

SUMMARY
Cdc48 (p97/VCP) is a AAA-ATPase that can extract ubiquitinated proteins from their binding partners and can cooperate with the proteasome for their degradation. A fission yeast cdc48 mutant (cdc48-353) shows low levels of the cohesin protease, separase, and pronounced chromosome segregation defects in mitosis. Separase initiates chromosome segregation when its binding partner securin is ubiquitinated and degraded. The low separase levels in the cdc48-353 mutant have been attributed to a failure to extract ubiquitinated securin from separase, resulting in co-degradation of separase along with securin. If true, Cdc48 would be important in mitosis. In contrast, we show here that low separase levels in the cdc48-353 mutant are independent of mitosis. Moreover, we find no evidence of enhanced separase degradation in the mutant. Instead, we suggest that the cdc48-353 mutant uncovers specific requirements for separase translation. Our results highlight a need to better understand how this key mitotic enzyme is synthesized.
Graphical abstract
In brief
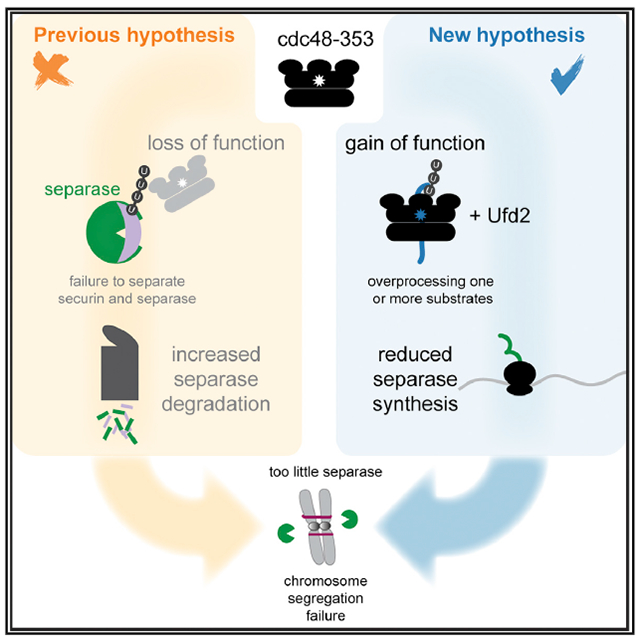
Vijayakumari et al. provide evidence against the long-standing hypothesis that low levels of separase in the S. pombe cdc48-353 mutant are caused by mitotic co-degradation of separase, along with its binding partner securin. Instead, separase protein synthesis may be defective in cdc48-353 cells.
INTRODUCTION
Separase is a protease that triggers chromosome separation in anaphase by cleaving the cohesin complex (Uhlmann et al., 2000). Prior to anaphase, separase is kept inactive by inhibitory binding of securin, as well as in some eukaryotes by inhibitory binding of cyclin B and Sgo2/Mad2 (Hellmuth et al., 2020; Stemmann et al., 2001; Uhlmann, 2001). Removal of securin from separase is initiated by the anaphase promoting complex (APC/C), an E3 ligase. The APC/C ubiquitinates securin, which marks it for proteasomal degradation. The APC/C also ubiquitinates cyclin B, whose degradation then leads to the inactivation of cyclin-dependent kinase (CDK1) and mitotic exit (Peters, 2006).
Cdc48 (also known as p97 or VCP in vertebrates) is a homohexameric AAA-ATPase that targets ubiquitinated proteins and is essential for cell viability. Cdc48 can unfold ubiquitinated proteins and promote their degradation by the proteasome (van den Boom and Meyer, 2018; Ye et al., 2017). Cdc48 has an N-terminal domain, two tandem ATPase domains (D1 and D2), and an unstructured C-terminal tail. In the hexameric complex, D1 and D2 form a double-ring structure with a central pore through which substrates can be threaded, which unfolds the polypeptide (Banerjee et al., 2016; Bodnar and Rapoport, 2017; Cooney et al., 2019; Ji et al., 2021; Twomey et al., 2019). Both the N-terminal domain and the C-terminal tail provide platforms for cofactor binding. Two of the best studied cofactors are the heterodimeric Ufd1/Npl4 complex and Shp1 (p47), which mediate substrate recognition (Buchberger et al., 2015; Schuberth et al., 2004; Ye et al., 2017). Other cofactors change the degree or type of ubiquitination of substrates (Jentsch and Rumpf, 2007; Ye et al., 2017). Ufd2 is one such cofactor, which in budding yeast (S. cerevisiae) binds to the C-terminal tail of Cdc48 and acts as E4 ubiquitin ligase, adding additional ubiquitin moieties to enhance proteasomal degradation (Böhm et al., 2011; Rumpf and Jentsch, 2006).
Central to the function of Cdc48 is its role as a segregase. Cdc48 separates ubiquitinated targets from their non-modified binding partners or from cellular structures (Stolz et al., 2011; Ye et al., 2017). For example, p97Ufd1/Npl4 extracts ubiquitinated Aurora B kinase from chromatin at mitotic exit to promote chromatin decondensation and nuclear envelope reformation (Ramadan et al., 2007), and yeast Cdc48 mobilizes the transcription factors Mga2 and Spt23 from the endoplasmic reticulum membrane (Rape et al., 2001; Shcherbik and Haines, 2007).
In fission yeast (S. pombe), a temperature-sensitive allele of cdc48 (cdc48-353) was isolated based on its characteristic of being suppressed by separase overexpression (Yuasa et al., 2004). The mutation in cdc48-353 resides in the D1 domain and replaces glycine 338 with aspartate (G338D) (Ikai and Yanagida, 2006). The cdc48-353 mutant has low levels of separase and displays a severe chromosome segregation defect, similar to separase mutants. This segregation defect is rescued by overexpression of separase (Ikai and Yanagida, 2006; Yuasa et al., 2004). To explain this phenotype, it has been proposed that Cdc48 targets polyubiquitinated securin and extracts it from separase (Yuasa et al., 2004). The low levels of separase in the cdc48-353 mutant could then be explained by a failure to extract securin from separase and subsequent co-degradation of separase, along with securin, during anaphase (Figure 1A). This is an attractive model that is consistent with findings that impairing Cdc48/p97 activity can lead to mitotic delays in S. cerevisiae and human cells (Chien and Chen, 2013; Wojcik et al., 2004), and that the Cdc48 ortholog p97 recognizes branched ubiquitin chains synthesized by the human APC/C (Meyer and Rape, 2014; Oh et al., 2020). If the hypothesis is true, Cdc48 would be a key player in the regulation of anaphase.
Figure 1. Mitotic phenotype of cdc48-353 cells and the previously proposed model.
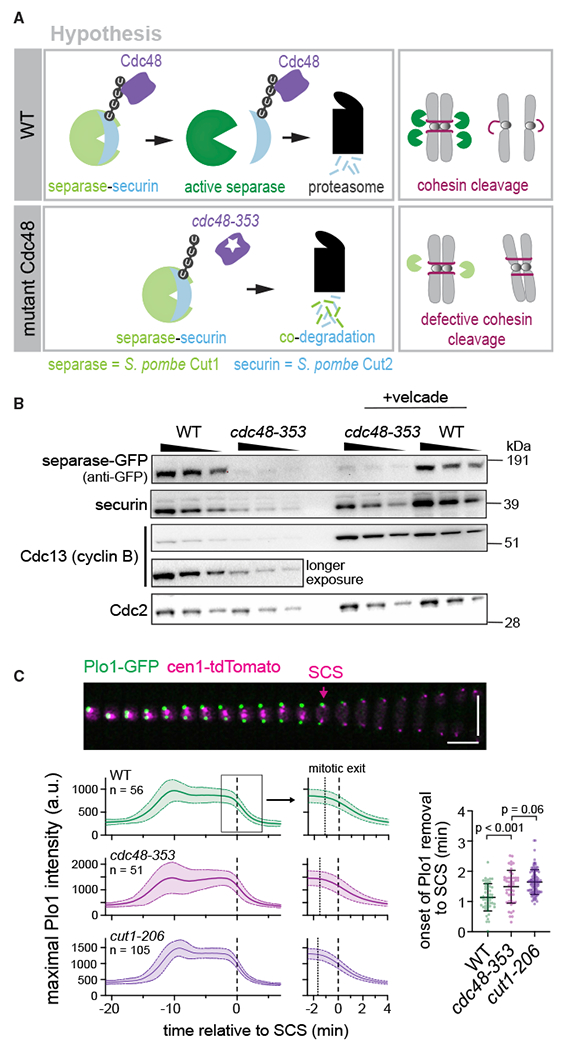
(A) The current hypothesis suggests failure of Cdc48 segregase activity in the cdc48-353 mutant, leading to co-degradation of separase and securin during mitosis.
(B) Immunoblot of extracts from asynchronously growing (30°C, minimal medium) wild-type (WT) or cdc48-353 mutant cells with and without 45-min treatment with the proteasome inhibitor Velcade. Wedges indicate a 1:1 dilution series for semi-quantitative comparison. Cdc2 (CDK1) serves as loading control.
(C) Top: representative kymograph of a WT cell during mitosis (30°C, minimal medium). Plo1-GFP localization to spindle pole bodies serves as marker for mitotic entry and exit; the centromere of chromosome 1 is labeled with tdTomato to visualize sister chromatid separation (SCS). Vertical scale bar: 10 μm; horizontal scale bar: 2 min. Bottom left: maximum Plo1 intensity in cells undergoing mitosis (central line = mean, area = standard deviation, n = number of cells). The maximum signal increases as Plo1 binds to spindle pole bodies (mitotic entry), decreases slightly when spindle pole bodies split (spindle formation), and decreases to baseline at the end of mitosis when CDK1 activity declines (mitotic exit). Curves are aligned to SCS. Panels on the right are zoomed in to mitotic exit. Dotted lines indicate the mean time of onset of Plo1 removal, as indicated in the scatterplot on the right. Bottom right: time from the onset of Plo1 removal from spindle pole bodies to SCS. Black bars: mean and standard deviation. Unpaired t tests.
See also Figure S1.
Here, we have tested—and falsify—this hypothesis. Using a combination of live-cell imaging, genetics, and biochemistry in fission yeast (S. pombe), we demonstrate that separase is not specifically degraded during mitosis in the cdc48-353 mutant. Moreover, we generally do not find evidence of enhanced separase degradation. Instead, we argue that the cdc48-353 mutant uncovers that separase has specific requirements for its translation, which is an aspect of separase regulation that has been little studied.
RESULTS
Securin mutants that lack the ability to bind separase are not toxic when overexpressed in the S. pombe cdc48-353 mutant
The levels of separase and of the key APC/C substrates securin and Cdc13 (S. pombe mitotic cyclin B) are reduced in the cdc48-353 mutant (Figures 1B and S1) (Ikai and Yanagida, 2006). As we will discuss in more detail below, treatment with the proteasome inhibitor Velcade (bortezomib) did not restore separase levels in the cdc48-353 mutant, although it increased securin and Cdc13 levels (Figure 1B). Consistent with the low levels of separase, we found that sister chromatid separation was delayed in cdc48-353 mutant cells relative to the decline in CDK1 activity at mitotic exit (Figure 1C). This strongly resembles the defects seen in a separase mutant (cut1-206) and thus suggests that the mitotic phenotype of the cdc48-353 mutant is caused by reduced separase activity.
Consistent with low separase activity in cdc48-353 mutant cells, overexpression of the separase inhibitor securin becomes lethal (Ikai and Yanagida, 2006; Yuasa et al., 2004). We found this to be the case even if securin levels were elevated to only about eight times the wild-type level (Kamenz et al., 2015) (Figure 2A). To better understand this toxicity, we screened for securin mutants whose overexpression is tolerated in cdc48-353 mutant cells (Figure 2B). Of 10 such mutants that we sequenced, six were truncations of securin that terminated before the separase-binding motif (SBM) (Boland et al., 2017; Lin et al., 2016; Luo and Tong, 2017; Nagao and Yanagida, 2006). The other four mutants had missense or frameshift mutations clustered around the SBM. The SBM of securin is known to bind into the separase catalytic site (Boland et al., 2017; Lin et al., 2016; Luo and Tong, 2017; Nagao and Yanagida, 2006). Overexpression of the previously characterized securin-AIA mutant (DIE in the SBM replaced with AIA; Nagao et al., 2004) or of a mutant that lacks the SBM and all sequences downstream (securin-N121), was also tolerated in the cdc48-353 mutant background. In contrast, the expression of wild-type securin from the same promoter was lethal (Figure 2C). Hence the SBM is crucial for the toxic effect. The levels of all overexpressed securin versions (both mutant and wild type) were drastically lowered in the cdc48-353 mutant background, suggesting that the mutants remain susceptible to the effects of the cdc48-353 mutant (Figure 2D).
Figure 2. Overexpression of securin mutants compromised for interaction with separase is tolerated in the cdc48-353 mutant.
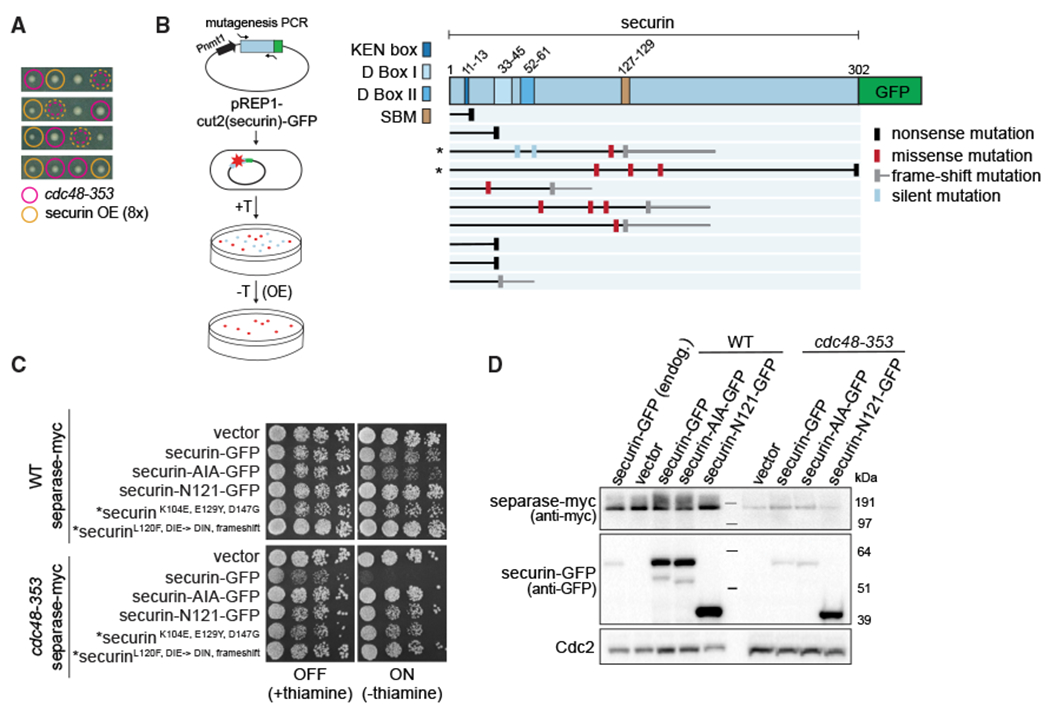
(A) Tetrads from a cross between cdc48-353 mutant and a strain overexpressing (OE) securin to about eight times the WT level (rich medium). Colored circles: colony genotype; dashed colored circles: lethality of presumed double mutants.
(B) Left: schematic of securin mutagenesis screen to identify mutations that are non-toxic when overexpressed in cdc48-353 cells. Securin expression from the plasmid is suppressed in the presence of thiamine (+T) and induced in its absence (−T). Right: positions and type of mutation identified in the non-toxic mutants. The length of the protein is indicated with a black or gray horizontal line. Mutants examined in (C) are marked by an asterisk.
(C) Growth assay of cells inducibly expressing the indicated securin mutants from the nmt81 promoter in the WT or cdc48-353 mutant background. Growth at 30°C on minimal medium in the presence or absence of thiamine. The latter induces expression. The strains marked with asterisks express mutants retrieved from the screen.
(D) Immunoblot of extracts from WT and cdc48-353 mutant strains overexpressing the indicated variants of securin or an empty vector, 15 h after induction. A strain expressing securin-GFP from the endogenous locus is included as reference in the first lane. Cdc2 serves as loading control.
SBM, separase-binding motif (also known as separase-interaction segment [SIS]).
The securin mutants that lacked toxicity when overexpressed presumably neither bind nor inhibit separase efficiently (Nagao et al., 2004; Nagao and Yanagida, 2006). Hence these mutants may lack toxicity because they are unable to mediate separase co-degradation with securin as a result of their failure to bind separase, or they may lack toxicity because they do not inhibit the small, remaining amount of separase in the cdc48-353 mutant. When we compared short-term overexpression of the toxic wild-type securin and the non-toxic securin-AIA in cdc48-353 mutant cells, we did not observe any difference in the amount of separase (Figure 2D). Wild-type securin and securin-AIA should have been separase binding and non-binding, respectively, and the result therefore indicates that overexpression of wild-type securin (able to bind separase) does not further enhance separase degradation. This casts doubt on whether separase indeed undergoes co-degradation with securin in cdc48-353 mutant cells.
Separase levels do not drop during mitosis in the cdc48-353 mutant
The proposed model suggests that Cdc48 extracts securin from separase and aids the proteasome-mediated mitotic degradation of securin (Figure 1A). When this fails in the cdc48-353 mutant, there may be two consequences: (1) securin degradation may become less efficient because Cdc48-mediated pre-processing fails, and (2) separase may be co-degraded with securin. To directly test this, we imaged cells expressing securin-GFP or separase-GFP as they underwent mitosis. The level of securin-GFP in the cdc48-353 mutant was lower than in the wild-type background (Figures 3A and S1), consistent with the finding for endogenous securin by immunoblot (Figure 1B). However, the cellular degradation kinetics of securin-GFP were indistinguishable in cdc48+ and cdc48-353 mutant cells after normalizing forthe reduced level (Figure 3A), suggesting that securin degradation was unaffected in the cdc48-353 mutant. Similar results were obtained for Cdc13-GFP (Figure S1). Hence securin and Cdc13 are still efficiently targeted for proteasomal degradation in the cdc48-353 mutant.
Figure 3. Separase levels do not drop during mitosis in the cdc48-353 mutant.
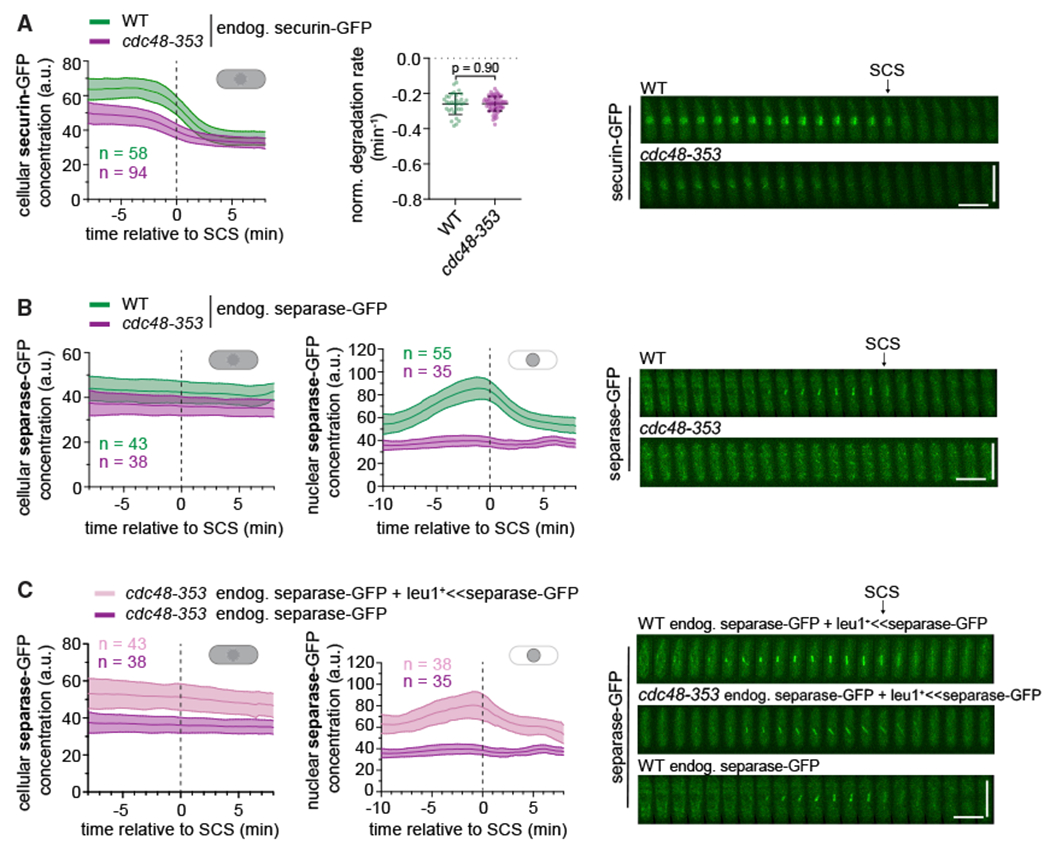
(A) Left: cellular concentrations of securin-GFP during mitosis (central line = mean, area = standard deviation, n = number of cells). Data are aligned to the time of SCS. Middle: normalized degradation rate of cellular securin during mitosis. Black bars: mean and standard deviation. Unpaired t test. Right: Representative kymographs of securin-GFP as cells undergo mitosis. SCS is indicated by an arrow. Horizontal scale bar: 2 min; vertical scale bar: 10 μm.
(B and C) Cellular (left) and nuclear (middle) concentrations of separase-GFP during mitosis. Right: representative kymographs of separase-GFP as cells progress through mitosis. In (C), leu1+ <<separase-GFP indicates that a second copy of separase-GFP is expressed from the exogenous leu1+ locus. The data for the strain with only endogenous separase-GFP are the same as in (B).
See also Figures S1–S3.
We next checked whether a decline in separase levels occurs during mitosis. Similar to securin-GFP, both the cellular and nuclear levels of separase-GFP were low in cdc48-353 mutant cells (Figures 3B and S1). However, we did not observe a drop in separase-GFP levels during mitosis, as would be expected if separase was co-degraded with securin (Figure 3B). Given the already low levels, a further drop may be difficult to detect. We therefore expressed a second copy of separase-GFP under its endogenous promoter from another genetic locus (leu1). A second copy of separase was sufficient to rescue growth of the cdc48-353 mutant cells, but only if it was catalytically active (Figure S2A). The cellular and nuclear separase levels now were indeed increased, but we were still unable to observe any drop during mitosis (Figures 3C and S2B).
To separately assess the effect of cdc48-353 on the two copies of separase, we used two different tags: GFP at the endogenous and myc at the exogenous locus. Separase-myc expressed from the exogenous locus remained susceptible to the cdc48-353 mutant because its levels were also lowered (Figure S2C). The presence of this extra copy did not change the level of separase-GFP expressed from the endogenous locus (Figures S2C and S2D). Hence the mechanism that lowers separase levels in the cdc48-353 mutant acts equally on separase expressed from the endogenous and the exogenous locus. Securin levels were moderately higher in cdc48-353 mutant cells with two copies of separase compared with one copy (Figure S2D), consistent with separase being able to stabilize securin (Kumada et al., 1998).
To further test the original model of securin and separase codegradation, we anchored Cdc48 away from the nucleus (Figure S2E) (Ding et al., 2014; Haruki et al., 2008). Despite successfully reducing the amount of Cdc48 in the nucleus, we did not observe an obvious decline of separase levels during mitosis (Figure S2F).
Taken together, the unaltered securin degradation kinetics and stable separase levels throughout mitosis in the cdc48-353 mutant suggest that the reduction in separase levels is not caused by mitotic co-degradation with securin. Supporting this notion, we did not find a convincing physical association between Cdc48 and separase in asynchronous cultures, or between securin and Cdc48 in cells undergoing mitosis (Figure S3).
Cdc48-353 is a dominant-negative gain-of-function allele
The cdc48-353 allele causes a single amino acid change (G338D) in the D1 domain of Cdc48 within a loop (loop 2) that faces the central pore (Ikai and Yanagida, 2006; Marinova et al., 2015). In the D2 domain, pore-facing loops contain aromatic residues that are used to translocate substrates (Twomey et al., 2019). Pore-facing D1 loops of archaeal Cdc48 homologs retain aromatic residues, but in eukaryotic Cdc48 they have been replaced by non-aromatic residues (Esaki et al., 2017; Twomey et al., 2019). In budding yeast (S. cerevisiae), re-inserting an aromatic residue into the pore-facing loop 1 of D1 (e.g., M288Y) led to lethality (Esaki et al., 2017). Overexpression of the S. cerevisiae Cdc48-M288Y mutant in the wild-type background was lethal as well, and this lethality was rescued by deletion of some, but not all, Cdc48 cofactors, by truncation of the Cdc48 C-terminal tail, or by impairing the ATPase activity of the D1 domain (Esaki et al., 2017). Interestingly, the S. pombe cdc48-353 mutant has also been shown to be rescued by deletion of the cofactor Ufd2, by a specific mutation in the cofactor Ufd1, by mutations in the Cdc48 C-terminal tail, or by mutations expected to lower the D1 ATPase activity (Marinova et al., 2015; xu et al., 2018). This suggested that the mechanism behind the toxicity of S. pombe Cdc48-G338D and S. cerevisiae Cdc48-M288Y may be similar.
It was previously reported that S. pombe strains overexpressing Cdc48-G338D retained viability (Ikai and Yanagida, 2006). In our hands, though, nmt1 promoter-driven overexpression of Cdc48-G338D, but not wild-type Cdc48, strongly reduced separase and securin levels, caused abnormal cell division, and was lethal (Figures 4 and S4). These phenotypes were rescued to a large extent by introducing an additional E325A mutation in the Walker B motif of the D1 domain (Figures 4 and S4), which is expected to lower ATPase activity (Bodnar and Rapoport, 2017). Because C-terminal tagging of Cdc48-G338D ameliorates the mutant phenotype (Marinova et al., 2015), these constructs were N-terminally tagged. The difference to previous results, where no lethality was observed (Ikai and Yanagida, 2006), may be because of different tagging of Cdc48 or different levels of overexpression. Based on our results, we conclude that Cdc48-G338D behaves as dominant negative.
Figure 4. cdc48-353 is a dominant-negative gain-of-function allele.
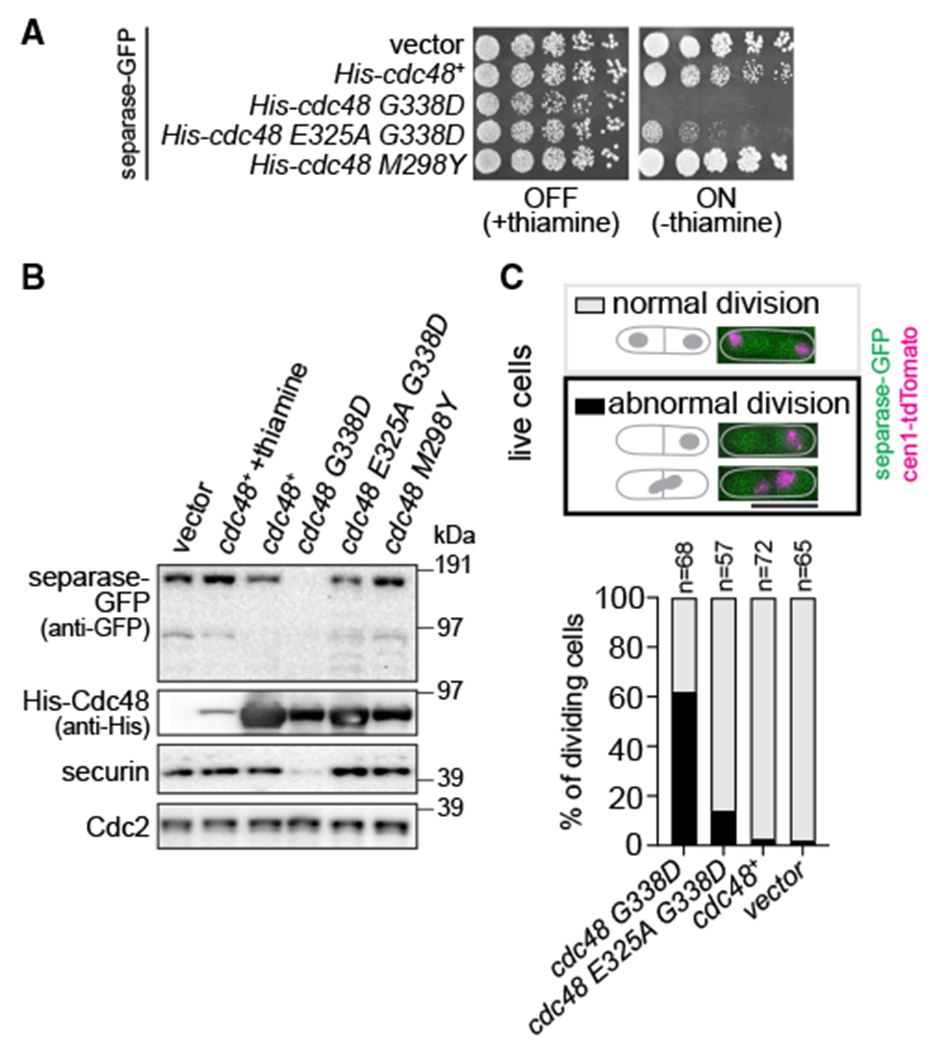
(A) Growth of WT strains transformed with vectors for conditional overexpression of cdc48+, cdc48 mutants, or the empty vector. Growth on minimal medium at 30°C in the presence or absence of thiamine; the latter induces expression.
(B) Immunoblot of extracts from the same strains as in (A). Exogenous Cdc48 probed with anti-His antibody. Growth in thiamine-depleted conditions for 15 h, except for lane 2, for which cells were grown in the presence of thiamine. Cdc2 serves as loading control.
(C) Quantification of cellular phenotypes when overexpressing the indicated Cdc48 protein. Abnormal division includes cells with “cut” phenotype, unequal nuclear division, or chromosome bridges. Scale bar: 10 μm.
See also Figure S4.
We also generated the mutant analogous to S. cerevisiae Cdc48-M288Y, S. pombe M298Y. However, overexpression of this mutant neither lowered separase levels nor was lethal (Figures 4A and 4B). Furthermore, some cofactor deletions (shp1/ubx3Δ or ubx4Δ) that rescued the lethality of Cdc48-M288Y overexpression in S. cerevisiae did not rescue S. pombe cdc48-353 mutant growth (Figure S5A). Altogether, this indicates that G338D and M298Y do not cause the same cellular phenotype, although it remains possible that they mechanistically cause similar defects in Cdc48 function. For example, both may cause misprocessing of Cdc48 substrates, but possibly of different substrates.
Because the cdc48-353 phenotype is rescued by impairing the D1 ATPase activity (Figure 4) or by mutating or deleting cofactors that cooperate with Cdc48 (Marinova et al., 2015; Xu et al., 2018), we conclude that cdc48-353 is a gain-of-function allele whose toxic effects are mitigated by making Cdc48 less active. These findings further conflict with the proposed model, which assumes cdc48-353 to be a loss-of-function allele (Figure 1A).
The rescue of the cdc48-353 mutant by deletion of the Ufd2 cofactor is independent of securin degradation during mitosis
Deletion of the Cdc48 cofactor ufd2+ strongly suppresses the cdc48-353 mutant growth phenotype (Figures 5A and 6A) (Xu et al., 2018). Note that tagging of separase alone impairs cdc48-353 growth (Figures 6A and S5B). So, all direct comparisons use the same version of separase. In accordance with the rescue of cdc48-353 growth by ufd2Δ, we found that separase, securin, and Cdc13 levels were restored in the double mutant (Figures 5A–5C and S5C), and the mitotic phenotype was rescued (Figure 7D). Ufd2 is an E4 ubiquitin ligase that in S. cerevisiae has been shown to add ubiquitin moieties to preformed ubiquitin conjugates to aid proteasomal degradation (Richly et al., 2005; Rumpf and Jentsch, 2006). The suppression observed in the cdc48-353 ufd2Δ double mutant has been proposed to arise from inefficient securin degradation rescuing separase from undergoing co-degradation (Xu et al., 2018). However, despite slightly higher securin and separase levels in the ufd2Δ mutant, we found no obvious change in securin degradation kinetics during mitosis (Figures 5D, S5C, and S5D). These results indicate that the rescue of separase levels seen in the cdc48-353 ufd2Δ double mutant is unlikely to be a consequence of impaired securin degradation during mitosis.
Figure 5. Deletion of ufd2 rescues separase levels in the cdc48-353 mutant.
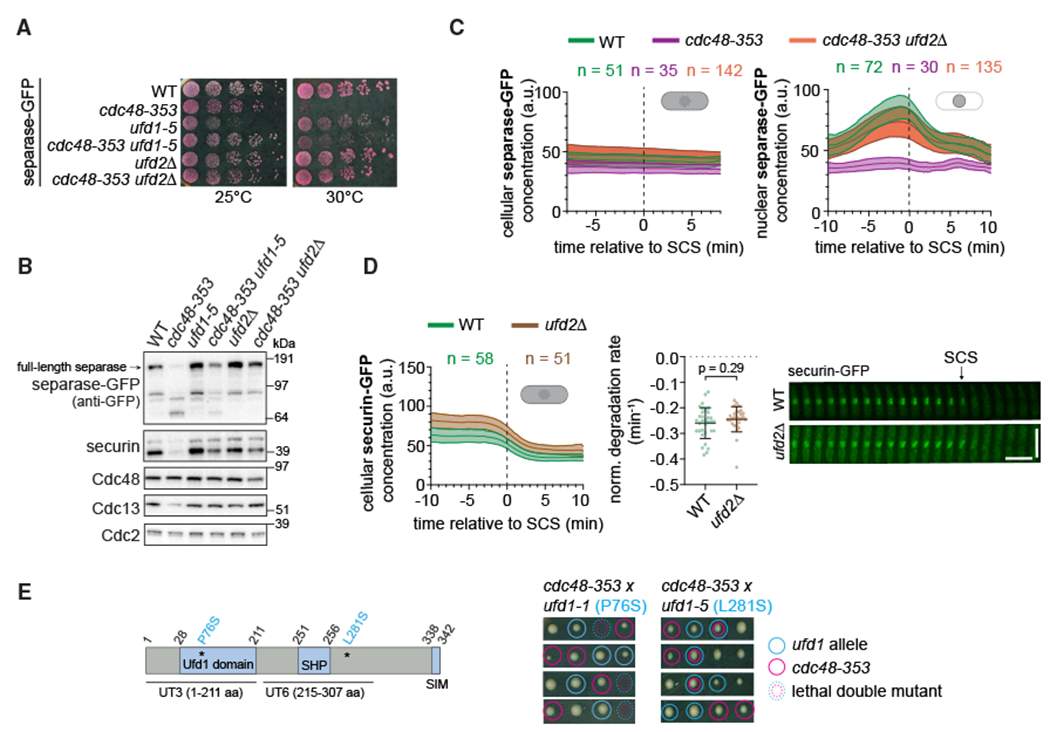
(A) Growth assay on rich medium containing Phloxine B, which stains dead cells.
(B) Immunoblot of extracts from asynchronously growing WT or mutant strains.
(C) Cellular (left) and nuclear (right) concentrations of separase-GFP during mitosis (central line = mean, area = standard deviation, n = number of cells). Data are aligned to the time of SCS.
(D) Left: cellular concentration of securin-GFP in the WT and ufd2Δ mutant during mitosis. Middle: normalized degradation rate of cellular securin during mitosis. Black bars: mean and standard deviation. Unpaired t test. The WT data are the same as in Figure 3. Right: kymographs of securin-GFP as cells progress through mitosis. Horizontal scale bar: 2 min; vertical scale bar: 10 μm.
(E) Left: domain organization of the Ufd1 protein. Asterisks indicate the mutations present in ufd1-1 and ufd1-5, respectively. Right: tetrads from crosses between cdc48-353 and ufd1 mutants (rich medium). Colored circles: colony genotype; dashed colored circles: lethality of presumed double mutants.
See also Figure S5.
Figure 6. Mutation of tor2 rescues both growth and mitotic defects in the cdc48-353 mutant without rescuing separase levels.
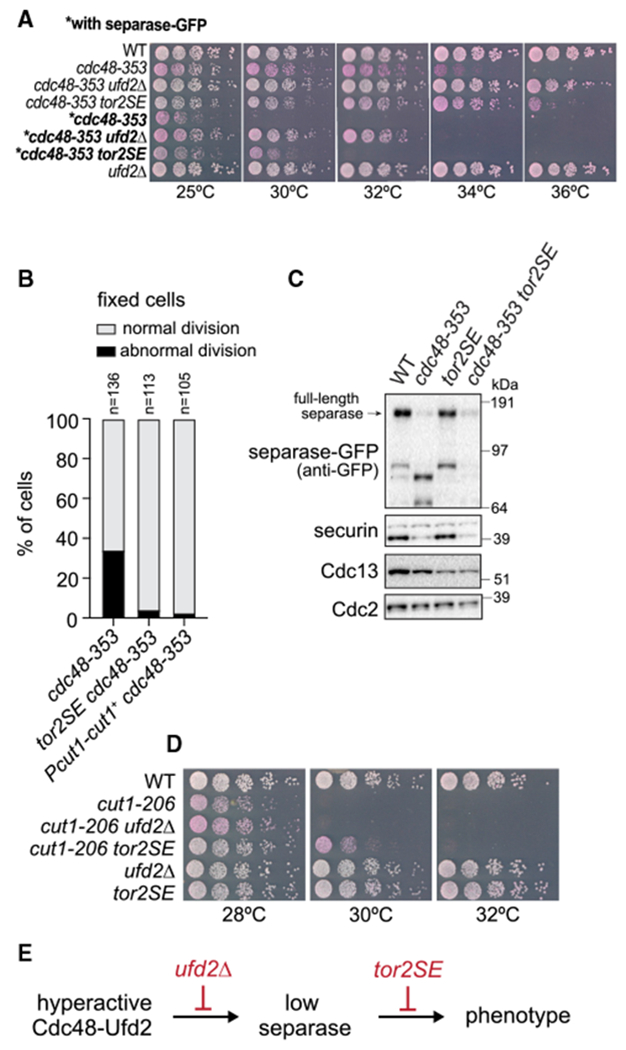
(A) Growth assay on rich medium containing Phloxine B, which stains dead cells.
(B) Phenotypic analysis by DNA staining of fixed cells. Abnormal division includes “cut” phenotype, unequally divided nucleus, or chromosome bridges. The cdc48-353 data are the same as in Figure 7D.
(C) Immunoblot of extracts from asynchronously growing WT and mutant strains.
(D) Growth assay on rich medium containing Phloxine B.
(E) Proposed point of action of the suppressor mutants.
See also Figure S6.
Figure 7. Inhibition of major degradation pathways fails to rescue separase levels in the cdc48-353 mutant.
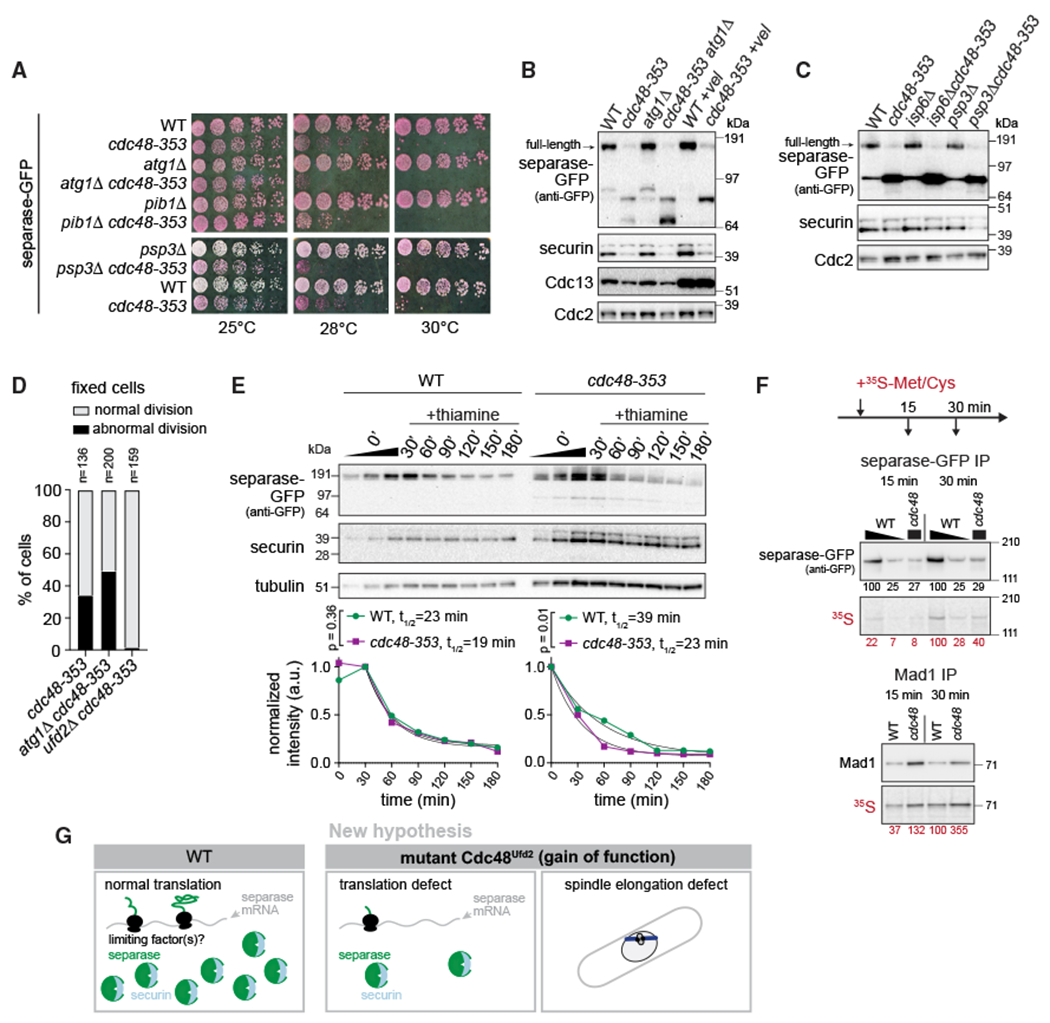
(A) Growth assay on rich medium containing Phloxine B, which stains dead cells.
(B) Immunoblot of extracts from asynchronously growing WT and cdc48-353 strains with or without inhibition of autophagy by atg1+ deletion or inhibition of the proteasome by Velcade (+vel) treatment. Higher levels of Cdc13 (cyclin B) upon Velcade treatment suggest efficacy of proteasome inhibition.
(C) Immunoblot of extracts from WT and cdc48-353 strains with or without deletion of the vacuolar protease genes isp6+ and psp3+.
(D) Phenotypic analysis by DNA staining of fixed cells. Abnormal division includes “cut” phenotype, unequally divided nucleus, or chromosome bridges. The cdc48-353 data are the same as in Figure 6B.
(E) Nmt81 promoter-driven expression of separase-GFP at an exogenous locus in WT and cdc48-353 mutant cells. Extracts were prepared after thiamine addition to shut off transcription. Approximately four times more cdc48-353 than WT extract was loaded for better comparability. Top: immunoblot with 1:1 dilution series for the 0-min time point. Bottom: quantification of two independent experiments. Fit of one phase decay and comparison of degradation rates by extra-sum-of-squares F test.
(F) Pulse labeling with radioactive 35S-methionine/cysteine added to culture medium for 15 and 30 min. Immunoblot and radiograph of separase-GFP and Mad1 immunoprecipitation (IP). Wedge indicates a 1:4 dilution of the separase IP from WT cells to match the protein level in the IP from cdc48-353 cells. Radioactivity was quantified relative to the band from WT at 30 min. Shown is one out of two experiments yielding the same result.
(G) New model proposing that low separase levels in the cdc48-353 mutant are a result of poor separase translation and that the cdc48-353 mutation impairs mitosis in additional ways.
See also Figure S7.
A cdc48-353 suppressor screen identified a frameshift mutation in ufd1+ (Marinova et al., 2015), another Cdc48 cofactor. Ufd1 in complex with Npl4 binds ubiquitinated substrates and helps Cdc48-mediated unfolding and processing (Ji et al., 2021; Meyer et al., 2002; Rape et al., 2001; Rumpf and Jentsch, 2006; Twomey et al., 2019). We tested the genetic interaction of two ufd1 mutants (ufd1-1 and ufd1-5) with cdc48-353. The first of these (ufd1-1, P76S) is expected to impair ubiquitin binding, the second (ufd1-5, L281S) is expected to impair Cdc48 binding (Burr et al., 2017; Hänzelmann and Schindelin, 2016; Nie et al., 2012; Twomey et al., 2019). The ufd1-1 mutant slightly raised separase-GFP levels (Figures S5E and S5F) but was synthetic lethal with cdc48-353, whereas the ufd1-5 mutant was not (Figure 5E) and rescued cdc48-353 growth in some conditions (Figures 5A and S5B). The rescue suggests that interaction of Ufd1 with Cdc48 is important for the toxic effect of cdc48-353. If securin degradation was indeed facilitated by Ufd1/Npl4-Cdc48, we would expect securin overexpression to be toxic in ufd1 mutants. However, securin overexpression was tolerated by all ufd1 mutants we tested, ufd1-1, ufd1-5, and ufd1-3 (N175S Y179N; Nie et al., 2012) (Figure S5G).
In summary, these results show that the detrimental effects of cdc48-353 can be alleviated by removing Cdc48 cofactors or impairing their interaction with Cdc48. This supports the idea that cdc48-353 is a gain-of-function allele. The fact that deletion of ufd2+ strongly rescues the cdc48-353 mutant phenotype, but does not impair securin degradation, further supports our conclusion that low separase levels are independent of mitotic securin degradation and not a consequence of co-degradation of separase with securin in the cdc48-353 mutant.
TORC1 mutation rescues cdc48-353 growth and mitotic defects without restoring separase levels
In addition to cofactor deletions, growth on minimal medium suppresses the temperature sensitivity of the cdc48-353 mutant, suggesting a link to nutrient availability (Ikai and Yanagida, 2006). The conserved TOR (target of rapamycin) pathway controls cell growth in response to nutrients (González and Hall, 2017). In S. pombe, Tor2 is the kinase subunit of TOR complex 1 (TORC1), and Fkh1 (Fkbp12 ortholog) is the direct target of rapamycin (Hayashi et al., 2007; Weisman et al., 2001). Interestingly, mutations in Tor2 and deletion of fkh1 have been shown to rescue the phenotype of separase mutants (Ikai et al., 2011). We therefore asked whether such mutants would also rescue the effects caused by cdc48-353. Both fkh1Δ and the Tor2-S1837E mutation (tor2SE; Laor et al., 2014; Nakashima et al., 2010) rescued growth of cdc48-353 mutant cells (Figures 6A and S6A). Rescue by tor2SE was stronger than by fkh1Δ. Furthermore, tor2SE rescued mitotic phenotypes of cdc48-353 cells, such as chromosome bridges and the “cut” phenotype (Figure 6B). Strikingly, though, separase and securin levels remained low (Figures 6C and S6B). Although surprising, this fits with the prior observation that growth of the separase mutant cut1-206 is rescued by the TOR inhibitor rapamycin, although separase levels are not restored (Ikai et al., 2011). The rescuing effect of tor2SE on cut1-206 and cdc48-353 was specific, because tor2SE did not rescue most other temperature-sensitive mutants that we tested, including other “cut” mutants, such as the condensin cut3-477 mutant (Figures S6C and S6D).
Because tor2SE suppresses the “cut” phenotype (Figure 6B), we considered whether the tor2SE mutation may prolong mitosis, so that cells with reduced separase activity have more time to cleave cohesin and separate chromosomes. However, by using Plo1 kinase localization to spindle pole bodies as a marker for mitosis (Figure S6E; see Figure 1B for an example image), we did not observe an obvious mitotic delay in the tor2SE mutant. Furthermore, the delay in sister chromatid separation relative to the removal of Plo1 from spindle pole bodies seen in cut1-206 and cdc48-353 mutant cells (Figure 1B) persisted in cdc48-353 tor2SE double mutants (Figure S6F). These results suggest that mitotic timing was not altered by tor2SE mutation.
Interestingly, ufd2Δ and tor2SE differ in their influence on cdc48-353 and the separase cut1-206 mutant. Whereas tor2SE rescues growth of both cdc48-353 and cut1-206 mutants, deletion of ufd2 rescues growth of the cdc48-353 mutant more efficiently than tor2SE does but does not rescue growth of the cut1-206 mutant (Figures 6A and 6D). This suggests that ufd2Δ rescues at the level of Cdc48 (consistent with restoring separase levels), whereas tor2SE rescues the phenotypic effect of low separase concentrations, without correcting the underlying problem of the Cdc48 malfunction (Figure 6E). This is corroborated by tor2SE not rescuing a short spindle phenotype that is seen in cdc48-353 mutant cells, but not in cut1-206 mutant cells (Figure S6G).
Low separase levels in the cdc48-353 mutant do not depend on any major degradation pathway
If low separase levels in the cdc48-353 mutant were a consequence of mitotic co-degradation with securin (Figure 1A), impairing proteasomal degradation would be expected to rescue separase levels. However, combining cdc48-353 with a proteasome mutant (mts3-1) (Ikai and Yanagida, 2006) or chemical inhibition of proteasome activity by Velcade (bortezomib) failed to rescue separase levels in the cdc48-353 mutant (Figures 1B and 7B). In contrast, the levels of securin and Cdc13 increased, indicating that different mechanisms are behind the low levels of these three proteins in the cdc48-353 mutant. The results suggest that the low separase levels are not a direct consequence of proteasomal degradation.
This prompted us to test alternative degradation pathways. Autophagy was a particularly promising candidate because Cdc48 can promote autophagy (Dargemont and Ossareh-Nazari, 2012; Franz et al., 2014). However, deletion of the core autophagy kinase atg1+ did not rescue growth of the cdc48-353 mutant or separase levels or abnormal mitosis (Figures 7A, 7B, and 7D). Similarly, deletion of the vacuolar ubiquitin ligase pib1+ or the vacuolar serine proteases isp6+ and psp3+ did not rescue growth or separase levels (Figures 7A and 7C).
We did observe two faster migrating bands in the GFP immunoblot of cdc48-353 cells expressing separase-GFP (Figures 5, 6, and 7). Unlike full-length separase-GFP, these bands were affected by interfering with the different degradation pathways (Figures 7B and 7C), and they disappeared on combination of cdc48-353 with the tor2SE mutant (Figure 6C). However, these bands seemed absent in cells overexpressing the mutant Cdc48-G338D (Figure 4B) and did not appear at corresponding sizes in cdc48-353 mutant cells expressing myc-tagged separase (Figure S2C). Hence their presence or absence does not seem to correlate with functional outcomes, and we did not investigate their origin further.
To more generally address whether separase is becoming destabilized in the cdc48-353 mutant, we assessed separase levels after promoter shut-off, using a second copy of separase integrated at the leu1 locus. Separase mRNA has a short half-life (3.6 min; Eser et al., 2016), so a transcriptional shut-off is expected to rapidly stop protein production. Although we observed a slightly shorter protein half-life of separase in cdc48-353 compared with wild-type cells (Figures 7E and S7A), the difference is insufficient to explain the large difference in separase levels (at least 4-fold; e.g., Figures 1B and 7F). This provides further evidence that the low separase level in the cdc48-353 mutant cannot be purely explained by degradation and instead suggests that separase is less efficiently produced in the cdc48-353 mutant.
Low separase levels may result from poor separase translation in the cdc48-353 mutant
Because we could not positively confirm any of the major cellular degradation pathways to be involved in lowering full-length separase levels in the cdc48-353 mutant, we asked whether synthesis is inefficient. Separase mRNA levels have been shown to be unaffected in the cdc48-353 mutant (Ikai and Yanagida, 2006), and we therefore hypothesized that translation is impaired. Based on recent genome-wide ribosome profiling in S. pombe (Rubio et al., 2021), separase has a very low translation efficiency (Figure S7B), and this may be exacerbated by the cdc48-353 mutation.
To assess separase synthesis, we pulse-labeled newly synthesized protein with radioactive methionine and cysteine and immunopurified separase-GFP, as well as a control protein, the stably expressed spindle assembly checkpoint protein Mad1 (Esposito et al., 2021). Much less radiolabeled separase was obtained from a cdc48-353 extract than from the same volume of wild-type extract (Figure 7F, lane 1 versus 3 and lane 4 versus 6), and this was not the case for Mad1. When comparing approximately the same amount of immunopurified separase protein (labeled and unlabeled) from wild-type or cdc48-353 extracts (Figure 7F, lane 2 versus 3 and lane 5 versus 6), roughly the same proportion was radiolabeled. Along with the similar degradation rates (Figure 7E), this suggests that separase synthesis is less efficient in the cdc48-353 mutant, which may explain the lower separase level. This does not necessarily mean that translation initiation is impaired. It is possible that some problem during translation leads to co-translational degradation, which would not be detected in our assessment of degradation rates (Figure 7E) and would also lead to less nascent protein.
Of interest with respect to the hypothesis that translation may be impaired, the overexpression of components of the translation initiation complex eIF3 (Int6, Moe1) has been reported to rescue cdc48-353 growth and, vice versa, cdc48-353 defects are aggravated by int6+ or moe1+ deletion (Otero et al., 2010). We therefore overexpressed Int6 in wild-type and cdc48-353 mutant strains. However, separase levels remained unchanged upon Int6 overexpression in the cdc48-353 mutant, and we could not confirm the rescue of growth defects upon Int6 overexpression from its endogenous promoter or from a strong nmt1 promoter (Figure S7C–S7E).
Hence the cause for inefficient separase synthesis remains uncertain, but our results argue that separase has specific requirements for its translation that are impaired in the cdc48-353 mutant (Figure 7G).
DISCUSSION
In contrast to the long-standing hypothesis that separase undergoes mitotic co-degradation with securin in the S. pombe cdc48-353 mutant, which would have implicated Cdc48 (VCP/p97) in the regulation of anaphase (Figure 1A), we show here that low separase levels in this cdc48 mutant occur independent of securin degradation in anaphase, and our data suggest that Cdc48 does not play a major role in securin degradation and separase activation during mitosis. Experiments in Xenopus laevis extracts and more recently in human cells reached similar conclusions (Heubes, 2007; Wang et al., 2021). The unfoldase activity of Cdc48 is thought to be critical for proteasomal degradation when substrates are well folded and lack a flexible region (Beskow et al., 2009; Olszewski et al., 2019). Securin, in contrast, is a largely unstructured protein whose ubiquitinated N terminus is natively unfolded (Cox et al., 2002; Csizmok et al., 2008; Sánchez-Puig et al., 2005). It is therefore plausible that securin is efficiently degraded without the help of Cdc48. Separase becomes activated not only during mitosis, but also locally in interphase upon DNA damage (Hellmuth et al., 2018; McAleenan et al., 2013; Nagao et al., 2004). Cdc48 is known to be involved in DNA repair (Franz et al., 2016; Torrecilla et al., 2017). Whether Cdc48 plays a role in temporarily and locally removing securin from separase in this situation remains an open question.
We show that the previously known rescue of separase mutants by tor2 (TORC1) mutations (Ikai et al., 2011) extends to a rescue of cdc48-353 (Figure 6). However, separase levels are not restored (Figure 6C), and a short spindle phenotype observed in the cdc48-353, but not in the separase cut1-206, mutant is not rescued either (Figure S6G). Hence the tor2SE mutation specifically alleviates the effects of low separase activity. So far, we have been unable to pinpoint the mechanism of action. We were not able to entirely delete separase in the cdc48-353 tor2SE double mutant (data not shown), which suggests that some separase activity remains necessary for successful mitosis in tor2SE cells. We did not find evidence that tor2SE prolongs mitosis, which could have given cells more time for chromosome separation (Figures S6E and S6F).
Instead, it is possible that rescue by tor2SE is related to lipid metabolism. Nuclear division in S. pombe cells requires nuclear membrane expansion, and some mutations or drugs that impair lipid synthesis also cause “cut” phenotypes, similar to separase mutants (Saitoh et al., 1996; Takemoto et al., 2016; Yam et al., 2011; Zach and Prevorovsky, 2018). Such mutants are rescued by minimal medium, more specifically by the ammonium present in standard minimal medium (Zach et al., 2018). A similar rescue by standard minimal medium is seen for cdc48-353 (Ikai and Yanagida, 2006). Growth of rapamycin-treated cells, in contrast, is worsened by ammonium, which in these cells limits amino acid uptake, including that of leucine (Weisman et al., 2005). We have also seen that leucine auxotrophy affects growth of cdc48-353 (Figure S5B). Amino acids are an important regulator of TOR signaling, and leucine and TOR signaling can influence lipid metabolism (González and Hall, 2017; Lamming and Sabatini, 2013; Madeira et al., 2015; Neinast et al., 2019). Altogether, this suggests that separase mutants may not only cause the “cut” phenotype because of deficient cohesin cleavage, but possibly also because of effects on lipid metabolism, and the latter may be rescued by ammonium or reduced TORC1 signaling.
With respect to the influence of cdc48-353 on cellular physiology, our data suggest that cdc48-353 is a gain-of-function mutation (Figures 4 and 5), and we propose that the mutant Cdc48-G338D, along with the cofactors Ufd1 and Ufd2, over-processes one or more natural substrates, thereby impairing their function. Because not all aspects of the cdc48-353 mitotic phenotype can be attributed to the reduction in separase activity (Figure S6G), Cdc48-G338D may misprocess multiple substrates or a single substrate with multiple downstream effects. The involvement of the Ufd1 and Ufd2 cofactors suggests that a relevant substrate is ubiquitinated. A comparative proteome analysis or comparative Cdc48 immunoprecipitations between cdc48-353 and wild-type cells may identify the relevant substrates in the future. As of now, there is no evidence that Cdc48 acts directly on securin and separase (Figure S3), and cdc48-353 may therefore lower separase levels indirectly. In contrast with the previously prevailing hypothesis that separase has become unstable in the cdc48-353 mutant, our data suggest that separase is inefficiently produced.
Separase requires securin for its stability and full activity (Hornig et al., 2002; Jallepalli et al., 2001; Rosen et al., 2019), and securin starts to associate with separase during separase translation (Hellmuth et al., 2015). Hence a failure to establish this interaction may lead to reduced separase translation. However, we did not find a defect in securin-separase interaction in the cdc48-353 mutant (Figure S3A). It remains possible, though, that co-translational association is inefficient and leads to a defect in separase translation, but that the few successfully formed securin-separase complexes are fully translated and only those are evaluated in our assay. Given the stabilizing effect of securin, we also considered whether the low separase levels could be a consequence of a failure to sustain proper concentrations of securin in the cdc48-353 mutant. However, this seems unlikely given that expression of additional securin is toxic and does not rescue separase levels (Figure 2) (Ikai and Yanagida, 2006; Yuasa et al., 2004). It remains possible that cdc48-353 has an influence on securin, independent of separase, because the expression of securin mutants that should be unable to bind to separase (AIA and N121) is still lowered in the cdc48-353 mutant (Figure 2).
The lower level of separase in the cdc48-353 mutant may also result from other defects in separase translation. In addition to the canonical translation machinery, non-canonical trans-acting proteins and cellular tRNA composition affect the translation of specific subsets of mRNAs (Baltz et al., 2012; Harvey et al., 2018; Rak et al., 2018). The previously established genetic interaction between cdc48 and components of the eIF3 translation initiation complex (Otero et al., 2010) and findings that these eIF3 components promote proper chromosome segregation (Yen and Chang, 2000) were exciting, but we have so far been unable to verify this connection (Figure S7). Cdc48 has also been shown to promote the disassembly and subsequent degradation of defective or stalled RNA polymerase III complexes (Wang et al., 2018). Hence it is possible that cdc48-353 shapes the tRNA pool with consequences on the translation of separase and likely other proteins.
Although it remains unknown how mutant Cdc48 impairs separase, our study suggests that separase production has specific requirements that are impaired by dysfunctional Cdc48, leading to catastrophic cellular outcomes. It will be interesting to address what these requirements are and what purpose they serve in wild-type cells. This may also shed light on the causes of the separase overexpression that is often seen in cancer cells (Meyer et al., 2009).
Limitations of the study
Our study does not unravel how cdc48-353 affects separase levels. Both the relevant Cdc48 substrate(s) and the downstream consequences remain unclear. Overall, the data favor a defect in separase synthesis, but we did And slightly accelerated protein degradation (Figure 7E), and simple pulse labeling (Figure 7F) cannot clearly distinguish between the individual contributions of reduced synthesis and enhanced degradation; an extended and costly kinetic analysis would be required. The lack of mechanistic understanding also makes it hard to gauge whether wild-type Cdc48 influences separase in unperturbed cells. The gain-of-function nature of cdc48-353 may have caused a “freak accident,” where the mutated Cdc48 processesone or more non-physiologic substrates with catastrophic consequences. Regardless of these uncertainties, this and earlier studies suggest that separase has special requirements for its protein homeostasis that are not shared by other proteins.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Silke Hauf (silke@vt.edu).
Materials availability
Yeast strains and vectors generated in this study are available from the lead contact without restriction.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
S. pombe strains
Tagging or deletion of genes at the endogenous locus was performed by PCR-based gene targeting (Bähler et al., 1998). For strains expressing a second copy of separase, a pDUAL vector (Matsuyama et al., 2004) containing Pcut1-cut1+-GFP or Pcut1-cut1+-(myc)13 was linearized with NotI and integrated at the leu1 locus. 905 bp of cut1 region upstream of its start codon were used. For strains expressing cut1+-GFP under the regulatable nmt81 promoter, pDUAL-Pnmt81-cut1+-GFP was linearized with NotI and integrated at the leu1 locus. For plasmids expressing cdc48+ and mutant versions, a (His)6 tag was introduced at the N-terminus of cdc48 by PCR and the entire fragment was introduced into Ndel/BamHI digested pREP1-Pnmt1 vector. To generate the Pint6-int6+ expression construct, full-length int6+ including 1105 bp upstream of the int6+ start codon was inserted into the pREP1 vector replacing the nmt1 promoter. For Pnmt1 driven overexpression of int6+, full-length int6+ was fused to a dual Flag tag at the C-terminus in a pDUAL vector which was linearized with NotI and integrated at the leu1 locus. For the Cdc48 anchor away strains, Cdc48 was C-terminally tagged with either FRB-GFP or only FRB (for strains expressing Cut1-GFP). Ribosomal protein Rpl13 tagged with 2 tandem copies of FKBP12 at the C-terminus was integrated at the leu1 locus and expressed from the Pnmt1 promoter.
Cell culture and growth assays
For all immunoprecipitations, immunoblots and imaging assays, cells were cultured in Edinburgh minimal medium (EMM, MP Biomedicals, 114110022) with supplements for auxotrophic strains as needed (0.2 g/L L-leucine, 0.15 g/L adenine). To suppress expression from nmt1 or nmt81 promoters, 16 μM thiamine was used. For growth assays with or without thiamine, cultures were grown in EMM media containing 16 μM thiamine, followed by thiamine washout before spotting onto EMM plates with or without thiamine. For growth assays on rich media, cells were grown in yeast extract with adenine (YEA: 5% (w/v) yeast extract (IBI Scientific, IB49160), 20 g/L glucose, 0.15 g/L Adenine) and spotted onto YEA plates (5% (w/v) yeast extract, 30 g/L glucose, 0.15 g/L adenine, 2% (w/v) agar) including 2 μg/mL Phloxine B (Sigma, P4030). Growth assays have approx. 4,000 cells in the first spot and a 1:5 dilution series in the following. To inhibit proteasome activity, bortezomib (Velcade, LC Laboratories, B-1408) was added at a final concentration of 1 mM to cultures in exponential growth phase, and cultures were incubated at 30°C for 45 min in the presence of the drug. A small volume of the culture was fixed with methanol to check for proteasomal inhibition by scoring for cells arrested at metaphase. Rapamycin (LC Laboratories, R-5000) was added to medium at 2.5 μg/mL for 45 min prior to the start of imaging.
METHOD DETAILS
Cut2 mutagenesis
The cut2+ coding sequence was mutagenized by PCR using Taq polymerase with conditions that further compromise fidelity of the polymerase (Wilson and Keefe, 2001). The mutant library was introduced into the pREP1-Pnmt1-GFP vector backbone by Gibson assembly (New England Biolabs, E2611). In this vector, cut2 was fused to GFP at its C-terminus and expressed under the nmt1 promoter (Pnmt1). The validation of the cut2 mutagenesis screen was performed by introducing the indicated cut2 variants into a pREP81 vector where they are expressed under the weaker nmt81 promoter (a mutated version of the nmt1 promoter).
Live-cell imaging
For live-cell imaging, cells were mounted in lectin-coated (40 μg/mL; Sigma L1395) culture dishes (μ-Slide 8-well, glass bottom; Ibidi, 80827) at a density of 1.5 × 106 cells/mL and pre-incubated on the microscope stage at 30°C for 30 min. For temperature-sensitive strains, the culture was grown at 25°C and cells were shifted to 30°C when pre-incubating on the microscope stage for 30 min prior to imaging at 30°C for 90 min. Images were acquired on a DeltaVision Elite system (Applied Precision/GE Healthcare) equipped with a pco.edge 4.2 (sCMOS) camera, Olympus 60X/1.42 (UIS2, 1-U2B933) objective, Trulight fluorescent illumination module, and an EMBL environmental chamber for temperature control. Images were acquired using the optical axis integration (OAI) mode available with the SoftWoRx software using a range of 3.6 μm. Images were acquired every 15 s for 90 min to follow the levels of GFP-tagged separase, securin, Cdc13, or Plo1 in cells undergoing mitosis. Kymographs were assembled using Matlab (Mathworks), and kymographs in the same panel use the same intensity scaling.
Pnmt81 promoter shut down of separase expression
Wild-type and cdc48-353 mutant strains expressing Pnmt81-cut1+-GFP from the leu1 locus were grown in EMM medium lacking thiamine for 15 h to induce Cut1-GFP expression; 1 × 108 cells were harvested from both WT and mutant cultures, 16 μM thiamine was added to both remaining cultures and 1 × 108 cells were harvested at every 30-min interval for 3 h. Harvested cells were washed with 20% trichloroacetic acid, spun down at 1,150 rcf and snap-frozen in liquid nitrogen. Protein samples were prepared using the denatured extract preparation described below.
Radioactive pulse-labelling
Cells were grown in EMM at 25°C and shifted to 30°C when reaching approx. 1.1 × 107 cells/mL. After 30 min incubation at 30°C, 3.5 mCi of a mix of 35S-L-methionine and 35S-L-cysteine (Perkin Elmer, NEG 772007MC) was added to 100 mL of cells. Half of the culture was harvested after 15 min, the other half after 30 min. In a replicate experiment, 1 mCi of 35S-L-methionine/cysteine mix was added, and cultures were harvested after 10 min and 20 min. Cell pellets were washed with cold (4°C) buffer (20 mM Tris pH 7.5, 150 mM NaCl), resuspended in 150 μL of the same buffer, and dropped into liquid nitrogen. Frozen droplets were kept at −80°C before processing for extract and immunoprecipitation as below.
Cell extracts
For denatured extracts, protein samples were prepared using trichloroacetic acid-based extraction: 1 × 108 cells were resuspended in 500 μL water. To this suspension, 75 μL of 1.85 M sodium hydroxide/1 M beta-mercaptoethanol was added and the mixture incubated on ice for 15 min; 75 μL of 55% trichloroacetic acid were added and incubation was continued on ice for another 15 min. The samples were spun down at 16,000 rcf for 15 min at 4°C and supernatant was completely removed from the pellet. The pellets were resuspended in 180 μL of sample buffer (500 μL of HU buffer [8 M urea, 5% SDS (w/v), 200 mM Tris-HCl pH 6.8, 20% glycerol (v/v), 1 mMEDTA, 0.1% (w/v) bromophenol blue], 400 μL of 2M Tris-HCl pH 6.8, 100 μL of 1 M DTT). 150 μL of acid-washed beads (Sigma, G8772) were added to the resuspended pellet and tubes agitated in a ball mill (Retsch, Mixer mill MM400) for 2 min at 30 Hz. Tubes were pierced at the bottom and eluates collected into fresh tubes by centrifugation at 2,400 rcf for 1 min. The extracts were heated at 75°C for 5 min. Lysate corresponding to 5 × 106 cells was loaded per lane.
For native extracts, cell pellets were resuspended in 100-250 μL of sterile water or buffer and frozen as droplets in liquid nitrogen. Cells were disrupted under cryogenic conditions for 15–30 s at 30 Hz in a ball mill (Retsch MM400) using 10 mL or 25 mL grinding jars with a fitting metal ball (Retsch). Powder was stored at −80°C until needed. A small sample of the protein powder was resuspended in lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 5% glycerol, and 0.1% NP-40) to determine the protein concentration by BCA assay (Pierce). For experiments, frozen powder was resuspended to a final concentration of 10–20 mg/mL in lysis buffer supplemented with protease inhibitors (either Halt, Pierce, or cOmplete) and phosphatase inhibitors (either Halt or PhosSTOP). Protease inhibitors were used at 5× concentration. Extracts were spun down at 16,000 rcf for 10 min at 4°C and the supernatant was collected. For immunoblotting, the supernatant was mixed with an equal volume of sample buffer (900 μL HU buffer +100 μL of 1 M DTT) and heated to 75°C for 5 min. Typically, 30 μg of protein was loaded into one lane.
Immunoblotting
Immunoblotting used either denatured extracts or native extracts. Proteins were separated by SDS-PAGE (NuPAGE 4–12% Bis-Tris or 3–8% Tris-Acetate, Invitrogen) and the resolved proteins were transferred onto a PVDF membrane (Immobilon-P, Millipore) using a semi-dry blotting assembly (Amersham Biosciences) with Tris-glycine buffer (39 mM glycine, 48 mM Tris base) containing 10% methanol, 0.01% SDS, and 0.1% NuPAGE Antioxidant (Invitrogen, NP0005).
Primary antibodies were goat anti-Cdc48 (Abcam, ab206320), mouse anti-myc (Sigma, M4439), mouse anti-Cdc13 (cyclin B, Novus, NB200-576), rabbit anti-Cdc2 (CDK1,Santa Cruz, SC-53), mouse anti-tubulin (Sigma, T5168), mouse anti-GFP (Roche, 11814460001), rabbit anti-Cut2 (Kamenz and Hauf, 2014), and rabbit anti-Mad1 (Heinrich et al., 2013). Secondary antibodies were anti-mouse, anti-rabbit, or anti-goat HRP conjugates (Jackson 115-035-003, 111-035-003; Abcam, ab6741). Antibodies were diluted in 4% (w/v) milk in TBS-T (150 mM NaCl, 20 mM Tris, 0.05% Tween-20); membranes were washed with TBS-T. Blots were developed using an enhanced chemiluminescent substrate (SuperSignal West Pico Plus, ThermoFisher). Radioactive immunoblots were dried, exposed to a phosphorimager screen (GE) for several days, and read out on a Typhoon Trio (GE).
Immunoprecipitation
Immunoprecipitations were performed using native extracts prepared as above. Mouse anti-GFP monoclonal antibody (Roche, 11814460001) or rabbit anti-Mad1 antibody (Heinrich et al., 2013) were covalently coupled to Dynabeads Protein G (ThermoFisher, 10003D) at 12–16 μg antibody per 100 μL bead slurry for anti-GFP, and 8 μg antibody per 100 μL bead slurry for anti-Mad1. Around 1.5–2.5 mg of cell lysate and 10–25 μL bead slurry was used per sample. Immunoprecipitates were eluted using 7–15 μL of 100 mM citric acid pH 2; pH of the sample was adjusted with 1.5 M Tris pH 9.2; either HU buffer (8 M urea, 5% SDS, 200 mM Tris HCl pH 6.8, 1mMEDTA, 20% glycerol, and 0.01% bromophenol blue) or 4× LDS-sample buffer (Invitrogen, NP0007) with beta-mercaptoethanol (3% (v/v) final conc.) were added and samples incubated at 85°C for 3 min.
QUANTIFICATION AND STATISTICAL ANALYSIS
Calculating translation efficiency
Translation efficiency was calculated and plotted in R studio as the ratio between previously reported raw counts of ribosome-protected RNA fragments and RNA-seq of wild-type untreated cells from different experiments (Rubio et al., 2021). Only non-mitochondrial, non-dubious protein-coding genes were included, which resulted in a list of 5,057 genes. From this, an additional 30 genes were excluded because they had no RNA-seq raw counts or data were only available from four or fewer experiments out of a total of ten experiments.
Quantification of fluorescent signals
Fiji/ImageJ (Schindelin et al., 2012) was used to quantify nuclear and cellular concentrations of the protein analyzed. Nuclear and cellular region of interest (ROI) at each time point were defined using TetR-tdTomato- and brightfield-based thresholding, respectively. The mean background outside of cells was subtracted. The degradation kinetics of securin and Cdc13 were determined in Matlab (MathWorks). A cubic spline was fit to the degradation curve. The onset of degradation was defined as the point when the degradation rate first reached 10% of the maximal degradation rate; the end of degradation was defined as the point when the curve first became flat. Data were normalized to the intensities at these two points. For the normalized degradation rate, the region of the curve between 30 and 70% of degradation was used to determine the slope. Plo1 binding to spindle pole bodies was monitored by recording the maximum signal within the cellular ROI. The onset of Plo1 removal from spindle pole bodies was defined as the point when the loss in maximum signal first reached 20% of the maximal loss rate. The time point of sister chromatid separation (SCS) was determined by splitting of the dh1L<<tetO/TetR-tdTomato signal (Sakuno et al., 2009), and all fluorescence intensity curves were aligned to SCS. Spindle length was determined by manually measuring the distance between Plo1-GFP at spindle pole bodies in Fiji/ImageJ.
Quantification of immunoblots
Immunoblots were quantified in ImageLab (Bio-Rad) on images were none of the relevant pixels was saturated. The dilution series was used to create a standard curve by linear regression, with the exception of separase-GFP blots in the promoter shut-off experiment. In these experiments, the protein level dropped below the lowest amount in the dilution series. Therefore, a lane without sample was included in the quantification, and a hyperbola was fit using Prism (GraphPad). A one phase exponential decay was fit to the data to determine protein half-lives.
Quantification of radiographs
Radiographs were quantified in ImageLab (Bio-Rad) on images were none of the relevant pixels was saturated. A lane without sample was measured as background, and all values were determined relative to the immunoprecipitation from wild-type extract at 30 min.
Statistical analysis
The number of cells (n) is shown in the figure; error bars and statistical tests are described in the figure legend.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Goat anti-Cdc48 | Abcam | Cat#ab206320; RRID: AB_2747420 |
| Mouse anti-myc | Sigma | Cat#M4439; RRID: AB_439694 |
| Mouse anti-Cdc13 | Novus | Cat#NB200-576; RRID: AB_10003103 |
| Rabbit anti-Cdc2 | Santa-Cruz | Cat#sc-53; RRID: AB_2074908 |
| Mouse anti-tubulin | Sigma | Cat#T5168; RRID: AB_477579 |
| Mouse anti-GFP | Roche | Cat#11814460001; RRID: AB_390913 |
| Rabbit anti-Cut2 | Kamenz and Hauf, 2014, PMID: 24583014 | N/A |
| Rabbit anti-Mad1 | Heinrich et al., 2013, PMID: 24161933 | N/A |
| Goat anti-mouse HRP | Jackson ImmunoResearch Labs | Cat#115-035-003; RRID: AB_10015289 |
| Goat anti-rabbit HRP | Jackson ImmunoResearch Labs | Cat#111-035-003; RRID: AB_2313567 |
| Rabbit anti-goat HRP | Abcam | Cat#ab6741; RRID: AB_955424 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Bortezomib | LC Laboratories | Cat#B-1408 |
| Rapamycin | LC Laboratories | Cat#R-5000 |
| Lectin | Sigma | Cat#L1395 |
| Dynabeads™ Protein G | Thermo Fisher Scientific | Cat#10003D |
| Halt™ Protease Inhibitor Cocktail, EDTA-Free (100X) | Thermo Fisher Scientific | Cat#87785 |
| Pierce™Protease Inhibitor Tablets, EDTA-free | Thermo Fisher Scientific | Cat#A32965 |
| cOmplete™, EDTA-free Protease Inhibitor Cocktail | Roche | Cat#04693132001 |
| Halt™ Phosphatase Inhibitor Cocktail | Thermo Fisher Scientific | Cat#78420 |
| PhosSTOP™ | Roche | Cat#04906837001 |
| EasyTag™ EXPRESS 35S Protein Labeling Mix, [35S] | Perkin Elmer | Cat#NEG772007MC |
| EMM (Edinburgh’s Minimal Medium) | MP Biomedicals | Cat# 114110022 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Pierce™ BCA Protein Assay Kit | Thermo Fisher Scientific | Cat#23225 |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | Thermo Fisher Scientific | Cat#34580 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| See Table S1 for S. pombe strains | This study | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| See Table S2 for vectors | This study | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Fiji/ImageJ | Schindelin et al., 2012, PMID: 22743772 | https://imagej.net/Using_Fiji; RRID: SCR_002285 |
| SoftWoRx | Applied Precision, GE Healthcare | http://incelldownload.gehealthcare.com/bin/download_data/SoftWoRx/6.5.2/SoftWoRx.html; RRID: SCR_019157 |
| ImageLab | Bio-Rad Laboratories | Version 6.0.1 build 34 |
| Matlab | Mathworks | https://www.mathworks.com; RRID: SCR_001622 |
| Prism 9 | GraphPad Software, Inc | https://www.graphpad.com; RRID: SCR_002798 |
| R | Cran.R | https://cran.r-project.org; RRID: SCR_001905 |
| R studio | N/A | https://www.rstudio.com; RRID: SCR_000432 |
| tidyverse package | Cran.R | https://tidyverse.tidyverse.org; RRID:SCR_019186 |
| ggplot2 package | Cran.R | https://ggplot2.tidyverse.org/; SCR_014601 |
|
| ||
| Other | ||
|
| ||
| Mixer mill MM400 | Retsch | Cat#20.745.0001 |
| Grinding jar 10 mL | Retsch | Cat#01.462.0236 |
| Grinding jar 25 mL | Retsch | Cat#01.462.0213 |
| Adapter for reaction vials | Retsch | Cat#22.008.0008 |
| Glass beads, acid-washed | Sigma | Cat#G8772 |
| μ-Slide 8-well, glass bottom | Ibidi | Cat#80827 |
| Invitrogen™ NuPAGE™ 4 to 12%, Bis-Tris, 20-well | Invitrogen | Cat#WG1402BOX |
| Invitrogen™ NuPAGE™ 4 to 12%, Bis-Tris, 12-well | Invitrogen | Cat#NP0322BOX |
| Invitrogen™ NuPAGE™ 3 to 8%, Tris-Acetate, 12-well | Invitrogen | Cat#EA03752BOX |
| Immobilon-P PVDF membrane | Millipore Sigma | Cat#IPVH00010 |
Highlights.
Separase is not degraded during mitosis in cdc48-353 cells
Securin and Cdc13 degradation during mitosis are not impaired in cdc48-353 cells
cdc48-353 is a dominant-negative gain-of-function allele
cdc48-353 may impair separase protein synthesis rather than increase degradation
ACKNOWLEDGMENTS
We thank Tatiana Boluarte for help with yeast strain construction; Michael Boddy, Peter Espenshade, Yoshinori Watanabe, and Mitsuhiro Yanagida for providing yeast strains; Susan Forsburg for providing plasmids; Shiv Grewal for generously providing resources to establish radioactive labeling; Andrea Ciliberto, Julia Kamenz, and all members of the Hauf Lab for critical reading of the manuscript; as well as the Virginia Tech Open Access Subvention Fund for subsidizing article processing charges. This work was supported by the NIH/National Institute of General Medical Sciences under award R35GM119723. J.M. acknowledges support by the German Academic Exchange Service (DAAD).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110554.
REFERENCES
- Bähler J, Wu JQ, Longtine MS, Shah NG, McKenzie A 3rd, Steever AB, Wach A, Philippsen P, and Pringle JR (1998). Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14, 943–951. 10.1002/(sici)1097-0061(199807)14:10<943::Aid-yea292>3.0.Co;2-y. [DOI] [PubMed] [Google Scholar]
- Baltz AG, Munschauer M, Schwanhäusser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, et al. (2012). The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell 46, 674–690. 10.1016/j.molcel.2012.05.021. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Bartesaghi A, Merk A, Rao P, Bulfer SL, Yan Y, Green N, Mroczkowski B, Neitz RJ, Wipf P, et al. (2016). 2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science 351, 871–875. 10.1126/science.aad7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beskow A, Grimberg KB, Bott LC, Salomons FA, Dantuma NP, and Young P (2009). A conserved unfoldase activity for the p97 AAA-ATPase in proteasomal degradation. J. Mol. Biol 394, 732–746. 10.1016/j.jmb.2009.09.050. [DOI] [PubMed] [Google Scholar]
- Bodnar NO, and Rapoport TA (2017). Molecular mechanism of substrate processing by the Cdc48 ATPase complex. Cell 169, 722–735.e729. 10.1016/j.cell.2017.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhm S, Lamberti G, Fernández-Sáiz V, Stapf C, and Buchberger A (2011). Cellular functions of Ufd2 and Ufd3 in proteasomal protein degradation depend on Cdc48 binding. Mol. Cell Biol 31, 1528–1539. 10.1128/mcb.00962-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland A, Martin TG, Zhang Z, Yang J, Bai XC, Chang L, Scheres SH, and Barford D (2017). Cryo-EM structure of a metazoan separase-securin complex at near-atomic resolution. Nat. Struct. Mol. Biol 24, 414–418. 10.1038/nsmb.3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchberger A, Schindelin H, and Hänzelmann P (2015). Control of p97 function by cofactor binding. FEBS Lett. 589, 2578–2589. 10.1016/j.febslet.2015.08.028. [DOI] [PubMed] [Google Scholar]
- Burr R, Ribbens D, Raychaudhuri S, Stewart EV, Ho J, and Espenshade PJ (2017). Dsc E3 ligase localization to the Golgi requires the ATPase Cdc48 and cofactor Ufd1 for activation of sterol regulatory elementbinding protein in fission yeast. J. Biol. Chem 292, 16333–16350. 10.1074/jbc.M117.802025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien CY, and Chen RH (2013). Cdc48 chaperone and adaptor Ubx4 distribute the proteasome in the nucleus for anaphase proteolysis. J. Biol. Chem 288, 37180–37191. 10.1074/jbc.M113.513598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooney I, Han H, Stewart MG, Carson RH, Hansen DT, Iwasa JH, Price JC, Hill CP, and Shen PS (2019). Structure of the Cdc48 segregase in the act of unfolding an authentic substrate. Science 365, 502–505. 10.1126/science.aax0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CJ, Dutta K, Petri ET, Hwang WC, Lin Y, Pascal SM, and Basavappa R (2002). The regions of securin and cyclin B proteins recognized by the ubiquitination machinery are natively unfolded. FEBS Lett. 527, 303–308. 10.1016/s0014-5793(02)03246-5. [DOI] [PubMed] [Google Scholar]
- Csizmok V, Felli IC, Tompa P, Banci L, and Bertini I (2008). Structural and dynamic characterization of intrinsically disordered human securin by NMR spectroscopy. J. Am. Chem. Soc 130, 16873–16879. 10.1021/ja805510b. [DOI] [PubMed] [Google Scholar]
- Dargemont C, and Ossareh-Nazari B (2012). Cdc48/p97, a key actor in the interplay between autophagy and ubiquitin/proteasome catabolic pathways. Biochim. Biophys. Acta 1823, 138–144. 10.1016/j.bbamcr.2011.07.011. [DOI] [PubMed] [Google Scholar]
- Ding L, Laor D, Weisman R, and Forsburg SL (2014). Rapid regulation of nuclear proteins by rapamycin-induced translocation in fission yeast. Yeast 31, 253–264. 10.1002/yea.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esaki M, Islam MT, Tani N, and Ogura T (2017). Deviation of the typical AAA substrate-threading pore prevents fatal protein degradation in yeast Cdc48. Sci. Rep 7, 5475. 10.1038/s41598-017-05806-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eser P, Wachutka L, Maier KC, Demel C, Boroni M, Iyer S, Cramer P, and Gagneur J (2016). Determinants of RNA metabolism in the Schizosaccharomyces pombe genome. Mol. Syst. Biol 12, 857. 10.15252/msb.20156526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito E, Weidemann DE, Rogers JM, Morton CM, Baybay EK, Chen J, and Hauf S (2021). Mitotic checkpoint gene expression is tuned by coding sequences. Preprint at bioRxiv. 10.1101/2021.04.30.442180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz A, Ackermann L, and Hoppe T (2014). Create and preserve: proteostasis in development and aging is governed by Cdc48/p97/VCP. Biochim. Biophys. Acta 1843, 205–215. 10.1016/j.bbamcr.2013.03.031. [DOI] [PubMed] [Google Scholar]
- Franz A, Ackermann L, and Hoppe T (2016). Ring of change: CDC48/p97 drives protein dynamics at chromatin. Front Genet. 7, 73. 10.3389/fgene.2016.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González A, and Hall MN (2017). Nutrient sensing and TOR signaling in yeast and mammals. Embo j 36, 397–408. 10.15252/embj.201696010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänzelmann P, and Schindelin H (2016). Characterization of an additional binding surface on the p97 N-terminal domain involved in bipartite cofactor interactions. Structure 24, 140–147. 10.1016/j.str.2015.10.027. [DOI] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, and Laemmli UK (2008). The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol. Cell 31, 925–932. 10.1016/j.molcel.2008.07.020. [DOI] [PubMed] [Google Scholar]
- Harvey RF, Smith TS, Mulroney T, Queiroz RML, Pizzinga M, Dezi V, Villenueva E, Ramakrishna M, Lilley KS, and Willis AE (2018). Transacting translational regulatory RNA binding proteins. Wiley Interdiscip. Rev. RNA 9, e1465. 10.1002/wrna.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Hatanaka M, Nagao K, Nakaseko Y, Kanoh J, Kokubu A, Ebe M, and Yanagida M (2007). Rapamycin sensitivity of the Schizosaccharomyces pombe tor2 mutant and organization of two highly phosphorylated TOR complexes by specific and common subunits. Genes Cells 12, 1357–1370. 10.1111/j.1365-2443.2007.01141.x. [DOI] [PubMed] [Google Scholar]
- Heinrich S, Geissen EM, Kamenz J, Trautmann S, Widmer C, Drewe P, Knop M, Radde N, Hasenauer J, and Hauf S (2013). Determinants of robustness in spindle assembly checkpoint signalling. Nat. Cell Biol 15, 1328–1339. 10.1038/ncb2864. [DOI] [PubMed] [Google Scholar]
- Hellmuth S, Gómez HL, Pendás AM, and Stemmann O (2020). Securin-independent regulation of separase by checkpoint-induced shugoshin-MAD2. Nature 580, 536–541. 10.1038/s41586-020-2182-3. [DOI] [PubMed] [Google Scholar]
- Hellmuth S, Gutiérrez-Caballero C, Llano E, Pendás AM, and Stemmann O (2018). Local activation of mammalian separase in interphase promotes double-strand break repair and prevents oncogenic transformation. EMBO J. 37, e99184. 10.15252/embj.201899184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmuth S, Pöhlmann C, Brown A, Böttger F, Sprinzl M, and Stemmann O (2015). Positive and negative regulation of vertebrate separase by Cdk1-cyclin B1 may explain why securin is dispensable. J. Biol. Chem 290, 8002–8010. 10.1074/jbc.M114.615310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heubes S (2007). The AAA-ATPase P97 in Mitosis and Fertilization. PhD (LMU München; ). [Google Scholar]
- Hornig NC, Knowles PP, McDonald NQ, and Uhlmann F (2002). The dual mechanism of separase regulation by securin. Curr. Biol 12, 973–982. 10.1016/s0960-9822(02)00847-3. [DOI] [PubMed] [Google Scholar]
- Ikai N, Nakazawa N, Hayashi T, and Yanagida M (2011). The reverse, but coordinated, roles of Tor2 (TORC1) and Tor1 (TORC2) kinases for growth, cell cycle and separase-mediated mitosis in Schizosaccharomyces pombe. Open Biol. 1, 110007. 10.1098/rsob.110007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikai N, and Yanagida M (2006). Cdc48 is required for the stability of Cut1/separase in mitotic anaphase. J. Struct. Biol 156, 50–61. 10.1016/j.jsb.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Jallepalli PV, Waizenegger IC, Bunz F, Langer S, Speicher MR, Peters JM, Kinzler KW, Vogelstein B, and Lengauer C (2001). Securin is required for chromosomal stability in human cells. Cell 105, 445–457. 10.1016/s0092-8674(01)00340-3. [DOI] [PubMed] [Google Scholar]
- Jentsch S, and Rumpf S (2007). Cdc48 (p97): a “molecular gearbox” in the ubiquitin pathway? Trends Biochem. Sci 32, 6–11. 10.1016/j.tibs.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Ji Z, Li H, Peterle D, Paulo JA, Ficarro SB, Wales TE, Marto JA, Gygi SP, Engen JR, and Rapoport TA (2021). Translocation of polyubiquitinated protein substrates by the hexameric Cdc48 ATPase. Mol. Cell 82, 570–584.e8. 10.1016/j.molcel.2021.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenz J, and Hauf S (2014). Slow checkpoint activation kinetics as a safety device in anaphase. Curr. Biol 24, 646–651. 10.1016/j.cub.2014.02.005. [DOI] [PubMed] [Google Scholar]
- Kamenz J, Mihaljev T, Kubis A, Legewie S, and Hauf S (2015). Robust ordering of anaphase events by adaptive thresholds and competing degradation pathways. Mol. Cell 60, 446–459. 10.1016/j.molcel.2015.09.022. [DOI] [PubMed] [Google Scholar]
- Kumada K, Nakamura T, Nagao K, Funabiki H, Nakagawa T, and Yanagida M (1998). Cut1 is loaded onto the spindle by binding to Cut2 and promotes anaphase spindle movement upon Cut2 proteolysis. Curr. Biol 8, 633–641. 10.1016/s0960-9822(98)70250-7. [DOI] [PubMed] [Google Scholar]
- Lamming DW, and Sabatini DM (2013). A Central role for mTOR in lipid homeostasis. Cell Metab. 18, 465–469. 10.1016/j.cmet.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laor D, Cohen A, Pasmanik-Chor M, Oron-Karni V, Kupiec M, and Weisman R (2014). Isp7 is a novel regulator of amino acid uptake in the TOR signaling pathway. Mol. Cell Biol 34, 794–806. 10.1128/mcb.01473-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Luo X, and Yu H (2016). Structural basis of cohesin cleavage by separase. Nature 532, 131–134. 10.1038/nature17402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S, and Tong L (2017). Molecular mechanism for the regulation of yeast separase by securin. Nature 542, 255–259. 10.1038/nature21061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira JB, Masuda CA, Maya-Monteiro CM, Matos GS, Montero-Lomeli M, and Bozaquel-Morais BL (2015). TORC1 inhibition induces lipid droplet replenishment in yeast. Mol. Cell Biol 35, 737–746. 10.1128/MCB.01314-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinova IN, Engelbrecht J, Ewald A, Langholm LL, Holmberg C, Kragelund BB, Gordon C, Nielsen O, and Hartmann-Petersen R (2015). Single site suppressors of a fission yeast temperature-sensitive mutant in cdc48 identified by whole genome sequencing. PLoS One 10, e0117779. 10.1371/journal.pone.0117779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama A, Shirai A, Yashiroda Y, Kamata A, Horinouchi S, and Yoshida M (2004). pDUAL, a multipurpose, multicopy vector capable of chromosomal integration in fission yeast. Yeast 21, 1289–1305. 10.1002/yea.1181. [DOI] [PubMed] [Google Scholar]
- McAleenan A, Clemente-Blanco A, Cordon-Preciado V, Sen N, Esteras M, Jarmuz A, and Aragón L (2013). Post-replicative repair involves separase-dependent removal of the kleisin subunit of cohesin. Nature 493, 250–254. 10.1038/nature11630. [DOI] [PubMed] [Google Scholar]
- Meyer HH, Wang Y, and Warren G (2002). Direct binding of ubiquitin conjugates by the mammalian p97 adaptor complexes, p47 and Ufd1-Npl4. EMBO J. 21, 5645–5652. 10.1093/emboj/cdf579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer HJ, and Rape M (2014). Enhanced protein degradation by branched ubiquitin chains. Cell 157, 910–921. 10.1016/j.cell.2014.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer R, Fofanov V, Panigrahi A, Merchant F, Zhang N, and Pati D (2009). Overexpression and mislocalization of the chromosomal segregation protein separase in multiple human cancers. Clin. Cancer Res 15, 2703–2710. 10.1158/1078-0432.CCR-08-2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao K, Adachi Y, and Yanagida M (2004). Separase-mediated cleavage of cohesin at interphase is required for DNA repair. Nature 430, 1044–1048. 10.1038/nature02803. [DOI] [PubMed] [Google Scholar]
- Nagao K, and Yanagida M (2006). Securin can have a separase cleavage site by substitution mutations in the domain required for stabilization and inhibition of separase. Genes Cells 11, 247–260. 10.1111/j.1365-2443.2006.00941.x. [DOI] [PubMed] [Google Scholar]
- Nakashima A, Sato T, and Tamanoi F (2010). Fission yeast TORC1 regulates phosphorylation of ribosomal S6 proteins in response to nutrients and its activity is inhibited by rapamycin. J. Cell Sci 123, 777–786. 10.1242/jcs.060319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neinast M, Murashige D, and Arany Z (2019). Branched chain amino acids. Annu. Rev. Physiol 81, 139–164. 10.1146/annurev-physiol-020518-114455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie M, Aslanian A, Prudden J, Heideker J, Vashisht AA, Wohlschlegel JA, Yates JR 3rd, and Boddy MN (2012). Dual recruitment of Cdc48 (p97)-Ufd1-Npl4 ubiquitin-selective segregase by small ubiquitin-like modifier protein (SUMO) and ubiquitin in SUMO-targeted ubiquitin ligase-mediated genome stability functions. J. Biol. Chem 287, 29610–29619. 10.1074/jbc.M112.379768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh E, Mark KG, Mocciaro A, Watson ER, Prabu JR, Cha DD, Kampmann M, Gamarra N, Zhou CY, and Rape M (2020). Gene expression and cell identity controlled by anaphase-promoting complex. Nature 579, 136–140. 10.1038/s41586-020-2034-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszewski MM, Williams C, Dong KC, and Martin A (2019). The Cdc48 unfoldase prepares well-folded protein substrates for degradation by the 26S proteasome. Commun. Biol 2, 29. 10.1038/s42003-019-0283-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero JH, Suo J, Gordon C, and Chang EC (2010). Int6 and Moe1 interact with Cdc48 to regulate ERAD and proper chromosome segregation. Cell Cycle 9, 147–161. 10.4161/cc.9.1.10312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM (2006). The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat. Rev. Mol. Cell Biol 7, 644–656. 10.1038/nrm1988. [DOI] [PubMed] [Google Scholar]
- Rak R, Dahan O, and Pilpel Y (2018). Repertoires of tRNAs: the couplers of genomics and proteomics. Annu. Rev. Cell Dev. Biol 34, 239–264. 10.1146/annurev-cellbio-100617-062754. [DOI] [PubMed] [Google Scholar]
- Ramadan K, Bruderer R, Spiga FM, Popp O, Baur T, Gotta M, and Meyer HH (2007). Cdc48/p97 promotes reformation of the nucleus by extracting the kinase Aurora B from chromatin. Nature 450, 1258–1262. 10.1038/nature06388. [DOI] [PubMed] [Google Scholar]
- Rape M, Hoppe T, Gorr I, Kalocay M, Richly H, and Jentsch S (2001). Mobilization of processed, membrane-tethered SPT23 transcription factor by CDC48(UFD1/NPL4), a ubiquitin-selective chaperone. Cell 107, 667–677. 10.1016/s0092-8674(01)00595-5. [DOI] [PubMed] [Google Scholar]
- Richly H, Rape M, Braun S, Rumpf S, Hoege C, and Jentsch S (2005). A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell 120, 73–84. 10.1016/j.cell.2004.11.013. [DOI] [PubMed] [Google Scholar]
- Rosen LE, Klebba JE, Asfaha JB, Ghent CM, Campbell MG, Cheng Y, and Morgan DO (2019). Cohesin cleavage by separase is enhanced by a substrate motif distinct from the cleavage site. Nat. Commun 10, 5189. 10.1038/s41467-019-13209-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio A, Ghosh S, Mülleder M, Ralser M, and Mata J (2021). Ribosome profiling reveals ribosome stalling on tryptophan codons and ribosome queuing upon oxidative stress in fission yeast. Nucleic Acids Res. 49, 383–399. 10.1093/nar/gkaa1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpf S, and Jentsch S (2006). Functional division of substrate processing cofactors of the ubiquitin-selective Cdc48 chaperone. Mol. Cell 21, 261–269. 10.1016/j.molcel.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Saitoh S, Takahashi K, Nabeshima K, Yamashita Y, Nakaseko Y, Hirata A, and Yanagida M (1996). Aberrant mitosis in fission yeast mutants defective in fatty acid synthetase and acetyl CoA carboxylase. J. Cell Biol 134, 949–961. 10.1083/jcb.134.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuno T, Tada K, and Watanabe Y (2009). Kinetochore geometry defined by cohesion within the centromere. Nature 458, 852–858. 10.1038/nature07876. [DOI] [PubMed] [Google Scholar]
- Sánchez-Puig N, Veprintsev DB, and Fersht AR (2005). Human full-length Securin is a natively unfolded protein. Protein Sci. 14, 1410–1418. 10.1110/ps.051368005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuberth C, Richly H, Rumpf S, and Buchberger A (2004). Shp1 and Ubx2 are adaptors of Cdc48 involved in ubiquitin-dependent protein degradation. EMBO Rep. 5, 818–824. 10.1038/sj.embor.7400203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbik N, and Haines DS (2007). Cdc48p(Npl4p/Ufd1p) binds and segregates membrane-anchored/tethered complexes via a polyubiquitin signal present on the anchors. Mol. Cell 25, 385–397. 10.1016/j.molcel.2007.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmann O, Zou H, Gerber SA, Gygi SP, and Kirschner MW (2001). Dual inhibition of sister chromatid separation at metaphase. Cell 107, 715–726. 10.1016/s0092-8674(01)00603-1. [DOI] [PubMed] [Google Scholar]
- Stolz A, Hilt W, Buchberger A, and Wolf DH (2011). Cdc48: a power machine in protein degradation. Trends Biochem. Sci 36, 515–523. 10.1016/j.tibs.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Takemoto A, Kawashima SA, Li JJ, Jeffery L, Yamatsugu K, Elemento O, and Nurse P (2016). Nuclear envelope expansion is crucial for proper chromosomal segregation during a closed mitosis. J. Cell Sci 129, 1250–1259. 10.1242/jcs.181560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrecilla I, Oehler J, and Ramadan K (2017). The role of ubiquitin-dependent segregase p97 (VCP or Cdc48) in chromatin dynamics after DNA double strand breaks. Philos. Trans. R. Soc. Lond. B Biol. Sci 372, 20160282. 10.1098/rstb.2016.0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey EC, Ji Z, Wales TE, Bodnar NO, Ficarro SB, Marto JA, Engen JR, and Rapoport TA (2019). Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science 365, eaax1033. 10.1126/science.aax1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann F (2001). Secured cutting: controlling separase at the metaphase to anaphase transition. EMBO Rep. 2, 487–492. 10.1093/embo-reports/kve113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann F, Wernic D, Poupart MA, Koonin EV, and Nasmyth K (2000). Cleavage of cohesin by the CD clan protease separin triggers anaphase in yeast. Cell 103, 375–386. 10.1016/s0092-8674(00)00130-6. [DOI] [PubMed] [Google Scholar]
- van den Boom J, and Meyer H (2018). VCP/p97-Mediated unfolding as a principle in protein homeostasis and signaling. Mol. Cell 69, 182–194. 10.1016/j.molcel.2017.10.028. [DOI] [PubMed] [Google Scholar]
- Wang F, Li S, Houerbi N, and Chou TF (2021). Temporal proteomics reveal specific cell cycle oncoprotein downregulation by p97/VCP inhibition. Cell Chem. Biol 10.1016/j.chembiol.2021.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wu C, Aslanian A, Yates JR 3rd, and Hunter T (2018). Defective RNA polymerase III is negatively regulated by the SUMO-Ubiquitin-Cdc48 pathway. Elife 7, e35447. 10.7554/eLife.35447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisman R, Finkelstein S, and Choder M (2001). Rapamycin blocks sexual development in fission yeast through inhibition of the cellular function of an FKBP12 homolog. J. Biol. Chem 276, 24736–24742. 10.1074/jbc.M102090200. [DOI] [PubMed] [Google Scholar]
- Weisman R, Roitburg I, Nahari T, and Kupiec M (2005). Regulation of leucine uptake by tor1+ in Schizosaccharomyces pombe is sensitive to rapamycin. Genetics 169, 539–550. 10.1534/genetics.104.034983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DS, and Keefe AD (2001). Random mutagenesis by PCR. Curr. Protoc. Mol. Biol 8, Unit8.3. 10.1002/0471142727.mb0803s51. [DOI] [PubMed] [Google Scholar]
- Wojcik C, Yano M, and DeMartino GN (2004). RNA interference of valosin-containing protein (VCP/p97) reveals multiple cellular roles linked to ubiquitin/proteasome-dependent proteolysis. J. Cell Sci 117, 281–292. 10.1242/jcs.00841. [DOI] [PubMed] [Google Scholar]
- Xu X, Wang L, and Yanagida M (2018). Whole-genome sequencing of suppressor DNA mixtures identifies pathways that compensate for chromosome segregation defects in Schizosaccharomyces pombe. G3 (Bethesda) 8, 1031–1038. 10.1534/g3.118.200048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam C, He Y, Zhang D, Chiam KH, and Oliferenko S (2011). Divergent strategies for controlling the nuclear membrane satisfy geometric constraints during nuclear division. Curr. Biol 21, 1314–1319. 10.1016/j.cub.2011.06.052. [DOI] [PubMed] [Google Scholar]
- Ye Y, Tang WK, Zhang T, and Xia D (2017). A mighty “protein extractor” of the cell: structure and function of the p97/CDC48 ATPase. Front Mol. Biosci 4, 39. 10.3389/fmolb.2017.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen HC, and Chang EC (2000). Yin6, a fission yeast Int6 homolog, complexes with Moe1 and plays a role in chromosome segregation. Proc. Natl. Acad. Sci. U S A 97, 14370–14375. 10.1073/pnas.97.26.14370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa T, Hayashi T, Ikai N, Katayama T, Aoki K, Obara T, Toyoda Y, Maruyama T, Kitagawa D, Takahashi K, et al. (2004). An interactive gene network for securin-separase, condensin, cohesin, Dis1/Mtc1 and histones constructed by mass transformation. Genes Cells 9, 1069–1082. 10.1111/j.1365-2443.2004.00790.x. [DOI] [PubMed] [Google Scholar]
- Zach R, and Prevorovsky M (2018). The phenomenon of lipid metabolism “cut” mutants. Yeast 35, 631–637. 10.1002/yea.3358. [DOI] [PubMed] [Google Scholar]
- Zach R, Tvaruzkova J, Schatz M, Tupa O, Grallert B, and Prevorovsky M (2018). Mitotic defects in fission yeast lipid metabolism ‘cut’ mutants are suppressed by ammonium chloride. FEMS Yeast Res 18, foy064. 10.1093/femsyr/foy064. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.