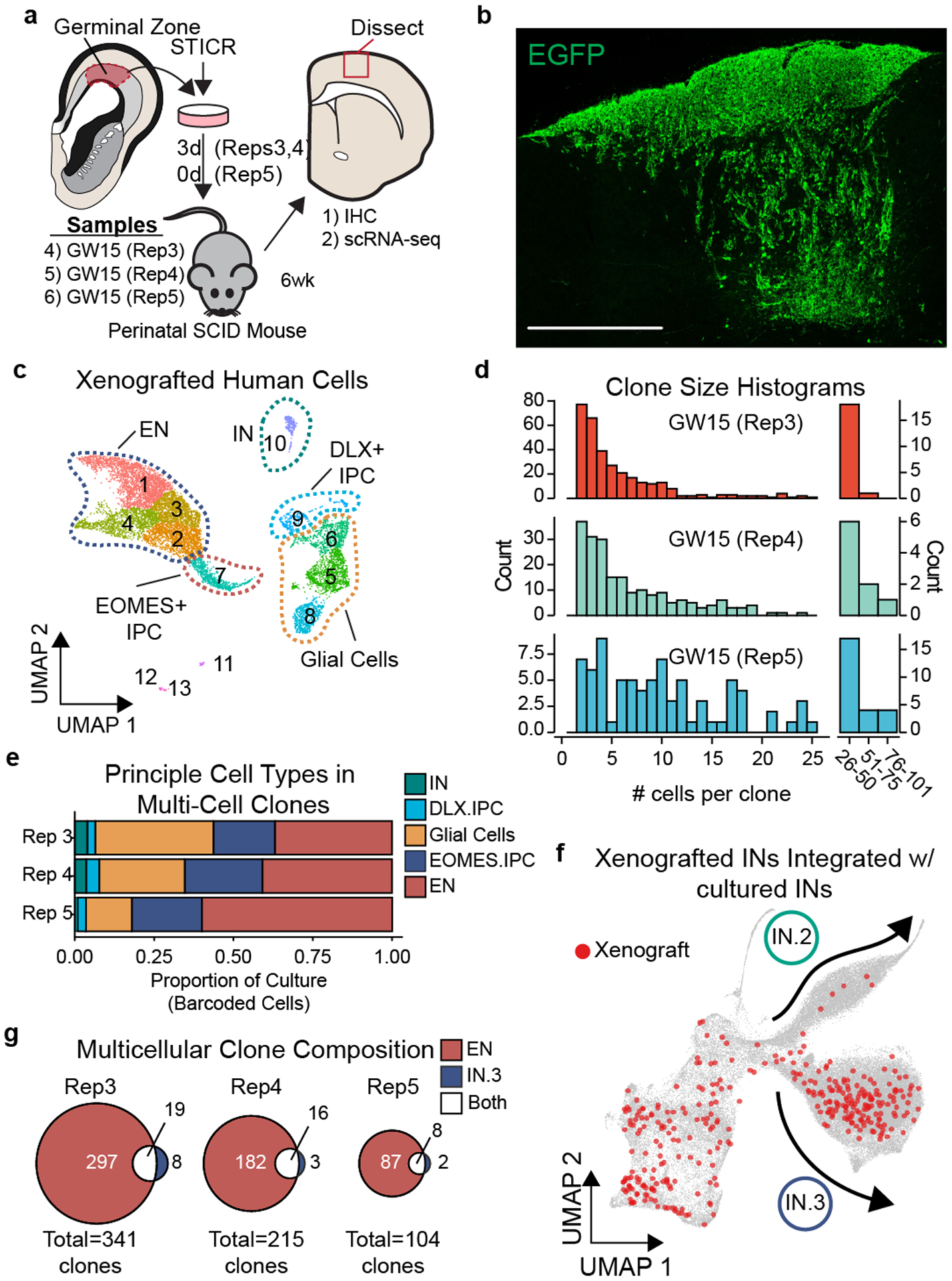
a) Schematic depicting experimental design and analysis of STICR-labeled progenitors by IHC and scRNA-seq following transplantation into the postnatal murine cortex. b) Representative image of transplanted human cortical cells. EGFP expression from STICR depicted in green. Scale bar, 500um. c) UMAP embedding and Louvain clustering of xenografted cells following scRNA-seq. d) Histogram of clone sizes within each xenograft sample. Left, clone sizes from 1–55 cells. Right, clone sizes of >25 cells in 25 cell bins. e) Stacked barplot depicting relative proportion of principal cell types within multi-cellular clones of each sample. f) UMAP embedding of both cultured and xenograft-derived INs. Xenograft-derived cells that are members of multicellular clones highlighted in red. IN.2 and IN.3 trajectories depicted with arrows. g) Venn diagram showing number of multi-cell clones containing excitatory neurons and/or IN.3 neurons.
We recovered STICR barcodes from 76±10% (n=3 libraries) of xenografted cells and identified 660 multicellular clones that ranged in size from 2 to 101 cells with a median clone size of 5 cells (Fig. 3d). Within multicellular clones, the proportions of principal cortical cell types were highly similar across biological samples (Fig. 3e). We then further analyzed cells categorized as either INs or DLX2+ IPCs to determine their transcriptional identities and clonal relationships. Subclustering analysis revealed several distinct groups of GABAergic cells (Extended Data Fig. 8a), including one that was enriched for genes found in cortical interneuron-like IN.3 cells – such as NR2F1, KLHL35, NFIX, and SCGN – and was similar to reference GABAergic inhibitory neurons (Extended Data Fig. 8b,c). In order to directly compare the transcriptomic identities of GABAergic neurons from xenografts to those observed following in vitro culture, we integrated scRNA-seq data of GABAergic neurons from both sets of experiments using the in vitro cultured cells as a reference. GABAergic neurons from the xenografts clustered well with their in vitro counterparts (Fig. 3f, Extended Data Fig 8d) and integrated entirely within the previously-defined transcriptional trajectories (Fig. 2f). Consistent with their marker gene expression, 85% (211 of 249) of GABAergic xenograft cluster 1 cells clustered within the IN.3 trajectory (Extended Data Fig. 8d), suggesting that they had the transcriptional identity of cortical interneurons. Few (4%, 9 of 249 cells) GABAergic xenograft cluster 1 cells clustered within the IN.2 trajectory, (Extended Data Fig. 8d) consistent with previous transplantation studies which found that the cortical environment is not conducive for the generation of OB interneurons.27 Xenograft-derived GABAergic IN.3 neurons were found in 56 multicellular clones, 43 (77%) of which also contained excitatory neurons (Fig. 3g, Extended Data Fig 8e–g, Supplementary Table 2). Mixed IN.3/EN clones were found in all 3 xenografted samples including GW15 Rep5, which was transplanted immediately following transduction with STICR and was never cultured (see methods). Thus, as we had previously observed in vitro, cortical progenitors are capable of generating mixed excitatory/inhibitory neuron clones when xenografted into the perinatal mouse brain, and these inhibitory neurons are transcriptionally similar to cortical interneurons.
Finally, to determine whether human cortical progenitor-derived GABAergic inhibitory neurons had additional features consistent with cortical interneurons, we characterized their morphology and distribution in two additional xenograft experiments from GW17 samples. For these experiments, we used a FACS-isolation strategy to enrich for human progenitors prior to transducing with STICR in order to prevent labeling post-mitotic cortical interneurons that already existed in the specimen (Fig 4a, Extended Data Fig 9). Given the protracted time scale of human brain development, we waited for 12 weeks after transplantation before analyzing the xenografts by IHC. We observed STICR-labeled cells with well-elaborated processes in the olfactory bulbs well as distributed throughout the cortical layers (Fig 4b, Extended Data Fig 9). 8.3% (89 of 1071) of STICR-labeled cells located in the cortical plate expressed GABA, and these were also broadly dispersed across the cortical layers (Fig. 4c,d, Extended Data Fig 10). While the majority of STICR-labeled GABA+ cells were located at a similar rostral-caudal level as the transplantation site, we did observe cells more distant from the transplantation site in both directions. Together, these morphological characteristics of cortically-derived GABAergic inhibitory neurons are consistent with their classification as cortical interneurons. Thus, individual human cortical progenitors are capable of generating both excitatory neurons and cortical GABAergic inhibitory neurons (Fig. 4e).
Figure 4. Xenografted human cortical progenitors generate GABAergic inhibitory neurons that distribute across the cortical laminae.
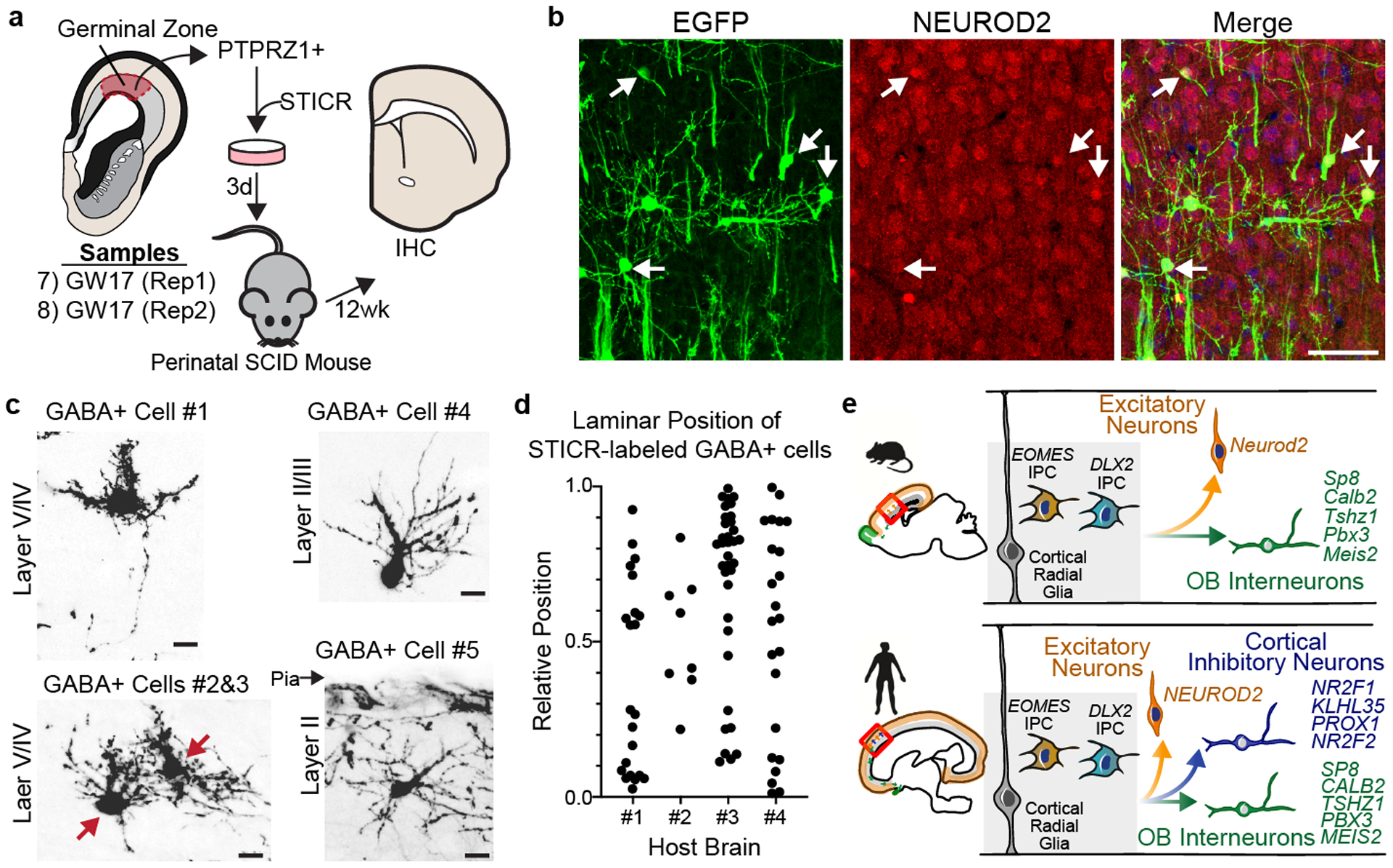
a) Schematic depicting experimental design and analysis of PTPRZ1-enriched, STICR-labeled progenitors by IHC following transplantation into the postnatal murine cortex. c) Representative images of transplanted human cortical cells analyzed by IHC. EGFP expression from STICR depicted in green, NEUROD2 in red, and DAPI counterstain in blue. Arrows indicate NEUROD2+/STICR+ double positive cells. Scale bars, 50um d) Representative images of STICR-labeled GABA+ cells throughout the cortical plate. Red arrows depict soma of two cells in the same image. Scale bar, 10um. e) Relative laminar position of GABA+/STICR+ cells in the cortical plate from corpus callosum (value of 0.0) to pial surface (value of 1.0) in each mouse host brain. Host brains #1 and #2 transplanted with GW17 (Rep2), Host brains #3 and #4 transplanted with GW17 (Rep3). f) Schematic depicting difference in the developmental potential between human and mouse cortical progenitors.
DISCUSSION:
By performing high-throughput lineage tracing of ~1900 human cortical progenitors, we were able to definitively demonstrate a lineage relationship between human excitatory neurons and putative cortical interneurons, two populations that had been widely believed to arise from distinct progenitor populations. While current scRNA-seq informatic tools have been used to describe the transcriptional trajectories between progenitors and mature cell types (i.e., pseudotime analysis28), extensive analysis of the developing human brain at this time has not uncovered this lineage relationship29, nor would further analysis likely do so given the degree to which the transcriptional trajectories of these cell types differ. Furthermore, pseudotime analysis itself does not provide direct evidence of clonal relationships and the lineages of transcriptionally similar progenitors can differ. Approaches have retrospectively infer developmental lineage relationships from somatic mutations predicted to have arisen during development30–32 currently lack the resolution to confidently assign daughter cells to individual progenitors and thus would not have revealed this relationship.
Previous studies suggested that dorsal telencephalic radial glia generate cortical interneurons in primates, but were limited by their use of a small set of putative “marker” genes which do not directly link progenitors to their progeny.6,7 In these studies, differences in the expression of GABAergic marker genes between different germinal zones were presumed to persist across differentiation from progenitors to inhibitory neurons. These markers were thus used to retrospectively infer the origin of cortical interneurons. However, this type of approach cannot account for potential changes in gene expression that can occur over development, or for potentially unknown sources of cells expressing the same putative “marker” genes. Additionally, while Letinic and colleagues used a gammaretroviral labeling approach to identify interneurons born in the dorsal forebrain7, their approach did not determine whether these cells were cortical interneurons or OB interneurons, which this study and others have found to be produced from progenitors in the cortex11–13. The prospective approach of clonal labeling by STICR combined with scRNA-seq based transcriptome-wide analysis performed in our current study allowed us to distinguish between these two types of interneurons, and we surprisingly found that both types of interneurons can be clonally related to excitatory neurons (Figs. 2–4). As with any prospective labeling study performed in humans, we cannot definitively rule out the possibility that our methodologies influenced progenitor behavior. However, the observation of clonally-related excitatory and inhibitory neurons in all our experimental contexts provides strong evidence that human cortical progenitors have the capacity to generate both types of neurons.
In this study, we investigated the clonal relationships of cells born from GW15-GW18 human cortical progenitors over a six-week developmental window. A previous 5-bromo-2’-deoxyuridine (BrdU) birth-dating study by Hansen and colleagues8 quantified BrdU+;DLX2+ cells in organotypic slice culture derived from GW17.5-GW20.5 human cortex. After ~8–10 days of BrdU labeling, the study did not find a substantial amount of BrdU+;DLX2+ cells from cortical progenitors. Given the longer window of our experiments, these data suggest that the production of interneurons by cortical progenitors does not occur throughout the entire duration of corticogenesis, but instead begins at some point after midgestation. Consistent with this interpretation, six weeks after labeling we observed a higher proportion of inhibitory neurons within clones derived from GW18 progenitors than within those from GW15 progenitors (Supplementary Table 1). Furthermore, extensive chains of migrating interneurons have been observed in the perinatal human cortex.33 Thus, production of cortical interneurons from cortical progenitors may extend beyond the period in which excitatory neurons are born. It is also possible that the different experimental systems used in these studies (i.e. in vitro cell culture and xenograft in this study and ex vivo organotypic slice culture by Hansen and colleagues8) might contribute to some of the observed incongruencies between these three systems. Future studies aimed at detailing the output of cortical progenitors over a broader developmental period and throughout different cortical regions will help to further elucidate the contribution of this phenomenon to human brain development.
In a companion study by Bandler et al., STICR was used to perform in vivo clonal labeling of embryonic mouse forebrain progenitors and then analyzed STICR-labeled cells by scRNA-seq postnatally. While they recovered both glutamatergic excitatory neurons and cortical interneurons, these never occurred within the same clone. The lineage relationship we observe between cortical excitatory and inhibitory neurons in this study raises new questions regarding the development of the human cerebral cortex. First, what are the implications of a single progenitor producing both excitatory neurons and cortical interneurons? Evolutionary expansion of the primate neocortex has been attributed to the increased proliferative capacity of cortical neural progenitors. Adaptations in cortical progenitor competence to produce both principal types of cortical neurons could help ensure appropriate inhibitory/excitatory balance despite the dramatic increase in the cortical excitatory neuron pool.34 Recent studies have revealed that while the inhibitory/excitatory balance increases from mouse to human, the relative composition of cortical interneuron types remains relatively constant across evolution. While current technical limitations prevent us from confidently estimating the precise cellular contributions of cortical progenitors to the mature human brain, future studies quantifying the relative contributions of progenitors from the cortex and ganglionic eminences will be helpful in understanding the cellular basis of how normal human cortical function is achieved.
This study opens many important questions for future investigation. The molecular mechanisms that regulate the production of locally-born cortical inhibitory neurons are currently unknown. In mice, sonic hedgehog signaling35 is required for individual cortical progenitors to undergo a GABAergic “switch” and generate inhibitory neurons36 that migrate to the OB13. Does a similar molecular mechanism govern the production of cortically-derived cortical interneurons in humans? Furthermore, what molecular markers, if any, can distinguish them from cortical INs born in the CGE or MGE? Previous studies have found that NR2F1/2 are expressed not only in the CGE but also in cortical progenitors.9,10 Given the transcriptional similarity of cortically-derived IN.3 cells observed in this study to CGE-derived interneurons, it is possible that a similar developmental program is used. Several such important questions are raised by this new understanding of human cortical lineage, and future studies addressing these will help further decipher the origins and mechanisms underlying human brain development.
METHODS
STICR Barcode Design
STICR barcode fragment sequences were generated using the Barcode Generator script written by Luca Comai and Tyson Howell (http://comailab.genomecenter.ucdavis.edu/index.php/Barcode_generator) using a sequence length of 15bp and a minimum hamming distance of 5. Sequences containing the restriction enzyme sites matching STICR’s multicloning site (MCS) or homopolymer repeats longer than 4bp were excluded. In total, 3 sets of 500 sequences meeting these design criteria were selected (Supplementary Table 3).
STICR Library Creation
STICR barcode libraries were created using a modified pSico lentiviral plasmid (addgene, #11578) in which the sequence between the cPPT/CTS and 3’ LTR was replaced with a DNA fragment containing the hybrid CMV/chicken β-actin promoter (CAG) promoter, EGFP transgene, WPRE, MCS, and bGH polyadenylation signal. The MCS consisted of three adjacent pairs of restriction enzyme sites (EcoRI-BamHI-NheI-XhoI) between which the STICR barcode fragments would be added. Each dsDNA STICR barcode fragment was synthesized as a pair of ssDNA oligonucleotides (Genewiz), annealed, and then pooled together with all the other barcode fragments belonging to its set. Each set of barcode fragments were added into the STICR MCS individually over three rounds of restriction enzyme cloning. After each round of barcode fragment addition, a “negative selection” digest was performed using a restriction enzyme that targeted the stuffer sequence in the MCS that should have been replaced with a barcode fragment so that un-digested/barcoded molecules were removed from the library. Following each “negative selection”, the resulting STICR barcode plasmid library was amplified by transformation into MegaX DH10B Electrocompetent E. Coli (Thermo, C640003) and grown overnight on LB agar plates at 37C. In order to maintain even sequence distribution, transformed bacteria were plated at high density (~75 million colonies per large format plate) which helped restrict colony size and make them grow more uniformly. In order to maximize the diversity of barcode sequences, we transformed enough barcoded plasmid to get at least 10 times as many colonies as there were potential barcode sequences. Additionally, the STICR plasmid contained a sequencing primer site upstream of the STICR barcode. Each STICR library contained a 3 base pair viral “index” immediately downstream of the sequencing primer binding site. The viral index is unique to each library and allowed us to differentiate STICR barcodes from different libraries.
Generation of NGS Libraries for Estimation of STICR Plasmid and Lentiviral Diversity
To generate a STICR plasmid library for next generation sequencing, we first digested 1ug of each library with XhoI and then ligated a PCR adapter containing a UMI to this site (Supplementary Table 4). Ligation products were amplified by PCR using Q5 Hot Start High Fidelity 2x Master Mix (NEB, #M0494) using primers targeting to the STICR sequencing primer site and the adapter sequence (Supplementary Table 4) using the following program: 1) 98C, 30sec, 2) 98C, 10sec, 3) 62C, 20 sec ,4) 72C, 10 sec, 5) Repeat steps 2–4 15x, 6) 72C, 2 min, 7) 4C, hold. Following PCR amplification, a 0.8–0.6X dual-sided size selection was performed using Ampure XP beads (Beckman Coulter, #A63881). The resulting libraries were sequenced to the depth of ~30 million reads.
To generate a STICR lentiviral library for next generation sequencing, we performed an RNA extraction from 1/20 of the total lentiviral prep using 300 uL Trizol (Thermo Fisher, #15596026). After incubating for 5 minutes, 60 uL chloroform was added, incubated for 3 minutes, then centrifuged at 12,000×g at 4C for 15 minutes. The aqueous phase was extracted and mixed with equal volume of 100% ethanol, which was then loaded onto a Zymo Direct-zol RNA Microprep (Zymo, #R2061) column. The Zymo protocol was followed from there to bind and rinse the RNA, which was eluted in a final volume of 7uL. cDNA was generated from 5ug of template RNA with the SuperScript IV kit (Thermo Fisher, #18090010) using 1uL of 2uM STICR viral library RT primer (Table 1). To add a UMI and a primer-binding handle to the individual cDNA molecules, one-cycle PCR was performed with cDNA and STICR viral library cDNA UMI primer (Supplementary Table 4) with 25uL Q5 High Fidelity 2X MasterMix (NEB, #M0492S), 2.5uL primer (10uM), 2.5uL H2O, and 20uL cDNA with the following settings: 98C 40s, 62C 20s, 72C 2:10, 4C hold. Primers were removed with a left-sided 0.8X SPRISelect cleanup (Beckman Coulter, #B23318). Finally, cDNA was amplified with the same methods as the lentiviral library described above and the library was sequenced to a depth of ~100 million reads.
Diversity and Collision Modeling of STICR Plasmid and Lentiviral Libraries
STICR barcode sequences were extracted from fastq files using custom scripts that removed PCR duplicate reads using the UMI (see below in scRNA-seq Analysis and STICR Barcode Analysis for a general description). Since it is prohibitively expensive to sequence high diversity libraries to saturation, we extrapolated the total number of unique STICR barcodes using the Preseq37 command lc_extrap using default settings. Together with the measured relative barcode abundances, we used the extrapolated STICR barcode library size to model barcode collisions using the R (v4.0.1) programing language. Using base R functions, we simulated the labeling of a starting population of cells with a range of sizes from 101 to 106 and repeated each simulation 20000. We then quantified the mean number of unique barcodes chosen for each starting cell population size. The difference between the starting cell population size and the number of unique barcodes present represented the number of collisions that had happened at that population size.
Cell Lines
NIH/3T3 cells (ATCC) and Lenti-X HEK293T cells (Takara Bio) were used in this study. We did not test them for mycoplasma nor authenticate their authenticity.
Lentivirus production
STICR lentivirus was produced using a third-generation lentivirus packaging system: pMDLg/pRRE (Addgene, #12251), pRSV-Rev (Addgene, #12253), and VSVG envelope (Addgene, #12259). Plasmids were transfected into Lenti-X HEK293T cells (Takara Bio, #632180) using jetPRIME (Polyplus ,#114–15). In order to improve the viral titer which is reduced due in part to the reverse orientation of the STICR EGFP-barcode transcript relative to the external lentiviral promoter, we also co-transfected pcDNA3.1 puro Nodamura B2 plasmid (Addgene, #17228) along with the other plasmids. Lenti-X 293T cells were grown and transfected in in DMEM (Fisher, #MT10017CV) supplemented with 10% FBS (Hyclone, # SH30071.03) and 1% penicillin streptomycin (Fisher, # 15070063). 24 hours after transfection, media was replaced with Ultraculture media (Lonza, #BE12–725F) supplemented with sodium pyruvate (0.11mg/ml final concentration, Sigma #P2256–25G) and sodium butyrate (0.005M final molarity, Sigma #B5887–1G) and penicillin streptomycin. 72 hours after transfection, media was collected, passed through a 0.45μM filter (corning, #431220), and then ultracentrifuged at 22,000 × g for 2 hours. Pellet was resuspended in 100ul of sterile PBS (Thermo, #14190250) overnight at 4C and then aliquoted and stored at −80C.
Tissue Procurement and STICR Transduction
De-identified tissue samples were collected with previous patient consent in strict observance of the legal and institutional ethical regulations. Protocols were approved by the Human Gamete, Embryo, and Stem Cell Research Committee (institutional review board) at the University of California, San Francisco. In order to visualize tissue for microdissection, samples were embedded in 3% low melting point agarose (Fisher, # BP165–25) and then cut into 300 μm sections perpendicular to the ventricle on a Leica VT1200S vibrating blade microtome in oxygenated artificial cerebrospinal fluid containing 125 mM NaCl, 2.5 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 1.25 mM NaH2PO4. The germinal zone was then isolated by microdissection using a scalpel and fine forceps under a Leica MZ10F dissecting microscope. In order to dissociate cells into a single cell suspension, microdissected tissue was incubated in 200μl of 0.25% trypsin (Fisher, # 25200056) and 2000 units/mL of DNase I (NEB, #M0303) for 20 minutes at 37C and then gently mechanically triturated with an 1000ul pipetteman 10 times. 800ul of DMEM supplemented with 10% FBS was added to the sample to neutralize the papain and the trypsin/DNAse/FBS solution was removed by centrifuging the sample for 5 min at 300×g. The sample was rinsed in DPBS (Thermo, #14190250) and then centrifuged again for 5 min at 300×g to remove rinse.
Cells were then immediately resuspended in NES media previously defined by Onorati and colleagues38, supplemented with rock inhibitor at a final concentration of 10 uM to reduce cell death and 1% penicillin streptomycin (Fisher, # 15070063), and then plated on a 24 well tissue culture dish coated with 0.01% poly-L-ornithine (Sigma, #P4957), 5ug/ml laminin (Invitrogen, #23017–015), and 1ug/ml fibronectin (Corning, #354008) at a density of 500,000 cells per well. STICR lenitivirus was added to culture media at ~1:250 to 1:500 dilution so that ~ 30% of cells were infected. After 24 hours, virus-containing media was removed and replaced with fresh media. 72 hours after infection, cultures were dissociated using papain (Worthington, # LK003163), EGFP+ cells were isolated by FACS and then used in a 1) barnyard assay or 2) in vitro culture assay. In order to maximize cell viability in the in vivo mouse transplantation assay, EGFP+ cells were not FACS-isolated prior to transplantation, but instead the entire culture (containing both EGFP− and EGFP+ cells was used). See below for descriptions of each assay. In an effort to label subclones, the GW15 Rep2 in vitro sample was initially infected with a STICR viral library derived from a fully-barcoded STICR plasmid encoding a GFP-T2a-TVA transgene and a viral index “E” and then plated on mouse astrocytes as above. This culture was then subsequently infected 2- and 4-weeks later with EnvA-pseudotyped (Addgene # 74420) STICR libraries with viral index “1” (2 weeks) and “3” (4 weeks) with a 1:100 dilution of virus/media. During analysis, we found that labeling with EnvA-pseudotyped libraries was poor, so STICR barcodes with a “1” and “3” index were not considered during analysis. Therefore, clonal analysis of GW15 Rep2 was performed using only the initial “E” index STICR barcodes transduced 6 weeks prior to analysis, as done for all the other in vitro libraries.
Barnyard Experiment
To confirm that transcribed STICR barcodes can be accurately recovered using scRNA-seq, we performed a “barnyard experiment” where we infected separate cultures of human cortical cells (GW18 sample) and mouse 3T3 cells (ATCC) with different STICR libraries. These libraries could be distinguished from each other by a constant sequence unique to each library (“viral index”) (Fig. 1a). After three days, we dissociated cultures with papain and FACS-isolated EGFP+ cells. EGFP+ cells from both species were then mixed together and loaded into a 10X Genomics Chromium Single Cell 3 prime kit (10x Genomics, #PN-100007). Following sequencing (see below), transcript libraries were aligned with Cell Ranger (version 3.0.2) to a hybrid mouse/human genome and droplets were determined to be either a mouse cell, human cell, or multiplet. Quantification of recovered STICR viral index (see below) for mouse, human, and multiplate droplets is included in Extended Data Fig. 1f.
In Vitro Culture Assay
Long-term in vitro experiments were performed using an astrocyte co-culture system. Primary mouse cortical astrocytes were isolated from CD-1 postnatal day 1 mice and cultured as previously described39, but with additional subdissection to remove the subventricular zone surrounding the lateral ventricles. Astrocytes were plated at a density of 400k cells/3.5cm2 in 12 well cell culture plates containing DMEM (Fisher, #MT10017CV) supplemented with 10% FBS (Hyclone, # SH30071.03) and 1% penicillin streptomycin (Fisher, # 15070063) 3–5 days prior to the addition of human cells.
For each experiment, ~1000–2000 STICR labeled (EGFP+) cells were added to a 12 well plate already containing mouse cortical astrocytes and cultured in DMEM, 1% B-27 supplement (Invitrogen, #12587–010), 1% N-2 supplement (Invitrogen, #17501–048), and 1% penicillin streptomycin. Media was half-changed every 3–4 days for 6 weeks. Cultures were then dissociated into single cell solution using papain and EGFP cells were isolated by FACS. Following FACS isolation, EGFP+ cells were concentrated by centrifugation (300xg for 10 min), and prepared for scRNA-seq.
PTPRZ1 FACS
For enrichment of progenitors prior to xenograft transplantation, we used an adaptation of the protocol developed by Crouch and Doetsch40 in order to isolate cells that express the cell surface protein PTPRZ1 that is enriched on the surface of cortical progenitors15,41. Cortical tissue was dissociated to a single cell suspension as described above, then resuspended in a solution of 1% BSA (Sigma Aldrich, A7979–50ML) and 0.1% glucose in HBSS (Life Technologies, 14175–095) for staining. Cells were incubated with mouse anti-PTPRZ1 primary antibody (Santa Cruz Biotechnology, sc-33664) at 1:50 dilution for 20 minutes on ice, washed with HBSS/BSA/glucose, incubated in goat anti-mouse IgM 488 secondary antibody (Thermo Fisher Scientific, A-21042) at 1:500 dilution for 20 minutes on ice, washed, and resuspended in HBSS/BSA/glucose. PTPRZ1+ cells were then isolated via FACS, plated, labelled with STICR, and cultured as above. Representative FACS plots shown in Extended Data Fig. 9a.
Xenograft Transplantation Assay
Mouse transplantation assays were performed in CB17.Cg-PrkdcscidLystbg-J/Crl mice (Envigo) at postnatal day 3–5. STICR labeled cultures were dissociated with papain, centrifuged at 300xg for 5 min, rinsed once with DPBS, and then resuspended in ice-cold L15 media (Fisher, #11-415-064) with 180 Kunitz/ml of DNAse (Fisher, #50-100-3290). Following anesthetization, 100nl of cell mixture (~40–80k cells) was injected through a beveled glass needle using a stereotactic rig into L - 1, A 2.5, D 0.-8 mm from lamda. At least 5 mice of were injected with cells from each human specimen. Both male and female mice were used. In order to minimize clumping of xenograft cells, 20mM EGTA (Sigma, #E4378) was added to cell mixture for GW17 Rep1 and Rep2 samples as well as GW15 Rep3.
Following 6 weeks, mice were euthanized and one brain from each set of transplantations was cut into 1mm coronal sections using a brain mold (Stoelting, #51386). EGFP+ regions of cortex were dissected from slices using a fluorescent dissecting scope and then dissociated into a single cell solution using papain. EGFP+ cells were then isolated by FACS, concentrated by centrifugation (300×g for 10 min), and prepared for scRNA-seq. Mice were housed in a barrier facility with 12hr light/12hr dark cycle and temperature and humidity control (70F, 50% rack humidity). All protocols and procedures followed the guidelines of the Laboratory Animal Resource Center at the University of California, San Francisco and were conducted with IACUC approval.
Immunohistochemistry of Xenograft Transplantation Assay
At the experimental end point, transcardiac perfusion of sterile PBS followed by 4% PFA (Fisher, # 50-980-487) was used to rinse and then fix the specimens. Brains were dissected out and drop-fixed overnight in 4% PFA at 4C. Brains used for cryosections (Fig 3, Extended Data Fig 7) were then cryopreserved in a 1:1 solution of OCT (VWR, # 25608–930) and 30% sucrose, embedded in cryomolds containing the same solution, frozen on dry ice, and stored at −80 °C. Brains were then cryosectioned at 12 μm onto glass slides and stored at −80 °C. Blocking and permeabilization were performed using a blocking solution consisting of 10% normal donkey serum, 1% Triton X-100, and 0.2% gelatin in PBS for 1 hour. Primary and secondary antibodies were diluted and incubated in this same blocking solution. Cryosections were incubated with primary antibodies at 4°C overnight, washed 3×10 min with washing buffer (0.1% Triton X-100 in PBS), incubated with secondary antibodies for 2 hours at room temperature, washed 3×10min with washing buffer, and then coverslips (Azer Scientific, 1152460) were mounted using Prolong Gold Antifade Reagent (Invitrogen, P36930).
Brains used for morphological analysis of GABAergic cells (Fig 4, Extended Data Figs 9–10) were fixed as above but stored in PBS at 4°C. Brains were then sectioned on a Leica VT1000 S vibrating blade microtome to 40um and slices were stored in PBS. Slices were incubated in blocking solution composed of 10% normal donkey serum and 0.1% Triton-X in PBS at room temperature for 2 hours and then incubated in primary antibodies diluted in blocking solution overnight at 4°C, washed 5×30 mins in 0.1% PBST, incubated in secondary antibodies in blocking solution overnight at 4°C, washed 5×30 mins in 0.1% PBST, mounted on glass slides, and coverslipped as above using ProLong Gold Antifade reagent.
In vitro cultures comprised of human cortical cells co-cultured with mouse astrocytes were prepared as described above on 8 well chamber slides (Thermo Scientific, 154534PK). After 6 weeks, cultures were fixed with 4% PFA for 1 hour at 4°C. Cultures were then washed 3 times and stored in PBS. Immunohistochemistry was performed as described for the 40um mouse brain sections but with 10 minute washes. Slides were coverslipped with ProLong Gold Antifade Reagent.
The antibodies used in this study include: chicken anti-GFP (Aves, GFP-1020; 1:1000), mouse anti-human nuclear antigen (Novus, NBP2–34342; 1:100), rabbit anti-GABA (Millipore Sigma, A2052–100ul; 1:250), rabbit anti-NEUROD2 (Abcam, ab104430; 1:500), guinea pig anti-DCS (Millipore Sigma, AB2253; 1:200), rabbit anti-GFAP (abcam, ab7260; 1:1500), rabbit anti-SOX9 (Abcam, ab104430; 1:250), and mouse anti-OLIG2 (Millipore Sigma, MABN50; 1:200). Secondaries used include AlexaFluor anti-chicken 488 (Jackson Immunoresearch 703-545-155; 1:500), anti-mouse 488 (ThermoFisher A-21042; 1:500), anti-rabbit 594 (ThermoFisher A-21207; 1:500) , anti-guinea pig 647 (Jackson Immunoresearch 706-605-148; 1:500), anti-mouse IgG1 488 (ThermoFisher A-21121; 1:500) , and anti-mouse IgG2a 647 (ThermoFisher A-21241; 1:500).
Confocal imaging was performed using a Leica SP8 confocal microscope with either a 10x or 20x air objective. 2um optical z-step was used for all images. Images were processed using ImageJ/Fiji. For quantification of the major cell types in xenografted mice (Fig 3, Extended Data Fig 7), tilescans of the transplanted region were z-projected with average intensity, channel intensity was normalized across images, and cells expressing EGFP and/or human nuclear antigen in addition to cell type markers were counted manually using the CellCounter plugin for ImageJ/Fiji. Quantification of GABA+ STICR-labeled cells in the cortex of host mice as depicted in Fig 4 was performed by imaging 4×40um thick brain slices derived from within 400um of the transplantation site of each animal. Two mice for each of the two GW17 specimens were analyzed in total. GFP+ cells in the cortex were counted using CellCounter plugin for ImageJ/Fiji. The relative laminar position of GABA+/GFP+ double-positive cells was measured using the Measure tool from ImageJ/Fiji to draw a line from the top of the corpus callosum straight up to the pial surface through the soma of each cell. The relative location of the cell’s soma to the top of the corpus callosum was then divided by the total length of the line drawn from the corpus callosum to pial surface.
scRNA-seq Library Preparation
scRNA-seq library preparation was performing using the 10X Genomics Chromium Single Cell ‘3 prime kit (10x Genomics, #PN-100007). Library preparation was performed according to manufacturer’s protocol.
STICR Barcode Library Recovery
STICR barcodes were sub-amplified from each 10X cDNA library using Q5 Hot Start High Fidelity 2x Master Mix (NEB, #M0494). In brief, 10ul of cDNA was used as template in a 50ul PCR reaction containing primers (0.1μM) targeting the region immediately upstream of the STICR viral index/barcode as well as the partial Illumina Read1 sequence added during cDNA library preparation (Table 1) using the following program: 1) 98C, 30sec, 2) 98C, 10sec, 3) 62C, 20 sec ,4) 72C, 10 sec, 5) Repeat steps 2–4 11x, 6) 72C, 2 min, 7) 4C, hold. Following PCR amplification, a 0.8–0.6 dual-sided size selection was performed using Ampure XP beads (Beckman Coulter, #A63881).
Sequencing
10X transcriptomic libraries and STICR barcode libraries were sequenced using Illumina Novaseq 6000 or Illumina HIseq 4000 machines. 10X transcriptomic libraries were sequenced to the average depth of 53,191 reads per cell (2,280 genes per cell). STICR barcode libraries from lineage tracing experiments were sequenced to the depth of ~30 million reads per library. STICR plasmid libraries from lineage tracing experiments were sequenced to the depth of ~30 million reads per library.
scRNA-seq Analysis and STICR Barcode Analysis
10X transcriptomic libraries were aligned to the hg38 genome using CellRanger (v3.0.2). Aligned cell/transcript counts were processed by Seurat42 (v3.2.0.9014 for initial in vitro cultures and v4.0 for subsequent xenografted cultures and integration of xenograft data into in vitro data) to remove cells containing fewer than 1000 genes, fewer than 1250 transcripts, or a high abundance of mitochondrial reads (>7% total transcripts). Cells passing these thresholds were then processed with Cellbender43(v.0.1) in order to identify and remove background reads and instances of barcode swapping. We then identified and remove multiplets that arose during cell capture using Solo44(v.0.1). Additionally, xenograft libraries were aligned to a chimeric hg38/mm10 genome using CellRanger (v.3.0.2) in order to identify potential cross-species multiplets—cells identified as such were removed from analysis. Libraries were integrated using Seurat’s SCTransform and FindIntegrationAnchors functions, to identify integration features. First, in vitro cultured STICR experiments were integrated together. Subsequently, transcriptomic libraries form xenograft experiments were integrated with in vitro cells using integration anchors identified from in vitro cultures. Leiden cell clustering, pseudotime analysis, and data visualizations (i.e. creation of UMAP images) were performed using Monocle345 (v0.2.1.9). Pseudotime analysis of inhibitory neuron (IN) trajectory cells was performed by setting the root node within mitotic IN clusters. IN.1, IN.2, and IN.3 trajectories were defined as major branches of the principal graph that led to distinct sets of sets of clusters. Within subclustered IN cells, cluster 17 appeared to be at the beginning of both IN.2 and IN.3 trajectory cells in pseudotime analysis and was thus termed “IN.early”.
Differential gene expression analysis was conducted using Seurat FindMarkers/FindAllMarkers functions. With the exception of cluster 34 from the in vitro culture data (Fig. 1c), we identified marker gene expression consistent with previously described cell types. Cluster 34 expressed markers of multiple cell types and did not show strong cluster correlation with reference data (Extended Data Fig. 2d). Thus, we refer to cells in this cluster as “Unknown” in Supplementary Tables 1–2.
Iterative subclustering of EN and EOMES+ IPC trajectory cells from in vitro STICR cultures revealed “upper-like” (subcluster 3,4,7,9), “deep-like” (subcluster 1,5), “newborn EN” (subcluster 6), “EOMES+ IPC” (subcluster 2), and “EN.other” (subcluster 8) subgroups based on gene expression. EN.other group did not have a strong correlation with excitatory neurons and thus cells in this category were removed from further clonal analysis. Within xenografted EN and EOMES+ IPC trajectory cells, iterative subclustering revealed “EN” and “EOMES.IPC” subclusters. STICR barcode analysis was performed using custom scripts. First, BBMap (BBMap – Bushnell B. – sourceforge.net/projects/bbmap/) was used to remove low quality reads and then extract reads containing STICR barcode sequences. Then, BBMap was used to extract individual STICR barcode fragments which were then aligned to our pre-defined fragment reference sets using Bowtie46 (v5.2.1), allowing for up to 2 mismatches per fragment. Aligned STICR barcodes were compiled into a file containing their corresponding 10X cell barcode and 10X UMI sequences using Awk. Finally, UMI-tools47 (v.0.5.1) was used to remove duplicate STICR barcode/cell barcode reads by UMI, allowing for 1bp mistmatch in the UMI. STICR barcodes/CBC pairings with at least 5 distinct UMIs were retained. Cells with a single STICR barcode meeting this criteria were retained for clonal analysis. Possible instances of STICR barcode superinfection (multiple STICR barcodes per starting progenitor) were identified by calculating jaccard similarity indexes of all STICR barcodes pairings found to co-occur within a single cell. Those pairings with a jaccard similarity index of ≥ 0.55 that occurred in ≥10 cells were considered to be a valid superinfection clone and retained for clonal analysis. Cells that contained multiple STICR barcodes that were not determined to be valid superinfections were further analyzed for the relative abundance of individual STICR barcodes. Cells that contained a “dominant” STICR barcode with ≥ 5 times the number of barcode counts compared to the next most abundance STICR barcode as determined by UMI were retained and assigned that dominant barcode. Those cells that didn’t contain barcodes meeting these criteria were not considered for clonal analysis.
Validation of Marker Gene Expression Analysis with Allen Brainspan Dataset
Expression data (Z-score) for IN.2 and IN.3 marker genes were downloaded from the Allen Brainspan database (https://www.brainspan.org). Biological samples from the cortex, basal ganglia, and RMS/OB were retained for further analysis. Hierarchical clustering was performed using the R package pheatmap(v.1.0.12), with the ward.D clustering method. For visualization purposes, the data ranges were divided into 20 quantiles.
Transcriptional Cluster correlation
Marker genes from each cluster in the Nowakowski primary human brain reference atlas14 were calculated using Seurat FindAllMarkers, restricting genes to those present in at least 25% of cells of that cluster. The top 100 marker genes by fold-expression for each cluster were then retained for further analysis. The average expression for each cluster’s top marker genes were calculated for each cluster in the Nowakowski reference atlas as well as in each cluster with the STICR transcriptional datasets. We then calculated pairwise Pearson correlations between every reference and STICR cluster and depicted the result using a heatmap.
Statistics and Reproducibility
Images shown in figures were representative of results found in multiple replicates: Figure 3b (2 specimens each xenografted into 3 mice), Figure 4b&c (2 specimens each xenografted into 2 mice), Extended Data Figure 4d&e (4 specimens), and Extended Data Figure 9&10 (2 specimens each xenografted into 2 mice).
Extended Data
Extended Data Fig. 1. Validation of the STICR Barcode Design.
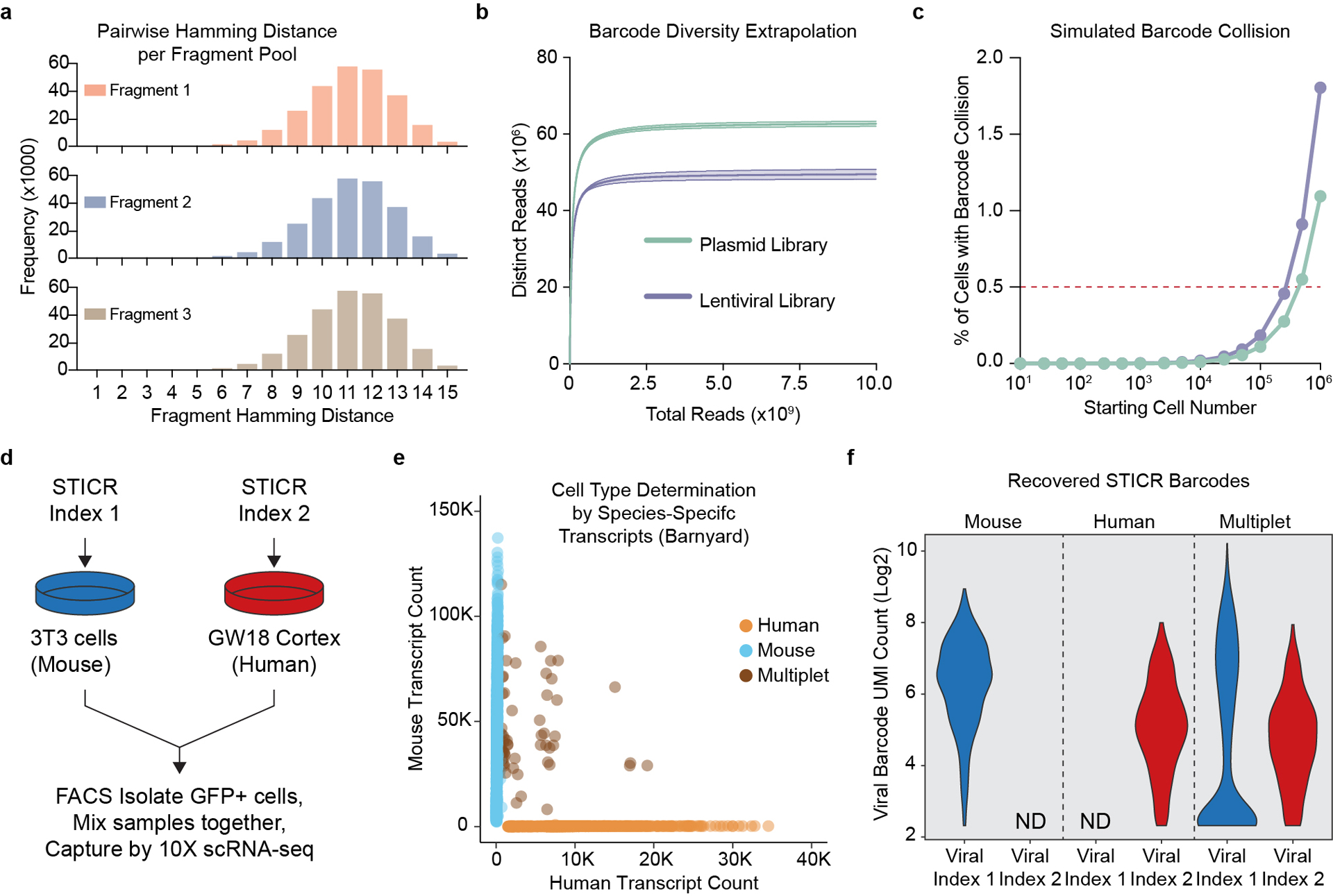
a) Histogram of pairwise hamming distances between every sequence in each STICR fragment pool. b) Barcode diversity extrapolations derived from sequencing ~30 million reads of a representative STICR plasmid or lentiviral library. Mean±95% confidence range for each library shown. c) Simulated barcode collision frequencies (mean±SD) for a range of starting cell numbers based on barcode diversity estimated in (b). Barcode sampling performed with replacement using measured proportions of barcodes within representative plasmid library depicted in panel b. Each simulation performed 20,000 times. Most error bars (depicting standard deviation) are not visible as they are smaller than the dots (depicting mean value). d) Schematic of “barnyard” species mixing experiment. e) Plot depicting species specific transcript counts from barnyard experiment. Each dot depicts a single cell and the dot color indicates whether the cell was determined to be a 3T3 cell (mouse), Cortex cell (human), or mixed droplet (multiplet). f) Violin plots depicting the number of unique STICR barcode molecules recovered from droplets identified as either mouse, human, or multiplet. ND, not detected.
Extended Data Fig. 2. Cluster Analysis of In Vitro STICR Datasets.
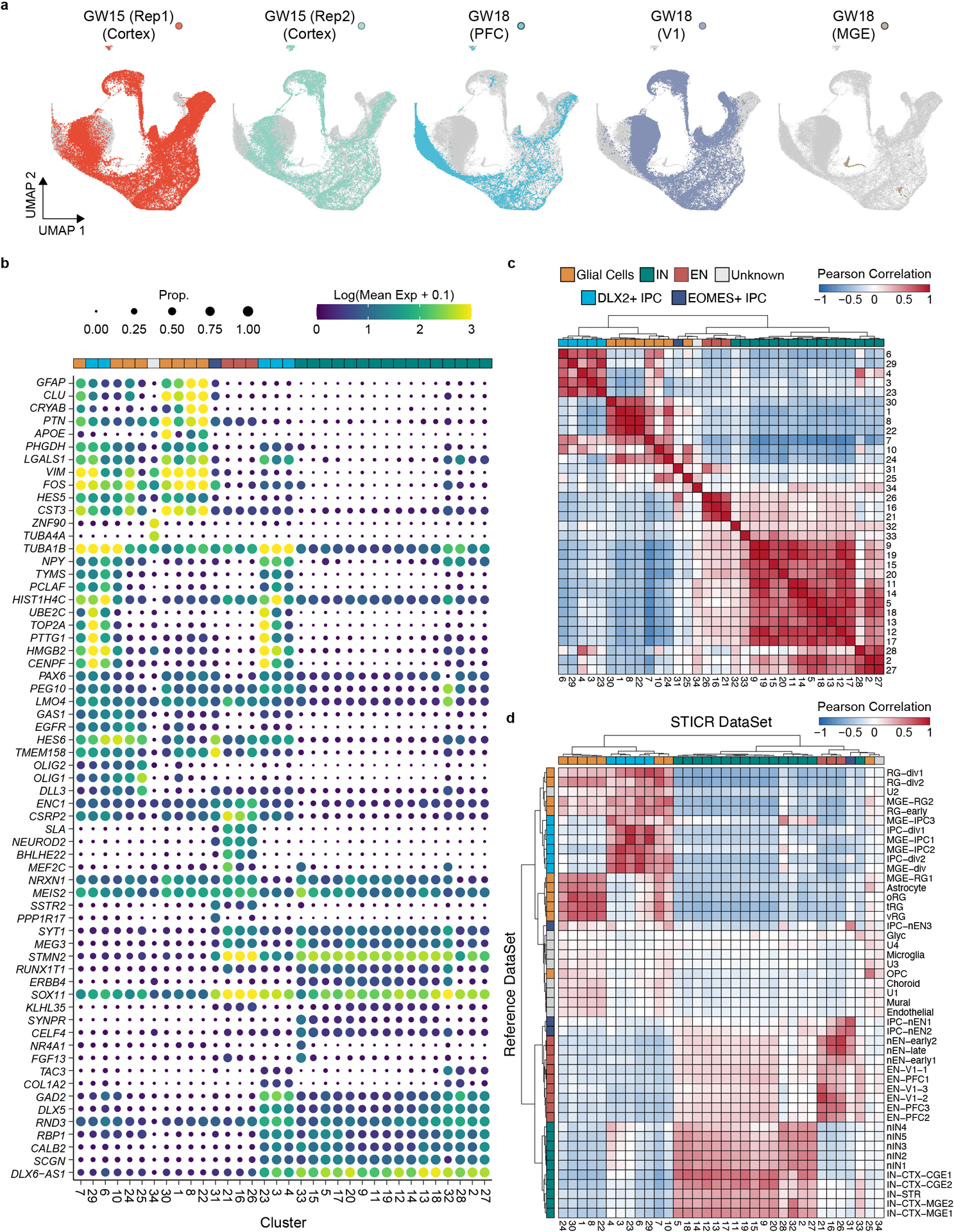
a) UMAP plots of each individual biological sample highlighted. b) Top marker gene expression for each cluster. Size of dot corresponds to the proportion of cells in cluster expressing gene while the color of dot corresponds to the average expression level per cluster. c-d) Heatmap depicting pairwise transcriptional cluster correlation of in vitro cultured cells with c) self and d) 2017 Nowakowski et al scRNA-seq atlas. Principal cell type designation depicted next to each column and row. Dendrogram depicts hierarchical clustering distance.
Extended Data Fig. 3. Transcriptional Analysis of In Vitro STICR Datasets.
a-f) Feature plots depicting expression of genes corresponding to (a) cell cycle, (b) glia, (c) oligodendroglia, (d) excitatory neurons, (e) interneurons, (f) regional markers. g) Bar plot depicting the proportion of cells within each cluster with a recovered STICR barcode. h) Heatmap depicting percentage of STICR barcodes shared between biological samples. GW15 (Rep1), and all GW18 samples were labeled with same viral stock, while GW15 (Rep2) was labeled with a different stock (methods). i) Stacked barplot depicting relative proportions of principal cell types within each sample, restricted to cells that are members of multi-cellular clones.
Extended Data Fig. 4. Clonal Analysis of EN-Containing Cortical Clones.
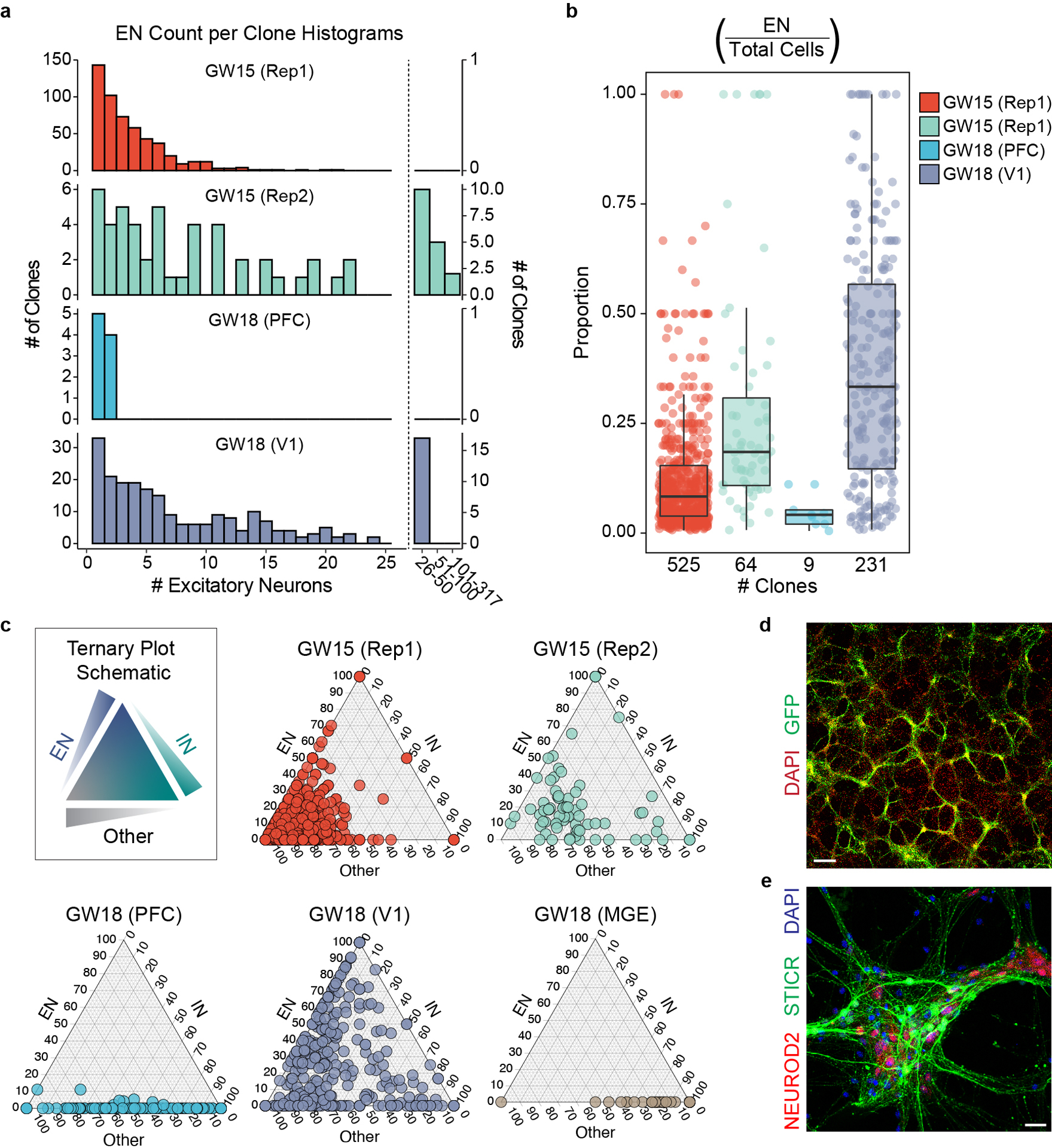
a) Histogram of excitatory neuron counts within each multi-cell cortical clone. Left, clone sizes from 1–25 cells in single cell bins. Right, clone sizes of >25 cells in indicated bin sizes. b) Box and whisker plot depicting the proportion of EN cells within individual multi-cell clones for each biological sample. Maxima and minima of box depicts 3rd and 1st quartiles, while center of box depicts the median. Whiskers depict 1.5x the interquartile distance. Individual clone value shown as dots. Number of clones is listed below each sample group. c) Ternary plots depicting the relative proportions of interneurons, excitatory neurons, and all other cell types (“Other”) within individual clones. d-e) Immunohistochemistry of in vitro cultures derived from GW15 germinal zone cells labeled with STICR. d) Low-magnification image to show distribution, Scale bar; 25μm. e) High-magnification image showing cluster of ENs, Scale bar; 250μm.
Extended Data Fig. 5. Clonal and Transcriptional Analysis of In Vitro INs and DLX2+ IPCs.
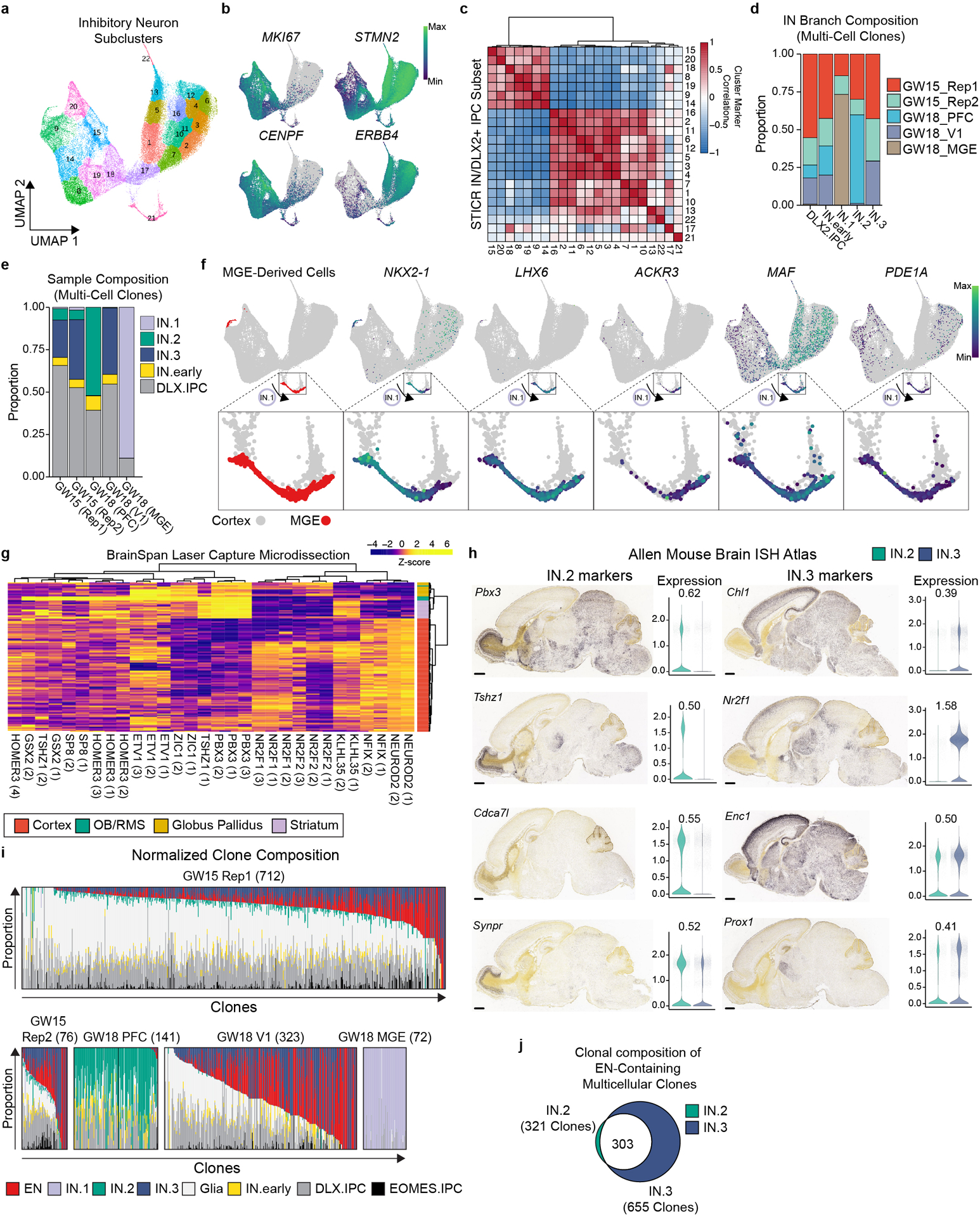
a) UMAP embedding and Louvain sub-clustering of GABAergic inhibitory neuron trajectory cells. b) Feature plots depicting expression of MKI67, STMN2, CENPF, and ERBB4. c) Heatmap depicting pairwise transcriptional cluster correlation of this dataset with itself d) Stacked barplot depicting relative proportion of multicellular clones from each sample that comprise each IN trajectory. e) Feature plots depicting MGE-derived cells (red) and expression of NKX2–1, LHX6, ACKR3, MAF, and PDE1A. Enlarged inset shown below shows IN.1 trajectory cells. f) Stacked barplot depicting relative proportion of different IN trajectory cells within multi-cell clones of each sample. g) Heat-plot depicting differential expression of IN.2 and IN.3 marker genes in developing human cortex, OB/rostral migratory stream, and basal ganglia. Data derived from the Allen BrainSpan Laser Capture Microdissection database. Color of each cell corresponds to quantile-normalized z-score. Dendrograms reflect hierarchical clustering of genes and samples while colors represent quantile-normalized z-score. h) Paired violin plots and ISH images of P60 mouse brains from the Allen Brain Atlas for select genes. Log2 fold difference between IN.2 (OB-like) and IN.3 (cortical interneuron-like) cells depicted above each violin plot. i) Stacked bar-plots depicting relative proportions of IN.1, IN.2, IN.3, excitatory neuron, and glia trajectory cells within multi-cell clones. Number of clones listed below each sample. j) Venn diagram depicting the number of EN-containing multicellular cortical clones that also contain IN.2 and/or IN.3 cells.
Extended Data Fig. 6. Clonal and Transcriptional Analysis of In Vitro ENs and EOMES+ IPCs.
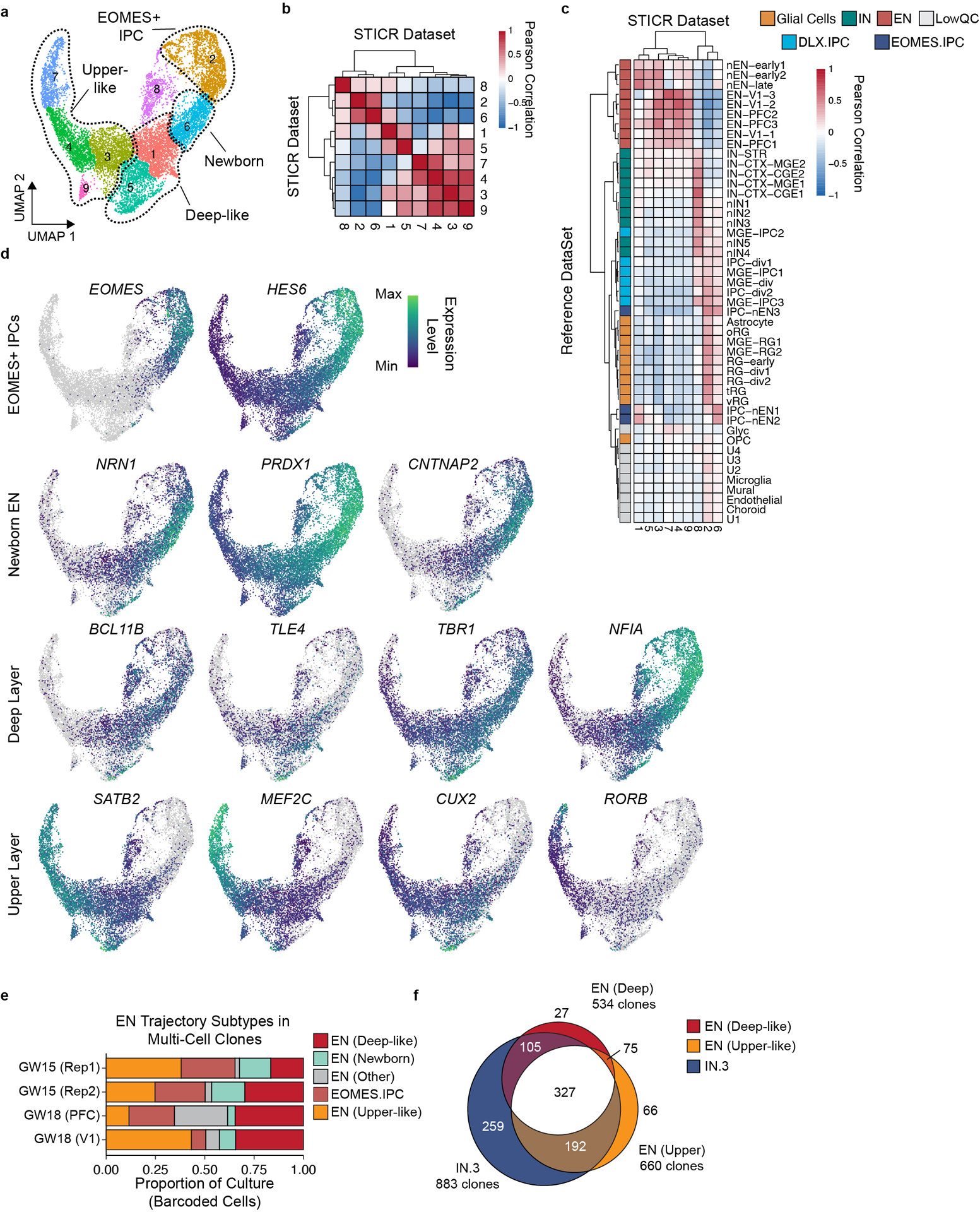
a) UMAP embedding and Louvain subclustering of EN trajectory cells. b-c) Heatmap depicting pairwise transcriptional cluster correlation of subclustered EN trajectory cells with b) self and c) 2017 Nowakowski et al. developing human brain scRNA-seq atlas. d) Feature plots depicting expression of genes corresponding to labelled subclustered EN trajectory subtypes. e) Stacked barplot depicting relative proportions of EN subtypes within EN trajectory cells of multicellular clones. f) Venn diagram depicting the number of multicellular cortical clones containing Deep-like ENs, Upper-like ENs, and IN.3 cells.
Extended Data Fig. 7. Characterization of Human Cortical Progenitor Xenografts at 6 Weeks.
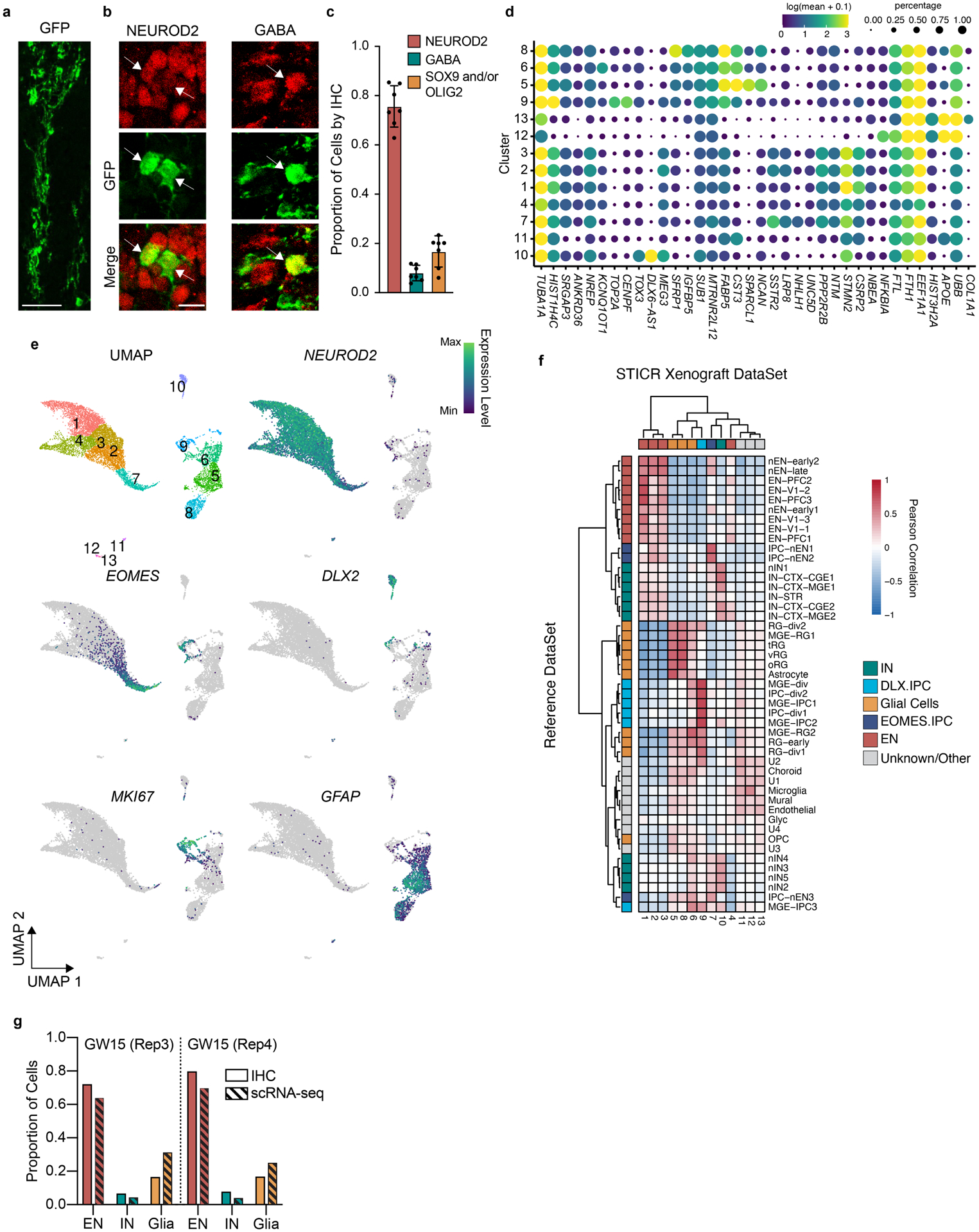
a-b) Representative images of transplanted human cortical cells analyzed by IHC for principal cell type markers 6 weeks after transplantation. EGFP expression from STICR in green, NEUROD2 or GABA shown in red. Scale bars: a, 50μm; b,10μm. c) Bar-plot depicting proportion (mean±SD) of transplanted cells expressing principal cell type markers as assessed by IHC. n=7 sections derived from 6 xenografted mice, 3 of which were transplanted with donor cells from GW15 Rep1 and 3 GW15 Rep2. d) Top marker gene expression for each cluster from xenografted cells. Size of dot corresponds to the proportion of cells in cluster expressing the gene while the color of each dot corresponds to the average expression level per cluster. e) UMAP embedding of xenografted cells and feature plots depicting expression of NEUROD2, EOMES, DLX2, MKI67, and GFAP. f) Heatmap depicting pairwise transcriptional cluster correlation of subclustered EN trajectory cells with 2017 Nowakowski et al. developing human cortex scRNA-seq atlas g) Comparison of principal cell type quantification (mean) in transplanted cells by analysis method (IHC vs scRNA-seq) and biological replicate.
Extended Data Fig. 8. Transcriptional analysis of ENs and INs from Xenografts.
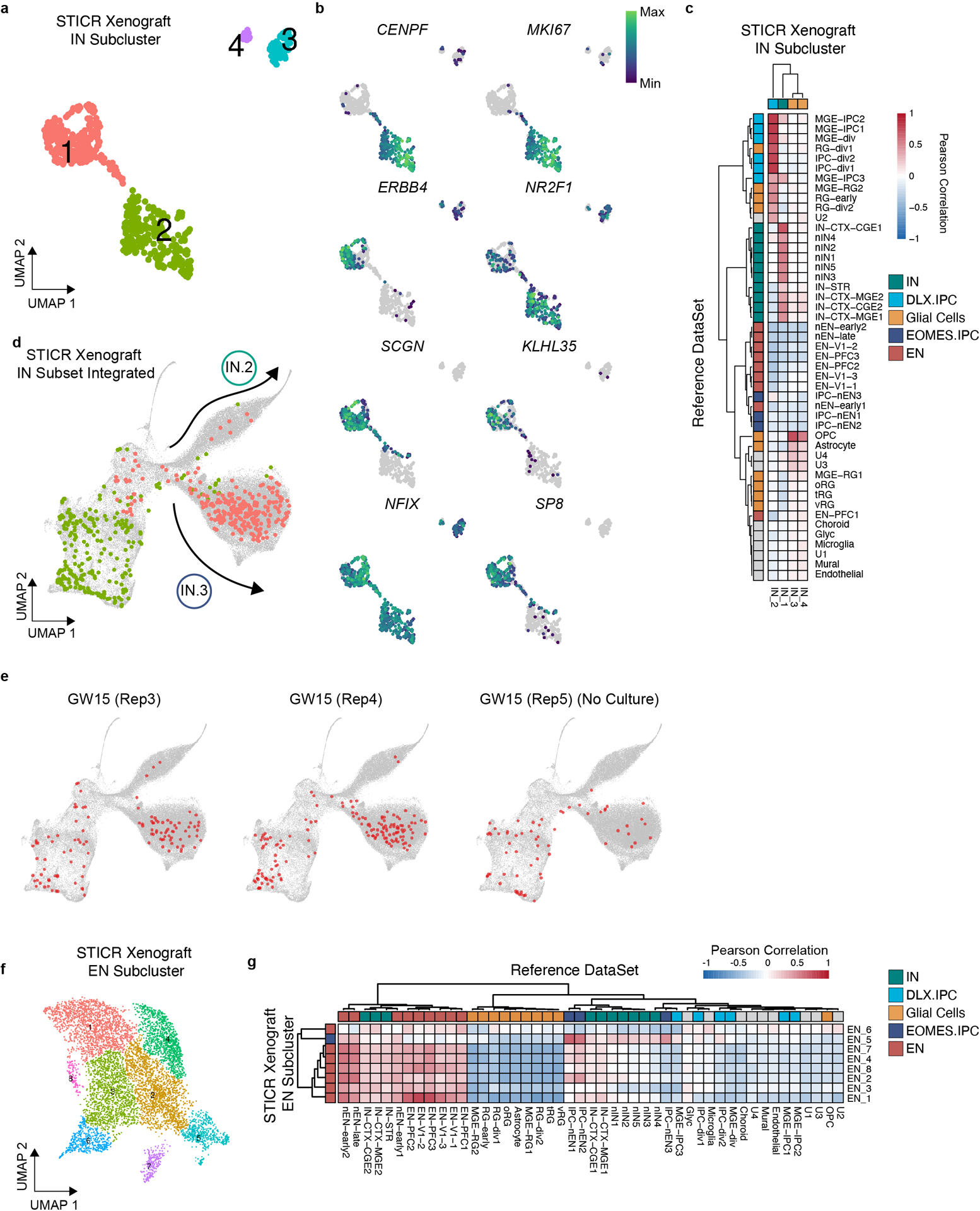
a) UMAP embedding and Louvain subclustering of IN trajectory cells from xenografts. b) Feature plots depicting expression of CENPF, MKI67, ERBB4, NR2F1, NFIX, SP8, SCGN, and KLHL35. c) Heatmap depicting pairwise transcriptional cluster correlation of subclustered xenograft IN and DXL2+ IPC trajectory cells with 2017 Nowakowski et al. developing human cortex scRNA-seq atlas. d) UMAP embedding depicting cells in multicellular clones from xenograft IN subclusters 1 (salmon) and 2 (lime) integrated with in vitro cultured STICR IN subset. e) UMAP embedding depicting individual multicellular clones from xenograft IN subset cells integrated with in vitro cultured STICR IN subset, split by biological replicate. Members of multicellular clones highlighted in red. f) UMAP embedding and Louvain subclustering of excitatory neuron and EOMES+ IPC trajectory cells from xenografts. g) Heatmap depicting pairwise transcriptional cluster correlation of subclustered xenograft EN and EOMES+ IPC trajectory cells with 2017 Nowakowski et al. developing human cortex scRNA-seq atlas.
Extended Data Fig. 9. Analysis of PTPRZ1-Sorted STICR+ Cells in Ctx, SVZ, RMS, and OB at 12 weeks.
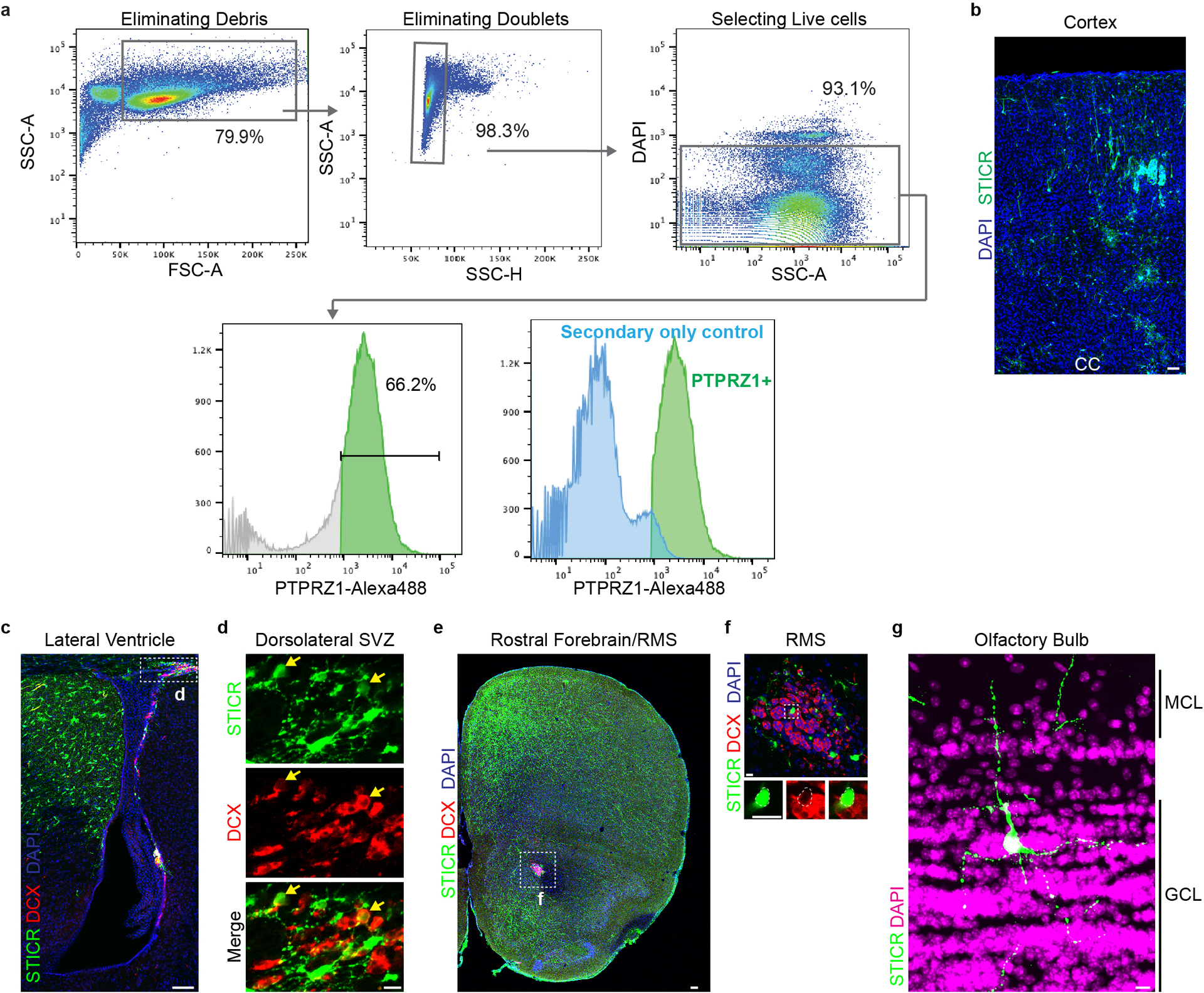
a) Representative FACS plots depicting isolation of PTPRZ1+ cells from cortical germinal zone. b) Representative image of transplanted human cortical cells cells in cortex of 12 week old host mouse. EGFP expression from STICR depicted in green, DAPI in blue. Scale bar, 50μm. CC, corpus callosum. c) Representative images of PTPRZ1-sorted, STICR-labeled cells in the dorsolateral corner of the lateral ventricle of a 12 week old host mouse analyzed by IHC. EGFP from STICR in green, DCX in red, and DAPI in blue. Scale bar, 100um. d) High magnification inset of region boxed in panel c. Scale bar, 10um. e) Representative images of PTPRZ1-sorted, STICR-labeled cells in the rostral forebrain analyzed by IHC. RMS outlined by white box labeled f. GFP expression in green, DCX expression in red, and DAPI in blue. Scale bar, 100um. f) High magnification insets of RMS depicted in white box in panel e. Scale bar, 10um. Cell outlined by white box magnified below. g) Representative images of PTPRZ1-sorted, STICR-labeled cells that migrated from transplantation site in cortex to the olfactory bulb analyzed by IHC. EGFP expression in green and DAPI in magenta. MCL= Mitral cell layer, GCL= glomerular cell layer. Scale bar, 10um.
Extended Data Fig. 10. Immunohistochemistry of STICR-Labeled Cortical INs from Xenografts at 12 weeks.
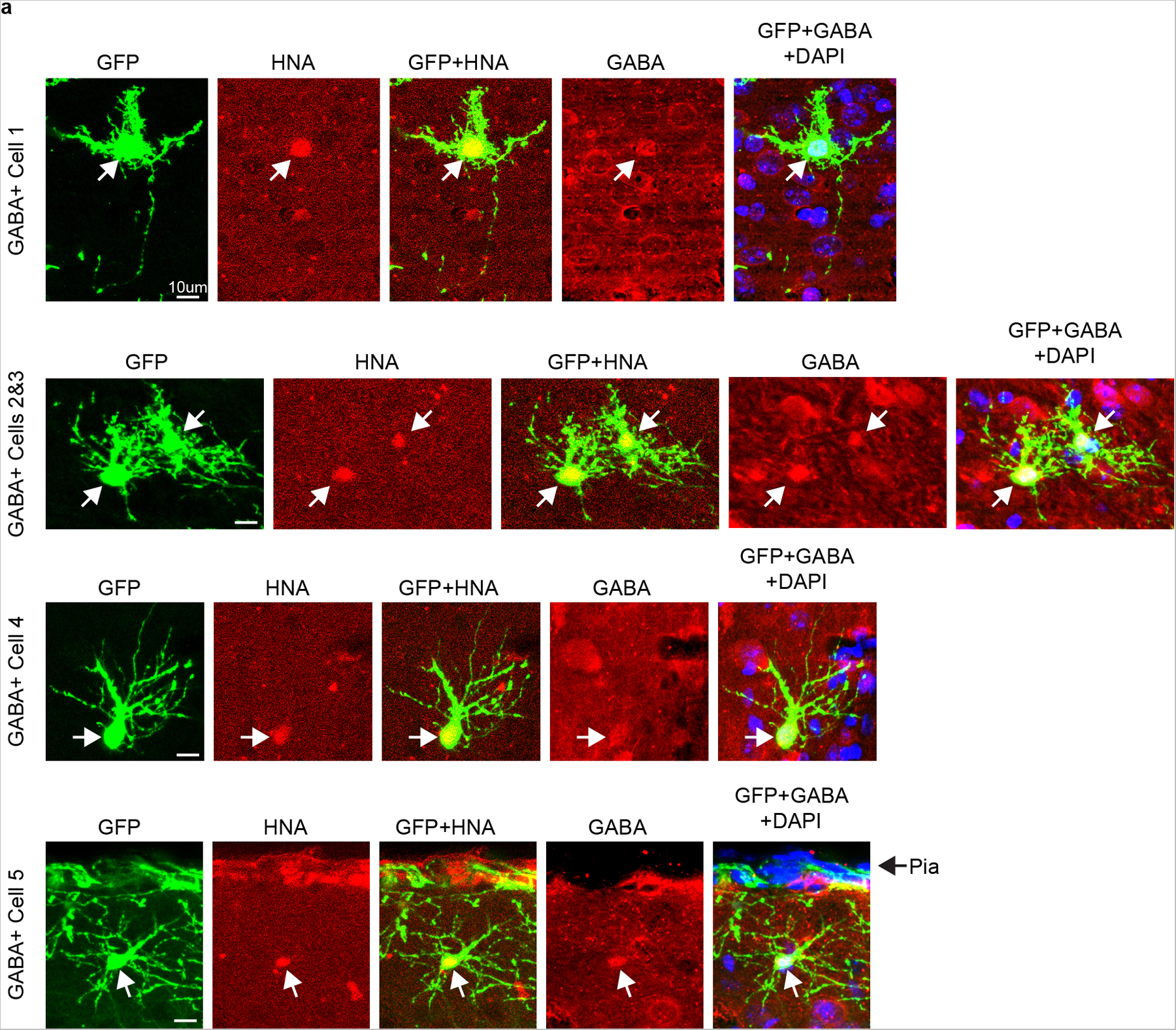
a) Representative images of STICR-labeled GABA+ cells throughout the cortical plate analyzed by IHC. EGFP from STICR in green, GABA in red, and DAPI in blue. Same cells from Fig 4d. Arrows point to STICR-labeled GABA+ cells. Scale bar, 10μm.
Supplementary Material
ACKNOWLEDGEMENTS
We thank A. Bhaduri for helpful discussion regarding scRNA-seq analysis; B. Rabe and C. Cepko for helpful discussion regarding viral vectors and sharing of reagents; C. Cadwell and M. Paredes for discussions regarding interneuron morphology; J. Rubenstein and R. Andersen for reading of manuscript; M. Speir and B. Wick for data wrangling at UCSC single cell browser. This study was supported by the Psychiatric Cell Map Initiative Convergence Neuroscience award U01MH115747, an Innovation Award from the Broad Foundation (to T.J.N.), a New Frontiers Research Award from the Sandler Program for Breakthrough Biomedical Research (PBBR) (to T.J.N.), NSF GRFP (to D.E.A.), an Autism Speaks Predoctoral Fellowship (11874 to R.S.Z.), gifts from Schmidt Futures and the William K. Bowes Jr Foundation (to T.J.N.). Work in the Alvarez-Buylla laboratory is supported by NIH grants R01NS028478 and R01EY025174, and a generous gift from the John G. Bowes Research Fund. A.A.-B. is the Heather and Melanie Muss Endowed Chair and Professor of Neurological Surgery at UCSF.
Footnotes
Code Availability
Custom codes used in this study are available at the following GitHub repository: https://github.com/NOW-Lab/STICR.
COMPETING INTERESTS
A.A.-B. is Co-founder and on the Scientific Advisory Board of Neurona Therapeutics.
SUPPLEMENTARY INFORMATION
This file contains the guide for Supplementary Tables 1–4.
Data Availability
scRNA-seq transcriptomic data and STICR barcode data are available at dbGAP under accession number phs002624.v1.p1, at GEO under accession number GSE187875. An interactive browser of single-cell data and raw and processed count matrices can be found at the UCSC cell browser.48 Publicly available reference genomes hg38 and mm10 used for analysis.
REFERENCES
- 1.Anderson SA, Eisenstat DD, Shi L & Rubenstein JL Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science 278, 474–476 (1997). [DOI] [PubMed] [Google Scholar]
- 2.Sussel L, Marin O, Kimura S & Rubenstein JL Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development 126, 3359–3370 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Gorski JA et al. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. The Journal of neuroscience : the official journal of the Society for Neuroscience 22, 6309–6314, doi:20026564 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu Q, Tam M & Anderson SA Fate mapping Nkx2.1-lineage cells in the mouse telencephalon. The Journal of comparative neurology 506, 16–29, doi: 10.1002/cne.21529 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Anderson SA, Marin O, Horn C, Jennings K & Rubenstein JL Distinct cortical migrations from the medial and lateral ganglionic eminences. Development 128, 353–363 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Petanjek Z, Berger B & Esclapez M Origins of cortical GABAergic neurons in the cynomolgus monkey. Cerebral cortex 19, 249–262, doi: 10.1093/cercor/bhn078 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Letinic K, Zoncu R & Rakic P Origin of GABAergic neurons in the human neocortex. Nature 417, 645–649, doi: 10.1038/nature00779 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Hansen DV et al. Non-epithelial stem cells and cortical interneuron production in the human ganglionic eminences. Nature neuroscience 16, 1576–1587, doi: 10.1038/nn.3541 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alzu’bi A et al. Distinct cortical and sub-cortical neurogenic domains for GABAergic interneuron precursor transcription factors NKX2.1, OLIG2 and COUP-TFII in early fetal human telencephalon. Brain Struct Funct 222, 2309–2328, doi: 10.1007/s00429-016-1343-5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alzu’bi A et al. The Transcription Factors COUP-TFI and COUP-TFII have Distinct Roles in Arealisation and GABAergic Interneuron Specification in the Early Human Fetal Telencephalon. Cerebral cortex 27, 4971–4987, doi: 10.1093/cercor/bhx185 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kohwi M et al. A subpopulation of olfactory bulb GABAergic interneurons is derived from Emx1- and Dlx5/6-expressing progenitors. The Journal of neuroscience : the official journal of the Society for Neuroscience 27, 6878–6891, doi: 10.1523/JNEUROSCI.0254-07.2007 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Young KM, Fogarty M, Kessaris N & Richardson WD Subventricular zone stem cells are heterogeneous with respect to their embryonic origins and neurogenic fates in the adult olfactory bulb. The Journal of neuroscience : the official journal of the Society for Neuroscience 27, 8286–8296, doi: 10.1523/JNEUROSCI.0476-07.2007 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuentealba LC et al. Embryonic Origin of Postnatal Neural Stem Cells. Cell 161, 1644–1655, doi: 10.1016/j.cell.2015.05.041 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nowakowski TJ et al. Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323, doi: 10.1126/science.aap8809 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pollen AA et al. Molecular identity of human outer radial glia during cortical development. Cell 163, 55–67, doi: 10.1016/j.cell.2015.09.004 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bystron I, Blakemore C & Rakic P Development of the human cerebral cortex: Boulder Committee revisited. Nature reviews. Neuroscience 9, 110–122, doi: 10.1038/nrn2252 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Bandler RC, Mayer C & Fishell G Cortical interneuron specification: the juncture of genes, time and geometry. Current opinion in neurobiology 42, 17–24, doi: 10.1016/j.conb.2016.10.003 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mayer C et al. Developmental diversification of cortical inhibitory interneurons. Nature 555, 457–462, doi: 10.1038/nature25999 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J et al. Transcription Factors Sp8 and Sp9 Coordinately Regulate Olfactory Bulb Interneuron Development. Cerebral cortex 28, 3278–3294, doi: 10.1093/cercor/bhx199 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo T et al. Dlx1/2 are Central and Essential Components in the Transcriptional Code for Generating Olfactory Bulb Interneurons. Cerebral cortex 29, 4831–4849, doi: 10.1093/cercor/bhz018 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Batista-Brito R et al. The cell-intrinsic requirement of Sox6 for cortical interneuron development. Neuron 63, 466–481, doi: 10.1016/j.neuron.2009.08.005 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stumm RK et al. CXCR4 regulates interneuron migration in the developing neocortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 23, 5123–5130 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez-Bendito G et al. Chemokine signaling controls intracortical migration and final distribution of GABAergic interneurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 28, 1613–1624, doi: 10.1523/JNEUROSCI.4651-07.2008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tripodi M, Filosa A, Armentano M & Studer M The COUP-TF nuclear receptors regulate cell migration in the mammalian basal forebrain. Development 131, 6119–6129, doi: 10.1242/dev.01530 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Azim E, Jabaudon D, Fame RM & Macklis JD SOX6 controls dorsal progenitor identity and interneuron diversity during neocortical development. Nature neuroscience 12, 1238–1247, doi: 10.1038/nn.2387 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lein ES et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176, doi: 10.1038/nature05453 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Herrera DG, Garcia-Verdugo JM & Alvarez-Buylla A Adult-derived neural precursors transplanted into multiple regions in the adult brain. Ann Neurol 46, 867–877, doi: 10.1002/1531-8249(199912)46:6<867::aid-ana9>3.0.co;2-z (1999). [DOI] [PubMed] [Google Scholar]
- 28.Qiu X et al. Reversed graph embedding resolves complex single-cell trajectories. Nature methods 14, 979–982, doi: 10.1038/nmeth.4402 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhaduri A et al. An atlas of cortical arealization identifies dynamic molecular signatures. Nature 598, 200–204, doi: 10.1038/s41586-021-03910-8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lodato MA et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 350, 94–98, doi: 10.1126/science.aab1785 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ludwig LS et al. Lineage Tracing in Humans Enabled by Mitochondrial Mutations and Single-Cell Genomics. Cell 176, 1325–1339 e1322, doi: 10.1016/j.cell.2019.01.022 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lareau CA et al. Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling. Nat Biotechnol, doi: 10.1038/s41587-020-0645-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paredes MF et al. Extensive migration of young neurons into the infant human frontal lobe. Science 354, doi: 10.1126/science.aaf7073 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rakic P Evolution of the neocortex: a perspective from developmental biology. Nature reviews. Neuroscience 10, 724–735, doi: 10.1038/nrn2719 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y et al. Cortical Neural Stem Cell Lineage Progression Is Regulated by Extrinsic Signaling Molecule Sonic Hedgehog. Cell reports 30, 4490–4504 e4494, doi: 10.1016/j.celrep.2020.03.027 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cai Y, Zhang Y, Shen Q, Rubenstein JL & Yang Z A subpopulation of individual neural progenitors in the mammalian dorsal pallium generates both projection neurons and interneurons in vitro. Stem cells 31, 1193–1201, doi: 10.1002/stem.1363 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Daley T & Smith AD Modeling genome coverage in single-cell sequencing. Bioinformatics 30, 3159–3165, doi: 10.1093/bioinformatics/btu540 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Onorati M et al. Zika Virus Disrupts Phospho-TBK1 Localization and Mitosis in Human Neuroepithelial Stem Cells and Radial Glia. Cell reports 16, 2576–2592, doi: 10.1016/j.celrep.2016.08.038 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schildge S, Bohrer C, Beck K & Schachtrup C Isolation and culture of mouse cortical astrocytes. Journal of visualized experiments : JoVE, doi: 10.3791/50079 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crouch EE & Doetsch F FACS isolation of endothelial cells and pericytes from mouse brain microregions. Nature protocols 13, 738–751, doi: 10.1038/nprot.2017.158 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Bhaduri A et al. Outer Radial Glia-like Cancer Stem Cells Contribute to Heterogeneity of Glioblastoma. Cell stem cell 26, 48–63 e46, doi: 10.1016/j.stem.2019.11.015 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821, doi: 10.1016/j.cell.2019.05.031 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fleming SJ, Marioni JC & Babadi M CellBender remove-background: a deep generative model for unsupervised removal of background noise from scRNA-seq datasets. bioRxiv, 791699, doi: 10.1101/791699 (2019). [DOI] [Google Scholar]
- 44.Bernstein NJ et al. Solo: Doublet Identification in Single-Cell RNA-Seq via Semi-Supervised Deep Learning. Cell Syst 11, 95–101 e105, doi: 10.1016/j.cels.2020.05.010 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Cao J et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502, doi: 10.1038/s41586-019-0969-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Langmead B, Trapnell C, Pop M & Salzberg SL Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology 10, R25, doi: 10.1186/gb-2009-10-3-r25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith T, Heger A & Sudbery I UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res 27, 491–499, doi: 10.1101/gr.209601.116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Speir ML et al. UCSC Cell Browser: Visualize Your Single-Cell Data. Bioinformatics, doi: 10.1093/bioinformatics/btab503 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
scRNA-seq transcriptomic data and STICR barcode data are available at dbGAP under accession number phs002624.v1.p1, at GEO under accession number GSE187875. An interactive browser of single-cell data and raw and processed count matrices can be found at the UCSC cell browser.48 Publicly available reference genomes hg38 and mm10 used for analysis.