

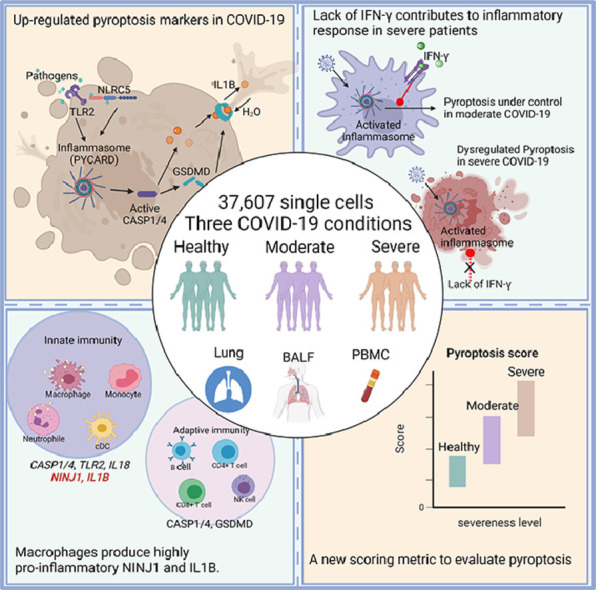
Abstract
Cytokine storm and inflammatory cytokine release syndrome are often found to be associated with severe instances of the 2019 coronavirus disease (COVID-19). However, factors that contribute to the development of the COVID-19-associated cytokine storm and intensify the hyperinflammatory response are not well known. Here, we integratively analyzed scRNAseq data of 37,607 immune cells of eight different cell types from four studies involving COVID-19 patients in either moderate or severe conditions. Our analysis showed that pyroptosis—a lytic, inflammatory type of programmed cell death—may play a critical role in the SARS-CoV-2-induced cytokine storm. The expression of the key markers of pyroptosis, such as pro-inflammatory cytokine genes IL1B and IL18, is significantly higher in moderate and severe COVID-19 patients than in healthy controls. The pattern is more pronounced in macrophages and neutrophils than in adaptive immune cells such as T cells and B cells. Furthermore, the lack of interferon-gamma (IFN-γ) and overexpression of ninjurin 1 (NINJ1) in macrophages may exacerbate the systemic inflammation, as shown in severe COVID-19 patients. Finally, we developed a scoring metric to quantitatively assess single cell's pyroptotic state and demonstrated the use of this pyroptosis signature score to scRNAseq data. Taken together, our study underscores the importance of the pyroptosis pathway and highlights its clinical relevance, suggesting that pyroptosis is a cellular process that can be a potential target for the treatment of COVID-19 associated diseases.
Keywords: Systems immunology, COVID-19, Pyroptosis score, Single-cell RNA-seq, IFN-γ, Marker genes
Graphical abstract
Introduction
Coronavirus disease 2019 (COVID-19) is an unprecedented disease caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). COVID-19 usually causes mild cold-like symptoms, but in certain cases, serious illness with deadly consequences is developed. Cytokine storm can develop at a systemic level in COVID-19 patients, especially in severe cases, according to clinical research. Cytokine blockade such as glucocorticoids can improve the clinical outcome of most severe COVID-19 patients who are at risk of respiratory collapse [1]. However, the selection of patients and the timing of the treatment are critical [2]. Thus, a better understanding of the initiation of the systemic cytokine storm is essential to determine the disease state and choose effective treatments.
Pyroptosis, a regulated form of cell death induced by pathogens, is known to intensify the cytokine storm in severe COVID-19 [3,4]. The secretion of cytokines during the pyroptosis process is triggered by inflammasome assembly and activation. The inflammasome is a multiprotein complex recruited during the innate immune response. The inflammasome comprises of three basic protein units: (1) a sensor molecule such as a nucleotide oligomerization domain (NOD)-like receptor (NLR) or an absent in melanoma 2 (AIM2)-like receptor (ALR), (2) the adaptor PYCARD, often referred to as apoptosis-associated speck-like protein containing a caspase recruitment domain, and (3) pro-caspase 1, the cysteine protease that can initiate pyroptosis. The activation of the inflammasome leads to cell membrane damage and rapid release of pro-inflammatory cytokines, which induce the formation of the cytokine storm.
Several lines of evidence have been accumulated to support the activation of inflammasome and pyroptosis, and their critical roles, in severe COVID-19 cases [5,6]. Pyroptosis is found to be associated with caspase-1 activation, GSDMD cleavage, and enhanced pro-inflammatory cytokine levels in primary monocytes and macrophages of COVID-19 patients [1,6]. Other studies also reported elevated IL1B levels, which is an indicator of pyroptosis, in COVID-19 cases [7]. However, it is unclear whether pyroptosis occurs and contributes to the cytokine storm in other cell types, such as B cells, CD4+ T cells, dendritic cells, and neutrophils. It is also unclear which cell type contributes most to the inflammatory niche. To address these questions, we integrated four publicly available single-cell RNA-seq (scRNAseq) datasets, which contain a total of 37,607 cells spanning eight different cell types from healthy individuals and moderate, severe cases. Our analysis revealed pyroptosis marker gene expression increases in moderate and severe COVID-19 patients, including CASP1, CASP4, TLR2, IL18, IL1B and GSDMD. By looking into different pyroptosis marker genes’ expression patterns in different cell types and disease conditions, we found that not all the cell types play equal roles in the initiation of pyroptosis. Phagocytes, especially macrophages and neutrophils, play a more important role in the pyroptosis-related cytokine storm in comparison to lymphocytes such as B cells and natural killer (NK) cells. Results suggested that the innate immune system is a major player in patients’ responses to the virus infection. In addition, we found that a lack of IFN-γ in macrophages is accompanied by an excessive expression level of NINJ1, the encoder of a membrane damage protein, from severe patients. Our findings indicated that pyroptosis is closely related to the clinical outcome of COVID-19, and the cellular process may be exploited as a target for therapeutic use.
Methods
Datasets
Data used in the analysis of this study was gathered from four previous studies [8], [9], [10], [11] and comprised four datasets, namely the peripheral blood mononuclear cell dataset (PBMC), the bronchoalveolar lavage fluid dataset 1 (BALF1), the bronchoalveolar lavage fluid dataset 2 (BALF2), and the control dataset (Healthy lung) (Supplementary Fig. S1). The PBMC dataset [8] contains scRNAseq data from six healthy donors and eight COVID-19 patients, three of whom developed acute respiratory distress syndrome. The BALF1 dataset from the COVID-19 Cell Atlas [10] contains scRNAseq data of three patients with moderate COVID-19 and three patients with severe/critical infection. The BALF2 dataset from [11] contains scRNAseq data of two patients with moderate and 20 with severe COVID-19. The Healthy lung dataset [9] contains scRNAseq data of five healthy donors. The curation and analysis of the datasets are depicted in Fig. 1 . We combined the datasets and divided the samples into three categories: healthy, moderate, and severe. Patients who did not require mechanical ventilation or were discharged alive were designated as moderate patients. Patients that required mechanical ventilation, intubation or died were designated as severe patients. The datasets, which included samples from different organs and tissues, were merged and integrated to form the described categories. The cell type makeup of the COVID-19 patient groups and healthy controls was visualized using uniform manifold approximation and projection (UMAP) [12]. The final combined dataset contains 37,607 single cells from eight different cell types.
Fig. 1.

Multi-tissue and multi-stage scRNAseq data integration of COVID-19 patients and healthy controls. The PBMC dataset was collected from six healthy donors and eight patients separated into moderate and severe COVID-19 conditions, BALF1 and BALF2 from moderate and severe conditions, and the healthy lung dataset from healthy donors. cDC, dendritic cells. A. Overview of the cell types in the integrated single-cell transcriptomes of 37,607 cells derived from COVID-19 patients and healthy controls. Cells are colored by the cell type. B. The cell types in the healthy lung group, PBMC dataset, BALF dataset, and BALF2 dataset. C. The COVID-19 disease conditions in the integrated and D. Separate datasets. Cells are colored according to their COVID-19 condition. The orange color stands for the severe condition where the patients received mechanical ventilation or died from the SARS-CoV-2. The purple color stands for patients diagnosed as COVID-19-positive but only showed mild symptoms. The green color stands for the healthy group. E. The natural log-normalized expression levels of the eight pyroptosis marker genes across cell types, i.e., genes for two caspases: CASP1 and CASP4, two inflammatory cytokines: IL1B and IL18, two membrane damage proteins: NINJ1 and GSDMD, and two pattern recognition receptors: TLR2 and NLRC5.
Single-cell data analysis
The data analysis was carried out using Seurat (v4.0.2) [13] in R software environment (v4.1.0). Individual expression matrices were used to construct Seurat objects. The number of unique molecular identifiers (UMIs) in each cell was adjusted using the “NormalizeData” function, which first divides UMIs by the total counts for that cell, then multiplied by the default scale factor of 10,000, and then natural log-transformed using log1p. The “ScaleData” function was used to scale the normalized data such that the mean expression across cells was 0 and the variance 1. To group comparable cells from each dataset, principal component analysis [14] was employed by the “RunPCA” function in Seurat to reduce the dimensionality of the data. We performed UMAP using the “RunUMAP” function in Seurat to display high-dimensional cellular data. The “RunHarmony” function in Harmony (v0.1.0) [15] was used to remove the batch effect across the four datasets. The clustering was computed using the smart local moving (SLM) algorithm [16] in Seurat's “FindClusters” function.
Cell type annotation
We used the cell type annotation of each dataset from their original papers. We also verified each cell type annotation for all datasets by finding differentially expressed (DE) genes for each cluster using Seurat's implementation of the MAST [17] test and comparing those markers to known cell-type-specific genes from previous datasets [18], [19], [20], [21], [22]. The marker gene expressions are shown with dot plots in Supplementary Fig. S2.
Gene set enrichment analysis
The fast gene set enrichment analysis (fgsea) package (v1.18.0) [23] was used to perform the gene set enrichment analysis (GSEA) with the pre-ranked DE genes. To obtain the ranked gene list, DE genes were sorted according to the signed fold change. The function “fgseaMultilevel” in the fgsea package was then used with default settings to query the Bioplanet database [24] and identify enriched signaling pathways. Permutation test with Benjamini–Hochberg multiple test correction [25] was used to determine the statistical significance of enriched pathways.
Single-cell pyroptosis signature scoring
We calculated pyroptosis signature scores for single cells based on the expression of eight pyroptosis marker genes: CASP1, CASP4, IL18, IL1B, NINJ1, GSDMD, TLR2, and NLRC5, in each cell. The marker genes were selected according to their roles during the pyroptosis process. If a given cell shows a high level of expression of the eight marker genes collectively, then the pyroptosis signature score is high. We used the difference between the total expression of the eight marker genes and the total expression of eight selected random genes as a scoring metric in order to quantify the collective expression level, which we hereafter call single-cell pyroptosis signature scoring.
Specifically, given a gene expression matrix of dimension , where is the number of genes and the number of cells, we first calculated the mean µ and standard deviation for each gene and then calculated the squared coefficients of variation: . Next, we randomly sampled genes with similar µ and to the selected pyroptosis markers, and calculated the proptosis score for a cell k using the formula: , where is the expression level of the i th gene in the pyroptosis pathway and the expression level of the corresponding, pre-selected random gene.
The corresponding random gene for the i th marker gene was selected from a pool of random genes with similar natural log-transformed µ and . For a random gene to be in the pool, its µ and need to be in the ranges defined as follows: and , where and are expression means, and and are squared coefficients of variation of the i th marker gene and random gene, respectively. The range-defining cutoffs (i.e., 0.02 and 1.0 for log-transformed µ and , respectively) were selected so that we had random gene pools of 50 up to 150 genes for different marker genes. We sampled one random gene for each pyroptosis marker out of its corresponding random gene pool. We used all the eight pyroptosis marker genes and their corresponding random genes to calculate one pyroptosis score for each cell.
Comparison of pyroptosis scores between cell groups
The pyroptosis scores were compared between phagocytes and lymphocytes, and between cells of different patient groups, to characterize the pyroptotic states of cell groups. The comparisons were made using two-sample t-tests. The reliability of the comparison results was further assessed using permutation tests when the numbers of cells in two groups differ substantially. Permutation tests were performed as follows: 1000 cells were randomly sampled from each group with replacement. The pyroptosis score was then calculated for every sampled cell in each group. Two-sample t-tests were performed to compare the mean of pyroptosis scores between different groups. A positive t-statistic indicated a higher mean pyroptosis score in the first group than the second. For each paired comparison between two groups, the procedure was repeated 10,000 times to evaluate the significant level of the difference.
Data source and code availability
The PBMC dataset was obtained from the Gene Expression Omnibus (GEO) database using accession GSE150728. The BALF dataset was downloaded using the link https://covid19.cog.sanger.ac.uk/submissions/release2/vento_pbmc_processed.h5ad. The BALF2 dataset was downloaded using the link http://covid19.lambrechtslab.org. The Healthy lung dataset was downloaded using the link https://cellgeni.cog.sanger.ac.uk/tissue-stability/lung.cellxgene.h5ad. The source code for calculating the proptosis score is provided for download at https://github.com/qianxu05172019/Pyroptosis_COVID19.
Results
Increased expression of pyroptosis marker genes in moderate and severe COVID-19 patients
Monocytes infected by SARS-CoV-2 undergo pyroptosis and release pro-inflammatory cytokines, implying that monocytes may be the major driver of cytokine storm [6]. However, it is less clear whether other cell types such as neutrophils, macrophages, and NK cells in the lung and peripheral blood also undergo pyroptosis, thus propagating systemic cytokine storm. To address this question, we collected 37,607 cells spanning eight cell types from four publicly available datasets and integrated them into a single dataset (Fig. 1A). The cell types included four lymphocytes (B cells, CD4+ T cells, CD8+ T cells, and NK cells), which are key components of the adaptive immune system, and four phagocytes (dendritic cells, macrophages, monocytes, and neutrophils), which are key components of the innate immune system. Each dataset contained a slightly different cell type composition (Fig. 1B). For example, neutrophil infiltration in the three datasets which contain COVID-19 patients was higher than the Healthy lung dataset, which is consistent with a previous study [26]. The integrated dataset contained cells from healthy, moderate and severe COVID-19 patients (Fig. 1C). The moderate group consisted of patients who were tested positive for COVID-19 but did not require ventilation or were fully recovered after treatment. The severe group consisted of patients who required ventilation or eventually died. Fig. 1D shows three group conditions for each dataset.
The eight pyroptosis marker genes showed an overall elevated expression level in moderate and severe COVID-19 patients compared with the healthy group (Fig. 1E). The pyroptosis process consists of the protein products encoded by those maker genes. Specifically, two caspases, CASP1 and CASP4, are the initiators of pyroptosis. Gasdermin D (GSDMD) is a pore-forming protein and a well-known trigger of pyroptosis. NINJ1 is a membrane protein essential for pyroptosis-related plasma membrane rupture [27]. The inflammatory cytokine IL1B (IL-1β), a key mediator of the inflammatory response, is processed by CASP1 and released during pyroptosis. IL1B also exacerbates damage during chronic diseases and acute tissue injuries [28]. IL18 is processed and released after inflammasome activation. Additionally, IL18 is a strong indicator of pyroptosis and is used as a predictive biomarker for clinical outcomes of COVID-19 [6]. Our analysis found an overall elevated expression of pyroptosis-associated marker genes, more specifically higher in the severe group of cells.
TLR2 and NLRC5 may be involved in inflammasome activation in COVID-19
Inflammasome activation occurs when molecules from pathogens or released by damaged cells bind to pattern recognition receptors (PRRs), such as NOD-like receptors (NLRs) and toll-like receptors (TLRs) [29]. Subsequently, inflammasome activation induces the production of pro-IL-1 and pro-IL-18 in an NF-β-dependent way [29]. In fact, inflammasomes are categorized and named based on the sensor protein that initiates their activation [30]. NLRs such as NLRP1b, NLRC4, and NLRP3 have well-established roles during inflammasome assembly and pyroptosis initiation [31]. Other NLRs, such as NLRP6, NLRP7, and NLRP12, are known to be involved in the inflammasome signaling pathway.
Additionally, NLRP3 inflammasome is known to be activated in response to SARS-CoV-2 infection [3]. However, it is unclear whether other PRRs are also involved in SARS-CoV-2 inflammasome activation. To address this question, we systematically examined all genes of 23 NLRs and 10 TLRs in the human genome (Supplementary Fig. S3). The examination identified another member of the NLR family, NLRC5, that may participate in the activation of SARS-CoV-2 associated inflammasome. NLRC5 interacts with NLRP3 to cooperatively activate the inflammasome [32]. However, NLRC5 is not well studied for inflammasome function in COVID-19 associated diseases. In our study, NLRC5 showed a significantly higher expression level in the severe condition group than healthy and moderate groups, especially in macrophages (P < 0.0001, t-test) (Supplementary Fig. S4). Collectively, our analysis suggested for the first time that NLRC5 may be involved in the COVID-19 associated inflammasome activation.
We identified TLR2 as another PRR gene that may participate in pyroptosis activation during SARS-CoV-2 infection. In our analysis, TLR2 showed a higher expression in the macrophages, monocytes, and neutrophils of the severe COVID-19 group than in other cell types and the healthy and moderate groups (Supplementary Fig. S3). TLR stimulation can induce the production of pro-inflammatory cytokines and chemokines, such as IL1B and IL6, in the lung macrophages [33]. In addition, TLR2 is reported to be required for inflammasome activation in infection by cytosolic bacterial pathogens [27] and in asthma [34]. In summary, our analysis indicated that TLR2 might play a critical role in the pyroptosis activation during SARS-CoV-2 infection, especially in the triggering cells such as macrophages and neutrophils.
Pyroptotic macrophages stimulate the SARS-CoV-2-associated cytokine storm
Macrophages showed a higher expression of pyroptosis marker genes among all the cells investigated (Fig. 1E, Supplementary Figs. S4, S5), inspiring us to examine macrophages from different COVID-19 conditions. We randomly sampled macrophages and made the numbers of cells from the three conditions more comparable (Fig. 2 A). We performed clustering analysis and DE analysis in order to compare gene expression between cells of different condition groups.
Fig. 2.

Analysis of macrophages for different conditions of COVID-19 patients. A. The clusters of macrophages are colored according to the COVID-19 condition. B. The macrophages are clustered into five groups. For example, cluster #0 mainly consists of cells from the healthy condition, clusters #2 and #3 of cells from the severe condition, and cluster #1 of cells from the moderate condition. C. The joint density of DE genes in macrophages from three COVID-19 conditions (healthy group on the right, moderate group in the middle, and severe group on the left). D. Dot plot showing the expression level of genes in each cluster. E. GSEA analysis of DE genes for cluster #3 (left and middle) and cluster #1 (right).
Macrophages were clustered into five different groups (Fig. 2B). In cluster #3, macrophages (123 cells) from the severe condition showed high expression of three pyroptosis genes (CASP1, IL1B, and NINJ1) (Fig. 2C, left). IL1B is a downstream pro-inflammatory cytokine that is released after CASP1 activation [35]. NINJ1 is a gene encoding a membrane damage protein (Fig. 2C, left). Moreover, NINJ1 is a critical player which mediates plasma membrane rupture during lytic cell death [27]. Thus, macrophages in cluster #3 are likely to induce pyroptosis-associated cell rupture.
Then, DE genes were identified for each cluster (Fig. 2D). The collective function of DE genes from cluster #3 was studied using GSEA analysis, revealing significantly enriched pathways (P < 0.05, permutation test). In brief, the enriched pathways include NF-κB signaling (Fig. 2E, left), NOD signaling, IL-1R signal transduction (Fig. 2E, middle), and MAPK signaling pathways. These pathways play essential roles in pyroptosis. For example, the NF-κB signaling pathway is a common signaling event that follows the PRR activation and also is responsible for transcriptional induction of the pro-inflammatory cytokines [36]. IL-1R signaling pathway can initiate caspase-1 cleavage, pyroptosis, and secretion of pre-synthesized IL-18 [37]. Our analysis suggested that the macrophages from the severe group, compared with the moderate and healthy groups, showed higher pyroptosis activity, which is more likely to lead to cell membrane rupture. These features of macrophages in the severe condition may propagate systemic inflammation and augment immune cell depletion.
Lack of interferons in macrophages may intensify the COVID-19 severity
We then focused on macrophages from the moderate group and performed the DE analysis between cells in cluster #1 (443 cells) and cells in other clusters. GSEA analysis for genes differentially expressed in cluster #1 revealed several pathways, including IFN-α/β and IFN-γ signaling pathways (Fig. 2E, right). IFN-α/β and IFN-γ are structurally unrelated type I and type II interferons (IFNs), respectively, grouped according to receptor specificity and sequence homology. IFNs may be produced and released by host cells in response to viral infection [38]. IFN-α/β can initiate cell-mediated immune responses at the early infection stage [39]. More importantly, IFNs also have anti-inflammation effects as evidenced by inhibiting the transcription of pro-IL-1β as well as IL-1β maturation [40]. IFN-γ is an immunomodulator that can be effective while not inducing pro-inflammatory effects [41], [42], [43].
Unlike the moderate group, DE genes of the macrophages from the severe group were not enriched in the same IFN pathways. Moreover, on the monocyte-derived macrophages subtype, there was a downregulation of both IFN-α/β and IFN-γ signaling pathways in the severe COVID-19 patients (Supplementary Fig. S6). Monocyte-derived macrophages showed increased pyroptosis signatures compared with tissue-resident alveolar macrophages (Supplementary Fig. S5). Thus, our analysis suggested that IFNs in the macrophages from the moderate group might help to reduce tissue damage in the lung. On the other hand, the lack of IFNs in the macrophages of severe COVID-19 patients, specifically the lack of IFN-γ, might be a factor that intensifies the disease by causing uncontrolled inflammation.
Single-cell pyroptosis signature scoring
We found a positive association between the level of pyroptosis and the symptomatic severeness of patients, thus suggesting the pyroptosis is clinically relevant. Therefore, a scoring system based on pyroptosis was proposed to indicate the developmental stage of COVID-19. We developed such a scoring system using single-cell pyroptosis signatures in order to evaluate the level of pyroptosis. We selected the same eight pyroptosis markers as in Fig. 1, which are associated with the severity of COVID-19. The expression levels of these eight marker genes were used to evaluate the progress of pyroptosis in specific single cells. Details for scoring metric calculation are presented in Methods. Briefly, we first calculated the sum of all pyroptosis marker genes’ expressions. Then, we randomly subsampled genes with similar mean and variation in gene expression level across cells to the marker genes, regardless of their biological functions. Random genes’ expression levels were considered as the baselines and were compared with the expression levels of marker genes in each cell. The comparison is computed for each cell as the difference between the total expression of pyroptosis marker genes and corresponding random genes (see Methods). This difference is used as a metric to evaluate the pyroptosis state of certain single cells.
Each cell type's pyroptosis score is computed for cells in different COVID-19 conditions using our pyroptosis scoring metric. We found a significantly greater average pyroptosis score in phagocytes of the severe COVID-19 group than the healthy group (P < 0.0001, permutation test), while no difference was observed in lymphocytes (P = 0.16, permutation test). The moderate and severe COVID-19 groups had higher average pyroptosis scores than the healthy control group. This is true for both phagocytes and lymphocytes (Fig. 3 A) but more evident for phagocytes than lymphocytes. The more pronounced pattern for phagocytes was mainly due to the contribution of neutrophils and macrophages (Fig. 3B). For lymphocytes, merely CD8+ T cells showed slightly higher average pyroptosis scores in the moderate and severe COVID-19 groups than the healthy control group (Fig. 3C). We changed the box plot grouping to further illustrate the positive correlation between pyroptosis score and COVID-19 severeness in phagocytes (Fig. 3D). Taken together, our analysis revealed the central role of myeloid-lineage phagocytes, especially the macrophages and neutrophils, in COVID-19 associated hyperinflammation, suggesting the innate immune system has a more critical role than the adaptive immune system in the COVID-19 pathogenesis.
Fig. 3.

Comparison of single-cell pyroptosis scores of different cell types and disease conditions. A. Box plots showing single-cell pyroptosis score comparison between the two types of white blood cells, namely phagocytes and lymphocytes. In the moderate and severe groups, phagocytes (dendritic cells, monocytes, neutrophils, and macrophages) have higher pyroptosis scores than those in the healthy group. In contrast, lymphocytes (B cells, NK, CD8+ T-cells, and CD4+ T-cells) in moderate, severe, and healthy groups have comparatively similar scores. B. Box plots showing single-cell pyroptosis score for phagocytes. Neutrophils and macrophages show an increased level of score across the COVID-19 disease conditions. H, healthy group. M, moderate group. S, Severe group. C. Single-cell pyroptosis score for lymphocytes, also colored as in B. CD8+ T cells show more considerable score difference among different COVID-19 conditions when compared with other lymphocytes, such as B cells. D. Single-cell pyroptosis score comparison among three COVID-19 conditions. The cells are colored by being either lymphocytes or phagocytes.
Discussion
COVID-19 caused over 4.5 million deaths worldwide and cast a shadow across the global industries and economy in the 21st century. The most life-threatening complication in severe COVID-19 patients is the cytokine storm, accompanied by other symptoms such as acute respiratory distress syndrome and subsequent lung fibrosis. According to current data, seriously sick individuals have a higher level of pro-inflammatory cytokines, such as IL-1B, than those who are moderately ill [44]. In addition, lymphopenia is another common characteristic of severe COVID-19 [31] that often indicates a poor prognosis [45]. Our study showed that pyroptosis—an intensely inflammatory form of programmed cell death in which pro-inflammatory cytokines are released—might be the link between lymphopenia and hyperinflammation in the severe COVID-19, which constitute a pathogenic cycle. In this cycle, pyroptosis causes lymphocyte death and inflammatory cytokines release. At the same time, the release of the excessive cytokines attracts more inflammatory cells such as macrophages and neutrophils. The cytokines release and the gathering of inflammatory cells can then propagate inflammation and lymphopenia. In fact, pyroptosis has been considered a culprit behind more than 95% of the death of lymphoid CD4+T cells in HIV-infected patients [46]. Likewise, our results highlighted the importance of the pyroptosis pathway in COVID-19, as evidenced by increased pyroptosis marker gene expression.
This study found an unbalanced activation and inhibition of pyroptosis among different immune cells. Notably, macrophages may intensify the uncontrolled tissue response. Macrophages are reported to initiate the hyperinflammation and cytokine storm in COVID-19 patients [31]. However, it is unclear what causes patients to show diverse macrophage activation stages and disease severity. We found macrophages from the severe group show an increment of pyroptosis signs and plasma membrane rupture over the healthy and moderate groups. The major difference between severe and moderate COVID-19 patients is the lack of interferons in the macrophages of severe COVID-19 patients.
Hence, we developed a single-cell pyroptosis scoring metric in order to evaluate each cell type's pyroptosis state. The scoring metric supports the elevated pyroptosis shown in COVID-19 patients with severe and moderate symptoms. Pyroptosis may propagate the uncontrolled release of pro-inflammatory cytokines and the subsequent lymphopenia. Additionally, we showed that innate immune cells such as macrophages, monocytes, and neutrophils have higher pyroptosis scores. Thus, these innate immune cells should be considered as primary therapeutic targets. Indeed, a potential usage for our scoring metric is to predict the clinical outcome of COVID-19 patients having single-cell data available.
Our study is limited due to the lack of protein-level confirmation of the pyroptosis state. Future studies may focus on evaluating pyroptosis signatures at the protein level, including caspase activation and GSDMD cleavage. Nevertheless, our study provided single-cell transcriptomic evidence for overactivated pyroptosis in severe COVID-19 patients. The pyroptosis-associated pathway may be a therapeutic target to alleviate the cytokine storm, especially in severe COVID-19 patients.
Author contributions
QX conceived the study, performed data analysis, and wrote the manuscript. YY provided critical feedback and contributed to the writing of the manuscript. XZ supervised the study and contributed to the writing of the manuscript. JJC helped shape the workflow, verified the analytical methods, supervised the data analysis, and contributed to the writing of the manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This research was partially funded by Texas A&M University 2019 X-Grant for XZ and JJC and the DoD grant GW200026 and NIH grant R21-AI152050 for JJC.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.immuno.2022.100013.
Appendix. Supplementary materials
References
- 1.Zhang J., et al. Pyroptotic macrophages stimulate the SARS-CoV-2-associated cytokine storm. Cell Mol Immunol. 2021;18(5):1305–1307. doi: 10.1038/s41423-021-00665-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cron R.Q., Caricchio R., Chatham W.W. Calming the cytokine storm in COVID-19. Nat Med. 2021:1–2. doi: 10.1038/s41591-021-01500-9. [DOI] [PubMed] [Google Scholar]
- 3.Rodrigues T.S., et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J Exp Med. 2020;218(3) doi: 10.1084/jem.20201707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freeman T.L., Swartz T.H. Targeting the NLRP3 inflammasome in severe COVID-19. Front Immunol. 2020;11:1518. doi: 10.3389/fimmu.2020.01518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Rivero Vaccari J.C., et al. The inflammasome in times of COVID-19. Front Immunol. 2020;11:2474. doi: 10.3389/fimmu.2020.583373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferreira A.C., et al. SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discov. 2021;7(1):1–12. doi: 10.1038/s41420-021-00428-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chua R.L., et al. COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis. Nat Biotechnol. 2020;38(8):970–979. doi: 10.1038/s41587-020-0602-4. [DOI] [PubMed] [Google Scholar]
- 8.Wilk A.J., et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med. 2020;26(7):1070–1076. doi: 10.1038/s41591-020-0944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Madissoon E., et al. scRNA-seq assessment of the human lung, spleen, and esophagus tissue stability after cold preservation. Genome Biol. 2020;21(1):1–16. doi: 10.1186/s13059-019-1906-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ballestar E., et al. Single cell profiling of COVID-19 patients: an international data resource from multiple tissues. medRxiv. 2020 2020.11.20.20227355. [Google Scholar]
- 11.Wauters E., et al. Discriminating mild from critical COVID-19 by innate and adaptive immune single-cell profiling of bronchoalveolar lavages. Cell Res. 2021;31(3):272–290. doi: 10.1038/s41422-020-00455-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McInnes L., Healy J., Melville J. 2018. Umap: uniform manifold approximation and projection for dimension reduction. arXiv preprint arXiv:1802.03426. [Google Scholar]
- 13.Stuart T., et al. Comprehensive integration of single-cell data. Cell. 2019;177(7):1888–1902. doi: 10.1016/j.cell.2019.05.031. e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wold S., Esbensen K., Geladi P. Principal component analysis. Chemometr Intell Lab Syst. 1987;2(1–3):37–52. [Google Scholar]
- 15.Korsunsky I., et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Method. 2019;16(12):1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waltman L., Van Eck N.J. A smart local moving algorithm for large-scale modularity-based community detection. Eur Phys J B. 2013;86(11):1–14. [Google Scholar]
- 17.Finak G., et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16(1):1–13. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gutierrez-Arcelus M., et al. Lymphocyte innateness defined by transcriptional states reflects a balance between proliferation and effector functions. Nat Commun. 2019;10(1):1–15. doi: 10.1038/s41467-019-08604-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Villani A.-.C., et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. 2017;356(6335):eaah4573. doi: 10.1126/science.aah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang H.M., et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat Biotechnol. 2018;36(1):89–94. doi: 10.1038/nbt.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ravetch J. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 22.Palmer C., et al. Cell-type specific gene expression profiles of leukocytes in human peripheral blood. BMC Genom. 2006;7(1):1–15. doi: 10.1186/1471-2164-7-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korotkevich G., et al. 2021. Fast gene set enrichment analysis. BioRxiv. [Google Scholar]
- 24.Huang R., et al. The NCATS BioPlanet–an integrated platform for exploring the universe of cellular signaling pathways for toxicology, systems biology, and chemical genomics. Front Pharmacol. 2019:445. doi: 10.3389/fphar.2019.00445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Statist Soc. 1995;57(1):289–300. [Google Scholar]
- 26.Park J.H., Lee H.K. Re-analysis of single cell transcriptome reveals that the NR3C1-CXCL8-neutrophil axis determines the severity of COVID-19. Front Immunol. 2020:2145. doi: 10.3389/fimmu.2020.02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kayagaki N., et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature. 2021;591(7848):131–136. doi: 10.1038/s41586-021-03218-7. [DOI] [PubMed] [Google Scholar]
- 28.Lopez-Castejon G., Brough D. Understanding the mechanism of IL-1β secretion. Cytokine Grow Fact Rev. 2011;22(4):189–195. doi: 10.1016/j.cytogfr.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kigerl K.A., et al. Pattern recognition receptors and central nervous system repair. Exp Neurol. 2014;258:5–16. doi: 10.1016/j.expneurol.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Angosto-Bazarra D., et al. Techniques to Study Inflammasome Activation and Inhibition by Small Molecules. Molecules. 2021;26(6):1704. doi: 10.3390/molecules26061704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ragab D., et al. The COVID-19 cytokine storm; what we know so far. Front Immunol. 2020;11:1446. doi: 10.3389/fimmu.2020.01446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davis B., et al. NLRC5 dependent activation of the inflammasome in response to bacteria (114.8) Am Assoc Immnol. 2012 [Google Scholar]
- 33.Grassin-Delyle S., et al. The role of toll-like receptors in the production of cytokines by human lung macrophages. J Innate Immun. 2020;12(1):63–73. doi: 10.1159/000494463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu H.-.M., et al. TLR2-melatonin feedback loop regulates the activation of NLRP3 inflammasome in murine allergic airway inflammation. Front Immunol. 2020;11:172. doi: 10.3389/fimmu.2020.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lamkanfi M., Dixit V.M. Mechanisms and functions of inflammasomes. Cell. 2014;157(5):1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 36.Liu T., et al. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2(1):1–9. doi: 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xing Y., et al. Cutting edge: TRAF6 mediates TLR/IL-1R signaling–induced nontranscriptional priming of the NLRP3 inflammasome. J Immunol. 2017;199(5):1561–1566. doi: 10.4049/jimmunol.1700175. [DOI] [PubMed] [Google Scholar]
- 38.De Andrea M., et al. The interferon system: an overview. Eur J Paed Neurol. 2002;6:A41–A46. doi: 10.1053/ejpn.2002.0573. [DOI] [PubMed] [Google Scholar]
- 39.McNab F., et al. Type I interferons in infectious disease. Nat Rev Immunol. 2015;15(2):87–103. doi: 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guarda G., et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34(2):213–223. doi: 10.1016/j.immuni.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 41.Prokunina-Olsson L., et al. COVID-19 and emerging viral infections: the case for interferon lambda. J Exp Med. 2020;217(5) doi: 10.1084/jem.20200653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robinson B.A., Nice T.J. You can breathe easy: iFNλ handles flu without triggering a damaging inflammatory response. Immunity. 2017;46(5):768–770. doi: 10.1016/j.immuni.2017.04.027. [DOI] [PubMed] [Google Scholar]
- 43.Bach E.A., Aguet M., Schreiber R.D. The IFNγ receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol. 1997;15(1):563–591. doi: 10.1146/annurev.immunol.15.1.563. [DOI] [PubMed] [Google Scholar]
- 44.Tang Y., et al. Cytokine storm in COVID-19: the current evidence and treatment strategies. Front Immunol. 2020;11:1708. doi: 10.3389/fimmu.2020.01708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang I., Pranata R. Lymphopenia in severe coronavirus disease-2019 (COVID-19): systematic review and meta-analysis. J Intensive Care. 2020;8(1):1–10. doi: 10.1186/s40560-020-00453-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doitsh G., et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. 2014;505(7484):509–514. doi: 10.1038/nature12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.