

Cells respond to their internal and external environment by changing their gene expression. Clones of cells can arise that persistently display such changes even after the initial trigger is removed. Most instances of heritable changes in cell state do not involve changes in DNA sequence and are thus termed ‘epigenetic’[1]. In humans, epigenetic mechanisms specify cell type, silence repeated sequences, program parent-of-origin expression of monoallelic genes, and go awry in certain diseases such as cancer. Many molecular mechanisms can underpin such processes, ranging from positive-feedback loops mediated by transcription factors, DNA methylation, and histone modification[2–4]. Most work in this area has focused on mammalian systems. However, many remarkable mechanisms have been unearthed elsewhere on the evolutionary tree. Evolutionary innovations involving fungal epigenetics are the focus of this piece. These findings, some of which seem unbelievable, include chromatin prions, centromeric nucleosome erasure leading to kinetochore inactivation, and the Darwinian evolution of DNA methylation over million-year timescales[5–8].
There are numerous reasons to study processes in fungi. One is experimental tractability, which enables facile testing of hypotheses. The brewer’s yeast Saccharomyces cerevisiae has been pivotal to much of our understanding of the eukaryotic cell. Nonetheless, it is increasingly appreciated that not all highly conserved aspects of eukaryotic cell behavior can be modelled in this one species, in part due to ancestral evolutionary loss events. Mechanistic studies in a relatively small number of other fungi have yielded insights and true surprises. A celebrated example is the finding that mutations in the RNAi machinery of Schizosaccharomyces pombe impacts repressive histone H3 lysine 9 methylation[9]. However, the fungal kingdom is vast with perhaps over a million species[10]. Thus, research into this ancient kingdom has barely scratched the surface of its secrets. While the genomes of fungi can now be easily obtained through high throughput DNA sequencing, a mechanistic understanding of their biology still requires experimental investigations, which in turn requires communities of scientists willing to do the painstaking work of tool development and discovery.
Introduction to the players
S. cerevisiae and S. pombe are members of the Ascomycota phylum, which harbors a major portion of the fungal kingdom[11]. Many species in this phylum are not yeast, defined as single-celled fungi. Rather, the majority are either dimorphic fungi (that alternate between yeast and filamentous mycelial forms) or strictly filamentous[11]. The filamentous fungi Aspergillus nidulans and Neurospora crassa have served as dominant models because of their long history, strong research communities, and the development of powerful tools and resources. Neurospora in particular has been used to investigate DNA methylation and DNA methylation-coupled processes[12]. More recently, it has served as a model for Polycomb silencing[13]. Another ascomycete, Candida albicans, is the most common fungal pathogen of humans as well as a normal commensal[14]. As will be detailed below, it has evolved a chromatin-programmed system for regulated chromosome destabilization.
The other major fungal phylum is Basidiomycota. Ascomycota and Basidiomycota together form a subphylum called the Dikarya[11]. Basidiomycota include mushrooms and many other species, including several plant pathogens[11] and the human pathogens, Cryptococcus neoformans, Cryptococcus deneoformans and Cryptococcus gattii[15, 16]. Like S. cerevisiae, the human pathogens grow as single-cell budding yeasts, maintain stable haploid genomes, and display a complete sexual cycle. They can also be transformed with DNA and display efficient homologous recombination[17]. In particular, Cryptococcus harbors all of the chromatin gene silencing systems identified in mammalian cells, including a Polycomb system[18], an H3K9 methylation system[18], and DNA methylation[19].
A chromatin prion enables bet hedging.
First identified as the infectious agent of Scrapie, a neurodegenerative disease of sheep, prions (proteinaceous infectious agents) are protein-only infective agents[20]. Prions are now known to be widespread in biology and have been studied extensively in S. cerevisiae, where such non-chromosomally heritable states of protein activity are widespread[21]. While prions display epigenetic inheritance, they do so by multiple mechanisms, including the formation of protein amyloid aggregates[22]. Employing the well-developed toolbox of S. cerevisiae, Jarosz and colleagues took advantage of a property of many prions to identify new candidates in a systemic fashion: Many prions are induced by overexpression of the monomer form of the protein that forms the prion. In some cases, this triggers the assembly of protein aggregates (for example, amyloids) that are the prion form of the protein. This form is self-propagating, can persist in the absence of the initial overexpression triggers, is heritable both mitotically and meiotically, and is often inactive. Thus, transient overexpression of protein can trigger an epigenetic change mediated by prion formation.
These investigators systematically transiently overexpressed nearly every yeast protein and then used a series of stress conditions to identify cases in which the transient overexpression altered cellular fitness. This bold approach yielded dozens of new candidate prions. Subsequently, the authors followed up on a single hit from the screen (Figure 1), a prion dubbed [ESI+]. [ESI+] formation is triggered by transient overexpression of SNT1, which encodes a subunit of the Set3 histone deacetylase complex, a yeast ortholog of the human NCOR-SMRT deacetylase complex[8]. Transient overexpression of SNT1 triggers resistance to zinc ion stress that lasts for hundreds of generations. As with many yeast prions, overexpression of a protein chaperone, Hsp90, can cure cells of this phenotype [8].
Figure 1. Key Figure. A Stress-Induced Chromatin Prion in Saccharomyces cerevisiae.
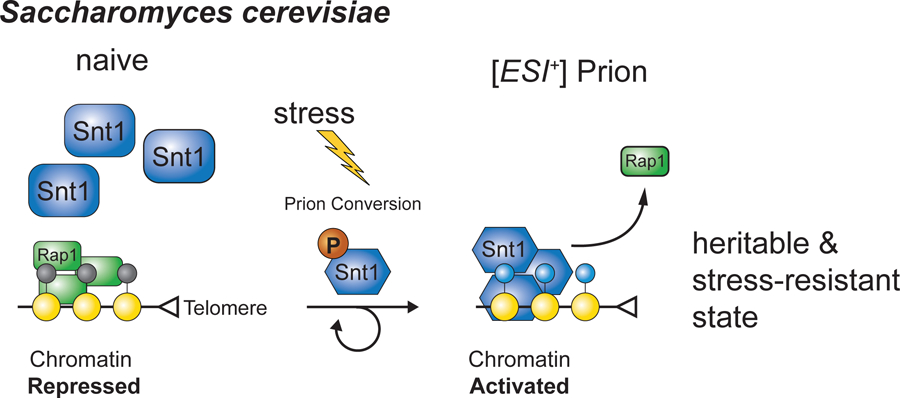
Depicted is a model for how repressed subtelomeric heterochromatin is activated via the formation of a prion involving a subunit of the Set3 histone deacetylase complex called Snt1.
Following mating, the [ESI+] prion state of a haploid parent can be transmitted to all four meiotic progeny, even when the second parent lacks the prion[8]. In addition, using an experimental trick called cytoduction in which cytoplasm and organelles but not nuclei are introduced from one cell to another, it was shown that the [ESI+] state can be transferred from cell to cell in this manner[8]. To definitively demonstrate protein-only inheritance, Jarosz and colleagues introduced an aggregated form of the protein (produced by recombinant expression in E. coli followed by purification) into yeast lacking the prion phenotype and found that it was sufficient to trigger the [ESI+] phenotype in a significant fraction of transformants[8]. Thus, transient SNT1 overexpression can trigger prion activity that can mediate zinc ion tolerance.
Further work demonstrated that SNT1 harbors several predicted disordered regions, which may help to promote the formation of the prion state. In addition, the protein is phosphorylated on multiples sites and phosphomimic mutations promote prion formation[8]. Although not proposed by the authors, these properties raise the possibility that the prion form of SNT1 is a coacervate, that is, a droplet formed by liquid-liquid phase separation[8]. Genome-wide transcript profiling revealed that the prion form of SNT1 results in increased expression of subtelomeric genes. Strikingly, binding of the Set3 complex increases at subtelomeric domains when SNT1 is in the prion form [8]. Moreover, subtelomeric binding of Set3 anticorrelates with binding of a nucleator of subtelomeric silencing, the sequence-specific DNA-binding protein Rap1. Thus, a prion form of a histone deacetylase complex associates with chromatin and activates subtelomeric gene expression by antagonizing gene silencing [8].
Much remains to be learned here: How precisely does the prion form lead to increased gene expression? How does the prion form interfere with DNA binding by Rap1? Is the deacetylase activity of the complex important? Are histones the relevant target for this activity? Do other chromatin-modifying complexes act by forming chromatin-associated prions? Is phase-separation involved in this mechanism? The chromatin-associated prion identified by Jarosz and colleagues seems may portend a new principle and mechanism in chromatin biology.
Centromeric nucleosome erasure and a newly evolved defective histone H2A triggers adaptive aneuploidy.
Candida albicans is part of a group of human pathogenic fungi that have several shared characteristics including a nonuniversal genetic code and a sexual cycle that does not involve meiosis [23]. C. albicans cells are diploid. Like diploid S. cerevisiae, they do not mate because they are heterozygous at the mating type locus, displaying a MATa/MATα genotype[24]. However, loss of one copy of the chromosome harboring the mating type locus, Chromosome V, results in C. albicans cells that are sexually analogous to haploid a and α cells of S. cerevisiae[24]. These are capable of differentiating into mating-competent cells called opaque cells[24]. Mating between MATa and MATα C. albicans cells results in tetraploid cells [23]. However, this is where things get weird: rather undergoing meiosis, under certain conditions, C. albicans undergoes massive, apparently random chromosome loss, which results in massive cell death and ultimately the selection of a new diploid cell population [23]. The ability of C. albicans cells to lose chromosomes easily under particular conditions suggested that it harbored a mechanism to destabilize its chromosome segregation machinery.
Likewise, multiple studies have shown that C. albicans can develop resistance to an antifungal drug by evolving an aneuploidy of a chromosome encoding the target of the drug (the drug being fluconazole and the target being the ERG11 gene, encoding lanosterol 14-demethylase). Studies of this process led to the model that fluconazole stress itself induces chromosome instability, suggesting a Lamarckian-like mode of evolution [25, 26]. However, the chromosome-destabilizing mechanisms underlying such an unconventional mechanism remained unclear.
Noble and coworkers identified two underlying mechanisms that appear to act in parallel to enable induced chromosome destabilization (Figure 2). In the process of characterizing the histone gene of C. albicans, these authors noticed that homozygous deletions of either of two distinct histone H2A genes yielded distinct colony phenotypes[5]. They further noticed that one of the H2A genes displayed an amino acid substitution on a universally conserved phosphorylation site for the Bub1 mitotic kinase[5]. Phylogenetic analysis revealed that this change occurred in a shared ancestor of C. albicans and two of the related Candida species that share a noncanonical genetic code and a nonmeiotic sexual cycle [5]. Both the noncanonical and canonical H2A genes are expressed and, as expected, replacement of the canonical with the noncanonical coding sequences result in decreased chromosome stability while replacement of the noncanonical H2A coding sequence with the canonical resulted in increased chromosome stability[5]. Thus, it is clear that C. albicans has evolved to have nonoptimal chromosome segregation, a striking finding. The impact of H2A was demonstrated to be relevant to drug resistance as cells harboring only the noncanonical H2A displayed increased rates of fluconazole tolerance as do mutations in the phosphorylation site in the canonical H2A as well as knockouts of Bub1 or its downstream effector Shugoshin[5]. Likewise, efficient chromosome loss after mating in tetraploids also required the noncanonical H2A[5].
Figure 2. Adaptive Induction of Mitotic Chromatin Destabilization in Candida albicans.
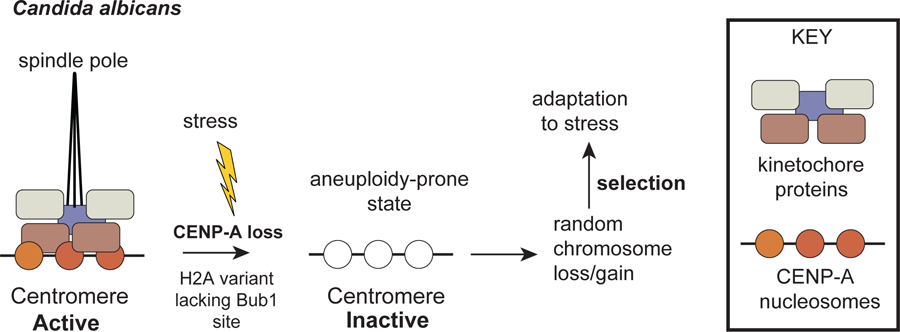
Depicted is the stress-induced kinetochore inactivation enabled by loss of CENP-A (centromeric histone H3 variant) in the context of a newly evolved histone H2A variant that lacks the otherwise conserved phosphorylation site for the Bub1 mitotic kinase.
Because the two H2A genes appear to be constitutively expressed, these authors sought chromosome-destabilizing mechanisms that would cooperate with the noncanonical H2A that would respond to conditions thought to trigger chromosome loss[5]. CENP-A is a centromeric histone H3 variant that forms the foundation of the kinetochore in nearly all eukaryotes. Thus, Noble and colleagues examined CENP-A deposition in C. albicans using ChIP-seq[5]. These studies revealed that CENP-A is significantly depleted at the kinetochores of tetraploid cells compared to diploids, which correlates with the reduced chromosome stability of tetraploids [5]. Strikingly, the condition that triggers chromosome loss in tetraploids (“pre-Spo” medium) triggers the near elimination of CENP-A at centromeres[5], similar to the result in diploid cells exposed to fluconazole [5]. As CENP-A is the platform of the eukaryotic kinetochore, its loss provides a plausible explanation for the inducibility of chromosome loss in C. albicans. It is also striking that lineages can evolve mechanisms to eliminate a highly conserved cellular feature to enhance evolvability.
These findings suggest the existence of a signal-regulated mechanism to control centromere activity via CENP-A. What programs the loss of CENP-A? Is the noncanonical H2A required for CENP-A removal? What is the mechanism for CENP-A depletion at centromeres? Other key questions include the following: What is the signal transduction pathway upstream of CENP-A removal? How is CENP-A re-deposited in the correct place after nonstress conditions have returned? Is there natural variation in this process? It is relevant to virulence and commensalism?
Darwinian evolution of DNA methylation over million-year timescales
DNA methylation in mammals and plants is a critical genome defense and regulatory mechanism involving two types of enzymes: de novo enzymes that can methylated unmethylated DNA and maintenance enzymes that prefer hemimethylated DNA[27]. The latter recognize sequences in palindromic contexts (e.g. CG or CHG) which, after DNA replication, harbor methylcytosine on the parental strand and unmethylated cytosine on the newly-synthesized daughter strains[27]. Maintenance methylases restore the original state enabling epigenetic propagation of DNA methylation patterns that were established by the prior action of de novo methylases[28]. This paradigm in widespread in animals and plants. In fungi, the best characterized DNA methylation system occurs in the ascomycete filamentous fungus Neurospora crassa, known colloquially as bread mold. Foundational work by Selker and colleagues demonstrated that all cytosine methylation in Neurospora is mediated by a single enzyme, DIM-2 [29]. DIM-2 acts downstream of a repressive histone modification H3-K9 methylation[30]. Although DIM-2 has not been purified in active form, there does not appear to be epigenetic memory in Neurospora – methylation appears is continuously established[29]. Studies of another ascomycete, Ascobolus immersus, demonstrated that memory systems exist in fungi: in this species, duplicated DNA sequences become methylated after meiosis, in a process called methylation-induced premeiotically (MIP)[31].
Cryptococcus neoformans is opportunistic meningitis pathogen responsible for ~200,000 deaths annually. We recently investigated cytosine DNA methylation, an epigenetic mechanism involved in genome defense, in this organism[6]. CpG methylation in C. neoformans is mediated by Dnmt5, the only DNMT in C. neoformans. Dnmt5 is a member of a clade of cytosine methyltransferases that harbor a Snf2 ATPase domain in addition to a DNMT domain. They are widespread in the fungal and protist kingdoms. Our analysis of CG methylation in C. neoformans revealed that, as in Neurospora crassa, it acted downstream of H3K9 methylation via two different readers of this chromatin mark: the chromodomain of Dnmt5 itself and the C. neoformans ortholog of HP1/Swi6[6]. Biochemical analysis revealed the purified C. neoformans Dnmt5 to be an exquisitely-specific maintenance type enzyme that only recognizes hemimethylated DNA[6]. In vitro it requires ATP for activity, an unprecedented requirement for a cytosine DNA methylase [6]. In vivo, methylation is not globally restored once the gene for Dnmt5 is removed from cells and then re-introduced. This was shown in three ways: 1) using a regulated promoter, 2) deleting the catalytic domain of DMT5 gene and re-introducing it by homologous recombination and 3) by re-introducing the wild-type gene through a genetic cross [6]. Moreover, it was shown that integration of a fragment of foreign DNA that had been methylated in vitro using the HpaII methylase resulted in maintenance of DNA methylation at HpaII sites without detectable spread to nearby CG sites[6]. This indicates that Dnmt5 acts as a maintenance enzyme in cells. In other words, Dnmt5 requires preexisting methylation to propagate methylation.
If Dnmt5 is the only DNMT in C. neoformans, how was methylation ever established in the first place? Our evolutionary studies revealed that the ancestor of C. neoformans harbored both a gene for Dnmt5 but also a gene for a second Dnmt, DnmtX[6]. Introduction of DnmtX from extant species triggered de novo methylation that could then be maintained by Dnmt5, indicating that DnmtX has de novo activity in vivo[6]. The gene for DnmtX was lost between 50 and 150 million years ago, suggesting the startling conclusion that DNA methylation has been maintained in this species for millions of years without a de novo enzyme (Figure 3).
Figure 3. Epigenetic Evolution of Cryptocccus neoformans.
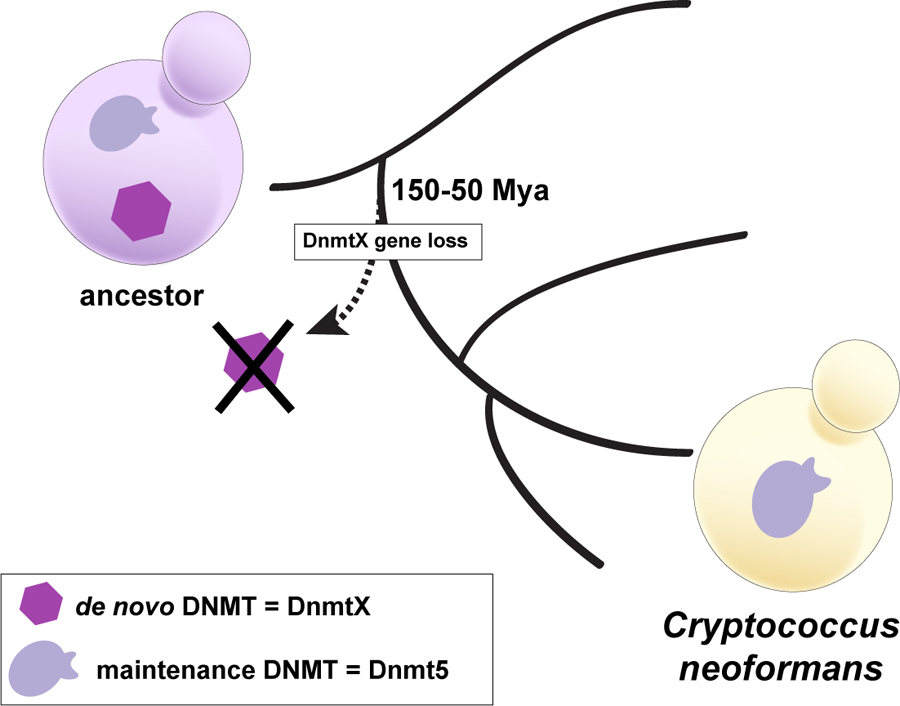
Loss of a gene encoding a de novo DNA cytosine methyltransferase (DNMT) resulting in a long-lived lineage harboring only a maintenance-type DNA cytosine methyltransferase. This lineage gave rise of the human pathogenic yeast, Cryptococcus neoformans. Cytosine methylation patterns have been undergoing Darwinian evolution that is mediated by rare random loss events, rarer random gain events and the action of natural selection on the silencing of transposable elements.
As it seems unlikely that an original pattern imparted by DnmtX could be propagated without change for millions of years, we performed two types of experiments aimed at understanding how methylation might evolve over such long time periods without the agency of a dedicated de novo system. The first set of experiments consisted of experimental evolution experiments in which a culture was propagated for ~140 generations and then the methylation patterns of two colonies were examined and compared to the parental strains using whole-genome bisulfite sequencing (WGBS). This analysis revealed apparently random losses of methylation at a rate of about 10−4/ generation per site[6]. Considerably rarer gain events were observed at a 20-fold lower rate. They also appeared to be random[6]. These very rare gain events may reflect a trace of de novo activity produced by Dnmt5. Because the rate of loss was considerably greater than the rate of gain, the system is not in equilibrium. We concluded from this analysis that selection for methylation levels is required to maintain the steady state[6].
To investigate the evolution of methylation over longer time periods, we analyzed across a phylogeny of C. neoformans isolates that shared a common ancestor ~5 Mya. This required the long-read assembly of eight C. neoformans genomes using Nanopore sequencing. By combining whole genome bisulfite sequencing and methylation information obtained from the Nanopore platform, the methylation landscapes of the eight genomes was obtained[6]. A standard phylogenetic approach to evaluate methylation evolution was not feasible as rearrangement of the methylated sequences precluded multiple sequence alignment. Nonetheless, local pairwise alignment revealed higher sharing of methylation patterns than expected by chance[6]. Moreover, annotation of putative full-length transposons revealed that they displayed a considerably higher fraction of methylated CGs than other centromeric sequences[6].
Taken together these studies indicate that methylation has been maintained for millions of years by a Darwinian process in which random rare losses and rarer gains of methylation occur and are then maintained by natural selection for methylation of transposable elements. This Darwinian mode of methylation evolution indicates that DNA sequence is not the only substrate for evolution.
Numerous questions remain. What are the determinants of accurate maintenance methylation? What determines the exquisite specificity of Dnmt5? Why does Dnmt5 require ATP hydrolysis to function? How do cofactors such as Uhrf1 function with Dnmt5? How is de novo methylation controlled in species that harbor both DnmtX and Dnmt5? Does maintenance methylation also evolve via a Darwinian process in species harboring DnmtX?
Microbial evolution: less constrained?
Multicellular systems may be constrained in ways that are not the case for microbes, particularly single-cell organisms. Thus, it may be that processes that are thought to be highly conserved such as chromatin modifications may not in fact be static in many lineages, enabling innovations that are unanticipated from work in more complex organisms. Devising ways of identifying and investigating such evolutionary surprises is a challenge for the future. A major limitation remains our ability to develop the experimental toolboxes for unstudied organisms.
Supplementary Material
ACKNOWLEDGEMENTS
I thank to Suzanne Noble for discussions and critical reading of the manuscript. I am also grateful to Dan Jarosz (Stanford University) for providing the template for Figure 1 and Sandra Catania for Figure 3. Work in the Madhani lab is supported by grants from the National Institutes of Health. H.D.M. is an Investigator of the Chan-Zuckerberg Biohub.
REFERENCES
- 1.Deans C and Maggert KA (2015) What do you mean, “epigenetic”? Genetics 199 (4), 887–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allis CD and Jenuwein T (2016) The molecular hallmarks of epigenetic control. Nat Rev Genet 17 (8), 487–500. [DOI] [PubMed] [Google Scholar]
- 3.Ferrell JE Jr. and Ha SH (2014) Ultrasensitivity part III: cascades, bistable switches, and oscillators. Trends Biochem Sci 39 (12), 612–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moris N et al. (2016) Transition states and cell fate decisions in epigenetic landscapes. Nat Rev Genet 17 (11), 693–703. [DOI] [PubMed] [Google Scholar]
- 5.Brimacombe CA et al. (2019) A natural histone H2A variant lacking the Bub1 phosphorylation site and regulated depletion of centromeric histone CENP-A foster evolvability in Candida albicans. PLoS Biol 17 (6), e3000331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Catania S et al. (2020) Evolutionary Persistence of DNA Methylation for Millions of Years after Ancient Loss of a De Novo Methyltransferase. Cell 180 (2), 263–277 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dumesic PA et al. (2020) ATP Hydrolysis by the SNF2 Domain of Dnmt5 Is Coupled to Both Specific Recognition and Modification of Hemimethylated DNA. Mol Cell 79 (1), 127–139 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harvey ZH et al. (2020) A Prion Epigenetic Switch Establishes an Active Chromatin State. Cell 180 (5), 928–940 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Volpe TA et al. (2002) Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 297 (5588), 1833–7. [DOI] [PubMed] [Google Scholar]
- 10.Hawksworth DL and Lucking R (2017) Fungal Diversity Revisited: 2.2 to 3.8 Million Species. Microbiol Spectr 5 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spatafora JW et al. (2017) The Fungal Tree of Life: from Molecular Systematics to Genome-Scale Phylogenies. Microbiol Spectr 5 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aramayo R and Selker EU (2013) Neurospora crassa, a model system for epigenetics research. Cold Spring Harb Perspect Biol 5 (10), a017921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ridenour JB et al. (2020) Polycomb Repression without Bristles: Facultative Heterochromatin and Genome Stability in Fungi. Genes (Basel) 11 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noble SM et al. (2017) Candida albicans cell-type switching and functional plasticity in the mammalian host. Nat Rev Microbiol 15 (2), 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun S et al. (2019) The Evolution of Sexual Reproduction and the Mating-Type Locus: Links to Pathogenesis of Cryptococcus Human Pathogenic Fungi. Annu Rev Genet 53, 417–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kozubowski L and Heitman J (2012) Profiling a killer, the development of Cryptococcus neoformans. FEMS Microbiol Rev 36 (1), 78–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chun CD and Madhani HD (2010) Applying genetics and molecular biology to the study of the human pathogen Cryptococcus neoformans. Methods Enzymol 470, 797–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dumesic PA et al. (2015) Product binding enforces the genomic specificity of a yeast polycomb repressive complex. Cell 160 (1–2), 204–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Catania S et al. (2020) Evolutionary Persistence of DNA Methylation for Millions of Years after Ancient Loss of a De Novo Methyltransferase. Cell 180 (4), 816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Colby DW and Prusiner SB (2011) Prions. Cold Spring Harb Perspect Biol 3 (1), a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wickner RB (2016) Yeast and Fungal Prions. Cold Spring Harb Perspect Biol 8 (9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jarosz DF and Khurana V (2017) Specification of Physiologic and Disease States by Distinct Proteins and Protein Conformations. Cell 171 (5), 1001–1014. [DOI] [PubMed] [Google Scholar]
- 23.Bennett RJ (2015) The parasexual lifestyle of Candida albicans. Curr Opin Microbiol 28, 10–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lohse MB and Johnson AD (2010) Temporal anatomy of an epigenetic switch in cell programming: the white-opaque transition of C. albicans. Mol Microbiol 78 (2), 331–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Selmecki A et al. (2006) Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science 313 (5785), 367–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Selmecki AM et al. (2009) Acquisition of aneuploidy provides increased fitness during the evolution of antifungal drug resistance. PLoS Genet 5 (10), e1000705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du J et al. (2015) DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol 16 (9), 519–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng S et al. (2010) Epigenetic reprogramming in plant and animal development. Science 330 (6004), 622–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kouzminova E and Selker EU (2001) dim-2 encodes a DNA methyltransferase responsible for all known cytosine methylation in Neurospora. EMBO J 20 (15), 4309–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tamaru H and Selker EU (2001) A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature 414 (6861), 277–83. [DOI] [PubMed] [Google Scholar]
- 31.Rossignol JL and Faugeron G (1995) MIP: an epigenetic gene silencing process in Ascobolus immersus. Curr Top Microbiol Immunol 197, 179–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.