

Abstract
The NMDA subtype glutamate receptors (NMDARs) play important roles in both physiological and pathologic processes in the brain. Compared with their critical roles in synaptic modifications and excitotoxicity in excitatory neurons, much less is understood about the functional contributions of NMDARs to the inhibitory GABAergic neurons. By using selective NMDAR inhibitors and potentiators, we here show that NMDARs bidirectionally modulate the intrinsic excitability (defined as spontaneous/evoked spiking activity and EPSP-spike coupling) in inhibitory GABAergic neurons in adult male and female mice. This modulation depends on GluN2C/2D- but not GluN2A/2B-containing NMDARs. We further show that NMDAR modulator EU1794-4 mostly enhances extrasynaptic NMDAR activity, and by using it we demonstrate a significant contribution of extrasynaptic NMDARs to the modulation of intrinsic excitability in inhibitory neurons. Together, this bidirectional modulation of intrinsic excitability reveals a previously less appreciated importance of NMDARs in the second-to-second functioning of inhibitory GABAergic neurons.
SIGNIFICANCE STATEMENT NMDA subtype of glutamate receptors (NMDARs) have important roles in brain functions, including both physiological and pathologic ones. The role of NMDARs in inhibitory neurons has been less elucidated compared with that in excitatory neurons. Our results demonstrate the importance of GluN2C/GluN2D-containing but not GluN2A/GluN2B-containing extrasynaptic NMDARs in modulating the intrinsic excitability of inhibitory neurons. These results further suggest distinct contributions of subsynaptic locations and subunit compositions of NMDARs to their functions in excitatory and inhibitory neurons. The above findings have implications for better understanding of brain diseases, such as schizophrenia.
Keywords: excitability, extrasynaptic NMDARs, NMDAR modulators, schizophrenia
Introduction
The NMDA subtype of glutamate receptors (NMDARs) are important for both physiological and pathologic processes. Their adequate activation is required for certain critical brain functions, such as learning and memory, and developmental refinement of neuronal connections. On the other hand, their excessive activation during certain pathologic conditions is believed to induce synapse loss and even neuronal loss (Hardingham and Bading, 2010; Traynelis et al., 2010; Paoletti et al., 2013; Ge et al., 2020). However, these conclusions are largely based on studies of NMDARs in excitatory neurons, while the contribution of NMDARs in inhibitory GABAergic neurons to brain physiology and pathology is far less understood (Moreau and Kullmann, 2013; Akgul and McBain, 2016; Pelkey et al., 2017).
Inhibitory neurons, although accounting for only ∼20% neurons in the cerebral cortex, play key roles in information processing, oscillations, tuning, and proper balance between excitation and inhibition (Markram et al., 2004; Isaacson and Scanziani, 2011). NMDARs in inhibitory neurons have received much attention for their important contributions to pathology and treatment in many psychiatric disorders, including schizophrenia and depression (Lewis et al., 2005; Paoletti et al., 2013; Zhou and Sheng, 2013; Cohen et al., 2015). However, detailed properties of the participating NMDARs, such as their subcellular location, synaptic versus extrasynaptic, are poorly understood, compared with the vast literature on the excitatory neurons (Cohen et al., 2015; Pal, 2018).
Previous studies have revealed important contributions of NMDARs to synaptic plasticity and neuronal intrinsic properties, both critical to the activity-dependent remodeling of neural circuits (Maccaferri and Dingledine, 2002; Hunt and Castillo, 2012; Bannon et al., 2020). Although the contribution of NMDARs to synaptic plasticity has been widely studied and well understood, how they modulate the intrinsic neuronal excitability is less understood (Sah et al., 1989; Pagadala et al., 2013). Intrinsic excitability, defined as neuronal excitability in the absence of synaptic inputs, is determined by the expression of ion channels and receptors contributing to the electrical properties of neurons (Zhang and Linden, 2003; Beck and Yaari, 2008). To investigate this in inhibitory neurons, we have used spontaneous spiking, injected current-induced spiking, and EPSP-spike coupling (E-S coupling) as readouts (Daoudal et al., 2002; Malik and Chattarji, 2012). Spontaneous spiking is essential for tuning the function of neuronal networks, whereas E-S coupling has significant contributions to the integration of synaptic excitation for generating action potentials (Daoudal et al., 2002).
Subcellular localization and subunit composition are two important contributors to NMDAR functions, and both have been extensively explored in the context of synaptic plasticity and excitotoxicity in excitatory neurons (Oliet et al., 2004; Hardingham and Bading, 2010; Paoletti et al., 2013; Zhou and Sheng, 2013). It is well established that NMDARs are located at both synaptic and extrasynaptic regions, with extrasynaptic NMDARs activated by spillover of synaptic glutamate or glutamate released from glia (Kullmann and Asztely, 1998; Haydon and Carmignoto, 2006), whereas synaptic NMDARs are activated by synaptically released glutamate but not by glutamate in the extracellular space. While the above conclusions are true for excitatory neurons, we recently showed that ambient glutamate could readily activate synaptic NMDARs in inhibitory neurons because of the low presence of glutamate transporter GLT-1 (Yao et al., 2018). As for subunit composition, studies support a general conclusion that GluN2A and GluN2B subunits are highly expressed in excitatory neurons, whereas GluN2C and GluN2D subunits are more concentrated in inhibitory GABAergic neurons (Paoletti et al., 2013; von Engelhardt et al., 2015; Riebe et al., 2016). In addition, GluN2A-containing NMDARs are more likely to be concentrated at synapses, whereas GluN2B- and GluN2C/2D-containing NMDARs are more enriched at extrasynaptic sites in excitatory neurons (Zhou and Sheng, 2013). Limited evidence suggested that extrasynaptic NMDAR-mediated currents might contribute to the excitability of inhibitory neurons (Riebe et al., 2016; Garst-Orozco et al., 2020). It needs to be noted that the contributions of NMDARs to brain physiology and pathology are likely more complex than a simple dichotomy between GluN2A- and GluN2B-containing NMDARs, or between synaptic and extrasynaptic NMDARs. Thus, a more thorough understanding of the functional contribution of NMDARs on the inhibitory neurons to neuronal excitability will provide a deeper understanding the function of inhibitory neuron and the involved pathologic processes.
In this study, we used electrophysiological recording in vitro and in vivo to address how NMDARs may influence the excitability of inhibitory neurons, and the subunits and subcellular localization of participating NMDARs. We found bidirectional modulation of intrinsic excitability by NMDARs, which are likely be mediated at least partially by the GluN2C/2D-containing extrasynaptic NMDARs.
Materials and Methods
Animals
Mice were housed under standard conditions with free access to food and water. All experiments were conducted in accordance with the animal protection law and were approved by the Guangzhou University of Chinese Medicine, Peking University Shenzhen Graduate School Animal Care and Use Committee, and the Animal Care and Use Committee at the Southern Medical University. Male and female mice 6–10 weeks of age, including WT (C57BL/6J), GAD67-GFP, PV-Cre, Ai14 and Ai32, and GluN2D-Flox transgenic mice were used. Mice were housed in a 12 h light-dark cycle with free access to food and water.
Electrophysiological recordings and analysis in vitro
Methods of brain slicing and patch-clamp recording were published previously (Yao et al., 2018). In brief, coronal frontal sections (400 μm) were cut on a VT1200S vibratome (Leica Microsystems). Recording started at least 1 h after slicing on an upright microscope (Eclipse FN1, Nikon) at room temperature (23°C-26°C) with oxygenated aCSF (4–5 ml/min). Recordings were made from layer 2/3 PFC neurons, with GABAergic inhibitory neurons identified using GFP fluorescence in the GAD67-GFP transgenic mice. Signals were acquired at a sampling rate of 10 kHz and filtered at 2 kHz. To facilitate spontaneous spiking in the whole-cell configuration, most recorded neurons were held at − 45 ± 5 mV. For E-S coupling experiments, the resting membrane potential (RMP) of the majority of recorded neurons was near −70 mV. The intensity of electrical stimulation through a glass pipette was adjusted to trigger spikes with ∼50% probability for the first EPSP. E-S coupling test using 5 pulses was given at 20 Hz unless mentioned otherwise.
NMDAR EPSCs were analyzed using Clampfit software (MDS Analytical Technologies). The average of NMDAR EPSCs in a 2-5 ms window centered on the peak response was taken as peak amplitude. For measuring area, NMDAR EPSCs were integrated from the start of responses to the time point with responses decayed to baseline (prestimulation) level. Stimulus artifacts were not subtracted in these measurements. For measuring the ambient NMDAR responses, slices were bathed in aCSF containing NBQX (10 μm), picrotoxin (50 μm), and low Mg2+ (0.5 mm), with neurons held at 40 mV. D-APV (100 μm) was added into the recording chamber via a thin tube positioned at the edge of the recording chamber.
To examine the potential activity-dependent property of EU1794-4, baseline NMDAR-mediated synaptic responses were acquired using 1 Hz synaptic stimulation, for which 1 Hz is a typical low-frequency stimulation that used to test basal responses before a manipulation (e.g., high-frequency stimulation). Following 1 Hz stimulation, 20 Hz stimulation (for 50 s) was given after sufficient time (∼15 min) was allowed for EU1794-4 to take effect (see Fig. 5E). To examine the contribution of GluN2C/2D-containing NMDARs to the EU1794-4's effect, changes in holding current at 40 mV were measured by applying EU1794-4 (300 μm) directly onto slices in the presence of Veh or NAB-14 (see Fig. 6E). To mimic physiological conditions, neurons were clamped at −60 mV (see Fig. 6I), to examine the contribution of GluN2C/2D-containing NMDARs to ambient NMDAR responses. With neurons recorded at −60 mV, we measured changes in the holding current induced by NAB-14, or Pip18, or DMSO, normalized to the responses during subsequent perfusion of D-APV. In these experiments, slices were bathed in NMDA (3 μm) to enhance ambient NMDAR responses, and all response were normalized to the responses after application of D-APV (see Fig. 6I).
Figure 5.
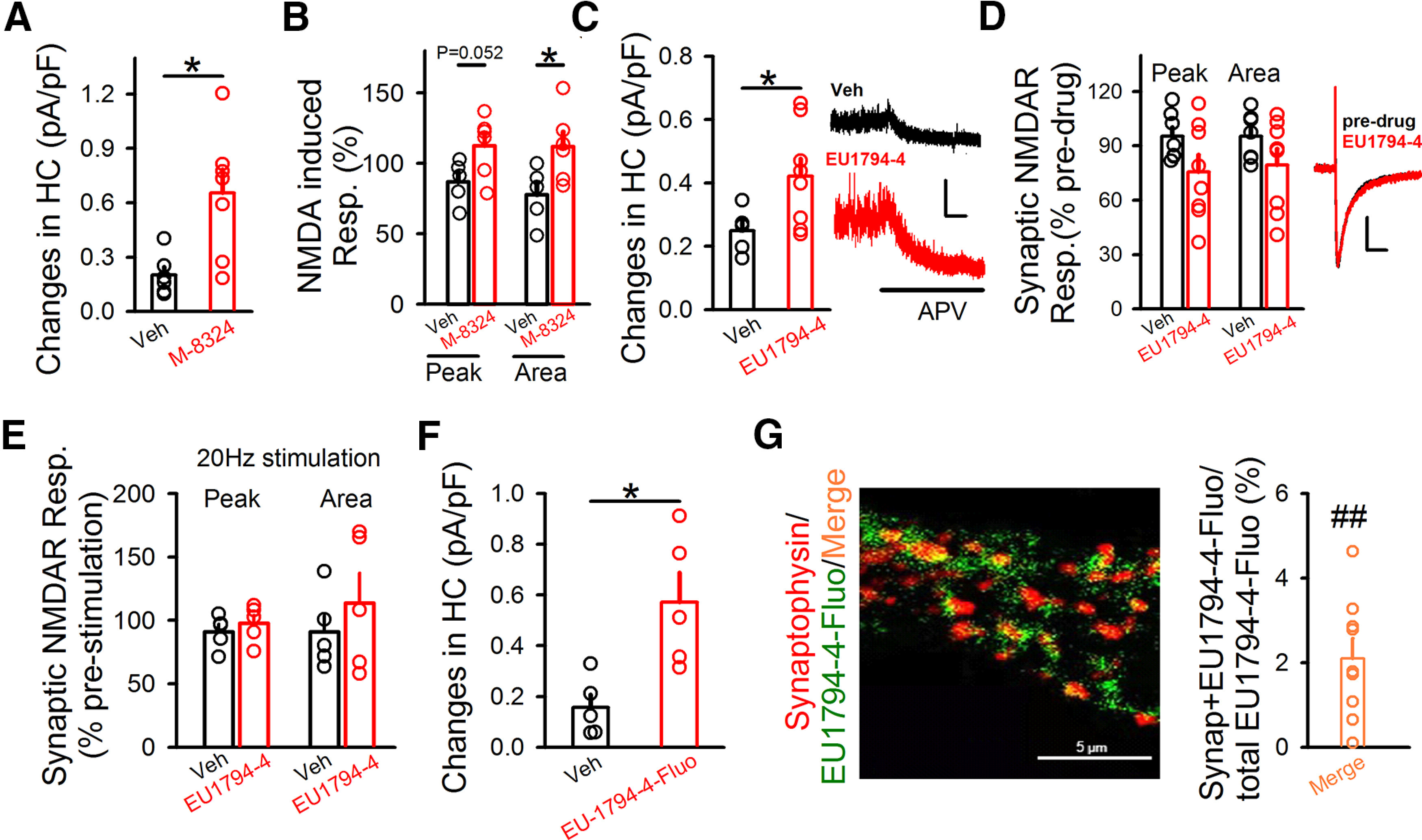
EU1794-4 mostly acts on extrasynaptic NMDARs in GABAergic neurons. A, Ambient NMDAR responses in the presence of M-8324 or vehicle. HC, Holding current with neurons clamped at 40 mV. Veh, 0.20 ± 0.046 pA/pF; N (cells) = 6. M-8324, 0.65 ± 0.13 pA/pF; N (cells) = 7. *p<0.05 (unpaired t test). B, M-8324 increased extrasynaptic NMDAR responses induced by puffed NMDA. Peak and area of puffed NMDA-induced response in the presence of M-8324 or Veh. Veh, Peak, 87 ± 6.77%; Area, 112.62 ± 8.79%; N (cells) = 5. M-8324, Peak, 77.81 ± 8.92%; Area, 112.03 ± 10.28%; N (cells) = 6. *p<0.05 (unpaired t test). C, Ambient NMDAR responses in the presence of EU1794-4 (30 μm) or vehicle, with sample traces on the right. Veh, 0.24 ± 0.03 pA/pF; N (cells) = 5. EU1794-4, 0.42 ± 0.05 pA/pF; N (cells) = 8. Calibration: 10 s, 50 pA. *p<0.05 (unpaired t test). D, EU1794-4's impact on evoked NMDAR-EPSCs, compared with vehicle, with sample traces on the right. Calibration: 100 ms, 20 pA. Peak amplitude, Veh, 95.22 ± 5.01%; EU1794-4, 75.71 ± 9.43%; p = 0.10. Area, Veh, 95.24 ± 4.95%; EU1794-4, 79.46 ± 8.92%; p = 0.16. N (cells) = 7 (Veh), 8 (EU1794-4). E, Activity dependence of EU1794-4 on NMDAR-EPSCs. Neither peak nor area was altered after 20 Hz synaptic stimulation in the presence of EU1794-4 (30 μm). Veh, Peak, 91.17 ± 5.87%; Area, 90.98 ± 13.4%; N (cells) = 5; EU1794-4, Peak, 97.84 ± 6.5%; Area, 113.7 ± 23.58%; N (cells) = 5. F, EU1794-4-Fluo increased extrasynaptic NMDAR responses. Ambient NMDAR responses were significantly larger in the presence of EU1794-4-Fluo (300 μm), compared with the Veh. Veh, 0.15 ± 0.05; N (cells) = 5; EU1794-4-Fluo, 0.57 ± 0.11; N (cells) = 5. *p<0.05 (unpaired t test). G, Left, Staining of EU1794-4-Fluo and synaptophysin on cultured neurons. Representative images of synaptophysin (red), EU1794-4-Fluo (green), and merge between them. Right, Percentage of (synaptophysin + EU1794-4-Fluo)/total EU1794-4-Fluo; 2.09 ± 0.47%, N (cells) = 9. ##p<0.01, compared with fluorescent density of the total EU1794-4-Fluo (paired t test). Data are mean ± SEM.
Figure 6.
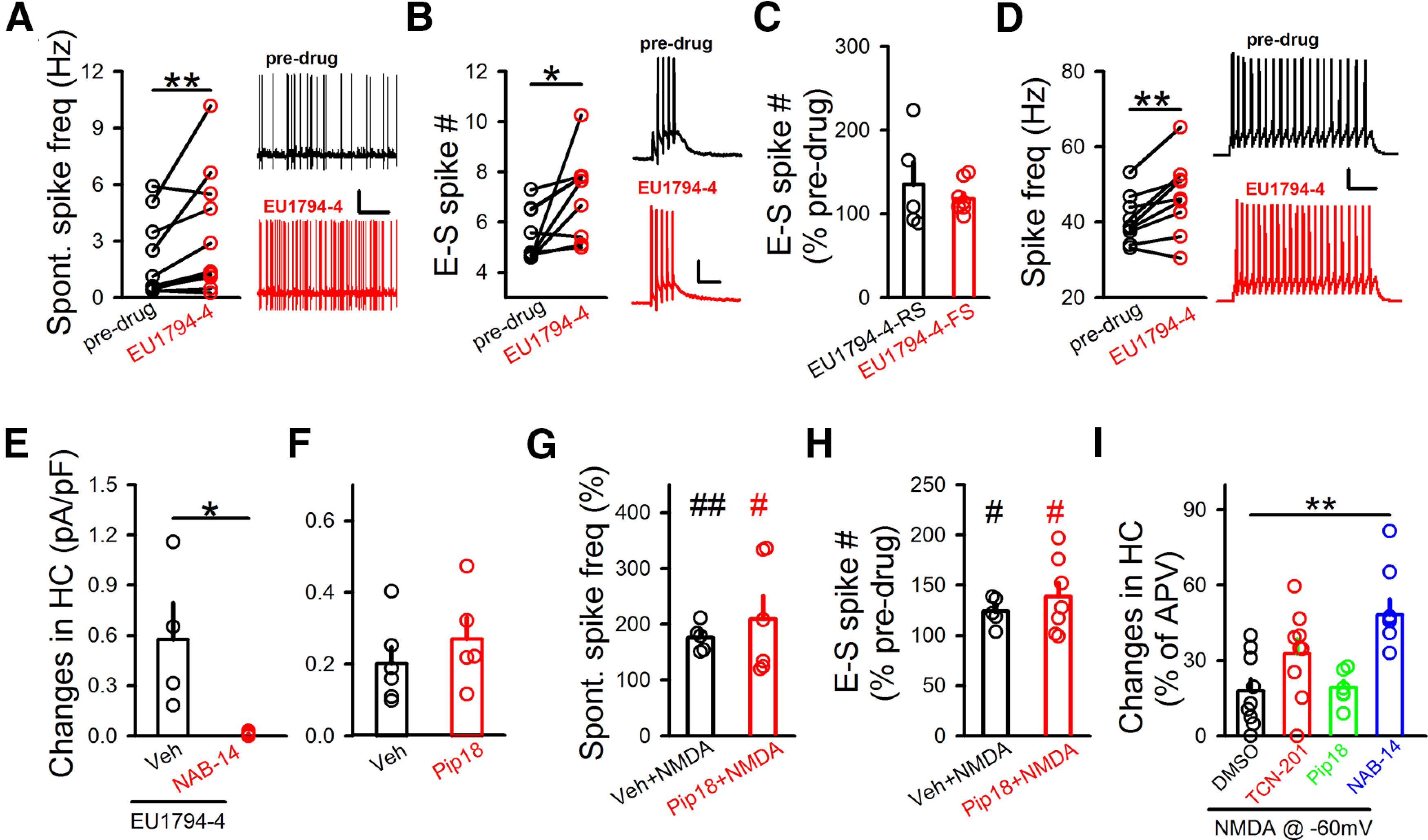
Contribution of extrasynaptic NMDARs to the intrinsic excitability of GABAergic neurons. A, EU1794-4's impact on spontaneous spike frequency, with sample traces on the right. Calibration: 10 s, 20 mV. Predrug, 2.04 ± 0.66 Hz; EU1794-4, 3.43 ± 1.02 Hz; RMP, −68.27 ± 1.73 mV; N (cells) = 11. **p<0.01 (paired t test). B, EU1794-4's impact on E-S coupling, with sample traces on the right. Calibration: 200 ms, 20 mV. Predrug, 5.47 ± 0.35; EU1794-4, 7.05 ± 0.56; RMP, −68.10 ± 1.95 mV; N (cells) = 9. *p<0.05 (paired t test). C, Impact of EU1794-4 on E-S coupling did not differ between RS- and FS-inhibitory neurons. RS, 135.38 ± 25.82%; N (cells) = 5. FS, 118.14 ± 5.94%; N (cells) = 9. D, Bath application (5-10 min) of EU1794-4 on spikes triggered by current injection, with sample traces on the right. Calibration: 500 ms, 10 mV. Predrug, 40.76 ± 2.12 Hz; EU1794-4, 46.76 ± 3.35 Hz; RMP, −68.96 ± 0.78 mV; N (cells) = 9. **p<0.01 (unpaired t test). E, Impact of NAB-14 (20 μm) on EU1794-4 (300 μm)-induced changes in holding current (HC) at 40 mV. Veh, 0.57 ± 0.21 pA/pF, N (cells) = 4; NAB-14, 0.01 ± 0.0001 pA/pF, N (cells) = 5. *p<0.05 (unpaired t test). F, Impact of Pip18 on the ambient NMDAR responses in GABAergic neurons. Veh, 0.20 ± 0.046 pA/pF; N (cells) = 6. Pip18, 0.29 ± 0.082 pA/pF; N (cells) = 5. G, Pip18 (1 μm) did not affect the NMDA (3 μm)-induced increase of spontaneous spiking. Spontaneous spiking, Veh+NMDA, 175.6 ± 11.03%; N (cells) = 5. Pip18+NMDA, 209.21 ± 41.82%; N (cells) = 6. #p<0.05, ##p<0.01, compared with predrug (paired t test). H, Pip18 did not affect the NMDA-induced increase in E-S coupling. E-S coupling, Veh+NMDA, 123.88 ± 6.37%; N (cells) = 5. Pip18+NMDA, 138.60 ± 14.20%; N (cells) = 7. #p<0.05, compared with predrug (paired t test). I, Impacts of various GluN2-NMDAR antagonists on isolated NMDAR responses in the presence of NMDA (3 μm) at −60 mV. Changes in holding current (HC) to bath perfusion of antagonists were normalized to changes in holding currents after subsequent D-APV application. DMSO, 17.9 ± 4.78%; N (cells) = 9. TCN-201, 32.80 ± 5.83%; N (cells) = 9. Pip18, 19.20 ± 2.82%; N (cells) = 6. NAB-14, 48.21 ± 6.3%; N (cells) = 8. **p<0.01 (one-way ANOVA with Bonferroni test). Data are mean ± SEM.
Western blot
The GluN2D-flox transgenic mice were injected with CMV-cre virus (BrainVTA) into the PFC (AP: 1.94 mm anterior to bregma; ML: ±0.4 mm lateral to midline; DV: 2.65 ventral from the cortical surface). After viral expression for 21 d, mice were anesthetized and perfused with PBS through the heart. Freshly dissected mouse brains were treated with RIPA lysis and homogenized using S10 High Speed Homogenizer (Xinzhi Biotechnology), and then centrifuged at 12,000 RPM for 15 min at 4°C. Protein concentration was measured using a BCA Protein Assay Kit (Pierce). Protein samples were run on 10% SDS-PAGE using a Bio-Rad gel system and transferred onto PVDF membranes. Loading controls (GAPDH) were run on the same gel. Membranes were then probed with antibodies of proper dilutions, including anti-GRIN2D (1:1000; Abcam, catalog #DF4086) and anti-GADPH (1: 3000; Abcam, catalog #AB0101). ImageJ was used for densitometric analysis. Experiments were performed in a double-blind manner, with the person performing the analysis blinded to the experimental conditions.
Neuronal culture
Cortical neurons were cultured from newborn mice as described previously (Chanda et al., 2017). Dissected cortices were digested for 30 min with 10 U/ml papain in HBSS buffer supplemented with 2 mm Ca2+ and 0.5 mm EGTA in an incubator, washed with HBSS buffer, dissociated in plating media (MEM supplemented with 0.5% glucose, 0.02% NaHCO3, 0.1 mg/ml transferrin, 10% FBS, 2 mm l-glutamine, and 0.025 mg/ml insulin), and seeded on coverslips placed inside 24-well dishes. After 24 h (DIV1), 75% of the plating media was replaced with neuronal growth media (MEM supplemented with 0.5% glucose, 0.02% NaHCO3, 0.1 mg/ml transferrin, 5% FBS, 2% B27 supplement, and 0.5 mm l-glutamine).
Immunolabeling
Mouse cortical neurons were fixed in 4% PFA and permeabilized with 0.2% Triton X-100 and 10% goal serum, stained with anti-synaptophysin primary antibody (1:200, Abcam, catalog #ab32594) in PBS, and visualized using AlexaFluor-546 goat anti-rabbit secondary antibody (1:500, Invitrogen, catalog #A-11 010). Then samples were bathed in 30 µM EU1794-4-Fluo (labeled as green) for 30 min. Images were acquired using a Nikon confocal microscope. Identical settings were used for all samples in each experiment. Colocalization analysis was performed using ImageJ software.
In vivo single-cell recording
Animal preparation for in vivo recording
Male and female WT (C57BL/6J) and transgenic (PV-Cre, Ai14 and Ai32) mice 2-3 months of age were used. The method was described in previous studies (Li et al., 2019, 2021; Liang et al., 2019). Mice were anesthetized with isoflurane (1.5%, v/v), and a head post for fixation was mounted on top of the skull with dental cement. Skull over A1 was cleaned and protected from being covered by dental cement (Li et al., 2019, 2021; Liang et al., 2019). Afterward, the mouse was injected subcutaneously with 0.1 mg/kg buprenorphine and returned to its home cage. During the recovery period, the mouse was trained to be accustomed to the head fixation on the recording setup. To keep the mouse head in place, a screw was tightly clamped by a custom-made post holder. Recordings were performed in a sound attenuation booth. During recording sessions, the mouse was briefly anesthetized with isoflurane and a craniotomy was performed over A1. Dura was removed to make the drug seep into A1. The mouse was then allowed to recover from isoflurane for at least 2 h. Recording experiments started after the mouse exhibited normal running behavior. Each recording session lasted for ∼4 h. The mouse was given drops of 5% sucrose (w/v) through a pipette every hour. Between sessions, the mouse was returned to its home cage for a break of at least 2 h.
Auditory stimulation
Software for sound stimulation and data acquisition was custom-made using LabVIEW (National Instruments). Stimuli include pure tones (2 −64 kHz spaced at 0.1 octave, 50 ms duration, 3 ms ramp, 0-70 dB sound pressure level [SPL] spaced at 10 dB, in pseudorandom sequence, 3 repetitions, 0.5 s interstimulus interval), characterize frequency and noise (broadband white noise, 50 ms duration, 3 ms ramp, 60 dB SPL, 1 s interstimulus interval). The loudspeaker (Brüel and Kjaer 4138, 4135) and an amplifier (Brüel and Kjaer 2610) were placed 10 cm away from the mouse's head and facing the unsealed ear.
Pharmacological effect and optogenetically guided loose-patch recordings
Neurons were recorded in the heterozygous offspring crossed between a Cre-dependent ChR2-EYFP (Ai32) line and a PV-tomato-Cre line (heterozygous offspring of a cross between PV-Cre line and Ai14 line). Loose-patch recordings using pipettes of small openings (pipette impedance, ∼10 mΩ) were performed. An optic fiber and a drug infusion tube were positioned close to the cortical surface of the recording site. PV neurons were identified by their responses to 10 pulses (50 ms duration, 50 ms interstimulus interval) of blue LED stimulation (470 nm, Thorlabs). Identified PV neurons responded to 50 ms light pulses with a reliable, short-latency burst of spikes and showed sustained spiking responses (see Fig. 7B). The pharmacological effect was verified by comparing spontaneous and evoked spiking responses before and after drug infusion. We found that spiking activity was altered within 1 min from the start of the infusion, and these changes may last 15 min. This method allowed us to temporally and reversibly manipulate A1 when recording from one neuron, but also to separate inhibitory and excitatory neurons. After experiments, the mouse brain was sectioned and imaged with a fluorescence microscope to confirm PV+ expression.
Figure 7.

Impact of EU1794-4 on neuronal spiking in vivo. A, Experimental setup. A mouse was head-fixed via a headpost (P) but could run freely on a rotatable plate. Sound (S) was applied to one ear, and patch recording (R) was performed in the contralateral A1. Blue light (L) and drug infusion tube (I) were positioned next to recording site in A1. B, Left, Confocal images represent tdTomato-labeled PV neurons (red) and expression of ChR2-YFP (green) in a representative brain section. Scale bar, 500 μm. Right top, Raster plot of spikes in a representative PV neuron to pulses of blue LED light stimulation (blue bars, 50 ms each pulse). Right bottom, Corresponding poststimulus spike time histogram. C, Peak/trough amplitude ratio plotted against trough-to-peak (T–P) interval of spike waveform. Each data point represented an individual neuron. Solid symbols represent mean ± SD. Inset, Spike waveforms of a representative PV neuron. Black traces were 20 superimposed spikes. Red dotted vertical lines indicate the timing of trough and peak. Blue arrows point to peak and trough. Peak/trough ratio: Pyr, 0.19 ± 0.08; PV, 0.69 ± 0.16. Trough-peak interval: Pyr, 0.81 ± 0.16; PV, 0.24 ± 0.05. Scale bar, 0.5 ms. D, Noise-evoked responses (raster plots) of PV neurons in A1 before and after EU1794-4 injection (red arrow). Dashed lines indicate the onset and offset of acoustic stimulation. Inset, Twenty randomly selected superimposed spike waveforms. Noise-evoked (E) and spontaneous (F) spike frequency of recorded PV neurons before, 15 min, and recovery after EU1794-4 infusion. N (cells) = 16. **p < 0.01 (one-way repeated-measures ANOVA with Bonferroni test). G-I, Similar to D-F, but for pyramidal neurons. N (cells) = 18. **p< 0.01, *p < 0.05, (one-way repeated-measures ANOVA with Bonferroni test). Data are mean ± SEM.
Histology and imaging
To verify the PV+ expression, mice were deeply anesthetized with urethane (25%) and transcardially perfused with 4% PFA in PBS. The brain tissue was sliced into coronal sections (150 μm thickness) by using a vibratome (Leica Microsystems). Slices were imaged using a confocal microscope (Olympus FluoView FV1000).
Quantification and analysis
We performed data analysis with custom-developed software (MATLAB, The MathWorks). Data from all recorded neurons were first pooled together for a randomized batch processing without categorizing neurons according to their specific identity (e.g., age, condition, laminar location, etc.). In loose-patch recordings, spikes could be detected without ambiguity because their amplitudes were normally >50 pA, whereas the baseline fluctuation was <5 pA. Tone- and noise-evoked spikes were counted within a 100 ms window after the onset of stimuli. Evoked firing rate (FR) was calculated after subtracting the average baseline FR. Evoked responses were defined as FR higher than the average baseline FR by 3 SDs. Neurons that did not exhibit evoked spiking responses were excluded from the analysis.
Multichannel single-unit recording
Experimental methods were described previously (Deng et al., 2020). Briefly, mice were anesthetized with isoflurane (∼ 1% in a gas mixture) on a stereotaxic apparatus (RWD Life Science). A craniotomy was made over the lateral ventricle (AP: −0.5; ML: −1.0; DV: 2.0). A hole was drilled through the skull. Infusion kit was slowly lowered with a gripper, until the pedestal touched the skull. Electrode array was unilaterally implanted aiming at mPFC (1.94 mm anterior to bregma; 0.4 mm lateral to midline; 2.65 ventral from the cortical surface), and sealed to the skull with dental cement. After surgery, mice were allowed to recovery for 7–14 d. During recording of spontaneous spiking, mice moved freely inside a recording box (390 × 180 × 180 mm). Baseline spiking of 30 min was collected before injection of M-8324 (100 μm, 5 µl, i.c.v.), and recording was continued for 120 min afterward. Individually recorded neurons were sorted using Plexon Offline Sorter and analyzed using Neuro Explorer (Nex Technologies). Classification of excitatory and inhibitory neurons and calculation of E/I ratio were performed as described previously (Deng et al., 2020). Briefly, the principal component analysis was used to calculate the principal component score of the unsorted neuronal waveforms, and scores were plotted in a three-dimensional principal component space.
Statistical analysis
Details on the numbers of animal are indicated in the figure legends for each experiment. Data are shown as mean ± SEM. Quantitative results were analyzed (IBM SPSS Statistics, version 20) using unpaired or paired t test, and one-way repeated-measures ANOVA followed by Bonferroni test was made in the drug application and drug washout experiments. The statistical outliers were excluded using the Stem-and-Leaf display. Normalized values were calculated as percentage change over the baseline. A p value of < 0.05 was considered significant.
Reagents
D-APV, NBQX, and QX-314 chloride were obtained from Tocris Bioscience; picrotoxin, EU1794-4, NMDA, and TCN-201 were from Sigma-Aldrich; NAB-14, EU1794-4-Fluo, and M-8324 were synthesized at Peking University Shenzhen Graduate School. Pip18 was provided by Genentech.
Results
NMDARs bidirectionally modulate the intrinsic excitability of inhibitory neurons in vitro
Previous studies have suggested that NMDARs may contribute to the excitability of inhibitory neurons (Jackson et al., 2004; Homayoun and Moghaddam, 2007; Xue et al., 2011; Geoffroy et al., 2022). To test whether this is the case, we examined the effect of bath-applied NMDAR modulators on GFP-labeled GABAergic neurons in PFC slices from GAD-67 GFP mice. The spontaneous spiking rate was used for measuring excitability (Turrigiano, 2011), and normal aCSF Mg2+ (1 mm) was used to mimic physiological conditions. We first tested bath perfusion of a competitive NMDAR antagonist D-APV (100 μm), and observed that spontaneous spiking was reversibly reduced in most of the recorded GABAergic neurons held at ∼−45 mV 5-10 min after application (Fig. 1A,B). We then examined the impact of enhancing NMDAR activity using a NMDAR-PAM M-8324 which we have shown previously to selectively enhance the activity of NMDARs on the inhibitory neurons (Deng et al., 2020). Bath perfusion of M-8324 (30 μm) for 10–15 min led to significantly enhanced spontaneous spiking in GABAergic neurons (Fig. 1C,D), which occurred to a similar degree in both fast spiking (FS) and non-FS (regular spiking [RS]) GABAergic neurons (Fig. 1E), suggesting that this modulation by NMDARs is bidirectional. In addition, the effect of M-8324 was independent on the initial spontaneous spike activity (data not shown). This impact of M-8324 on excitability was not affected by GABAergic synaptic transmission since it was not affected by GABAAR blocker (Fig. 1F).
Figure 1.
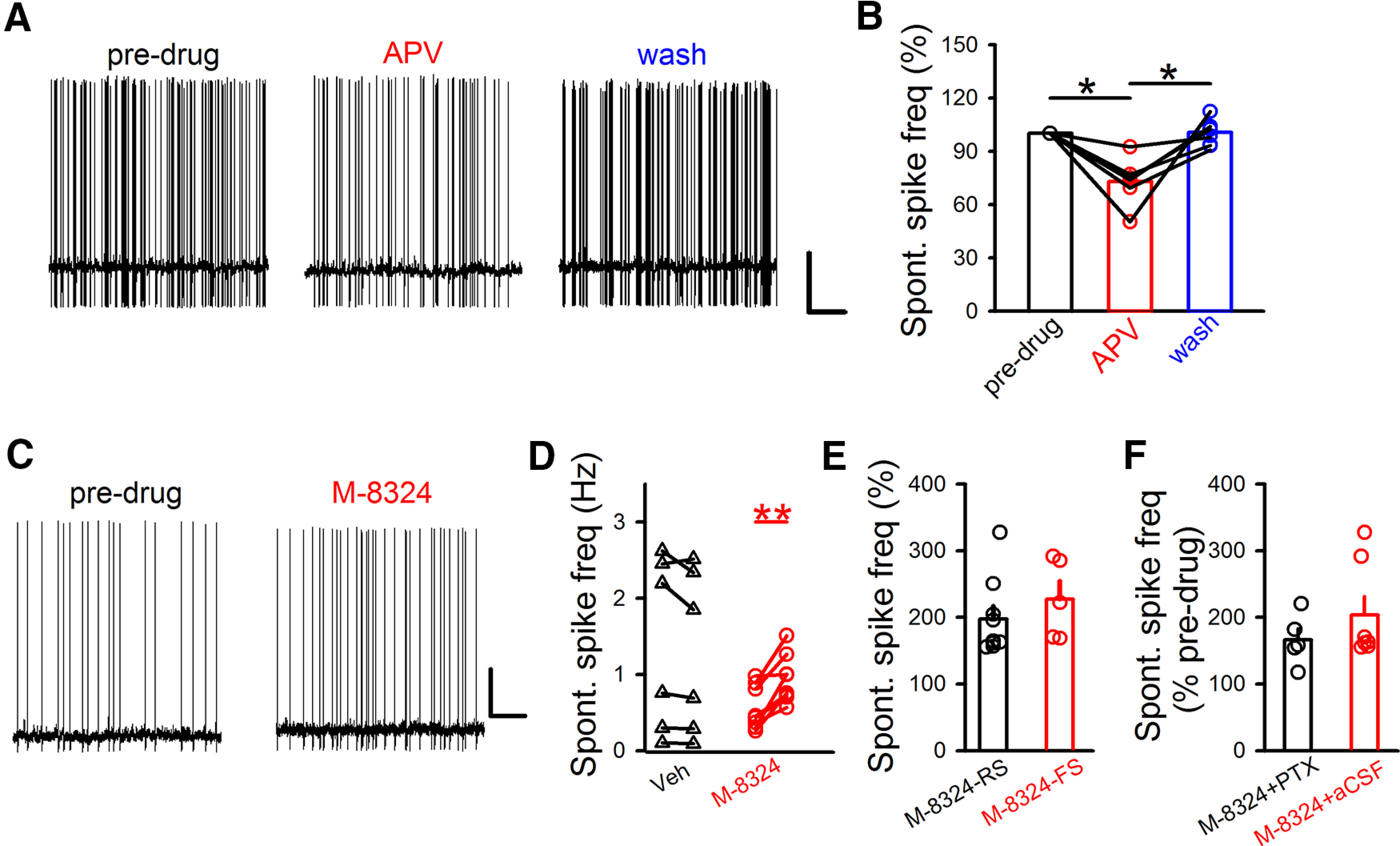
Bidirectional modulation of spontaneous spiking in PFC GABAergic/inhibitory neurons by NMDAR modulators. A, Sample traces of spontaneous spiking in identified GABAergic/inhibitory neurons before, during, and after bath application of D-APV (100 μm) in PFC slices. Calibration: 0.5 s, 20 mV. B, Spontaneous spiking frequency was reversibly reduced by bath application of D-APV (RMP: −70 ± 0.74 mV). Predrug, 100%; APV (5-10 min after application), 72.92 ± 5.55%; wash (∼10 min after wash), 100.03 ± 3.23%. N (cells) = 6. Points connected with lines came from the same neurons. *p<0.05 (one-way repeated-measures with Bonferroni test). C, Sample traces represent the impact of M-8324 (30 μm) on spontaneous spiking in GABAergic neurons. Calibration: 5 s, 20 mV. D, Quantification of Veh (Vehicle) and M-8324's impact on spontaneous spiking in GABAergic neurons 10-15 min after the application. Predrug, 1.14 ± 0.38 Hz, Veh, 1.05 ± 0.35 Hz, RMP, −69.52 ± 0.46 mV; predrug, 0.56 ± 0.1, M-8324, 0.94 ± 0.11 Hz, RMP, −69.69 ± 0.17 mV. N (cells) = 8 (Veh), N (cells) = 8 (M-8324). **p<0.01 (paired t test). E, To explore a potential difference between inhibitory neuronal subtypes, changes in spontaneous spiking frequency were measured in either RS or FS GABAergic neurons. M-8324 (RS), 197.5 ± 19.29%, RMP, −70.10 ± 0.60 mV, N (cells) = 9; M-8324 (FS), 227.35 ± 26.62%, RMP, −69.19 ± 0.56 mV, N (cells) = 5. F, Picrotoxin (100 μm) did not affect M-8324's impact on spontaneous spiking in GABAergic neurons. M-8324 (PTX), 167.47 ± 18.01%; RMP, −69.69 ± 0.17 mV; N (cells) = 5. M-8324 (aCSF), 203.5 ± 27.68%; RMP, −69.83 ± 0.59 mV; N (cells) = 7. Data are mean ± SEM.
The above results suggest that NMDARs may modulate the intrinsic excitability of inhibitory neurons. Another method to measure the intrinsic excitability is to inject depolarizing current through the recording patch pipette. Significantly higher spike frequency was seen in the presence of M-8324 in GABAergic neurons compared with that treated with vehicle (Fig. 2A), and this increase occurred to a similar degree in FS and RS neurons (Fig. 2B). In addition, spike frequency induced by current injection was significantly and reversibly reduced in the presence of D-APV (Fig. 2C). Thus, NMDAR modulation of current-induced spiking in GABAergic neurons is also bidirectional. To better mimic the natural trigger of spikes, we used depolarizing ramp current and observed that M-8324 significantly enhanced the probability of spiking (Fig. 2D). To understand what may underlie the observed higher excitability, we measured a few parameters that contribute to excitability. The rheobase current, defined by the amount of current needed to evoke a single spike, was significantly reduced in the presence of M-8324 (Fig. 2E). We then measured the threshold for spiking, defined as the membrane potential reached by injection of rheobase current, and we found that spikes occurred at a significantly more negative (hyperpolarized) Vm in the presence of M-8324 (Fig. 2F). Perfusion of M-8324 did not significantly alter RMP of the recorded neurons compared with baseline (Fig. 2G). To determine whether changes in the passive properties of neurons might also contribute to NMDAR modulator-induced alteration in excitability, we measured input resistance (Rin), spike height (amplitude), afterhyperpolarization, or spike half-width (Fig. 2H), and found none of them was altered by M-8324. Together, altered intrinsic excitability is likely a major contributor to the NMDAR modulator-induced changes in spiking which is likely mediated at least partially by altered spiking threshold.
Figure 2.
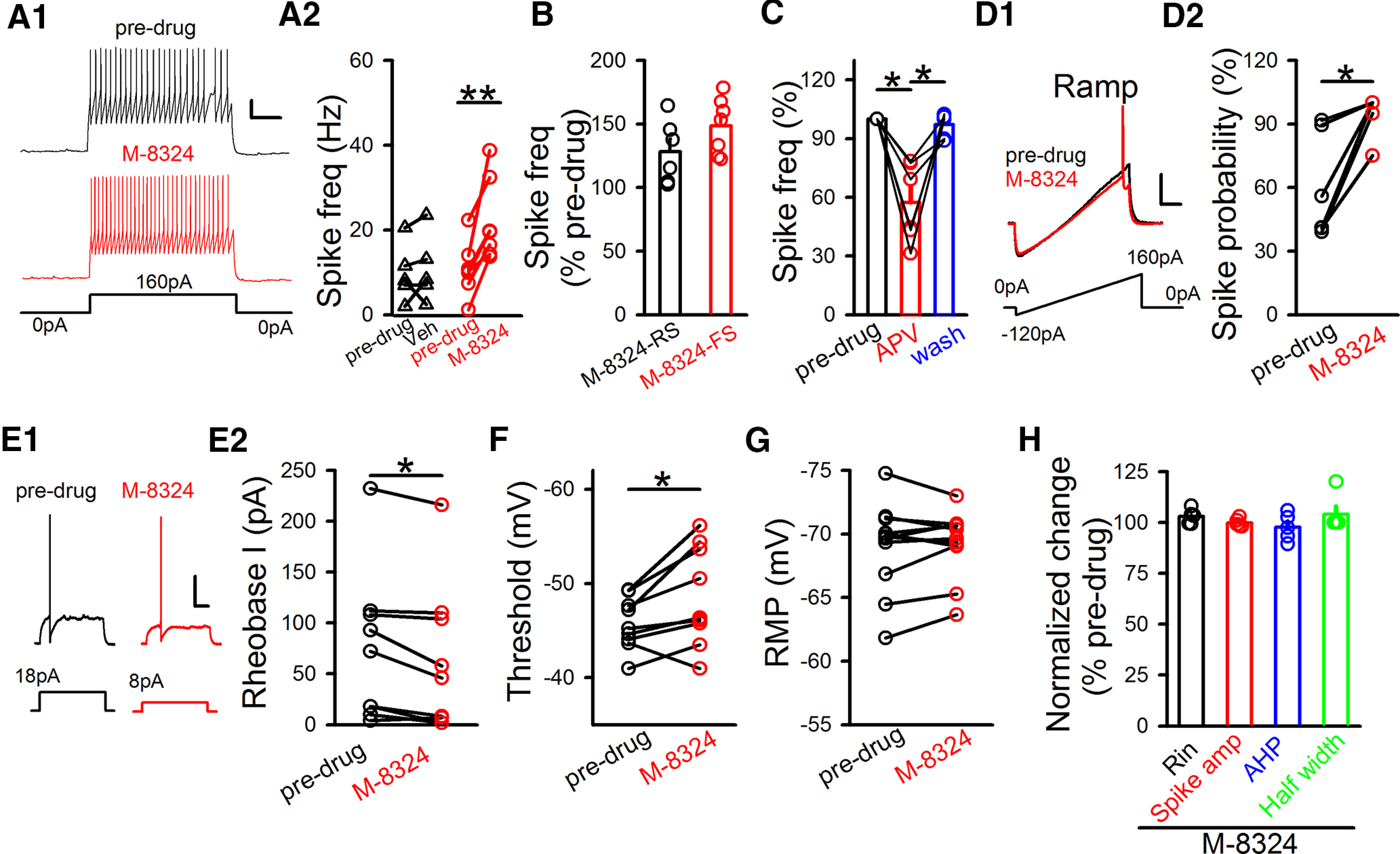
Impact of M-8324 on current injection-triggered spikes. A1, Sample traces represent M-8324's impact on spiking triggered by current injection in GABAergic neurons. Calibration: 200 ms, 20 mV. A2, Quantification of Veh and M-8324's impact after its application for 10-15 min. Predrug, 9.81 ± 3.07 Hz; Veh, 10.87 ± 3.58 Hz; RMP, −69.70 ± 0.21 mV, N (cells) = 5. Predrug, 10.84 ± 2.42 Hz; M-8324, 22.12 ± 3.64 Hz; RMP, −70.31 ± 0.43 mV, N (cells) = 7. **p<0.01 (paired t test). B, M-8324's impact on spike frequency in RS- and FS-GABAergic neurons. M-8324 (RS), 128.19 ± 10.13%; RMP, −71.71 ± 0.43 mV; N (cells) = 6. M-8324 (FS), 148.39 ± 8.34%; RMP, −70.5 ± 0.38 mV; N (cells) = 7. C, Impact of D-APV (5-10 min after application) on spike frequency on the same neurons. APV, 57.4 ± 8.2%; wash, 97.08 ± 2.44%; RMP, −68.96 ± 3.26 mV; N (cells) = 6. *p<0.05 (one-way repeated-measures ANOVA with Bonferroni test). D, M-8324 increased spiking probability measured using a ramp test. D1, Sample traces. Calibration: 100 ms, 20 mV. D2, Population data. Predrug, 59.7 ± 10.07%; M-8324, 95.4 ± 4.08%; N (cells) = 6. *p<0.05 (paired t test). E, Reduced rheobase current in the presence of M-8324 in the same GABAergic neurons. E1, Sample traces. Calibration: 100 ms, 20 mV. E2, Population data. Predrug, 74.11 ± 24.49 pA; M-8324, 61.44 ± 23.92 pA; N (cells) = 9. *p<0.05 (paired t test). F, Threshold to spike was reduced by M-8324 application. Predrug, −45.77 ± 0.92 mV; M-8324, −48.59 ± 1.76 mV; N (cells) = 9. *p<0.05 (paired t test). G, M-8324 did not change RMP. Predrug, −69.03 ± 1.05 mV; M-8324, −69.24 ± 0.79 mV; N (cells) = 11. H, M-8324 did not affect basic neuronal properties in GABAergic neurons. Input resistance (Rin), 101.60 ± 2.21%, N (cells) = 5. Spike amplitude (spike amp), 99.90 ± 0.91%; afterhyperpolarization, 97.72 ± 0.91%; spike half-width, 105.41 ± 3.56%. Data are mean ± SEM.
Spontaneous spiking is triggered by either synaptic inputs (EPSPs) or fluctuations in membrane potentials (Stern et al., 1997; Graupner and Reyes, 2013). Under in vivo conditions, a large portion of spikes are driven by the preceding EPSPs. This coupling between EPSPs and spikes is termed E-S coupling and measures the probability of spiking triggered by similarly sized EPSPs. E-S coupling is also considered a measure of intrinsic excitability (Malik and Chattarji, 2012). We found that the degree of E-S coupling, as measured by the number of spikes generated by five depolarization pulses, was significantly higher in GABAergic neurons in the presence of M-8324 compared with that with vehicle (Fig. 3A). We also found a significant increase in the spiking probability by M-8324 using one test pulse (Fig. 3B). This enhancement was present in both FS and RS neurons (Fig. 3C), suggesting that it occurs in GABAergic neurons in general. There was no significant difference between RMPs in the presence of M-8324 and vehicle (Fig. 3D), suggesting no alteration of RMP by M-8324. A reversible reduction in the EPSP-triggered spike numbers was observed with bath perfusion of D-APV (Fig. 3E). In addition, M-8324 significantly shortened the latency to spike compared with the predrug baseline (Fig. 3F), consistent with higher excitability. To exclude the possibility that changes in EPSPs underlies the enhanced E-S coupling, we examined EPSPs in the trials without spikes, in the presence and absence of M-8324. We found no significant difference in EPSP area, time to peak, or slope of rise phase (Fig. 3G). Together, our results indicate a bidirectional modulation of intrinsic excitability by NMDARs in inhibitory neurons.
Figure 3.
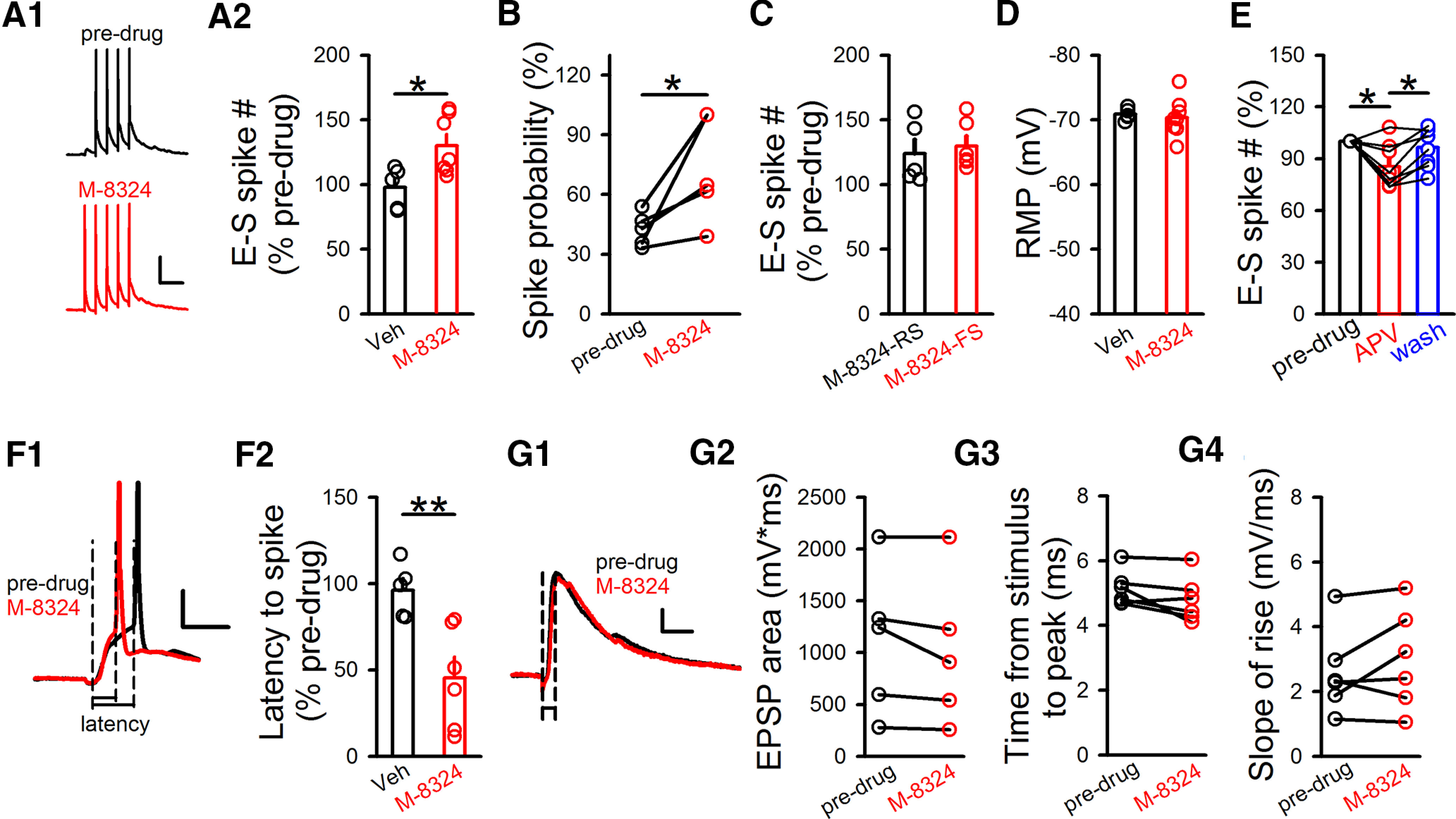
Enhanced E-S coupling by M-8324 in the PFC GABAergic neurons in vitro. A, Impact of M-8324 on E-S coupling in GABAergic neurons held near −70 mV. Sample traces (A1) and population data (A2). Calibration: 100 ms, 20 mV. Veh, 97.79 ± 7.13%, N (cells) = 5; M-8324, 130.2 ± 8.58%; N (cells) = 7. *p<0.05 (unpaired t test). B, M-8324's impact on the probability of single EPSP-induced spike. Predrug, 42.48 ± 3.71%; M-8324, 73.02 ± 11.87%; N (cells) = 5. *p<0.05 (paired t test). C, Impact of M-8324 on E-S coupling in RS- and FS-GABAergic neurons. M-8324 (RS), 124.08 ± 10.65%; N (cells) = 5; M-8324 (FS), 129.8 ± 7.53%; N (cells) = 6. D, No significant difference in RMP between Veh and M-8324 groups. Veh, −70.84 ± 0.41 mV; N (cells) = 5. M-8324, −70.36 ± 1.04 mV; N (cells) = 8. E, Impact of D-APV on E-S coupling in the same neurons. APV, 85.36 ± 4.51%; wash, 96.45 ± 4.02%; both normalized to predrug level. N (cells) = 8. *p<0.05 (one-way repeated-measures ANOVA with Bonferroni test). F, M-8324 shortened the latency to spike. Sample traces (F1) and population data (F2). Calibration: 10 ms, 20 mV. Veh, 96.11 ± 6.92%; N (cells) = 5. M-8324, 45.47 ± 12.03%; N (cells) = 6. **p<0.01 (unpaired t test). G, M-8324 did not affect EPSPs in GABAergic neurons. Sample traces (G1) and population data (G2-G4). Calibration: 50 ms, 5 mV. Time to peak (G2), slope of rise time (G3), or EPSP area (G4) was not altered by M-8324. N (cells) = 5. Data are mean ± SEM.
GluN2 subunits have distinct contributions to NMDAR's modulation of excitability of GABAergic neurons
Distinct contributions of GluN2-NMDAR subunits to various important NMDAR functions have been shown previously (Traynelis et al., 2010; Paoletti et al., 2013; Ge et al., 2020). Thus, we examined the possibility that GluN2 subunits have distinct contributions in modulating of the intrinsic excitability of GABAergic neurons. To do so, we tested the impacts of modulators selectively targeting different GluN2-containing NMDARs on spontaneous spiking and E-S coupling. Significant increase in spontaneous spiking or E-S coupling induced by M-8324 still occurred in the presence of TCN-201 (10 μm), a GluN2A-selective NMDAR antagonist (Hansen et al., 2012) (Fig. 4A). Selective GluN2B-NMDAR antagonist piperidine-18 (Pip18, 1 μm) also did not block the increase of spontaneous spiking or E-S coupling induced by M-8324 (Fig. 4B). No enhancement of spontaneous spiking or E-S coupling by M-8324 was observed in the presence of selective GluN2C/2D antagonist NAB-14 (20 μm) (Fig. 4C), while GluN2C/2D-selective NMDAR enhancer CIQ (20 μm) significantly increased spontaneous spiking or E-S coupling (Fig. 4D). The above results suggest that GluN2C/2D-NMDARs play pivotal roles in modulating the intrinsic excitability of GABAergic neurons, while GluN2A- or GluN2B-NMDARs have minimal contributions. To further examine the contribution of GluN2C/2D-NMDARs to the excitability of inhibitory neurons, we induced conditional GluN2D knockdown by injecting virus in the GluN2D-Loxp transgenic mice. First, to confirm the efficacy of GluN2D-Loxp, CMV-Cre virus was injected to target all types of neurons to maximize the reduction. Western blot analysis showed significant reduction in the GluN2D protein level in the PFC (Fig. 4E). Then, for the electrophysiological experiments, to selectively induce GluN2D-NMDAR knockdown in the GABAergic neurons, vGAT-Cre virus was injected in the PFC. We found no change in either spontaneous spiking or E-S coupling to bath application of M-8324 (Fig. 4F), supporting the preferential contribution of GluN2C/2D-NMDARs to spontaneous spiking and E-S coupling.
Figure 4.
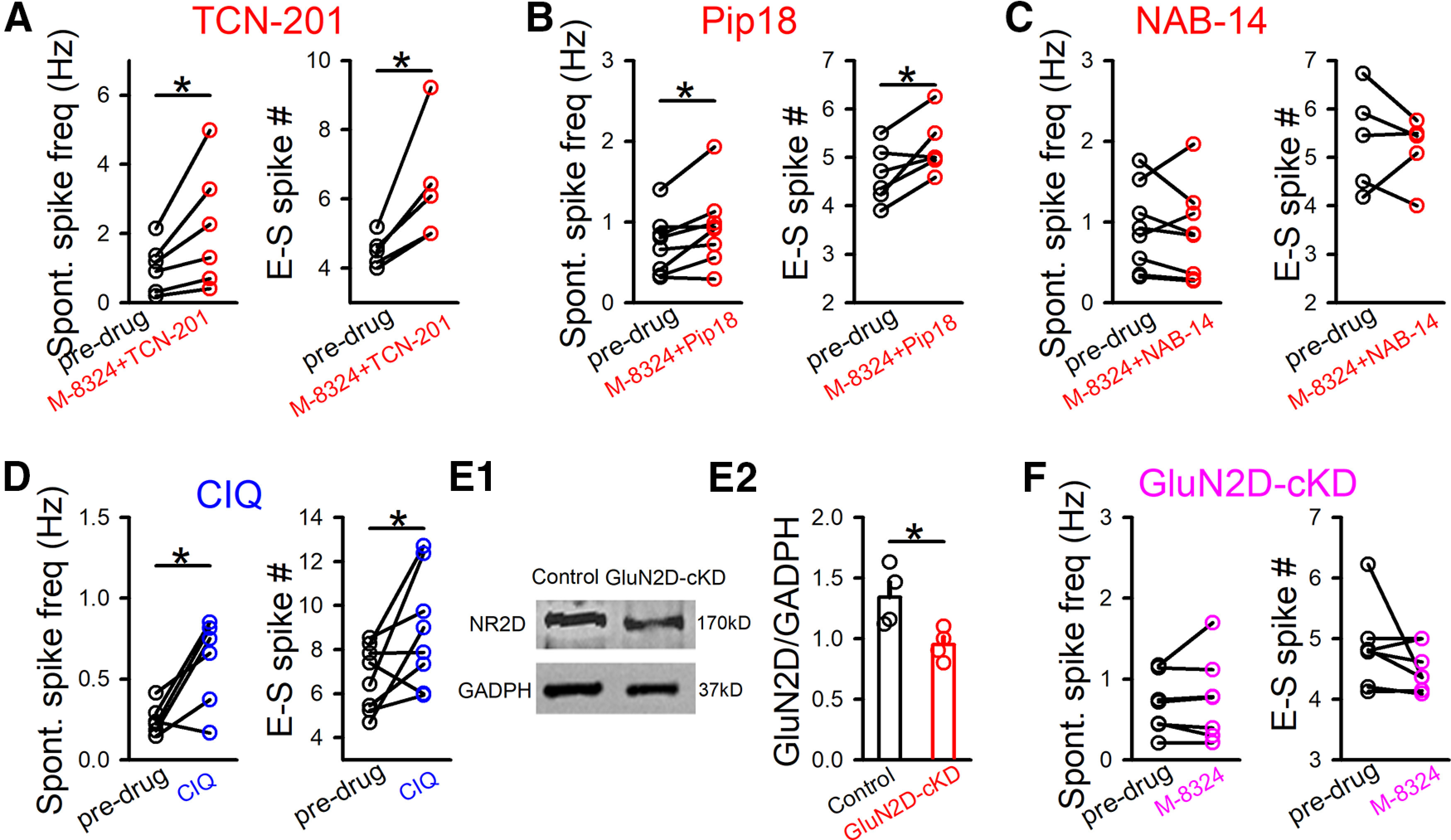
Contributions of GluN2-NMDARs to M-8324's impact on spontaneous spiking and E-S coupling in GABAergic neurons in vitro. A, Impact of TCN-201 (10 μm) on M-8324's effect on spontaneous spiking and E-S coupling. Spontaneous spiking, predrug, 1.01 ± 0.29 Hz; M-8324+TCN-201, 2.15 ± 0.71 Hz; RMP, −66.77 ± 2.12; N (cells) = 6. E-S coupling, predrug, 4.49 ± 0.20; M-8324+TCN-201, 6.34 ± 0.77; RMP, −67.09 ± 1.37 mV; N (cells) = 5. *p<0.05 (paired t test). B, Impact of Pip18 (1 μm) on M-8324's effect on spontaneous spiking and E-S coupling. Spontaneous spiking, predrug, 0.71 ± 0.12 Hz; M-8324+Pip18, 0.93 ± 0.17 Hz; RMP, −69.88 ± 2.97 mV; N (cells) = 8. E-S coupling, predrug, 4.63 ± 0.24; M-8324+Pip18, 5.21 ± 0.23; RMP, −65.62 ± 2.06 mV; N (cells) = 6. *p<0.05 (paired t test). C, Impact of NAB-14 (20 μm) on M-8324's effect on spontaneous spiking and E-S coupling. Spontaneous spiking, predrug, 0.91 ± 0.18 Hz; M-8324+NAB-14, 0.86 ± 0.20 Hz; RMP, −69.81 ± 1.56 mV; N (cells) = 8. E-S coupling, predrug, 7.32 ± 1.80; M-8324+NAB-14, 6.85 ± 1.41; RMP, −73.84 ± 2.38 mV; N (cells) = 5. *p<0.05 (paired t test). D, Impact of CIQ (10 μm, 5-10 min bath application) on spontaneous spiking and E-S coupling. Spontaneous spiking, predrug, 0.24 ± 0.03 Hz; CIQ, 0.60 ± 0.11 Hz; RMP, −73.12 ± 1.56 mV; N (cells) = 6. E-S coupling, predrug, 1.22 ± 0.70; CIQ, 2.26 ± 1.30; RMP, −69.52 ± 1.87 mV; N (cells) = 8. *p<0.05 (paired t test). E, Sample images showing the GluN2D protein expression level in the GluN2D-KD (E1). The population data (E2). Control, 1.34 ± 0.12; N (mice) = 4; GluN2D-KD, 0.95 ± 0.06; N (mice) = 4. F, Potentiation of M-8324 was absent in inhibitory neurons from GluN2D-NMDAR cKD mice. Spontaneous spiking, predrug, 0.69 ± 0.13 Hz; M-8324+NAB-14, 0.75 ± 0.19 Hz; RMP, −68.30 ± 0.94 mV; N (cells) = 7. E-S coupling, predrug, 4.84 ± 0.26; M-8324+NAB-14, 4.51 ± 0.14; RMP, −66.78 ± 1.9 mV; N (cells) = 7. Data are mean ± SEM.
Extrasynaptic GluN2C/2D-NMDARs have major contribution to modulating the intrinsic excitability of GABAergic neurons
In the above experiments, the participating NMDARs may be located at either synaptic or extrasynaptic regions of GABAergic neurons. We have shown previously that M-8324 enhances the activity of synaptic NMDARs (Deng et al., 2020); and here we examined whether it can also enhance the activity of extrasynaptic NMDARs. To do so, we isolated NMDAR responses induced by ambient glutamate present in the extracellular space (Le Meur et al., 2007; Povysheva and Johnson, 2012; Yao et al., 2018). Bath application of D-APV (100 μm) resulted in a larger reduction in the NMDAR responses (measured as changes in holding current) in the presence of M-8324 than with vehicle (Fig. 5A), indicating a significant impact of M-8324 on the activity of extrasynaptic NMDARs. In addition, NMDAR responses showed larger noise in the presence of M-8324 than in vehicle (data not shown), consistent with larger NMDAR responses (Yao et al., 2018). We also used another widely accepted method to activate extrasynaptic NMDARs, namely, puffing NMDA onto dendrites (Papouin et al., 2012). Both peak and area of NMDAR responses were larger in the presence of M-8324 than with vehicle (Fig. 5B), supporting enhanced activation of extrasynaptic NMDARs by M-8324.
To address whether enhancing the activity of extrasynaptic NMDARs may increase the intrinsic excitability of GABAergic neurons, we need to modulate extrasynaptic NMDARs selectively. An NMDAR modulator named EU1794-4 has been suggested to act selectively on the extrasynaptic NMDARs (Perszyk et al., 2018). We observed significantly larger ambient NMDAR responses in the presence of EU1794-4 (30 μm) than that in the presence of vehicle in GABAergic neurons (Fig. 5C), suggesting that EU1794-4 activates/enhances extrasynaptic NMDARs. For the isolated NMDAR-EPSCs in GABAergic neurons, there was no significant difference between EU1794-4 and vehicle (Fig. 5D), indicating minimal effect of EU1794-4 on synaptic NMDAR responses. An alternative explanation for the differential impact of EU1794-4 on synaptic versus extrasynaptic NMDARs is its use dependence (Perszyk et al., 2018), which we did not observe using 20 Hz synaptic stimulation (Fig. 5E). To further confirm that EU1794-4 selectively binds to the extrasynaptic NMDARs using a different method, we synthetized an EU1794-4 compound with a fluorophore attached (named EU1794-4-Fluo, see Materials and Methods). This EU1794-4-Fluo also enhanced extrasynaptic NMDAR responses (Fig. 5F). We then incubated EU1794-4-Fluo with primary neuronal culture, and costained with a synapse marker synaptophysin. We found that there was not much overlapping between the fluorescence of these two markers, with the fluorescence of EU1794-4-Fluo mostly surrounding that of synaptophysin (Fig. 5G). The percentage of spots stained with both synaptophysin and EU1794-4-Fluo was a very small portion of the total EU1794-4-positive spots (2.09 ± 0.47), supporting that EU1794-4 mostly binds to the extrasynaptic NMDARs. This result is consistent with EU1794-4 selectively binding to extrasynaptic NMDARs.
We next used EU1794-4 to examine whether extrasynaptic NMDARs play a major role in modulating the intrinsic excitability of GABAergic neurons. We found that EU1794-4 significantly enhanced spontaneous spiking (Fig. 6A), E-S coupling (Fig. 6B,C), and current-injection induced spiking (Fig. 6D), indicating a significant contribution of extrasynaptic NMDARs. The increase by EU1794-4 on E-S coupling did not differ between RS- and FS-GABAergic neurons (Fig. 6C). Since GluN2C/2D-NMDARs but not GluN2A- or GluN2B-NMDARs play a prominent role in modulating the intrinsic excitability of the GABAergic neurons (Fig. 4), we examined whether EU1794-4 shows any selectivity on GluN2-NMDARs. Compared with vehicle, NAB-14 abolished changes in holding current induced by bath perfusion of EU1794-4 (Fig. 6E). Considering that it is widely accepted that extrasynaptic NMDARs in excitatory neurons contain GluN2B subunits (Zhou and Sheng, 2013; Lai et al., 2014), we further tested whether GluN2B-NMDARs contribute to EU1794-4-activated ambient NMDAR responses. Pip18 did not significantly affect changes in holding current induced by EU1794-4 (Fig. 6F). Furthermore, Pip18 did not affect the increase in spontaneous spiking or E-S coupling induced by bath perfusion of NMDA (Fig. 6G,H). Although there was a trend toward reduction of NMDAR-EPSCs in inhibitory neurons in the presence of EU1794-4, this reduction cannot explain the enhancement of excitability by EU1794-4 caused by its potentiation of extrasynaptic NMDARs. Put together, GluN2C/2D-NMDARs but not GluN2B-NMDARs have significant contributions to EU1794-4's effect on the extrasynaptic NMDARs. Since the experiments on E-S coupling or spontaneous spiking were performed in normal [Mg2+] and at −60 mV (Fig. 6A–H), different from the EU1794-4 experiments shown above (at 40 mV; Fig. 5C), we conducted additional experiments to test whether extrasynaptic NMDARs activated under the above physiological conditions also contain GluN2C/2D- but not GluN2A- or GluN2B-NMDARs. To do so, slices were bathed in NMDA (3 μm) and changes in holding current in responses to D-APV were used as a measure of extrasynaptic NMDAR responses, at −60 mV and in 1 mm Mg2+. NAB-14 induced a significantly larger reduction on the holding current compared with that induced by DMSO, while there was no significant difference between TCN-201 and DMSO or between Pip18 and DMSO (Fig. 6I). This result suggests that GluN2C/GluN2D-, but not GluN2A- or GluN2B-NMDARs have significant presence at the extrasynaptic regions on the GABAergic neurons, consistent with their contribution to the intrinsic excitability in these neurons.
Modulation of spiking in GABAergic neurons by NMDAR modulators in vivo
Previous studies, including our own, have shown that NMDAR inhibitors lead to reduced spiking of inhibitory neurons in vivo (Homayoun and Moghaddam, 2007; Yao et al., 2019). We recently showed that NMDAR-PAMs elevate spiking of the inhibitory neurons in the auditory cortex in vivo (Deng et al., 2020). Our current results indicate that the excitability of GABAergic neurons is modulated bidirectionally by NMDARs, especially those at the extrasynaptic locations. To test whether this modulation also occurs in vivo and involves extrasynaptic NMDARs, we used in vivo cell-attached recording to record the sound-induced spiking and spontaneous spiking in PV neurons in the primary auditory cortex (A1; Fig. 7A). These neurons in ChR2-EYFP/PV-Cre mice were confirmed positive for parvalbumin (PV) based on their responses to opto-stimulation (see Materials and Methods) (Fig. 7A–C). In addition, their spike waveforms were characteristic for PV neurons (Fig. 7C). Both sound-evoked and spontaneous spiking frequency were significantly and reversibly enhanced after local infusion of EU1794-4 (Fig. 7D–F). PV neurons, abundantly present in the cerebral cortex among inhibitory neurons, play key roles in controlling the excitability of pyramidal neurons (Li et al., 2021). Significant and reversible reduction in both sound-evoked and spontaneous spiking was observed in pyramidal/excitatory neurons in A1, likely as a consequence of enhanced activation of PV neurons (Fig. 7G–I). Together, we have observed enhanced spiking in inhibitory neurons to application of EU1794-4 in vivo, in a manner similar to that of in vitro, suggesting that activation of extrasynaptic NMDAR in vivo also significantly enhances the activity of inhibitory neurons.
We then examined the impact of M-8324 on neuronal spiking in the PFC in vivo (Fig. 8A) since PFC is a key brain region for many critical brain functions (e.g., working memory and executive functions) and various psychiatric disorders (including schizophrenia and depression). By using the well-established criteria to distinguish between excitatory and inhibitory neurons (Fig. 8B), we observed significantly enhanced spiking in inhibitory neurons and reduced spiking in excitatory neurons after infusion of M-8324 (Fig. 8C,D), consistent with our previous finding in the auditory cortex (Deng et al., 2020). The reduced spiking level in excitatory neurons is likely caused by the enhanced spiking in inhibitory neurons (Hackos et al., 2016; Yao et al., 2018; Deng et al., 2020). Our in vivo results collectively showed that modulating NMDARs, including the extrasynaptic NMDARs, leads to a robust modulation of inhibitory neuron activity, likely as a consequence of altering excitability in these neurons.
Figure 8.

Modulation of spontaneous spiking in PFC neurons in vivo. A, The location of recording electrodes in the PFC. Each dot represents the electrode position from each experiment. Only data from those positioned in the PrL (Prelimbic) and IL (Infralimbic) were included for further analysis. B, Criteria for distinguishing FS neurons (inhibitory neurons [Inh]) and non-FS neurons (excitatory neurons [Ext]) based on their spike waveforms. C, Sample raster plots showing effect of M-8324 (intracerebroventricular [i.c.v.] injection, 100 μm) on spontaneous spiking of excitatory (Ext) and inhibitory (Inh) neurons. Total duration, 10 s. D, Quantification of M-8324's impact on spontaneous spiking in excitatory and inhibitory neurons normalized to baseline (before drug infusion). For excitatory neurons (Ext), Veh, 96.97 ± 4.12%; M-8324, 49.54 ± 6.2%; N = 34 units/5 mice (M-8324, 100 μm, i.c.v.). For inhibitory neurons (Inh), Veh, 102 ± 2.88%; M-8324, 230.8 ± 31.81%; N = 9 units/5 mice.
Discussion
In this study, we have revealed important contributions of extrasynaptic NMDARs to the intrinsic excitability of inhibitory/GABAergic neurons: (1) NMDARs bidirectionally modulate this excitability in that activation enhances while inhibition reduces it; (2) GluN2C/2D-containing NMDAR subunits have a significant contribution to this modulation; and (3) extrasynaptic NMDARs play a critical role in this modulation. These new findings have far-reaching implications for the contributions of NMDARs to both physiology of brain functions and pathology of brain diseases.
Mostly based on studies on the excitatory neurons, extrasynaptic NMDARs have been shown to play a few distinct functions: (1) their excessive activation leads to synapse loss and neuronal loss, especially in diseases, such as stroke (Hardingham and Bading, 2010; Ge et al., 2020); (2) their activation can modulate neuronal excitability in developing excitatory neurons (Sah et al., 1989; Wu et al., 2012); and (3) their suppression by ketamine (especially via GluN2B-contaning NMDARs) mediates enhanced glutamatergic synaptic transmission and is proposed to underlie the anti-depressant effect of ketamine (Monteggia et al., 2013; Miller et al., 2014; Suzuki and Monteggia, 2020). Although inhibition of NMDARs on the inhibitory neurons has also been suggested to mediate ketamine's anti-depression effect, the location of these NMDARs has not been examined (Miller et al., 2016; Zanos and Gould, 2018; Fogaca and Duman, 2019). Here we provide strong evidence that extrasynaptic NMDARs play an important role in modulating the intrinsic excitability of inhibitory neurons in a bidirectional manner. This finding emphasizes the role of NMDARs in the second-to-second routine functions of inhibitory neurons, in contrast to the much-emphasized role of NMDARs in synaptic plasticity. There is some evidence that NMDARs are required for the induction of synaptic plasticity in inhibitory neurons (Moreau and Kullmann, 2013), but the subcellular localization of these NMDARs is unclear.
Subunit composition and subcellular localization of NMDARs participating in the modulation of intrinsic excitability
We have previously characterized M-8324 and shown that M-8324 has higher potency than GNE-8324 and potentiates synaptic NMDAR responses selectively on the inhibitory neurons (Deng et al., 2020). We do not have information on the subunit selectivity of M-8324, although it is likely similar to that of GNE-8324. Since extrasynaptic NMDARs are mainly activated by the extracellular or ambient glutamate under the physiological conditions, their activation may be subjected to modulations distinct from that of synaptic NMDARs, such as low-activity dependence and no input specificity (Cathala et al., 2003). This point can be appreciated for depolarization-induced spiking or spontaneous spiking in that this modulation likely involves most or all extrasynaptic NMDARs on an entire neuron and is affected by the concentration of extracellular glutamate surrounding the entire neuron. For E-S coupling, however, the situation can be complex since synaptically released glutamate may affect the extrasynaptic NMDARs surrounding the active synapses in inhibitory neurons (Chaudhry et al., 1995; Yao et al., 2018).
NMDARs with different GluN2 subunits have been shown to be present at distinct synaptic localizations and serve distinct functions, although this conclusion has been debated (Paoletti et al., 2013; Zhou and Sheng, 2013; Lai et al., 2014). More specially, extrasynaptic NMDARs are shown to be enriched with GluN2B-, GluN2C-, or GluN2D-NMDARs in excitatory neurons, while GluN2C/2D-containing NMDARs are present at extrasynaptic regions in inhibitory neurons and mostly in young animals (Le Meur et al., 2007; Riebe et al., 2016; Hanson et al., 2019). Since their activation requires spillover of synaptic glutamate or elevated extracellular glutamate, extrasynaptic NMDARs on the excitatory neurons show more prominent activation under pathologic condition or at hyperactivity brain states (Lai et al., 2014). Pharmacological evidence suggests that GluN2C/2D-containing NMDARs can modulate the activity of both GABAergic neurons and excitatory neurons (Swanger et al., 2015; Hanson et al., 2019; Garst-Orozco et al., 2020). Our results strongly support the conclusion of GluN2C/2D-NMDARs on the inhibitory neurons and further indicate the absence of GluN2A-, GluN2B-containing NMDARs at the extrasynaptic regions in inhibitory neurons, distinct from the excitatory neurons. This lack of extrasynaptic GluN2A-, GluN2B-containing NMDARs is also consistent with our finding that modulation of intrinsic excitability occurs in the presence of normal extracellular Mg2+ concentration and near RMPs, and consistent with the low sensitivity of GluN2C/2D-NMDARs to Mg2+ block at negative membrane potentials (Mullasseril et al., 2010; Paoletti et al., 2013). Since FS and non-FS inhibitory neurons do not show significant difference on NMDAR's impacts on the intrinsic excitability, it is thus likely that extrasynaptic NMDARs on these neurons do not differ substantially, such as on GluN2-subunit composition. This is in contrast to the less contribution of synaptic NMDARs in FS than in non-FS inhibitory neurons (Wang and Gao, 2009; Povysheva and Johnson, 2012), and further supports potentially different contributions between synaptic and extrasynaptic NMDARs to the inhibitory neuronal functions. Nonetheless, various subtypes of inhibitory neurons have been well documented, including those containing PV, somatostatin, vasoactive intestinal peptide, calretinin, or other markers (Kepecs and Fishell, 2014). This heterogeneity of the inhibitory neurons may account for the high variability in responses we observed in Figure 4 and synaptic NMDAR-EPSC responses to EU1794-4 application in Figure 5D.
With extracellular glutamate level playing a major role in their activation, this mode of modulation of extrasynaptic NMDARs is distinct from the activity/input-dependent modulation that typically occurs during synaptic plasticity. The modulation of intrinsic excitability by extrasynaptic NMDARs is influenced by the level of extracellular/ambient glutamate, and/or the density and/or composition of extrasynaptic NMDARs. Significant alterations in the extracellular/ambient glutamate levels occur during development and under pathologic conditions, including epilepsy, stroke, and Alzheimer's disease (Herman and Jahr, 2007; Moussawi et al., 2011; Soni et al., 2014; Hanson et al., 2019). The density/activity of extrasynaptic NMDARs has been shown to be altered in Alzheimer's disease, Huntington diseases, and stroke (Parsons and Raymond, 2014; Pallas-Bazarra et al., 2019). In theory, changes in the subunit composition of extrasynaptic NMDARs will affect the activation of these receptors, such as switching from GluN2C/2D-NMADRs to GluN2A/2B-NMDARs. How the above factors may interact to influence the activation of extrasynaptic NMDARs to affect the intrinsic excitability of inhibitory neurons and brain functions requires further exploration.
Pathologic contribution of extrasynaptic NMDARs in schizophrenia
Our finding of modulation of intrinsic excitability by extrasynaptic NMDAR may also have important implications for the hypofunction of NMDARs in schizophrenia. Evidence from both human postmortem and animal models indicates that reduced activity/expression of NMDARs in inhibitory neurons (especially PV neurons) plays key roles in the pathogenesis and/or pathology of schizophrenia (Lewis et al., 2005; Moreau and Kullmann, 2013; Cohen et al., 2015; Nakazawa et al., 2017). Because of the low presence of synaptic NMDARs on certain subtypes of adult inhibitory neurons (especially PV neurons) (Goldberg et al., 2003; Wang and Gao, 2009; Gonzalez-Burgos and Lewis, 2012; Gonzalez-Burgos et al., 2015), it is not apparent how this hypofunction might occur or cause pathology (Moreau and Kullmann, 2013). Current findings suggest a potential solution to puzzle: reduced presence/activity of extrasynaptic NMDARs mediates this hypofunction in inhibitory neurons (especially PV neurons), leading to reduced neuronal excitability and imbalanced excitation/inhibition in the brain, which is generally assumed to be critical for the pathogenesis/pathology in schizophrenia. An implication of this model is that alterations and consequences of reduced NMDAR function occur in an input-nonspecific manner and hence may affect a wider range of functions than those associated with selective inputs onto inhibitory neurons (Garcia-Munoz et al., 2015).
Potential mechanisms underlying the modulation of intrinsic excitability by NMDARs
We here showed similar impacts of NMDAR modulators on three measurements of intrinsic excitability (spontaneous spiking, depolarization-induced spiking, and E-S coupling) in inhibitory neurons. As shown in Figure 2H, the input resistance is not altered by M-8324 application. This lack of change might be accounted for by a few potential possibilities: (1) the experimental condition used. To mimic physiological conditions, normal ACSF and at/near RMP were used under which may be difficult to detect subtle changes in input resistance. (2) An amplification cascade between NMDAR activation and alteration in intrinsic excitability may exist, which means that subtle changes in NMDAR activation can be amplified and result in large change in neuronal excitability.
Previous studies in excitatory neurons have demonstrated an interaction between Kv4.2 channels and NMDARs. Kv4.2 channels are present at extrasynaptic sites in excitatory neurons (Shibasaki et al., 2004; Kaufmann et al., 2013), and they appear to interact with both synaptic and extrasynaptic NMDARs. Activation of extrasynaptic NMDARs results in the reduction/inhibition of Kv4.2 channels and increases neuronal excitability, which is blocked by the NMDAR antagonist (Lei et al., 2008, 2010; Mulholland and Chandler, 2010). This signaling process is mediated by protein kinase A, protein kinase C, extracellular signal-regulated kinase, and CaMKII (Adams et al., 2000; Schrader et al., 2006; Naskar and Stern, 2014; Kline, 2017). In addition, GSK3β has been shown to directly interact with Kv4.2 channels via phosphorylation of Ser-616 site of Kv4.2 channels in medium spiny neurons of NAc (Aceto et al., 2020). Thus, modulation of IA/Kv4.2 channels may mediate the impacts of NMDARs on intrinsic excitability in inhibitory neurons, which is worthy of further examination. Other mechanisms mediating NMDAR modulation of excitability might also exist, such as via NMDAR-dependent regulation of hyperpolarization-activated cyclic nucleotide-gated channels and Akt/mTOR signaling (Pirbhoy et al., 2016; Hou and Zhang, 2017). In addition, inhibition/ablation of extrasynaptic NMDARs has been shown to increase the neuronal excitability via repression of Kv2.1 or Kv1.1 channels (Mulholland et al., 2008; Frangeul et al., 2017), suggesting that linking NMDAR activity with K+ channels might be a general mechanism in the modulation of neuronal excitability. Whether this mechanism may differ between excitatory and inhibitory neurons is worthy of further exploration.
NMDAR-PAMs
We have used NMDAR-PAMs to enhance the activation of NMDARs to understand their contributions to the excitability of inhibitory neurons. Compared with NMDAR antagonists, there is no upper limit for the enhancement induced by PAMs; hence, their impact can be revealed as long as some residual activation of NMDARs exists. This strategy is especially useful for in vitro analysis since it is likely that the level of NMDAR activation is low in brain slices compared with in vivo. Although a large number of NMDAR-PAMs have been developed (Geoffroy et al., 2022), for treating diseases with reduced inhibitory neuron activity, NMDAR-PAMs that selectively enhance the activity of inhibitory neurons (e.g., GNE-8324 and M-8324) (Hackos et al., 2016; Yao, 2018; Deng et al., 2020) will likely be more useful by avoiding potential counteraction and excitotoxicity because of enhanced activation of excitatory neurons (Hanson et al., 2019).
In conclusion, our findings provide strong support for NMDARs having important impacts on the second-by-second neuronal functions in inhibitory neurons by modulating the intrinsic excitability bidirectionally. Since excitability influence many critical functions of neurons and brain states, we suggest that NMDARs may play wider and more important roles in inhibitory neurons than we have previously recognized.
Footnotes
This work was supported by Shenzhen Bay Laboratory SZBL2019062801003; Youth Program of the National Natural Science Foundation of China 82004469; Shenzhen-Hong Kong Institute of Brain Science-Shenzhen Fundamental Research Institutions 2019SHIBS0004; and Opening Operation Program of Key Laboratory of Acupuncture and Moxibustion of Traditional Chinese Medicine in Guangdong 2017B030314143. All data generated and codes created during the current study are available from the lead contact upon reasonable request. We thank Jiahui Shi for performing the western blot experiment.
The authors declare no competing financial interests.
References
- Aceto G, Colussi C, Leone L, Fusco S, Rinaudo M, Scala F, Green TA, Laezza F, D'Ascenzo M, Grassi C (2020) Chronic mild stress alters synaptic plasticity in the nucleus accumbens through GSK3beta-dependent modulation of Kv4.2 channels. Proc Natl Acad Sci USA 117:8143–8153. 10.1073/pnas.1917423117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JP, Anderson AE, Varga AW, Dineley KT, Cook RG, Pfaffinger PJ, Sweatt JD (2000) The A-type potassium channel Kv4.2 is a substrate for the mitogen-activated protein kinase ERK. J Neurochem 75:2277–2287. 10.1046/j.1471-4159.2000.0752277.x [DOI] [PubMed] [Google Scholar]
- Akgul G, McBain CJ (2016) Diverse roles for ionotropic glutamate receptors on inhibitory interneurons in developing and adult brain. J Physiol 594:5471–5490. 10.1113/JP271764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannon NM, Chistiakova M, Volgushev M (2020) Synaptic plasticity in cortical inhibitory neurons: what mechanisms may help to balance synaptic weight changes? Front Cell Neurosci 14:204. 10.3389/fncel.2020.00204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck H, Yaari Y (2008) Plasticity of intrinsic neuronal properties in CNS disorders. Nat Rev Neurosci 9:357–369. 10.1038/nrn2371 [DOI] [PubMed] [Google Scholar]
- Cathala L, Brickley S, Cull-Candy S, Farrant M (2003) Maturation of EPSCs and intrinsic membrane properties enhances precision at a cerebellar synapse. J Neurosci 23:6074–6085. 10.1523/JNEUROSCI.23-14-06074.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda S, Hale WD, Zhang B, Wernig M, Sudhof TC (2017) Unique versus redundant functions of neuroligin genes in shaping excitatory and inhibitory synapse properties. J Neurosci 37:6816–6836. 10.1523/JNEUROSCI.0125-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry FA, Lehre KP, van Lookeren Campagne M, Ottersen OP, Danbolt NC, Storm-Mathisen J (1995) Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 15:711–720. 10.1016/0896-6273(95)90158-2 [DOI] [PubMed] [Google Scholar]
- Cohen SM, Tsien RW, Goff DC, Halassa MM (2015) The impact of NMDA receptor hypofunction on GABAergic neurons in the pathophysiology of schizophrenia. Schizophr Res 167:98–107. 10.1016/j.schres.2014.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daoudal G, Hanada Y, Debanne D (2002) Bidirectional plasticity of excitatory postsynaptic potential (EPSP)-spike coupling in CA1 hippocampal pyramidal neurons. Proc Natl Acad Sci USA 99:14512–14517. 10.1073/pnas.222546399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng D, Masri S, Yao L, Ma X, Cao X, Yang S, Bao S, Zhou Q (2020) Increasing endogenous activity of NMDARs on GABAergic neurons increases inhibition, alters sensory processing and prevents noise-induced tinnitus. Sci Rep 10:11969. 10.1038/s41598-020-68652-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogaca MV, Duman RS (2019) Cortical GABAergic dysfunction in stress and depression: new insights for therapeutic interventions. Front Cell Neurosci 13:87. 10.3389/fncel.2019.00087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangeul L, Kehayas V, Sanchez-Mut JV, Fievre S, Krishna KK, Pouchelon G, Telley L, Bellone C, Holtmaat A, Graff J, Macklis JD, Jabaudon D (2017) Input-dependent regulation of excitability controls dendritic maturation in somatosensory thalamocortical neurons. Nat Commun 8:2015. 10.1038/s41467-017-02172-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Munoz M, Lopez-Huerta VG, Carrillo-Reid L, Arbuthnott GW (2015) Extrasynaptic glutamate NMDA receptors: key players in striatal function. Neuropharmacology 89:54–63. 10.1016/j.neuropharm.2014.09.013 [DOI] [PubMed] [Google Scholar]
- Garst-Orozco J, Malik R, Lanz TA, Weber ML, Xi H, Arion D, Enwright JF 3rd, Lewis DA, O'Donnell P, Sohal VS, Buhl DL (2020) GluN2D-mediated excitatory drive onto medial prefrontal cortical PV+ fast-spiking inhibitory interneurons. PLoS One 15:e0233895. 10.1371/journal.pone.0233895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Chen W, Axerio-Cilies P, Wang YT (2020) NMDARs in cell survival and death: implications in stroke pathogenesis and treatment. Trends Mol Med 26:533–551. 10.1016/j.molmed.2020.03.001 [DOI] [PubMed] [Google Scholar]
- Geoffroy C, Paoletti P, Mony L (2022) Positive allosteric modulation of NMDA receptors: mechanisms, physiological impact and therapeutic potential. J Physiol 600.2:233–259. [DOI] [PubMed] [Google Scholar]
- Goldberg JH, Yuste R, Tamas G (2003) Ca2+ imaging of mouse neocortical interneurone dendrites: contribution of Ca2+-permeable AMPA and NMDA receptors to subthreshold Ca2+ dynamics. J Physiol 551:67–78. 10.1113/jphysiol.2003.042598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Lewis DA (2012) NMDA receptor hypofunction, parvalbumin-positive neurons, and cortical gamma oscillations in schizophrenia. Schizophr Bull 38:950–957. 10.1093/schbul/sbs010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Cho RY, Lewis DA (2015) Alterations in cortical network oscillations and parvalbumin neurons in schizophrenia. Biol Psychiatry 77:1031–1040. 10.1016/j.biopsych.2015.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graupner M, Reyes AD (2013) Synaptic input correlations leading to membrane potential decorrelation of spontaneous activity in cortex. J Neurosci 33:15075–15085. 10.1523/JNEUROSCI.0347-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackos DH, Lupardus PJ, Grand T, Chen Y, Wang TM, Reynen P, Gustafson A, Wallweber HJ, Volgraf M, Sellers BD, Schwarz JB, Paoletti P, Sheng M, Zhou Q, Hanson JE (2016) Positive allosteric modulators of GluN2A-containing NMDARs with distinct modes of action and impacts on circuit function. Neuron 89:983–999. 10.1016/j.neuron.2016.01.016 [DOI] [PubMed] [Google Scholar]
- Hansen KB, Ogden KK, Traynelis SF (2012) Subunit-selective allosteric inhibition of glycine binding to NMDA receptors. J Neurosci 32:6197–6208. 10.1523/JNEUROSCI.5757-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson E, Armbruster M, Lau LA, Sommer ME, Klaft ZJ, Swanger SA, Traynelis SF, Moss SJ, Noubary F, Chadchankar J, Dulla CG (2019) Tonic activation of GluN2C/GluN2D-containing NMDA receptors by ambient glutamate facilitates cortical interneuron maturation. J Neurosci 39:3611–3626. 10.1523/JNEUROSCI.1392-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H (2010) Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 11:682–696. 10.1038/nrn2911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G (2006) Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev 86:1009–1031. 10.1152/physrev.00049.2005 [DOI] [PubMed] [Google Scholar]
- Herman MA, Jahr CE (2007) Extracellular glutamate concentration in hippocampal slice. J Neurosci 27:9736–9741. 10.1523/JNEUROSCI.3009-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homayoun H, Moghaddam B (2007) NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci 27:11496–11500. 10.1523/JNEUROSCI.2213-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou G, Zhang ZW (2017) NMDA receptors regulate the development of neuronal intrinsic excitability through cell-autonomous mechanisms. Front Cell Neurosci 11:353. 10.3389/fncel.2017.00353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt DL, Castillo PE (2012) Synaptic plasticity of NMDA receptors: mechanisms and functional implications. Curr Opin Neurobiol 22:496–508. 10.1016/j.conb.2012.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Scanziani M (2011) How inhibition shapes cortical activity. Neuron 72:231–243. 10.1016/j.neuron.2011.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson ME, Homayoun H, Moghaddam B (2004) NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc Natl Acad Sci USA 101:8467–8472. 10.1073/pnas.0308455101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann WA, Matsui K, Jeromin A, Nerbonne JM, Ferraguti F (2013) Kv4.2 potassium channels segregate to extrasynaptic domains and influence intrasynaptic NMDA receptor NR2B subunit expression. Brain Struct Funct 218:1115–1132. 10.1007/s00429-012-0450-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepecs A, Fishell G (2014) Interneuron cell types are fit to function. Nature 505:318–326. 10.1038/nature12983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD (2017) Tuning excitability of the hypothalamus via glutamate and potassium channel coupling. J Physiol 595:4583–4584. 10.1113/JP274446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM, Asztely F (1998) Extrasynaptic glutamate spillover in the hippocampus: evidence and implications. Trends Neurosci 21:8–14. 10.1016/s0166-2236(97)01150-8 [DOI] [PubMed] [Google Scholar]
- Lai TW, Zhang S, Wang YT (2014) Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol 115:157–188. 10.1016/j.pneurobio.2013.11.006 [DOI] [PubMed] [Google Scholar]
- Le Meur K, Galante M, Angulo MC, Audinat E (2007) Tonic activation of NMDA receptors by ambient glutamate of non-synaptic origin in the rat hippocampus. J Physiol 580:373–383. 10.1113/jphysiol.2006.123570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Z, Deng P, Xu ZC (2008) Regulation of Kv4.2 channels by glutamate in cultured hippocampal neurons. J Neurochem 106:182–192. 10.1111/j.1471-4159.2008.05356.x [DOI] [PubMed] [Google Scholar]
- Lei Z, Deng P, Li Y, Xu ZC (2010) Downregulation of Kv4.2 channels mediated by NR2B-containing NMDA receptors in cultured hippocampal neurons. Neuroscience 165:350–362. 10.1016/j.neuroscience.2009.10.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW (2005) Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci 6:312–324. 10.1038/nrn1648 [DOI] [PubMed] [Google Scholar]
- Li H, Liang F, Zhong W, Yan L, Mesik L, Xiao Z, Tao HW, Zhang LI (2019) Synaptic mechanisms for bandwidth tuning in awake mouse primary auditory cortex. Cereb Cortex 29:2998–3009. 10.1093/cercor/bhy165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wang J, Liu G, Xu J, Huang W, Song C, Wang D, Tao HW, Zhang LI, Liang F (2021) Phasic Off responses of auditory cortex underlie perception of sound duration. Cell Rep 35:109003. 10.1016/j.celrep.2021.109003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F, Li H, Chou XL, Zhou M, Zhang NK, Xiao Z, Zhang KK, Tao HW, Zhang LI (2019) Sparse representation in awake auditory cortex: cell-type dependence, synaptic mechanisms, developmental emergence, and modulation. Cereb Cortex 29:3796–3812. 10.1093/cercor/bhy260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccaferri G, Dingledine R (2002) Control of feedforward dendritic inhibition by NMDA receptor-dependent spike timing in hippocampal interneurons. J Neurosci 22:5462–5472. 10.1523/JNEUROSCI.22-13-05462.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik R, Chattarji S (2012) Enhanced intrinsic excitability and EPSP-spike coupling accompany enriched environment-induced facilitation of LTP in hippocampal CA1 pyramidal neurons. J Neurophysiol 107:1366–1378. 10.1152/jn.01009.2011 [DOI] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C (2004) Interneurons of the neocortical inhibitory system. Nat Rev Neurosci 5:793–807. 10.1038/nrn1519 [DOI] [PubMed] [Google Scholar]
- Miller OH, Yang L, Wang CC, Hargroder E, Zhang Y, Delpire E, Hall BJ (2014) GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. Elife 3:e03581. 10.7554/eLife.03581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller OH, Moran JT, Hall BJ (2016) Two cellular hypotheses explaining the initiation of ketamine's antidepressant actions: direct inhibition and disinhibition. Neuropharmacology 100:17–26. 10.1016/j.neuropharm.2015.07.028 [DOI] [PubMed] [Google Scholar]
- Monteggia LM, Gideons E, Kavalali ET (2013) The role of eukaryotic elongation factor 2 kinase in rapid antidepressant action of ketamine. Biol Psychiatry 73:1199–1203. 10.1016/j.biopsych.2012.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau AW, Kullmann DM (2013) NMDA receptor-dependent function and plasticity in inhibitory circuits. Neuropharmacology 74:23–31. 10.1016/j.neuropharm.2013.03.004 [DOI] [PubMed] [Google Scholar]
- Moussawi K, Riegel A, Nair S, Kalivas PW (2011) Extracellular glutamate: functional compartments operate in different concentration ranges. Front Syst Neurosci 5:94. 10.3389/fnsys.2011.00094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland PJ, Chandler LJ (2010) Inhibition of glutamate transporters couples to Kv4.2 dephosphorylation through activation of extrasynaptic NMDA receptors. Neuroscience 165:130–137. 10.1016/j.neuroscience.2009.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland PJ, Carpenter-Hyland EP, Hearing MC, Becker HC, Woodward JJ, Chandler LJ (2008) Glutamate transporters regulate extrasynaptic NMDA receptor modulation of Kv2.1 potassium channels. J Neurosci 28:8801–8809. 10.1523/JNEUROSCI.2405-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullasseril P, Hansen KB, Vance KM, Ogden KK, Yuan H, Kurtkaya NL, Santangelo R, Orr AG, Le P, Vellano KM, Liotta DC, Traynelis SF (2010) A subunit-selective potentiator of NR2C- and NR2D-containing NMDA receptors. Nat Commun 1:90. 10.1038/ncomms1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa K, Jeevakumar V, Nakao K (2017) Spatial and temporal boundaries of NMDA receptor hypofunction leading to schizophrenia. NPJ Schizophr 3:7. 10.1038/s41537-016-0003-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naskar K, Stern JE (2014) A functional coupling between extrasynaptic NMDA receptors and A-type K+ channels under astrocyte control regulates hypothalamic neurosecretory neuronal activity. J Physiol 592:2813–2827. 10.1113/jphysiol.2014.270793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Piet R, Poulain DA, Theodosis DT (2004) Glial modulation of synaptic transmission: insights from the supraoptic nucleus of the hypothalamus. Glia 47:258–267. 10.1002/glia.20032 [DOI] [PubMed] [Google Scholar]
- Pagadala P, Park CK, Bang S, Xu ZZ, Xie RG, Liu T, Han BX, Tracey WD Jr, Wang F, Ji RR (2013) Loss of NR1 subunit of NMDARs in primary sensory neurons leads to hyperexcitability and pain hypersensitivity: involvement of Ca(2+)-activated small conductance potassium channels. J Neurosci 33:13425–13430. 10.1523/JNEUROSCI.0454-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal B (2018) Involvement of extrasynaptic glutamate in physiological and pathophysiological changes of neuronal excitability. Cell Mol Life Sci 75:2917–2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallas-Bazarra N, Draffin J, Cuadros R, Esteban JA, Avila J (2019) Tau is required for the function of extrasynaptic NMDA receptors. Sci Rep 9:9116. 10.1038/s41598-019-45547-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q (2013) NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 14:383–400. 10.1038/nrn3504 [DOI] [PubMed] [Google Scholar]
- Papouin T, Ladepeche L, Ruel J, Sacchi S, Labasque M, Hanini M, Groc L, Pollegioni L, Mothet JP, Oliet SH (2012) Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 150:633–646. 10.1016/j.cell.2012.06.029 [DOI] [PubMed] [Google Scholar]
- Parsons MP, Raymond LA (2014) Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 82:279–293. 10.1016/j.neuron.2014.03.030 [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Chittajallu R, Craig MT, Tricoire L, Wester JC, McBain CJ (2017) Hippocampal GABAergic inhibitory interneurons. Physiol Rev 97:1619–1747. 10.1152/physrev.00007.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perszyk R, Katzman BM, Kusumoto H, Kell SA, Epplin MP, Tahirovic YA, Moore RL, Menaldino D, Burger P, Liotta DC, Traynelis SF (2018) An NMDAR positive and negative allosteric modulator series share a binding site and are interconverted by methyl groups. Elife 7:e34711. 10.7554/eLife.34711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirbhoy PS, Farris S, Steward O (2016) Synaptic activation of ribosomal protein S6 phosphorylation occurs locally in activated dendritic domains. Learn Mem 23:255–269. 10.1101/lm.041947.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povysheva NV, Johnson JW (2012) Tonic NMDA receptor-mediated current in prefrontal cortical pyramidal cells and fast-spiking interneurons. J Neurophysiol 107:2232–2243. 10.1152/jn.01017.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riebe I, Seth H, Culley G, Dosa Z, Radi S, Strand K, Frojd V, Hanse E (2016) Tonically active NMDA receptors: a signalling mechanism critical for interneuronal excitability in the CA1 stratum radiatum. Eur J Neurosci 43:169–178. 10.1111/ejn.13128 [DOI] [PubMed] [Google Scholar]
- Sah P, Hestrin S, Nicoll RA (1989) Tonic activation of NMDA receptors by ambient glutamate enhances excitability of neurons. Science 246:815–818. 10.1126/science.2573153 [DOI] [PubMed] [Google Scholar]
- Schrader LA, Birnbaum SG, Nadin BM, Ren Y, Bui D, Anderson AE, Sweatt JD (2006) ERK/MAPK regulates the Kv4.2 potassium channel by direct phosphorylation of the pore-forming subunit. Am J Physiol Cell Physiol 290:C852–C861. 10.1152/ajpcell.00358.2005 [DOI] [PubMed] [Google Scholar]
- Shibasaki K, Nakahira K, Trimmer JS, Shibata R, Akita M, Watanabe S, Ikenaka K (2004) Mossy fibre contact triggers the targeting of Kv4.2 potassium channels to dendrites and synapses in developing cerebellar granule neurons. J Neurochem 89:897–907. 10.1111/j.1471-4159.2004.02368.x [DOI] [PubMed] [Google Scholar]
- Soni N, Reddy BV, Kumar P (2014) GLT-1 transporter: an effective pharmacological target for various neurological disorders. Pharmacol Biochem Behav 127:70–81. 10.1016/j.pbb.2014.10.001 [DOI] [PubMed] [Google Scholar]
- Stern EA, Kincaid AE, Wilson CJ (1997) Spontaneous subthreshold membrane potential fluctuations and action potential variability of rat corticostriatal and striatal neurons in vivo. J Neurophysiol 77:1697–1715. 10.1152/jn.1997.77.4.1697 [DOI] [PubMed] [Google Scholar]
- Suzuki K, Monteggia LM (2020) The role of eEF2 kinase in the rapid antidepressant actions of ketamine. Adv Pharmacol 89:79–99. 10.1016/bs.apha.2020.04.005 [DOI] [PubMed] [Google Scholar]
- Swanger SA, Vance KM, Pare JF, Sotty F, Fog K, Smith Y, Traynelis SF (2015) NMDA receptors containing the GluN2D subunit control neuronal function in the subthalamic nucleus. J Neurosci 35:15971–15983. 10.1523/JNEUROSCI.1702-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62:405–496. 10.1124/pr.109.002451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G (2011) Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu Rev Neurosci 34:89–103. 10.1146/annurev-neuro-060909-153238 [DOI] [PubMed] [Google Scholar]
- von Engelhardt J, Bocklisch C, Tonges L, Herb A, Mishina M, Monyer H (2015) GluN2D-containing NMDA receptors-mediate synaptic currents in hippocampal interneurons and pyramidal cells in juvenile mice. Front Cell Neurosci 9:95. 10.3389/fncel.2015.00095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HX, Gao WJ (2009) Cell type-specific development of NMDA receptors in the interneurons of rat prefrontal cortex. Neuropsychopharmacology 34:2028–2040. 10.1038/npp.2009.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YW, Grebenyuk S, McHugh TJ, Rusakov DA, Semyanov A (2012) Backpropagating action potentials enable detection of extrasynaptic glutamate by NMDA receptors. Cell Rep 1:495–505. 10.1016/j.celrep.2012.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue JG, Masuoka T, Gong XD, Chen KS, Yanagawa Y, Law SK, Konishi S (2011) NMDA receptor activation enhances inhibitory GABAergic transmission onto hippocampal pyramidal neurons via presynaptic and postsynaptic mechanisms. J Neurophysiol 105:2897–2906. 10.1152/jn.00287.2010 [DOI] [PubMed] [Google Scholar]
- Yao L, Grand T, Hanson J, Paoletti P, Zhou Q (2018) Higher ambient synaptic glutamate at inhibitory versus excitatory neurons differentially impacts NMDA receptor activity. Nat Commun 9:4000. 10.1038/s41467-018-06512-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L, Wang Z, Deng D, Yan R, Ju J, Zhou Q (2019) The impact of D-cycloserine and sarcosine on in vivo frontal neural activity in a schizophrenia-like model. BMC Psychiatry 19:314. 10.1186/s12888-019-2306-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanos P, Gould TD (2018) Mechanisms of ketamine action as an antidepressant. Mol Psychiatry 23:801–811. 10.1038/mp.2017.255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Linden DJ (2003) The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat Rev Neurosci 4:885–900. 10.1038/nrn1248 [DOI] [PubMed] [Google Scholar]
- Zhou Q, Sheng M (2013) NMDA receptors in nervous system diseases. Neuropharmacology 74:69–75. 10.1016/j.neuropharm.2013.03.030 [DOI] [PubMed] [Google Scholar]