

Abstract
Medulloblastoma (MB) consists of four core molecular subgroups with distinct clinical features and prognoses. Treatment consists of surgery, followed by radiotherapy and cytotoxic chemotherapy. Despite this intensive approach, outcome remains dismal for patients with certain subtypes of MB, namely MYC-amplified Group 3 and TP53-mutated SHH. Using high-throughput assays, six human MB cell lines were screened against a library of 3208 unique compounds. We identified 45 effective compounds from the screen and found that inhibition cell cycle checkpoint kinases (CHK1 and 2) synergistically enhanced the cytotoxic activity of clinically-used chemotherapeutics cyclophosphamide, cisplatin, and gemcitabine. To identify the best-in-class inhibitor, multiple CHK1/2 inhibitors were assessed in mice bearing intracranial MB. When combined with DNA-damaging chemotherapeutics, CHK1/2 inhibition reduced tumor burden and significantly increased survival of animals with high-risk MB, across multiple different models. In total, we tested 14 different models, representing distinct MB subgroups, and data were validated in three independent laboratories. Pharmacodynamics studies confirmed central nervous system penetration. In mice, combination treatment significantly increased DNA damage and apoptosis compared to chemotherapy alone, and studies with cultured cells showed that CHK inhibition disrupted chemotherapy-induced cell cycle arrest. Our findings indicated CHK1/2 inhibition, specifically with LY2606368 (prexasertib), has strong chemosensitizing activity in MB that warrants further clinical investigation. Moreover, these data demonstrated that we developed a robust and collaborative preclinical assessment platform that can be used to identify potentially effective new therapies for clinical evaluation for pediatric MB.
One Sentence Summary:
The CHK1/2 inhibitor LY2606368 (prexasertib) sensitizes high-risk medulloblastoma to chemotherapy and improves survival in multiple in vivo models.
Introduction
Arising below the tentorium in the cerebellum or fourth ventricle of the brain, medulloblastoma (MB) is the most common malignant childhood brain cancer (1). Maximal safe tumor resection followed by craniospinal irradiation (CSI) and chemotherapy represent the standard treatment regimen for MB (2). Despite improvements in survival rates over the past 50 years, these treatment modalities cause severe side effects, including secondary malignancies, intellectual loss, endocrine problems, ototoxicity, myelosuppression, cardiotoxicity, hepatotoxicity, and renal toxicity (3).
Historically, MBs have been clinically managed as a single entity, stratified on the clinical criteria of age, evidence of metastases, histopathological subtypes, and the extent of surgical resection. However, extensive molecular analyses uniformly show that MB is a heterogeneous disease consisting of four core subgroups termed WNT, SHH, Group 3 (G3), and Group 4 (G4). Integrating multiple molecular data sources further refined stratification into as many as 13 subtypes: WNT; SHHα, β, γ, or δ; and G3 or G4 subtypes I through VIII (4). These subtypes exhibit diverse responses to current treatments, with the poorest survival rates for MYC-amplified G3 (G3γ or G3 or G4 subtype II, named herein as G3-II) and TP53-mutated SHH (SHHα) or excellent survival for WNT MB. This enhanced knowledge provides needed insight to modify and stratify therapy. Careful reductions of conventional therapy may be explored in patients whose tumors have the lowest risk of relapse to reduce treatment-related morbidities. Conversely, escalation or modification of treatment can be considered for patients with the highest risk of relapse. Such approaches are currently being explored in three international clinical trials: SJMB12 (NCT01878617), SIOP-PNET5 (NCT02066220) and ACNS1422 (NCT02724579).
To develop more rational treatments for discrete MB subtypes, we and others are generating preclinical models of each disease subtype, using either patient-derived cells or transformed murine cells. Paired with high-throughput drug screening, such a preclinical testing setup enables the assessment of many more drugs than could be evaluated in the clinic. A previous drug screen using murine G3 MB cells identified effective chemotherapeutics that are currently being explored clinically in an first-line setting (5). Another screen, also using murine G3 MB cells, unveiled potential efficacy of combining histone deacetylase (HDAC) inhibitors with inhibitors of phosphoinositide-3 kinase (PI3K) (6). In addition, extensive molecular interrogation of human MB has yielded therapeutic leads for these cancers, including inhibitors of the chromatin-remodeling enzymes EZH2 and BRD4 (7,8). Such leads are intriguing; however, as single agents, they will predictably fail if tested in an early phase clinical trial offered to patients after they fail all proven therapies, because MB becomes highly treatment-resistant at relapse (9).
We designed an experimental strategy to identify drugs that enhance the efficacy of currently used MB chemotherapies to discover compounds with the potential for inclusion in first-line clinical trials, rather than at disease relapse. This led to the development of an evaluation pipeline that includes screening drug libraries for agents active against patient-derived MB cells, prioritizing compounds predicted to cross the blood-brain barrier (BBB), confirming BBB penetration, and then identifying those that enhance the activity of current clinical treatments. To verify efficacy, optimal combinations are tested in multiple different models to ensure we only recommend the most promising for future clinical trials. G3-II MB patients with MYC overexpression or amplification and SHHα patients with mutations in TP53 have the worst prognosis, so we focused on these subtypes. Here, we demonstrated that the rigorous preclinical evaluation of drug combinations that pair rationally selected compounds with conventional chemotherapies identified regimens that enhance treatment response in preclinical animal models and thereby have potential to increase survival rates in patients.
Results
High-throughput drug screening identifies MB-selective agents with diverse mechanisms of action
G3-II MB patients have poor survival outcomes; therefore, we evaluated this subtype first using our pipeline (Fig. 1A). Using a panel of 6 patient-derived G3-II cell lines, we screened several compound libraries, including US Food and Drug Administration (FDA)-approved drugs, oncology drugs currently under clinical investigation (NCI Oncology Set IV), and a kinase inhibitor library (fig. S1A). Of the FDA-approved pharmaceuticals (n = 2888), only 137 drugs inhibited 50% growth in all cell lines when tested at 10 μM (fig. S1B). Dose-response testing identified 21 FDA-approved compounds with potent antiproliferative activity at biologically feasibly concentrations (< 1 μM) against all cell lines (fig. S1B). From the NCI Oncology Set (n = 110), there were 15 compounds with submicromolar ED50 (effective dose resulting in 50% growth inhibition) values for reducing viability by 50% (fig. S1B–C); from the kinase inhibitor library (n = 210), 9 inhibitors exhibited submicromolar efficacy across all cell lines (fig. S1B).
Figure 1. High-throughput screening identifies drugs effective against human G3 MB.

(A) Schematic illustrating the principles of our preclinical research pipeline. (B) The most effective drugs against G3-II MB cell lines grouped according to mechanism of action. FDA-approved drugs are indicated by blue squares, other drugs by red circles. (C) Results from the kinase inhibitor screen. The ED50s for 60 inhibitors tested across all cell lines (rows) are illustrated by the color scale. Each column represents a single drug with the target of the inhibitor illustrated at top. MAPK, mitogen-activated protein kinase; mTOR, mammalian target of rapamycin; PKC, protein kinase C; RSK, ribosomal S6 kinase; EGFR/HER, members of the epidermal growth factor receptor family; VEGFR, vascular endothelial growth factor receptor.
Ultimately, we identified 45 (1.4% of the total number tested) compounds consistently potent at less than 1 μM in all MB cell lines tested (Fig. 1B and fig. S1D). The effective drugs have diverse modes of action compared to each other and to the current clinical treatments, which are predominantly DNA-damaging agents. Of note, we identified several agents, including gemcitabine (GEM), PI3K pathway inhibitors, and HDAC inhibitors (Fig. 1B), in this current screen, validating results from previous independent drug screens and showing the consistency of our approach with the previous studies (5,10). Inhibitors of the cell cycle machinery emerged as a common theme from our data: These included kinase inhibitors targeting checkpoint kinases 1 and 2 (CHK1/2), polo-like kinases (PLKs), cyclin-dependent kinases (CDKs), and Aurora (AUR) kinases (Fig. 1C).
Targeting cell cycle checkpoints for cancer treatment
Several MB therapies currently in first-line clinical use, including cyclophosphamide (CPM), cisplatin (CDDP), GEM, or radiotherapy, promote cancer cell death by inducing DNA damage, suggesting that sensitizing the cells to DNA-damaging agents could be one strategy to improve outcomes (11). Of note, the most effective group of kinase inhibitors that we identified target kinases involved in the regulation of cell cycle arrest following DNA damage, such as CHK1/2, PLK, and CDKs, and have proven chemosensitizing activity in other cancers (12). Several of these have been investigated as single agents and in combination with DNA-damaging chemotherapies in different adult cancers (13). However, such combinations have not been previously reported in pediatric cancer. Dose-response testing confirmed drugs targeting CHK1/2, WEE1, and PLK1 were the most effective, inhibiting proliferation at nanomolar concentrations (Fig. 2A and fig. S2A), whereas the CDK4/6 inhibitor ribociclib was less effective with micromolar ED50 values (fig. S2A).
Figure 2. CHK1/2 inhibitors synergize with DNA-damaging chemotherapies in MB cells.
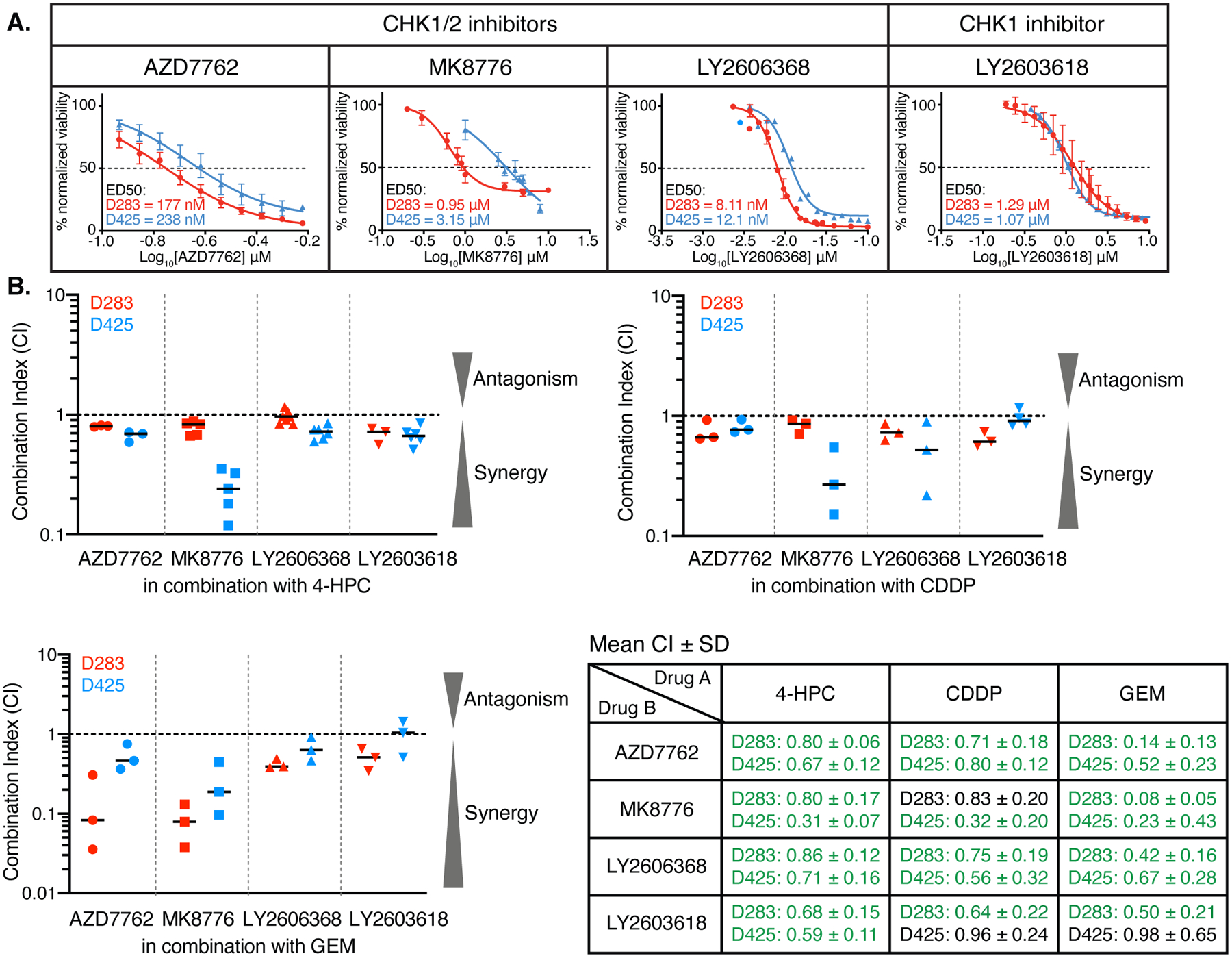
(A) Dose-response analyses of the indicated CHK1/2 or CHK1 inhibitors in D283 (red) and D425 (blue) cells. The ED50 is indicated. Mean ± SEM were calculated from 3–4 independent experiments. (B) Results from assays to determine the interactions between the four CHK inhibitors shown and three MB chemotherapies: 4-HPC, CDDP, and GEM. The mean combination index (CI, horizontal bar) was determined from the indicated pairwise drug combinations using D283 and D425 cells, with 3–5 independent experiments performed. Result for each experiment is indicated by the symbols. Table provides the summary of data shown in B (mean CI ± SD). Synergistic interactions are in green; additive interactions are indicated in black.
We performed drug interaction assays with two MB cell lines (D283 and D425) to identify the compounds that are most effective at enhancing the activity of conventional treatments. These experiments used alamar blue assays to measure the combined effect on cell viability of each cell cycle kinase inhibitor with currently used MB chemotherapeutics (CPM and CDDP), as well as GEM that is being tested [SJMB12 (NCT01878617)]. CPM is a pro-drug; therefore, we used the activated form 4-hydroperoxycyclophosphamide (4-HPC). The Chou-Talalay method was used to evaluate drug:drug interactions (14). With this method, combination index (CI) values less than 1 are synergistic, equal to 1 are additive, and greater than 1 are antagonistic.
The three CHK1/2 inhibitors (AZD7762, MK8776 and LY2606368) and the CHK1-specific inhibitor (LY2603618) exhibited consistent synergistic action with the DNA-damaging agents (Fig. 2B and table S1). These data showed that blocking CHK activity significantly reduces the concentrations of DNA-damaging drugs required to kill MB cells. In contrast, combining 4-HPC or GEM with CDK4/6, WEE1, or PLK inhibitors resulted in mostly additive interactions, with some combinations suggesting antagonism (fig. S2B–D and table S1).
CHK1/2 inhibition improves MB median survival in vivo with LY2606368 the best-in-class inhibitor
In the past, new drugs have frequently failed in the clinic because accurate laboratory models did not exist, and these therapies were not appropriately tested in the preclinical setting (15). We reasoned that an efficient approach to facilitate the clinical translation of optimally effective drugs would be to first identify the “best-in-class” agent through rigorous preclinical testing of multiple different drugs targeting the same protein.
Because we found that CHK1/2 inhibitors enhanced the effects of the three DNA-damaging MB drugs – CPM, CDDP, and GEM – and two phase I trials indicated that CHK1/2 inhibitors are well tolerated in adults with cancer, albeit with some manageable hematological toxicity (16,17), we sought to validate these findings in vivo. The BBB is a major impediment in brain tumor chemotherapy. To confirm that the CHK1/2 inhibitors penetrate into MB in vivo, mice with G3-II D425 MB orthotopic xenografts were treated with AZD7762, MK8776, or LY2606368 and tissue was harvested for immunohistochemistry (fig. S3A). Within 18 h after treatment with any of the inhibitors, CHK1 Ser296 phosphorylation, a marker of kinase activity (18), was undetectable in the tumor tissue (fig. S3B, upper). In addition, phosphorylation of CDC2 Tyr15, a downstream target of CHK signaling, was significantly reduced by treatment with any of the CHK1/2 inhibitors (fig. S3B, lower). These data confirmed successful penetration into MB and inhibition of the DNA damage response pathway, consistent with cerebral microdialysis studies of LY2606368 in mice (19,20).
To identify the optimal CHK inhibitor for MB, four CHK inhibitors (AZD7762, MK8776, LY2606368 and LY2603618) were comprehensively assessed using the D425 orthotopic xenograft model (Fig. 3A). No CHK inhibitor was effective as a single agent either for reducing tumor growth or prolonging survival (Fig. 3B). Based on our observed in vitro synergistic interactions, mice were administered a combination of CPM with each different CHK inhibitor, using previously reported maximum tolerated dosing schedules for mice (21–24). CPM treatment alone improved the median survival of mice, and survival was further improved when combined with any of the dual CHK1/2 inhibitors (Fig. 3B). In combination with CPM, AZD7762 enhanced median survival from 27 days with CPM alone to 54 days, MK8776 enhanced median survival from 31 days with CPM alone to 43 days, and LY2606368 extended median survival from 27 days with CPM alone to 64 days. Improved survival was not observed when CPM was combined with the CHK1-specific inhibitor LY2603618 (Fig. 3B). These effects were confirmed in a second orthotopic model, in which mice harboring D283 G3-II MB were treated with either AZD7762 or LY2606368 alone or in combination with CPM (fig. S3C). As observed in the D425 model, combining either AZD7762 or LY2606368 with CPM extended survival compared to CPM alone. Median survival for mice treated with AZD7762 or LY2606368 combined with CPM was 78 or 100 days, respectively, compared to mice treated with CPM alone (40 days median survival).
Figure 3. CHK inhibitors have significant anti-MB efficacy when combined with CPM.
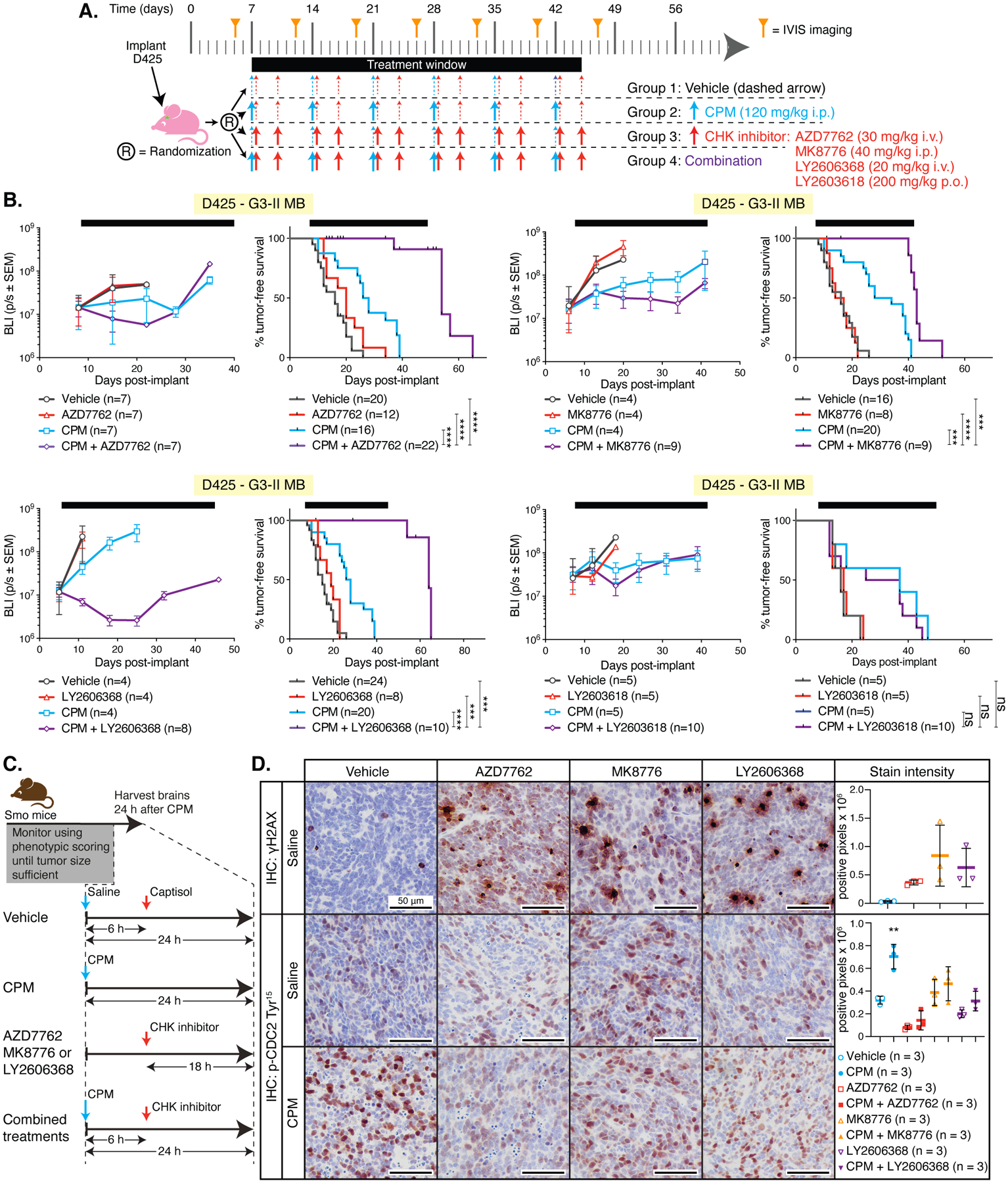
(A) Preclinical mouse treatment protocol for data shown in B. (B) Mice harboring orthotopic D425 MB xenografts were treated as indicated. Left graphs in each pair show bioluminescence (BLI) measurements from a single representative experiment; right graphs show survival curves pooled from multiple independent experiments (except for the experiment with LY26063618, which was only performed once). Number of mice tested per group (n) and comparisons between the combination-treated groups with each other group using Log-rank tests are shown: P < 0.001, ***; P < 0.0001, ****; not significant, (ns). (C) Schematic representation of drug administration in Smo mice and timing of tissue harvest for data shown in D. (D) Representative images of immunohistochemistry (IHC) for γH2AX or phosphorylated (p) CDC2 Tyr15 in MB tissue from Smo mice treated with vehicle, AZD7762, MK8776 or LY606268 with saline or with CPM. Mean staining intensity was determined using Image J software using 3 fields of view from 3 individual mice. Significant differences in γH2AX and p-Cdc2 Tyr15 staining between CPM-treated mice and all other groups was determined by an ordinary one-way ANOVA with Tukey’s multiple comparisons test. P < 0.01, **’; P < 0.001, ***.
In patients, the BBB is disrupted during surgical resection of the tumor; however, metastatic cells may reside at sites distant from the primary disease. Injection of the MB cells in the orthotopic models may cause a similar disruption. Therefore, to determine if CHK1/2 inhibitors penetrate MB and actively inhibit CHK1/2 activity in another mouse model, we used the homozygous ND2-SmoA1 (Smo) transgenic model (25). Smo mice harboring tumors were identified by phenotypic monitoring, and then treated with a single dose of AZD7762, MK8776, or LY2606368 alone, or in combination with CPM (Fig. 3C). Each of the CHK inhibitors caused the accumulation of γH2AX, although this was not statistically significant for MK8776 (Fig. 3D, upper row). The phospho-CHK1 Ser296 antibody is not cross reactive with murine Chk1; therefore, we analyzed phosphorylation of the Chk1 substrate Cdc2 as an indicator of Chk1/2 activity. As expected, CPM treatment increased Cdc2 phosphorylation in Smo MBs, indicating activation of the DNA damage response pathway, and all three CHK inhibitors tested suppressed this response (Fig. 3D, bottom row). During this study, clinical development of AZD7762 was ceased; therefore, we prioritized LY2606368, because it was more likely an option for future clinical studies.
Different salt forms of LY2606368 are available, with the hydrochloride salt typically used in preclinical testing and the mesylate salt (called prexasertib) used clinically. To determine if these salt forms exhibited different effects in vivo, we used the D425 orthotopic xenograft model to compare the formulations and investigate different routes of administration. Inhibition of CHK1 results in inhibition of PP2A phosphatase, which normally dephosphorylates Ser345 on CHK1, thus increased Ser345 phosphorylation indicates CHK1 inhibition in vivo (18). Phospho-CHK1 Ser345 immunohistochemistry showed cytoplasmic staining in vehicle-treated MBs, whereas treatment with either prexasertib or LY2606368 re-localized phospho-CHK1 Ser345 to the nucleus and increased stain intensity (fig. S3D–E). Both salts were equipotent, and no difference in median survival between intravenous (i.v.) or subcutaneous (s.c.) administration was observed (fig. S3F). To confirm that i.v. administration of LY2606368 enables sufficient concentration of the drug to reach the brain, we performed immunohistochemistry on MB tissue from Smo mice. Phospho-CHK1 Ser345 staining was increased following treatment with LY2606368 (fig. S3G–H). Based on these data and the fact that the pediatric phase I clinical trial was undertaken using i.v. prexasertib (NCT02808650) (20), we chose i.v. administration for further experiments.
Overall, our data indicated that CHK1/2 inhibition enhanced the efficacy of CPM in multiple models of MB and that LY2606368 in either salt form was the most effective CHK1/2 inhibitor. Given that this drug is already in early phase clinical trials for adults and children and has high translational potential, we sought to better understand its mechanism of action in MB.
LY2606368 blocks DNA damage-induced cell cycle checkpoint signaling in MB and induces apoptosis
DNA damage induced by chemotherapy results in cell cycle arrest and activation of DNA repair pathways; however, if the extent of DNA damage is too substantial to be repaired, apoptotic pathways are activated (26). To elucidate the mechanisms by which LY2606368 elicits effects in combination with CPM, G3-II MB cells were exposed to 4-HPC with or without LY2606368 and we used immunoblot analysis to detect changes in proteins and phosphorylated proteins indicative of DNA damage and the DNA damage response pathway (fig. S4A–C). As expected, 4-HPC induced DNA damage response signaling as indicated by increased phosphorylated CHK1 Ser296 and CDC2 Tyr15 in 3 MB cell lines compared to DMSO-exposed controls (lane 6 compared to lane 1): D283, D425 and SU-MB002 (fig. S4A–C). LY2606368 blocked CHK1 activity at all concentrations tested as shown by the lack of detectable or reduction in detected phosphorylated CHK1 Ser296 and an apparent increase in CHK1 Ser345 phosphorylation in D283 and D425 cells (fig. S4A–C, lanes 2–5 and 7–10). We did not observe a consistent effect of LY2606368 on CHK2 phosphorylation, either at T68 (the site phosphorylated by the kinase ATM) or the autophosphorylated site Ser516 [a marker of CHK2 activity (27)] (fig. S4A–C), suggesting that LY2606368 predominantly acts through CHK1 inhibition.
Following activation, CHK1/2-mediated signaling inhibits CDKs to arrest or delay the cell cycle, which is necessary for DNA repair and the subsequent restart of cellular replication. The major relevant targets are the phosphatases of the CDC25 family, which dephosphorylate cyclins to activate CDK complexes (28). For example, CDC25-mediated dephosphorylation of CDC2 (also known as CDK1) activates the CDC2 and cyclin B complex. In MB cells exposed to 4-HPC, phosphorylated CDC2 Tyr15 increased, indicating CDC2/Cyclin B complex inhibition and stalled G2/M progression (fig. S4A–C, lanes 1 and 6). However, combining CPM with LY2606368 caused an apparent dose-dependent reduction in CDC2 phosphorylation, suggesting impairment of CPM-induced cell cycle arrest. This corresponded with increased Ser139-phosphorylated histone H2A (γH2AX), indicating that the cells harbor more DNA damage (fig. S4A–C). Even in the absence of 4-HPC, LY2606368 treatment stabilized p53 and increased the abundance of γH2AX (fig. S4A–C, lanes 2–5), suggesting that CHK inhibition affects both the G1/S and G2/M cell cycle checkpoints.
Immunoblotting for cleaved PARP was used to detect apoptosis. In D283 and D425 cell lines, PARP cleavage was induced in cells treated with LY2606368 in the presence or absence of CPM (fig. 4A–B, lanes 2–5 and 7–10). For SU-MB002 cells, cleaved PARP was only detected in cells exposed to the higher concentrations of LY2606268 (fig. S4C, lanes 5, 9, 10).
Figure 4. LY2606368 abrogates cell cycle checkpoint signaling in MB cells and alters cell cycle arrest following treatment with DNA-damaging chemotherapy.

(A, B) D425 cells were treated with the indicated agents (A) or D283 and D425 cells were treated with the indicated agents (B). Cells were examined using flow cytometry for cell cycle phase or apoptosis. Data are mean ± SD from three independent experiments. Significance was determined using unpaired t tests. P < 0.05, *; P < 0.01, **; P < 0.001, ***; P < 0.0001, ****.
Similar findings were obtained with D425 cells treated with AZD7762 (fig. S4D), validating that the results are specific for CHK1/2 inhibition, rather than the result of non-specific effects of LY2606368. Importantly, these findings are consistent with the drug interaction assays and suggested that the synergy between LY2606368 and CPM may be a consequence of disrupting cell cycle checkpoint control following DNA damage. To determine if these effects of LY2606368 were specific for CPM-induced DNA damage or occurred with other DNA-damaging agents, we investigated this CHK inhibitor with GEM, a DNA-damaging drug in a trial for MB (NCT01878617). D283 and D425 MB cells were treated with DMSO or GEM in combination with increasing concentrations of LY2606368 and proteins were analyzed by immunoblotting (fig. S4E–F). Immunoblotting revealed similar results to those obtained with CPM: In cells exposed to LY2606368, phosphorylated CHK1 Ser296 was undetectable even in the presence of GEM. We also observed variably decreased CDC2 phosphorylation and increased PARP cleavage in cells exposed to both GEM and LY2606368, although overall the results supported the synergism between GEM and LY2606368 in MB cells observed in Figure 2.
Given that the immunoblotting results indicated that LY2606368 acted by disruption of DNA damage-induced cell cycle checkpoints, we used flow cytometry to examine cell cycle phase distribution and the induction of apoptosis in MB cells following CPM-induced DNA damage. D425 cells were exposed to DMSO, LY2606368, 4-HPC or a combination of both LY2606368 and 4-HPC. At multiple time points following addition of drug(s), cell cycle distribution was analyzed using EdU to identify cells in S phase, phospho-histone H3 for cells in mitosis, and DAPI for DNA content (Fig. 4A, fig. S5A). No significant differences in the proportion of cells in the various cell cycle phases were observed with LY2606368 treatment compared to DMSO controls. As expected, 4-HPC resulted in rapid cessation of DNA synthesis, as indicated by a reduction in EdU-labeled cells at 24 h, and a marked decrease in mitotic cells by 30h. Thus, 4-HPC-induced DNA damage caused cell cycle arrest of cultured MB cells, resulting in the accumulation of cells in G2 (identified as phospho-histone H3-negative cells with 4n DNA content) by 48 h post-treatment (Fig. 4A). In contrast, when 4-HPC was combined with LY2606368, cells continued incorporating EdU, suggesting that LY2606368 prevented the 4-HPC-induced cessation of DNA synthesis and enabled the cells to progress through to M phase and not arrest in G2 (Fig. 4A). These data corroborated the immunoblotting results suggesting that CHK inhibition disrupts chemotherapy-induced cell cycle checkpoint control.
We also evaluated cultured MB cells for cleaved PARP, as an indicator of apoptotic cells, in response to LY2606368, 4-HPC, or GEM, or combinations of LY2606368 with each of these DNA-damaging agents (fig. S6). 4-HPC alone or in combination with LY2606368 induced similar proportions of apoptotic D425 cells by 48 h (Fig. 4A). GEM alone did not induce apoptosis of D425 cells but did induce apoptosis of D283 cells by 48 h; however, the combination of GEM and LY2606368 induced apoptosis in both cell lines (Fig. 4B).
The mechanisms by which GEM induces DNA damage are different from those of 4-HPC. Conversion of 4-HPC to phosphoramide mustard induces inter- and intra-chromosomal crosslinking of DNA strands (29). In contrast, GEM is a deoxynucleoside analog that disrupts the elongation of new DNA strands during S phase (30). We examined the proportion of cells in mitosis, using phospho-histone H3 as the marker, in both D283 and D425 cells treated with LY2606368, GEM, or a combination of both. We observed reduced proportion of mitotic cells in GEM-treated D425 cells or D283 cells at 8 h, and this returned to control amounts by 48h (Fig. 4B). In contrast, for cells treated with GEM and LY2606368, the proportion of cells in mitosis was either similar to control (D283) or higher than control (D425) by 8 h with fewer cells dividing by 48 h. These results with both 4-HPC and GEM indicated, that although different MB chemotherapeutics induce DNA damage in different ways, LY2606368 sensitizes MB cells to the DNA-damaging effects of chemotherapy through disruption of cell cycle checkpoint control, resulting in induction of apoptosis.
LY2606368 increases CPM-induced DNA damage and enhances MB apoptosis in vivo
The effects of LY2606368 treatment on DNA damage accumulation and apoptosis in vitro were validated in vivo by immunohistochemical analysis of tissue from G3-II MB patient-derived xenograft (PDX) models. In addition, for these and subsequent in vivo experiments we investigate LY2606368 drug combinations in the SHHα subgroup, which is a high-risk molecular subgroup of MB. Patients with this disease subtype exhibit poor prognosis and frequently have Li-Fraumeni syndrome (31). Modelling SHH MBs in vitro is challenging (32–35), thus we opted to assess the efficacy of LY2606368 treatment only using mouse models. Moreover, there is no single PDX model that accurately reflects the G3-II or SHHα MB subtypes. Therefore, we tested treatment efficacy in multiple models to ensure the reproducibility of our data. Mice harboring G3-II or SHHα PDXs were treated with CPM, LY2606368, or the combination and tumors were harvested for immunohistochemistry (Fig. 5A). Tumors were examined for CHK1 inhibition (phospho-CHK1 Ser345), DNA damage accumulation (γH2AX), and tumor cell apoptosis (cleaved caspase 3). This analysis was performed in five G3 and three SHH models, and we present the results from selected examples of each subtype in Fig. 5 (G3: SU-MB002 and SHH: BT084) as our findings from each model were concordant amongst each subtype (remaining models are shown in fig. S6).
Figure 5. Combining LY2606368 with CPM inhibits CHK1 leading to increased DNA damage and apoptosis in G3-II and SHHα MB.
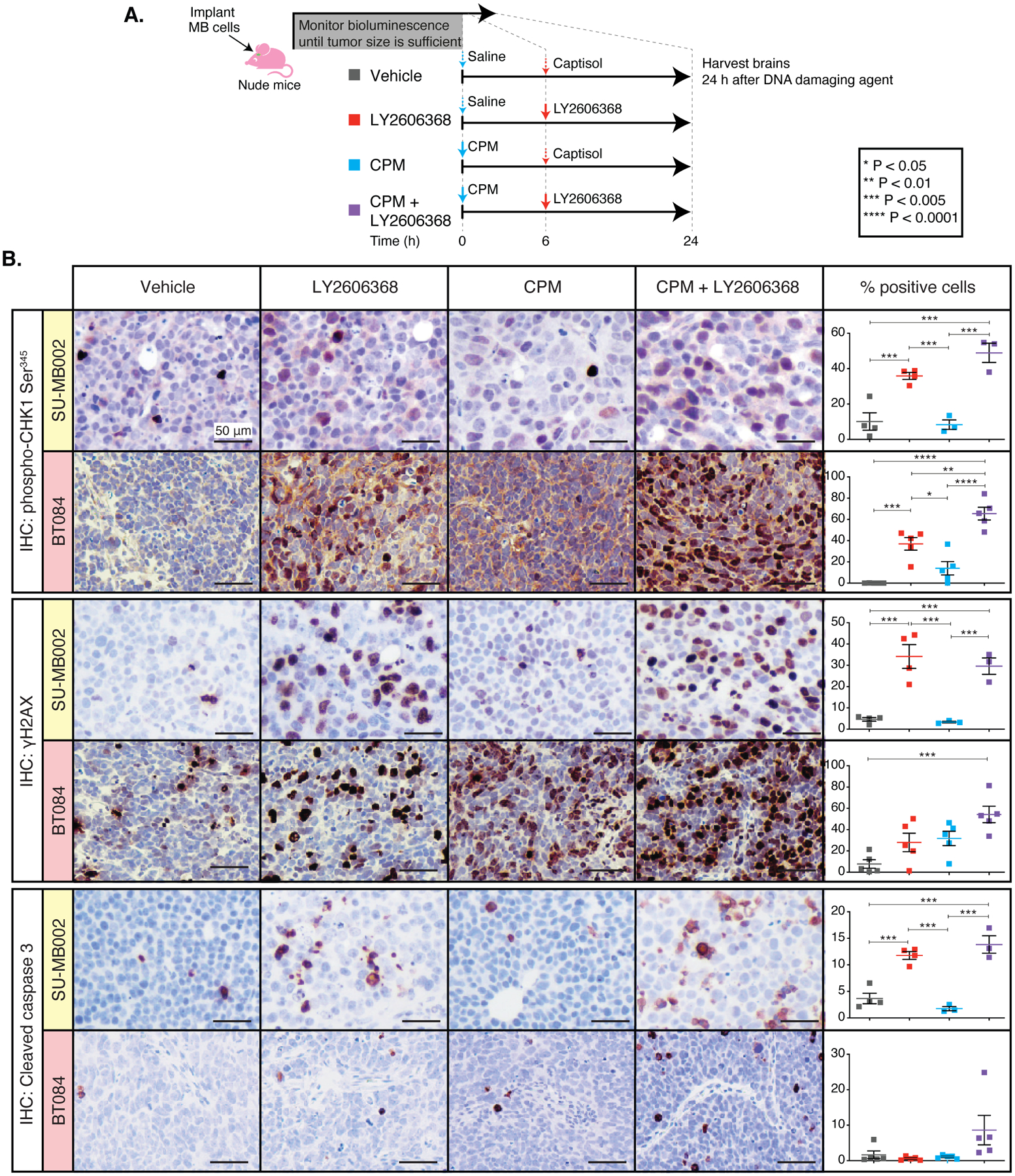
(A) Mouse treatment protocol for data shown in B. (B) Immunohistochemistry (IHC) of G3-II (SU-MB002, yellow shading) or SHHα (BT084, red shading) PDXs treated as indicated. Tumors were stained for phospho-CHK1 Ser345, γH2AX, or cleaved caspase 3. Scale = 50 μm for all images. The percentage of cells with strong nuclear-staining was quantified from 4 high-powered fields of view from multiple animals (n = 3 – 5 per group), and significant differences determined using an ordinary one-way ANOVA with Tukey’s multiple comparisons test. P values are indicated in (A).
Changes in the intensity and subcellular localization of phospho-CHK1 Ser345 confirmed LY2606368 successfully penetrated the tumors and inhibited CHK1 activity. Quantitation of staining intensity revealed this was significant in 4 of the 6 models assessed (p < 0.01 for LY2606368 compared to vehicle control, Fig. 5B and fig. 6B). LY2606368, whether used alone in some models or in combination with CPM in all models shown, increased γH2AX staining, and the combination also increased apoptosis in 5 of the 6 G3-II MB models tested (Fig.5B and fig. S6A–B). In summary, these in vivo results indicated that combining LY2606368 and CPM increases G3-II MB apoptosis and thus has potential to be a more effective therapeutic approach.
Figure 6. Combination therapy consisting of LY2606368 and CPM significantly improves survival in G3-II and SHHα MB.
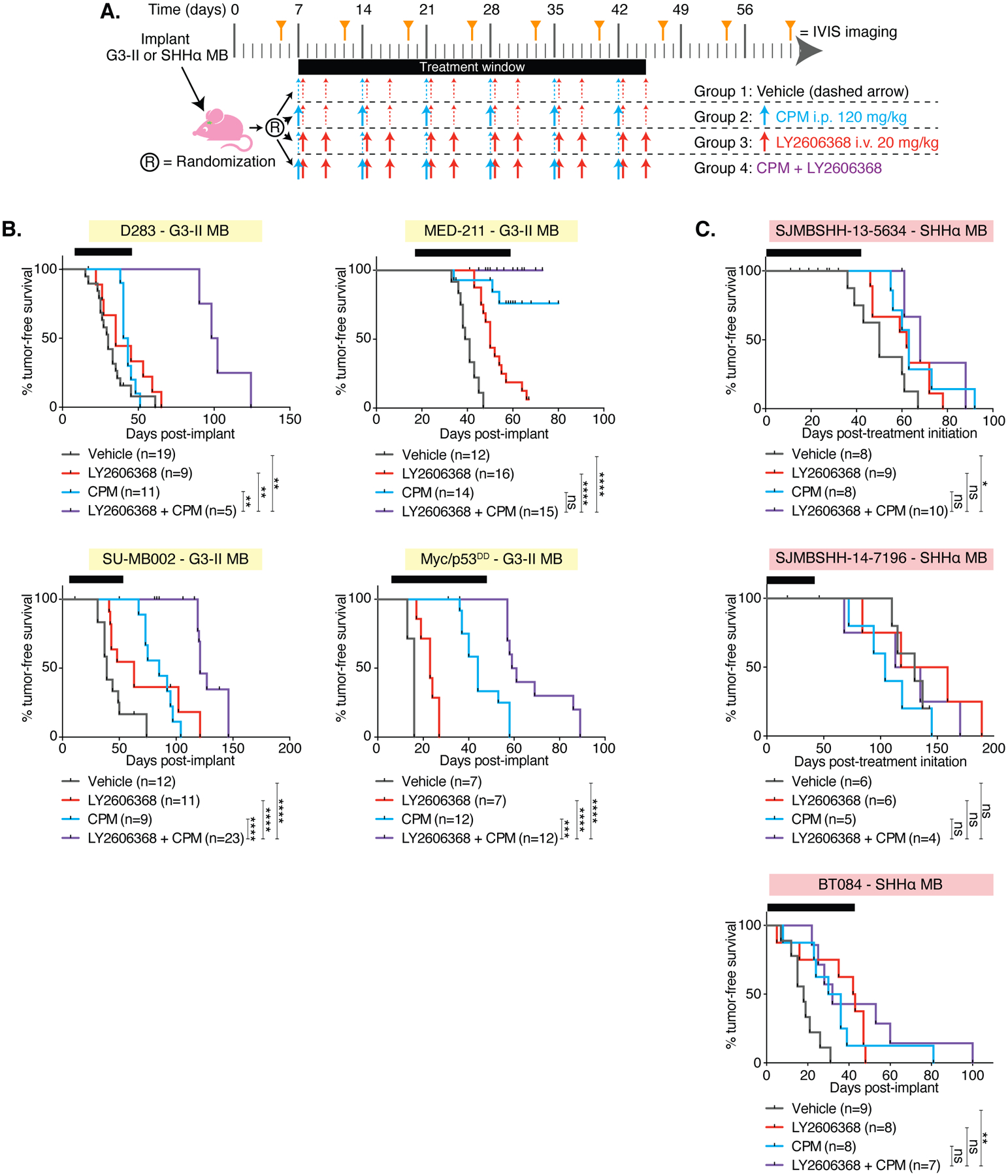
(A) Preclinical mouse treatment protocol for mice with orthotopic G3-II or SHHα MB for data shown in B and C. (B) Survival curves of the G3-II (yellow shading) and (C) SHHα (red shading) models indicated. Log-rank tests compared survival of combination-treated mice with each other group: P < 0.01667, *; P < 0.005, **; P < 0.001, ***; P < 0.0001, ****; not significant (ns), P ≧ 0.01667. The number of mice per group (n) is shown.
Multi-center validation of results using multiple models of MB to validate results
Non-reproducibility in preclinical cancer research is widely recognized as a major concern (36). For robustness ahead of clinical translation, we examined the survival benefits of combination therapy using four additional G3-II and three SHHα PDXs. To provide further validation, these experiments were performed in three different laboratories (two US and one Australian laboratory) using molecularly well-characterized models (table S1). Mice were treated according to a common regimen (Fig. 6A). Results from the D425 G3 model (Fig. 3) were corroborated in these additional orthotopic models of G3-II MB, confirming that the combination of LY2606368 and CPM delayed tumor growth and significantly extended survival compared to CPM alone (Fig. 6B and fig. S7A). Delayed tumor growth was observed in two (SJMBSHH-13-5634 and BT084) out of three SHHα PDX models, which also demonstrated a significant survival benefit with CPM/LY2606368 treatment compared with vehicle-treated mice (P < 0.0143 and P < 0.003, respectively, Fig. 6C and fig. S7B), although this was modest compared to the G3-II models. However, in the SHHα models SJMBSHH-13-5634 and BT084, there was no statistically significant difference in survival between CPM treatment alone and mice administered combination CPM/LY2606368 treatment. A summary of all survival data is provided (fig. S8A–B)
Tumor re-growth following treatment cessation was not due to the development of intrinsic resistance
Despite the significant survival benefits of combined CPM/LY2606368 treatment, with the exception of MED-211 mice, all mice with MB succumbed to disease, raising the question of whether drug resistance had developed. The MED-211 were euthanized for non-tumor-related reasons and were censored in the analysis. To determine if sensitivity to CPM or LY2606368 was altered by treatment in vivo, mice with D283 or D425 tumors were treated with both CPM and LY2606368 (fig. S9A) and monitored until the development of tumor-related morbidity, after which MB cells were extracted and cultured in vitro. The sensitivity of these cells to both 4-HPC and LY2606368 was re-assessed and compared to pre-implant cells that were treatment naïve. Drug treatment in vivo did not alter the sensitivity of these cells to either compound in vitro: The ED50 values for each drug before implantation or after extraction from treated mice were similar (fig. S9B–C). Thus, the morbidity associated with the combination may relate to adverse effects of CPM treatment, which is known to cause serious hematological side effects (37).
Combination therapy using LY2606368 and CDDP or GEM significantly improves median survival in G3 MB
Typical clinical treatment protocols for MB do not just use CPM as the sole chemotherapeutic agent; instead, treatment consists of multi-agent chemotherapy. A common approach combines CPM with vincristine and another DNA-damaging drug CDDP (38). Our goal was to identify agents to combine with existing MB treatment protocols. Because our in vitro data indicated that LY2606368 synergized with CDDP and GEM (Fig. 2), we evaluated the efficacy of combining LY2606368 with these agents in vivo.
Previous studies have successfully treated MB models with a combination of CPM and CDDP to more closely mimic clinical protocols (5). Although this treatment was possible at one institution (SJCHR, table S1), the mouse strains required for several other PDX models did not tolerate the side effects of combined CPM and CDDP treatment, so evaluation of simultaneous CPM, CDDP, and LY2606368 treatment was not possible. Therefore, we tested the pairwise combination of LY2606368 with CDDP in G3-II MB. Mice implanted with D283, D425, SU-MB002, or Myc/p53DD were treated with LY2606368 or CDDP alone or in combination (Fig. 7A) and survival was assessed. Across all models, CDDP alone had little survival benefit (Fig. 7B), and LY2606368 monotherapy was similar to our data reported in Figures 3 and 6 for the same models (fig. S8A). Mice treated with CDDP and LY2606368 demonstrated a significant survival benefit compared to CDDP alone (in 1 out of 4 models) or vehicle controls (in 4 out of 4 models) (fig. S8A). Median survival in all treatment groups generally reflected the tumor growth rates measured throughout each experiment using bioluminescence (fig. S10A), suggesting that these mice succumb to the cancer and not drug-induced side effects.
Figure 7. Combined therapy consisting of LY2606368 and cisplatin significantly improves survival in G3 MB but to a lesser extent than cyclophosphamide.

(A) Preclinical mouse treatment protocol for mice with orthotopic G3-II for data shown in B. (B) Survival curves are shown for the G3-II MB models indicated. Log-rank tests compared the combination-treated group with each other group: p < 0.016667, *; P < 0.005, **; P < 0.001, ***; P < 0.0001, ****; not significant (ns), P ≧ 0.01667. The number of mice per group (n) is shown. (C) Treatment protocol for mice with SU-MB002 or D425 G3 MBs for data shown in D. (D) Immunohistochemistry (IHC) for phospho-CHK1 Ser345, γH2AX, and cleaved caspase 3 was performed and positive nuclei were quantified from 4 sections from 4–5 mice/group (mean ± SEM) with colors corresponding to (C). Significant differences were determined using a one-way ANOVA with Tukey’s multiple comparisons test with p values indicated in (D).
We evaluated the effects of single and combination therapy with CDDP and LY2606368 on CHK1 activity, DNA damage, and apoptosis by immunohistochemistry on tumors from mice with SU-MB002, D425 or D283 MBs (Fig. 7C and fig. S10B). LY2606368 monotherapy or combination therapy with CDDP induced CHK1 Ser345 phosphorylation and altered its subcellular localization in D425 MB only (Fig. 7D and fig. S10B–C). The combination of LY2606368 and CDDP induced DNA damage accumulation in all models; however, this was insufficient to induce apoptosis by 24h in 2 out of the 3 models tested (Fig. 7D and fig. S10B–C). Thus, as with the results for CPM and the combination of CPM and LY2606368, different MB models respond differently to CDDP and the combination of CDDP and LY2606368.
Lastly, we investigated the effects of combining LY2606368 with GEM in vivo. We monitored tumor growth and survival of mice with tumors derived from D283, D425, SU-MB002, Myc/p53DD, or TK-MB870 cells (all G3-II MB models) and treated with GEM, LY2606368, or the combination (Fig. 8A). Delayed tumor growth and extended survival was seen in all five G3-II MB models when LY2606368 was combined with GEM compared to vehicle-treated mice and survival was longer than treatment with GEM alone in three out of five models tested (Fig. 8B and fig S11A). Immunohistochemistry was performed on tumors from mice with G3-II (D425 and TK-MB870) or SHHα (Smo and SJMBSHH-13-5634) MB treated with GEM, LY2606368, or the combination (fig. S11B). In G3-II MB, only tumors from combination-treated mice consistently showed DNA damage accumulation (γH2AX) with concurrently increased apoptosis (fig. S11C). Compared to the G3-II models, no delay in tumor growth and minimal survival benefit was observed for the SHHα models (fig. S12A). DNA damage accumulation was observed in SHHα MBs after GEM/LY2606368 treatment, but this did not result in any marked increase in apoptosis (fig. S12B), which may explain the lack of survival benefit in mice with this subtype of MB.
Figure 8. Therapy combining LY2606368 and GEM significantly improves survival in G3, but not SHH, MB.
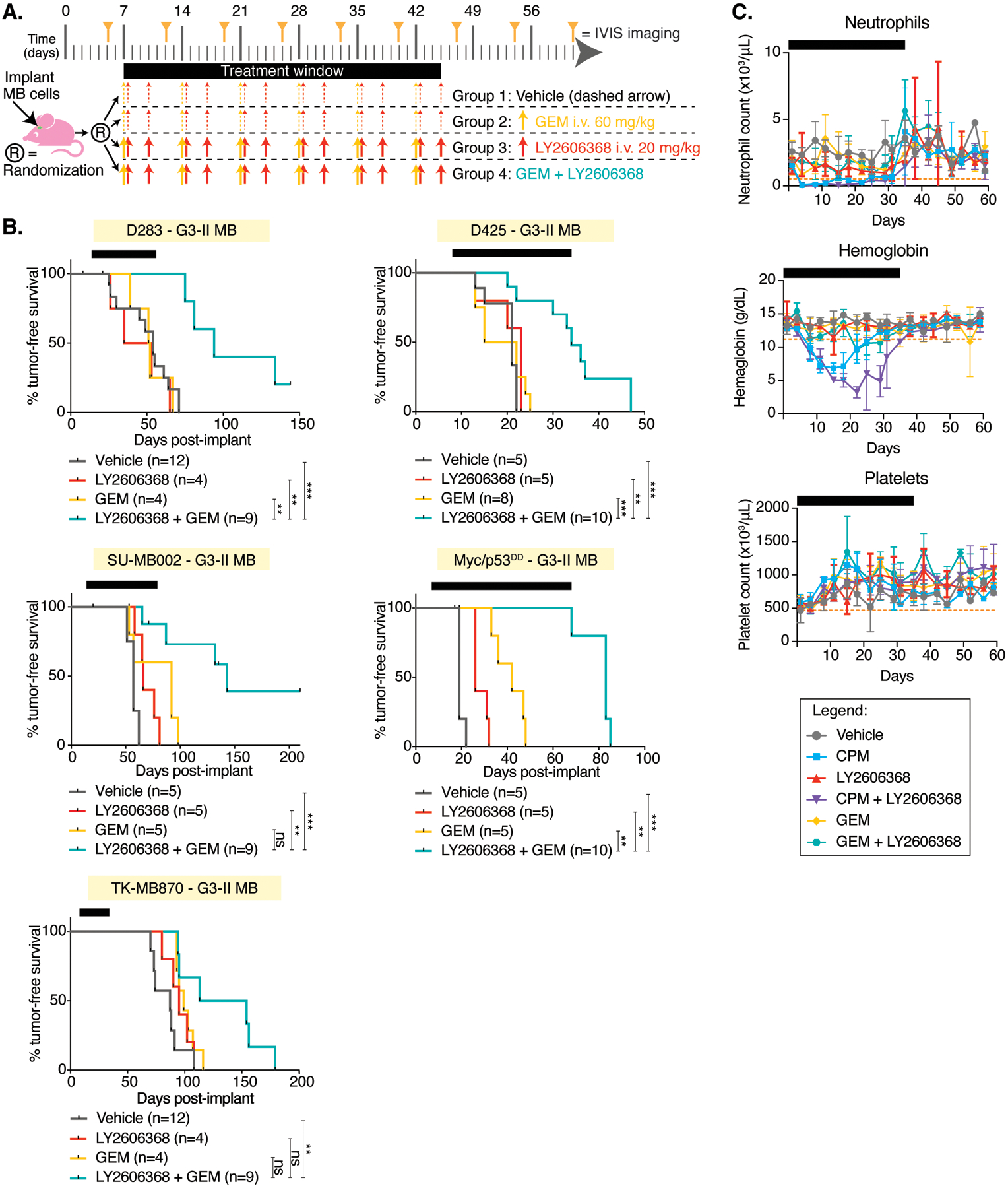
(A) Preclinical treatment protocol for mice with orthotopic G3 or SHHα MB xenografts for the data shown in B. (B) Mice harboring orthotopic G3 (yellow) or SHHα (red) MB xenografts were treated with vehicle (gray), LY2606368 (red), GEM (aqua), or a combination of LY2606368 with GEM (green). Bioluminescence measurements (left) and survival curves (right) were assessed. Comparisons of survival between combination-treated mice with each other group using Log-rank tests are shown with the number (n) of mice per group below each graph: P < 0.01667, *; P < 0.01, **; P < 0.005, ***; not significant (ns). (E-G) Toxicity was monitored twice weekly from the treatment groups as shown (legend applies to all graphs). Treatment duration is indicated by black bars. Blood counts (n=4 per group) from whole blood with ethylenediamine tetra-acetic acid were measured using a Forcyte Hematology Analyzer (Oxford Scientific). Shown are (E) neutrophils, (F) hemoglobin, and (G) platelets. The dotted orange line indicates an average normal murine parameter.
The combination of LY2606368 and CPM, but not LY2606368 and GEM, causes hematologic toxicity
To examine potential side effects of these treatment combinations, blood from treated mice was analyzed. Only mild effects on blood parameters were observed in mice receiving LY2606368 alone. CPM treatment alone or when combined with LY2606368 caused mild and reversible anemia, whereas GEM with or without LY2606368 was well tolerated (Fig. 8C).
Discussion
MB, together with high-grade glioma, is the most common malignant brain cancer in children, yet there remain only four chemotherapeutics commonly used worldwide in clinical protocols (39). Overall survival rates have plateaued in recent decades (40), indicating that conventional therapy has effectively maximized its potential. G3-II and SHHα are especially aggressive disease subtypes and the most likely to be refractory to current therapy (4). Addressing the need for new therapies, we report here the establishment of a preclinical research pipeline to rigorously assess new agents for MB. This involves the establishment of MB models and genetic confirmation of disease reproducibility; in vitro drug screening and drug interaction testing with cultured MB cell lines; studies to confirm tumor penetration of the candidate drugs; and assessment of biological mechanisms of drug action and survival benefits using preclinical models that represent specific disease subtypes. For additional robustness, experiments were performed across multiple independent laboratories to ensure reproducibility. Furthermore, to identify the most effective drug worthy of translation to clinical trial, we directly compared different compounds targeting the same protein(s), different salt formulations, and multiple routes of administration.
The two published high-throughput drug screens reported for MB both utilized genetically-modified murine G3 MB cells (5,6); whereas our study used a collection of human G3 MB cell lines. Although there is debate regarding the validity of cancer cell lines and how well they represent their original tumors, we carefully considered the cells used in our screen and used molecular techniques to select cell cultures that most closely resembled G3 disease. Overall, this methodical approach identified many drugs already known to have clinical effects in MB, such as the vinca alkyloids, as well as multiple compounds identified across the other reports (GEM, HDAC, and PI3K inhibitors (5,6)). Indeed, Morfouace et al. showed the combination of GEM and pemetrexed prolonged survival of MB-bearing mice to a similar extent as conventional chemotherapeutics CPM and CDDP (5). These findings were translated into the SJMB12 first-line clinical trial currently recruiting patients (NCT01878617). The consistency observed in these screens performed in three independent laboratories, using murine or human cells, provides strong evidence that collectively we can identify compounds that can be rapidly translated into clinical trials. Moreover, it shows that the results are not significantly influenced by different culture conditions (serum-free in the case of murine cells or serum-containing media used for human cells).
Because G3 MB cells were particularly sensitive to inhibition of kinases involved in cell cycle control, we focused on these drugs. With cultured cell lines, CHK1/2, WEE1, PLK1, and CDK4/6 inhibitors were effective as single agents. However, in pediatric cancer, it is well established that chemotherapies are most effective when given in combination (41), thus our goal was to identify drugs with potential to be combined with first-line conventional MB chemotherapies. Previously, in vitro assays to detect synergistic interactions have been successfully translated clinically. For example, CDDP and GEM were observed to act synergistically in several adult carcinoma cell lines, with a subsequent clinical trial showing that combination treatment provided superior response rates in patients compared to the historical use of either agent alone (42). Relevant to our study, CHK inhibition following chemotherapy-induced DNA damage increases apoptosis across a broad range of cancer types including adult glioblastoma (22,28,43). Indeed, our drug interaction studies consistently revealed that CHK inhibitors synergized with all three MB chemotherapies tested. Even though CDK4/6, WEE1, and PLK1 act downstream of ATM- and ATR-mediated detection of DNA damage (12), synergistic interactions with CPM or GEM were not observed with these inhibitors. CHK1/2 are also downstream of ATM and ATR in the DNA damage response pathway; however, CHK1/2 are upstream of CDK4/6, WEE1, and PLK1 in regulating the cell cycle (12). Previously, we reported drug interaction studies between the pan-ERBB inhibitor dacomitinib with CPM or vincristine using MB models (44). That study found that drug interactions predicted to be either additive or antagonistic in vitro were antagonistic when evaluated in vivo; thus, we chose to not investigate WEE1, PLK1 or CDK4/6 inhibitors further and instead pursued the clinical potential of CHK inhibitors. In addition to our unbiased screen, several independent studies had previously implicated CHK1 or CHK2 or both as viable targets in MB (45,46). Of note, there is promising clinic data emerging for PLK1 and WEE1 inhibitors (21,47); therefore, it remains of value to determine their effectiveness in vivo as MB therapies.
A key aspect of this work was the comprehensive analysis of combining CHK inhibitors with other MB therapies that are used in a first-line setting, either as standard of care (CPM and CDDP) or in clinical trial (GEM). This is a meaningful translational approach, as our overarching aim was to identify new agents that can be incorporated into existing clinical treatment protocols. Our data with multiple models in mice indicated that combining CHK inhibition with CPM or GEM is effective in G3-II MB. Immunoblotting suggested LY2606368 predominantly acts through CHK1 in MB cells because LY2606368-treatment reduced the amount of phosphorylation of CHK1 and not CHK2 at sites associated with activity. However, in preclinical survival studies, the dual CHK1/2 inhibitors were superior compared to a CHK1-specific inhibitor when used in combination with CPM, with LY2606368 appearing the most effective. Although this may suggest that CHK2 inhibition is important for combination treatment efficacy, it should be recognized that MK8776 is predominantly CHK1-specific with an in vitro inhibitory concentration of 3 nM against CHK1 versus 1.5 μM for CHK2 (48). As such, it is likely that treatment efficacy relies more on CHK1 inhibition; however, CHEK1 gene expression across a panel of nearly 400 cell lines does not appear to correlate with response to LY2606368 (16). Immunohistochemical analysis of orthotopic MB implants and Smo MBs showed that AZD7762, MK8776, and LY2606368 affected intratumoral CHK activity similarly; therefore, it is possible that LY2606368 is simply pharmacodynamically superior to the other compounds at the concentrations tested. This highlights a fundamental hurdle to overcome for brain cancer treatment, which is ensuring that adequate concentrations of the drug reach the tumor. Indeed several studies corroborate our data and confirm LY2606368 penetrates the central nervous system at clinically relevant dosages (19,20).
Mechanistically, flow cytometry revealed that LY2606368 prevented cell cycle arrest following chemotherapy-induced DNA damage and subsequently enhanced apoptosis. Our observation that combination CPM/LY2606368 treatment induced apoptosis in vivo across multiple G3-II MB models, including those with defective p53 function is noteworthy, because loss of p53 function disrupts apoptosis (49); thus, LY2606368 may help overcome p53-mediated treatment resistance. We did not investigate the mechanisms of apoptosis in this study. CHK inhibition causes mitotic catastrophe due to the presence of unresolved DNA breaks (50). CHK1 also has important roles in replication fork stabilisation during S phase (30). Therefore, together with the failure of cells to cease proliferating when DNA damage is present, LY2606368 may enhance DNA damage accumulation due to fork collapse. Indeed, we repeatedly and consistently observed increased γH2AX positivity in MBs treated with LY2606368. The G3 models used here overexpress MYC, a known driver of replication stress (51), and MYC amplification is currently used to select adult cancer patients for a LY2606368 basket trial (NCT02873975). Our finding that G3-II MBs were more responsive to therapy compared to SHHα may be due in part to MYC amplification. Furthermore, the reduced sensitivity of SHHα MB to combination treatment may be due to a disconnect between chemotherapy-induced DNA damage and the induction of apoptosis as a consequence of p53 loss. This is supported by the marked difference in apoptosis (indicated by cleaved caspase 3 staining) induced in BT084 xenografts following CPM/LY2606368 treatment compared to the extent of DNA damage (γH2AX staining).
Limitations of our study include that all immunohistochemical analyses were performed at a single timepoint for each model. This could limit our ability to detect time sensitive effects of treatment such as apoptosis. In addition, the duration of CPM treatment was limited by toxicity. We observed that CPM caused hematological side effects in mice, as it does in children (37); thus, continuation of treatment beyond six weeks was not possible. In addition, the NSG mice used with the MED-211 PDX are particularly susceptible to the hematological side effects of DNA-damaging agents due to the scid mutation (52). It is possible that due to toxicity, this therapy may not be well-tolerated clinically. Alternatively, if hematological side effects can be managed and treatment continued for longer, we speculate that improved disease control might have been observed, especially given the fact that the cells did not develop intrinsic resistance to the inhibitor.
Our preclinical data consistently showed that CHK inhibition enhances the effects of DNA-damaging chemotherapy in MB resulting in improved animal survival. Importantly, our study identified two novel treatment combinations that are more likely to translate into true clinical benefit over others. Specifically, the LY2606368/CPM combination was effective in both G3-II and SHHα medulloblastoma, whereas the LY2606368/GEM combination was only effective in G3-II disease. The lack of efficacy of GEM in SHH MB is consistent with our previous data revealing that GEM combined with pemetrexed significantly increased survival in G3-II MB models but was not efficacious in SHH MB (5). Despite the weaker response observed in SHH MB, the finding that LY2606368/CPM therapy induced a survival benefit over vehicle in several models was sufficient evidence to recommend this treatment given that patients with these aggressive tumors have very poor outcomes. Future research is still required to identify more effective agents for this MB subtype. Of note, LY2606368 combined with CDDP also enhanced survival of mice G3-II MB with minimal systemic toxicity, which is an important finding given that CDDP is used concurrently with CPM in upfront clinical protocols. Lastly, our comparison of different CHK inhibitors indicated that LY2606368 was the superior CHK inhibitor, further validating the capability of our pipeline to select optimal agents for new clinical trials. The level of preclinical evidence that accurately predicts response in the clinic has not yet been established for MB and the ultimate validation of whether studies in mouse models do in fact inform clinical outcomes can only be determined in clinical trial. As such, we have applied these data to rationally inform the development of a new and precise clinical trial for children with MB. Encouragingly, a phase I study assessing LY2606368 monotherapy in adults observed an objective clinical response in two patients and stable disease in several others (16). Our results enabled the development of a phase I/Ib clinical trial for patients with refractory or recurrent G3 or SHH MB named SJ-ELiOT (NCT04023669) to test LY2606368/CPM or LY2606368/GEM combination therapy. A small proportion of relapsed/recurrent patients have G4 MB; however, we have not tested LY2606368 in this molecular subgroup. Because diagnostically it remains challenging to separate G3 and G4 tumors, we have recommended inclusion of patients with G4 MB in this trial. SJ-ELiOT will reveal both safety and efficacy data to justify taking LY2606368 combined with the chemotherapies CPM and GEM to a phase II first-line clinical trial for children with the worst prognosis MB molecular subgroups (SHHα and G3-II) who currently have the least favorable outcomes with current gold standard therapy.
Materials & Methods
Study design
The main objective of this study was to evaluate the efficacy of combining LY2606368 with first-line MB treatments using preclinical models. Sample size calculations were performed based on the known mean (and standard deviation) survival for orthotopic implant models. These defined with 4 mice per group, we would be able to detect a true difference in the mean response of treated and control mice of −9.76 or 9.76 with probability (power) 0.80. The Type I error probability associated with this test of the null hypothesis that the population means of the treated and control groups are equal was 0.05. Individual experimental cohorts were repeated multiple times to ensure reproducibility. Overall animal numbers used are depicted on all figures. For survival studies, tumor size was monitored by bioluminescence using an IVIS Spectrum (Caliper). Once tumors were established, mice were randomized into groups based on bioluminescence intensity prior to initiation of treatment as indicated in the text. Blinding during the conduct of experiments was not feasible due to the route and timing of drug administration. An event was counted when mice required euthanasia due to tumor-related morbidity. Mice requiring euthanasia for non-tumor-related reasons (weight loss, infection, physical trauma) were censored. Animal experiments were approved by the Animal Ethics Committee of the Telethon Kids Institute and performed in accordance with Australia’s Code for the Care and Use of Animals for Scientific Purposes. Additional animal studies were conducted according to the NIH guidelines and approved by the Animal Care and Usage Committees of St Jude Children’s Research Hospital and the Sanford-Burnham-Prebys Medical Discovery Institute.
MB cell lines and culture conditions
PER547 cells were a gift from Prof Ursula Kees of Telethon Kids Institute (53). D425, D283, D341, and D458 were gifted from Prof Darell Bigner of Duke University (54), and SU-MB002 cells from Dr Yoon-Jae Cho of Oregon Health and Science University (7). STR analysis and sequencing of previously reported genetic alterations confirmed the identity of all cell lines. DNA methylation array, MYC expression or TP53 mutation were used to determine molecular subgroup (55). Of note, D283 MB cells have been reported as both G3 and G4 (56–58) but our data classifies them as G3-II. Mycoplasma-free cells were transduced to express luciferase using the retroviral expression construct MSCV-ires-pacLuc2 (D283), or lentiviral expression construct pCL20-MSCV-GFP-ires-Luc2 (PER547, MB002, and D425). Constructs were kindly provided by Drs Suzanne Baker, Arthur Nienhuis, and Richard Williams of St Jude Children’s Research Hospital. Cells were cultured at 37°C/5% CO2 in antibiotic-free medium supplemented with Glutamax (all Gibco) as follows: PER547: RPMI/10% fetal bovine serum (FBS, Cell Sera), D283: DMEM/10% FBS, D425: MEM-alpha/HEPES/10% FBS, and D341: MEM-alpha/20% FBS. SU-MB002 and D458 cells were cultured as previously described (7).
Drug screen
The screen was performed at the Australian Cancer Research Foundation Drug Discovery Centre at Children’s Cancer Institute, Australia. MB cells (1,000/50 μL) in exponential growth phase were seeded into each well of 384-well plates containing drugs. After 72 h, cell viability was determined by alamar blue assay (0.6 mM resazurin, 1 mM potassium hexacyanoferrate(II)trihydrate, 1 mM potassium hexacyanoferrate(III), 2.5% methylene blue, all Sigma). Resorufin was detected after 6 h with 570 nm excitation and 590 nm emission. Drug libraries were acquired from commercial and academic sources and consisted of the NIH/NCI Approved Oncology Drugs Set IV (101 compounds) and Selleck Chemicals kinase inhibitor library (210 compounds). FDA-approved drugs were sourced from (a) Prestwick Chemical Library (1200 compounds), (b) LOPAC Library (1120 compounds), and (c) Tocris Library (1280 compounds). The percentage overlap between these three libraries was: Prestwick/Tocris (8.5%), LOPAC/Tocris (22%), Prestwick/LOPAC (26%), and crude analysis of structures showed that there were 2888 unique compounds. Drugs were initially evaluated at 10 μM, then selected compounds were tested in a 4 or 10-point dose response curve.
Compounds
4-HPC was purchased from Toronto Research Chemicals, and GEM from Medchem Express. For in vitro assays, drugs were dissolved in DMSO and stored at −80°C, with the exception of CDDP, which was purchased as a 1 mg/mL solution in saline (Hospira). For in vivo administration, CPM (Endoxan, Baxter) was dissolved in saline and 120 mg/kg delivered intraperitoneally (i.p.) once weekly. CDDP and GEM were diluted in phosphate-buffered saline (PBS) and 3 mg/kg (i.p.) or 60 mg/kg (i.v.), respectively, were delivered once weekly. CHK inhibitors were purchased from Medchem Express except for prexasertib (MedKoo Biosciences). AZD7762 was dissolved in 11% cyclodextrin (Sigma) and 30 mg/kg delivered i.v. (22), MK8776 was dissolved in 20% hydroxypropyl-β-cyclodextrin (Sigma) and 40 mg/kg delivered i.p., and LY2606368-hydrochloride and prexasertib were dissolved in 20% Captisol (Medchem Express) and 20 mg/kg delivered i.v. once per day, or 10 mg/kg delivered s.c. twice in one day 10–12 h apart. LY2603618 was formulated for daily oral dosing (p.o.) at 200 mg/kg in 16.7% Captisol in 25 mM phosphate buffer, pH 4 (59). Treatment schedules are shown in figures and were repeated weekly for 6 weeks unless tumor-related morbidity or other ill health was observed.
Dose-response assays
Cells (1,000/well) were plated in black-walled 384-well plates (Costar) using a Microlab NIMBUS (Hamilton), and drugs were added using a Tecan HP300 digital dispenser maintaining <0.1% DMSO. Cells were treated for 72 h and incubated with alamar blue for the final 6 h of treatment. The reduction of resazurin was assessed as described above. The effect of drug treatment was measured by calculating the percent growth inhibition compared to DMSO-treated wells. Non-linear regression was performed using Graphpad Prism using a four-parameter variable point equation with the minimum and maximum values constrained at >0 or <100, respectively, from which the ED50 was calculated. Experiments were performed in triplicate at a minimum and data shown as mean ± SEM.
Drug interaction assays
Cells were seeded as above with each plate containing dose-response curves for each individual drug alone, at least 30 wells containing drug:drug combinations of varying concentrations, DMSO (0.1%), and media controls. Cells were treated for 72 h, and growth inhibition was measured using alamar blue assay as described above. A minimum of three independent experiments were performed. The Chou–Talalay median effect model (14) was used to classify whether the two drugs interacted in an antagonistic, additive, or synergistic manner. Combination index (CI) > 1 demonstrates antagonism, CI = 1 demonstrates additivity, and CI < 1 indicates synergistic interactions.
Protein extraction and immunoblotting
Cells were cultured in the presence or absence of drugs as described. DMSO (0.1%) remained consistent across all samples. Cells were lysed 24 h after addition of the DNA-damaging agent in radioimmunoprecipitation buffer (150 mM sodium chloride, 50 mM Tris.HCl pH 8, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) containing protease and phosphatase inhibitors (Roche). Protein (20 μg) was separated using 4–12% gradient Bis-Tris gels (Nupage) and transferred to nitrocellulose (BioRad). Antibodies were purchased from Cell Signaling and used at 1:1000: phospho-CHK1 Ser345 (#2348), phospho-CHK1 Ser296 (#2349), phospho-CHK2 Ser516 (#2669), phospho-CHK2 Thr68 (#2661), phospho-CDC2 Tyr15 (#4539), γH2AX (#9718), CHK1 (#2360), CHK2 (#6334), CDC2/CDK1 (#9116), and p53 (#2527). β-actin (#A1978, 1:5000, Sigma Aldrich) was used as a loading control. Antibodies were detected using SuperSignal West Dura (Pierce), and images collected using a BioRad Chemidoc and Image Lab software.
Flow cytometry
Cell cycle distribution was analyzed using EdU (added 45 minutes before harvest) to label cells undergoing DNA synthesis and DAPI to label DNA content. D425 cells were treated with DMSO, 15 nM LY2606368, 7 μM 4-HPC, 20 nM GEM, or drug combinations as indicated in the text. D283 cells were treated with DMSO, 12 nM LY2606368, 20 nM GEM, or a combination of both. DMSO was 0.1% across all samples. Time of harvest is indicated in the text. Cells were stained using the Click-iT EdU AlexaFluor488 kit (Invitrogen). In addition, cells were stained with AlexaFluor647-conjugated cleaved PARP (Cell Signaling, 68975) and PE-conjugated phospho-histone H3 Ser10 (Cell Signaling, 5764) to identify apoptotic cells and cells in mitosis, respectively. Samples were analyzed using an LSRFortessa X20 (BD) with FlowJo software. Data are pooled from three independent experiments and show the mean with standard deviation (SD).
Preclinical models
Homozygous ND2:SmoA1 (Smo) transgenic mice were gifted from James Olson (Fred Hutchinson Cancer Research Centre, USA) (25). Models of G3 MB consisted of the cell lines described above implanted in the brain of immunodeficient mice. In addition, an allograft model of murine G3-II MB (Myc/p53DD) generated by overexpressing Myc and the p53 dimerization domain (p53DD) in cerebellar stem cells was used (10). Several PDX models were generated using human MB cells obtained through informed consent of a parent/legal guardian prior to surgery according to institutional regulatory standards. G3 MB PDXs: TK-MB913 and TK-MB870 were generated at Telethon Kids Institute (Australia), MED211 was generated by the J. Olson Lab (Fred Hutchinson Cancer Research Center, USA) (60). SHHα PDXs: BT084 was established at DKFZ (Germany), SJMBSHH-13-5634 and SJMBSHH-14-7196 were generated at St Jude Children’s Research Hospital (USA) (61).
For implantation, cells were harvested from culture or donor mice, dissociated, suspended in matrigel (BD Biosciences), and 100,000 – 500,000 cells were implanted into the brains of 6 – 12-week-old mice as previously described (5,62,63). Experiments using D283, D425, MB002, Myc/p53DD, BT084, TK-MB913, and TK-MB870 were performed using BALB/c nude mice (Animal Resources Centre). MED211 xenografts were grown in NOD-SCID-IL2RγKO (NSG) mice (Jackson Laboratory), whereas SJMBSHH-13-5634 and SJMBSHH-14-7196 xenografts were grown in CD-1 nude mice (Charles River Laboratories).
For SJMBSHH-13-5634 and SJMBSHH-14-7196 PDXs, treatment was started once the average radiance exceeded 106 photons per second per centimeter squared per steradian (abbreviated as p/s). For all other models, treatment started 7 or 14 days post-implant as marked in the figures.
Immunohistochemistry
Mouse brain tissue was fixed in 4% paraformaldehyde in PBS overnight at 4°C and embedded in paraffin. Tissue sections (5 μm) underwent antigen retrieval in a citrate buffer before immunostaining with the following antibodies: Ki-67 (Leica, #NCL-Ki67p, 1:5000), cleaved caspase-3 (BD, #559565, 1:500), phospho-CHK1 Ser345 (Cell Signaling, #2348, 1:200), phospho-CHK1 Ser296 (Cell Signaling, #2349, 1:200), phospho-CDC2 Tyr15 (Cell Signaling, #4539, 1:200), γH2AX (Cell Signaling, #9718S, 1:500). Antibodies were detected using Elite ABC kit and NovaRED substrate, then counterstained with Gill’s hematoxylin (all Vector Laboratories). Positively-stained cells were quantified using a Nuance spectral unmixing camera and InForm software (Perkin Elmer), or Image J software (64) was used to apply a threshold limit to a minimum of three images per tumor and the average number of pixels above the threshold was determined from at least three independent animals per group.
Statistical analyses
Unpaired two-tailed Student’s t tests were used to evaluate the statistical significance of differences between treatment groups for flow cytometry data. IHC images were compared using ordinary one-way ANOVA with Tukey’s multiple comparisons test using GraphPad Prism software, where a P value <0.05 was considered significant. Kaplan-Meier survival curves were compared using the log-rank test. In each survival experiment, the combination-treated group was compared to each other group. To account for the three repeated comparisons, the Bonferroni-corrected threshold for statistical significance was set at p<0.0166667. Values of significance are indicated by asterisks and described in each figure legend where appropriate.
Supplementary Material
Fig.S1. Summary of drug screening results for G3 MB.
Fig.S2. Multiple kinases that control cell cycle checkpoints were effective against G3 MB.
Fig.S3. All CHK1/2 inhibitors penetrate MB and behave similarly in multiple models.
Fig.S4. CHK1/2 inhibition abrogates cell cycle checkpoint signaling in MB cells following treatment with 4-HPC or GEM.
Fig.S5. LY2606368 abrogates cell cycle arrest following chemotherapy-induced DNA damage in MB cells.
Fig.S6. Combined CPM and LY2606368 treatment induced apoptosis in additional G3 MB PDX models.
Fig.S7. LY2606368 and CPM combination therapy delays tumor growth of G3-II and SHHα MB.
Fig.S8. Summary of mean and median survival of mouse models treated with LY2606368 combination therapy.
Fig S9. No evidence of intrinsic drug resistance following combination therapy with CPM and LY2606368.
Fig.S10. Bioluminescence and immunohistochemistry data from G3-II models treated with LY2606368 and CDDP.
Fig.S11. Combination therapy using GEM and LY2606368 is effective in other models of G3 MB and increases DNA damage and apoptosis in vivo
Fig.S12. Combination therapy using GEM and LY2606368 increases DNA damage in SHH MB in vivo.
Table S1. Summary of in vitro drug sensitivity data
Table S2. Summary of different MB models used and the location of experiments performed.
Acknowledgements
We acknowledge Ursula Kees (Telethon Kids Institute), Darell Bigner (Duke University), Yoon-Jae Cho (Oregon Health and Science University), James Olson (Fred Hutchinson Cancer Research Centre), Suzanne Baker, Arthur Nienhuis, and Richard Williams (all from St Jude Children’s Research Hospital) for providing reagents. We appreciate input from Greg Arndt, Tim Failes and Anna Mariana (Australian Cancer Research Foundation (ACRF) Drug Discovery Centre for Childhood Cancer) for the drug screen. Thank you to the Bioresources teams of TKI, SJCRH, and SBP for the care of our animals. We thank Kimberly Mercer, Sarah Robinson and the Center for In Vitro Imaging and Therapeutics for the xenografting and amplification of primary human MB and help with the preclinical studies at SJCRH. Editorial services were provided by Nancy R. Gough (BioSerendipity, LLC, Elkridge, MD). We thank all members of the Telethon Kids Institute Brain Tumour Research team for their advice, suggestions and discussions during the course of this project. Funding for this work was provided by Cancer Council of Western Australia (APP1129386), Cancer Australia (APP1101390, APP1147153), Telethon-Perth Children’s Hospital Research Fund, The Kids Cancer Project, Cure Brain Cancer Foundation, Perth Children’s Hospital Foundation, and with generous support from the Pirate Ship Foundation. Experiments executed at SJCRH were funded in part by NIH/NCI grants CA-09832, Core Grant CA-02165 and the American Lebanese Syrian Associated Charities (MRF). Experiments at SBP were supported by funding from the NCI (R01-CA159859 and P30-CA30199), William’s Superhero Fund and the McDowell Charity Trust (RJW-R). RE has support from the John Lillie Cancer Research Fellowship and Brainchild Fellowship, NGG is supported by a Cancer Council of Western Australia Fellowship and the Stan Perron Chair of Paediatric Haematology and Oncology, and TJ holds a National Health and Medical Research Council Principal Research Fellowship. MA and CG were each supported by an Australian Postgraduate Award from the Australian Government.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
All PDX models reported here are available through an MTA from Telethon Kids Institute or St Jude Children’s Research Hospital. Methylation array results and RNAseq data are available (https://ega-archive.org/studies/EGAS00001004748). All data associated with this study are available in the main text or the supplementary materials.
References and notes
- 1.Siegel RL, Miller KD, Jemal A, Cancer statistics, 2016. CA. Cancer J. Clin 66, 7–30 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Martin AM, Raabe E, Eberhart C, Cohen KJ, Management of pediatric and adult patients with medulloblastoma. Curr. Treat. Options Oncol 15, 581–594 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Armstrong GT, Liu Q, Yasui Y, Huang S, Ness KK, Leisenring W, Hudson MM, Donaldson SS, King AA, Stovall M, Krull KR, Robison LL, Packer RJ, Long-term outcomes among adult survivors of childhood central nervous system malignancies in the Childhood Cancer Survivor Study. J. Natl. Cancer Inst 101, 946–958 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hovestadt V, Ayrault O, Swartling FJ, Robinson GW, Pfister SM, Northcott PA, Medulloblastomics revisited: biological and clinical insights from thousands of patients. Nat. Rev. Cancer 20, 42–56 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morfouace M, Shelat A, Jacus M, Freeman BB 3rd, Turner D, Robinson S, Zindy F, Wang YD, Finkelstein D, Ayrault O, Bihannic L, Puget S, Li XN, Olson JM, Robinson GW, Guy RK, Stewart CF, Gajjar A, Roussel MF, Pemetrexed and gemcitabine as combination therapy for the treatment of Group3 medulloblastoma. Cancer Cell 25, 516–529 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pei Y, Liu KW, Wang J, Garancher A, Tao R, Esparza LA, Maier DL, Udaka YT, Murad N, Morrissy S, Seker-Cin H, Brabetz S, Qi L, Kogiso M, Schubert S, Olson JM, Cho YJ, Li XN, Crawford JR, Levy ML, Kool M, Pfister SM, Taylor MD, Wechsler-Reya RJ, HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC-Driven Medulloblastoma. Cancer Cell 29, 311–323 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, Bolin S, Schumacher SE, Zeid R, Masoud S, Yu F, Vue N, Gibson WJ, Paolella BR, Mitra SS, Cheshier SH, Qi J, Liu KW, Wechsler-Reya R, Weiss WA, Swartling FJ, Kieran MW, Bradner JE, Beroukhim R, Cho YJ, BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin. Cancer Res 20, 912–925 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dubuc AM, Remke M, Korshunov A, Northcott PA, Zhan SH, Mendez-Lago M, Kool M, Jones DT, Unterberger A, Morrissy AS, Shih D, Peacock J, Ramaswamy V, Rolider A, Wang X, Witt H, Hielscher T, Hawkins C, Vibhakar R, Croul S, Rutka JT, Weiss WA, Jones SJ, Eberhart CG, Marra MA, Pfister SM, Taylor MD, Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol 125, 373–384 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hill RM, Kuijper S, Lindsey JC, Petrie K, Schwalbe EC, Barker K, Boult JK, Williamson D, Ahmad Z, Hallsworth A, Ryan SL, Poon E, Robinson SP, Ruddle R, Raynaud FI, Howell L, Kwok C, Joshi A, Nicholson SL, Crosier S, Ellison DW, Wharton SB, Robson K, Michalski A, Hargrave D, Jacques TS, Pizer B, Bailey S, Swartling FJ, Weiss WA, Chesler L, Clifford SC, Combined MYC and P53 Defects Emerge at Medulloblastoma Relapse and Define Rapidly Progressive, Therapeutically Targetable Disease. Cancer Cell 27, 72–84 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pei Y, Moore CE, Wang J, Tewari AK, Eroshkin A, Cho YJ, Witt H, Korshunov A, Read TA, Sun JL, Schmitt EM, Miller CR, Buckley AF, McLendon RE, Westbrook TF, Northcott PA, Taylor MD, Pfister SM, Febbo PG, Wechsler-Reya RJ, An animal model of MYC-driven medulloblastoma. Cancer Cell 21, 155–167 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Begg AC, Stewart FA, Vens C, Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 11, 239–253 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Luo Y, Leverson JD, New opportunities in chemosensitization and radiosensitization: modulating the DNA-damage response. Expert Rev. Anticancer Ther 5, 333–342 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Carrassa L, Damia G, DNA damage response inhibitors: Mechanisms and potential applications in cancer therapy. Cancer Treat. Rev 60, 139–151 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Chou TC, Talalay P, Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul 22, 27–55 (1984). [DOI] [PubMed] [Google Scholar]
- 15.Franshaw L, Tsoli M, Byrne J, Mayoh C, Sivarajasingam S, Norris M, Marshall GM, Ziegler DS, Predictors of Success of Phase II Pediatric Oncology Clinical Trials. Oncologist 24, e765–e774 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong D, Infante J, Janku F, Jones S, Nguyen LM, Burris H, Naing A, Bauer TM, Piha-Paul S, Johnson FM, Kurzrock R, Golden L, Hynes S, Lin J, Lin AB, Bendell J, Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J. Clin. Oncol 34, 1764–1771 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Infante JR, Hollebecque A, Postel-Vinay S, Bauer TM, Blackwood EM, Evangelista M, Mahrus S, Peale FV, Lu X, Sahasranaman S, Zhu R, Chen Y, Ding X, Murray ER, Schutzman JL, Lauchle JO, Soria JC, LoRusso PM, Phase I Study of GDC-0425, a Checkpoint Kinase 1 Inhibitor, in Combination with Gemcitabine in Patients with Refractory Solid Tumors. Clin. Cancer Res 23, 2423–2432 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Parsels LA, Qian Y, Tanska DM, Gross M, Zhao L, Hassan MC, Arumugarajah S, Parsels JD, Hylander-Gans L, Simeone DM, Morosini D, Brown JL, Zabludoff SD, Maybaum J, Lawrence TS, Morgan MA, Assessment of chk1 phosphorylation as a pharmacodynamic biomarker of chk1 inhibition. Clin. Cancer Res 17, 3706–3715 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhong B, Maharaj A, Davis A, Roussel MF, Stewart CF, Development and validation of a sensitive LC MS/MS method for the measurement of the checkpoint kinase 1 inhibitor prexasertib and its application in a cerebral microdialysis study. J. Pharm. Biomed. Anal 156, 97–103 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campagne O, Davis A, Maharaj AR, Zhong B, Stripay J, Farmer D, Roussel MF, Stewart CF, CNS penetration and pharmacodynamics of the CHK1 inhibitor prexasertib in a mouse Group 3 medulloblastoma model. Eur. J. Pharm. Sci 142, 105106 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Russell MR, Levin K, Rader J, Belcastro L, Li Y, Martinez D, Pawel B, Shumway SD, Maris JM, Cole KA, Combination therapy targeting the Chk1 and Wee1 kinases shows therapeutic efficacy in neuroblastoma. Cancer Res. 73, 776–784 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zabludoff SD, Deng C, Grondine MR, Sheehy AM, Ashwell S, Caleb BL, Green S, Haye HR, Horn CL, Janetka JW, Liu D, Mouchet E, Ready S, Rosenthal JL, Queva C, Schwartz GK, Taylor KJ, Tse AN, Walker GE, White AM, AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol. Cancer Ther 7, 2955–2966 (2008). [DOI] [PubMed] [Google Scholar]
- 23.King C, Diaz H, Barnard D, Barda D, Clawson D, Blosser W, Cox K, Guo S, Marshall M, Characterization and preclinical development of LY2603618: a selective and potent Chk1 inhibitor. Invest. New Drugs 32, 213–226 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Lowery CD, VanWye AB, Dowless M, Blosser W, Falcon BL, Stewart J, Stephens J, Beckmann RP, Bence Lin A, Stancato LF, The Checkpoint Kinase 1 Inhibitor Prexasertib Induces Regression of Preclinical Models of Human Neuroblastoma. Clin. Cancer Res 23, 4354–4363 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Hatton BA, Villavicencio EH, Tsuchiya KD, Pritchard JI, Ditzler S, Pullar B, Hansen S, Knoblaugh SE, Lee D, Eberhart CG, Hallahan AR, Olson JM, The Smo/Smo model: hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res. 68, 1768–1776 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Branzei D, Foiani M, Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol 9, 297–308 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Schwarz JK, Lovly CM, Piwnica-Worms H, Regulation of the Chk2 protein kinase by oligomerization-mediated cis- and trans-phosphorylation. Mol. Cancer Res 1, 598–609 (2003). [PubMed] [Google Scholar]
- 28.Pitts TM, Davis SL, Eckhardt SG, Bradshaw-Pierce EL, Targeting nuclear kinases in cancer: development of cell cycle kinase inhibitors. Pharmacol. Ther 142, 258–269 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Ewig RA, Kohn KW, DNA damage and repair in mouse leukemia L1210 cells treated with nitrogen mustard, 1,3-bis(2-chloroethyl)-1-nitrosourea, and other nitrosoureas. Cancer Res. 37, 2114–2122 (1977). [PubMed] [Google Scholar]
- 30.Zegerman P, Diffley JF, DNA replication as a target of the DNA damage checkpoint. DNA Repair (Amst) 8, 1077–1088 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Zhukova N, Ramaswamy V, Remke M, Pfaff E, Shih DJ, Martin DC, Castelo-Branco P, Baskin B, Ray PN, Bouffet E, von Bueren AO, Jones DT, Northcott PA, Kool M, Sturm D, Pugh TJ, Pomeroy SL, Cho YJ, Pietsch T, Gessi M, Rutkowski S, Bognar L, Klekner A, Cho BK, Kim SK, Wang KC, Eberhart CG, Fevre-Montange M, Fouladi M, French PJ, Kros M, Grajkowska WA, Gupta N, Weiss WA, Hauser P, Jabado N, Jouvet A, Jung S, Kumabe T, Lach B, Leonard JR, Rubin JB, Liau LM, Massimi L, Pollack IF, Shin Ra Y, Van Meir EG, Zitterbart K, Schuller U, Hill RM, Lindsey JC, Schwalbe EC, Bailey S, Ellison DW, Hawkins C, Malkin D, Clifford SC, Korshunov A, Pfister S, Taylor MD, Tabori U, Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J. Clin. Oncol 31, 2927–2935 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neve A, Santhana Kumar K, Tripolitsioti D, Grotzer MA, Baumgartner M, Investigation of brain tissue infiltration by medulloblastoma cells in an ex vivo model. Sci Rep 7, 5297 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sasai K, Romer JT, Lee Y, Finkelstein D, Fuller C, McKinnon PJ, Curran T, Shh pathway activity is down-regulated in cultured medulloblastoma cells: implications for preclinical studies. Cancer Res. 66, 4215–4222 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Zhao X, Ponomaryov T, Ornell KJ, Zhou P, Dabral SK, Pak E, Li W, Atwood SX, Whitson RJ, Chang AL, Li J, Oro AE, Chan JA, Kelleher JF, Segal RA, RAS/MAPK Activation Drives Resistance to Smo Inhibition, Metastasis, and Tumor Evolution in Shh Pathway-Dependent Tumors. Cancer Res. 75, 3623–3635 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng Y, Franco-Barraza J, Wang Y, Zheng C, Zhang L, Qu Y, Long Y, Cukierman E, Z.-j. Yang, Sustained hedgehog signaling in medulloblastoma tumoroids is attributed to stromal astrocytes and astrocyte-derived extracellular matrix. Lab. Invest 100, 1208–1222 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ioannidis JP, Acknowledging and Overcoming Nonreproducibility in Basic and Preclinical Research. JAMA 317, 1019–1020 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Emadi A, Jones RJ, Brodsky RA, Cyclophosphamide and cancer: golden anniversary. Nature Reviews Clinical Oncology 6, 638–647 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Packer RJ, Gajjar A, Vezina G, Rorke-Adams L, Burger PC, Robertson PL, Bayer L, LaFond D, Donahue BR, Marymont MH, Muraszko K, Langston J, Sposto R, Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J. Clin. Oncol 24, 4202–4208 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Gottardo NG, Gajjar A, Current therapy for medulloblastoma. Curr. Treat. Options Neurol 8, 319–334 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Leary SE, Olson JM, The molecular classification of medulloblastoma: driving the next generation clinical trials. Curr. Opin. Pediatr 24, 33–39 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DeVita VT, Chu E, A History of Cancer Chemotherapy. Cancer Res. 68, 8643–8653 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Bergman AM, Ruiz van Haperen VW, Veerman G, Kuiper CM, Peters GJ, Synergistic interaction between cisplatin and gemcitabine in vitro. Clin. Cancer Res 2, 521–530 (1996). [PubMed] [Google Scholar]
- 43.Bucher N, Britten CD, G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br. J. Cancer 98, 523–528 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Endersby R, Whitehouse J, Hii H, Greenall SA, Johns TG, Gottardo NG, A Pre-Clinical Assessment of the Pan-ERBB Inhibitor Dacomitinib in Pediatric and Adult Brain Tumors. Neoplasia 20, 432–442 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krüger K, Geist K, Stuhldreier F, Schumacher L, Blümel L, Remke M, Wesselborg S, Stork B, Klöcker N, Bormann S, Roos WP, Honnen S, Fritz G, Multiple DNA damage-dependent and DNA damage-independent stress responses define the outcome of ATR/Chk1 targeting in medulloblastoma cells. Cancer Lett. 430, 34–46 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Prince EW, Balakrishnan I, Shah M, Mulcahy Levy JM, Griesinger AM, Alimova I, Harris PS, Birks DK, Donson AM, Davidson N, Remke M, Taylor MD, Handler MH, Foreman NK, Venkataraman S, Vibhakar R, Checkpoint kinase 1 expression is an adverse prognostic marker and therapeutic target in MYC-driven medulloblastoma. Oncotarget 7, 53881–53894 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cook Sangar ML, Genovesi LA, Nakamoto MW, Davis MJ, Knobluagh SE, Ji P, Millar A, Wainwright BJ, Olson JM, Inhibition of CDK4/6 by Palbociclib Significantly Extends Survival in Medulloblastoma Patient-Derived Xenograft Mouse Models. Clin. Cancer Res 23, 5802–5813 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guzi TJ, Paruch K, Dwyer MP, Labroli M, Shanahan F, Davis N, Taricani L, Wiswell D, Seghezzi W, Penaflor E, Bhagwat B, Wang W, Gu D, Hsieh Y, Lee S, Liu M, Parry D, Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening. Mol. Cancer Ther 10, 591–602 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Brosh R, Rotter V, When mutants gain new powers: news from the mutant p53 field. Nat. Rev. Cancer 9, 701–713 (2009). [DOI] [PubMed] [Google Scholar]
- 50.King C, Diaz HB, McNeely S, Barnard D, Dempsey J, Blosser W, Beckmann R, Barda D, Marshall MS, LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol. Cancer Ther 14, 2004–2013 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Kotsantis P, Petermann E, Boulton SJ, Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov 8, 537–555 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanaka T, Yamagami T, Oka Y, Nomura T, Sugiyama H, The scid mutation in mice causes defects in the repair system for both double-strand DNA breaks and DNA cross-links. Mutat. Res 288, 277–280 (1993). [DOI] [PubMed] [Google Scholar]
- 53.Holthouse DJ, Dallas PB, Ford J, Fabian V, Murch AR, Watson M, Wong G, Bertram C, Egli S, Baker DL, Kees UR, Classic and desmoplastic medulloblastoma: complete case reports and characterizations of two new cell lines. Neuropathology : official journal of the Japanese Society of Neuropathology 29, 398–409 (2009). [DOI] [PubMed] [Google Scholar]
- 54.Friedman HS, Burger PC, Bigner SH, Trojanowski JQ, Wikstrand CJ, Halperin EC, Bigner DD, Establishment and characterization of the human medulloblastoma cell line and transplantable xenograft D283 Med. J. Neuropathol. Exp. Neurol 44, 592–605 (1985). [DOI] [PubMed] [Google Scholar]
- 55.Rusert JM, Juarez EF, Brabetz S, Jensen J, Garancher A, Chau LQ, Tacheva-Grigorova SK, Wahab S, Udaka YT, Finlay D, Seker-Cin H, Reardon B, Gröbner S, Serrano J, Ecker J, Qi L, Kogiso M, Du Y, Baxter PA, Henderson JJ, Berens ME, Vuori K, Milde T, Cho YJ, Li XN, Olson JM, Reyes I, Snuderl M, Wong TC, Dimmock DP, Nahas SA, Malicki D, Crawford JR, Levy ML, Van Allen EM, Pfister SM, Tamayo P, Kool M, Mesirov JP, Wechsler-Reya RJ, Functional precision medicine identifies new therapeutic candidates for medulloblastoma. Cancer Res, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ivanov DP, Coyle B, Walker DA, Grabowska AM, In vitro models of medulloblastoma: Choosing the right tool for the job. J. Biotechnol 236, 10–25 (2016). [DOI] [PubMed] [Google Scholar]
- 57.Sengupta R, Dubuc A, Ward S, Yang L, Northcott P, Woerner BM, Kroll K, Luo J, Taylor MD, Wechsler-Reya RJ, Rubin JB, CXCR4 activation defines a new subgroup of Sonic hedgehog-driven medulloblastoma. Cancer Res. 72, 122–132 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Snuderl M, Batista A, Kirkpatrick ND, Ruiz de Almodovar C, Riedemann L, Walsh EC, Anolik R, Huang Y, Martin JD, Kamoun W, Knevels E, Schmidt T, Farrar CT, Vakoc BJ, Mohan N, Chung E, Roberge S, Peterson T, Bais C, Zhelyazkova BH, Yip S, Hasselblatt M, Rossig C, Niemeyer E, Ferrara N, Klagsbrun M, Duda DG, Fukumura D, Xu L, Carmeliet P, Jain RK, Targeting placental growth factor/neuropilin 1 pathway inhibits growth and spread of medulloblastoma. Cell 152, 1065–1076 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barnard D, Diaz HB, Burke T, Donoho G, Beckmann R, Jones B, Barda D, King C, Marshall M, LY2603618, a selective CHK1 inhibitor, enhances the anti-tumor effect of gemcitabine in xenograft tumor models. Invest. New Drugs 34, 49–60 (2016). [DOI] [PubMed] [Google Scholar]
- 60.Girard E, Ditzler S, Lee D, Richards A, Yagle K, Park J, Eslamy H, Bobilev D, Vrignaud P, Olson J, Efficacy of cabazitaxel in mouse models of pediatric brain tumors. Neuro Oncol 17, 107–115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith KS, Xu K, Mercer KS, Boop F, Klimo P, DeCupyere M, Grenet J, Robinson S, Dunphy P, Baker SJ, Ellison DW, Merchant TE, Upadayaya SA, Gajjar A, Wu G, Orr BA, Robinson GW, Northcott PA, Roussel MF, Patient-derived orthotopic xenografts of pediatric brain tumors: a St. Jude resource. Acta Neuropathol, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Endersby R, Zhu X, Hay N, Ellison DW, Baker SJ, Nonredundant functions for akt isoforms in astrocyte growth and gliomagenesis in an orthotopic transplantation model. Cancer Res. 71, 4106–4116 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brabetz S, Leary SES, Grobner SN, Nakamoto MW, Seker-Cin H, Girard EJ, Cole B, Strand AD, Bloom KL, Hovestadt V, Mack NL, Pakiam F, Schwalm B, Korshunov A, Balasubramanian GP, Northcott PA, Pedro KD, Dey J, Hansen S, Ditzler S, Lichter P, Chavez L, Jones DTW, Koster J, Pfister SM, Kool M, Olson JM, A biobank of patient-derived pediatric brain tumor models. Nat. Med 24, 1752–1761 (2018). [DOI] [PubMed] [Google Scholar]
- 64.Schneider CA, Rasband WS, Eliceiri KW, NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig.S1. Summary of drug screening results for G3 MB.
Fig.S2. Multiple kinases that control cell cycle checkpoints were effective against G3 MB.
Fig.S3. All CHK1/2 inhibitors penetrate MB and behave similarly in multiple models.
Fig.S4. CHK1/2 inhibition abrogates cell cycle checkpoint signaling in MB cells following treatment with 4-HPC or GEM.
Fig.S5. LY2606368 abrogates cell cycle arrest following chemotherapy-induced DNA damage in MB cells.
Fig.S6. Combined CPM and LY2606368 treatment induced apoptosis in additional G3 MB PDX models.
Fig.S7. LY2606368 and CPM combination therapy delays tumor growth of G3-II and SHHα MB.
Fig.S8. Summary of mean and median survival of mouse models treated with LY2606368 combination therapy.
Fig S9. No evidence of intrinsic drug resistance following combination therapy with CPM and LY2606368.
Fig.S10. Bioluminescence and immunohistochemistry data from G3-II models treated with LY2606368 and CDDP.
Fig.S11. Combination therapy using GEM and LY2606368 is effective in other models of G3 MB and increases DNA damage and apoptosis in vivo
Fig.S12. Combination therapy using GEM and LY2606368 increases DNA damage in SHH MB in vivo.
Table S1. Summary of in vitro drug sensitivity data
Table S2. Summary of different MB models used and the location of experiments performed.
Data Availability Statement
All PDX models reported here are available through an MTA from Telethon Kids Institute or St Jude Children’s Research Hospital. Methylation array results and RNAseq data are available (https://ega-archive.org/studies/EGAS00001004748). All data associated with this study are available in the main text or the supplementary materials.