

Abstract
CAPN1‐associated hereditary spastic paraplegia (SPG76) is a rare and clinically heterogenous syndrome due to loss of calpain‐1 function. Here we illustrate a translational approach to the case of an 18‐year‐old patient who first presented with psychiatric symptoms followed by spastic gait, intention tremor, and neurogenic bladder dysfunction, consistent with a complex form of HSP. Exome sequencing showed compound‐heterozygous missense variants in CAPN1 (NM_001198868.2: c.1712A>G (p.Asn571Ser)/c.1991C>T (p.Ser664Leu)) and a previously reported heterozygous stop‐gain variant in RCL1. In silico analyses of the CAPN1 variants predicted a deleterious effect and in vitro functional studies confirmed reduced calpain‐1 activity and dysregulated downstream signaling. These findings support a diagnosis of SPG76 and highlight that the psychiatric symptoms can precede the motor symptoms in HSP. Our results also suggest that multiple genes can potentially contribute to complex neuropsychiatric diseases.
Introduction
The hereditary spastic paraplegias (HSPs) are a group of more than 80 different neurogenetic disorders. 1 While pure and complex forms share the clinical feature of progressive spasticity due to corticospinal tract dysfunction, complex HSP presents with additional neurological symptoms such as cognitive impairment, ataxia, neuropathy, seizures, and psychiatric symptoms. 1 , 2 In some cases, nonmotor symptoms precede the onset of motor symptoms and signs of corticospinal tract dysfunction can be subtle initially. Next‐generation sequencing has enabled an early diagnosis of many forms of HSP 3 ; however, interpretation of novel missense variants remains challenging. Functional studies in patient‐derived cells assist the interpretation of molecular findings and can support a diagnosis.
SPG76 is a complex form of HSP, caused by bi‐allelic mutations in CAPN1, encoding the calcium‐activated protease calpain‐1. 4 Heterozygous variants in RCL1 are associated with early‐onset psychosis. 5 Here we further characterize a patient with novel CAPN1 variants and a previously reported heterozygous stop‐gain mutation in RCL1, 5 who presented in adolescence with psychiatric symptoms, later followed by lower limb spasticity, cerebellar signs, and neurogenic bladder dysfunction.
Methods
Clinical characterization
This study was approved at the Boston Children's Hospital (IRB‐P00033016). Written consent was obtained. Trio‐exome sequencing was performed at GeneDx (Gaithersburg, MD, USA).
In silico analyses
Information on the human CAPN1 protein sequence was obtained from the Universal Protein Resource (ID: P07384) and AlphaFold Protein Structure (DeepMind Technologies) databases, as well as published data. 6 Protein tertiary structures were visualized using Pymol (v2.5.0, Schroedinger, LLC, New York, NY, USA). CADD PHRED scores (v1.6) of all possible base substitutions of the CAPN1 transcript were computed and mapped to the corresponding protein sequence. 7 All variants were harmonized to NM_001198868.2/GRCh38/hg38. Modeling of the CAPN1 primary protein structure and variants was done in R version 4.1.0 (2021‐05‐18) and RStudio (version 1.4.1103; RStudio, Inc., Boston, MA, USA).
Functional assays
Primary skin fibroblasts were cultured according to established protocols. 8 , 9 , 10 Calpain enzyme activity was assessed using a fluorogenic enzyme assay (Sigma‐Aldrich, St. Louis, MO, USA; Cat#QIA120). Calpain activity was quantified in two biological replicates and normalized to protein concentrations. Western blotting was performed as described, and each experiment was performed in at least three biological replicates. 8 Antibodies and reagents are listed in Table S1. For treatment with PHLPP1 inhibitor fibroblasts were seeded at 500 × 103 in 6‐well‐plates and 24 h postplating media containing NSC117079 or 0.5% DMSO was added. After 24 h of treatment, cells were harvested. Statistical analysis was performed with Prism (GraphPad Software, San Diego, CA, USA). Groups were compared using Mann–Whitney U test or two‐way ANOVA with Tukey's post hoc analysis. p < 0.05 was considered significant.
Clinical Presentation
The proband's initial presentation has been described by Brownstein et al. 5 Briefly, at age 14‐years the proband presented with subacute‐onset catatonia, and several months of anxiety, auditory and visual hallucinations, disorganized thoughts, paranoia, and aggression, requiring hospitalization. Prior to this, there had been no neurological or psychiatric concerns. He was born at full term to nonconsanguineous parents of mixed European ancestry, achieved all developmental milestones appropriately, and was an average student with no history of learning difficulties. Given the above constellation of symptoms, he underwent a broad diagnostic evaluation. Neurological examination at age 14‐years was negative for pyramidal or cerebellar signs. EEG and brain MR imaging showed no significant abnormalities. CSF studies were notable for a persistently elevated protein (initially 80 mg/dL, remained between 50 and 65 mg/dL on repeat measurements), but serum and CSF autoantibody panels were negative. He received immunomodulatory treatment for presumed antibody‐negative autoimmune encephalitis without improvement. His psychiatric symptoms stabilized on treatment with clozapine. Serial neuropsychological examinations showed deficits in attention and executive function. At age 15‐years, he developed urinary urgency and incontinence. Clozapine can lead to urinary incontinence and may have been contributing to these symptoms, 11 however, urodynamic studies confirmed neurogenic bladder dysfunction. At the age of 16‐years, his gait was noted to decline, he complained about muscle cramps and his legs being stiff and started having difficulties with handwriting. At the age of 18‐years, he was found to have an intention tremor of the hands, mild spasticity in his distal legs, and a positive Babinski sign. At age 20 years, his exam was notable for dysarthria and a spastic gait. Between the age of 16 and 20 years, his spastic paraplegia was found to have progressed leading to a Spastic Paraplegia Rating Scale score of 10 at last follow‐up.
Molecular Findings and In Silico Analyses
Trio‐exome‐sequencing demonstrated two novel missense variants in CAPN1 and a paternally inherited heterozygous stop‐gain mutation in RCL1 (Table 1). The paternally inherited CAPN1 variant (c.1712A>G (p.Asn571Ser)) showed an allele frequency of 0.000017 in gnomAD exomes with a homozygous allele count of 0 and is predicted to result in a conservative amino acid exchange in the penta‐EF‐hand (PEF) Ca2+ binding domain. The maternally inherited CAPN1 variant (c.1991C>T (p.Ser664Leu)) showed an allele frequency of 0.000012 and a homozygous allele count of 0 and leads to a nonconservative amino acid exchange also affecting the PEF domain. CADD PHRED scores were 27.7 and 24.1, respectively. Analysis of sequence conservation suggests the c.1712A>G variant to be highly and the c.1991C>T variant to be moderately conserved (Table 1). No tertiary structure has been published for the PEF domain of human calpain‐1. We, therefore, used the AlphaFold Protein Structure Database 12 , 13 to predict interactions of the Asn571 and Ser664 residues. Asn571 is predicted to form two hydrogen bonds with Phe585 and Ser664 forms a hydrogen bond with Ile660. Based on these predictions, amino acid exchanges at positions 571 and 664 likely lead to a change in tertiary structure (Fig. 1A). An additive model of CADD PHRED scores for all possible missense across the linear protein structure identified no clear mutational hotspots with low tolerability to missense variation across the entire protein (Fig. 1B). The RCL1 variant has been discussed previously. 5
Table 1.
Variants discovered in the proband.
| Gene | Genomic location (hg38) | Inheritance | Variant impact | ACMG classification | Conservation prediction (GERP/PhyloP100way/PhastCons100way) | Allele count gnomAD (homozygous/total) | Allele frequency gnomAD (exomes/genomes) |
|---|---|---|---|---|---|---|---|
| CAPN1 |
NC_000011.10: g.64975716A>G NM_001198868.2: c.1712A>G NP_001185797.1: p.Asn571Ser |
Paternal | Missense, conservative amino acid exchange | Likely pathogenic | 5.23/9.318/1.000 | 0/4 | ƒ = 0.0000167/0.0000319 |
| CAPN1 |
NC_000011.9: g.64977855C>T NM_001198868.2: c.1991C>T NP_001185797.1: p.Ser664Leu |
Maternal | Missense, nonconservative amino acid exchange | Likely pathogenic | 4.21/7.555/0.961 | 0/3 | ƒ = 0.0000121/0 |
| RCL1 |
NC_000009.11: g.4827019C>T NM_005772.5: c.370C>T NP_005763.3: p.Gln124Ter |
Paternal | Stop‐gain | Pathogenic | 5.92/7.542/1.000 | 0/0 | ƒ = 0/0 |
Figure 1.
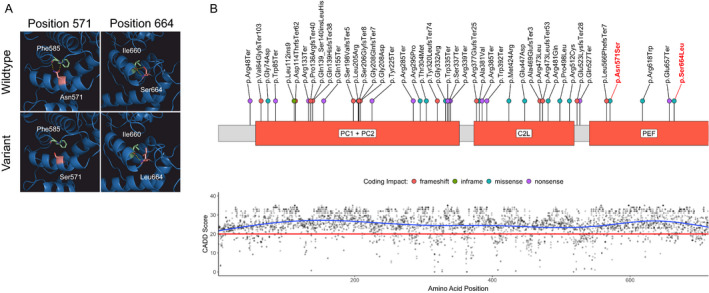
Calpain‐1 structure with novel variants, variant distribution, and CADD PHRED scores. (A) The sites of predicted amino acid exchange in the PEF domain in the proband compared to wildtype. Mutated amino acids are colored in light red, interaction partners in light green, and hydrogen bonds in yellow. (B, upper panel) Schematic of the calpain‐1 primary protein structure. Disease‐associated variants identified in the literature are annotated along the protein with colored dots representing coding impacts. Novel variants identified in this report are labeled in red. (B, lower panel) CADD PHRED scores for all possible missense variants aligned to the CAPN1 protein structure. The recommended cut‐off for deleteriousness (20) is depicted as a red line. C2L, C2‐like domain; PC, protease core domains; PEF, large subunit of the penta‐EF‐hand domains. [Colour figure can be viewed at wileyonlinelibrary.com]
Functional Assessment of Novel CAPN1 Variants
Considering the possible pathogenicity of the CAPN1 missense variants implied by in silico predictions and their localization in the PEF domain, we next performed experiments in patient‐derived fibroblasts to examine the impact on calpain‐1 activity and downstream signaling. Figure 2A shows an overview of the calpain pathway. Pan‐calpain activity was reduced by 30.4 ± 11.5% in the proband's fibroblasts compared to the control (Fig. 2B). To determine whether this was caused by altered protein abundance, levels of calpain‐1 and calpain‐2 were quantified, with no changes identified (Fig. 2C). Next, we measured protein levels of the calpain‐1 substrate PHLPP1 and the calpain‐2 substrate PTEN. 14 , 15 PHLPP1, but not PTEN, was significantly increased in the proband's cells (Fig. 2D and E). PHLPP1 is an important negative regulator of the Akt signaling pathway through specific dephosphorylation of the active phospho‐Akt at Ser473 (pAkt) (Fig. 2A). 16 In agreement with increased PHLPP1 levels present in the proband's cells, levels of pAkt were reduced (Fig. 2F). To further investigate whether this was a result of increased PHLPP1 activity, we treated fibroblasts with increasing concentrations of NSC117079, a potent inhibitor of PHLPP1, and determined pAkt levels after 24 h of treatment. Treatment with 50 μmol/L NSC117079 increased pAkt in the proband's fibroblasts (36.2% vs. 23.4% in the control, p = 0.14, Fig. 2G). Taken together, these results provide evidence for a partial loss of calpain‐1 function in the proband's cells, leading to disinhibition of PHLPP1 activity and subsequent reduction of pAkt levels. This supports a diagnosis of CAPN1‐related SPG76.
Figure 2.
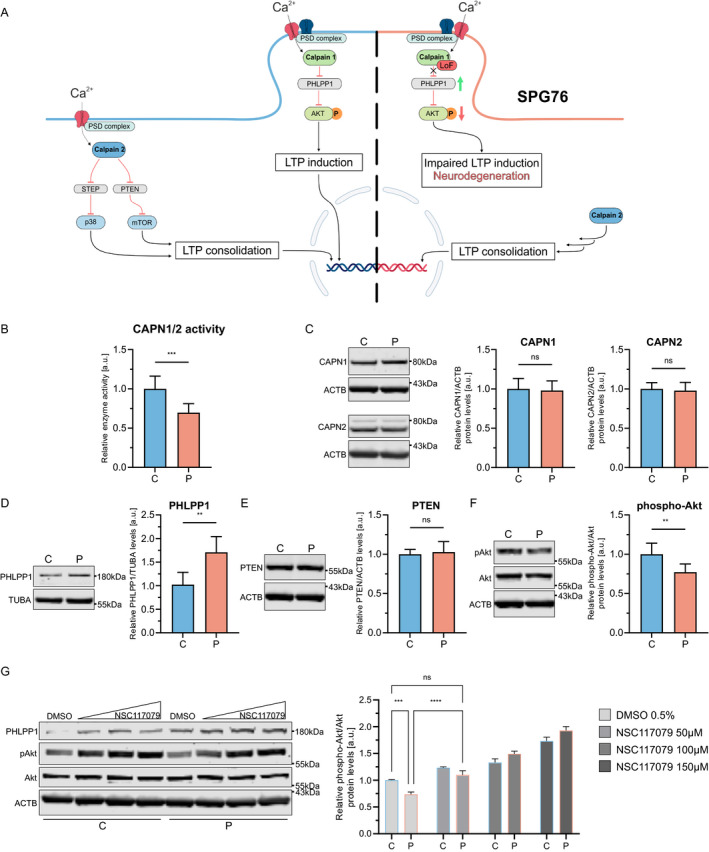
Calpain‐1 pathway and results of functional studies. (A) Schematic of the calpain‐1 and calpain‐2 pathways. The left half depicts physiological calpain signaling. The right half shows the impact of the loss of calpain‐1 function and associated dysregulation of downstream signaling in SPG76. (B) Calpain‐1 and calpain‐2 activity are measured in fibroblast lysates using a fluorogenic assay (p = 0.0004). (C) Western blot for calpain‐1 and calpain‐2 (p = 0.67, p = 0.93). (D) Western blot for calpain‐1 substrate PHLPP1 (p = 0.0087). (E) WB for calpain‐2 substrate PTEN (p > 0.99). (F) Western blot for Akt and phospho‐(Ser473)‐Akt (p = 0.004). (G) Western blot of NSC117079‐ or DMSO‐treated fibroblasts for Akt and phospho‐Akt. Whole‐cell fibroblast lysates were used for all WB experiments. Molecular weights are provided in kilodaltons (kDa). C, control; CAPN1, calpain‐1; CAPN2, calpain‐2; P, proband. Values are shown as mean ± SD, (ns = not significant, **p < 0.01, ***p < 0.001, ****p < 0.0001; [B–F] Mann–Whitney U test; [G] two‐way ANOVA with multiple comparisons and Tukey's post hoc analysis). [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
We here report a patient with significant psychiatric symptoms during adolescence and subsequent development of a spastic gait, intention tremor and neurogenic bladder dysfunction. The proband was previously reported as an index case for RCL1‐associated psychiatric syndrome 5 , 17 but his progressive motor and urinary symptoms remained unexplained. Exome sequencing showed compound‐heterozygous missense variants in CAPN1. Calpain‐1 is an important regulator of neuronal survival, axonal homeostasis and synaptic plasticity, 18 and loss of calpain‐1 function is known to cause SPG76. 4 In light of the proband's evolving neurological syndrome, consistent with a complex form of HSP, we pursued functional assays to assess calpain‐1 function in his cells.
Our in silico analyses predicted a potential impact of both CAPN1 variants on protein tertiary structure. In support of this, we found that, although our patient's CAPN1 variants do not alter protein expression and stability, the resultant amino acid exchanges in the PEF domain lead to reduced enzyme activity. This results in reduced degradation of PHLPP1 and subsequently reduced Akt‐signaling. Further studies are needed to separate the impact of each variant on enzyme activity. Another important consideration are cell‐type‐specific differences in the calpain‐1 pathway. Although primary fibroblasts are an important tool for functional studies, patient‐derived neuronal cells will provide a more relevant model system to understand the molecular mechanisms of CAPN1‐associated HSP.
Both RCL1 haploinsufficiency and bi‐allelic variants in CAPN1 have been associated with a variety of psychiatric symptoms. 5 , 6 , 19 SPG76 shows a wide phenotypic spectrum with pure and complex forms and significant inter‐ and intra‐familial variability. 4 , 6 , 20 , 21 , 22 , 23 Our patient's current symptoms and age at presentation align with the reported spectrum of SPG76 6 while his early‐onset psychosis may be attributable to the additional effect of his RCL1 variant. Further studies are needed to evaluate if psychiatric symptoms precede motor symptoms in other patients with SPG76 and if RCL1 variants can act as a genetic modifier in SPG76 or other neuropsychiatric diseases. The existing literature might be biased toward diagnoses based on motor symptoms and psychiatric symptoms, that is, anxiety or mood disorders, are likely underrecognized. Several genes known to cause spastic paraplegia or spastic‐ataxia can lead to psychiatric symptoms, including psychosis, 24 , 25 but no common molecular or clinical signature has yet been identified.
In conclusion, we reported a patient with CAPN1‐associated SPG76 and expand the clinical and molecular spectrum associated with this rare form of complex HSP. The present case highlights the need to carefully characterize the functional impact of novel variants and that multiple genes can contribute to complex neuropsychiatric diseases.
Conflict of Interest
D. E. F. received a speaker honorarium from the Movement Disorder Society, publishing royalties from the Cambridge University Press and reports research funding through a joint research agreement with Astellas Pharmaceuticals Inc.
Supporting information
Table S1. Primary antibodies and reagents.
Acknowledgments
The authors thank the patient and his family who participated in this study. J. E. A. is supported by the Deutsche Forschungsgemeinschaft (German Research Foundation, 270949263/GRK2162), the German National Academic Foundation, the Max Weber‐Program of the State of Bavaria, and the German Academic Exchange Service (DAAD). A. S. is funded by the Deutsche Forschungsgemeinschaft (German Research Foundation, SA 4171/1‐1). M. Z. received a scholarship from the German National Academic Foundation. D. E. F. received support from the CureAP4 Foundation, the CureSPG50 Foundation, the Spastic Paraplegia Foundation, the Manton Center for Orphan Disease Research and the National Institute of Health/National Institute of Neurological Disorders and Stroke (1K08NS123552‐01). The BCH Intellectual and the Developmental Disabilities Research Center is supported by the National Institutes of Health (BCH IDDRC, 1U54HD090255).
Funding Information
J. E. A. is supported by the Deutsche Forschungsgemeinschaft (German Research Foundation, 270949263/GRK2162), the German National Academic Foundation, the Max Weber‐Program of the State of Bavaria, and the German Academic Exchange Service (DAAD). A. S. is funded by the Deutsche Forschungsgemeinschaft (German Research Foundation, SA 4171/1‐1). M. Z. received a scholarship from the German National Academic Foundation. D. E. F. received support from the CureAP4 Foundation, the CureSPG50 Foundation, the Spastic Paraplegia Foundation, the Manton Center for Orphan Disease Research and the National Institute of Health/National Institute of Neurological Disorders and Stroke (1K08NS123552‐01). The BCH Intellectual and the Developmental Disabilities Research Center is supported by the National Institutes of Health (BCH IDDRC, 1U54HD090255).
Funding Statement
This work was funded by CureAP4 Foundation; CureSPG50 Foundation; Deutsche Forschungsgemeinschaft grants 270949263/GRK2162 and SA 4171/1‐1; German Academic Exchange Service (DAAD) ; German National Academic Foundation ; Manton Center for Orphan Disease Research ; National Institute of Neurological Disorders and Stroke grant 1K08NS123552‐01; National Institutes of Health grant 1U54HD090255; Program of the State of Bavaria; Spastic Paraplegia Foundation ; Max Weber‐Program of the State of Bavaria.
References
- 1. Shribman S, Reid E, Crosby AH, Houlden H, Warner TT. Hereditary spastic paraplegia: from diagnosis to emerging therapeutic approaches. Lancet Neurol. 2019;18(12):1136‐1146. [DOI] [PubMed] [Google Scholar]
- 2. Erfanian Omidvar M, Torkamandi S, Rezaei S, et al. Genotype‐phenotype associations in hereditary spastic paraplegia: a systematic review and meta‐analysis on 13,570 patients. J Neurol. 2021;268(6):2065‐2082. [DOI] [PubMed] [Google Scholar]
- 3. Saputra L, Kumar KR. Challenges and controversies in the genetic diagnosis of hereditary spastic paraplegia. Curr Neurol Neurosci Rep. 2021;21(4):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gan‐Or Z, Bouslam N, Birouk N, et al. Mutations in CAPN1 cause autosomal‐recessive hereditary spastic paraplegia. Am J Hum Genet. 2016;98(5):1038‐1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brownstein CA, Smith RS, Rodan LH, et al. RCL1 copy number variants are associated with a range of neuropsychiatric phenotypes. Mol Psychiatry. 2021;26(5):1706‐1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mereaux JL, Firanescu C, Coarelli G, et al. Increasing involvement of CAPN1 variants in spastic ataxias and phenotype‐genotype correlations. Neurogenetics. 2021;22(1):71‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Neuser S, Brechmann B, Heimer G, et al. Clinical, neuroimaging, and molecular spectrum of TECPR2‐associated hereditary sensory and autonomic neuropathy with intellectual disability. Hum Mutat. 2021;42(6):762‐776. [DOI] [PubMed] [Google Scholar]
- 8. Behne R, Teinert J, Wimmer M, et al. Adaptor protein complex 4 deficiency: a paradigm of childhood‐onset hereditary spastic paraplegia caused by defective protein trafficking. Hum Mol Genet. 2020;29(2):320‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ebrahimi‐Fakhari D, Alecu JE, Brechmann B, et al. High‐throughput imaging of ATG9A distribution as a diagnostic functional assay for adaptor protein complex 4‐associated hereditary spastic paraplegia. Brain Commun. 2021;3(4):fcab221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ebrahimi‐Fakhari D, Wahlster L, Bartz F, et al. Reduction of TMEM97 increases NPC1 protein levels and restores cholesterol trafficking in Niemann‐pick type C1 disease cells. Hum Mol Genet. 2016;25(16):3588‐3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fuller MA, Borovicka MC, Jaskiw GE, Simon MR, Kwon K, Konicki PE. Clozapine‐induced urinary incontinence: incidence and treatment with ephedrine. J Clin Psychiatry. 1996;57(11):514‐518. [DOI] [PubMed] [Google Scholar]
- 12. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583‐589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Varadi M, Anyango S, Deshpande M, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein‐sequence space with high‐accuracy models. Nucleic Acids Res. 2022;50(D1):D439‐D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Briz V, Hsu YT, Li Y, Lee E, Bi X, Baudry M. Calpain‐2‐mediated PTEN degradation contributes to BDNF‐induced stimulation of dendritic protein synthesis. J Neurosci. 2013;33(10):4317‐4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shimizu K, Phan T, Mansuy IM, Storm DR. Proteolytic degradation of SCOP in the hippocampus contributes to activation of MAP kinase and memory. Cell. 2007;128(6):1219‐1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell. 2007;25(6):917‐931. [DOI] [PubMed] [Google Scholar]
- 17. Amin N, de Vrij FMS, Baghdadi M, et al. A rare missense variant in RCL1 segregates with depression in extended families. Mol Psychiatry. 2018;23(5):1120‐1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baudry M, Bi X. Calpain‐1 and calpain‐2: the yin and yang of synaptic plasticity and neurodegeneration. Trends Neurosci. 2016;39(4):235‐245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Amini M, Ma CL, Farazifard R, et al. Conditional disruption of calpain in the CNS alters dendrite morphology, impairs LTP, and promotes neuronal survival following injury. J Neurosci. 2013;33(13):5773‐5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kocoglu C, Gundogdu A, Kocaman G, et al. Homozygous CAPN1 mutations causing a spastic‐ataxia phenotype in 2 families. Neurol Genet. 2018;4(1):e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lai LL, Chen YJ, Li YL, et al. Novel CAPN1 mutations extend the phenotypic heterogeneity in combined spastic paraplegia and ataxia. Ann Clin Transl Neurol. 2020;7(10):1862‐1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Travaglini L, Bellacchio E, Aiello C, Pro S, Bertini E, Nicita F. Expanding the clinical phenotype of CAPN1‐associated mutations: a new case with congenital‐onset pure spastic paraplegia. J Neurol Sci. 2017;378:210‐212. [DOI] [PubMed] [Google Scholar]
- 23. Shetty A, Gan‐Or Z, Ashtiani S, et al. CAPN1 mutations: expanding the CAPN1‐related phenotype: from hereditary spastic paraparesis to spastic ataxia. Eur J Med Genet. 2019;62(12):103605. [DOI] [PubMed] [Google Scholar]
- 24. Soares IFZ, Ciarlariello VB, Feder D, Carvalho AAS. Cognitive dysfunction and psychosis: expanding the phenotype of SPG7. Neurocase. 2021;27(3):253‐258. [DOI] [PubMed] [Google Scholar]
- 25. McMonagle P, Hutchinson M, Lawlor B. Hereditary spastic paraparesis and psychosis. Eur J Neurol. 2006;13(8):874‐879. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primary antibodies and reagents.