

Abstract
Mutation of profilin 1 (PFN1) can cause amyotrophic lateral sclerosis (ALS). To assess how PFN1 mutation causes the disease, we created transgenic rats with human genomic DNA that harbors both the coding and the regulatory sequences of the human PFN1 gene. Selected transgenic lines expressed human PFN1 with or without the pathogenic mutation C71G at a moderate and a comparable level and in the similar pattern of spatial and temporal expression to rat endogenous PFN1. The artificial effects of arbitrary transgene expression commonly observed in cDNA transgenic animals were minimized in PFN1 transgenic rats. Expression of the mutant, but not the wild type, human PFN1 in rats recapitulated the cardinal features of ALS including the progressive loss of motor neurons and the subsequent denervation atrophy of skeletal muscles. Detergent-insoluble PFN1 inclusions were detected as the first pathology in otherwise asymptomatic transgenic rats expressing mutant human PFN1. The findings suggest that protein aggregation is involved in the neurodegeneration of ALS associated with PFN1 mutation. The resulting rat model is useful to mechanistic study on the ALS.
Keywords: aggregation, ALS, neurodegeneration, PFN1, rat
1 ∣. INTRODUCTION
Amyotrophic lateral sclerosis (ALS) results from the progressive degeneration of upper and lower motor neurons and subsequent denervation atrophy of skeletal muscles. At present, no effective treatment is available to cure ALS or even to significantly relieve its symptoms. Whereas the majority of ALS cases are sporadic without a clear cause, about 10% of the cases are attributable to pathogenic mutations in a heterogenous group of genes (Nguyen, Van Broeckhoven, & van der Zee, 2018). Genetic clues not only can help define the molecular pathways leading to neurodegeneration but also can help identify biomarkers for monitoring disease progression and therapeutic effectiveness. Pathogenic mutations of the profilin 1 gene (PFN1) have been identified as a cause of familial ALS (Smith et al., 2015; Wu et al., 2012). In mammals, four profilin paralogs (PFN1-PFN4) are expressed (Di Nardo, Gareus, Kwiatkowski, & Witke, 2000; Witke, 2004). PFN1 deficiency in mouse embryos cannot be compensated for by the remaining PFN paralogs (Witke, Sutherland, Sharpe, Arai, & Kwiatkowski, 2001), suggesting a distinct role (at least during development) for PFN paralogs. PFN1 is expressed substantially throughout the lifetime of mammals and plays a critical role in regulating neuronal function with and without interactions with actin (Di Nardo et al., 2000). PFN1 interacts with actin to regulate its assembly and thus to regulate cytoskeletal dynamics, axonal integrity, and cell growth (Gau et al., 2018; Michaelsen et al., 2010; Romero et al., 2004). PFN1 is also known to interact with many other proteins (e.g., phosphatidylinositol 3 kinase) to regulate signaling transduction (Ding et al., 2014; Fan, Arif, & Gong, 2012). PFN1 plays an essential role in cellular functions, but how PFN1 mutation causes neurodegeneration remains to be determined.
The effects of pathogenic mutations on PFN1 functions have been examined using both in vivo and in vitro models. Along with other ALS-associated genes, such as tubulin A4A, dynactin, and neurofilament, PFN1 regulates cytoskeletal dynamics (Taylor, Brown, & Cleveland, 2016). PFN1 sequesters actin from the pool of polymerizable actin and converts poorly polymerizable ADP-actin monomers into readily polymerizable ATP-actin monomers (Witke, 2004). A study using cultured neurons showed that a pathogenic mutation of PFN1 altered its binding affinity for actin (Wu et al., 2012), suggesting a functional loss in mutant PFN1. However, PFN1 is not required in mature neurons because deletion of Pfn1 in adult mice did not cause appreciable neuropathology (Gorlich et al., 2012). Rather than resulting in loss-of-function, PFN1 mutation is more likely to result in gain-of-function pathogenesis because over-expression of mutant PFN1 (i.e., C71G and G118V substitutions), but not wild-type (WT), in transgenic mice caused neuronal dysfunction and a paralysis phenotype (Fil, DeLoach, & Yadav, 2017; Yang et al., 2016). Because these mouse models were created with PFN1 cDNA driven by artificial promoters, it is not surprising that the patterns of spatial and temporal transgene expression did not agree with endogenous Pfn1 expression patterns (Fil et al., 2017; Yang et al., 2016). Findings in transgenic mice support gain-of-functions with PFN1 mutations, but the pathomechanisms of PFN1 mutations are largely unknown.
A pathogenic mutation of PFN1 increases its propensity for aggregation in cultured cells (Boopathy et al., 2015; Wu et al., 2012), but how this aggregation is involved in PFN1 pathogenesis is not known. Protein aggregation is a shared feature of neurodegenerative diseases including ALS (Kwong, Neumann, Sampathu, Lee, & Trojanowski, 2007; Kwong, Uryu, Trojanowski, & Lee, 2008; Neumann et al., 2006), Alzheimer's disease (Bruggink, Muller, Kuiperij, & Verbeek, 2012), and Parkinson's disease (Goedert, Spillantini, Del Tredici, & Braak, 2012). Protein aggregation per se could be toxic, and protein-aggregate intermediates are thought to be cytotoxic (Bucciantini et al., 2002; Walsh et al., 2002). Protein aggregation could disrupt the cellular protein degradation machinery or entrap key proteins to compromise cellular functions. Although the exact mechanisms by which protein aggregation can cause neurodegeneration are not clear, protein aggregation involvement in disease pathogenesis is being studied intensely (Luk, Kehm, Carroll, et al., 2012; Luk, Kehm, Zhang, et al., 2012; Rodriguez, Ivanova, & Sawaya, 2015). To study how PFN1 mutation causes neurodegeneration, we created novel PFN1 transgenic rats using genomic DNA such that the patterns of transgene expression accorded with the endogenous PFN1 gene. Expression of human PFN1 with a pathogenic mutation recapitulated the cardinal features of ALS in transgenic rats at the middle ages. Pathogenic mutation afforded PFN1 a propensity to form protein inclusion that was detected as the first pathology in otherwise non-symptomatic rats. This novel rat model is useful to mechanistic study on the ALS.
2 ∣. MATERIALS AND METHODS
2.1 ∣. Generation and genotyping of PFN1 transgenic rats
Animal use was in accord with NIH guidelines and the animal use protocol was approved by the Institutional Animal Care and Use Committees at the University of Central Florida College of Medicine (protocol No. 16-28).
The study was not preregistered and no sample calculation was performed. In total, 200 rats were used in this study and no animal was excluded from the study. No randomization was performed to allocate subjects and no blinding was performed.
PFN1 transgenic rats will be deposited to a public resource (RRRC.us) for free distribution to academic researchers after appropriate documentations are completed.
Rats were housed at the animal facility of the University of Central Florida and two rats of the same sex were housed in a ventilated cage with free access to food and water. Transgenic rats were created using a Sprague–Dawley strain that was established in the laboratory (Zhou et al., 2009). The 10.5-kb mini human PFN1 gene was extracted from a BAC clone (BACPAC Resources Cat. No. RP11-115H24), and the C71G substitutional mutation was introduced into the mini PFN1 gene by homologous recombination in Escherichia coli (Warming, Costantino, Court, Jenkins, & Copeland, 2005). Normal and mutant mini PFN1 transgenes were linearized by restriction digestion, purified from agarose gels, and injected into the pronuclei of fertilized rat eggs for creating transgenic rats using an established protocol in the laboratory (Huang, Tong, Bi, Wu, et al., 2012; Huang, Tong, Bi, Zhou, & Xia, 2012; Huang et al., 2011). Transgenic founders were identified by analyzing rat tail DNA with the primers 5’-TTTTGGGCCATTACCCCAT (forward) and 5’-CCTTGTTAGTAGAATCTTTT (reverse). PCR with these primers amplified the rat Pfn1 gene and the human PFN1 transgene of different sizes. Copies of the mini PFN1 transgene were estimated by quantitative PCR with the primers 5’-CACGGTGGTTTGATCAACAAGA (forward) and 5’-TGGTTTTGGCAGCAATAAGGG (reverse) that amplified the rat and the human PFN1 gene fragments of similar sizes. Stable transmission lines were selected and examined for transgene expression of mRNA using quantitative PCR, and protein expression by immunoblotting. Expression levels of rat and human PFN1 mRNA were estimated by quantitative PCR with the primers 5’-CACGGTGGTTTGATCAACAAGA (forward) and 5’-AGCCCATGTGGTTTTGGCAG (reverse). Rat lines with stable gene expression were selected for phenotypic expression and characterization.
2.2 ∣. Behavioral testing and disease staging
The Open-Field Activity assay (Med Associates) was established in the laboratory as a standard method for reliably detecting mobility impairment in transgenic rats (Huang, Tong, Bi, Zhou, et al., 2012; Tong et al., 2013). PFN1 transgenic rats were monitored with this assay to record distances traveled within 10 min. Disease onset was defined by an unrecoverable reduction in distance-traveled recorded by the assay, and paralysis was defined by leg-dragging or an inability for leg retraction. End-stage disease was defined as the inability to retract two or more legs, as well as the inability for self-righting when a rat was placed on its side. Rats were asphyxiated by inhaling carbon dioxide. Tissue was harvested from killed rats, and mortality was determined when rats reached end-stage disease.
2.3 ∣. Cresyl violet staining and stereological cell counting
Lower motor neurons in the lumbar spinal cord (L3-L5) were examined using cresyl violet staining, and were quantified by unbiased stereological cell counting. The lumbar cords (L3–L5) of transgenic and littermate control rats were cut into 30 μm cross sections. Every 10th spinal cord section was assessed for motor neurons on both sides. A total of 15–20 sections per rat were assessed. Cresyl violet staining is a routine method established in the lab (Huang, Tong, Bi, Zhou, et al., 2012; Huang et al., 2011; Tong et al., 2013).
2.4 ∣. Antibody information
The following primary antibodies were used: mouse monoclonal anti-GAPDH (RRID:AB_2107448), rabbit anti-GFAP (RRID:AB_94844), rabbit anti-Iba-1 (RRID:AB_839504), rabbit polyclonal antibody against p62 (RRID:AB_2810880), and rabbit anti-PFN1 (ThermoFisher cat. No. PIPA522991). Primary antibodies were diluted at 1:1,000 for immunoblotting and were used for immunostaining at the lowest dilutions recommended by the manufacturers.
2.5 ∣. Histology and immunostaining
Gastrocnemius muscle structure was assessed by hematoxylin and eosin (H&E) staining in transgenic rats using an established method in the laboratory (Huang et al., 2011; Zhou et al., 2010). Fresh muscle was snap-frozen in liquid nitrogen and cut into cross sections (10 μm) using a cryostat. Muscle sections were stained with H&E and then examined using a light microscope. Degenerating neurons in the spinal cords of PFN1 transgenic rats were detected by Bielschowski silver staining of paraffin-embedded sections (10 μm) (Zhou et al., 2010). The structure of axons in the ventral and dorsal roots of transgenic rats was assessed by toluidine blue staining. Rats were deeply anesthetized with ketamine (10 mg/ml)/xylazine (1 mg/ml) mixture and were then perfused with a mixture of 4% paraformaldehyde and 2% glutaraldehyde. Ventral and dorsal nerve roots were dissected and post-fixed in the same fixative overnight at 4°C. Fixed tissues were embedded in Epon 812 and were cut into semi-thin (1 μm) sections. These sections were stained with 1% toluidine blue, and then axons were assessed using a light microscope.
Immunohistochemistry and immunofluorescence staining were both performed on the cross sections of rat spinal cord. Tissue sections were assessed for gene expression by immunohistochemistry using a Nikon microscope or by immunofluorescence staining using a confocal microscope (Nikon confocal system). Single-layer images were scanned and then image Z-stacks (at 1 mm intervals) were projected to reconstruct cell structures.
2.6 ∣. Cell culture and immunoblotting
HEK293 cells were purchased from ATCC (CRL-11268TM) and were not found on the list of commonly misidentified cell lines by ICLAC. The cells were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS) and were used for experiments between 10 and 12 passages. Cells were transiently transfected with plasmids premixed with Lipofectamine 2000 using an established method in the laboratory (Wu et al., 2015). Cells were transfected in the absence of FBS for 4 hr and were supplemented with 10% FBS thereafter. Plasmids expressing human PFN1 with or without a pathogenic mutation were sequence-verified before transfection. Control cells were sham-transfected with a carrier plasmid expressing no mammalian-cell genes. Cells were harvested for biochemistry assays 48 hr after transfection. HEK293 cells were transiently transfected with plasmids and were not modified for stable transgene expression. No authentication was performed to characterize the transfected cells.
For immunoblotting, animal tissue and HEK293 cells were homogenized in RIPA buffer and proteins in the lysates were resolved using SDS-PAGE (Zhou et al., 2010). Immunoreactivity for a protein was detected with specified primary antibodies after the resolved proteins were transferred to nitrocellulose membranes.
2.7 ∣. Protein solubility assays
The solubility of PFN1 in rat tissues and transfected cells was examined (Wu et al., 2015). Tissues and cells were sonicated in RIPA buffer containing 1% Triton X-100 and 0.1% SDS and the lysates were centrifuged at 100,000 g for 10 min to produce the Triton X-100 soluble fraction. The resulting pellets containing protein aggregates were washed twice in cold PBS and centrifuged again at 100,000 g for 10 min to remove soluble proteins. Final pellets were sonicated in lysis buffer containing 1% SDS and were boiled to fully dissolve precipitated proteins. A mixture of protease inhibitors was added to lysates during this procedure. The soluble and insoluble fractions were then examined for specific proteins using standard SDS–PAGE.
2.8 ∣. Statistics
Motor neuron counts were compared between groups of rats with different genotypes. Unpaired t tests were used to analyze any differences in counts between groups using Graphpad Instat software. A p value <.05 was considered statistically significant. The data were not assessed for normality and no test for outliers was conducted.
3 ∣. RESULTS
3.1 ∣. Transgenic rats carrying the human mini PFN1 gene expressed human PFN1 in a similar pattern to rat endogenous Pfn1
To create an appropriate animal model for ALS associated with PFN1 mutation, we used genomic DNA rather cDNA so that both the coding and regulatory sequences of the transgene were introduced into the host genome ( Figure 1a ). The human PFN1 transgene was driven by its own promoter and was expected to be regulated mainly by its own regulatory sequence in transgenic rats. We chose rats over mice for expressing PFN1 mutations because rats have been found to model neurodegenerative diseases better than mice under certain circumstances (Creed & Goldberg, 2018; Dave et al., 2014). The C71G substitution was selected as an example of PFN1 mutations for transgenic studies (Figure 1a). Three expressing lines were established for WT and mutant mini-PFN1 transgenes (Figure S1a). One WT and one mutant rat lines expressed PFN1 mRNA at comparable levels determined by quantitative PCR (Figure S1b), and the remaining mutant line expressed less PFN1 mRNA. Regardless of expression levels, all three expressing lines expressed human PFN1 proteins in similar patterns to endogenous rat PFN1 (Figure S1c-f).
FIGURE 1.

Expression of mutant human PFN1 induces progressive paralysis in transgenic rats. (a) A schematic showing the structure of the human PFN1 gene with (hPFN1-C71G) or without (hPFN1-WT) the pathogenic mutation (C71G substitution) used for creating transgenic (TG) rats. (b) Immunoblotting revealed that human PFN1 was moderately expressed and was superimposed on rat PFN1. TG rats carrying the wild-type (WT) or the mutant (C71G) human PFN1 transgene were examined of PFN1 expression. The ratio of PFN1 to GAPDH was calculated, and the ratio for non-transgenic (NT) rats was arbitrarily set to 1. (c) A representative image showing hindlimb paralysis in a TG rat expressing mutant PFN1. (d) Open-field testing revealed a progressive loss of mobility in mutant PFN1 TG rats (C71G) compared to NT or WT rats. Data represent mean ± SD (n = 10, 5 males and 5 females). (e and f) Probabilities of weakness onset (e) and rat mortality (f). Onset was determined by a non-recoverable decrease in mobility during the open-field test, and full paralysis determined mortality
3.2 ∣. Expression of the PFN1 mutation in transgenic rats recapitulated the cardinal features of ALS
Transgenic rats expressing the PFN1-C71G mutation developed leg weakness by 240 days of age, and full paralysis in two or more legs by 300 days of age (Figure 1a-f). Consistent with findings in TDP-43 transgenic rats(Huang, Tong, Bi, Zhou, et al., 2012), open-field testing readily detected a reduction in mobility in PFN1 transgenic rats (Figure 1d). Both the mutant and WT transgenic rat lines expressed human PFN1 at comparable levels (Figure 1b), but no abnormality was detected in the WT PFN1 transgenic rats by 600 days old (the oldest rats examined), suggesting that the PFN1 mutation rather than transgene over-expression caused neurotoxicity (Figure 1). Patients carrying the PFN1-C71G mutation have been reported to develop ALS in the middle of their fifth decade (Wu et al., 2012). The rat strain used in our studies lives up to 2 years. Our PFN1 transgenic rats developed paralysis by 290 days and thus recapitulated the human middle-age onset of the disease. Pathological assays revealed a progressive loss of motor neurons in the spinal cord of PFN1-C71G transgenic rats and an aggressive activation of microglia and astrocytes in the nerve tissues affected (Figure 2). Quantification using unbiased stereological cell counting confirmed the loss of spinal motor neurons in diseased rats (Figure 2f). Histological analyses revealed degeneration of motor axons in the ventral roots and denervation atrophy of skeletal muscles in PFN1-C71G transgenic rats (Figure 3). Bielschowski silver staining revealed degenerating neurons in PFN1-C71G transgenic rats compared to the control rats (Figure S2). Similar, but more moderate, pathologies were also detected in the low-expressing line of PFN1-C71G transgenic rats (Figure S3), indicating that pathogenic PFN1 caused the disease in a dose-dependent manner. All the abnormalities detected by behavioral tests and pathology assays were not identified in WT PFN1 transgenic rats expressing normal human PFN1 at comparable levels (Figures 1-3). Our PFN1-C71G transgenic rats reproduced the cardinal features of the ALS caused by PFN1 mutation and would be useful for mechanistic studies of PFN1-related neurodegenerative diseases.
FIGURE 2.

Transgenic rats expressing mutant PFN1 developed a progressive loss of motor neurons and a robust activation of glial cells in the spinal cord. (a–e) Cresyl violet staining of lumbar spinal cords revealed progressive motor neuron loss in mutant PFN1 transgenic rats (C71G) both at disease onset (d) and at disease end-stage (e) compared to non-transgenic rats (a, NT), wild-type (WT) PFN1 transgenic rats (b, PFN1-WT), and presymptomatic C71G rats (c). (f) Stereological cell counting estimated the total number of motor neurons (>25 μm in diameter) in the L3-L5 lumbar segments of cords from C71G TG rats at onset and during paralysis stages, as well as NT and WT rats matched to paralyzed C71G rat ages. Data represent mean ± SD (n = 8, 4 males and 4 females). (g–p) Immunofluorescent staining for the microglial marker Iba-1 (g–k) and the astrocytic marker GFAP (i–p) revealed the focal activation of glial cells in spinal cord ventral horns. Immunostaining was done on two sections from each animal and the representative images were reproduced in two animals of each genotype. Scale bar: 100 μm (a–e and g–p)
FIGURE 3.

Histology assays revealed progressive loss of motor axons and denervation atrophy in skeletal muscles. (a–j) Toluidine blue staining revealed the structures of ventral roots (a–e) and dorsal roots (f–j) in mutant PFN1 TG rats (C71G) as well as in non-transgenic (NT) and wild-type (WT) PFN1 TG (PFN1-WT) rats age-matched to completely paralyzed C71G TG rats. Arrows point to degenerating axons. (k–o) Hematoxylin and eosin (H&E) staining revealed denervation atrophy in gastrocnemius muscles of C71G TG rats with partial (onset) or full paralysis, but not in age-matched NT or PFN1-WT rats. One typical section of ventral dorsal nerve roots and gastrocnemius muscles was stained for each rat. Histology was done for four rats of equal sex representation at a defined disease stage or matched age. Scale bars: 30 μm (a–j) and 60 pm (k–o)
3.3 ∣. PFN1 mutations gained a propensity for aggregation in cultured cells and in transgenic rats
ALS-causing mutations have been reported to destabilize the native conformation of PFN1, and increase the tendency of PFN1 to aggregate in test tube and in cell culture (Boopathy et al., 2015; Del Poggetto, Bemporad, Tatini, & Chiti, 2015; Rennella, Sekhar, & Kay, 2017). In our transgenic rats, over-expression of mutant, but not the WT, human PFN1 rats induced protein inclusions in affected neurons (Figure 4). As the disease progressed in PFN1-C71G transgenic rats, both PFN1 and p62 aggressively formed inclusions in affected spinal motor neurons as detected by immunohistochemistry and confocal microscopy (Figure 4d, h and j). Biochemistry assays revealed that the C71G mutant form, but not the WT form, aggressively accumulated in the detergent-insoluble fraction of tissue extracts from the spinal cords of transgenic rats (Figure 4). We examined four PFN1 mutations (i.e., A20T, C71G, M114T, and G118V) in HEK293 cells and observed an increased tendency for aggregation in all the PFN1 mutants examined, but variable insolubility was observed (Figure 5), indicating that aggregation is a common feature of pathogenic PFN1 mutations. Detergent-insoluble PFN1 was detected in transgenic rats by 150 days of age, long before disease onset in these rats by 240 days of age. PFN1 aggregation was detected as the first pathology in otherwise nonsymptomatic rats. Protein aggregation appears to be an important contributor rather than a bystander in neurodegeneration.
FIGURE 4.

Mutant PFN1 aggressively accumulated as protein inclusions in transgenic rats. (a–h) Immunohistochemistry revealed that PFN1 (a–d) and p62 (e–h) accumulated in spinal motor neurons of mutant PFN1 transgenic (TG) rats (C71G) compared to age-matched wild-type PFN1 TG (WT) and non-transgenic (NT) rats (d = days). Arrows point to motor neurons with enhanced staining for accumulated PFN1 (d) and p62 (h). Scale bars: 60 μm (a–h). (i–j) Confocal microscopy revealed PFN1 aggregates in spinal motor neurons of mutant PFN1 TG rats compared to age-matched NT rats. (k) A schematic showing the strategy of extracting detergent-insoluble PFN1 from mutant PFN1 TG rats. (l) Biochemical analyses revealed that mutant, but not WT, human PFN1 aggressively accumulated in the detergent-insoluble fractions of spinal cord extracts that were precipitated by ultracentrifugation. Immunostaining was done for 4 rats (2 males and 2 females) at each defined age and two tissue sections from each animal were stained to verify the consistence of pathological changes
FIGURE 5.
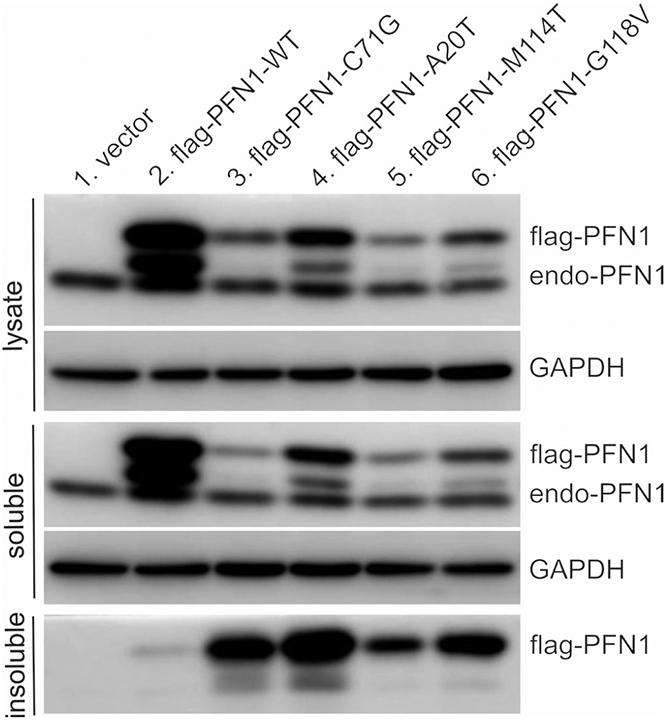
Pathogenic mutations reduced PFN1 solubility. HEK293 cells were transfected with plasmids expressing FLAG-tagged human PFN1 with or without pathogenic mutation. Cell lysates were fractionated into detergent-soluble and -insoluble fractions and PFN1 immunoreactivity in each fraction was assessed by immunoblotting. Equal loading of total proteins for both cell lysates and soluble fractions was confirmed by probing the same membrane with an antibody to GAPDH. Two batches of cells were transfected at different times and the immunoblotting was reproduced for the two separate transfections
4 ∣. DISCUSSION
We reported a novel rat model that expressed human PFN1 with a disease-linked mutation and developed the cardinal features of ALS. The transgenic rats expressing mutant PFN1-C71G progressively developed limb paralysis, resulting from the substantial loss of motor neurons and subsequent denervation atrophy of skeletal muscle. Mutant PFN1 transgenic rats also recapitulated the middle-age onset of the ALS in patients carrying the PFN1-C71G mutation (Wu et al., 2012). The two independent PFN1-C71G transgenic rat lines developed similar pathologies, but their phenotypes varied by degree and displayed a dose-dependent effect of the mutant PFN1 on neuronal function. This confirms that the rat disease phenotypes resulted from transgene expression and not from unexpected gene modifications such as transgene-insertional mutation. We obtained one mutant line and one WT PFN1 line that both expressed human PFN1 at comparable levels, but ALS-reminiscent phenotypes were only detected in the mutant PFN1-C71G transgenic rats. The differing phenotypes observed in PFN1 transgenic rats indicate that the neurotoxicity of PFN1-C71G resulted from the pathogenic mutation rather than from transgene over-expression. The transgenic rats expressed human PFN1 at a moderate level that was approximately twice the endogenous level of rat PFN1, and was much lower than other reports of expression levels of mutant PFN1 in transgenic mice (Fil et al., 2017; Yang et al., 2016). This difference in the level of mutant PFN1 transgene expression required for phenotypic expression in our rats and in mice is probably due to the different patterns of spatial and temporal transgene expression achieved in the two rodent models. Whereas the two reported PFN1 mouse models were created with artificial promoters driving human PFN1 cDNA transgenes (Fil et al., 2017; Yang et al., 2016), we used genomic DNA rather than cDNA to make transgenic rats so that both the coding and the regulatory sequences of the human PFN1 gene were introduced into the host genome. In our PFN1 transgenic rats, the transgene expression patterns were similar to those of the host Pfn1 gene. Mutant PFN1 was likely expressed in the right cells at the right time throughout the rat life cycle, minimizing any artificial effects of arbitrary transgene expression. Only a minimal level of transgene expression was thus required to induce disease phenotypes in PFN1 transgenic rats. The effects of transgene expression patterns on disease induction are exemplified in our study on TDP-43 transgenic rats. When mutant TDP-43 was expressed in one selected cell type, a higher level of transgene expression was required to induce a disease phenotype compared to mutant TDP-43 expression in multiple cell types (Huang, Tong, Bi, Zhou, et al., 2012; Tong et al., 2013; Zhou et al., 2010). In addition to the available transgenic PFN1 mouse models (Fil et al., 2017; Yang et al., 2016), these PFN1 transgenic rats may be useful for mechanistic studies of PFN1-related diseases.
The presence of detergent-insoluble PFN1 inclusions was a predominant pathology preceding disease onset in mutant PFN1 transgenic rats. PFN1 transgenic rats carried minimal copies of the mini human PFN1 gene and expressed human PFN1 at a moderate level. This moderate expression of mutant PFN1 caused late-onset ALS phenotypes and a slow progression of the disease in these transgenic rats. On average, disease onset occurred by the 240 days of age, and full paralysis occurred by 290 days. Long before the onset of the ALS phenotype, detergent-insoluble PFN1 was detected in the spinal cords of mutant PFN1 transgenic rats by 150 days of age. Appreciable PFN1 inclusions were detected by immunostaining only in symptomatic transgenic rats, and the low amount of insoluble PFN1 in asymptomatic rats was likely due to concentration by ultracentrifugation that reached threshold for biochemical detection. This biochemical approach was more sensitive than immunohistochemistry for the detection of insoluble PFN1. Pathogenic mutation of PFN1 alters its conformation and destabilizes the protein (Boopathy et al., 2015), increasing its propensity for aggregation. The cellular balance between mutant PFN1 production and degradation may initially be maintained but fails with time. The formation of protein inclusions indicates that the protein degradation machinery has become overloaded or is functionally impaired (Ito & Suzuki, 2009; Sifers, Brashears-Macatee, Kidd, Muensch, & Woo, 1988). Protein aggregation per se may also produce cytotoxic intermediate products, and the accumulation of misfolded or unfolded proteins may overwhelm protein degradation machinery and thus compromise cellular functions (Bucciantini et al., 2002; Cheroni et al., 2009; Walsh et al., 2002). Abnormal protein aggregation appears to be actively involved in neurodegeneration in the ALS.
Supplementary Material
ACKNOWLEDGEMENT
This work is supported by the National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (NS110455 and NS089701 to X.G.X. and H.Z., NS095962 to X.G.X. and NS07829 to H.Z.) and NIH/National Institute on Aging (AG064822 to X.G.X. and H.Z.). The content is the author's responsibility and does not necessarily represent the official view of the NIH institutes.
All experiments were conducted in compliance with the ARRIVE guidelines.
Abbreviations:
- ALS
amyotrophic lateral sclerosis
- FBS
fetus bovine serum
- PFN1
profilin 1
- RRID
research resource identifier
- WT
wildtype
Footnotes
CONFLICT OF INTEREST
Xugang Xia is an editor for Journal of Neurochemistry.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
REFERENCES
- Boopathy S, Silvas TV, Tischbein M, Jansen S, Shandilya SM, Zitzewitz JA, … Bosco DA (2015). Structural basis for mutation-induced destabilization of profilin 1 in ALS. Proceedings of the National Academy of Sciences of the United States of America, 112, 7984–7989. 10.1073/pnas.1424108112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruggink KA, Muller M, Kuiperij HB, & Verbeek MM (2012). Methods for analysis of amyloid-beta aggregates. Journal of Alzheimer's Disease, 28, 735–758. [DOI] [PubMed] [Google Scholar]
- Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J Stefani M (2002). Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature, 416, 507–511. 10.1038/416507a [DOI] [PubMed] [Google Scholar]
- Cheroni C, Marino M, Tortarolo M, Veglianese P, De Biasi S, Fontana E, Bendotti C (2009). Functional alterations of the ubiquitin-proteasome system in motor neurons of a mouse model of familial amyotrophic lateral sclerosis. Human Molecular Genetics, 18, 82–96. 10.1093/hmg/ddn319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creed RB, & Goldberg MS (2018). New Developments in genetic rat models of Parkinson's disease. Movement Disorders, 33, 717–729. 10.1002/mds.27296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave KD, De Silva S, Sheth NP, Ramboz S, Beck MJ, Quang C, … Frasier MA (2014). Phenotypic characterization of recessive gene knockout rat models of Parkinson's disease. Neurobiology of Diseases, 70, 190–203. 10.1016/j.nbd.2014.06.009 [DOI] [PubMed] [Google Scholar]
- Del Poggetto E, Bemporad F, Tatini F, & Chiti F (2015). Mutations of Profilin-1 associated with amyotrophic lateral sclerosis promote aggregation due to structural changes of its Native State. ACS Chemical Biology, 10, 2553–2563. 10.1021/acschembio.5b00598 [DOI] [PubMed] [Google Scholar]
- Di Nardo A, Gareus R, Kwiatkowski D, & Witke W (2000). Alternative splicing of the mouse profilin II gene generates functionally different profilin isoforms. Journal of Cell Science, 113(Pt 21), 3795–3803. [DOI] [PubMed] [Google Scholar]
- Ding Z, Joy M, Bhargava R, Gunsaulus M, Lakshman N, Miron-Mendoza M, … Roy P (2014). Profilin-1 downregulation has contrasting effects on early vs late steps of breast cancer metastasis. Oncogene, 33, 2065–2074. 10.1038/onc.2013.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Arif A, Gong Y et al. (2012). Stimulus-dependent phosphorylation of profilin-1 in angiogenesis. Nature Cell Biology, 14, 1046–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fil D, DeLoach A, Yadav S et al. (2017). Mutant profilin1 transgenic mice recapitulate cardinal features of motor neuron disease. Human Molecular Genetics, 26, 686–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gau D, Lewis T, McDermott L, Wipf P, Koes D, & Roy P (2018). Structure-based virtual screening identifies a small-molecule inhibitor of the profilin 1-actin interaction. Journal of Biological Chemistry, 293, 2606–2616. 10.1074/jbc.M117.809137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Del Tredici K, & Braak H (2012). 100 years of Lewy pathology. Nature Reviews. Neurology, 9, 13–24. 10.1038/nrneurol.2012.242 [DOI] [PubMed] [Google Scholar]
- Gorlich A, Zimmermann AM, Schober D, Bottcher RT, Sassoe-Pognetto M, Friauf E, … Rust MB (2012). Preserved morphology and physiology of excitatory synapses in profilin1-deficient mice. PLoS One, 7, e30068. 10.1371/journal.pone.0030068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Tong J, Bi F, Wu Q, Huang B, Zhou H, & Xia XG (2012). Entorhinal cortical neurons are the primary targets of FUS mislocalization and ubiquitin aggregation in FUS transgenic rats. Human Molecular Genetics, 21, 4602–4614. 10.1093/hmg/dds299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Tong J, Bi F, Zhou H, & Xia XG (2012). Mutant TDP-43 in motor neurons promotes the onset and progression of ALS in rats. J Clin Invest, 122, 107–118. 10.1172/JCI59130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Zhou H, Tong J, Chen H, Liu Y-J, Wang D, … Xia X-G (2011). FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLOS Genetics, 7, e1002011. 10.1371/journal.pgen.1002011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito D, & Suzuki N (2009). Seipinopathy: A novel endoplasmic reticulum stress-associated disease. Brain, 132, 8–15. 10.1093/brain/awn216 [DOI] [PubMed] [Google Scholar]
- Kwong LK, Neumann M, Sampathu DM, Lee VM, & Trojanowski JQ (2007). TDP-43 proteinopathy: The neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathologica, 114, 63–70. 10.1007/s00401-007-0226-5 [DOI] [PubMed] [Google Scholar]
- Kwong LK, Uryu K, Trojanowski JQ, & Lee VM (2008). TDP-43 proteinopathies: Neurodegenerative protein misfolding diseases without amyloidosis. Neurosignals, 16, 41–51. 10.1159/000109758 [DOI] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, & Lee VM (2012). Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science, 338, 949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, & Lee VM (2012). Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. Journal of Experimental Medicine, 209, 975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelsen K, Murk K, Zagrebelsky M, Dreznjak A, Jockusch BM, Rothkegel M, & Korte M (2010). Fine-tuning of neuronal architecture requires two profilin isoforms. Proceedings of the National Academy of Sciences, 107, 15780–15785. 10.1073/pnas.1004406107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, … Lee V-M-Y (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science, 314, 130–133. 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- Nguyen HP, Van Broeckhoven C, & van der Zee J (2018). ALS Genes in the Genomic Era and their Implications for FTD. Trends in Genetics, 34, 404–423. 10.1016/j.tig.2018.03.001 [DOI] [PubMed] [Google Scholar]
- Rennella E, Sekhar A, & Kay LE (2017). Self-assembly of human profilin-1 detected by Carr-Purcell-Meiboom-Gill nuclear magnetic resonance (CPMG NMR) spectroscopy. Biochemistry 56, 692–703. 10.1021/acs.biochem.6b01263 [DOI] [PubMed] [Google Scholar]
- Rodriguez JA, Ivanova MI, Sawaya MR et al. (2015). Structure of the toxic core of alpha-synuclein from invisible crystals. Nature, 525, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero S, Le Clainche C, Didry D, Egile C, Pantaloni D, & Carlier MF (2004). Formin is a processive motor that requires profilin to accelerate actin assembly and associated ATP hydrolysis. Cell, 119, 419–429. 10.1016/j.cell.2004.09.039 [DOI] [PubMed] [Google Scholar]
- Sifers RN, Brashears-Macatee S, Kidd VJ, Muensch H, & Woo SL (1988). A frameshift mutation results in a truncated alpha 1-antitrypsin that is retained within the rough endoplasmic reticulum. Journal of Biological Chemistry, 263, 7330–7335. [PubMed] [Google Scholar]
- Smith BN, Vance C, Scotter EL, Troakes C, Wong CH, Topp S, … Shaw CE (2015). Novel mutations support a role for Profilin 1 in the pathogenesis of ALS. Neurobiology of Aging, 36(1602), 1602.e17–1602.e27. 10.1016/j.neurobiolaging.2014.10.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JP, Brown RH Jr, & Cleveland DW (2016). Decoding ALS: From genes to mechanism. Nature, 539, 197–206. 10.1038/nature20413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J, Huang C, Bi F, Wu Q, Huang B, Liu X, … Xia XG (2013). Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO Journal, 32, 1917–1926. 10.1038/emboj.2013.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, … Selkoe DJ (2002). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature, 416, 535–539. [DOI] [PubMed] [Google Scholar]
- Warming S, Costantino N, Court DL, Jenkins NA, & Copeland NG (2005). Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Research, 33, e36. 10.1093/nar/gni035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witke W (2004). The role of profilin complexes in cell motility and other cellular processes. Trends in Cell Biology, 14, 461–469. 10.1016/j.tcb.2004.07.003 [DOI] [PubMed] [Google Scholar]
- Witke W, Sutherland JD, Sharpe A, Arai M, & Kwiatkowski DJ (2001). Profilin I is essential for cell survival and cell division in early mouse development. Proceedings of the National Academy of Sciences of the United States of America, 98, 3832–3836. 10.1073/pnas.051515498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C-H, Fallini C, Ticozzi N, Keagle PJ, Sapp PC, Piotrowska K, … Landers JE (2012). Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature, 488, 499–503. 10.1038/nature11280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Liu M, Huang C, Liu X, Huang B, Li N, … Xia XG (2015). Pathogenic Ubqln2 gains toxic properties to induce neuron death. Acta Neuropathologica, 129, 417–428. 10.1007/s00401-014-1367-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Danielson EW, Qiao T, Metterville J, Brown RH Jr, Landers JE, & Xu Z (2016). Mutant PFN1 causes ALS phenotypes and progressive motor neuron degeneration in mice by a gain of toxicity. Proceedings of the National Academy of Sciences, 113, E6209–E6218. 10.1073/pnas.1605964113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Huang C, Chen H, Wang D, Landel CP, Xia PY, … Xia XG (2010). transgenic rat model of neurodegeneration caused by mutation in the TDP gene. PLOS Genetics, 6, e1000887. 10.1371/journal.pgen.1000887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Huang C, Yang M, Landel CP, Xia PY, Liu YJ, & Xia XG (2009). Developing tTA transgenic rats for inducible and reversible gene expression. International Journal of Biological Sciences, 2, 171–181. 10.7150/ijbs.5.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.