

SUMMARY
The development of the apical dendrite from the leading process of the bipolar pyramidal neuron might be directed by spatially organized extrinsic cues acting on localized intrinsic determinants. The extracellular cues regulating apical dendrite polarization remain elusive. We show that leading process and apical dendrite development are directed by class III Semaphorins and mediated by a localized cGMP-synthesizing complex. The scaffolding protein Scribble that associates with the cGMP-synthesizing enzyme soluble guanylate cyclase (sGC) also associates with the Semaphorin3A (Sema3A) co-receptor PlexinA3. Deletion or knockdown of PlexinA3 and Sema3A or disruption of PlexinA3-Scribble association prevents Sema3A-mediated cGMP increase and causes defects in apical dendrite development. These manipulations also impair bipolar polarity and leading process establishment. Local cGMP elevation or sGC expression rescues the effects of PlexinA3 knockdown or PlexinA3-Scribble complex disruption. During neuronal polarization, leading process and apical dendrite development are directed by a scaffold that links Semaphorin cue to cGMP increase.
Graphical Abstract
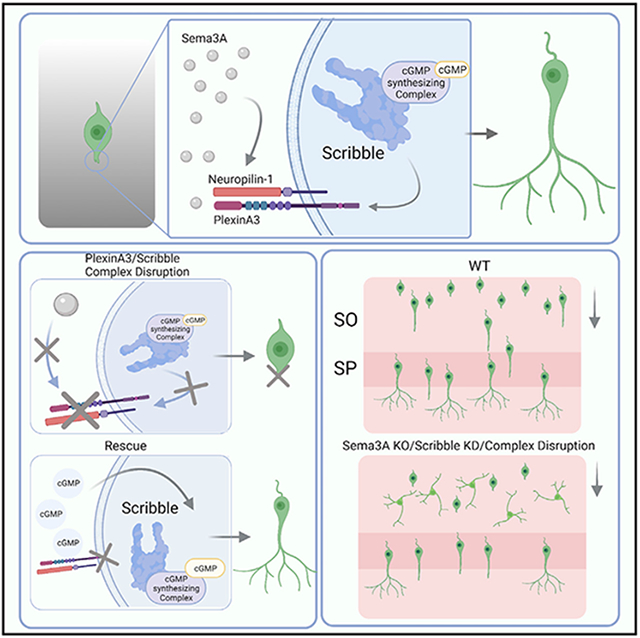
In brief
Szczurkowska et al. show that spatially directed Sema3A may promote development of the leading process and the apical dendrite via the co-receptor PlexinA3 by orchestrating localized cGMP increase on the scaffold protein, Scribble, at the leading edge of developing pyramidal neurons.
INTRODUCTION
Upon completion of mitosis, neocortical and CA1 pyramidal neuron progenitors lose apico-basal polarity of their neuroepithelial ancestors and assume an unpolarized multipolar (multineurite) morphology. The event that marks apical dendrite/axon polarity occurs upon acquisition of a bipolar morphology, with two neurites having a leading and trailing orientation. The apical dendrite develops from the leading process whereas the trailing process becomes the axon. The events leading to polarity establishment remain inconclusive. It is unclear whether axon formation is essential for and absolutely precedes polarity establishment at the multipolar stage. Furthermore, it remains largely unknown whether specific mechanisms are necessary for apical dendrite development.
Our own as well as other findings suggest that directed events determine apical dendrite development during polarization. Axon formation in multipolar neurons precedes polarization in ~60% of pyramidal neurons (Namba et al., 2014). However, in ~30% of the other neurons, leading process and polarity establishment precede trailing process formation (Gartner et al., 2012; Namba et al., 2014). More importantly, knockdown of critical determinants that result in abolishment of axon formation does not prevent leading process polarity or subsequent apical dendrite development (Barnes et al., 2007; Kishi et al., 2005; Shelly et al., 2007; Yi et al., 2010). Precisely organized extracellular cues acting on localized intrinsic mechanisms may thus direct leading process and apical dendrite polarization independently of axon formation. Nevertheless, the underlying mechanisms and the extracellular cues directing apical dendrite development remain largely unknown.
Semaphorin3A (Sema3A) signal is mediated via a complex of the Neuropilin-1 (NP-1) receptor that binds Sema3A and Plexin co-receptors that transmit the intracellular signaling (Cheng et al., 2001; Takahashi et al., 1999; Tran et al., 2009; Yaron et al., 2005). In the developing cortex, Sema3A is expressed in a descending gradient with the highest concentration at the pia (Polleux et al., 2000), and orients axon and apical dendrite guidance. Sema3A is also robustly expressed in the developing hippocampus where it regulates the pathfinding of major axonal projections (Bagri et al., 2003; Chedotal et al., 1998; Giger et al., 2000; Mata et al., 2018; Nakamura et al., 2009; Rohm et al., 2000). Among Plexin co-receptors, PlexinA3 is prominently expressed in the hippocampus throughout embryonic and early postnatal development, including the CA1 (Cheng et al., 2001; Murakami et al., 2001; Tran et al., 2009). Among other Plexin co-receptors, PlexinA4 and PlexinD1 are also expressed in the developing hippocampus (Cheng et al., 2001; Mata et al., 2018; Murakami et al., 2001; Tran et al., 2009). PlexinA3 can partner with either NP-2 or NP-1 (Yaron et al., 2005) and is a critical regulator of class III Semaphorin effects on axon pathfinding and pruning (Bagri et al., 2003; Cheng et al., 2001; Polleux et al., 1998, 2000; Yaron et al., 2005). In the CA1, PlexinA3 preferentially associates with NP-1 to mediate pruning of pyramidal axons downstream of Sema3A (Bagri et al., 2003). Previous studies that examined Sema3A or PlexinA3 deletion did not report effects on axon/dendrite formation or their early morphogenesis (Tran et al., 2009). These studies found defects in late events of dendrite maturation and spine morphogenesis. However, we showed that acute knockdown of NP-1, the direct receptor of Sema3A, impaired bipolar polarity of cortical progenitors upstream of the cyclic guanosine monophosphate (cGMP)-synthesizing enzyme soluble guanylate cyclase (sGC) and cGMP activities (Shelly et al., 2011). The seeming discrepancy between these studies remains unexplained.
Sema3A regulates several aspects of neuronal development in the embryonic cortex and hippocampus. Despite extensive work in the field, it is unknown whether Sema3A regulates early apical dendrite morphogenesis (Danelon et al., 2020; Tran et al., 2009). Moreover, little is known about Sema3A’s downstream actions or how its signal is transduced to sGC. We showed that the scaffolding protein Scribble recruited sGC and that the complex was localized to developing apical dendrites (Szczurkowska et al., 2020). Scribble was required for preferential cGMP generation in dendrites, and its association with sGC was necessary for CA1 apical dendrite development. In this study, we find that the Sema3A co-receptor PlexinA3 also associates with Scribble. We show that the associations within the complex are necessary for Sema3A-mediated cGMP increase in dendrites and CA1 apical dendrite development. We further show that Sema3A, Scribble, and the PlexinA3-Scribble association are necessary for leading process and bipolar polarity establishment in CA1 pyramidal progenitors. Together, our findings demonstrate that apical dendrite development is promoted by the extrinsic Sema3A/PlexinA3 cue and mediated by a localized cGMP-synthesizing complex.
RESULTS
Sema3A/PlexinA3 cue is linked to cGMP increase via the Scribble complex
To study whether Sema3A mediates apical dendrite development via cGMP, we asked whether Sema3A is linked to and regulates cGMP increase via the sGC-Scribble complex, which mediates cGMP increase in dendrites (Szczurkowska et al., 2020). Co-immunoprecipitation (co-IP) from embryonic day 18 (E18) rat brain lysates showed that Scribble associated with PlexinA3 (Figures 1A and S1A), a major co-receptor in class III Semaphorin signaling in the hippocampus (Bagri et al., 2003; Cheng et al., 2001; Tran et al., 2009; Yaron et al., 2005). We did not observe Scribble association with other Plexin co-receptors, including PlexinA4 or PlexinD1 (Figures 1A and S1A). PlexinA3-Scribble association was confirmed by co-IP of PlexinA3 cytoplasmic domain (FLAG-PlexinA3-CD) and HA-Scribble in HEK-293 cells (Figure 1C) and was mediated via the Scribble LRR domain (Figures 1B and 1C) (Scribble contains N-terminal leucine-rich repeats [LRR] and four PDZ domains separated by an intermediate region [IMR]; Figure 1B). PlexinA4 or PlexinD1 did not interact with the LRR domain of Scribble (Figures S1B and S1C). Together with our previous findings (Szczurkowska et al., 2020), we show that Scribble associated with PlexinA3 and sGC via discrete LRR and IMR domains, respectively (Figure 1E).
Figure 1. Sema3A mediates cGMP increase and dendrite development via PlexinA3-Scribble association.

(A) Co-immunoprecipitation (co-IP) of Scribble with PlexinA2, PlexinA3, or PlexinA4 from rat E18 embryonic brain lysates. Immunoprecipitation (IP) with antibody (Ab) to PlexinA2, PlexinA3, or PlexinA4, and immunoblotting (IB) with Ab to Scribble. IB also with Abs to PlexinA2, PlexinA3, or PlexinA4 to detect their expression. IP with non-specific IgG, control (normal IgG). “Brain lysate,” total brain lysate (n = 3). PlexinA3 co-precipitated Scribble. IB with PlexinA Abs confirmed that absence of Scribble association with PlexinA2 or PlexinA4 was not due to lack of protein expression. IB for PlexinA2, PlexinA3, or PlexinA4 showed cross-reactivity among Abs for PlexinA2 and PlexinA4, but not PlexinA3, reinforcing specificity of Scribble-PlexinA3 association (see Figure S1A).
(B) Schematics of wild-type (WT) Scribble and deletion mutants. WT-Scribble contains 16 N-terminal leucine-rich repeats (LRRs) followed by four PDZ domains, separated by an intermediate region (IMR). Deletion mutants included those in which LRR (ΔLRR), LRR and IMR (PDZ/C), or all three regions, LRR, IMR, and PDZ (C-term) were deleted; variants of isolated LRR (LRR), IMR (IMR), or PDZ domains (PDZs). Scribble proteins fused to HA tag (Szczurkowska et al., 2020).
(C) Co-IP of PlexinA3 cytoplasmic domain (PlexinA3-CD) with WT or mutant Scribble variants from HEK-293 cell lysates co-expressing FLAG-PlexinA3-CD with HA-Scribble mutants (B). IP, FLAG; IB, HA (right two panels). “Total Lysates,” total cell lysates (left two panels): IB, FLAG, or HA. Association of FLAG-PlexinA3-CD with HA-Scribble mutants quantified as fold change (n = 3, one-way ANOVA, Dunnett’s multiple comparison test: **p ≤ 0.01; ***p ≤ 0.001) relative to WT-Scribble, normalized to total respective protein levels (see Figures S1B, S1C, and S2C).
(D) Co-IP of PlexinA3-CD with sGC-β1 in presence of Scribble from HEK-293 cell lysates co-expressing FLAG-PlexinA3-CD and GFP-sGC-β1, with WT-HA-Scribble, in presence of increasing concentration (0 up to 5 μg/μL) of dTom-LRR, to test PlexinA3-sGC-β1 association in presence of WT-Scribble, and its disruption in presence of increasing LRR concentration. IP, GFP (sGC-β1); IB, FLAG (PlexinA3-CD). Total cell lysates, IB, FLAG (PlexinA3-CD), GFP (sGC-β1), HA (Scribble), or Tom (LRR), to check protein expression. FLAG-PlexinA3-CD IP with GFP-sGC-β1, in presence of dTom-LRR, quantified as fold change (n = 3, one-way ANOVA; Dunnett’s multiple comparison test: **p ≤ 0.01; ***p ≤ 0.001) relative to cells not expressing dTom-LRR, normalized to total FLAG-PlexinA3-CD levels. β-Actin, loading control.
(E) Schematics of Sema3A signaling mediated by NP-1 that binds Sema3A and Plexin co-receptors mediating intracellular signaling. Depicted are PlexinA3 and sGC-β1 binding sites in Scribble, LRR, and IMR, respectively.
(F) Representative images of normalized somatic FRET (YFP-FRET/YFP-total)for control, Scribble-shRNA (shScrib#4), or LRR-expressing cultured hippocampal neurons, co-expressing cGMP FRET probe cGi-500, upon recombinant Sema3A (5 nM) treatment. Control, not treated with Sema3A. Scale bar, 10 μm. Right: higher-magnification images of boxed regions on left. Scale bar, 5 μm. FRET decrease upon Sema3A treatment reflects cGMP increase.
(G and H) Summary of difference in normalized somatic FRET (ΔYFP-FRET/YFP-total) compared with average of control for shScrib#4 (G) or dTom-LRR (H) transfected cells upon Sema3A treatment. Control, not treated with Sema3A (n = 3–4 cultures, 20–30 cells each; one-way ANOVA, Šídák’s multiple comparison test: *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001) (see Figure S2).
(I and K) Representative images of normalized dendritic FRET (YFP-FRET/YFP-total), for control, PlexinA3-shRNA (I; shPlexinA3), or LRR (K) transfected neurons, co-expressing cGi-500, upon Sema3A treatment. Control, not treated with Sema3A. Scale bar, 10 μm. Bottom: higher-magnification images of individual dendrites (boxed regions in top panels). Scale bar, 5 μm.
(J and L) Summary of difference in normalized dendritic FRET (ΔYFP-FRET/YFP-total) compared with average of control for shPlexinA3 (J) or dTom-LRR (L) transfected neurons upon Sema3A treatment. Control, not treated with Sema3A (n = 3–4 cultures, 20–30 cells each; one-way ANOVA, Šídák’s multiple comparison test: **p ≤ 0.01; ****p ≤ 0.0001; L, LRR + Sema3A, **p = 0.0164).
(M) Representative images of cultured hippocampal neurons transfected with shRNAs for NP-1 (shNP-1), PlexinA3 (shPlexinA3), Scribble (shScrib#4), control-shRNA, or EGFP-LRR or control vector, or co-expressing shPlexinA3 and LRR, plated on Sema3A stripes, alone or together with membrane-permeable fluorescent analog of cGMP (F-cGMP), following immunostaining with Tuj-1 at 5 DIV. Shown are neurons with soma located at stripe boundary. Scale bar, 20 μm.
(N) Quantification of preferential dendrite initiation “on” Sema3A stripe, patterned alone or together with F-cGMP, presented as preference index (PI): [(% dendrites initiated on stripe) – (% dendrites initiated off stripe)]/100, for neurons with soma located on stripe boundary, examined at 5 DIV, transfected as in (M). PI value of 0 indicates equal preference for dendrite formation “on” or “off” stripe (n = 3–5 cultures, 35–75 cells each; one-way ANOVA, Dunnett’s multiple comparison test: ****p ≤ 0.0001; rescue with F-cGMP, one-way ANOVA, Tukey’s multiple comparison test: ****p ≤ 0.0001; shPlexinA3 versus shPlexinA3 + LRR, unpaired t test, **p = 0.0159) (see Figure S3).
*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001. Error bars represent SEM.
By associating with both PlexinA3 and sGC, Scribble might link the Sema3A signal to cGMP. In support, we found that FLAG-PlexinA3-CD and GFP-sGC-β1, the main sGC subunit in the brain (Koesling et al., 1991; Mergia et al., 2003; Russwurm et al., 1998), interact following their co-expression with Scribble (Figure 1D). Overexpressing dTom-LRR decreased FLAG-PlexinA3-CD and GFP-sGC-β1 interaction in a dose-dependent manner (Figure 1D), showing that LRR competitively interrupted their association. Our data show that LRR is the necessary interface in PlexinA3-Scribble-sGC-β1 complex formation. In our subsequent experiments we used overexpression of the LRR domain to competitively disrupt endogenous PlexinA3-Scribble association.
We inquired whether PlexinA3-Scribble association was necessary for Sema3A-mediated cGMP increase in dendrites. We quantified Sema3A-mediated cGMP elevation in the soma and dendrites of cultured hippocampal neurons using a cGMP fluorescence resonance energy transfer (FRET) sensor, cGi-500 (FRET signal from cGi-500 decreases as cGMP increases [Russwurm et al., 2007]; Figure S2A). Following Sema3A treatment, normalized somatic FRET signal (YFP-FRET/YFP-total) was scattered below that of control (Figure S2B, black circles) and the difference in somatic FRET showed a decrease, reflecting a cGMP increase (Figures 1F-1H and S2B). Quantification of normalized FRET in each dendrite showed that Sema3A promoted a FRET decrease, reflecting cGMP increase, in dendrites (Figures 1I-1L). To test whether Sema3A mediates cGMP elevation via PlexinA3-Scribble association, we disrupted their interaction by knocking down Scribble or PlexinA3, or overexpressing LRR. We found an increase in somatic FRET, reflecting a cGMP decrease, in Sema3A-treated cells transfected with Scribble-short hairpin (sh)RNA (shScrib#4 [Szczurkowska et al., 2020]) (Figures 1F, 1G, and S2B) or LRR (Figures 1F, 1H, and S2B). Furthermore, knocking down PlexinA3 (Figures 1I and 1J) or overexpressing LRR (Figures 1K and 1L) prevented cGMP increase in dendrites following Sema3A treatment (Figure S3, shRNA knockdown efficiency). Our FRET measurements showed that Sema3A/PlexinA3 signaling mediates cGMP elevation in soma and dendrites via Scribble association, likely by engaging sGC. We examined the ability of Sema3A to promote Scribble-PlexinA3 association by co-IP in HEK-293 cells co-expressing wild-type (WT)-PlexinA3 and NP-1 together with WT-HA-Scribble, following Sema3A treatment. Sema3A promoted Scribble-PlexinA3 interaction in a concentration-dependent manner (Figure S2C). Together, our data show that the PlexinA3-Scribble complex was necessary for increased cGMP generation in dendrites, suggesting that localized increase in Sema3A can promote localized cGMP increase in dendrites via the Scribble complex.
We determined the role of PlexinA3-Scribble association in Sema3A-mediated dendrite development in cultured hippocampal neurons plated on substrate patterned with stripes of Sema3A. We tested the effects of disrupting PlexinA3-Scribble association by knocking down PlexinA3, NP-1, or Scribble, or overexpressing LRR. Dendrite formation was quantified at 5 days in vitro (DIV) in polarized neurons with their soma located on the stripe boundary according to their initiation “on” or “off” stripe regions. Dendrite formation occurred more “on” Sema3A stripe relative to “off stripe” in control neurons (Figures 1M and 1N). In contrast, transfection of PlexinA3- or NP-1-shRNAs, Scribble-shRNA (shScrib#4), or LRR, or combining PlexinA3 knockdown with LRR overexpression, increased dendrite formation “off stripe” (Figures 1M and 1N). To test whether the critical role of PlexinA3-Scribble association in dendrite development is attributed to Sema3A-mediated cGMP increase, we attempted to rescue the effects of PlexinA3 knockdown or LRR overexpression by local presentation of a cGMP analog. Neurons transfected with shPlexinA3 or LRR were plated on stripes of Sema3A together with a membrane-permeable cGMP analog (F-cGMP). The localized increase in cGMP restored the preference for dendrite formation “on” Sema3A stripe in neurons following PlexinA3 knockdown or LRR overexpression or their combined manipulation (Figures 1M and 1N). These findings showed that PlexinA3-Scribble association was necessary for Sema3A-mediated dendrite formation and that this effect was mediated via cGMP increase.
Sema3A/PlexinA3 association with the Scribble-cGMP complex is necessary for apical dendrite development
To inquire whether Sema3A is necessary for apical dendrite development, we determined the expression of Sema3A and NP-1 in the hippocampus using alkaline phosphatase (AP)-NP-1 or AP-Sema3A ectodomain fusion protein binding, respectively, to hippocampal sections at postnatal day 0 (P0) and P7. Sema3A was found mainly in the CA1 pyramidal cell layer (Figure S4; NP-1(ecto)-AP). NP-1 had a complementary expression in the stratum radiatum (SR) and stratum oriens (SO) (Figure S4; Sema3A-AP), consistent with expression in apical and basal dendrites. Notably, Sema3A also appeared to be expressed at the bottom stratum lacunosum-moleculare (SLM) (Figure S4, P7, arrowhead), where apical dendrites of CA1 pyramidal neurons terminate.
We determined whether Sema3A and PlexinA3, and their association with the Scribble complex, are necessary for apical dendrite development in the embryonic CA1. We examined apical dendrite development in PlexinA3 (Tran et al., 2009) and Sema3A (Taniguchi et al., 1997) null mice. Neurons following PlexinA3 (Figures 2A-2C) or Sema3A (Figures 2D-2F) deletion from homozygous (plexinA3−/−, sema3A−/−) embryos showed a reduction in apical dendrite length and branching at P7 compared with WT littermates, whereas heterozygotes (plexinA3+/−, sema3A+/−) had an intermediate phenotype. These findings are consistent with the effects of Scribble deletion in apical dendrite development (Szczurkowska et al., 2020) and suggest that Sema3A mediates CA1 apical dendrite development via PlexinA3 and its association with Scribble.
Figure 2. PlexinA3 and Sema3A are necessary for CA1 apical dendrite development.

(A and D) Representative images of CA1 pyramidal neurons, P7, from WT (PlexinA3+/+), heterozygous (PlexinA3+/−), or homozygous (PlexinA3−/−) (A), or WT (Sema3A+/+), heterozygous (Sema3A+/−), or homozygous (Sema3A−/−) (D) mouse littermate brains, transfected in utero at E15.5 with dTomato marker. SO, stratum oriens; SP, stratum pyramidale; SR, stratum radiatum. Scale bar, 100 μm. Bottom: sample tracings of neuritic arbor of representative neurons. Scale bar, 20 μm.
(B and E) Quantification of average total apical dendrite length per cell for CA1 pyramidal neurons from PlexinA3+/+, PlexinA3+/−, or PlexinA3−/− (B), or Sema3A+/+, Sema3A+/−, or Sema3A−/− (E) littermate brains (B, n = 20–35 cells; E, n = 20–22 cells; one-way ANOVA, Dunnett’s multiple comparison test: *p ≤ 0.05; ***p ≤ 0.001).
(C and F) Quantification of average total apical dendrite branch points per cell for neurons from PlexinA3+/+, PlexinA3+/−, or PlexinA3−/− (C), or Sema3A+/+, Sema3A+/− , or Sema3A−/− (F) littermate brains; same dataset as (B) and (E). One-way ANOVA, Dunnett’s multiple comparison test: *p ≤ 0.05; ***p ≤ 0.001.
*p ≤ 0.05; ***p ≤ 0.001. Error bars represent SEM.
To test whether the interaction of PlexinA3 with the Scribble-cGMP complex is necessary for apical dendrite development, we disrupted the complex by overexpressing LRR. Expressing LRR impaired CA1 apical dendrite length and branching (Figures 3A-3C). Knockdown of PlexinA3 caused defects in apical dendrite branching without affecting growth (Figures 3A-3C), likely resulting from incomplete downregulation with the shRNA (Figure S3). Combining PlexinA3 knockdown with LRR overexpression decreased dendrite length compared with PlexinA3 knockdown (Figures 3D-3F). Knockdown of NP-1 caused severe defects in apical dendrite length and branching (Figures 3A-3C). Importantly, knockdown of PlexinA4 or PlexinD1 did not impair apical dendrite development (Figures 3A-3C), consistent with their lack of association with Scribble (Figures 1A and S1). Co-expressing WT-PlexinA3 with PlexinA3-shRNA rescued apical dendrite branching defects following PlexinA3 knockdown, whereas co-expressing ΔCT-PlexinA3, a PlexinA3 mutant with the entire cytoplasmic tail deleted, did not rescue these defects (Figures 3D-3F). Furthermore, supporting the idea that LRR competitively disrupts PlexinA3-Scribble interaction, co-expressing WT-PlexinA3 fully rescued apical dendrite defects following LRR overexpression (Figures 3D-3F).
Figure 3. PlexinA3-Scribble association is necessary for CA1 apical dendrite development.

(A and D) Representative images of rat CA1 pyramidal neurons, P7, transfected in utero at E17.5 with shRNA for PlexinA3 (shPlexinA3), NP-1 (shNP-1), PlexinA4 (shPlexinA4), or PlexinD1 (shPlexinD1), or LRR, or control vector (A). Neurons co-transfected with WT-PlexinA3, PlexinA3-ΔC2 (associated Scribble; Figure S5), or PlexinA3-ΔCT (did not associate Scribble; Figure S5), or functional sGC (co-expressing sGC-α1 and -β1 subunits) (D). Neurons also co-transfected with shPlexinA3 and LRR (shPlexinA3 + LRR) (D). Neurons co-transfected with dTom marker. SO, SP, and SR layers as in Figure 2A. Scale bar, 100 μm. Bottom: sample tracings of neuritic arbor of representative neurons. Scale bar, 20 μm (see Figures S5 and S3).
(B and E) Quantification of average total apical dendrite length per cell for CA1 pyramidal neurons transfected as in (A) and (D) (n = 20–52 cells; data for control, shPlexinA3, and LRR were pooled among B and E). Control versus LRR or shPlexinA3 + LRR, one-way ANOVA, Dunnett’s multiple comparison test: ***p ≤*** 0.001; all other conditions, one-way ANOVA, Tukey’s multiple comparison test: ***p ≤ 0.001; ****p ≤ 0.0001. Data for shPlexinA3, shPlexinA4, and shPlexinD1 were not significantly different (ns) from control.
(C and F) Quantification of average total apical dendrite branch points per cell; same dataset as in (B) and (E). Data for Control, shPlexinA3, and LRR were pooled among (C) and (F). Control versus shPlexinA3, LRR, or shPlexinA3 + LRR, one-way ANOVA, Dunnett’s multiple comparison test: **p ≤ 0.01; ***p ≤ 0.001; all other conditions, one-way ANOVA, Tukey’s multiple comparison test: **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001; shPlexinA3 versus shPlexinA3 + WT-PlexinA3, Student’s t test, **p ≤ 0.01; data for shPlexinA4 or shPlexinD1 were not significantly different (ns) from control.
**p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001. Error bars represent SEM.
To determine the specificity of PlexinA3-Scribble association, we mapped the Scribble binding site in the cytoplasmic region of PlexinA3. The cytoplasmic tail of PlexinA receptors contains C1 and C2 domains with similarity to guanosine triphosphatase (GTPase)-activating proteins, which are separated by a Hinge/Rho GTPase binding domain (H/RBD) (Hota and Buck, 2012). Using Myc-tagged WT or mutant PlexinA3 constructs in which the different cytoplasmic domains were internally deleted (ΔC1; ΔH/RBD; ΔC2) or expressed singly (C1; H/RBD; C2) (Figure S5A), we found that PlexinA3-LRR association was mediated mainly via the PlexinA3 C1 domain (Figure S5B) and was further promoted in the presence of both C1 and H/RBD domains (Figure S5B; ΔC2-PlexinA3). Co-expressing LRR with - ΔC2-PlexinA3 in which both the C1 and H/RBD domains were present rescued apical dendrite length and branching (Figures 3D-3F) like the co-expression of WT-PlexinA3. These rescue experiments showed that C1 and H/RBD domains mediated the association of PlexinA3 with the Scribble-sGC complex and their subsequent.
effects on apical dendrite development. Our findings on defective apical dendrite development following PlexinA3 or Sema3A deletion (Figure 2) or disruption of their association with Scribble (Figure 3) are consistent with those with Scribble or sGC manipulations (Szczurkowska et al., 2020), suggesting that Sema3A mediates CA1 apical dendrite development via PlexinA3-Scribble-sGC associations and the subsequent increase in cGMP. We found that increased cGMP production by overexpression of functional sGC (by co-expressing sGC-α1 and -β1 subunits) rescued apical dendrite development defects following PlexinA3 knockdown or LRR expression (Figures 3D-3F). Our findings support the idea that the critical role of PlexinA3-Scribble association in apical dendrite development is to elevate cGMP downstream of Sema3A.
Sema3A and PlexinA3-Scribble association are necessary for bipolar polarity and leading process development
The critical role of Sema3A/PlexinA3 in early apical dendrite development (Figures 2 and 3) prompted us to ask whether Sema3A and Scribble activities regulate bipolar polarity and leading process development. We knocked down Scribble (shScrib#4) or overexpressed LRR in rat CA1 progenitors by in utero electroporation at E17.5. We examined neurons at E21 (Figure 4A) and P1 (Figure 4F) as, according to our fluorescent birth-dating experiments, at this time neurons settle at the stratum pyramidale (SP). At E21, control, shScrib#4-expressing, or LRR-expressing neurons were similarly located at the SO or the SP (Figure 4A). We found no perturbation in neuron positioning from the subventricular zone (SVZ) to the SO. We analyzed neurons at the SO or the SP separately. At the SO, control neurons exhibited a polarized, bipolar morphology and had established a leading process (Figures 4A and 4B). In contrast, a large fraction of shScrib#4- or LRR-expressing neurons were unpolarized (Figures 4A and 4B) and lacked a leading neurite. The shScrib#4- or LRR-expressing neurons also had more primary neurites than the control (Figure 4C). At the SP, control or shScrib#4- or LRR-expressing neurons were polarized and had a leading process (Figures 4A and 4D). Quantification of primary neurite number showed no difference among control and Scribble knockdown and an increase upon LRR expression (Figure 4E). Neurites in LRR-expressing cells were shorter and simpler than in the control (Figure 4A; SP) (Figures S6A and S6B, DAPI-layer determination for images in Figures 4A and 4F, respectively). Our data show that unlike the control, most neurons upon Scribble knockdown or LRR overexpression at the SO are unpolarized at E21, whereas only polarized neurons are found at the SP. These data indicate a crucial role for Scribble-PlexinA3 association in the assumption of bipolar morphology at the SO before neurons settle at the SP.
Figure 4. Sema3A and Scribble are necessary for bipolar polarity and leading process establishment.

(A and F) Representative images of rat CA1 pyramidal neurons at E21 (A) or P1 (F), transfected in utero at E17.5 with Scribble-shRNA (shScrib#4), LRR, or control vector, with EGFP marker. SO, stratum oriens; SP, stratum pyramidale; Scale bar, 100 μm. Bottom: sample tracings of neuritic arbor of representative neurons at SO or SP. Scale bar, 20 μm (see Figures S6A and S6B, DAPI-layer determination for images in A and F, respectively).
(B, D, and G) Quantification of percentage of polarized bipolar neurons at SO or SP, at E21 (B, SO; D, SP) or P1, expressing shScrib#4, LRR, or control vector. Data represent percentage from total number of transfected cells (n = 20–30 cells; B, one-way ANOVA, Dunnett’s multiple comparison test, shScrib#4, **p = 0.0137; LRR, ***p = 0.0013; D, one-way ANOVA, Dunnett’s multiple comparison test, ns; G, one-way ANOVA, Dunnett’s multiple comparison test, **p ≤ 0.01). (C, E, and H) Quantification of percentage of primary neurite number for neurons at SO or SP, at E21 (C, SO; E, SP) or P1, expressing shScrib#4, LRR, or control vector; same dataset as in (B, D, and G). Data represent percentage of primary neurites directly extending from the soma, from total number of processes including all neurites and branches (C, one-way ANOVA, Dunnett’s multiple comparison test, ***p ≤ 0.001; E, one-way ANOVA, Dunnett’s multiple comparison test, ***p ≤ 0.001; H, one-way ANOVA, Dunnett’s multiple comparison test, ns).
(I) Quantification of average leading process length per cell for polarized neurons transfected with shScrib#4, LRR, or control vector, at SO or SP, and at P1; same dataset as in (G) (one-way ANOVA, Dunnett’s multiple comparison test: **p ≤ 0.01; ***p ≤ 0.001).
(J) Representative images of CA1 pyramidal neurons at E18, at SO, from Sema3A+/+, Sema3A+/−, or Sema3A−/− mouse littermate brains, transfected in utero at E15.5 with dTom marker. SO, SP, and SR layers as in Figure 2A. Scale bar, 100 μm. Bottom: sample tracings of neuritic arbor of representative neurons. Scale bar, 20 μm (see Figure S6C, DAPI-layer determination for images).
(K) Quantification of percentage of polarized bipolar neurons at SO for neurons from Sema3A+/+, Sema3A+/−, or Sema3A−/− littermate brains at E18. Data represent percentage of total number of transfected cells (n = 20–30 cells; Dunnett’s multiple comparison test, **p ≤ 0.01; ***p ≤ 0.001).
(L) Quantification of percentage of primary neurite number for neurons at SO from Sema3A+/+, Sema3A+/−, or Sema3A−/− littermate brains at E18, same dataset as in (K). Data represent percentage of primary neurites directly extending from the soma, from total number of processes including all neurites and branches (Dunnett’s multiple comparison test, *p ≤ 0.05).
*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. Error bars represent SEM.
To determine leading process development, we examined neurons at P1. At this stage, all control neurons were at the SP, whereas shScrib#4- or LRR-expressing neurons were at the SO and SP (Figure 4F). All neurons at the SP were polarized (Figures 4F and 4G) and had a leading process. Conversely, at the SO, a large fraction of shScrib#4-expressing neurons were unpolarized and lacked a leading neurite, whereas LRR-expressing neurons were mostly polarized, having a short and simple leading neurite (Figures 4F and 4G). Despite their unpolarized morphology, we found no difference in the number of primary neurites among shScrib#4 or LRR and control neurons at the SO or SP (Figure 4H), showing that considerable neurite remodeling occurred by P1 compared with E21. As we did not find perturbation prior to the SO and as only polarized neurons were at the SP, our findings suggest that lack of polarity preceded and caused the positioning defect at the SO at P1 following Scribble knockdown or LRR expression (Figure 4F). Polarized shScrib#4 expressing neurons at the SP had a shorter leading neurite than the control (Figure 4I). Polarized shScrib#4- or LRR-expressing neurons at the SO had a leading process with a similar length that was shorter than that of control neurons at the SP (Figures 4F and 4I). Nevertheless, LRR neurons at the SP had a leading neurite with a similar length to the control (Figure 4I). These data are largely consistent with defective development of the apical dendrite upon Sema3A, PlexinA3, or Scribble manipulations at P7 (Figures 2 and 3; Szczurkowska et al., 2020). Our data underlie the critical role of the PlexinA3-Scribble scaffold in bipolar polarity establishment at the SO and the subsequent development of the leading process and the apical dendrite likely occurring at both the SO and SP.
To test whether Sema3A is necessary for bipolar polarity establishment, we examined neurons in Sema3A null mice. CA1 pyramidal progenitors were labeled with dTomato at E15.5 (Figure 4J). Cells were analyzed at E18, a time before neurons settle at the SP as determined by fluorescent birth-dating. As predicted, at E18 WT, sema3A+/−, or sema3A−/− neurons were at the SO (Figure 4J). We found no perturbation in neuron positioning from SVZ to the SO. Most WT neurons had a polarized bipolar morphology and formed a leading process (Figure 4K). In contrast, many sema3A+/− or sema3A−/− neurons were unpolarized (Figure 4K) and had no leading neurite. Furthermore, sema3A+/− orsema3A−/− neurons had more primary neurites than WT (Figure 4L) (Figure S6C, DAPI-layer determination for images in Figure 4J). Together, our data underscore the critical role of Sema3A/PlexinA3 in bipolar polarity and leading process establishment and suggest that Sema3A acts via the Scribble-cGMP complex.
DISCUSSION
Studies from cultured neurons (Arimura and Kaibuchi, 2007; Barnes et al., 2007; Cheng et al., 2011a, 2011b; Da Silva et al., 2005; de Anda et al., 2005; Dotti and Banker, 1987; Dotti et al., 1988; Inagaki et al., 2001; Jacobson et al., 2006; Jiang et al., 2005; Kishi et al., 2005; Shelly et al., 2007, 2010; Shi et al., 2003; Toriyama et al., 2006; Yoshimura et al., 2005) that are supported by in vivo findings (Namba et al., 2014) proposed that axon specification precedes neuronal polarization and is necessary for dendrite development. However, spatially organized extrinsic cues may promote leading process polarity and apical dendrite development by orchestrating localized intrinsic signaling at the leading edge. Recent evidence support this notion and show that in many cortical pyramidal progenitors, leading process formation preceded trailing process development (Namba et al., 2014). Furthermore, this view is supported by findings showing that prevention of axon formation did not interfere with leading process polarity or apical dendrite development (Barnes et al., 2007; Kishi et al., 2005; Shelly et al., 2007; Yi et al., 2010), suggesting that these events can occur independently.
The extrinsic cues and mechanisms that promote leading process polarization and subsequent apical dendrite development have remained elusive. Deletion of key secreted factors and their receptors reportedly yielded only subtle defects on axon/ dendrite development in vivo (Crowley et al., 1994; Da Silva et al., 2005; Jones et al., 1994; Klein et al., 1993; Shelly et al., 2007; Smeyne et al., 1994; Sosa et al., 2006; Yoshimura et al., 2005). These include previous studies on Sema3A and PlexinA3. Global examination of mice with Sema3A gene deletion did not suggest effects on axon and dendrite formation (Behar et al., 1996; Polleux et al., 1998) or a major role for Sema3A in neuronal polarization in vivo. Studies specifically aimed at characterizing dendrite morphogenesis upon manipulations of class III Semaphorin signaling, including deletion of Sema3A, NP-1, or PlexinA3, showed defects in spine morphogenesis and dendrite maturation and arborization (Danelon et al., 2020; Tran et al., 2009). These studies found no other apparent defects in dendrite development, and no effects were observed in CA1 pyramidal neurons. It must be noted, however, that dendritic phenotypes in these genetic models were determined in adult neurons at P21, P45, or later, times that are not optimal for detecting effects on embryonic and early postnatal neuronal polarization. It is therefore perhaps not surprising that these studies did not reveal defects in bipolar polarity establishment and leading process polarization or apical dendrite development and its initial growth and branching. We found that the Sema3A cue linked to the assembly of a cGMP production machinery via the Scribble scaffold is required for early apical dendrite development. Our examinations in Sema3A and PlexinA3 null mice and following acute manipulations of PlexinA3 and disruption of its association with Scribble during embryonic and early postnatal development (up to P7) showed severe defects in CA1 apical dendrite development. This is consistent with our findings with Scribble or sGC manipulations or disruption of their association (Szczurkowska et al., 2020). Examination of axon/dendrite development with precise spatiotemporal resolution at embryonic and early postnatal stages in specific mouse models is required to further identify the extracellular cues that mediate neuronal polarization in vivo.
The PlexinA isoforms PlexinA3 and PlexinA4 were shown to be prominently expressed in the developing hippocampus and to mediate the effects of class III Semaphorins in pathfinding of major axonal projections (Bagri et al., 2003; Cheng et al., 2001; Tran et al., 2009). PlexinA3 and PlexinA4 interchangeably mediate the effects of Sema3A and Sema3F. In the developing hippocampus and in sensory and sympathetic neurons (Bagri et al., 2003; Cheng et al., 2001; Tran et al., 2009), the Plexin-Neuropilin complexes that form most efficiently are PlexinA3/NP-2 and PlexinA4/NP-1, which mediate the effects of Sema3F and Sema3A, respectively. Moreover, Sema3F via PlexinA3/NP-2 regulates spine morphogenesis in apical dendrites of dentate granule cells and layer V cortical pyramidal neurons, whereas Sema3A via PlexinA4/NP-1 regulates basal dendrite arborization in layer V cortical neurons (Danelon et al., 2020; Tran et al., 2009). This preference, however, is not absolute as in other brain regions, PlexinA3/NP-1 complex formation was preferred and shown to mediate the effects of Sema3A. This was particularly true in the hippocampal CA1 where PlexinA3 and not PlexinA4 partnered with NP-1 to mediate pruning of CA1 pyramidal axons projecting to the medial septum downstream of Sema3A (Bagri et al., 2003). This is consistent with our findings on the role of Sema3A/PlexinA3 in CA1 apical dendrite development. Our data regarding deletion and/or knockdown of Sema3A, PlexinA3, and NP-1 show that PlexinA3 mediates CA1 apical dendrite development downstream of Sema3A via Scribble association and cGMP activities. We show that PlexinA4 might not be involved in these early events, as PlexinA4 knockdown did not disrupt apical dendrite development. The more divergent isoform PlexinD1 is also unlikely to play a role in early CA1 apical dendrite morphogenesis. Nevertheless, we do not exclude the possibility that Scribble may associate with other Plexin co-receptors in the developing hippocampus. We also do not exclude the possibility that via PlexinA3 association, Scribble might mediate cGMP increase and apical dendrite development downstream of other secreted class III Semaphorins in the hippocampus. The specific co-receptors that mediate the effects of class III Semaphorins in the development of major brain regions is an important issue that remains to be further elucidated.
Subsequent to its role in early dendrite development, the Scribble scaffold likely also plays a role in dendrite maturation and spine morphogenesis (Moreau et al., 2010; Richier et al., 2010). Studies with the Scribble circletail mutant showed that Scribble regulates dendrite and spine morphogenesis by associating with the Rac-1 guanine nucleotide exchange factor (GEF), β-Pix (Moreau et al., 2010). Furthermore, Sema3A/PlexinA4 mediate basal dendrite elaboration and maturation in cortical pyramidal neurons by engaging the GEF protein FARP2 and Rac-1 signaling (Danelon et al., 2020; Tran et al., 2009). Whether both Rac-1 and cGMP pathways or an interaction between them via Scribble association are required for the Scribble effects in dendrite and spine morphogenesis remains for future investigation.
We show that deletion of Sema3A, knockdown of Scribble or interruption of its association with PlexinA3 caused defects in bipolar polarity and leading process establishment in CA1 pyramidal progenitors. Postmitotic CA1 pyramidal neurons assume unpolarized multipolar morphology (Kitazawa et al., 2014), and not much is known about the subsequent development of these neurons. Our findings demonstrate the critical role of Sema3A in bipolar polarity establishment at the SO before neurons settle at the SP and suggest that the Sema3A effects are mediated via the Scribble complex. First, we did not find perturbation in neuronal positioning from the SVZ to the SO following Sema3A deletion, Scribble knockdown, or LRR overexpression. Second, while most control neurons at the SO had polarized bipolar morphology, many neurons upon Sema3A deletion or disruption of PlexinA3-Scribble association appeared unpolarized, had multiple primary neurites, and did not properly develop a leading process. Lastly, only polarized bipolar neurons that had a leading process were found at the SP. Our findings underlie the role of Sema3A and the Scribble scaffold in bipolar polarity and development of the leading process/nascent apical dendrite and show that these likely occur at the SO and SP. We propose that the formation of the leading edge during bipolar polarity establishment and the subsequent development of the apical dendrite are tightly linked events. Although dedicated mechanisms that regulate these events independently of each other must exist, our study shows that cGMP-mediated mechanisms direct bipolar polarity and development of the apical dendrite downstream of Semaphorin signaling.
In the presence of spatially organized extracellular cues in developing pyramidal neurons in vivo, the signaling for apical dendrite development might assemble autonomously. Our findings suggest that Sema3A might promote PlexinA3-Scribble association. Whether the association with PlexinA3 is necessary for the dendritic localization of Scribble or vice versa, and whether Sema3A promotes Scribble-PlexinA3 association and thereby orchestrates their dendritic localization, remain to be further determined. Nevertheless, the assembly of Sema3A co-receptors and cGMP-synthesis enzymes on the Scribble scaffold might enable higher cGMP levels at the leading process/nascent apical dendrite to specifically promote its development.
Limitations of the study
In the embryonic brain, selectivity for cGMP elevation in apical dendrite development may be independently mediated by a gradient of Sema3A generated across the developing neuron. Our study suggests that the assembly of cGMP-synthesis enzymes and Sema3A co-receptors on the Scribble scaffold would generate a gradient of cGMP production in response to the Sema3A gradient, with the highest at the leading process/nascent apical dendrite, to specifically promote its development. The presence of a gradient of Sema3A and the gradient of cGMP across the developing neuron in vivo is not directly addressed in our study.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Maya Shelly (maya.shelly@stonybrook.edu).
Materials availability
All plasmids and generated materials in this study are available from the lead contact with a completed materials transfer agreement.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Rats and mice were used according to protocols approved by the Institutional Animal Care and Use Committees at Stony Brook University. Timed pregnant Sprague Dawley rats were from Envigo. Sema3A −/− mice were provided by Dr. Alex Kolodkin (Johns Hopkins University School of Medicine, Baltimore, MD). PlexinA3 −/− mice were provided by Dr. Tracy Tran (Rutgers University, Newark, NJ). As all manipulations were done in embryonic brains in utero, bias among male or female progeny from pregnancies, or data differences between males and females in FRET or morphological analyses, is not anticipated. Pregnancies and in utero transfection timings were carefully monitored. In genetically modified mice, data was acquired from littermates to prevent age variables.
Primary hippocampal culture and transfection
Cultures of dissociated hippocampal neurons were prepared from rat embryos as described previously (Dotti and Banker, 1987; Dotti et al., 1988). Neurons were plated onto a glass coverslip coated with poly-L-lysine (PLL), and cultured for 5 days. Amaxa nucleofection system (Lonza AG) was used to transfect expression vectors or shRNAs into primary neurons.
METHOD DETAILS
Antibodies and materials
Scribble, Myc, and HA antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies for PlexinA3, PlexinA4, PlexinA2, PlexinD1, and NP-1 were from Santa Cruz Biotechnology, from Abcam (Cambridge, MA), from Cell Signaling Technology (Danvers, MA), and from R&D Systems (Minneapolis, MN). β-actin antibody was from Cell Signaling Technology. FLAG®M2, peroxidase-conjugated anti-FLAG®M2 and peroxidase-conjugated anti-HA antibodies were from Sigma (St. Louis, MO). Neuronal Class III β-Tubulin (Tuj1) antibody was from Covance (Princeton, NJ). Fluorescently conjugated Bovine Serum Albumin (BSA) was from Invitrogen Corporation (Carlsbad, CA). Analogue of cGMP (F-cGMP) - (8-[(2-[(fluoresceinylthioureido)amino]ethyl]thio] guanosine-3′,5′-cyclic monophosphate (8-Fluo-cGMP (F-cGMP) was from BIOLOG (Life Science Institute, Bremen, Germany). Recombinant Sema3A was kindly provided by T. Tran (Rutgers University, Newark, NJ).
Plasmid constructs and shRNAs
To suppress endogenous rat Scribble, we generated three shRNA sequences, shScrib#1, shScrib#2, and shScrib#3, and shScrib#4, as previously described (Szczurkowska et al., 2020). For the FRET experiments, these shRNA sequences were cloned into PRNT U6.1 expression vector that also drives expression of dTom, otherwise, the vector expressed EGFP. The shRNAs targeting rat NP-1 or PlexinA3, were previously described (Chen et al., 2008). Their sequences are as follows: NP-1, 5′-AGAGAAGCCAACCAT TATA-3′; PlexinA3, 5′-GTGCGGGTTCGGCCTAATA-3′. The sequences of the shRNAs targeting rat PlexinA4 and PlexinD1 are as follows: PlexinA4, 5′-AACACCTCCTATTCCTATGAA-3′; PlexinD1, 5′-AAGATGCTTACCAACTGGATG-3′. Expression constructs for Scribble, including the LRR domain and other mutants, as well as constructs for sGC-β1 and sGC-α1, were previously described (Szczurkowska et al., 2020). The cGMP FRET Probe cGi-500 was a kind gift from Dr. Doris Koesling (Ruhr-University Bochum, Germany) and described previously (Russwurm et al., 2007). The pCAG-promoter driven vectors, including pCAG-dTomato were described previously (Shelly et al., 2007). N-terminal Myc-tagged full-length mouse PlexinA3 construct was kindly provided by Dr. Alex Kolodkin (Johns Hopkins University, Baltimore, Maryland), and cloned into pCAG vector. Mutant PlexinA3 isoforms were generated in which the different cytoplasmic domains, C1, C2 and H/RBD, were either internally deleted (C1, ΔC1; H/RBD, ΔH/RBD, C2, ΔC2) or expressed in isolation (C1; H/RBD; C2). Rat sequences for NP-1 (GenBank: AAH85689.1), PlexinD1 (GenBank: NP_001381981.1), PlexinA3 (GenBank: NP_001101051.2) and PlexinA4 (GenBank: XP_032762687.1), were generated (GenScript, Piscataway, NJ), and cloned into pCAG vector.
Micro-fabrication and substrate patterning
Poly(dimethylsiloxane) (PDMS) molds were generated and substrates were patterned in parallel stripes of 50 μm width separated by 50 μm gaps as previously described (Shelly et al., 2007, 2017). PDMS mold was reversibly sealed on poly-L-lysine-coated glass coverslip, and microchannels formed between the PDMS mold and coverslip were used for microfluidic patterning of recombinant Sema3A together with fluorescently conjugated BSA as a marker. To examine rescue with cGMP-activity, membrane permeable F-cGMP analog was patterned together with Sema3A. Recombinant Sema3A was kindly provided by T. Tran (Rutgers University, Newark, NJ). The stripe-coated coverslips were washed extensively before neuronal plating. Dendrite initiation “on” or “off” the stripe was defined for cells with their soma located on the stripe boundary, and quantified by using the Preference Index (PI = [(% dendrites initiated on stripe) - (% dendrites initiated off stripe)]/ 100%. Dendrite formation ‘on’ Sema3A stripe refers to the side of soma in direct contact with the stripe relative to ‘off-stripe’.
Transfection, immunoblotting and immunoprecipitation
HEK-293 cells were transfected with plasmid DNA using calcium-phosphate method. 1 to 2 days after transfection, the cells were lysed and processed for immunoprecipitation and immunoblotting. For immunoprecipitation and immunoblotting of endogenous proteins from embryonic brains, the brains were lysed in 0.3% CHAPS lysis buffer, and lysates subjected to immunoblotting or immunoprecipitation.
Immunohistochemistry and AP-binding assay
Cultured cells were fixed in 4% paraformaldehyde and permeabilized with 0.05% Triton x-100. Primary antibodies were incubated overnight at 4°C. Following extensive washes, cells were incubated with secondary antibodies for 2 h at room temperature. Brains were fixed in 4% paraformaldehyde, coronally sectioned at 30–60 μm thickness, and mounted.
The expression pattern of Sema3A and its direct receptor Neuropilin-1 (NP-1) in the mouse hippocampus was determined at P0 and P7. Alkaline phosphatase (AP)-NP-1 ectodomain or AP-Sema3A fusion proteins were bound to sagittal hippocampal sections of P0 or P7 WT mice, to detect endogenous Sema3A or NP-1 expression, respectively, in the hippocampus, using a colorimetric AP activity assay, as previously described (Tran et al., 2009).
FRET imaging and analysis
We designed measurement for determining changes in cGMP levels in cultured hippocampal neurons, under control conditions or following Sema3A treatment, resulting from genetic manipulations of Sema3A/PlexinA3 signaling or disruption of their interaction with Scribble, using a fluorescence resonance energy transfer (FRET) sensor for cGMP, cGi-500 (Russwurm et al., 2007). Our FRET assay and quantifications were previously described (Szczurkowska et al., 2020). The neurons, plated on glass-bottom dishes for live imaging (MatTek Corporation), were co-transfected prior to cell plating by electroporation with a construct encoding the FRET cGMP reporter cGi-500, together with shRNAs or DNA constructs. The FRET measurements were performed at 3 to 4 DIV, a time when neuron polarization is complete. Neurons were imaged with EMCCD camera (Andor Technology PLC, UK), and epifluorescence microscope (Olympus, Center Valley, PA), equipped with a 75-W Xenon lamp and a 60× NA 1.42 objective (Olympus). FRET measurements were performed by CFP excitation with a band pass (440/20 nm) excitation filter, and simultaneous measurement of the fluorescence emission of CFP and YFP, using a dual view beam splitter equipped with a band pass (480/40 nm) emission filter for CFP and a band pass (530/40 nm) emission filter for YFP. The intensity of the light source and the exposure times were kept constant for all measurements for somatic FRET, or FRET measurements in dendrites, respectively. FRET signal was determined by YFP fluorescence measured upon CFP excitation (YFP-FRET). Because the FRET signal (YFP-FRET) in each cell would depend on the level of probe expression in that cell, FRET values were normalized to total YFP levels, determined upon YFP excitation (YFP-total), with the assumption that total YFP levels (YFP-total) reflect the level of probe expression in the cell. To measure YFP-total, YFP was excited using a band pass (515/20 nm) excitation filter. Importantly, the cGi-500 sensor relaxes its conformation upon cGMP binding, resulting in FRET reduction, whereas reduction in cGMP levels is reflected in FRET increase (Russwurm et al., 2007).
For somatic FRET measurements, for each cell, we measured the mean fluorescence intensity over the cell area. For dendritic FRET measurements, we measured the mean fluorescence intensity over the entire dendritic length. We made sure that the imaged cell expressed the shRNA or the expression plasmid before FRET imaging. FRET imaging was performed for 15 min following Sema3A application. The imaging for each channel: YFP-total obtained upon YFP excitation and measurement of YFP emission, CFP donor obtained upon CFP excitation and measurement of CFP emission, and YFP acceptor obtained upon CFP excitation and measurement of YFP emission. All fluorescence intensities were background-subtracted with background intensity taken from a cell-free region, and were corrected for bleed-through. cGMP was determined by FRET signals in each control transfected cell, normalized to cGi-500 probe expression level in that cell, in the soma or dendrites (YFP-FRET / YFP-total), and presented for all cells with varying probe concentration (YFP-total). This analysis in control-transfected cells enabled to determine a range in which the FRET signals demonstrated a linear correlation with the levels of probe expression. This linear range of FRET response represents the baseline levels of cGMP in control-transfected cells, in the soma or dendrites. To determine the change in cGMP levels following different genetic manipulations, we examined the difference in FRET signals (ΔYFP-FRET / YFP-total) for experimental-transfected cells compared to respective control-transfected cells, at corresponding levels of probe expression (YFP-total), in the soma or dendrites.
In utero electroporation
In utero electroporation with three-electrode configuration for targeting the neurogenic epithelia of the hippocampus was performed as previously described (Dal Maschio et al., 2012; Szczurkowska et al., 2016). In brief, timed-pregnant Sprague Dawley rats at E17.5 or timed-pregnant mice at E15.5, were anesthetized with isoflurane and their uterine horns were exposed by laparotomy. The target DNA solution (0.5–1.5 mg/mL) together with a pCAG-dTomato construct as a marker, mixed with the Fast Green dye (0.3 mg/mL; Sigma) was injected (1–2 μL) through the uterine wall into one of the lateral ventricles of each embryo. Six electrical pulses were delivered during electroporation (Rat E17.5 embryos: amplitude, 50V; duration, 50ms; intervals, 100ms; Mouse E15.5 embryo: amplitude, 30V; duration, 50ms; intervals, 1s) with a square-wave electroporation generator (model ECM 830, BTX, Inc.). The uterine horns were then returned into the abdominal cavity, the wall and skin sutured, and the embryos allowed to continue their development. Control and experimental embryos/pups were obtained from the same litter. All animal protocols were approved by the Institutional Animal Care and Use Committees of Stony Brook University.
Image acquisition and morphometric analysis
In cultured hippocampal neurons, a primary neuritic process directly extending from the soma of polarized neurons was determined to be a dendrite based on MAP2 labeling. Only processes longer than 5 μm, and that were initiated within 5 μm from the soma when branching occurred, and that were positive for MAP2, were considered individual dendrites.
For image acquisition and morphological analysis of CA1 pyramidal neurons, brains were dissected at E18, E21, or P1, and fixed for 24 h in 4% paraformaldehyde (PFA) in PBS. Postnatal brains (P7) were fixed by transcardial perfusion with 4% PFA solution. Hippocampal coronal sections were obtained and the slices (Rat, 80μm; Mouse, 60μm) were mounted in Fluomount G (Southern Biotech, Inc.). For morphometric analysis of dendrite morphology and arborization, confocal images of isolated neurons were acquired with a 40× (N.A. 1.43) oil-immersion objective, and images of entire dendritic arbors were reconstructed from the Z-series stacks of the confocal images. Number of dendritic processes, their length and branching were measured and analyzed using ImageJ software. Furthermore, 3D images of entire dendritic arbors were reconstructed from the Z-series stacks of confocal images using IMRS 7.3.1 software (Bitplane). For analysis of dendrite number and length, total length and complexity of each process were measured and analyzed using the filament measurement in the software. Up to 30 neurons from multiple sections, from 3-5 brains, were analyzed for each condition. DAPI staining was used for layer determination.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of biochemical analyses of protein immunoblotting (IB) or immunoprecipitation (IP) are based on at least three independent experiments. Number of dendritic and neuritic processes, their length and branching were measured and analyzed using ImageJ software. Moreover, 3D images of entire dendritic arbors were reconstructed from the Z-series stacks of confocal images using IMRS 7.3.1 software (Bitplane). At least 20 neurons from multiple sections and brains were analyzed for each condition. FRET cGMP levels were quantified in 20–30 cells each from 3 to 4 cultures. Preferential dendrite initiation ‘on’ Sema3A stripe was quantified for neurons with soma located on the stripe boundary, from 35 to 75 cells each from 3 to 5 cultures. All data analyses were performed using GraphPad (Prism v8.1.2). One Way ANOVA was used to compare between multiple experimental conditions, Student’s t-test was used to compare between two different experimental conditions. Statistical significance was determined as: *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001. Error bars represent SEM. Additional quantification and statistical details can be found in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti PlexinA3 | Abcam | Cat#ab41564 |
| rabbit anti PlexinA3 | Santa Cruz | Cat#sc-25641 (H-90) |
| mouse anti PlexinA3 | Santa Cruz | Cat#sc-374662 (A-8) |
| mouse anti PlexinA4 | R&D Systems | Cat#MAB5856 |
| rabbit anti PlexinA4 | Abcam | Cat#ab39350 |
| rabbit anti PlexinA4 | Cell Signaling Technology | Cat#3816 |
| goat anti PlexinA2 | R&D Systems | Cat#AF5486 |
| rabbit anti PlexinA2 | Abcam | Cat#ab39357 |
| rabbit anti PlexinD1 | Abcam | Cat#ab96313 |
| goat anti PlexinD1 | R&D Systems | Cat#AF4160 |
| goat anti PlexinD1 | Santa Cruz | Cat#sc-46245 (E-13) |
| rabbit anti NP-1 | Santa Cruz | Cat#sc-5541 (H-286) |
| mouse anti NP-1 | Santa Cruz | Cat#sc-5307 (A-12) |
| rabbit anti Scribble (H-300) | Santa Cruz | Cat#sc-28737 |
| rabbit anti Tuj1 | Covance | Cat#PRB-435P |
| mouse anti-GFP | Roche | Cat#11814460001 |
| rabbit anti RFP | Rockland | Cat#600-401-379 |
| rabbit anti β-actin | Cell Signaling Technology | Cat#4967L |
| mouse anti-FLAG®M2 | Sigma-Aldrich | Cat#F1804 |
| mouse anti-HA | Santa Cruz | Cat#sc-7392 |
| mouse anti Myc | Santa Cruz | Cat#sc-41 (C-8) |
| Rabbit anti Myc | Santa Cruz | Cat#sc-789 (A-14) |
| Alexa Fluor® 488 AffiniPure Goat Anti-Mouse IgG | Jackson ImmunoResearch | RRID: AB_2338840 |
| Alexa Fluor® 594 AffiniPure Goat Anti-Mouse IgG | Jackson ImmunoResearch | RRID: AB_2338871 |
| Alexa Fluor® 594 AffiniPure Goat Anti-Rabbit IgG | Jackson ImmunoResearch | RRID: AB_2338059 |
| Alexa Fluor® 488 AffiniPure Goat Anti-Rabbit IgG | Jackson ImmunoResearch | RRID: AB_2338046 |
| Alexa Fluor® 647 AffiniPure Goat Anti-Rabbit IgG | Jackson ImmunoResearch | RRID: AB_2338072 |
| Alexa Fluor® 594 AffiniPure Goat Anti-Chicken | Jackson ImmunoResearch | RRID: AB_2337391 |
| Bacterial and virus strains | ||
| DH5α competent cells | Life Technologies | Cat#18265017 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Sema3A | Tracy Tran Lab | N/A |
| Fluorescently conjugated Bovine Serum Albumin (BSA) | Life Technologies | Cat#A34786; Cat#A13100 |
| Analogue of cGMP (F-cGMP) | BIOLOG | Cat#F 001 |
| Hoescht 33342 (bis-benzimide 33342) | Sigma-Aldrich | Cat#B2261 |
| Triton X-100 | Millipore Sigma | Cat#TX1568-1 |
| Fast Green | Sigma-Aldrich | Cat#F7252 |
| Fluoromount-G | SouthernBiotech | Cat#0100-01 |
| Phosphate buffered saline | Sigma-Aldrich | Cat#P4417 |
| Normal Goat Serum | Jackson ImmunoResearch | RRID: AB_2336990 |
| Amersham Hyperfilm ECL | GE Healthcare | Cat#28906839 |
| Amersham ECL Western Blotting Detection Reagents | GE Healthcare | Cat#RPN2209 |
| Protein G agarose | Roche | Cat#11719416001 |
| CHAPS hydrate | Millipore Sigma | Cat#9426 |
| Nitrocellulose Membranes | BIO RAD | Cat#1620115 |
| Tween 20 | BIO RAD | Cat#170-6531 |
| Paraformaldehyde | Milipore Sigma | Cat#441244 |
| 0.25% Trypsin-EDTA | Gibco | Cat#25200056 |
| GlutaMAX | Gibco | Cat#35050061 |
| Neurobasal Medium minus Phenol Red | Gibco | Cat#12348017 |
| Neurobasal Medium | Gibco | Cat#21103049 |
| Hibernate E minus CaCl2 | BrainBits | Cat#HECA |
| Hibernate E | BrainBits | Cat#HE |
| Papain | Worthington Biochemical Corporation | Cat#LK003178 |
| Poly-L-Lysine Hydrochloride | Millipore Sigma | Cat#P9404 |
| B-27 Supplement | Life Technologies | Cat#17504044 |
| Critical commercial assays | ||
| EndoFree Plasmid Maxi Kit | Qiagen | Cat#12362 |
| Experimental models: Cell lines | ||
| HEK-293 | ATCC | Cat# PTA-4488 |
| Experimental models: Organisms/strains | ||
| CD® (Sprague Dawley) rats | Charles River | RRID:RGD_734476 |
| Sema3A −/− mice | Alex Kolodkin Lab | N/A |
| PlexinA3 −/− mice | Tracy Tran Lab | N/A |
| Oligonucleotides | ||
| shPlexinA3 | Chen et al. (2008) | GATCCCGTATTAGGCCGAACCCGCAC TTGATATCCGGTGCGGGTTCGGCCTAA TATTTTTTCCAAA |
| shNP-1 | Chen et al. (2008) | GATCCCGTATAATGGTTGGCTTCTC TTTGATATCCGAGAGAAGCCAACCATTA TATTTTTTCCAAA |
| shPlexinA4 | This study | GATCCCGTTCATAGGAATAGGAGGTG TTTTGATATCCGAACACCTCCTATTCCTA TGAATTTTTTCCAAA |
| shPlexinD1 | This study | GATCCCGCATCCAGTTGGTAAGCATC TTTTGATATCCGAAGATGCTTACCAAC TGGATGTTTTTTCCAAA |
| shScrib#1 | Szczurkowska et al. (2020) | N/A |
| shScrib#2 | Szczurkowska et al. (2020) | N/A |
| shScrib#3 | Szczurkowska et al. (2020) | N/A |
| Recombinant DNA | ||
| PRNT U6.1 vector | GeneScript | |
| shPlexinA3 | Chen et al. (2008) | N/A |
| shNP-1 | Chen et al. (2008) | N/A |
| shPlexinA4 | This study | N/A |
| shPlexinD1 | This study | N/A |
| shScrib#1 | Szczurkowska et al. (2020) | N/A |
| shScrib#2 | Szczurkowska et al. (2020) | N/A |
| shScrib#3 | Szczurkowska et al. (2020) | N/A |
| pCAG-HA-WT-Scribble-cDNA | Szczurkowska et al. (2020) | N/A |
| pCAG-HA-ΔLRR | Szczurkowska et al. (2020) | N/A |
| pCAG-HA-PDZ/C | Szczurkowska et al. (2020) | N/A |
| pCAG-HA-C-term | Szczurkowska et al. (2020) | N/A |
| pCAG-HA-LRR | Szczurkowska et al. (2020) | N/A |
| pCAG-HA-IMR | Szczurkowska et al. (2020) | N/A |
| pCAG-HA-PDZs | Szczurkowska et al. (2020) | N/A |
| pCAG-dTom-LRR | Szczurkowska et al. (2020) | N/A |
| pCAG-sGC-β1 | Szczurkowska et al. (2020) | N/A |
| pCAG-sGC-α1 | Szczurkowska et al. (2020) | N/A |
| pCAG- cGi-500 | Dr. Doris Koesling (Ruhr-University Bochum, Germany) | N/A |
| pCAG-mouse-Myc-WT-PlexinA3 | Alex Kolodkin Lab/This study | N/A |
| pCAG-Myc-ΔCT-PlexinA3 | This study | N/A |
| pCAG-Myc-ΔC1-PlexinA3 | This study | N/A |
| pCAG-Myc-ΔH/RBD-PlexinA3 | This study | N/A |
| pCAG-Myc-ΔC2-PlexinA3 | This study | N/A |
| pCAG-Myc-C1-PlexinA3 | This study | N/A |
| pCAG-Myc- H/RBD-PlexinA3 | This study | N/A |
| pCAG-Myc-C2-PlexinA3 | This study | N/A |
| pCAG-Flag-PlexinA3-CD | This study | N/A |
| pCAG-dTomato | Szczurkowska et al. (2020) | N/A |
| pCAG-EGFP | Szczurkowska et al. (2020) | N/A |
| Software and algorithms | ||
| ImageJ Fiji software | RRID:SCR_002285 | |
| Prism v8.1.2 | GraphPad | RRID:SCR_002798 |
| IMRS 7.3.1 | Bitplane | RRID:SCR_007370 |
Highlights.
Sema3A co-receptor PlexinA3 interacts with the Scribble scaffold for cGMP synthesis
Sema3A/PlexinA3 association with Scribble mediates cGMP increase in dendrites
Sema3A/PlexinA3 mediate bipolar polarity and apical dendrite development via cGMP
Sema3A/PlexinA3-mediated localized cGMP increase may direct leading-edge polarity
ACKNOWLEDGMENTS
We thank Dr. S. Halegoua, Dr. D. Talmage, and Dr. Craig Evinger for critical comments on the manuscript. This work was supported by grants from NIH, NS084111, NS114914, and NS119512 (M.S.), and NSF, IOS1556968 and IOS2034864 (T.S.T.). We thank Dr. V. Nikolaev (University Medical Center Hamburg-Eppendorf) for kindly providing the cGMP FRET reporter cGi-500. We thank Dr. A. Kolodkin (Johns Hopkins University) for kindly providing the Sema3A−/− mice.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110483
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science.
REFERENCES
- Arimura N, and Kaibuchi K (2007). Neuronal polarity: from extracellular signals to intracellular mechanisms. Nat. Rev. Neurosci 8, 194–205. [DOI] [PubMed] [Google Scholar]
- Bagri A, Cheng HJ, Yaron A, Pleasure SJ, and Tessier-Lavigne M (2003). Stereotyped pruning of long hippocampal axon branches triggered by retraction inducers of the semaphorin family. Cell 113, 285–299. [DOI] [PubMed] [Google Scholar]
- Barnes AP, Lilley BN, Pan YA, Plummer LJ, Powell AW, Raines AN, Sanes JR, and Polleux F (2007). LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell 129, 549–563. [DOI] [PubMed] [Google Scholar]
- Behar O, Golden JA, Mashimo H, Schoen FJ, and Fishman MC (1996). Semaphorin III is needed for normal patterning and growth of nerves, bones and heart. Nature 383, 525–528. [DOI] [PubMed] [Google Scholar]
- Chedotal A, Del Rio JA, Ruiz M, He Z, Borrell V, de Castro F, Ezan F, Goodman CS, Tessier-Lavigne M, Sotelo C, et al. (1998). Semaphorins III and IV repel hippocampal axons via two distinct receptors. Development 125, 4313–4323. [DOI] [PubMed] [Google Scholar]
- Chen G, Sima J, Jin M, Wang KY, Xue XJ, Zheng W, Ding YQ, and Yuan XB (2008). Semaphorin-3A guides radial migration of cortical neurons during development. Nat. Neurosci 11, 36–44. [DOI] [PubMed] [Google Scholar]
- Cheng HJ, Bagri A, Yaron A, Stein E, Pleasure SJ, and Tessier-Lavigne M (2001). Plexin-A3 mediates semaphorin signaling and regulates the development of hippocampal axonal projections. Neuron 32, 249–263. [DOI] [PubMed] [Google Scholar]
- Cheng PL, Lu H, Shelly M, Gao H, and Poo MM (2011a). Phosphorylation of E3 ligase Smurf1 switches its substrate preference in support of axon development. Neuron 69, 231–243. [DOI] [PubMed] [Google Scholar]
- Cheng PL, Song AH, Wong YH, Wang S, Zhang X, and Poo MM (2011b). Self-amplifying autocrine actions of BDNF in axon development. Proc. Natl. Acad. Sci. U S A 108, 18430–18435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley C, Spencer SD, Nishimura MC, Chen KS, Pitts-Meek S, Armanini MP, Ling LH, McMahon SB, Shelton DL, Levinson AD, et al. (1994). Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell 76, 1001–1011. [DOI] [PubMed] [Google Scholar]
- Da Silva JS, Hasegawa T, Miyagi T, Dotti CG, and Abad-Rodriguez J (2005). Asymmetric membrane ganglioside sialidase activity specifies axonal fate. Nat. Neurosci 8, 606–615. [DOI] [PubMed] [Google Scholar]
- Dal Maschio M, Ghezzi D, Bony G, Alabastri A, Deidda G, Brondi M, Sato SS, Zaccaria RP, Di Fabrizio E, Ratto GM, et al. (2012). High-performance and site-directed in utero electroporation by a triple-electrode probe. Nat. Commun 3, 960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danelon V, Goldner R, Martinez E, Gokhman I, Wang K, Yaron A, and Tran TS (2020). Modular and distinct plexin-A4/FARP2/Rac1 signaling controls dendrite morphogenesis. J. Neurosci 40, 5413–5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Anda FC, Pollarolo G, Da Silva JS, Camoletto PG, Feiguin F, and Dotti CG (2005). Centrosome localization determines neuronal polarity. Nature 436, 704–708. [DOI] [PubMed] [Google Scholar]
- Dotti CG, and Banker GA (1987). Experimentally induced alteration in the polarity of developing neurons. Nature 330, 254–256. [DOI] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, and Banker GA (1988). The establishment of polarity by hippocampal neurons in culture. J. Neurosci 8, 1454–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartner A, Fornasiero EF, Munck S, Vennekens K, Seuntjens E, Huttner WB, Valtorta F, and Dotti CG (2012). N-cadherin specifies first asymmetry in developing neurons. EMBO J. 31, 1893–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giger RJ, Cloutier JF, Sahay A, Prinjha RK, Levengood DV, Moore SE, Pickering S, Simmons D, Rastan S, Walsh FS, et al. (2000). Neuropilin-2 is required in vivo for selective axon guidance responses to secreted semaphorins. Neuron 25, 29–41. [DOI] [PubMed] [Google Scholar]
- Hota PK, and Buck M (2012). Plexin structures are coming: opportunities for multilevel investigations of semaphorin guidance receptors, their cell signaling mechanisms, and functions. Cell Mol Life Sci. 69, 3765–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Chihara K, Arimura N, Menager C, Kawano Y, Matsuo N, Nishimura T, Amano M, and Kaibuchi K (2001). CRMP-2 induces axons in cultured hippocampal neurons. Nat. Neurosci 4, 781–782. [DOI] [PubMed] [Google Scholar]
- Jacobson C, Schnapp B, and Banker GA (2006). A change in the selective translocation of the Kinesin-1 motor domain marks the initial specification of the axon. Neuron 49, 797–804. [DOI] [PubMed] [Google Scholar]
- Jiang H, Guo W, Liang X, and Rao Y (2005). Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell 120, 123–135. [DOI] [PubMed] [Google Scholar]
- Jones KR, Farinas I, Backus C, and Reichardt LF (1994). Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell 76, 989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi M, Pan YA, Crump JG, and Sanes JR (2005). Mammalian SAD kinases are required for neuronal polarization. Science 307, 929–932. [DOI] [PubMed] [Google Scholar]
- Kitazawa A, Kubo K, Hayashi K, Matsunaga Y, Ishii K, and Nakajima K (2014). Hippocampal pyramidal neurons switch from a multipolar migration mode to a novel "climbing" migration mode during development. J. Neurosci 34, 1115–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Smeyne RJ, Wurst W, Long LK, Auerbach BA, Joyner AL, and Barbacid M (1993).Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell 75, 113–122. [PubMed] [Google Scholar]
- Koesling D, Bohme E, and Schultz G (1991). Guanylyl cyclases, a growing family of signal-transducing enzymes. FASEB J. 5, 2785–2791. [DOI] [PubMed] [Google Scholar]
- Mata A, Gil V, Perez-Clausell J, Dasilva M, Gonzalez-Calixto MC, Soriano E, Garcia-Verdugo JM, Sanchez-Vives MV, and Del Rio JA (2018). New functions of Semaphorin 3E and its receptor PlexinD1 during developing and adult hippocampal formation. Sci. Rep 8, 1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergia E, Russwurm M, Zoidl G, and Koesling D (2003). Major occurrence of the new alpha2beta1 isoform of NO-sensitive guanylyl cyclase in brain. Cell Signal. 15, 189–195. [DOI] [PubMed] [Google Scholar]
- Moreau MM, Piguel N, Papouin T, Koehl M, Durand CM, Rubio ME, Loll F, Richard EM, Mazzocco C, Racca C, et al. (2010). The planar polarity protein Scribble1 is essential for neuronal plasticity and brain function. J. Neurosci 30, 9738–9752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami Y, Suto F, Shimizu M, Shinoda T, Kameyama T, and Fujisawa H (2001). Differential expression of plexin-A subfamily members in the mouse nervous system. Dev. Dyn 220, 246–258. [DOI] [PubMed] [Google Scholar]
- Nakamura F, Ugajin K, Yamashita N, Okada T, Uchida Y, Taniguchi M, Ohshima T, and Goshima Y (2009). Increased proximal bifurcation of CA1 pyramidal apical dendrites in sema3A mutant mice. J. Comp. Neurol 516, 360–375. [DOI] [PubMed] [Google Scholar]
- Namba T, Kibe Y, Funahashi Y, Nakamuta S, Takano T, Ueno T, Shimada A, Kozawa S, Okamoto M, Shimoda Y, et al. (2014). Pioneering axons regulate neuronal polarization in the developing cerebral cortex. Neuron 81, 814–829. [DOI] [PubMed] [Google Scholar]
- Polleux F, Giger RJ, Ginty DD, Kolodkin AL, and Ghosh A (1998). Patterning of cortical efferent projections by semaphorin-neuropilin interactions. Science 282, 1904–1906. [DOI] [PubMed] [Google Scholar]
- Polleux F, Morrow T, and Ghosh A (2000). Semaphorin 3A is a chemoattractant for cortical apical dendrites. Nature 404, 567–573. [DOI] [PubMed] [Google Scholar]
- Richier L, Williton K, Clattenburg L, Colwill K, O’Brien M, Tsang C, Kolar A, Zinck N, Metalnikov P, Trimble WS, et al. (2010). NOS1AP associates with Scribble and regulates dendritic spine development. J. Neurosci 30, 4796–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohm B, Ottemeyer A, Lohrum M, and Puschel AW (2000). Plexin/neuropilin complexes mediate repulsion by the axonal guidance signal semaphorin 3A. Mech. Dev 93, 95–104. [DOI] [PubMed] [Google Scholar]
- Russwurm M, Behrends S, Harteneck C, and Koesling D (1998). Functional properties of a naturally occurring isoform of soluble guanylyl cyclase. Biochem. J 335 (Pt 1), 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russwurm M, Mullershausen F, Friebe A, Jager R, Russwurm C, and Koesling D (2007). Design of fluorescence resonance energy transfer (FRET)-based cGMP indicators: a systematic approach. Biochem. J 407, 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelly M, Cancedda L, Heilshorn S, Sumbre G, and Poo MM (2007). LKB1/STRAD promotes axon initiation during neuronal polarization. Cell 129, 565–577. [DOI] [PubMed] [Google Scholar]
- Shelly M, Cancedda L, Lim BK, Popescu AT, Cheng PL, Gao H, and Poo MM (2011). Semaphorin3A regulates neuronal polarization by suppressing axon formation and promoting dendrite growth. Neuron 71, 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelly M, Lee SI, Suarato G, Meng Y, and Pautot S (2017). Photolithography-based substrate microfabrication for patterning semaphorin 3A to study neuronal development. Methods Mol. Biol 1493, 321–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelly M, Lim BK, Cancedda L, Heilshorn SC, Gao H, and Poo MM (2010). Local and long-range reciprocal regulation of cAMP and cGMP in axon/dendrite formation. Science 327, 547–552. [DOI] [PubMed] [Google Scholar]
- Shi SH, Jan LY, and Jan YN (2003). Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI 3-kinase activity. Cell 112, 63–75. [DOI] [PubMed] [Google Scholar]
- Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira SA, and Barbacid M (1994). Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature 368, 246–249. [DOI] [PubMed] [Google Scholar]
- Sosa L, Dupraz S, Laurino L, Bollati F, Bisbal M, Caceres A, Pfenninger KH, and Quiroga S (2006). IGF-1 receptor is essential for the establishment of hippocampal neuronal polarity. Nat. Neurosci 9, 993–995. [DOI] [PubMed] [Google Scholar]
- Szczurkowska J, Cwetsch AW, dal Maschio M, Ghezzi D, Ratto GM, and Cancedda L (2016). Targeted in vivo genetic manipulation of the mouse or rat brain by in utero electroporation with a triple-electrode probe. Nat. Protoc 11, 399–412. [DOI] [PubMed] [Google Scholar]
- Szczurkowska J, Lee SI, Guo A, Cwetsch AW, Khan T, Rao S, Walz G, Huber TB, Cancedda L, Pautot S, et al. (2020). A localized scaffold for cGMP increase is required for apical dendrite development. Cell Rep. 31, 107519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Fournier A, Nakamura F, Wang LH, Murakami Y, Kalb RG, Fujisawa H, and Strittmatter SM (1999). Plexin-neuropilin-1 complexes form functional semaphorin-3A receptors. Cell 99, 59–69. [DOI] [PubMed] [Google Scholar]
- Taniguchi M, Yuasa S, Fujisawa H, Naruse I, Saga S, Mishina M, and Yagi T (1997). Disruption of semaphorin III/D gene causes severe abnormality in peripheral nerve projection. Neuron 19, 519–530. [DOI] [PubMed] [Google Scholar]
- Toriyama M, Shimada T, Kim KB, Mitsuba M, Nomura E, Katsuta K, Sakumura Y, Roepstorff P, and Inagaki N (2006). Shootin1: a protein involved in the organization of an asymmetric signal for neuronal polarization. J. Cell Biol 175, 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TS, Rubio ME, Clem RL, Johnson D, Case L, Tessier-Lavigne M, Huganir RL, Ginty DD, and Kolodkin AL (2009). Secreted semaphorins control spine distribution and morphogenesis in the postnatal CNS. Nature 462, 1065–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaron A, Huang PH, Cheng HJ, and Tessier-Lavigne M (2005). Differential requirement for Plexin-A3 and -A4 in mediating responses of sensory and sympathetic neurons to distinct class 3 Semaphorins. Neuron 45, 513–523. [DOI] [PubMed] [Google Scholar]
- Yi JJ, Barnes AP, Hand R, Polleux F, and Ehlers MD (2010). TGF-beta signaling specifies axons during brain development. Cell 142, 144–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, and Kaibuchi K (2005). GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120, 137–149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.