

Abstract
Biocatalysis, the application of enzymes to solve synthetic problems of human import, has blossomed into a powerful technology for chemical innovation. In the past decade, a threefold partnership, where nature provides blueprints for enzymatic catalysis, chemists introduce innovative activity modes with abiological substrates, and protein engineers develop new tools and algorithms to tune and improve enzymatic function, has unveiled the frontier of new-to-nature enzyme catalysis. In this perspective, we highlight examples of interdisciplinary studies which have helped to expand the scope of biocatalysis, including concepts of enzymatic versatility explored through the lens of biomimicry, to achieve both activities and selectivities that are not currently possible with chemocatalysis. We indicate how modern tools, such as directed evolution, computational protein design and machine learning-based protein engineering methods, have already impacted and will continue to influence enzyme engineering for new abiological transformations. A sustained collaborative effort across disciplines is anticipated to spur further advances in biocatalysis in the coming years.
Graphical Abstract
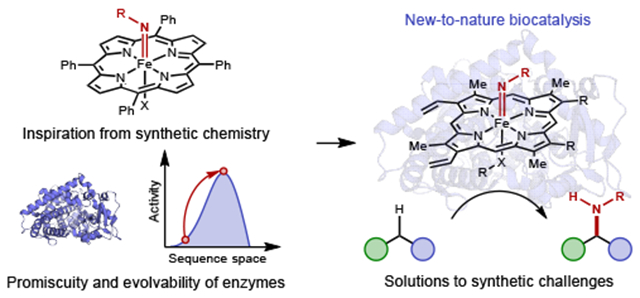
Exploration of catalysts to facilitate and refine reactivity is a central enterprise of organic chemistry. Over the past century, synthetic methods have been transformed by increasingly sophisticated catalytic systems that enable kinetic and stereoelectronic control over intermediates and reaction pathways. As a result, catalysis is used to synthesize medicines that cure once-lethal diseases, agrochemicals that allow food production to sustain a growing population, and many other essentials of modern society. However, these remarkable advances are only the latest punctuation in Earth’s chemical history. Billions of years before human chemists set out to harness the practical entirety of the periodic table, let alone formulate the rudimentary principles underlying modern chemistry, nature had already worked out a platform for executing catalysis for the processes of life in the form of amino acid-based polymers – enzymes – whose intricate and diverse three-dimensional structural space could be sampled by evolution to produce a potentially endless variety of catalysts for implementing a diverse array of chemistry.
Although common to both human and natural chemistry, catalyst development is approached differently by these parties. Enzymes are specialized macromolecules, evolved over time to catalyze specific transformations with high efficiency under defined environmental constraints. Assembled from a small set of building blocks and cofactors, they catalyze diverse processes ranging from ester hydrolysis and radical methylations to photosynthesis and nitrogen fixation. In contrast, human-developed catalysts capitalize on the entirety of the periodic table and perform myriad types of reactions, including many unknown in nature. While versatile, these catalysts are often unable to match the hallmark rates and selectivities of an enzyme. Biocatalysis is emerging as an interdisciplinary field that seeks to bridge these capabilities. Protein engineers have enabled us to steer enzymatic activities towards new functions to address synthetic problems facing chemists.1–3 By expanding the catalytic repertoire of nature through the discovery of enzymes for abiological reactions, it will be possible to exploit the robust rates and precise selectivities of enzymes for more transformations of human interest.4,5 Collaborations between chemists and protein engineers, with help from nature, have already achieved impressive successes, including establishing new chemical transformations. This Perspective highlights a few interdisciplinary investigations underlying advances in the field and identifies new directions that will accelerate the future development of enzymes for chemical synthesis.
Living systems have taught us much about synthesis through their diverse chemistry, and the study of enzyme mechanisms has been a cornerstone for developing principles and theories of chemical catalysis. Biomimetic catalysis, the development of human-made catalysts which seek to imitate life’s reactions and emulate enzymatic substrate activation, is the first of many important collaborations between synthetic chemistry and enzymatic catalysis, with origins in the 1950s.6,7 While unable to recapitulate the fast rates and unparalleled selectivities available to enzymes, biomimicry has continued to drive advances in synthetic chemistry, including the development of catalytic reactions which nature is not yet known to perform. Examples of applying the lessons of biology to catalysis include designing catalysts based on enzyme cofactors (e.g., N-heterocyclic carbene catalysis),8 repurposing elementary steps involved in biological redox processes (e.g., proton-coupled electron transfer),9 and capitalizing on supramolecular catalysis to raise effective concentrations (e.g., β-cyclodextrin-based catalysts).10 Conversely, nature has proven to be an adept learner, and abiological reactions of biomimetic catalysts have been a fruitful avenue to teach enzymes new functions.11
The remarkable manner in which biology and chemistry have mutually inspired, informed and advanced each other is apparent in a story of cytochrome P450 monooxygenases. The astonishing ability of heme-containing P450 enzymes to selectively oxygenate specific C–H bonds with molecular oxygen has captured the attention of the broader chemical community since their discovery in the 1960s. Biomimetic chemists played a critical role in elucidating the mechanism of these enzymes – pioneering studies by Groves using iron tetraphenylporphyrin complexes as synthetic model systems elucidated the P450 “radical rebound” mechanism12 – while numerous other metalloporphyrin complexes were discovered to perform oxygenation reactions.13,14 Inspired by these works and related studies for transition metal-catalyzed amination, Breslow and Gellman demonstrated in 1982 that iron tetraphenylporphyrin complexes could accept nitrene precursors and were competent to perform C–H amination reactions (Fig. 1a).15,16 They also speculated whether a cytochrome P450 itself could be induced to perform amidation in place of its normal oxygenation. Three years later, in collaboration with Svastits and Dawson, they demonstrated that rabbit liver microsomal cytochrome P450-LM3,4 could accept nitrene precursors as substrates to perform C–H amination reactions (Fig. 1b).17 Although the activity was far too low to be synthetically useful (2.2 turnovers observed for intramolecular sulfamidation), this line of inquiry provided two critical insights: enzymes could perform reactions outside of their natural purview, and chemists could use their knowledge to coax new chemistry out of nature’s catalytic machinery.
Figure 1. Biomimetic and enzymatic nitrene transfer for C-H insertion reactions.
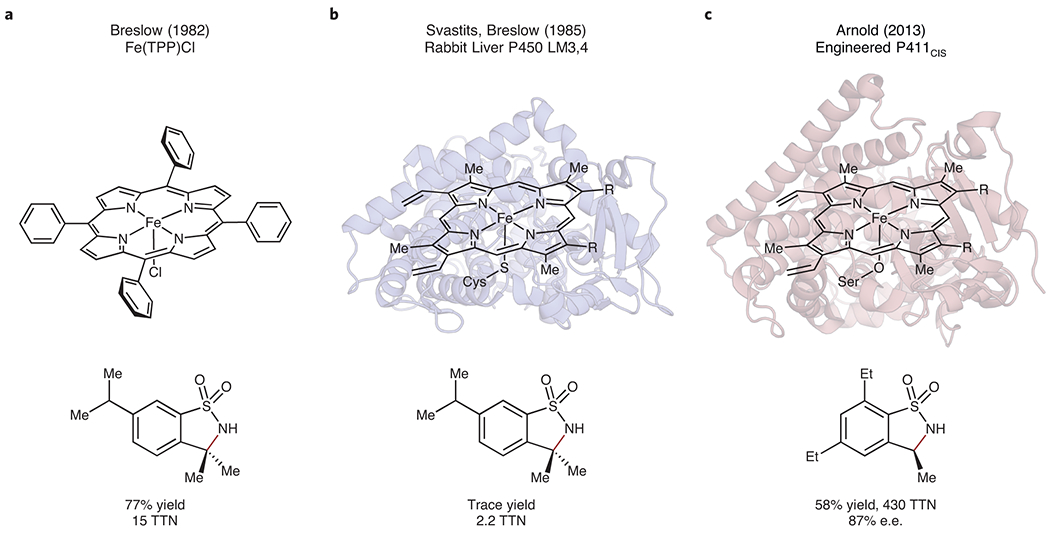
(a) Biomimetic intramolecular tosylamidation using Fe(TPP)Cl as a catalyst.15,16 (b) Enzyme-catalyzed intramolecular tosylamidation using rabbit liver P450 LM3,4 as a biocatalyst.17 (c) Engineered P411CIS-catalyzed asymmetric intramolecular tosylamidation.24 Bonds formed via nitrene C-H insertion are shown in red. e.e., enantiomeric excess; TPP, tetraphenylporphyrin; TTN, total turnover number.
The next step, elaboration of trace abiological activity into robust catalysis, would require protein engineering to train enzymes to perform new reactions with their hallmark rates and selectivities. Here, nature has as much to teach us about her catalyst engineering process, evolution, as she does about catalysis itself (vide supra). In his 1970 classic, ‘Natural Selection and the Concept of a Protein Space’, John Maynard Smith provided a theoretical foundation for how protein evolution by natural selection is operationally possible in a sequence space that is both vast and mostly devoid of functional sequences.18 He argued that for evolution to occur, among the set of possible single mutants of a functional protein there must exist at least one daughter variant which is also functional (in practice, many such variants exist). The same condition applies to the single-mutant neighborhood of the daughter variant, and so on. The inductive logic implies that the sequence space of functional proteins forms a connected network that can be accessed through single amino acid mutations. Advances in screening technology and genetic engineering, notably error-prone polymerase chain reaction (PCR),19 in the 1980s allowed ‘directed evolution’ to emerge as a tool for engineering proteins: Maynard Smith’s conjecture that natural evolution required a minimum density of functional proteins within a single mutational step was used by Arnold, who showed that enzymes could be evolved towards a user-defined goal by accumulating mostly single amino acid changes in a random uphill walk.20 Ever since the first demonstrations in the early 1990s, directed evolution has been a reliable strategy to engineer enzymes with improved properties such as thermostability, the ability to tolerate organic cosolvents, catalytic efficiency, increased substrate tolerance, and many others.21,22
But what about new reactivities? It was not immediately clear how such a conservative process could generate enzymes that catalyze new reactions. Here the understanding that enzymes selected by nature for specific transformations may nonetheless display other activities, perhaps following mechanistically related pathways, played an important role. Appreciation of this ‘catalytic promiscuity’ is often lost in the perception of enzymes as being highly specific for single transformations.23 While an enzyme’s biological function may be a product of natural selection, its promiscuous activities—unless deleterious to the organism—are unrestrained by selective pressures. Indeed, if conditions arise when a promiscuous activity becomes beneficial, natural selection might produce a new enzyme with that function as its mainstay. In fact, new enzymes appear, and they can do so in short time spans, for example, when a new food source or the ability to degrade a new antibiotic or pesticide provides an opportunity to outpace neighbors. This insight into how enzymes have evolved and diversified raised the tantalizing possibility that human-imposed conditions, unnatural reactants, or artificial protein modifications which are either inaccessible or irrelevant in nature might reveal promiscuous activities never seen in the natural world (just as Breslow and coworkers discovered). Provided an enzyme capable of performing a desired new-to-nature transformation can be identified, it should be possible to direct its evolution, elaborating low-yielding, promiscuous enzymatic activity into robust and efficient biocatalysis.
Because evolution on a human-friendly timescale can be applied to trace abiological activities, nearly three decades after Breslow and Gellman’s original inquiries into enzymatic nitrene transfer, engineered enzymes now perform highly stereoselective C–H amination reactions with hundreds to thousands of turnovers (Fig. 1c).24–27 The mechanistic analogy between biomimetic iron-porphyrin catalysts and hemoproteins led to the discovery of other non-natural reactivities supported by these proteins, such as carbene transfer, and even hemoproteins with no natural enzyme functions (for example, globins and cytochromes c) have been engineered for abiological carbene-transfer activity (Fig. 2a).28,29 Reactions discovered through this manifold include some not observed with chemocatalytic approaches, such as the formation of bicyclobutanes from alkynes.30,31
Figure 2: Representative examples of cofactor adaptation for abiological reactions with new-to-nature reactivity modes.
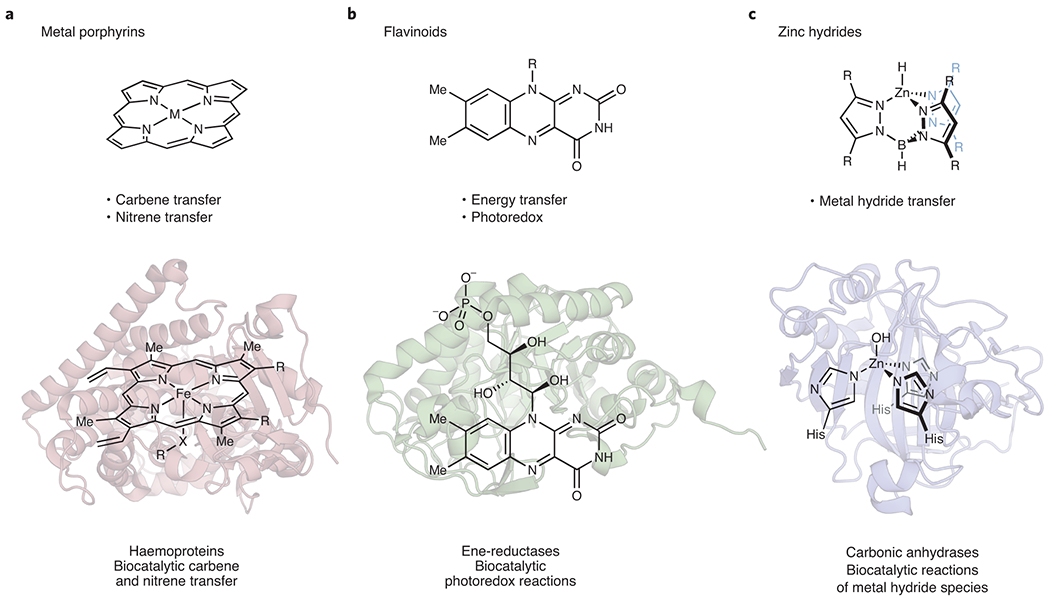
(a) The chemistry of metal porphyrins has been extended to hemoproteins for both carbene and nitrene transfer when presented with carbene and nitrene precursors.24–31 (b) Visible-light stimuli allows for flavoproteins to engage in biocatalytic photoredox transformations of alkyl halides.32–34 (c) Carbonic anhydrase uses silanes to generate an active zinc hydride intermediate for new reactions.38 His, histidine.
Imaginative chemists have now started to discover other promiscuous activities a protein may support when provided with non-natural reactants, external stimuli, or other abiological components. In addition to hemoprotein-catalyzed carbene and nitrene transfer discussed above, two other notable examples illustrate this creative exercise (Fig. 2). The wide class of flavin-dependent proteins encompasses enzymes that perform myriad redox chemistry. Drawing upon the rich literature of flavin photochemistry, Hyster demonstrated that flavin-dependent enzymes can effect a range of new-to-nature photoredox radical transformations within the active site (Figure 2b).32–34 Visible light excitation of the flavin cofactor allows radical generation from an appropriate precursor (typically, alkyl halides) for subsequent radical reactions, in effect converting an enzyme family known for two-electron reduction into biocatalysts for single-electron transfer reactivity. Stereocontrol imposed by the active site allows asymmetric radical transformations, including asymmetric hydrogen atom transfer – a longstanding challenge in synthetic chemistry.35 Most recently, Zhao and Hyster explored the mechanistic pliability of these enzymes, demonstrating their ability to perform asymmetric hydroalkylation of styrenes and other olefin acceptors.36,37 Hartwig has provided another example of how startling promiscuous activities can lurk within the active sites of enzymes performing even the simplest reactions. Carbonic anhydrase catalyzes the addition of water to carbon dioxide to generate carbonic acid via an active site, zinc-hydroxide intermediate. Hartwig and coworkers demonstrated that treating wild-type carbonic anhydrase with a silane can lead to an analogous zinc-hydride intermediate (Figure 2c).38 Although the generation of zinc-hydrides from silanes and zinc salts in anhydrous conditions is known,39,40 the formation of such a strongly hydridic species in an enzyme active site is striking. The intermediate could be harnessed for stereoselective reduction of aryl-methyl ketones to secondary carbinols. High activity was often observed in the above examples with just the wild-type (un-mutated) enzymes. Further improvement in enzyme parameters and performance, as well as substrate scope, is possible with directed evolution.41 The inherent mechanistic versatility of enzymes combined with protein engineering provides immense potential for discovering new biocatalysts.
Several reports of new-to-nature biocatalytic transformations highlight the potential of this collaborative effort among nature, synthetic chemistry, and protein engineering to overcome longstanding synthetic challenges (Figure 3). While the development of enzymatic cyclopropanation started with styrenyl alkenes as substrates, directed evolution has since expanded the scope to unactivated olefins. Notably, catalyst-controlled stereodivergent synthesis of all four diastereomeric cyclopropanation products could be achieved (Fig. 3a).28 An engineered cytochrome P450 that forms bicyclobutane products through two sequential carbene addition reactions across alkyne substrates demonstrates the capacity of enzymes to access highly strained carbocyclic cores with excellent efficiency (Figure 3b).31 Other engineered P450 enzymes catalyze unprecedented asymmetric nitrene C–H insertion reactions to access primary amines at allylic and benzylic positions (Fig. 3c).26 In this system, amination is believed to proceed through an “unprotected” iron-nitrenoid active site intermediate which can be considered as the nitrogen analogue of Compound I. In Hyster’s hydroalkylation reaction of α-acyl radicals and olefins (Fig. 3d), substrate activation in the active site is achieved through a rare quaternary charge-transfer complex between the radical precursor, olefin, flavin cofactor and protein scaffold, which enables stereoinduction over a challenging hydrogen-atom transfer step.42 These results are corroborated by mechanistic work performed by Zhao on a similar enzymatic hydroacylation reaction.36 Finally, Lewis and colleagues reported the enantioselective intramolecular halocyclization of carboxylic acids onto pendant olefins to form chiral, halogenated γ-lactones (Fig. 3e).43 The enzyme active site prevents racemization of chiral bromonium ion intermediates by limiting halenium ion transfer between olefin substrates, contributing to excellent stereoinduction.
Figure 3: Select examples of new-to-nature enzyme catalysis.
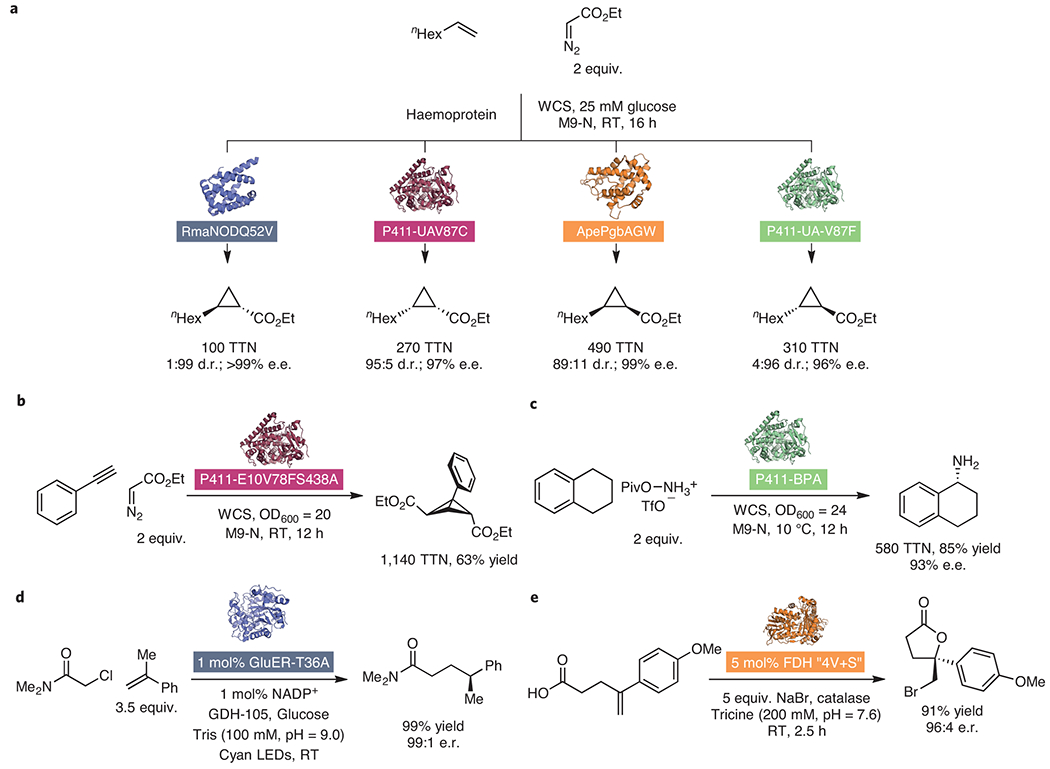
(a) Hemoprotein-catalyzed, stereodivergent cyclopropanation of unactivated olefin substrates.28 (b) P411-catalyzed bicyclobutane formation through sequential carbene additions across alkynes.31 (c) Asymmetric primary animation of benzylic C–H bonds catalyzed by P411s.26 (d) Biocatalytic, asymmetric hydroalkylation of styrenes via photochemical activation of flavins.42 (e) Asymmetric halolactonization catalyzed by halogenase enzymes.43 GDH, glucose dehydrogenase; d.r., diastereomeric ratio; e.e., enantiomeric excess; LEDs, light-emitting diodes; OD, optical density; Piv, pivaloyl; RT, room temperature; Tf, trifluoromethanesulfonyl; TTN, total turnover number; WCS, whole cell suspension.
The extraordinary new chemistry exhibited in these systems is amplified by the amenability of enzymes to operation in sequential reactions which generate tremendous molecular complexity in a one-pot operation, just as they do inside a living cell. Recent examples of biocatalytic cascade processes include Merck’s nine-enzyme, three-step route for the synthesis of the investigational HIV treatment islatravir44 and Deska’s one-pot total synthesis of angiopterlactone B using a five-enzyme cascade.45 Furthermore, new-to-nature transformations can be used to develop artificial pathways for synthetic metabolism in vivo.46 The ability to perform exquisite chemistry in the context of both in vitro and in vivo cascades demonstrates the unique synthetic opportunities available to biocatalysis, and suggests that the discovery of new-to-nature reactions could serve as versatile modules for “plug-and-play” synthetic cascades. In most of these examples, new activity is recognized using solely the genetically-encoded enzymatic machinery. The growing field of artificial metalloenzymes, where new transition metal cofactors are developed in order to imbue enzymes with novel activities,47 promises to further expand the reaction space of biocatalysts once robust methods for their assembly and directed evolution are developed.
Advances in our ability to engineer and design proteins have, concurrently with the chemical élan discussed above, facilitated the discovery of new abiological chemistry. Computational modeling has been a useful tool towards this end, and protein modeling, with the goal of designing new enzymes, has received considerable attention over the past two decades.48 In 2008, Houk and Baker computationally designed an enzyme that catalyzed an abiological Kemp elimination, and Tawfik evolved the minimally performing design to exhibit improved activity.49 Since this report, other enzymes have been computationally designed for reactions with limited representation in the natural world, including Diels-Alderases50 and Morita-Bayliss-Hillmanases.51 Initial activities from de novo enzymes generally have been very low, reflecting our limited understanding of the problem, but directed evolution can rescue the poor designs to achieve rates and selectivities closer to those of natural enzymes.52 Computational design can also guide the expansion of known enzymatic functions. For example, as part of their campaign to develop a biocatalytic process route for the synthesis of sitagliptin, researchers at Merck performed a computational redesign of a transaminase active site to accept the prositagliptin ketone, providing a critical starting point for a directed evolution campaign.53 The wealth of methods to reliably model proteins for both de novo generation of new enzymes and redesigning extant ones are invaluable for biocatalysis.
Moving forward, new tools for protein engineering which leverage computational protein design, next-generation sequencing and machine learning will accelerate our ability to engineer enzymes with new activities. Directed evolution, agnostic by nature, frequently improves enzyme performance by uncovering subtle mutations far away from the active site, which are non-intuitive for chemists and protein engineers. However, the effects of such changes may be predictable by computer algorithms that learn from data. Machine learning tools have enabled protein structure to be predicted from a sequence with much greater reliability; correlating enzyme sequences to their functions is the next big challenge. Nascent work in this field has demonstrated that machine-learning directed evolution (MLDE) can achieve higher-fitness variants more rapidly than the ‘greedy’ optimization strategies currently employed, reducing the experimental effort needed to achieve a highly functional biocatalyst.54 These approaches can help develop enzymes with complementary selectivities: MLDE guided the divergent evolution of a single enzyme for Si–H insertion to produce two biocatalysts with complementary enantioselectivities, enabling access to both antipodes of the desired product.55 The capacity to leverage MLDE to engineer enzymes for complementary stereoinduction in parallel could expedite the development of biocatalysts to access any desired stereoisomer. Such examples are only the beginning of what these technologies can do for biocatalysis: the exploration of machine learning to predict enzyme performance for reactions on non-natural substrates has already started,56 and we expect it to play a role in biocatalytic reaction discovery sooner rather than later. The acceleration of directed evolution, combined with the ability to optimize enzymes with complementary selectivities in parallel, dramatically amplifies the impact of discovering new-to-nature biocatalytic transformations.
In conclusion, biocatalysis is a sustained and fruitful collaboration among synthetic chemists, protein engineers, and nature. Just as nature has taught us chemistry and provided us with the blueprint to improve her catalysts systematically, her machinery has proven highly adaptable to acquiring new chemistry when presented with the right environment. The extent of new chemistry that enzymes can perform remains an open question. We believe, however, that scientists have just scratched the surface of chemical transformations that enzymes can achieve, and anticipate that the boundless surprises from biology, the increasing power and sophistication of protein engineering techniques, and continued chemical ingenuity will continue to power innovations in biocatalysis.
Acknowledgments
This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe on privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of the authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof.
D.C.M. was supported by a Ruth Kirschstein NIH Postdoctoral Fellowship (F32GM128247). This work was sponsored by the US Army Research Office and accomplished under cooperative agreement W911NF-19-2-0026 for the Institute of Collaborative Biotechnologies. This material is based upon work sponsored by the U.S. Department of Energy, Office of Basic Energy Sciences, under Award Number DE-SC0021141. The authors wish to acknowledge Dr. Edwin Alfonzo, Dr. Runze Mao and Kadina E. Johnston for helpful discussions. All protein structures were prepared with PyMOL (The PyMOL Molecular Graphics System, v.2.0 Schrödinger, LLC).
Footnotes
Competing Interests
The authors declare no competing interests.
References
- 1.Sun H, Zhang H, Ang EL & Zhao H Biocatalysis for the synthesis of pharmaceuticals and pharmaceutical intermediates. Bioorg. Med. Chem 26, 1275–1284 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Sheldon RA, Brady D & Bode ML The Hitchhiker’s guide to biocatalysis: recent advances in the use of enzymes in organic synthesis. Chem. Sci 11, 2587–2605 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Winkler CK, Schrittwieser JH & Kroutil W Power of biocatalysis for organic synthesis. ACS Cent. Sci 7, 55–71 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Renata H, Wang ZJ & Arnold FH Expanding the enzyme universe: accessing non-natural reactions by mechanism-guided directed evolution. Angew. Chem. Int. Ed 54, 3351–3367 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen K & Arnold FH Engineering new catalytic activities in enzymes. Nat Catal. 3, 203–213 (2020). [Google Scholar]
- 6.Breslow R Biomimetic chemistry: Biology as an Inspiration. J. Biol. Chem 284, 1337–1342 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Swiegers GF (ed.) Bioinspiration and biomimicry in chemistry: reverse-engineering nature. (Wiley, 2012). [Google Scholar]
- 8.Flanigan DM, Romanov-Michailidis F, White NA & Rovis T Organocatalytic reactions enabled by N-heterocyclic carbenes. Chem. Rev 115, 9307–9387 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller DC, Tarantino KT & Knowles RR Proton-coupled electron transfer in organic synthesis: fundamentals, applications, and opportunities. Top. Curr. Chem 374, 30 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Breslow R & Dong SD Biomimetic reactions catalyzed by cyclodextrins and their derivatives. Chem. Rev 98, 1997–2012 (1998). [DOI] [PubMed] [Google Scholar]
- 11.Prier CK & Arnold FH Chemomimetic biocatalysis: exploiting the synthetic potential of cofactor-dependent enzymes to create new catalysts. J. Am. Chem. Soc 137, 13992–14006 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Huang X & Groves JT Beyond ferryl-mediated hydroxylation: 40 years of the rebound mechanism and C–H activation. J. Biol. Inorg. Chem 22, 185–207 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kadish KM, Smith KM & Guilard R Bioinorganic and bioorganic chemistry. (Academic Press, 2003). [Google Scholar]
- 14.Mansuy D A brief history of the contribution of metalloporphyrin models to cytochrome P450 chemistry and oxidation catalysis. Comptes Rendus Chimie 10, 392–413 (2007). [Google Scholar]
- 15.Breslow R & Gellman SH Tosylamidation of cyclohexane by a cytochrome P-450 model. J. Chem. Soc., Chem. Commun 1400–1401 (1982). [Google Scholar]
- 16.Breslow R & Gellman SH Intramolecular nitrene carbon-hydrogen insertions mediated by transition-metal complexes as nitrogen analogs of cytochrome P-450 reactions. J. Am. Chem. Soc 105, 6728–6729 (1983). [Google Scholar]
- 17.Svastits EW, Dawson JH, Breslow R & Gellman SH Functionalized nitrogen atom transfer catalyzed by cytochrome P-450. J. Am. Chem. Soc 107, 6427–6428 (1985). [Google Scholar]
- 18.Maynard Smith J Natural selection and the concept of a protein space. Nature 225, 563–564 (1970). [DOI] [PubMed] [Google Scholar]
- 19.McCullum EO, Williams BAR, Zhang J & Chaput JC Random mutagenesis by error-prone PCR. in In Vitro Mutagenesis Protocols (ed. Braman J) vol. 634 103–109 (Humana Press, 2010). [DOI] [PubMed] [Google Scholar]
- 20.Arnold FH Innovation by evolution: bringing new chemistry to life (Nobel Lecture). Angew. Chem. Int. Ed 58, 14420–14426 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Kuchner O & Arnold FH Directed evolution of enzyme catalysts. Trends in Biotechnology 15, 523–530 (1997). [DOI] [PubMed] [Google Scholar]
- 22.Zeymer C & Hilvert D Directed evolution of protein catalysts. Annu. Rev. Biochem 87, 131–157 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Khersonsky O & Tawfik DS Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu. Rev. Biochem 79, 471–505 (2010). [DOI] [PubMed] [Google Scholar]
- 24.McIntosh JA et al. Enantioselective intramolecular C-H amination catalyzed by engineered cytochrome P450 enzymes in vitro and in vivo. Angew. Chem. Int. Ed 52, 9309–9312 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steck V, Kolev JN, Ren X & Fasan R Mechanism-guided design and discovery of efficient cytochrome P450-derived C-H amination biocatalysts. J. Am. Chem. Soc 142, 10343–10357 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jia Z-J, Gao S & Arnold FH Enzymatic primary amination of benzylic and allylic C(sp3)–H bonds. J. Am. Chem. Soc 142, 10279–10283 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Athavale S et al. Biocatalytic, intermolecular C- H bond functionalization for the synthesis of enantioenriched amides. Angew. Chem. Int. Ed anie.202110873 (2021) doi: 10.1002/anie.202110873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knight AM et al. Diverse engineered heme proteins enable stereodivergent cyclopropanation of unactivated alkenes. ACS Cent. Sci 4, 372–377 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bordeaux M, Tyagi V & Fasan R Highly diastereoselective and enantioselective olefin cyclopropanation using engineered myoglobin-based catalysts. Angew. Chem. Int. Ed 54, 1744–1748 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Y & Arnold FH Navigating the unnatural reaction space: directed evolution of heme proteins for selective carbene and nitrene transfer. Acc. Chem. Res 54, 1209–1225 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen K, Huang X, Kan SBJ, Zhang RK & Arnold FH Enzymatic construction of highly strained carbocycles. Science 360, 71–75 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emmanuel MA, Greenberg NR, Oblinsky DG & Hyster TK Accessing non-natural reactivity by irradiating nicotinamide-dependent enzymes with light. Nature 540, 414–417 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Biegasiewicz KF et al. Photoexcitation of flavoenzymes enables a stereoselective radical cyclization. Science 364, 1166–1169 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grosheva D & Hyster TK Light- driven flavin- based biocatalysis. in Flavin- Based Catalysis (eds. Cibulka R. & Fraaije M) 291–313 (Wiley, 2021). doi: 10.1002/9783527830138.ch12. [DOI] [Google Scholar]
- 35.Sandoval BA, Meichan AJ & Hyster TK Enantioselective hydrogen atom transfer: discovery of catalytic promiscuity in flavin-dependent ‘ene’-reductases. J. Am. Chem. Soc 139, 11313–11316 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Huang X et al. Photoenzymatic enantioselective intermolecular radical hydroalkylation. Nature 584, 69–74 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Fu H et al. Ground-state electron transfer as an initiation mechanism for biocatalytic C–C bond forming reactions. J. Am. Chem. Soc 143, 9622–9629 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ji P, Park J, Gu Y, Clark DS & Hartwig JF Abiotic reduction of ketones with silanes catalysed by carbonic anhydrase through an enzymatic zinc hydride. Nat. Chem 13, 312–318 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mukherjee D, Ellern A & Sadow AD Conversion of a zinc disilazide to a zinc hydride mediated by LiCl. J. Am. Chem. Soc 132, 7582–7583 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Sattler W & Parkin G Zinc catalysts for on-demand hydrogen generation and carbon dioxide functionalization. J. Am. Chem. Soc 134, 17462–17465 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Gao X, Turek-Herman J, Choi YJ, Cohen R & Hyster T Photoenzymatic synthesis of α-tertiary amines by engineered flavin-dependent ‘ene’-reductases. https://chemrxiv.org/engage/chemrxiv/article-details/61429406b817b40e781e701c (2021) doi: 10.33774/chemrxiv-2021-955hf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Page CG et al. Quaternary charge-transfer complex enables photoenzymatic intermolecular hydroalkylation of olefins. J. Am. Chem. Soc 143, 97–102 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mondal D, Fisher BF, Jiang Y & Lewis JC Flavin-dependent halogenases catalyze enantioselective olefin halocyclization. Nat Commun 12, 3268 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huffman MA et al. Design of an in vitro biocatalytic cascade for the manufacture of islatravir. Science 366, 1255–1259 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Kiefer A, Liu Y-C, Gummerer R, Jäger C & Deska J A fully biocatalytic approach to angiopterlactone b based on a chemoinspired artificial in vitro metabolism. https://chemrxiv.org/articles/preprint/A_Fully_Biocatalytic_Approach_to_Angiopterlactone_B_Based_on_a_Chemoinspired_Artificial_in_Vitro_Metabolism/14679738/1 (2021) doi: 10.26434/chemrxiv.14679738.v1. [DOI] [Google Scholar]
- 46.Cai T et al. Cell-free chemoenzymatic starch synthesis from carbon dioxide. Science 373, 1523–1527 (2021). [DOI] [PubMed] [Google Scholar]
- 47.Schwizer F et al. Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev 118, 142–231 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Huang P-S, Boyken SE & Baker D The coming of age of de novo protein design. Nature 537, 320–327 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Röthlisberger D et al. Kemp elimination catalysts by computational enzyme design. Nature 453, 190–195 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Siegel JB et al. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science 329, 309–313 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bjelic S et al. Computational design of enone-binding proteins with catalytic activity for the Morita—Baylis—Hillman reaction. ACS Chem. Biol 8, 749–757 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blomberg R et al. Precision is essential for efficient catalysis in an evolved Kemp eliminase. Nature 503,418–421 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Savile CK et al. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science 329, 305–309 (2010). [DOI] [PubMed] [Google Scholar]
- 54.Wittmann BJ, Yue Y & Arnold FH Informed training set design enables efficient machine learning-assisted directed protein evolution. Cell Systems S2405471221002866 (2021) doi: 10.1016/j.cels.2021.07.008. [DOI] [PubMed] [Google Scholar]
- 55.Wu Z, Kan SBJ, Lewis RD, Wittmann BJ & Arnold FH Machine learning-assisted directed protein evolution with combinatorial libraries. Proc. Natl. Acad. Sci 116, 8852–8858 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goldman S, Das R, Yang KK & Coley CW Machine learning modeling of family wide enzyme-substrate specificity screens. arXiv:2109.03900 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]