

Abstract
Ischemic preconditioning (IPC) is an experimental phenomenon in which a sub-threshold ischemic insult applied to the brain reduces damage caused by a subsequent more severe ischemic episode. Identifying key molecular and cellular mediators of IPC will provide critical information needed to develop novel therapies for stroke. Here we report that the transcriptomic response of acutely isolated preconditioned cortical microglia is dominated by marked up-regulation of genes involved in cell cycle activation and cellular proliferation. Notably, this transcriptional response occurs in the absence of cortical infarction. We employed ex vivo flow cytometry, immunofluorescent microscopy, and quantitative stereology methods on brain tissue to evaluate microglia proliferation following IPC. Using cellular co-localization of microglial (Iba1) and proliferation (Ki67 and BrdU) markers, we observed a localized increase in the number of microglia and proliferating microglia within the preconditioned hemicortex at 72, but not 24, hours post-IPC. Our quantification demonstrated that the IPC-induced increase in total microglia was due entirely to proliferation. Furthermore, microglia in the preconditioned hemisphere had altered morphology and increased soma volumes, indicative of an activated phenotype. Using transgenic mouse models with either fractalkine receptor (CX3CR1)-haploinsufficiency or systemic type I interferon signaling loss, we determined that microglial proliferation after IPC is dependent on fractalkine signaling but independent of type I interferon signaling. These findings suggest there are multiple distinct targetable signaling pathways in microglia, including CX3CR1-dependent proliferation, that may be involved in IPC-mediated protection.
Keywords: microglia, preconditioning, ischemia, interferon, fractalkine, proliferation
Graphical Abstract
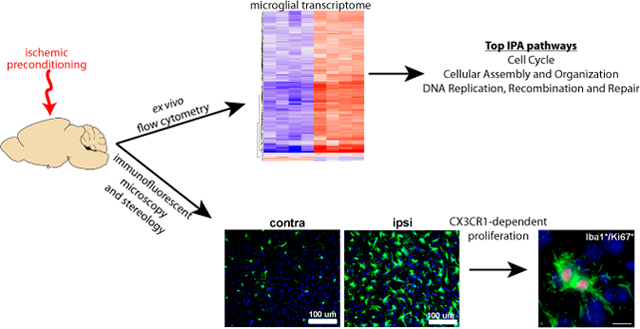
Introduction
Ischemic preconditioning (IPC) is an experimental phenomenon in which a brief ischemic event confers potent neuroprotection against a subsequent prolonged ischemic challenge (Gidday 2006; Kariko et al. 2004). IPC requires gene activation and de novo protein synthesis (Gidday 2006), in effect reprogramming the brain’s transcriptional response to ischemia (Stenzel-Poore et al. 2007; Stenzel-Poore et al. 2004). Genomic approaches to IPC have demonstrated a striking array of gene expression changes in mouse cortex following a preconditioning stimulus (Stenzel-Poore et al. 2004). Identifying key molecular and cellular mediators for IPC has important translational implications for progress in development of effective therapeutics for human stroke patients (Anrather and Iadecola 2016; Bahjat et al. 2017; McDonough and Weinstein 2016).
Microglia are the specialized tissue macrophages of the central nervous system (CNS) and play a significant role in the neuroinflammatory response of many neurological diseases including stroke (Garden and Moller 2006; Perry and Holmes 2014; Weinstein et al. 2010). Microglia derive from myeloid progenitor cells that infiltrate the CNS during early embryonic development (Alliot et al. 1999; Alliot et al. 1991; Ginhoux et al. 2010; Kierdorf et al. 2013). Early microglial cells arise from c-kit+ erythromyeloid cells in the extra-embryonic structures of the yolk sac at approximately embryonic day (E)7, enter the circulatory system, and colonize the developing brain at around E8-E10 (Ginhoux et al. 2010; Kierdorf et al. 2013). Maturation and development of microglia requires colony stimulating factor-1 receptor (CSF1R) (Ginhoux et al. 2010) and the transcription factors Pu.1 and Irf8 (Kierdorf et al. 2013). The existence of a microglia progenitor cell (Elmore et al. 2014) versus proliferation of existing or residual microglia (depending on experimental context) (Huang et al. 2018) is an ongoing debate. However, it is clear from multiple studies that microglia proliferate in response to injury (Tay et al. 2017), including prolonged cerebral ischemia (Denes et al. 2007; Moraga et al. 2015). Microglia respond to subtle alterations in their micro-environment and their activation precedes that of other cell types in the CNS (Hanisch and Kettenmann 2007; Kettenmann et al. 2011). Although microglial activation was once considered to be a stereotypic, relentlessly pro-inflammatory process, it is now clear that in certain pathologic conditions microglia can enhance and accelerate neural cell recovery and regeneration (Garden and Moller 2006; Hanisch and Kettenmann 2007; Weinstein et al. 2010). Several studies suggest that microglia could play a protective role in stroke (Lai and Todd 2006; Lalancette-Hebert et al. 2007) and/or facilitate protection in IPC (Hamner et al. 2015).
Recent transcriptomic and functional studies have demonstrated a key role for microglia and innate immune receptors in the establishment of IPC. Early studies of the global CNS response to IPC and lipopolysaccharide (LPS) preconditioning (cross tolerance) against cerebral ischemia identified a hallmark type I interferon (IFN) transcriptomal response (Marsh et al. 2009; Stenzel-Poore et al. 2004; Stenzel-Poore et al. 2003). Multiple preconditioning stimuli have converged on the requirement for type I interferon stimulated genes (ISGs) for establishing preconditioning (Stevens et al. 2011). We have demonstrated that type I IFN signaling specifically in myeloid cells is required for IPC-mediated axonal protection in white matter (Hamner et al. 2015), and in a separate study focusing on transcriptomal profiling of microglial gene expression following in vivo exposure to an IPC stimulus, we demonstrated that the cortical microglia transcriptome showed marked type I IFN receptor (IFNAR1)-dependent expression of ISGs (McDonough et al. 2017). These studies provide cell type specific context to earlier studies implicating type 1 IFN signaling in unsorted whole-brain tissue. However, a broader analysis of the microglia-specific transcriptomic response to transient ischemia/reperfusion has not been previously reported.
In addition to type I IFNs, there are other important molecular modulators of microglial activation in the context of ischemia/reperfusion. Chemokine (C-X3-C motif) ligand 1 (CX3CL1 or fractalkine) expression by neurons, and its receptor on microglia (C-X3-C chemokine receptor 1: CX3CR1), is a critical ligand/receptor system for neuron-glia crosstalk (Harrison et al. 1998; Nishiyori et al. 1998) and cellular responses to ischemic injury (He et al. 2019; Wu et al. 2016). One recent paper demonstrated that neuronal autophagy results in decreased CX3CL1 expression on neurons, which in turn results in activated (pro-inflammatory) microglia after ischemic stroke (He et al. 2019). This finding is in line with prior literature (Wolf et al. 2013) indicating that fractalkine signaling functions as a constitutive quiescence signal for microglia in the healthy CNS that can be disrupted by injury. Heterozygous Cx3cr1+/− mice are widely used in the neuroinflammation field and are valued for their ability to molecularly target and/or fate map microglia with a high degree of specificity (Goldmann et al. 2013; Wieghofer et al. 2015). Potential concerns about the experimentally confounding effects of Cx3cr1-haploinsufficiency on microglial function have been hinted at (Poniatowski et al. 2017; Wieghofer et al. 2015). However, these concerns have been allayed to an extent by reassuring findings in selective data sets. For example, Cx3cr1+/− mice were found to have similar post stroke infarct volumes and no significant change in motility of microglial processes compared with WT controls (Fumagalli et al. 2013). Other literature suggests that Cx3cr1 heterozygosity may have substantial effects on the innate immune response of microglia (Wendeln et al. 2018). The role of fractalkine signaling in the microglial response to ischemia, particularly in the context of IPC, remains to be determined, but may be critical in modulating microglial phenotype and function.
Characterizing changes in the microglial transcriptome in vivo and within the context of specific disease associated states is crucial to our understanding of innate immune responses in neurologic disorders (Butovsky and Weiner 2018; Crotti and Ransohoff 2016). Understanding the precise molecular signature of microglia in these settings also informs our approach to modulating the microglial response to CNS injury and disease (Wieghofer et al., Glia 2015). Here we present in vivo cell type specific microarray data demonstrating that up-regulation of proliferation and cell cycle genes is the dominant transcriptomal response in cortical microglia following an IPC stimulus. We also present immunohistological, cellular co-localization, and stereological findings characterizing the IPC-induced microglial proliferation phenomenon. Interestingly, others have reported that blocking cellular proliferation in the CNS with a DNA methylating agent attenuated IPC-mediated neuroprotection (Maysami et al. 2008). However, this study did not ascribe the salutary effects of cellular proliferation to any specific cell population. Other groups report that pharmacologic depletion of microglia (Jin et al. 2017; Szalay et al. 2016) or inhibition of their proliferation (Lalancette-Hebert et al. 2007) worsens stroke outcomes. We have observed via flow cytometry–based cell type specific quantification an increase in the numbers of both microglia and infiltrating peripheral immune cells in the preconditioned hemicortex (McDonough and Weinstein 2016). Building on these findings, we demonstrate here that microglia proliferate after IPC in the ipsilateral hemicortex in the absence of cortical infarction and provide evidence that this process is independent of type 1 IFN signaling, but dependent on signaling through the fractalkine receptor (CX3CR1).
Materials and Methods
Middle Cerebral Artery Occlusion Surgery
The experimental study was designed following National Institutes of Health (NIH) guidelines and with the approval of the University of Washington Institutional Animal Care and Use Committee. C57BL/6 male mice at 12–16 weeks of age were subjected to a transient (proximal) middle cerebral artery occlusion (tMCAO) surgery as published previously (McDonough et al. 2017; McDonough and Weinstein 2016; Su et al. 2014). In brief, the surgery was performed under isoflurane inhalational anesthesia using the intraluminal filament method (Longa et al. 1989) with laser doppler flowmetry for monitoring of cerebral blood flow (CBF), as we have previously described (McDonough et al. 2017; McDonough and Weinstein 2016; Su et al. 2014). We excluded mice with less than 70% reduction in CBF from further experiments. Following tMCAO, the filament was removed and reperfusion was confirmed by laser doppler. Incisions were sutured closed and mice were allowed to recover (McDonough et al. 2017; McDonough and Weinstein 2016; Su et al. 2014). For IPC surgeries, the period of occlusion was limited to 15 minutes. For confirmation of preconditioning, as shown in Figure 1, we performed a 15 minute tMCAO, allowed 72 hours of recovery and reperfusion, then performed a 60 minute tMCAO and collected tissue 48 hours later. This paradigm is well established and known to induce robust IPC-mediated neuroprotection (Stenzel-Poore et al. 2003; Zhang et al. 2008).
Figure 1.
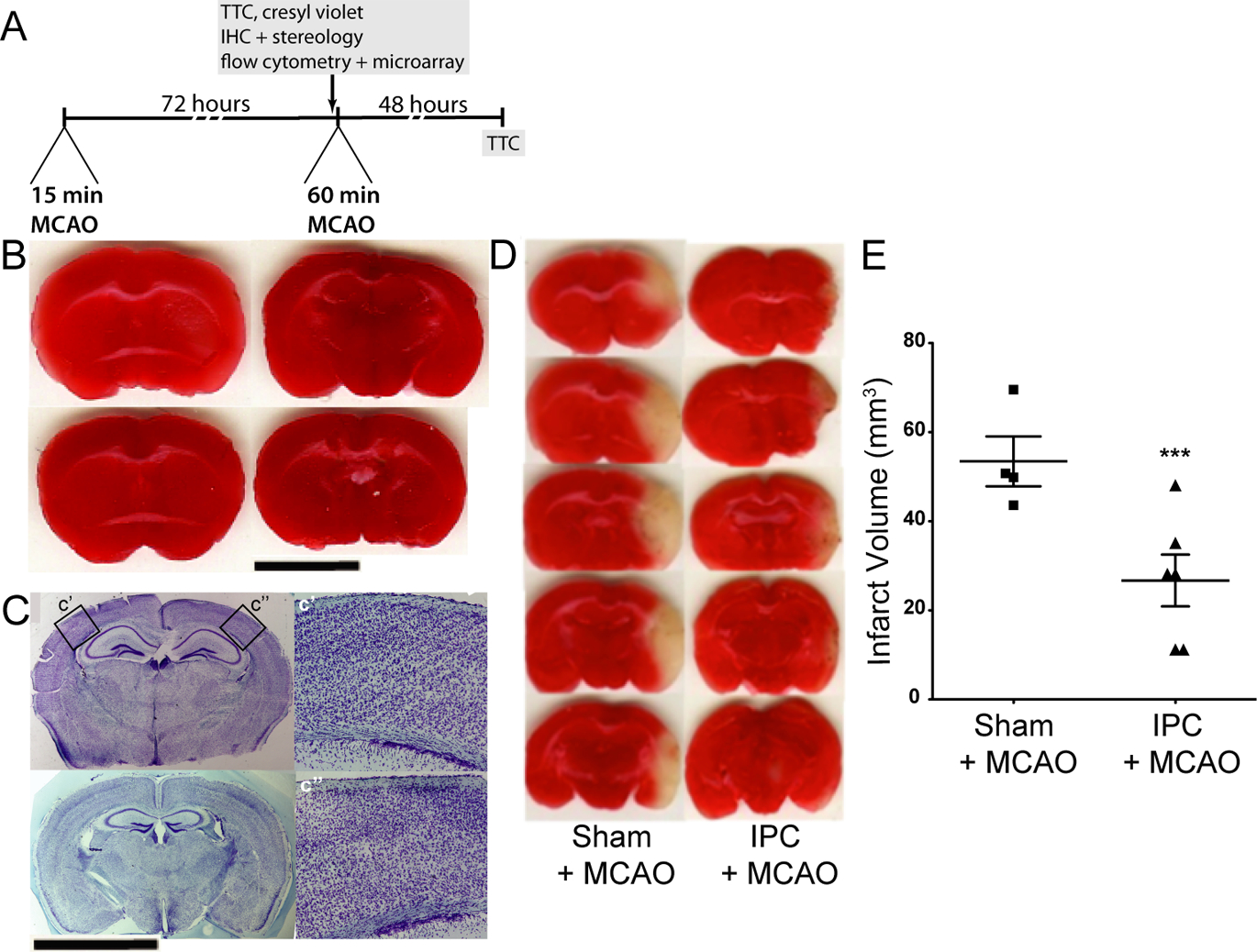
Ischemic preconditioning (IPC) does not result in cortical infarction but does protect against subsequent prolonged ischemia. A. Experimental paradigm describing the IPC stimulus and experimental end-points. B. Representative TTC-stained coronal brain tissue section images from four different animals that received a 15-minute tMCAO surgery 72 hours prior to perfusion/euthansia. Scale bar: 4 mm. C. Representative Cresyl violet-stained coronal brain tissue section images from two different animals that received a 15-minute tMCAO surgery 72 hours prior to perfusion/euthanasia. Scale bar: 4 mm. C’ is a 20x magnification view of the contralateral cortex within the MCA territory, and c” is the same view from the ipsilateral cortex. D. Representative TTC-stained coronal brain tissue section images of brains that received either a sham surgery or preconditioning surgery (15 minute tMCAO) 72 hours before a 60-minute tMCAO. E. Quantification of infarct volumes comparing brains that did (N = 6 animals) or did not (N = 4 animals) receive preconditioning 72 hours prior to a 60-minute tMCAO (t = 3.479, ***p = 0.0059; un-paired t-test).
Intracerebroventricular injection of IFNβ
2,000 U recombinant mouse IFNβ (R & D Systems) or sterile saline vehicle was injected in a volume of 2 μl over a period of 2 min into the right ventricle of 12 to 16 week-old old male mice as previously published (McDonough et al. 2017). The coordinates for the lateral ventricle location were determined based on the Paxinos and Franklin adult mouse brain atlas (−0.9 mm anteroposterior, 1.4 mm lateral to the bregma, and 2.0 mm from the skull surface). Twenty-four hours after injection of either IFNβ or vehicle, the mice were euthanized and their brains collected as described below for ex vivo flow cytometric sorting of microglia.
Ex vivo flow cytometry
Seventy-two hours after tMCAO (Figure 1A), mice were anesthetized with pentobarbital and perfused intracardially with ice cold Calcium- and Magnesium-free Hank’s Balance Salt Solution (HBSS) (Invitrogen) containing 1 mM HEPES. Ipsilateral and contralateral cortices were dissected out separately and then mechanically and enzymatically dissociated as previously described (McDonough et al. 2017; Su et al. 2014). Depletion of myelin and positive selection of myeloid cells with a CD11b microbead column (Miltenyi Biotec) were performed as described (McDonough et al. 2017; Su et al. 2014). Myeloid enriched cell suspension was stained with a panel of fluorochrome conjugated antibodies (BD Pharmingen, San Diego, CA; eBioscience Inc., San Diego, CA; Fisher Scientific, Kent, WA) against microglia, macrophage and neutrophil markers as well as 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) (Sigma-Aldrich, St. Louis, MO) and flow cytometrically sorted on a BD ARIA II cell sorter directly into TRIzol LS reagent (Invitrogen). Microglia were defined as the population of live (DAPInegative) cells that were Ly6Cnegative, Ly6Gnegative, F4/80positive, CD45low/intermediate as we have previously reported (McDonough et al. 2017; Su et al. 2014). Cellular identity of sorted cortical microglia was confirmed by our transcriptional analysis on extracted microglial RNA showing high levels of expression (>95% of all genes in microarray) for multiple known microglia specific genes (Butovsky et al. 2014) including P2ry12, Fcrls, Tmem119, Hexb and Tgfbr1 in both control (sham-operated) and experimental (tMCAO) WT mice. Cortical tissue from sham and tMCAO mice were isolated in parallel, and subjected to the same conditions of mechanical and enzymatic digestion. Thus, cellular changes in gene expression that may occur in the process of acute microglial isolation are reflected in both sham and tMCAO samples and should not confound true in situ biological changes induced by ischemia/reperfusion.
Microarray and bioinformatics analysis
Microglial RNA was extracted, analyzed and processed for cDNA synthesis, and cDNAs were hybridized to a Mouse Gene 1.0 ST Array (Affymetrix) as described previously (Anderson et al. 2011; McDonough et al. 2017). Raw data was processed, normalized and analyzed as previously described (Smyth 2004; Weinstein et al. 2010; Wu et al. 2004). Quality control (QC) analyses for microarrays included principal components analysis (PCA), using the prcomp function in R (r-project.org) comparing overall gene expression from each grouped ex vivo sorted microglial sample obtained from sham and tMCAO exposed mice and calculation of normalized unscaled standard errors (Brettschneider et al. 2008). Following QC analysis, absolute fold changes in individual genes were compiled. We selected genes based on an unadjusted p < 0.05 as well as a 1.5-fold difference in expression based on the recommendations of the MAQC consortium (Shi et al. 2006) and carried out bioinformatic analysis using Ingenuity Pathway Analysis, (IPA), a web-based application for data analysis in pathway context. IPA compilations included top gene networks, molecular and cellular functions, canonical pathways and upstream regulators. In order to better visualize and display differential categories of gene expression between our control and experimental samples, we used IPA to generate a heatmap of the individual genes that were specifically associated with the top five molecular and cellular function modules. For the heatmap, we subtracted out the mean expression, by gene, so the colors can be interpreted as the log fold change from the average gene expression. The transcriptomic data is available at the NCBI Gene Expression Omnibus at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE107983.
5-Bromodeoxyuridine labeling
To determine cell proliferation after MCAO, we injected mice immediately after tMCAO surgery with 50 mg/kg of body weight BrdU (Sigma, St. Louis, MO, USA) intraperitoneally and then administered follow-up injections of BrdU every 24 h until perfusion at 24 or 72 h after surgery.
Immunohistochemistry
At defined time points after surgery, mice were euthanized via intraperitoneal injection of a lethal dose of pentobarbital, then perfused intracardially with room temperature phosphate-buffered saline (PBS) followed by ice-cold 4% paraformaldehyde (PFA). Brains were removed immediately and post-fixed for 24 h in 4% PFA, then sectioned as 30-μm coronal slices on a vibratome (Leica, Wetzlar, Germany). Coronal sections of the brain within the bounds of the middle cerebral artery (MCA) vascular territory were taken for immunofluorescent staining using the dentate gyrus as a reference point. Every tenth section was taken at eight different levels, with one section being taken every 300 μm, accounting for a sampling area of 2,400 μm anterior to posterior. To allow BrdU antibodies to bind to their target in the cell nuclei, we post-fixed free-floating tissue in 4% PFA for 15 min, washed in PBS, and then incubated at 37°C in 2 N HCl for 30 min. We then blocked sections in 10% donkey serum (Jackson Immuno Research Labs) and 0.1% Triton X-100 (Sigma, St. Louis, MO, USA) and incubated tissue for 24 h at room temperature in primary antibodies which included: rat anti-BrdU (Abcam, Cambridge, MA, USA; 1:250), rabbit anti-Iba1 (Wako, Richmond, VA, USA; 1:500), and rabbit anti-Ki67 (BD Biosciences, San Jose, CA, USA; 1:500). Following incubation in the primary antibodies, we rinsed sections in PBS and incubated them for 24 h with appropriate secondary antibodies conjugated to fluorochromes Alexa-488 or Alexa-594 (Abcam, Cambridge, MA, USA; 1:500). We diluted all antibodies in incubation buffer containing 1% donkey serum (Jackson Immuno Research Labs) and 0.01% Triton X-100 (Sigma, St. Louis, MO, USA). As a control for background staining, we omitted the primary antibody for each experiment. We used the nuclear marker DAPI (Sigma, 1:1000) for cytoarchitectural reference.
Imaging and Stereology
Imaging and analysis was done with a Marianas imaging system from Intelligent Imaging Innovations, Inc. using a Zeiss Axiovert 200M microscope with an X, Y motorized stage, shuttered 175 W xenon lamp coupled with a liquid light guide and a Roper CoolSnap HQ digital camera. We used a Slidebook software package that allows multi-channel fluorescence and RGB DIC image capture, 3D imaging, deconvolution, montages, 3D segmentation, 3D co-localization, and stereology. We used 10–20x air, 63x water or 100x oil immersion objectives for all imaging, with stereological analysis performed on the 63x water immersion objective. Stereological analysis was accomplished by employing the optical dissector probe method. In each of the brains that were sectioned, eight sections were imaged with 8–12 cortical sampling sites in each section ipsilateral and contralateral to vessel occlusion. Each sampling site was 50 × 50 μm in the x- and y-axes, and 16 μm in the z-axis (2 μm guard zones on each extreme in the z-axis). Counting rules were established for our probe using standard stereological counting methods (Storkebaum et al. 2005). Cell volume was determined using the area fraction fractionator for estimating for volume on 50 × 50 × 16 μm grids to count points touching microglia. The volume was calculated using the formula (where P(ref) is points hitting the reference volume, Y is the sub-region, and P(Y) is points hitting sub-region) (Howard and Reed 2010). Coefficient of error for our stereology was less than 10% as determined from a pilot study that employed samplings in both naïve and preconditioned mouse brains.
Experimental Design and Statistical Analysis
Statistical evaluation was carried out using PRISM v5.0 software (GraphPad). Comparisons between multiple experimental groups were made using one or two-way ANOVA with Bonferroni’s or Dunnett’s post-test as appropriate. For comparisons between a single experimental group and a control group, we used student’s t-test. p<0.05 was considered to be significant. Data are given as mean ± SEM with each data point on a graph representing the mean for each individual animal determined following sampling rules described above. Details on statistical analyses and experimental design, including which tests were performed, F and/or t values as appropriate, exact p values, sample sizes (N) and replicates are provided within the Results describing each figure and/or within the legend of each figure. Data tables reporting fold regulation also include a separate column for p-values, with experimental conditions and replicates included to facilitate reproducibility of data.
Vertebrate animal numbers:
The number of animals used in each experiment (indicated in Methods, Results describing each Figure, and/or in figure legends) was the minimum required in order to obtain statistically and biologically meaningful results based on a modified power analysis with improved stopping rules for the design of efficient small sample experiments (Fitts 2010). This analysis identified a range (minimum to maximum) for the number of animals per experimental group sufficient to achieve a statistically significant difference (p<0.05, 90% power, variance <20% and anticipated effect size >25%) between the experimental and treatment groups, including multiple genotypes, for each in vivo experiment.
Results
Preconditioning does not result in cortical infarction
We verified that our model of IPC does not result in cortical infarction by examining sections of post-IPC brain within the middle cerebral artery (MCA) territory using both triphenyltetrazolium chloride (TTC) staining for live mitochondria (Figure 1B) and Cresyl violet staining of neuronal Nissl bodies (Figure 1C) to assess infarction and/or neuronal damage. Following a well-established protocol for induction of IPC after 15 minutes of tMCAO (Stenzel-Poore et al. 2003; Zhang et al. 2008), we observed mild striatal infarction in a few cases (data not shown), but not cortical infarction (Figure 1B). Similarly, in Cresyl violet stained tissue, we observed no cortical infarction (Figure 1C). These findings match previously published reports on similar mice with the same proximal occlusion model that indicate that MCA occlusion times of 15 min or less may result in death of striatal, but not cortical, neurons (Venna et al. 2012; Zhang et al. 2008). Furthermore, we verified that our model of a 15-min tMCAO was sufficient to induce profound neuroprotection against a 60 min tMCAO (stroke) 72 hours later. In animals that received preconditioning, infarct volumes were 20.03 ± 6.10 mm3 (N=6) compared to 53.46 ± 5.60 mm3 (N=4) in animals that received a sham surgery of identical duration, a significant reduction in infarct volume (t = 3.479, p = 0.0059; un-paired t-test) of approximately 60% (Figure 1D–E). The extent of IPC-mediated neuroprotection in our data was consistent with those of other groups that have used the same model and paradigm in mice (Stenzel-Poore et al. 2003; Zhang et al. 2008). It is important to note that studies on transient ischemic attacks (TIA) (Pedrono et al. 2010) and the response of spontaneously hypertensive rats to brief tMCAO (Ejaz et al. 2015a; Ejaz et al. 2015b) suggest that a 15 minute tMCAO (distal occlusion model) may be sufficient to induce delayed selective neuronal loss in some regions of the cortex, even in the absence of overt cortical infarction. However, the delayed selective neuronal loss effect is model-, species-, strain- (Rocha-Ferreira et al. 2015) and anesthesic drug choice- (Zhang et al. 2008) dependent and is unlikely to be critical for IPC-mediated protection (Zhang et al. 2008).
Transcriptome analysis of ex vivo sorted cortical microglia reveals that ischemic preconditioning induces robust up-regulation of cell cycle/proliferation genes
To investigate the effects of IPC on the microglial transcriptome we performed 15-minute tMCAO and then acutely isolated and analyzed gene expression in cortical microglia 72 hours following reperfusion at the peak of IPC-mediated neuroprotection in mice (Zhang et al. 2008). As previously described (McDonough et al. 2017; McDonough and Weinstein 2016; Su et al. 2014) we used ex vivo flow cytometry sorting to isolate microglia from the cerebral cortex ipsilateral or contralateral (Figure 2A) to the prior tMCAO (or sham) surgery for RNA extraction and subsequent microarray analysis (see Materials and Methods). Our microglial isolation protocol using ex vivo flow cytometry allows us to distinguish microglia from infiltrating macrophage subsets, neutrophils, and other resident CNS cell types based on a panel of cell surface markers (Figure 2A) (McDonough and Weinstein 2016). In our cortical microglial samples, we found that 2,173 genes were significantly regulated (p <0.05; absolute fold-change > 1.5) in preconditioned cortical microglia relative to non-preconditioned cortical microglia. The most striking finding in this microarray data set is a transcriptomic profile indicative of cell cycle activation and cell proliferation in the preconditioned cortical microglia. Of the 184 genes with significant and substantial (≥4-fold) increases in expression in preconditioned microglia 69 (38%) were identified by gene ontology analysis as being associated with either cell cycle or cellular proliferation (Table 1). Examples include genes encoding multiple cyclins or cyclin-dependent kinases (ccnb1, ccna2, ccnb2, ccne1, ccnf, cdk1, cdKn1a, cdkn2c), kinesins (kif11, kif4a, kif23, kif20b, kif2c, kif15, kif20a, kif14, kif22), topoisomerases (top2a), kinetochore components (nuf2, Ska3a, ndc80, zwilch, kntc1, spc25, dsn1), centromere proteins (cenpe, cenpf, cenpa, cenph, incenp, cenpi, cenpw), DNA repair genes (brca1 and brca2), DNA polymerases and ligases (pola1, lig1), and the gene for nuclear proliferation marker Ki67 (mki67). Increased expression of genes promoting cell proliferation in preconditioned cortical microglia was a consistent finding across all our individual experiments as depicted in the heatmap data shown in Figure 2B.
Figure 2.
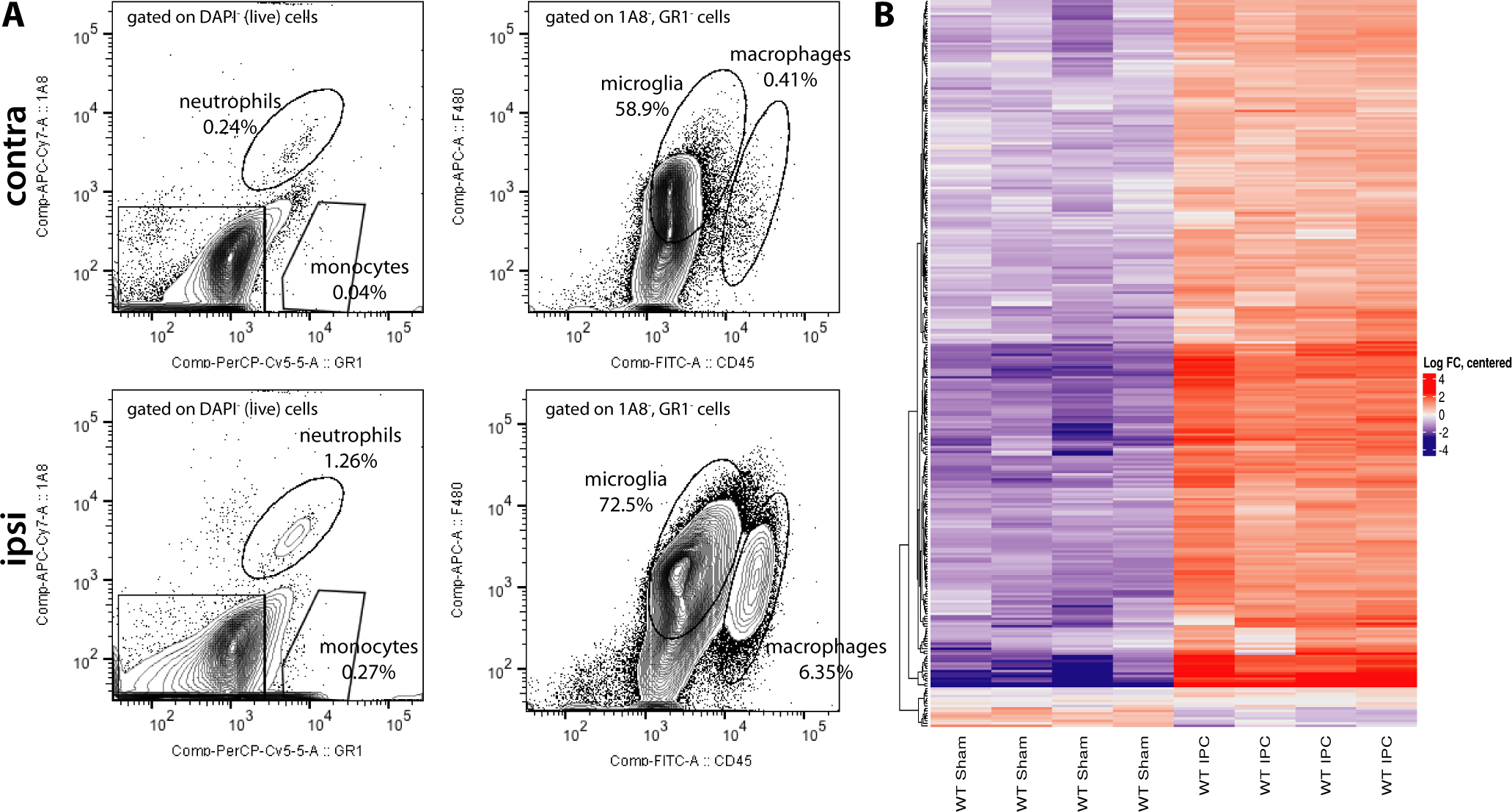
After IPC, sorted cortical microglia and peripheral immune cells are increased in the ipsilateral cortex and gene expression is altered in cortical microglia. A. Following mechanical and enzymatic digestion, microglia are sorted out from peripheral immune cells, including Ly6G+ (1A8+) neutrophils and Ly6C+ (GR-1+) monocytes, on the basis of being 1A8− and GR-1− (left panels). The resulting subpopulation of myeloid cells is differentiated based on CD45int and F4/80+ (microglia) vs CD45hi and F4/80+ (macrophages). B. Heat map comparison of cortical microglial gene expression in preconditioned and sham-operated mice within the gene ontology categories: cell cycle; cellular assembly and organization; DNA replication, recombination, and repair; cellular growth and proliferation; and cellular movement. The color gradations represent log fold changes from the average gene expression. Each column represents an individual experiment (N = 4 experiments per group, 3–4 pooled heimicortices per experiment).
Table 1:
Cell cycle genes induced in preconditioned cortical microglia
| Gene Name | Description | Fold increase | p-value |
|---|---|---|---|
| spp1 | secreted phosphoprotein 1 | 63.75 | 8.60E-15 |
| fn1 | fibronectin 1 | 46.36 | 2.48E-13 |
| bub1 | BUB1 mitotic checkpoint serine/threonine kinase | 27.25 | 1.86E-09 |
| cxcl10 | C-X-C motif chemokine ligand 10 | 19.43 | 9.23E-11 |
| nuf2 | NUF2, NDC80 kinetochore complex component | 14.35 | 1.27E-10 |
| ccna2 | cyclin A2 | 13.15 | 2.52E-08 |
| top2a | topoisomerase (DNA) II alpha | 13.12 | 4.91E-10 |
| ccnb2 | cyclin B2 | 12.73 | 2.11E-07 |
| kif11 | kinesin family member 11 | 12.56 | 4.53E-09 |
| mki67 | marker of proliferation Ki-67 | 10.68 | 9.21E-11 |
| cep55 | centrosomal protein 55 | 10.37 | 3.92E-09 |
| kif4a | kinesin family member 4A | 9.83 | 4.00E-10 |
| cenpe | centromere protein E | 9.77 | 2.84E-10 |
| tpx2 | TPX2, microtubule nucleation factor | 9.21 | 6.15E-08 |
| ttk | TTK protein kinase | 9.17 | 1.52E-11 |
| dlgap5 | DLG associated protein 5 | 9.02 | 9.28E-11 |
| bub1b | BUB1 mitotic checkpoint serine/threonine kinase B | 8.73 | 4.19E-09 |
| kif23 | kinesin family member 23 | 8.49 | 3.83E-10 |
| lgals1 | galectin 1 | 8.17 | 8.39E-09 |
| prc1 | protein regulator of cytokinesis 1 | 8.14 | 7.51E-08 |
| rad51 | RAD51 recombinase | 7.45 | 3.21E-08 |
| cdca8 | cell division cycle associated 8 | 7.23 | 1.78E-08 |
| ckap2 | cytoskeleton associated protein 2 | 7.21 | 8.96E-10 |
| ska3 | spindle and kinetochore associated complex subunit 3 | 7.10 | 2.98E-09 |
| nusap1 | nucleolar and spindle associated protein 1 | 7.09 | 1.47E-09 |
| ndc80 | NDC80, kinetochore complex component | 7.06 | 2.07E-08 |
| kif20b | kinesin family member 20B | 6.93 | 6.54E-10 |
| ncapg2 | non-SMC condensin II complex subunit G2 | 6.71 | 8.70E-08 |
| cks1b | CDC28 protein kinase regulatory subunit 1B | 6.57 | 2.08E-09 |
| kif2c | kinesin family member 2C | 6.44 | 3.35E-07 |
| aurkb | aurora kinase B | 6.41 | 2.02E-08 |
| ptgs2 | prostaglandin-endoperoxide synthase 2 | 6.40 | 1.62E-04 |
| zwilch | zwilch kinetochore protein | 6.33 | 1.53E-09 |
| mastl | microtubule associated serine/threonine kinase like | 6.30 | 1.33E-06 |
| e2f8 | E2F transcription factor 8 | 6.23 | 1.03E-10 |
| cdk1 | cyclin dependent kinase 1 | 6.20 | 2.11E-10 |
| kif15 | kinesin family member 15 | 6.16 | 7.16E-09 |
| pdcd1 | programmed cell death 1 | 6.12 | 5.72E-09 |
| stil | SCL/TAL1 interrupting locus | 6.11 | 7.51E-09 |
| kif20a | kinesin family member 20A | 6.07 | 3.75E-09 |
| plac8 | placenta specific 8 | 6.00 | 3.94E-05 |
| kntc1 | kinetochore associated 1 | 5.79 | 6.40E-11 |
| wee1 | WEE1 G2 checkpoint kinase | 5.70 | 2.51E-11 |
| esco2 | establishment of sister chromatid cohesion N-acetyltransferase 2 | 5.64 | 1.01E-09 |
| cdc25c | cell division cycle 25C | 5.63 | 1.94E-07 |
| plk1 | polo like kinase 1 | 5.43 | 1.02E-06 |
| smc4 | structural maintenance of chromosomes 4 | 5.18 | 3.89E-09 |
| cdkn1a | cyclin dependent kinase inhibitor 1A | 5.10 | 5.93E-09 |
| ube2c | ubiquitin conjugating enzyme E2 C | 5.01 | 4.00E-09 |
| mad2l1 | MAD2 mitotic arrest deficient-like 1 (yeast) | 4.99 | 4.42E-06 |
| racgap1 | Rac GTPase activating protein 1 | 4.98 | 5.56E-08 |
| foxm1 | forkhead box M1 | 4.96 | 9.93E-07 |
| chek1 | checkpoint kinase 1 | 4.96 | 1.93E-06 |
| kif14 | kinesin family member 14 | 4.88 | 5.32E-07 |
| tacc3 | transforming acidic coiled-coil containing protein 3 | 4.88 | 8.59E-08 |
| ccne2 | cyclin E2 | 4.68 | 9.03E-10 |
| cenpf | centromere protein F | 4.63 | 1.70E-08 |
| melk | maternal embryonic leucine zipper kinase | 4.63 | 4.37E-09 |
| spc25 | SPC25, NDC80 kinetochore complex component | 4.60 | 1.02E-08 |
| diaph3 | diaphanous related formin 3 | 4.45 | 2.06E-07 |
| smc2 | structural maintenance of chromosomes 2 | 4.41 | 2.46E-08 |
| cenpa | centromere protein A | 4.41 | 9.07E-09 |
| uhrf1 | ubiquitin like with PHD and ring finger domains 1 | 4.41 | 1.24E-07 |
| dtl | denticleless E3 ubiquitin protein ligase homolog | 4.20 | 9.88E-07 |
| brca1 | BRCA1, DNA repair associated | 4.09 | 9.61E-08 |
| cenph | centromere protein H | 3.96 | 5.05E-09 |
| cdca5 | cell division cycle associated 5 | 3.89 | 2.05E-06 |
| csf1 | colony stimulating factor 1 | 3.88 | 2.29E-07 |
| sgo1 | shugoshin 1 | 3.87 | 1.66E-08 |
| kif22 | kinesin family member 22 | 3.82 | 2.87E-06 |
| irf7 | interferon regulatory factor 7 | 3.78 | 1.05E-05 |
| mcm10 | minichromosome maintenance 10 replication initiation factor | 3.75 | 6.63E-08 |
| ncapd2 | non-SMC condensin I complex subunit D2 | 3.73 | 7.33E-08 |
| ercc6l | ERCC excision repair 6 like, spindle assembly checkpoint helicase | 3.69 | 1.40E-06 |
| incenp | inner centromere protein | 3.69 | 2.10E-08 |
| pola1 | DNA polymerase alpha 1, catalytic subunit | 3.57 | 4.22E-08 |
| tyms | thymidylate synthetase | 3.50 | 2.10E-06 |
| chaf1a | chromatin assembly factor 1 subunit A | 3.49 | 4.50E-07 |
| igf1 | insulin like growth factor 1 | 3.44 | 1.10E-06 |
| cdc45 | cell division cycle 45 | 3.41 | 9.23E-07 |
| ccnf | cyclin F | 3.41 | 5.64E-06 |
| aurka | aurora kinase A | 3.39 | 5.81E-08 |
| ezh2 | enhancer of zeste 2 polycomb repressive complex 2 subunit | 3.30 | 1.49E-07 |
| axl | AXL receptor tyrosine kinase | 3.28 | 8.05E-06 |
| gmnn | geminin, DNA replication inhibitor | 3.27 | 2.16E-06 |
| ddias | DNA damage induced apoptosis suppressor | 3.27 | 5.68E-04 |
| rrm1 | ribonucleotide reductase catalytic subunit M1 | 3.26 | 4.78E-06 |
| cdkn2c | cyclin dependent kinase inhibitor 2C | 3.22 | 4.55E-05 |
| rad54l | RAD54-like (S. cerevisiae) | 3.19 | 4.56E-07 |
| ahcy | adenosylhomocysteinase | 3.13 | 4.23E-05 |
| hgf | hepatocyte growth factor | 3.12 | 8.50E-08 |
| il1b | interleukin 1 beta | 3.11 | 1.82E-04 |
| cenpi | centromere protein I | 3.05 | 9.86E-07 |
| trip13 | thyroid hormone receptor interactor 13 | 3.05 | 8.80E-05 |
| asns | asparagine synthetase (glutamine-hydrolyzing) | 3.03 | 1.16E-06 |
| cd44 | CD44 molecule (Indian blood group) | 3.01 | 3.72E-08 |
| ccl2 | C-C motif chemokine ligand 2 | 2.82 | 7.17E-07 |
| fancd2 | Fanconi anemia complementation group D2 | 2.81 | 2.23E-07 |
| h2afx | H2A histone family member X | 2.80 | 4.63E-06 |
| mcm7 | minichromosome maintenance complex component 7 | 2.77 | 2.92E-07 |
| dbf4 | DBF4 zinc finger | 2.76 | 4.90E-08 |
| rad18 | RAD18, E3 ubiquitin protein ligase | 2.76 | 8.80E-09 |
| cdc20 | cell division cycle 20 | 2.75 | 3.75E-06 |
| fbxo5 | F-box protein 5 | 2.70 | 7.88E-06 |
| clspn | claspin | 2.69 | 1.92E-06 |
| plk4 | polo like kinase 4 | 2.69 | 7.30E-05 |
| exo1 | exonuclease 1 | 2.67 | 2.43E-05 |
| mcm2 | minichromosome maintenance complex component 2 | 2.66 | 1.24E-07 |
| lgals3 | galectin 3 | 2.64 | 1.90E-07 |
| brca2 | BRCA2, DNA repair associated | 2.60 | 3.61E-08 |
| cdc6 | cell division cycle 6 | 2.60 | 2.59E-05 |
| tnf | tumor necrosis factor | 2.59 | 4.67E-05 |
| rad51ap1 | RAD51 associated protein 1 | 2.59 | 3.28E-07 |
| chek2 | checkpoint kinase 2 | 2.59 | 1.28E-05 |
| id2 | inhibitor of DNA binding 2, HLH protein | 2.57 | 8.62E-06 |
| bard1 | BRCA1 associated RING domain 1 | 2.56 | 2.76E-06 |
| nasp | nuclear autoantigenic sperm protein | 2.53 | 1.61E-06 |
| mybl1 | MYB proto-oncogene like 1 | 2.51 | 7.35E-05 |
| ccne1 | cyclin E1 | 2.48 | 1.10E-06 |
| cenpw | centromere protein W | 2.45 | 3.01E-04 |
| sass6 | SAS-6 centriolar assembly protein | 2.44 | 3.31E-05 |
| tgfbi | transforming growth factor beta induced | 2.42 | 2.95E-06 |
| s100a4 | S100 calcium binding protein A4 | 2.40 | 5.05E-08 |
| eif2ak2 | eukaryotic translation initiation factor 2 alpha kinase 2 | 2.39 | 4.06E-05 |
| bhlhe40 | basic helix-loop-helix family member e40 | 2.38 | 9.14E-06 |
| mif | macrophage migration inhibitory factor (glycosylation-inhibiting factor) | 2.37 | 4.11E-05 |
| rad54b | RAD54 homolog B (S. cerevisiae) | 2.35 | 1.84E-04 |
| lig1 | DNA ligase 1 | 2.35 | 2.50E-06 |
| plaur | plasminogen activator, urokinase receptor | 2.32 | 4.62E-04 |
| cdc25b | cell division cycle 25B | 2.27 | 2.49E-05 |
| mms22l | MMS22 like, DNA repair protein | 2.26 | 2.32E-06 |
| cd274 | CD274 molecule | 2.25 | 4.65E-05 |
| blm | Bloom syndrome RecQ like helicase | 2.24 | 1.26E-07 |
| slfn1 | schlafen 1 | 2.23 | 4.66E-05 |
| pmf1/pmf1-bglap | polyamine modulated factor 1 | 2.22 | 4.59E-05 |
| dsn1 | DSN1 homolog, MIS12 kinetochore complex component | 2.22 | 6.35E-04 |
| capn1 | calpain 1 | 2.20 | 1.33E-05 |
| ska1 | spindle and kinetochore associated complex subunit 1 | 2.19 | 6.24E-05 |
| stat1 | signal transducer and activator of transcription 1 | 2.18 | 4.39E-06 |
| calm1 | calmodulin 1 | 2.18 | 8.62E-05 |
| nampt | nicotinamide phosphoribosyltransferase | 2.17 | 4.33E-06 |
| trim25 | tripartite motif containing 25 | 2.14 | 2.64E-05 |
| bora | bora, aurora kinase A activator | 2.12 | 2.86E-05 |
| polh | DNA polymerase eta | 2.12 | 2.20E-04 |
| txn | thioredoxin | 2.10 | 1.21E-06 |
| nme1 | NME/NM23 nucleoside diphosphate kinase 1 | 2.08 | 2.15E-04 |
| stat2 | signal transducer and activator of transcription 2 | 2.08 | 3.32E-04 |
| cit | citron rho-interacting serine/threonine kinase | 2.06 | 3.09E-05 |
| tex11 | testis expressed 11 | 2.02 | 3.39E-04 |
| tlr2 | toll like receptor 2 | 2.01 | 1.03E-06 |
| jun | Jun proto-oncogene, AP-1 transcription factor subunit | 0.49 | 2.99E-06 |
| hspa8 | heat shock protein family A (Hsp70) member 8 | 0.42 | 3.14E-04 |
Other specific proliferation associated genes of note that were markedly up-regulated in cortical microglia by IPC included the microglial/macrophage homeostasis factor/mitogen colony stimulating factor 1 (csf1) and the gene encoding the microglial/macrophage in vivo activation marker translocator protein (tspo). As we reported previously (McDonough et al. 2017), IPC also induces significant microglial expression of multiple type 1 interferon stimulated genes (ISGs) of which several are specifically associated with cellular proliferation, including chemokine cxcl10 and interferon regulator factor 7 (irf7) and which are found in Table 1. Bioinformatic gene ontology analysis revealed that the top molecular and cellular function categories for regulation were for genes associated with cell cycle, cellular assembly and organization, DNA replication/recombination/repair, and cellular growth/proliferation (Table 2). Top upstream regulators identified included E2F transcription factors 1 and 4 (e2f1, e2f4), both crucial regulators of the cell cycle. Other top upstream regulators included tumor suppressors p53 (tp53) and RB Transcriptional Corepressor (rb1) as well as the tumorigenesis promoting gene activator T-box 2 (tbx2).
Table 2:
Ingenuity pathway analysis on cortical microglia gene expression 72 hours post IPC
| Pathway Name | # of molecules |
|---|---|
|
| |
| Cell Cycle | 170 |
| Cellular Assembly and Organization | 93 |
| DNA Replication, Recombination and Repair | 146 |
| Cellular Growth and Proliferation | 201 |
| Cellular Movement | 128 |
Interestingly, expression levels of canonical microglial markers such as tgfbr1, tmem119, cx3cr1, p2ry12, Hexb and fcrls were significantly down-regulated in preconditioned cortical microglia relative to non-preconditioned cortical microglia (Table 3). This matches literature that suggests microglial activation or priming is associated with a decrease in these same genes (Hammond et al. 2018; Holtman et al. 2015), representing a shift in the microglial phenotype and transcriptome from surveilling to activated (Holtman et al. 2015). The conservation of certain responses, including downregulation of differentiation markers, may represent a core transcriptomic signature of microglial responses to injury.
Table 3:
expression levels of canonical microglial markers after IPC
| Gene Name | Description | fold change | p-value |
|---|---|---|---|
|
| |||
| Tgfbr1 | transforming growth factor, beta receptor I | 0.6610 | 0.0009 |
| Tmem119 | transmembrane protein 119 | 0.7045 | 0.0009 |
| Cx3cr1 | chemokine (C-X3-C) receptor 1 | 0.7452 | 0.0006 |
| P2ry12 | purinergic receptor P2Y, G-protein coupled 12 | 0.7885 | 0.0019 |
| Csf1r | colony stimulating factor receptor | 0.7674 | 0.0010 |
| Hexb | hexosaminidase B | 0.8181 | 0.0022 |
| C1qa | complement component 1, q subcomponent, alpha polypeptide | 0.8270 | 0.0752 |
| Fcrls | Fc receptor-like S, scavenger receptor | 0.8925 | 0.0189 |
| Trem2 | triggering receptor expressed on myeloid cells 2 | 0.9060 | 0.4723 |
Ischemic preconditioning increases the number of Iba1+ cells in the ipsilateral cortex
Using ex vivo flow cytometry we have observed that IPC induces significant increases in the number of both microglia and infiltrating peripheral immune cells in the ipsilateral cortex after a 15-minute tMCAO (IPC pulse) (McDonough and Weinstein 2016). However, the process of generating a single cell suspension for ex vivo flow cytometry involves enzymatic and mechanical digestion (McDonough et al. 2017; McDonough and Weinstein 2016; Su et al. 2014), which may select for preconditioned microglia that can better tolerate the multiple cellular stresses associated with ex vivo handling and cell sorting. To exclude this possibility, we utilized immunofluorescent microscopy to examine and quantify microglial/macrophage distribution in situ in the brain at the 72-hour time point following the IPC pulse. We chose this time point because it is the peak of IPC-induced neuronal (Stenzel-Poore et al. 2003; Zhang et al. 2008) and axonal (Hamner et al. 2015) protection. To assess microglial/macrophage cell number and volume we carried out immunofluorescent microscopy and quantitative stereology, as described in the Methods section, on coronal tissue sections within the rostral-caudal boundaries of the MCA vascular territory by immunostaining with an antibody against the microglial/macrophage marker Iba1. We observed a striking increase in the number of Iba1+ cells in the ipsilateral, but not contralateral, cerebral cortex (Figure 3A–C). Furthermore, we noticed that Iba1+ cells in the contralateral cortex displayed surveilling/quiescent ramified morphologies (Figure 3D) while Iba1+ cells in the ipsilateral cortex displayed complex amoeboid morphologies (Figure 3E) characteristic of classically activated microglia.
Figure 3.
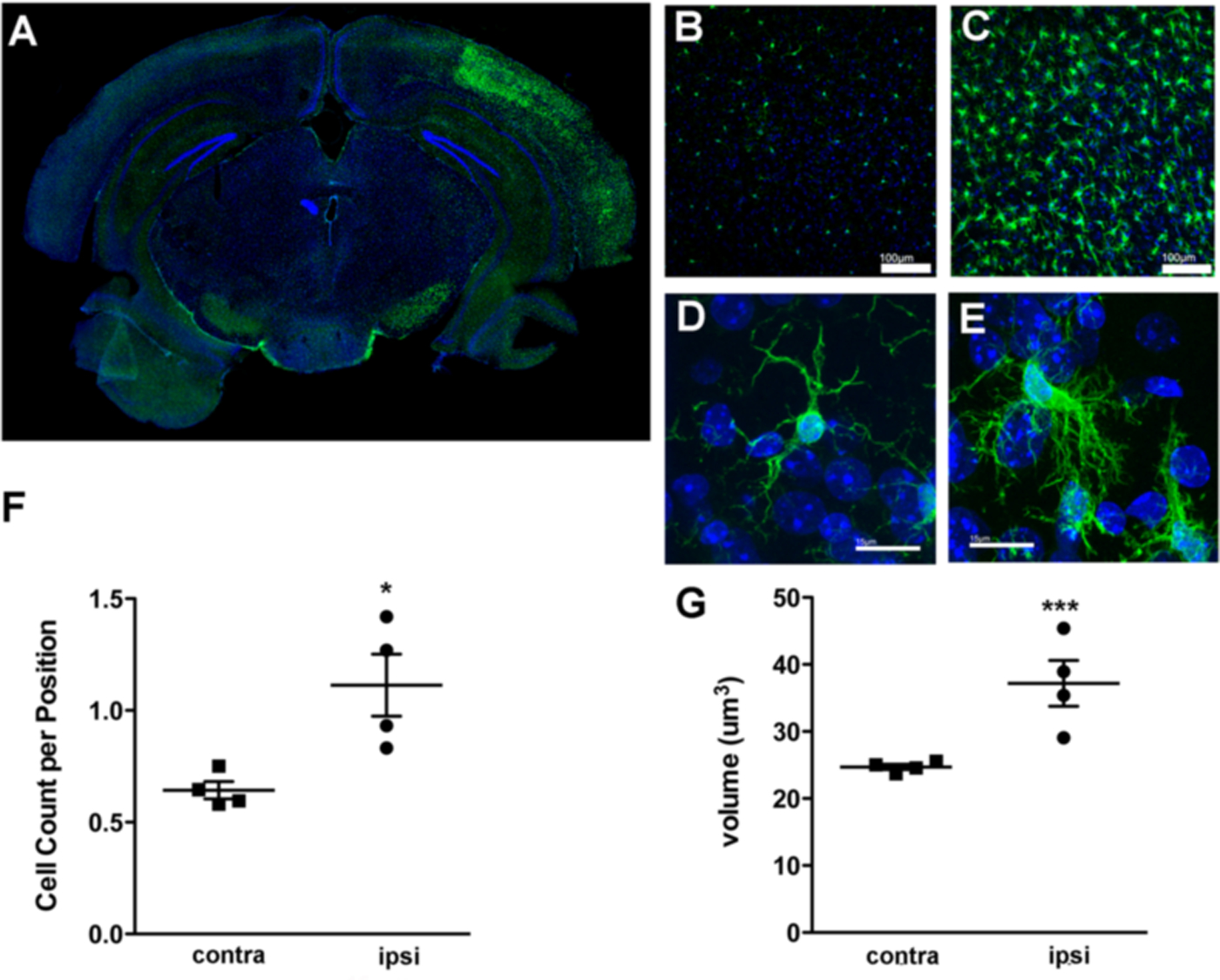
Ischemic preconditioning (IPC) induces an increase in the number of Iba1+ cells and alters the morphology of Iba1+ microglia in the ipsilateral cortex. A. Representative immunofluorescent microscopy image showing Iba1 (green) and DAPI (blue) stained coronal brain tissue section from wild-type mouse 72 h following a 15 min tMCAO (IPC). Iba1+ cells are present bilaterally, however their number and intensity of staining is increased in the ipsilateral (right) hemisphere of the cortex. B-C. Higher magnification images showing that Iba1+ microglia are present in the contralateral cortex (B), with substantially higher numbers of Iba1+ cells seen in the ipsilateral cortex (C). Iba1+ microglia display disparate morphologies within the contralateral and ipsilateral cortices. In the contralateral cortex (D), microglia display a quiescent or immunosurveilling morphology with fine ramified processes compared to the complex activated morphology of microglia in the ipsilateral cortex (E). F. Stereologic quantification of cell numbers confirm greater numbers of Iba1+ cells in the ipsilateral cortex (t = 3.272, *p = 0.0170, unpaired t-test; N = 4 animals), and G. Volumetric analysis of Iba1+ cells in both hemispheres reveals a larger cell volume in the ipsilateral cortex after IPC (t = 4.748, ***p < 0.0001, unpaired t-test; N = 4 animals). Scale bars: 100 μm B-C, 15 μm D-E.
We employed stereological methods to quantify the number of microglia within the ipsilateral and contralateral hemispheres of the brain after IPC. Our stereological findings confirmed our initial qualitative microscopy based observations and our flow cytometry results (Figure 2A and (McDonough and Weinstein 2016)); in the ipsilateral cortex the Iba1+ cell count per position mean was 1.113 ± 0.1385 (N=4 mice, 334 positions) and the contralateral cortex mean was 0.6430 ± 0.03831 (N=4 mice, 374 positions), a statistically significant 73% increase in cell density (t = 3.272, p = 0.0170, unpaired t-test) (Figure 3F). Additionally, we measured the volume of Iba1+ cells in both hemispheres using the area fractionator method and obtained means of 37.28 ± 2.32 μm3 (N = 4 mice, 640 positions) in the ipsilateral cortex and 24.71 ± 1.28 μm3 (N = 4 mice, 640 positions) in the contralateral cortex (Figure 3G), a significant increase of 51.7% (t = 4.748, p < 0.0001, unpaired t-test). This increase in cell volume is indicative of cellular hypertrophy and suggests the microglial phenotype is significantly altered in the ipsilateral cortex after IPC.
The increase in Iba1+ cortical cells following ischemic preconditioning is due entirely to cell proliferation
Since we observed a marked increase in the number of microglia in the ipsilateral cortex we next examined proliferative markers within Iba1+ cells. Using an antibody against Ki67, a well-described nuclear marker for proliferating cells, we observed many double-labeled Ki67+/Iba1+ cells in the ipsilateral, but not contralateral, cortex (Figure 4A–D). We observed several pairs of Ki67+/Iba1+ cells in close proximity to each other within the ipsilateral cortex that suggest recent cell division (Figure 4C–E). Stereological methods were employed to quantify the number of Ki67+ cells in both hemispheres (Figure 4F). In the ipsilateral cortex we counted a mean ± SEM of 0.7287 ± 0.2318 (N = 4 mice, 334 positions) Ki67+ cells per position and in the contralateral cortex we counted a mean of 0.005425 ± 0.005425 (N = 4 mice, 374 positions) Ki67+ cells per position, demonstrating that proliferation is greater in the ipsilateral cortex than in the contralateral (t = 3.120, p-value = 0.0206, unpaired t-test). We also quantified the number of Ki67+/Iba1+ cells (Figure 4G) and obtained a mean of 0.4506 ± 0.1074 (N = 4 mice, 334 positions) double-positive cells per position in the ipsilateral cortex. In none of the positions sampled in the contralateral cortex could we find double-positive cells (N = 4 mice, 374 positions). Strikingly, the difference observed between the total numbers of Iba1+ cells in the ipsilateral versus contralateral cortices can be entirely accounted for by the number of Ki67+/Iba1+ cells in the ipsilateral cortex (Figure 4H).
Figure 4.
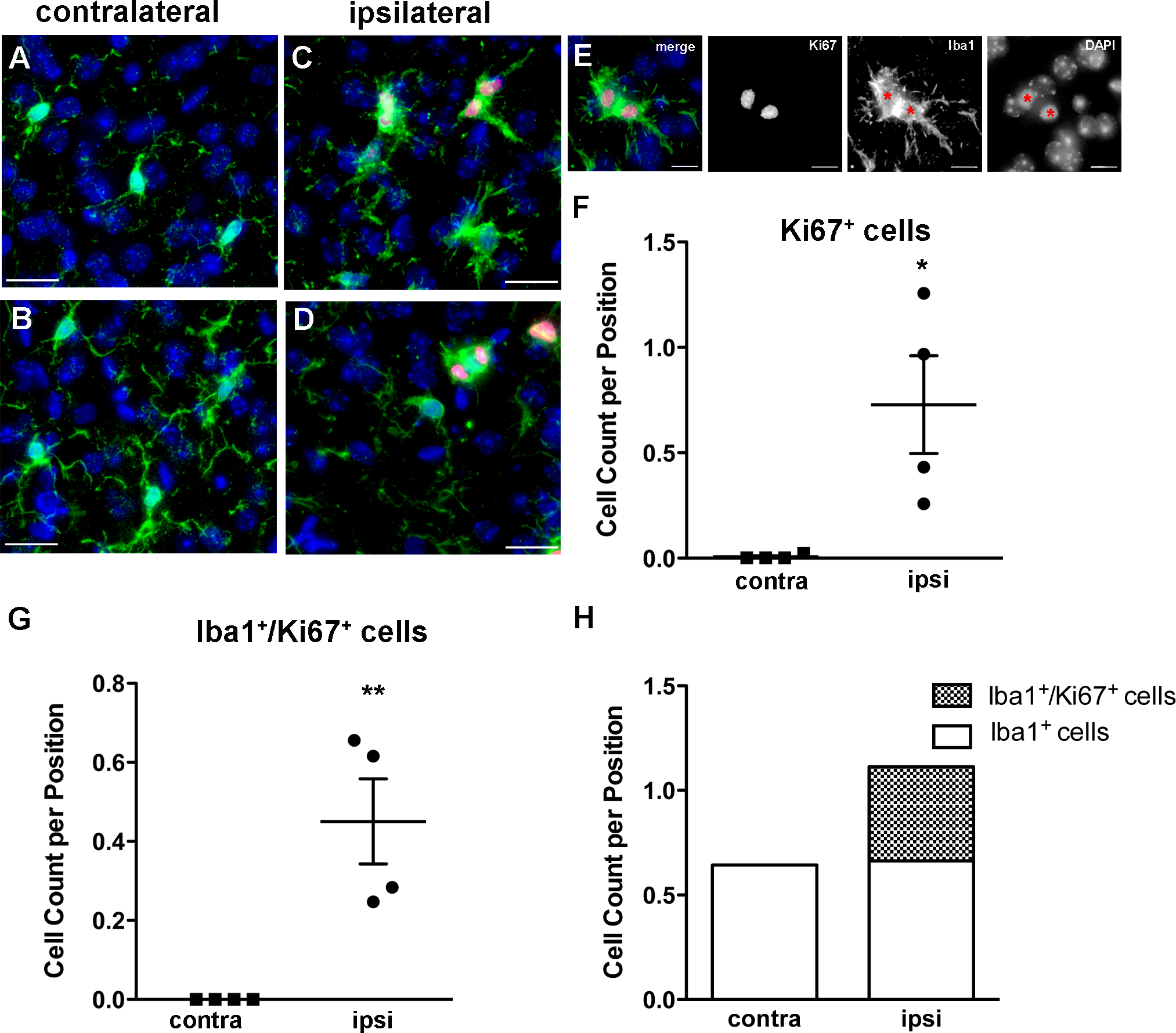
The number of proliferative cells is increased in the ipsilateral cortex after IPC. A-D. Representative images from the contralateral (A-B) and ipsilateral (C-D) cortex with immunostaining for Ki67 (red), Iba1 (green), and DAPI (blue) demonstrate a higher occurrence of double-positive cells in the ipsilateral cortex. E. Representative images of Iba1+/Ki67+ sister cells. We used stereological methods to quantify the number of Ki67+ cells (t = 3.120, *p-value = 0.0206, unpaired t-test; N = 4 animals) (F) and the number of Ki67+/Iba1+ cells (**p < 0.01, unpaired t-test; N = 4 animals) (G), which are both significantly higher in the ipsilateral cortex. H. The increase in Iba1+ cells seen in the ipsilateral relative to the contralateral cortex after IPC can be accounted for entirely by the population of Ki67+/Iba1+ cells. Scale bars: A-D, 20 um, E. 10 um.
Next we examined the timing of microglial proliferation after IPC and confirmed our Ki67 data by administering BrdU to label dividing cells and their progeny after a 15-minute tMCAO. We also examined an earlier time point, 24 hours after IPC, to see if we could detect earlier proliferation of microglia. At 24 hours after IPC we counted a mean of 0.00616 ± 0.003802 (N = 5 mice, 399 positions) BrdU+ cells in the ipsilateral cortex which did not differ significantly (t = 1.434, p-value = 0.6032, unpaired t-test) from the contralateral cortex mean of 0.0034 ± 0.0034 (N = 5, mice, 379 positions) cells per position (Figure 5F). At this time there were no BrdU+/Iba1+ cells in the cortex (data not shown). However, our stereological sampling occurred only within the MCA territory of the cortex, and we did observe some BrdU+ cells in non-cortical regions within the MCA territory, most notably within the ipsilateral hippocampus and subventricular zones, 24 hours after IPC. The latter finding is consistent with other published studies on the proliferation of neuroprogenitor cells in these brain regions after ischemia (Costello and Lynch 2013; Naylor et al. 2005). Seventy-two hours after IPC, we observed few BrdU+ cells in the contralateral cortex (Figure 5A) compared to a substantial increase of BrdU+ cells in the ipsilateral cortex (Figure 5B). A majority of the BrdU+ cells colocalized with Iba1. Double positive cells were frequently present in small clusters (Figure 5C–D) and occasionally as single cells (Figure 5E).
Figure 5.
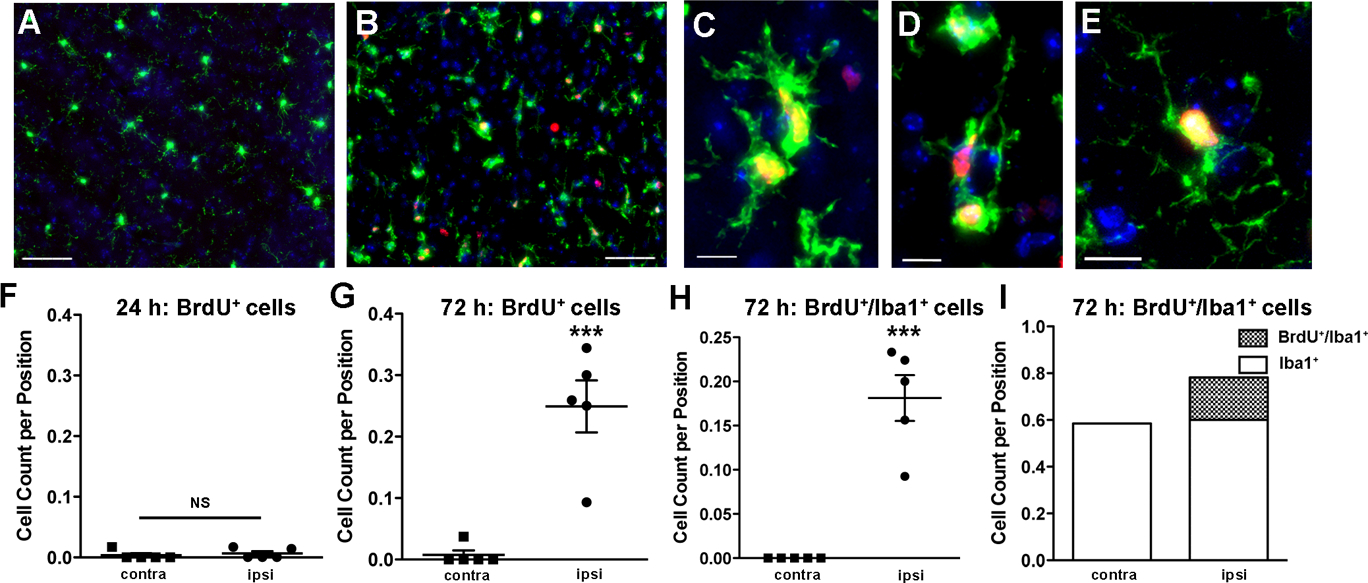
The number of BrdU-labeled microglia in the ipsilateral cortex increases after IPC. A-E. Representative images from the contralateral (A) and ipsilateral (B) cortex with immunostaining for BrdU (red), Iba1 (green), and DAPI (blue) demonstrate occurrence of BrdU+/Iba1+ cells only in the ipsilateral cortex at 72 hours after IPC. C-E. Higher magnification images of BrdU+/Iba1+ cells, occurring near each other (C-D) or as single diffuse cells (E). We used stereological methods to quantify the number of BrdU+ cells (F) 24 hours after IPC (N = 5 animals). There were very few BrdU+ cells in the cortex at this time, and no BrdU+/Iba1+ cells present (data not shown). 72 hours after IPC, we observed a significant increase in both BrdU+ cells (t = 5.610, ***p-value = 0.0005, unpaired t-test; N = 5 animals) (G) and BrdU+/Iba1+ cells (H) in the ipsilateral cortex (***p-value = 0.0005, unpaired t-test; N = 5 animals). I. The difference in the number of Iba1+ cells between the ipsilateral and contralateral cortex 72 hours after IPC was due to the number of BrdU+/Iba1+ cells.
Employing stereological methods we quantified a significant difference (t = 5.610, p-value = 0.0005, unpaired t-test) in the mean number of BrdU+ cells per position in the ipsilateral cortex (mean = 0.2492 ± 0.04246; N = 5 mice, 386 positions) compared with the contralateral cortex (mean = 0.0074 ± 0.0074; N = 5 mice, 420 positions) (Figure 5G). Also at this time we did not observe BrdU+/Iba1+ microglia in the contralateral cortex. We did observe BrdU+/Iba1+ cells in the ipsilateral cortex at 0.1813 ± 0.02586 double labeled cells per position sampled (Figure 5H). The increase in the number of Iba1+ cells 72 hours after IPC was due to the population of double positive proliferating (BrdU+/Iba1+) cells (Figure 5I). This data confirms that a high percentage of CNS resident microglia, and possibly also some infiltrating macrophages, are proliferating specifically in the ipsilateral cortex 72 hours after IPC. Thus, differences in the number of Iba1+ cells in the ipsilateral cortex are due almost entirely to cellular proliferation even in the absence of cortical infarction.
Cx3cr1-haploinsufficiency attenuates Iba1+ cellular response to IPC
CX3CR1 is the microglial receptor for neuronal CX3CL1 (fractalkine); loss or downregulation of fractalkine from neurons is an activation cue for microglia (Harrison et al. 1998; He et al. 2019; Kierdorf and Prinz 2013; Nishiyori et al. 1998). Studies on the consequences of CX3CR1-deficiency are mixed: some groups observed aberrant activation of microglia towards a neurotoxic profile in mice lacking CX3CR1 (Cardona et al. 2006; Fumagalli et al. 2013), one study suggests CX3CR1-deficiency is protective against ischemic stroke (Tang et al. 2014), and another suggests downregulation of CX3CL1 by neurons is responsible for microglial neurotoxicity after ischemic stroke (He et al. 2019). As pertains to heterozygosity of the gene locus, studies suggest that infarctions in Cx3cr1+/− mice were similar to WT controls after MCAO (Fumagalli et al. 2013). We first examined the impact of Cx3cr1-haploinsufficiency using the Cx3cr1-eGFP transgenic mouse, in which one allele of the Cx3cr1 gene has been replaced with the gene encoding enhanced green fluorescent protein (eGFP). This results in expression of eGFP in CX3CR1+ cells which include microglia, dendritic cells, and activated endothelial cells (Jung et al. 2000). This transgenic mouse line and related variants, such as the cell type specific knockdown Cx3cr1Cre and the conditional tamoxifen-inducible Cx3cr1CreER lines (Goldmann et al. 2013), are commonly used in studies that necessitate a reliable pan-microglial marker, especially as microglia may exhibit some degree of antigenic plasticity and markers for myeloid cells often overlap with those of microglia (Xavier et al. 2015).
We observed that the IPC-induced increase in the number of cortical Iba1+ cells was attenuated in the heterozygous Cx3cr1-eGFP animals relative to WT controls (Figure 6A). In an ANOVA analysis of WT and Cx3cr1+/eGFP animals we found significant effects of genotype (p-value = 0.0081, F = 9.50, DFn = 1, DFd = 14) and the expected hemisphere effect between ipsilateral and contralateral hemicortices (p-value = .0028, F = 13.06, DFn = 1, DFd = 14). In post-hoc tests we observed an increase in Iba1+ cells in the ipsilateral cortex of WT animals relative to the contralateral cortex (Figure 6A, p-value = 0.0247), but a non-significant difference in Iba1+ cell counts in the Cx3cr1+/eGFP animals (Figure 6A, p-value = 0.0899). Contralateral cell counts between the WT and Cx3cr1+/eGFP animals were significantly different (Figure 6A, p-value = 0.0123) and reduced in the transgenic animals compared to the WT animal, suggesting that there may be altered numbers of microglia in the Cx3cr1+/eGFP animals even in regions of the brain that had not been exposed to IPC. A similar trend was seen comparing ipsilateral cortices of WT and Cx3cr1+/eGFP animals, however this did not reach statistical significance (Figure 6A, p-value = 0.0596). We next sought to validate and further characterize our stereology findings using our established ex vivo flow cytometry protocol on preconditioned Cx3cr1-haploinsufficient mice. At the 72 hour time point following IPC, the same time point we previously found large increases in the number of microglia in WT animals (McDonough and Weinstein 2016), we found that Cx3cr1 heterozygous mice had no significant (t = 0.407, p-value = 0.7235, unpaired t-test) increase in the number of cortical microglia isolated from the ipsilateral versus contralateral hemicortices (Figure 6B). Based on these data, we conclude that Cx3cr1-haploinsufficiency substantially alters the proliferative response of microglia to IPC.
Figure 6.
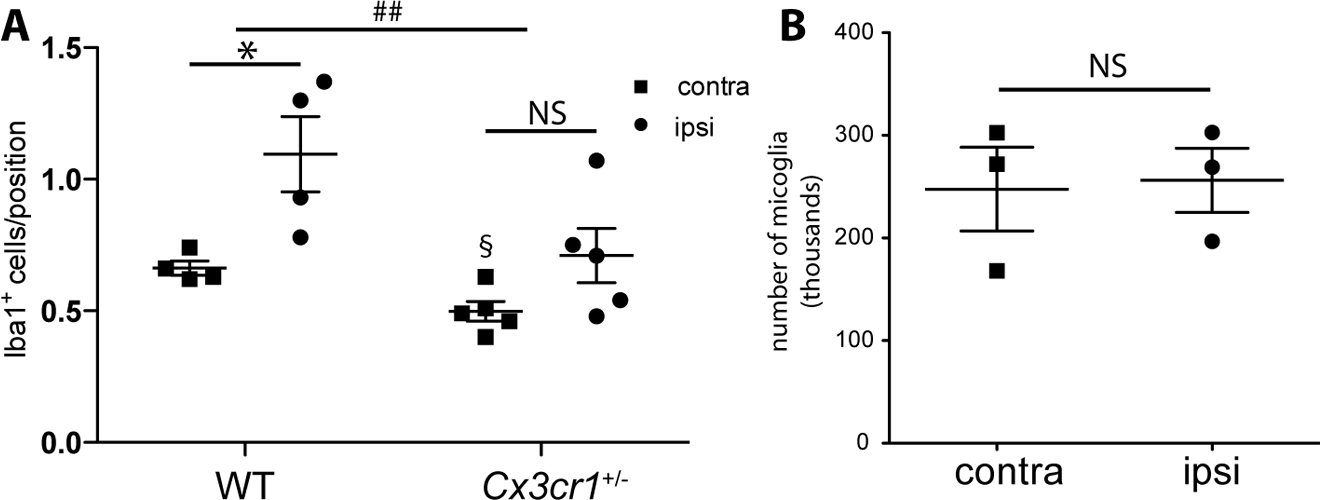
Cx3cr1-haploinsufficiency results in attenuated proliferative response to IPC. A. WT and Cx3cr1-eGFP mice were subjected to a 15 minute tMCAO and 72 hours later mice were anesthetized, brain tissue was perfused and fixed and immunofluorescent microscopy and stereology was performed. The counts in wild-type (WT) mice show an increased number of Iba1+ cells in the ipsilateral hemicortex relative to the contralateral hemicortex (*p-value = 0.0247; N = 4 animals), whereas in the Cx3cr1-eGFP heterozygous animals the Iba1+ cell counts between the ipsilateral and contralateral hemicortices displayed no significant differences (p-value = 0.0899; N = 5 animals); and overall a genotype effect was observed between WT and Cx3cr1-eGFP heterozygous animals (##p-value = 0.0081, F = 9.50, DFn = 1, DFd = 14; ANOVA). Intriguingly, when the contralateral hemicortices were compared by genotype the Cx3cr1-eGFP heterozygous animals had significantly fewer microglia than WT animals (§p-value = 0.0123), and the ipsilateral hemicortices were not significantly different in post-hoc tests (p-value = 0.0596). B. When ex vivo flow cytometry was performed on Cx3cr1CreER heterozygous animals 72 h after 15 min tMCAO, no difference was observed in the number of microglia collected from ipsilateral or contralateral hemispheres (t = 0.407, p-value = 0.7235, unpaired t-test; N = 3 experiments with 2–3 brains pooled per experiment).
Cortical microglial proliferation after IPC is independent of type I interferon signaling
Type I IFN signaling is necessary for multiple preconditioning scenarios including IPC (Gesuete et al. 2012; Leung et al. 2012; Stevens et al. 2011). In our previous studies we determined that type I IFN signaling in microglia was required for IPC-mediated axonal protection in a white matter model of IPC (Hamner et al. 2015) and in a whole brain / grey matter predominant model we identified significant ISG expression in cortical microglia after IPC (McDonough et al. 2017). Some of these ISGs, for example csf1, encode proteins that are proliferative cues for microglia (Ginhoux et al. 2010). Reduced proliferation of cultured microglia was also observed in interferon regulatory factor (IRF)8 deficiency (Horiuchi et al. 2012). Based on these reports, we hypothesized that animals deficient in type I interferon signaling (Ifnar1−/−) would have reduced proliferation after IPC. To test this hypothesis, we subjected Ifnar1−/− mice to our standard preconditioning paradigm (15-minute tMCAO followed by 72 hours reperfusion), administered BrdU daily, and performed immunohistochemistry, microscopy, and stereology following the same methods as described above in WT animals. Surprisingly, we observed no genotype effect for the mean total number of Iba1+ cells (p-value = 0.2337, F = 1.532), mean total number of BrdU+ cells (p-value = 0.2555, F = 1.391) or the mean total number of Iba1+/BrdU+ cells (p-value = 0.6933, F = 0.1613) (Figure 7A–C). In both genotypes the expected hemispheric effect was observed: ipsilateral numbers of Iba1+ and proliferative cells (BrdU+ and BrdU+/Iba1+) were increased relative to contralateral control hemicortices (Figure 7A–C). Furthermore, we observed no effect of intracerebroventricular injections of IFNβ on cortical microglial counts between animals treated with IFNβ or vehicle (t = 0.2061, p-value = 0.8468, unpaired t-test) (Figure 7D), although IFNβ injected mice do show up-regulation of ISG expression in cortical microglia (McDonough et al. 2017), which is a hallmark transcriptomic feature of preconditioning.
Figure 7.
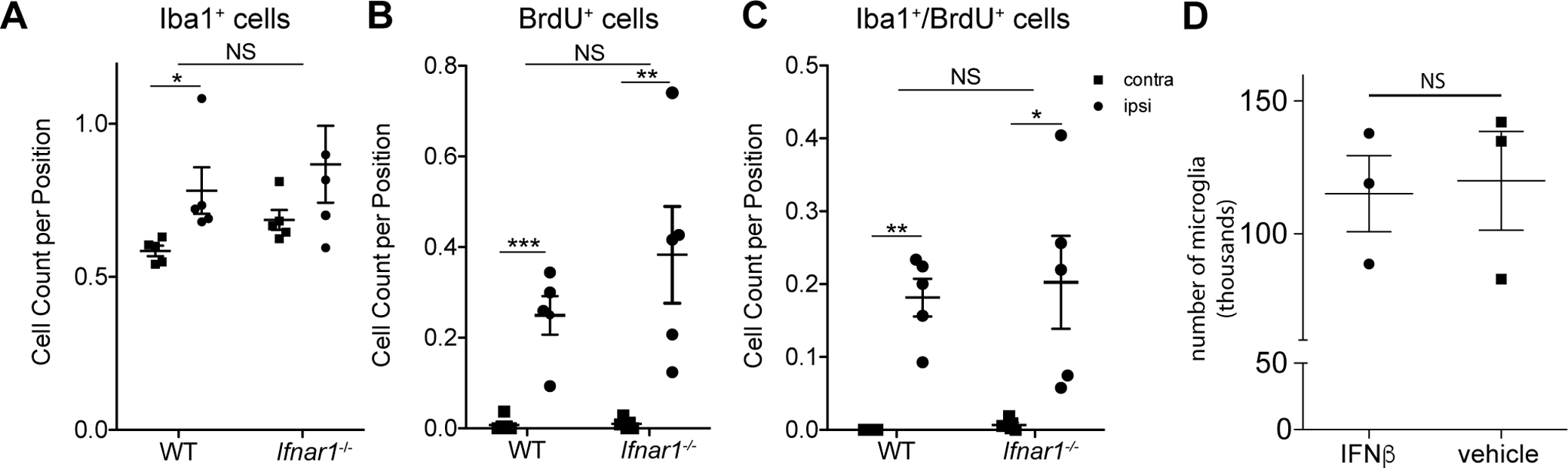
Loss of type I interferon signaling does not affect cortical microglia proliferation after IPC. A-C. WT and Ifnar1−/− mice were subjected to a 15 minute tMCAO and 72 hours later mice were anesthetized, brain tissue was perfused and fixed and immunofluorescent microscopy and stereology was performed (N = 5 animals per genotype). In both genotypes a hemispheric effect was observed: ipsilateral numbers of Iba1+ and proliferative cells (BrdU+ and BrdU+/Iba1+) were increased relative to contralateral control hemicortices according to post-hoc tests. A. There was no genotype effect observed for the total number of Iba1+ cells (p-value = 0.2337, F = 1.532, DFn = 1, DFd = 15; ANOVA), B. total number of BrdU+ cells (p-value = 0.2555, F = 1.391, DFn = 1, DFd = 15; ANOVA) or C. the total number of Iba1+/BrdU+ cells (p-value = 0.6933, F = 0.1613, DFn = 1, DFd = 15; ANOVA). D. 2,000 U of recombinant mouse IFNβ or a corresponding volume of saline was injected into the right lateral ventricle of WT mouse brains. Microglia were acutely isolated by ex vivo flow cytometry 24 hours later. There was no significant difference in the numbers of microglia isolated from animals treated with IFNβ or vehicle (t = 0.2061, p-value = 0.8468, unpaired t-test; N = 3 experiments with 3–4 brains pooled per experiment).
Discussion
Previous studies have demonstrated significant transcriptomic changes in response to preconditioning (Stenzel-Poore et al. 2007; Stenzel-Poore et al. 2004; Stenzel-Poore et al. 2003). However, these studies focused on RNA isolated from cortical brain tissue rather than specific cell types. We recently identified microglia as critical in establishing IPC-mediated protection (Hamner et al. 2015) and are interested in elucidating the microglial response to IPC as a means to identify signaling pathways and mechanisms that are important and characteristic of this biological phenomenon. We reported previously that a hallmark transcriptomic feature of microglia after IPC is significant expression of ISGs (McDonough et al. 2017). Here we report that a more potent cell type specific transcriptomic response to IPC is the up-regulation of cell cycle/proliferation genes in cortical microglia. This transcriptomic feature is accompanied by a decidedly unilateral response with microglia in the ipsilateral hemicortex proliferating and increasing cellular size after IPC. In contrast, microglia in the contralateral cortex displayed ramified morphologies indicative of a surveilling physiologic state and were rarely positive for markers of proliferation.
Although there has been significant progress in recent years characterizing the microglial transcriptome in healthy mice (Butovsky et al. 2014; Grabert et al. 2016; Hickman et al. 2013) there remains a major gap in our current knowledge with respect to disease-specific transcriptional signatures of microglia (Butovsky and Weiner 2018; Crotti and Ransohoff 2016). A recent paper examining single cell RNA sequencing data in microglia demonstrated expression of canonical microglial markers is down-regulated and some microglia express cell proliferation markers in a model of demyelinating injury (Hammond et al. 2018). However, apart from our recent publication (McDonough et al. 2017) there is little or no data available on the microglial transcriptome in the setting of ischemia/reperfusion. Here we report transcriptomic changes in microglia after IPC (Tables 1–3) that have significant commonalities with data published elsewhere in models of aging (Holtman et al. 2015) and demyelination (Hammond et al. 2018). The convergence of these findings in microglia isolated from disparate CNS disease models suggests a shared microglial gene expression response to injury. The common response invokes specific pathways in microglia including activation of type I IFN signaling, induction of cellular proliferation, and down-regulation of genes associated with microglial differentiation.
Significance of microglial proliferation in preconditioning
A previous study reports that microglia proliferate after ischemic stroke with the extent of proliferation affected by stroke severity (Denes et al. 2007). Another group ablated microglial cell proliferation after tMCAO and found that elimination of proliferating microglial cells exacerbated ischemic injury resulting in increased neuronal death, infarct volume, and apoptosis (Lalancette-Hebert et al. 2007). Based on these findings it is reasonable to conclude that there is protective potential in microglia in the context of ischemia/reperfusion injury. One prior study demonstrated that ablation of cellular proliferation abrogated ischemic tolerance in mice (Maysami et al. 2008) and other work indicates that preconditioning reprograms the genomic response of the brain to ischemic injury (Stenzel-Poore et al. 2007; Stenzel-Poore et al. 2003). However, none of these studies attributed primary effects to a particular cell subtype. We recently demonstrated that the type I IFN response in microglia is: (i) specifically required for preconditioning in the optic nerve (Hamner et al. 2015), and (ii) a major gene expression response observed following preconditioning (McDonough et al. 2017). In this study we demonstrate that microglial proliferation is a key feature of IPC and that the majority of proliferative cells in the cortex are microglia (62% of Ki67+ and 77% of BrdU+ cells). Our findings provide a new cellular framework through which to interpret prior tissue-based studies. Our data also suggest that attenuation of IPC-mediated protection in animals treated with anti-proliferative agents (Maysami et al. 2008) could be attributable to effects specifically on microglia. It is further notable that the IPC-induced microglial transcriptomic response is strikingly similar to the tissue-based transcriptomic response reported during the microglial proliferative phase that follows withdrawal of microglial-depleting CSF1R antagonists (Elmore et al. 2014). It remains to be seen if CSF1R antagonism (or withdrawal) prior to a preconditioning stimulus attenuates, or in other ways modulates, IPC.
Cx3cr1 haploinsufficiency attenuates IPC-induced microglial proliferation
In WT animals we consistently observe a strong unilateral response to IPC: microglia in the ipsilateral, but not contralateral, hemisphere proliferated and exhibited amoeboid morphology. Additionally, we obtain higher counts of microglia in the ipsilateral hemicortex using stereological (Figure 3) and flow cytometric (Figure 2A and (McDonough and Weinstein 2016)) methods. However, in two different heterozygous Cx3cr1 lines – both knock-in/knock-out lines in which one allele of Cx3cr1 is replaced with a construct encoding either eGFP (Figure 6A) or an inducible Cre recombinase (Figure 6B) – this hemisphere effect was attenuated. These findings suggest that fractalkine signaling is essential in mediating the microglial proliferative response to ischemia/reperfusion and even small perturbations of protein levels, in the case of haploinsufficiency, may skew the microglial response to acute injury. The finding here that CX3CR1 is important specifically for microglial proliferation is also interesting in light of previous literature demonstrating fractalkine-induced proliferation of human microglia in culture (Hatori et al. 2002).
However, the body of literature on CX3CR1 deficiency or haploinsufficiency is conflicted. Reports suggest that Cx3cr1−/− (homozygous) mice display significantly smaller infarcts, less severe neurological deficits, and fewer microglia in the ipsilateral hemisphere compared to WT controls 72 hours following prolonged MCAO (Fumagalli et al. 2013; Tang et al. 2014). Heterozygous Cx3cr1+/− mice are widely used in the neuroinflammation field and are valued for their ability to molecularly target and/or fate map microglia (Goldmann et al. 2013; Wieghofer et al. 2015). Potential concerns about the experimentally confounding effects of Cx3cr1-haploinsufficiency on microglial function have been noted (Poniatowski et al. 2017; Wieghofer et al. 2015). However, these concerns have been alleviated to an extent by reassuring findings in selective data sets as described in the Introduction. Our findings indicate that Cx3cr1-haploinsufficiency may profoundly impact aspects of microglial physiology in the setting of ischemia/reperfusion and preconditioning. Thus, experimental models dependent on Cx3cr1-haploinsufficient mice may be poorly suited to studies of acute CNS injury. It is also noteworthy that cortical microglial density differed between Cx3cr1+/− (heterozygous) and WT mice even on the contralateral side not exposed to transient ischemia/reperfusion (Figure 6A) suggesting partial disruption of fractalkine signaling may significantly influence microglial physiology even under naïve conditions. Intriguingly, one study suggests that microglial densities are attenuated during early postnatal development in Cx3cr1-deficient animals; in a study of Cx3cr1GFP/+ and Cx3cr1KO mice it was noted that microglial counts were significantly reduced from postnatal day (P)8 to P28, but normalized by P40, in the knockouts compared to the heterozygotes, however no wild-type data was provided for comparison to determine if there was a heterozygote phenotype (Paolicelli et al. 2011). These findings should prompt cautious re-interpretation of numerous studies that have made use of Cx3cr1-haploinsufficient mice and the perturbation of microglial responses due to Cx3cr1-haploinsufficiency warrants additional study.
Type I interferon signaling and proliferation
We previously reported that type I IFN signaling in microglia is robust following IPC (McDonough et al. 2017) and required for IPC-mediated axonal protection (Hamner et al. 2015). We also observed that Csf1, a potent regulator of microglial proliferation (Kierdorf and Prinz 2013) and a known ISG (www.interferome.org), is up-regulated in microglia after IPC (McDonough et al. 2017). We thus hypothesized that type 1 IFN signaling, potentially via induction of CSF1, may be critical for IPC-induced microglial proliferation. However, our stereology data show that the IPC-induced increase in microglial proliferation seen in WT animals was similar in Ifnar1−/− mice (Figure 7A–C). Intracerebroventricular injection of IFNβ did not influence cortical microglial numbers either (Figure 7D). Based on these results we conclude that type 1 IFN signaling in microglia does not appear to contribute directly to IPC-induced microglial proliferation. However, we identified numerous other genes encoding cell cycle regulators and proliferative factors that are induced by IPC (Table 1) and, as noted above, global inhibition of proliferation attenuates IPC-mediated neuroprotection (Maysami et al. 2008). Thus, although microglial proliferation and type I IFN signaling are both necessary for IPC-induced protection the mechanism of their effects appear to be independent of one another. Recent single cell RNA sequencing studies on acutely isolated microglia have identified distinct and separate clusters of microglial sub-populations including an interferon-related cluster and a proliferation cluster (Friedman et al. 2018; Hammond et al. 2018). Thus, the independence of the interferon and proliferation effects in the context of IPC may be emanating from disparate microglial sub-populations. In view of these findings, and considering earlier reports that blocking proliferation attenuates ischemic tolerance (Maysami et al. 2008), a combinatorial approach targeting multiple pathways may have synergistic therapeutic benefits for inducing preconditioning and/or promoting regeneration and repair after ischemic stroke.
Conclusions
Our in vivo cell-type specific microarray data demonstrate that following an IPC stimulus cortical microglia express a transcriptomic profile that is dominated by up-regulation of cellular proliferation genes at the time of peak neuroprotection in the mouse brain. Consistent with this, our stereology and flow cytometry results show that microglia proliferate focally within the middle cerebral artery territory of the cortex that experienced brief, non-infarcting ischemia followed by reperfusion. Cellular proliferation is critical for ischemic tolerance (Maysami et al. 2008) and myeloid cell proliferation is required for optimal protection against ischemic injury (Lalancette-Hebert et al. 2007). Our data indicate that more than two-thirds of the proliferating cells in preconditioned cortex are microglia and suggest microglial proliferation may be a necessary component of IPC-mediated protection. Although type I IFN signaling is required for IPC-mediated protection (Hamner et al. 2015; Stevens et al. 2011), we found that genetic deletion of type 1 IFN signaling did not attenuate IPC-induced microglial proliferation in the ipsilateral hemicortex. Thus, IPC-induced microglial proliferation is not dependent on type 1 IFN signaling and these two processes may be independent of each other. In contrast, Cx3cr1-haploinsufficiency attenuated IPC-induced microglial proliferation suggesting that fractalkine signaling is a critical mediator of the microglial response to IPC. Futures studies will focus on modulating multiple microglial processes (i.e. proliferation, interferon and/or fractalkine signaling) and determining how these manipulations: (i) influence the overall neuroimmune response to IPC, and (ii) modulate IPC-mediated protection.
Main Points:
Microglia-specific transcriptomic and immunohistological data highlights a proliferative response initiated by ischemic preconditioning.
This response is dependent on the fractalkine receptor CX3CR1 and independent of type 1 interferon signaling.
Acknowledgements:
We thank Anna Conti, Thu Le and Danielle Zierath for assistance with mouse middle cerebral artery occlusion surgery; Dallas Kramer and Cheryl Sung for assistance with immunofluorescent microscopy studies; Dr. Theo Bammler and Dr. James MacDonald at the University of Washington (UW) Functional Genomics and Proteomics Core for assistance with microarrays and bioinformatics analysis; Glen MacDonald and Kimberly Miller at the UW Digital Microscopy Center for assistance with microscopy and quantitative stereology; and UW Center on Human Development and Disability (CHDD) for their support of our work. Funding sources: NIH/NINDS grants NS076620 (Weinstein), NS065008 (Weinstein), NS100245 (McDonough), NS073848 (Garden), NS096334 (Garden) and HD083091 (UW CHDD).
Footnotes
Conflict of Interest: The authors have no conflicts of interest to disclose.
References
- Alliot F, Godin I, Pessac B. 1999. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res 117:145–52. [DOI] [PubMed] [Google Scholar]
- Alliot F, Lecain E, Grima B, Pessac B. 1991. Microglial progenitors with a high proliferative potential in the embryonic and adult mouse brain. Proc Natl Acad Sci U S A 88:1541–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson GD, Farin FM, Bammler TK, Beyer RP, Swan AA, Wilkerson HW, Kantor ED, Hoane MR. 2011. The effect of progesterone dose on gene expression after traumatic brain injury. J Neurotrauma 28:1827–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anrather J, Iadecola C. 2016. Inflammation and Stroke: An Overview. Neurotherapeutics 13:661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahjat FR, Alexander West G, Kohama SG, Glynn C, Urbanski HF, Hobbs TR, Earl E, Stevens SL, Stenzel-Poore MP. 2017. Preclinical Development of a Prophylactic Neuroprotective Therapy for the Preventive Treatment of Anticipated Ischemia-Reperfusion Injury. Transl Stroke Res 8:322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettschneider J, Collin F, Bolstad BM, Speed TP. 2008. Quality Assessment for Short Oligonucleotide Microarray Data. Technometrics 50:241–264. [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE and others. 2014. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 17:131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Weiner HL. 2018. Microglial signatures and their role in health and disease. Nat Rev Neurosci 19:622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R and others. 2006. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 9:917–24. [DOI] [PubMed] [Google Scholar]
- Costello DA, Lynch MA. 2013. Toll-like receptor 3 activation modulates hippocampal network excitability, via glial production of interferon-beta. Hippocampus 23:696–707. [DOI] [PubMed] [Google Scholar]
- Crotti A, Ransohoff RM. 2016. Microglial Physiology and Pathophysiology: Insights from Genome-wide Transcriptional Profiling. Immunity 44:505–15. [DOI] [PubMed] [Google Scholar]
- Denes A, Vidyasagar R, Feng J, Narvainen J, McColl BW, Kauppinen RA, Allan SM. 2007. Proliferating resident microglia after focal cerebral ischaemia in mice. J Cereb Blood Flow Metab 27:1941–53. [DOI] [PubMed] [Google Scholar]
- Ejaz S, Emmrich JV, Sawiak SJ, Williamson DJ, Baron JC. 2015a. Cortical selective neuronal loss, impaired behavior, and normal magnetic resonance imaging in a new rat model of true transient ischemic attacks. Stroke 46:1084–92. [DOI] [PubMed] [Google Scholar]
- Ejaz S, Williamson DJ, Jensen-Kondering U, Ahmed T, Sawiak SJ, Baron JC. 2015b. What is the Optimal Duration of Middle-Cerebral Artery Occlusion Consistently Resulting in Isolated Cortical Selective Neuronal Loss in the Spontaneously Hypertensive Rat? Front Neurol 6:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL and others. 2014. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82:380–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitts DA. 2010. Improved stopping rules for the design of efficient small-sample experiments in biomedical and biobehavioral research. Behav Res Methods 42:3–22. [DOI] [PubMed] [Google Scholar]
- Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, Cao Y, Lee SH, Haddick PCG, Ngu H and others. 2018. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep 22:832–847. [DOI] [PubMed] [Google Scholar]
- Fumagalli S, Perego C, Ortolano F, De Simoni MG. 2013. CX3CR1 deficiency induces an early protective inflammatory environment in ischemic mice. Glia 61:827–42. [DOI] [PubMed] [Google Scholar]
- Garden GA, Moller T. 2006. Microglia biology in health and disease. J Neuroimmune Pharmacol 1:127–37. [DOI] [PubMed] [Google Scholar]
- Gesuete R, Packard AE, Vartanian KB, Conrad VK, Stevens SL, Bahjat FR, Yang T, Stenzel-Poore MP. 2012. Poly-ICLC preconditioning protects the blood-brain barrier against ischemic injury in vitro through type I interferon signaling. J Neurochem 123 Suppl 2:75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidday JM. 2006. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci 7:437–48. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER and others. 2010. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330:841–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann T, Wieghofer P, Muller PF, Wolf Y, Varol D, Yona S, Brendecke SM, Kierdorf K, Staszewski O, Datta M and others. 2013. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat Neurosci 16:1618–26. [DOI] [PubMed] [Google Scholar]
- Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, Freeman TC, Summers KM, McColl BW. 2016. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci 19:504–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, Walker AJ, Gergits F, Segel M, Nemesh J and others. 2018. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamner MA, Ye Z, Lee RV, Colman JR, Le T, Gong DC, Ransom BR, Weinstein JR. 2015. Ischemic Preconditioning in White Matter: Magnitude and Mechanism. J Neurosci 35:15599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. 2007. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 10:1387–94. [DOI] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA and others. 1998. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A 95:10896–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatori K, Nagai A, Heisel R, Ryu JK, Kim SU. 2002. Fractalkine and fractalkine receptors in human neurons and glial cells. J Neurosci Res 69:418–26. [DOI] [PubMed] [Google Scholar]
- He HY, Ren L, Guo T, Deng YH. 2019. Neuronal autophagy aggravates microglial inflammatory injury by downregulating CX3CL1/fractalkine after ischemic stroke. Neural Regen Res 14:280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J. 2013. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16:1896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N, Wes PD, Moller T, Orre M, Kamphuis W and others. 2015. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol Commun 3:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi M, Wakayama K, Itoh A, Kawai K, Pleasure D, Ozato K, Itoh T. 2012. Interferon regulatory factor 8/interferon consensus sequence binding protein is a critical transcription factor for the physiological phenotype of microglia. J Neuroinflammation 9:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard CV, Reed MG. 2010. Unbiased Stereology, Second Edition. Liverpool, U.K.: QTP Publications. [Google Scholar]
- Huang Y, Xu Z, Xiong S, Sun F, Qin G, Hu G, Wang J, Zhao L, Liang YX, Wu T and others. 2018. Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat Neurosci 21:530–540. [DOI] [PubMed] [Google Scholar]
- Jin WN, Shi SX, Li Z, Li M, Wood K, Gonzales RJ, Liu Q. 2017. Depletion of microglia exacerbates postischemic inflammation and brain injury. J Cereb Blood Flow Metab:271678X17694185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. 2000. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20:4106–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariko K, Weissman D, Welsh FA. 2004. Inhibition of toll-like receptor and cytokine signaling--a unifying theme in ischemic tolerance. J Cereb Blood Flow Metab 24:1288–304. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. 2011. Physiology of microglia. Physiol Rev 91:461–553. [DOI] [PubMed] [Google Scholar]
- Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, Wieghofer P, Heinrich A, Riemke P, Holscher C and others. 2013. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci 16:273–80. [DOI] [PubMed] [Google Scholar]
- Kierdorf K, Prinz M. 2013. Factors regulating microglia activation. Front Cell Neurosci 7:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AY, Todd KG. 2006. Hypoxia-activated microglial mediators of neuronal survival are differentially regulated by tetracyclines. Glia 53:809–16. [DOI] [PubMed] [Google Scholar]
- Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. 2007. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci 27:2596–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung PY, Stevens SL, Packard AE, Lessov NS, Yang T, Conrad VK, van den Dungen NN, Simon RP, Stenzel-Poore MP. 2012. Toll-like receptor 7 preconditioning induces robust neuroprotection against stroke by a novel type I interferon-mediated mechanism. Stroke 43:1383–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. 1989. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20:84–91. [DOI] [PubMed] [Google Scholar]
- Marsh B, Stevens SL, Packard AE, Gopalan B, Hunter B, Leung PY, Harrington CA, Stenzel-Poore MP. 2009. Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. J Neurosci 29:9839–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maysami S, Lan JQ, Minami M, Simon RP. 2008. Proliferating progenitor cells: a required cellular element for induction of ischemic tolerance in the brain. J Cereb Blood Flow Metab 28:1104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough A, Lee RV, Noor S, Lee C, Le T, Iorga M, Phillips JLH, Murphy S, Moller T, Weinstein JR. 2017. Ischemia/Reperfusion Induces Interferon-Stimulated Gene Expression in Microglia. J Neurosci 37:8292–8308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough A, Weinstein JR. 2016. Neuroimmune Response in Ischemic Preconditioning. Neurotherapeutics 13:748–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraga A, Pradillo JM, Garcia-Culebras A, Palma-Tortosa S, Ballesteros I, Hernandez-Jimenez M, Moro MA, Lizasoain I. 2015. Aging increases microglial proliferation, delays cell migration, and decreases cortical neurogenesis after focal cerebral ischemia. J Neuroinflammation 12:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor M, Bowen KK, Sailor KA, Dempsey RJ, Vemuganti R. 2005. Preconditioning-induced ischemic tolerance stimulates growth factor expression and neurogenesis in adult rat hippocampus. Neurochem Int 47:565–72. [DOI] [PubMed] [Google Scholar]
- Nishiyori A, Minami M, Ohtani Y, Takami S, Yamamoto J, Kawaguchi N, Kume T, Akaike A, Satoh M. 1998. Localization of fractalkine and CX3CR1 mRNAs in rat brain: does fractalkine play a role in signaling from neuron to microglia? FEBS Lett 429:167–72. [DOI] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L and others. 2011. Synaptic pruning by microglia is necessary for normal brain development. Science 333:1456–8. [DOI] [PubMed] [Google Scholar]
- Pedrono E, Durukan A, Strbian D, Marinkovic I, Shekhar S, Pitkonen M, Abo-Ramadan U, Tatlisumak T. 2010. An optimized mouse model for transient ischemic attack. J Neuropathol Exp Neurol 69:188–95. [DOI] [PubMed] [Google Scholar]
- Perry VH, Holmes C. 2014. Microglial priming in neurodegenerative disease. Nat Rev Neurol 10:217–24. [DOI] [PubMed] [Google Scholar]
- Poniatowski LA, Wojdasiewicz P, Krawczyk M, Szukiewicz D, Gasik R, Kubaszewski L, Kurkowska-Jastrzebska I. 2017. Analysis of the Role of CX3CL1 (Fractalkine) and Its Receptor CX3CR1 in Traumatic Brain and Spinal Cord Injury: Insight into Recent Advances in Actions of Neurochemokine Agents. Mol Neurobiol 54:2167–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha-Ferreira E, Phillips E, Francesch-Domenech E, Thei L, Peebles DM, Raivich G, Hristova M. 2015. The role of different strain backgrounds in bacterial endotoxin-mediated sensitization to neonatal hypoxic-ischemic brain damage. Neuroscience 311:292–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Reid LH, Jones WD, Shippy R, Warrington JA, Baker SC, Collins PJ, de Longueville F, Kawasaki ES, Lee KY and others. 2006. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol 24:1151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. 2004. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3:Article3. [DOI] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Stevens SL, King JS, Simon RP. 2007. Preconditioning reprograms the response to ischemic injury and primes the emergence of unique endogenous neuroprotective phenotypes: a speculative synthesis. Stroke 38:680–5. [DOI] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Stevens SL, Simon RP. 2004. Genomics of preconditioning. Stroke 35:2683–6. [DOI] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, Mori M, Meller R, Rosenzweig HL, Tobar E, Shaw TE and others. 2003. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet 362:1028–37. [DOI] [PubMed] [Google Scholar]
- Stevens SL, Leung PY, Vartanian KB, Gopalan B, Yang T, Simon RP, Stenzel-Poore MP. 2011. Multiple preconditioning paradigms converge on interferon regulatory factor-dependent signaling to promote tolerance to ischemic brain injury. J Neurosci 31:8456–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storkebaum E, Lambrechts D, Dewerchin M, Moreno-Murciano MP, Appelmans S, Oh H, Van Damme P, Rutten B, Man WY, De Mol M and others. 2005. Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci 8:85–92. [DOI] [PubMed] [Google Scholar]
- Su W, Hopkins S, Nesser NK, Sopher B, Silvestroni A, Ammanuel S, Jayadev S, Moller T, Weinstein J, Garden GA. 2014. The p53 transcription factor modulates microglia behavior through microRNA-dependent regulation of c-Maf. J Immunol 192:358–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalay G, Martinecz B, Lenart N, Kornyei Z, Orsolits B, Judak L, Csaszar E, Fekete R, West BL, Katona G and others. 2016. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun 7:11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Gan Y, Liu Q, Yin JX, Liu Q, Shi J, Shi FD. 2014. CX3CR1 deficiency suppresses activation and neurotoxicity of microglia/macrophage in experimental ischemic stroke. J Neuroinflammation 11:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay TL, Mai D, Dautzenberg J, Fernandez-Klett F, Lin G, Sagar Datta M, Drougard A, Stempfl T, Ardura-Fabregat A and others. 2017. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat Neurosci 20:793–803. [DOI] [PubMed] [Google Scholar]
- Venna VR, Li J, Benashski SE, Tarabishy S, McCullough LD. 2012. Preconditioning induces sustained neuroprotection by downregulation of adenosine 5’-monophosphate-activated protein kinase. Neuroscience 201:280–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein JR, Koerner IP, Moller T. 2010. Microglia in ischemic brain injury. Future Neurol 5:227–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, Wagner J, Hasler LM, Wild K, Skodras A and others. 2018. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556:332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieghofer P, Knobeloch KP, Prinz M. 2015. Genetic targeting of microglia. Glia 63:1–22. [DOI] [PubMed] [Google Scholar]
- Wolf Y, Yona S, Kim KW, Jung S. 2013. Microglia, seen from the CX3CR1 angle. Front Cell Neurosci 7:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XM, Liu Y, Qian ZM, Luo QQ, Ke Y. 2016. CX3CL1/CX3CR1 Axis Plays a Key Role in Ischemia-Induced Oligodendrocyte Injury via p38MAPK Signaling Pathway. Mol Neurobiol 53:4010–4018. [DOI] [PubMed] [Google Scholar]
- Wu Z, Irizarry R, Gentleman RC, Murillo FM, Spencer F. 2004. A Model Based Background Adjustment for Oligonucleotide Expression Arrays. Journal of the American Statistical Association 99:909–917. [Google Scholar]
- Xavier AL, Lima FR, Nedergaard M, Menezes JR. 2015. Ontogeny of CX3CR1-EGFP expressing cells unveil microglia as an integral component of the postnatal subventricular zone. Front Cell Neurosci 9:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang ZJ, Klaus JA, Koehler RC, Huang J. 2008. Delayed tolerance with repetitive transient focal ischemic preconditioning in the mouse. Stroke 39:967–74. [DOI] [PMC free article] [PubMed] [Google Scholar]