

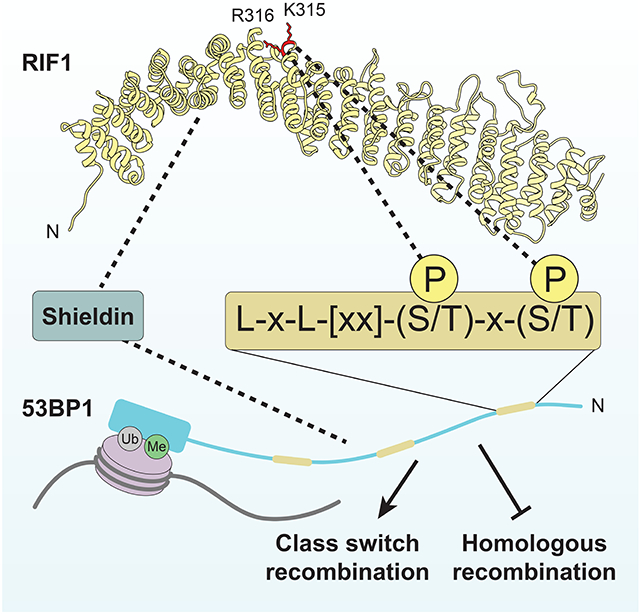
Summary
The chromatin-binding protein 53BP1 promotes DNA repair by orchestrating the recruitment of downstream effectors including PTIP, RIF1 and shieldin to DNA double-strand break sites. While we know how PTIP recognizes 53BP1, the molecular details of RIF1 recruitment to DNA damage sites remains undefined. Here, we report that RIF1 is a phosphopeptide-binding protein that directly interacts with three phosphorylated 53BP1 epitopes. The RIF1-binding sites on 53BP1 share an essential LxL motif followed by two closely apposed phosphorylated residues. Simultaneous mutation of these sites on 53BP1 abrogates RIF1 accumulation into ionizing radiation-induced foci, but surprisingly only fully compromises 53BP1-dependent DNA repair when an alternative mode of shieldin recruitment to DNA damage sites is also disabled. Intriguingly, this alternative mode of recruitment still depends on RIF1 but does not require its interaction with 53BP1. RIF1 therefore employs phosphopeptide recognition to promote DNA repair but also modifies shieldin action independently of 53BP1 binding.
Graphical Abstract
eTOC Blurb
53BP1 promotes DNA repair in part by recruiting RIF1 to DNA break sites. Setiaputra et al. show that RIF1 binds to 53BP1 by recognizing three related phosphorylated epitopes on 53BP1. The 53BP1-RIF1 interaction contributes to DNA repair in parallel to a second mode of 53BP1-dependent shieldin recruitment to DNA lesions.
Introduction
RIF1 (Rap1-interacting factor 1) is an evolutionarily conserved eukaryotic genome maintenance protein (Fontana et al., 2018). In vertebrates, RIF1 regulates the timing of DNA replication initiation, resolution of ultrafine bridges in mitosis and is a key mediator of 53BP1-dependent DNA double-strand break (DSB) repair (Fontana et al., 2018; Hengeveld et al., 2015). To regulate DNA replication, RIF1 recruits the PP1 protein phosphatase to replication origins where PP1 antagonizes the phosphorylation, and activation, of replicative helicase components such as MCM4 (Hiraga et al., 2014; Hiraga et al., 2017). PP1 binds linear RIF1 sequence motifs (RVxF/SILK) present at the N- and C-terminal regions of RIF1, but how this complex is recruited to origins has not yet been elucidated (Bollen et al., 2010; Hiraga et al., 2014). During DSB repair RIF1 similarly bridges the shieldin complex to 53BP1-bound chromatin at DSB sites (Dev et al., 2018; Findlay et al., 2018; Gao et al., 2018; Ghezraoui et al., 2018; Gupta et al., 2018; Mirman et al., 2018; Noordermeer et al., 2018; Setiaputra and Durocher, 2019). The RIF1-shieldin module shapes chromatin architecture at DSB sites (Ochs et al., 2019) and protects DNA ends from nucleolytic degradation (Callen et al., 2020; Chapman et al., 2013; Escribano-Diaz et al., 2013; Zimmermann et al., 2013). The molecular details by which RIF1 interacts with both 53BP1 and shieldin remain largely unknown. For example, it is not clear whether RIF1 binds directly to 53BP1 or via an as-yet unidentified partner, nor is it established whether this reported interaction is essential for DNA repair, even if it is widely assumed to be.
Human RIF1 is a 2472-residue protein with a highly structured N-terminal domain (NTD; residues 1-967) consisting mainly of α-helical HEAT repeats (Buonomo et al., 2009; Fontana et al., 2018; Xu and Blackburn, 2004). The RIF1 NTD is well conserved among eukaryotes, with the structure of the budding yeast Rif1 NTD being recently determined to reveal that it forms an elongated “shepherd’s crook” structure (Mattarocci et al., 2017). The C-terminal half of RIF1 is predicted to be largely unstructured with the exception of a multipartite C-terminal domain (CTD) at the extreme C-terminus consisting of a PP1-binding RVxF/SILK motif, a DNA-binding domain and an interaction site for the BLM helicase (Xu et al., 2010). The NTD is essential and sufficient for recruitment of RIF1 to DSB sites (Escribano-Diaz et al., 2013), indicating that this region is involved in 53BP1 binding.
53BP1 is composed of three main functional regions: an unstructured and highly phosphorylated N-terminus, a focus-forming region (FFR) that binds methylated and ubiquitinated chromatin, and a C-terminal tandem BRCT domains dispensable for DNA repair but involved in p53 regulation (Bothmer et al., 2011; Botuyan et al., 2006; Cuella-Martin et al., 2016; Fradet-Turcotte et al., 2013; Mirman and de Lange, 2020; Panier and Boulton, 2014). The 53BP1 N-terminus is rich in Ser/Thr-Gln (S/T-Q) sites, many of which are phosphorylated by ATM in response to DNA damage (Anderson et al., 2001; Jowsey et al., 2007; Rappold et al., 2001). Alanine substitutions of 28 S/T-Q sites (53BP128A) inactivates all known functions of 53BP1 in DNA repair such as the promotion of non-homologous end-joining (NHEJ) during immunoglobulin class switching, dysfunctional telomere fusion, and the formation of chromosome aberrations in BRCA1-deficient cells following poly(ADP-ribose) polymerase (PARP) inhibition (Bothmer et al., 2011; Dimitrova et al., 2008; Manis et al., 2004). 53BP1 also inhibits homologous recombination (HR) by suppressing the loading of RAD51 onto single-stranded DNA (ssDNA) in BRCA1-mutated cells (Bothmer et al., 2011; Callen et al., 2020). The 53BP1 S/T-Q sites promote at a minimum two key protein interactions that are essential for 53BP1 activity. First, phosphorylation of 53BP1 on Ser25 is directly recognized by PTIP via its tandem BRCT domains (Munoz et al., 2007), which is a known phosphopeptide-binding module (Manke et al., 2003; Yu et al., 2003), and second, through the phosphorylation of one or more ATM sites other than Ser25 that promote RIF1 binding and recruitment (Callen et al., 2013; Chapman et al., 2013; Di Virgilio et al., 2013; Escribano-Diaz et al., 2013; Zimmermann et al., 2013).
In this study, we sought to determine the basis of the RIF1-53BP1 interaction of RIF1 with 53BP1. We report that RIF1 is a phosphopeptide-binding protein that directly binds to three distinct phosphorylated epitopes on 53BP1. The combined mutation of these three motifs in 53BP1 abrogates RIF1 accumulation at DSB sites but results in a protein that retains DNA repair activity. The residual activity of this RIF1-binding mutant of 53BP1 was due to a hitherto unsuspected ability of 53BP1 to recruit shieldin to damaged chromatin in a manner that requires RIF1, but not the RIF1-53BP1 interaction per se. Combined inactivation of the RIF1 binding-dependent and - independent modes of shieldin recruitment abrogated RIF1- and 53BP1-dependent DNA repair to the same extent as the 53BP128A mutation. We therefore conclude that RIF1 is a phosphopeptide-binding protein, and that this phospho-recognition activity acts in parallel to a second mode of shieldin recruitment to mediate 53BP1 function.
Results
53BP1 recruits RIF1 through multiple sites in its N-terminus
To identify the phosphorylation sites responsible for the 53BP1-RIF1 interaction, we took advantage of the observation that the combined mutation of the 28 S/T-Q phosphorylation sites to alanine in the N-terminal region of 53BP1 (53BP128A) abolishes recruitment of RIF1 to DSBs (Bothmer et al., 2011; Chapman et al., 2013; Escribano-Diaz et al., 2013; Zimmermann et al., 2013). We reasoned that reverting individual phosphorylation sites from 53BP128A to their corresponding serine or threonine might identify critical residues responsible for the 53BP1-RIF1 interaction. We assessed RIF1 recruitment into ionizing radiation (IR)-induced foci in human U-2 OS (U2OS) cells that were co-transfected with 53BP1-targeting siRNAs and vectors expressing siRNA-resistant mRNA coding for 53BP1 or single-site Ala-to-Ser/Thr revertants derived from 53BP128A (Figure S1A-B). No single phosphoresidue was sufficient to restore RIF1 recruitment to DNA damage sites, indicating that RIF1 recruitment is dependent on more than one phosphorylation event (Figure S1A-B). As an alternative strategy, we searched for regions of the 53BP1 N-terminus that were sufficient to mediate recruitment of RIF1 to DSB sites when fused to the 53BP1 focus-forming region (FFR; residues 1220-1711) (Fradet-Turcotte et al., 2013) (Figure 1A-B). Three distinct 53BP1 segments could support RIF1 IRIF formation, consisting of residues 100-200, 400-550, and 650-800 (Figures 1A-B and S1C). In particular, the 53BP1 [100-200]-FFR fusion restored RIF1 IR-induced foci to wild-type levels, an activity which we further narrowed down to 53BP1 residues 145-182 (Figure 1A-B and S1C). Mutating all S/T-Q sites within the 53BP1 [100-200]-FFR construct ([100-200]-FFR 4A) completely abolished RIF1 accumulation at DSB sites (Figures 1C and S1D-E).
Figure 1. RIF1 directly binds an ATM-phosphorylated motif in the 53BP1 N-terminus.

(A) RIF1 IR-induced focus formation in U2OS cells depleted of 53BP1 by siRNA treatment and transfected with the indicated GFP-fusion constructs (FFR; 53BP1 residues 1220-1711). Cells were fixed 1 h post-IR (10 Gy) and evaluated by immunofluorescence. Representative micrographs are shown. Dashed lines indicate the nuclear area determined by DAPI staining (not shown).
(B) Quantitation of (A). Bar height corresponds to the mean ± s.d.; 53BP1 FL; N=7. 650-800; N=6. 1000-1220, 750-1050; N=4. 145-182; N=3, all else N=5.
(C) RIF1 focus formation in cells expressing the indicated GFP-[100-200]-FFR variants 1 h post-IR (10 Gy) was evaluated by immunofluorescence. Bar height corresponds to the mean ± s.d.; WT, 4A, A176S/A178S; N=3, all else N=4. Representative micrographs are shown in Figure S1D.
(D) RIF1 focus formation in cells expressing the indicated variants 1 h post-IR (10 Gy) was evaluated by immunofluorescence. Bar height corresponds to the mean ± s.d.; WT, S166A; N=5. S176A, S178A; N=3. All else N=4. Representative micrographs are shown in Figure S1G.
(E) Whole cell lysates of U2OS cells expressing the indicated GFP-[100-200]-FFR 53BP1 variants treated with 10 Gy IR were analyzed by immunoblotting with the indicated antibodies.
(F) RIF1 focus formation in cells expressing full-length 53BP1 variants 1 h post-IR (10 Gy) was evaluated by immunofluorescence. Bar height corresponds to the mean ± s.d.; WT; N=6. 28A; N=5. S166A; N=4. S176A/S178A; N=8. Representative micrographs are shown in Figure S1K.
(G) Streptavidin pulldown assay using biotinylated 53BP1 phosphopeptides incubated with HeLa nuclear extracts. Bound proteins were analyzed by immunoblotting with a RIF1 antibody.
(H-I) Fluorescence polarization assay measuring the binding of Cy5-labeled peptides to recombinant RIF1(1-980). To facilitate comparison, the data for RIF1(1-980) binding to 53BP1 pS176/pS178 peptides (*) are plotted twice in (H) and (I). Data are presented as the mean ± s.d; N=3 except for S176/S178 and pS176/pS178 where N=6. KD values ± 95% C.I. are also shown.
The 53BP1 [145-182] region contains three S/T-Q sites that lie in close proximity: S166, S176, and S178. Alanine scanning mutagenesis of this region revealed that S176 and S178 are essential for RIF1 IR-induced focus formation, while S166 is dispensable for this activity (Figure 1D and S1F). Consistent with this finding, phosphorylation of residues S176/S178 is induced in response to IR treatment whereas phosphorylation of S166 is not (Figure 1E). The combined reintroduction of these two putative ATM-targeted residues in the context of the 53BP1 [100-200]-FFR-4A fusion were sufficient to restore RIF1 accumulation at DSB sites (Figure 1C and S1D). The combined phosphomimetic mutations of S176 and S178 to either glutamic acid or aspartic acid (S176E/S178E or S176D/S178D) in 53BP1 [100-200]-FFR failed to support RIF1 focus formation whereas mutation of a single residue to Glu or Asp was tolerated (Figure S1H-I). Together, these results suggest that dual phosphorylation of S176 and S178 in 53BP1 promotes RIF1 recruitment to DSB sites. However, cells solely expressing a full-length 53BP1-S176A/S178A mutant still formed RIF1 IR-induced foci, albeit at lower levels (Figure 1F and S1J-K) indicating that sites other than S176 and S178 can recruit RIF1 to DSB sites.
The ability of 53BP1-pS176/pS178 to promote RIF1 accumulation at DSB sites suggests that RIF1 may directly recognize a phosphorylated epitope encompassing these residues. To test this possibility, we assessed whether biotinylated peptides corresponding to 53BP1 residues 145-182 could retrieve RIF1 from nuclear extracts. These experiments showed that a 53BP1 pS176/pS178 peptide, but not its unphosphorylated or singly phosphorylated counterparts, was able to bind RIF1 in nuclear extracts (Figures 1G and S1L).
To test whether RIF1 directly binds to 53BP1-derived phosphopeptides, we expressed and purified an N-terminal RIF1 protein fragment from insect cells, which encompassed the HEAT repeat-rich NTD domain (RIF11-980; Figure S1M) which is competent for DNA damage localization (Escribano-Diaz et al., 2013). We observed that RIF11-980 bound to fluorescently labeled peptides corresponding to 53BP1 [166-182] by fluorescence polarization (Figure 1H). In particular, the doubly phosphorylated peptide showed robust binding to RIF11-980, with a dissociation constant (KD) of 2.0 ± 0.3 μM, while the unphosphorylated peptide did not display any detectable binding to RIF1 (Figure 1H). Consistent with our earlier experiments, a single phosphomimetic mutation retains the interaction as long as one phosphoserine is present, whereas a double Ser-to-Glu variant had negligible binding (Figure 1H). We conclude that the NTD of RIF1 directly binds to ATM-phosphorylated 53BP1 epitopes.
The alanine scanning mutagenesis experiments described above (Figure 1D) also identified residues other than S176/S178 that were necessary to mediate RIF1 focus formation. Two leucine residues, L173 and L175, located immediately N-terminal of S176 were essential for RIF1 recruitment to DSB sites in the context of 53BP1 [100-200]-FFR (Figure 1D). Substitution of either of these leucine residues to alanine completely abrogated RIF1 IR-induced focus formation and blocked the interaction of RIF1 with the pS176/pS178 peptide (Figure 1I), suggesting that the LxL motif preceding S176/S178 plays a key role in ability of RIF1 to recognize phosphorylated epitopes. As a control, we also generated a pS176/pS178-derived peptide with the E174A substitution, which displayed wild type-level binding, as expected from the alanine scanning experiment (Figures 1D and 1I).
Equipped with this information, we identified similar LxL motifs preceding Ser/Thr residues in the other two 53BP1 segments (450-550 and 650-800) that are able to promote RIF1 focus formation when fused to the FFR (Figure 2A-B and S2A-B). Mutation of the LxL motifs in the context of the 53BP1 [400-550]- or [650-800]-FFR proteins abrogated RIF1 recruitment to DSB sites indicating that they were also important for recognition by RIF1 (Figure 2B and S2A-B). Two S/T-Q sites were present C-terminal to the LxL motif in the 650-800 segment, T696 and S698, and both have been identified as being phosphorylated in cells (Hornbeck et al., 2012; Sharma et al., 2014) (Figure 2A). These sites are critical for RIF1 recruitment promoted by the 53BP1 [650-800] segment (Figure 2B). Furthermore, pT696/pS698-derived peptides retrieved RIF1 from extracts (Figure 2C) and bound recombinant RIF1(1-980) HEAT repeat region with a KD of 1.3 ± 0.4 μM (Figure 2D). Mapping the phosphorylation sites responsible for the interaction between RIF1 and the 400-550 region proved more complicated owing to the presence of multiple phosphorylatable residues (S518, S520, S523) along with the acidic E521 residue (Figure 2A). While S523 is part of an S/T-Q motif that is phosphorylated in cells (Jowsey et al., 2007), pS523-derived peptides do not bind to RIF1 directly (Figure S2C). Instead, we found that peptides dually phosphorylated on S518 and S520, known 53BP1 phosphosites in cells (Hornbeck et al., 2012; Sharma et al., 2014), could retrieve RIF1 from nuclear extracts (Figure 2C) and were competent for binding to recombinant RIF1 (Figure 2D). Together, this work identifies three RIF1-binding phosphosites on 53BP1 that are anchored by a dileucine LxL motif and that conforms to a consensus LxL[xx](pS/pT)xpS motif where [xx] denotes the optional presence of two residues (Figure 2A).
Figure 2. Disrupting three RIF1-binding phosphopeptide motifs in 53BP1 abolishes RIF1 recruitment to sites of DNA damage.

(A) Sequence of the three 53BP1 sites sufficient to recruit RIF1 to IR-induced foci. Residues essential for RIF1 binding are highlighted. The consensus sequence is shown below.
(B) RIF1 focus formation in U2OS cells expressing the indicated 53BP1 constructs 1 h post-IR (10 Gy) were evaluated by immunofluorescence. Bar height corresponds to the mean ± s.d.; N=3, 3, 4, 4, 2, 2, 9, 5, 5, 3, 4, 5, 9, 5, 3, 3, 4, 6 in the order displayed. Representative micrographs are shown in Figure S2A.
(C) Streptavidin pulldown assay using the indicated biotinylated 53BP1 peptides incubated with HeLa nuclear extracts. Peptide-bound proteins were analyzed by immunoblotting with a RIF1 antibody.
(D) Fluorescence polarization binding assay of Cy5-labeleled phosphorylated 53BP1 peptides with recombinant RIF1(1-980). The data of RIF1 binding to the pS166/pS182 (S) peptide is the same as in Figure 1H (*). Data is presented as the mean ± s.d.; N=3 for pS166/pS182 and pT696/pS698 and N=2 pS518/pS520. KD values ± 95% C.I. are also shown.
(E) RPE1-hTERT p53−/− 53BP1-KO cells were transfected with the indicated 53BP1-FLAG constructs and assayed for RIF1 focus formation 1 h post-IR (10 Gy). Representative micrographs are shown in Figure S2E. Bar height corresponds to the mean value; N=2.
(F) The RIF1-53BP1 interaction was assessed by combining the indicated 53BP1-FLAG immunoprecipitates with HeLa nuclear extracts. Bound proteins were analyzed by immunoblotting with RIF1 and 53BP1 antibodies.
(G-H) U2OS 2-6-3 53BP1-KO cells were transfected with the indicated 53BP1 expression vectors. Localization of RIF1 at FokI-induced DSBs was determined by immunofluorescence. Representative micrographs are shown in (G). The fraction of cells containing RIF1-mCherry-LacR-FokI colocalizing foci is shown in (H). Bar height corresponds to the mean ± s.d.; N=3.
To test whether these identified binding elements were necessary for the recruitment of RIF1 to DSB sites in the context of a functional 53BP1, we mutated the RIF1-binding motifs in 53BP1 [1-1711], a variant of 53BP1 that lacks its C-terminal tandem BRCT domains that are largely dispensable for 53BP1 function in DSB repair (Bothmer et al., 2011). This truncated protein, which is more amenable to retroviral packaging, is hereafter referred to as 53BP1WT 53BP1WT is phosphorylated at S176/178 in response to IR (Figure S2D), and rescues IR-induced RIF1 focus formation when expressed in 53BP1 knockout (KO) cells (Figures 2E and S2E-F). In contrast, mutation of the three dileucine motifs to alanine (53BP13LA) or of the 6 phosphorylatable residues present in these motifs (53BP16STA), abolished RIF1 accumulation at DNA damage sites (Figures 2E and S2E-F) and abrogated the RIF1-53BP1 interaction as detected by co-immunoprecipitation (Figure 2F). The 53BP13LA variant still underwent IR-induced S176/S178 phosphorylation (Figure S2D), suggesting that disruption of the LxL motif impacts RIF1 binding rather than 53BP1 phosphorylation. Mutation of the PTIP-binding site (yielding 53BP1S25A) did not impact RIF1 IR-induced focus formation (Figures 2E and S2E-F), as expected (Callen et al., 2013). Similar results were obtained with targeted DSB formation by an mCherry-LacR-FokI fusion protein (Shanbhag et al., 2010) in a U2OS derivative (U2OS 2-6-3) that contains ~256 copies of the Lac operator (lacO256) sequence and biallelic 53BP1 inactivating mutations (Figures 2G-H and S2G). We conclude that the three LxL-containing phosphorylated epitopes of 53BP1 are responsible for mediating the 53BP1-RIF1 interaction and recruitment of RIF1 into IR-induced foci.
RIF1 focus formation is not on its own essential for DNA repair
The 53BP13LA and 53BP16STA mutations are separation-of-function mutations that allowed us to test the role of the 53BP1-RIF1 interaction in 53BP1-dependent DNA repair. Since the 53BP1- RIF1-shieldin pathway suppresses assembly of RAD51 filaments at DSB sites, we evaluated the ability of 53BP13LA to suppress RAD51 IR-induced focus formation in BRCA1-depleted cells. As shown previously (Nacson et al., 2018) , loss of 53BP1 partially rescues RAD51 IR-induced focus formation after siRNA-mediated depletion of BRCA1, while complementation with wild-type 53BP1 suppresses it (Figures 3A and S3A-C). To our surprise, we observed that 53BP13LA and 53BP16STA suppress RAD51 IR-induced focus formation as efficiently as 53BP1WT, suggesting that RIF1 accumulation into IR-induced foci is dispensable for the 53BP1 activity that suppresses HR (Figures 3A and S3A-C). This result was unexpected since RIF1 loss results in the formation of RAD51 foci in BRCA1-depleted cells (Escribano-Diaz et al., 2013).
Figure 3. RIF1 recruitment to sites of DNA damage is not essential for shieldin localization.

(A) Quantitation of RAD51 IR-induced foci in U2OS 53BP1-KO cells infected with retrovirus encoding the indicated 53BP1 variants and transfected with a non-targeting siRNA (siCTRL) or an siRNA targeting BRCA1. Cells were fixed 4 h post-IR (5 Gy) and processed for immunofluorescence with the indicated antibodies. Bar height corresponds to the mean ± s.d.; N=3 except for uninfected cells where N=4. Representative micrographs are shown in Figure S3C.
(B-C) Quantitation of REV7 and SHLD2 localization at FokI-induced DSBs in U2OS 2-6-3 53BP1-KO cells transfected with the indicated 53BP1-FLAG expression vectors in the presence (C) or absence (B) of a vector expressing GFP-SHLD2. 48 h post-transfection, cells were fixed and processed for immunofluorescence with the indicated antibodies. Colocalization of REV7 (B) and GFP-SHLD2 (C) with mCherry-LacR-FokI was quantitated. Bar height corresponds to the mean ± s.d.; N=5. Representative micrographs are shown in Figure S3D-E.
(D-E) U2OS 53BP1-KO cells expressing the indicated 53BP1-FLAG variants were UV laser microirradiated. RIF1 and REV7 colocalization was evaluated by immunofluorescence. Bar height corresponds to the mean ± s.d.; N=3.
(F) The RIF1-53BP1 interaction was assessed by combining the indicated 53BP1-FLAG immunoprecipitates with HeLa nuclear extracts. Bound proteins were analyzed by immunoblotting with RIF1 and 53BP1 antibodies. NE, nuclear extract; EV, empty vector.
(G) U2OS 53BP1-KO cells expressing the indicated 53BP1-FLAG variants were transfected with siRNA against RIF1 or a non-targeting siRNA (siCTRL) and UV laser microirradiated. REV7 colocalization was evaluated by immunofluorescence. Bar height corresponds to the mean ± s.d.; N=3. Representative micrographs are shown in Figure S3G.
We then tested whether the RIF1-binding deficient 53BP1 mutations impact the recruitment of shieldin, whose localization to DSB sites is dependent on both 53BP1 and RIF1. Shieldin recruitment to DSBs is sufficient to suppress formation of RAD51 IR-induced foci even in the absence of 53BP1 (Noordermeer et al., 2018). Using the U2OS 2-6-3 mCherry-LacR-FokI 53BP1-KO system, we found that REV7 and SHLD2 localization to DSBs were only partially reduced in cells expressing 53BP13LA (Figures 3B-C and S3D-E). In the course of these studies, we also assessed the impact of another phosphomutant of 53BP1, 53BP17A, which was initially reported to impact RIF1 recruitment to DSB sites in mouse cells (Callen et al., 2013). The sites mutated in 53BP17A (T302A, S452A, S523A, S543A, S625A, S784A, and S892A) do not overlap with the three RIF1-binding phosphoepitopes we characterized above but they do overlap with those altered in another 53BP1 mutant, 53BP1ΔRIF1, which also impairs RIF1 DNA damage localization in mouse cells (Sundaravinayagam et al., 2019). Human cells expressing 53BP17A have abundant recruitment of RIF1 (Figure 2H) but were impaired in REV7 and SHLD2 recruitment to FokI-induced DSBs (Figures 3B-C). To further investigate the differential localization of RIF1 and shieldin, we simultaneously measured RIF1 and REV7 recruitment to sites of UV laser microirradiation in complemented 53BP1-KO cells (Figures 3D-E). Consistent with our previous results, 53BP13LA is severely compromised in its ability to promote RIF1 localization but retains its ability to recruit REV7 to DSB sites, while the converse is true for 53BP17A. We therefore combined the 3LA and 7A mutations, which resulted in a protein (53BP13LA7A) that was unable to support either RIF1 or REV7 recruitment to UV laser-induced DNA lesions (Figures 3D-E and S3A). As expected from our analysis of RIF1 recruitment, 53BP13LA7A is unable to coimmunoprecipitate RIF1 from nuclear extracts, while the interaction of RIF1 with 53BP17A is indistinguishable from that observed with wild type 53BP1 (Figure 3F). We then evaluated the requirement for RIF1 in shieldin recruitment to UV laser-induced DNA damage in the context of the 53BP13LA mutation that results in loss of RIF1 recruitment. Surprisingly, RIF1 knockdown results in loss of REV7 recruitment, indicating that RIF1 is still required for shieldin localization despite its own defective recruitment (Figures 3G and S3F). Together our findings suggest that 53BP1-dependent shieldin recruitment to DSBs may occur by two distinct modes that each rely on different regions of 53BP1. One mode involves RIF1 accumulation at DSBs and is dependent upon the RIF1-binding motifs of 53BP1 identified in this study, whereas in the second mode RIF1 does not accumulate at DNA damage sites but this mode is dependent on the phosphorylated residues that are mutated in 53BP17A.
The two modes of RIF1 action in 53BP1-dependent DNA repair
Since the 3LA7A mutation disrupts both RIF1 and shieldin recruitment to sites of DNA damage, we investigated whether it also recapitulates 53BP1- and RIF1-null phenotypes in multiple assays that monitor 53BP1-dependent DNA repair. First, unlike the 3LA and 7A mutations, 53BP13LA7A could not suppress RAD51 focus formation in BRCA1-depleted cells (Figure 4A and S4A). We then tested the ability of the 53BP13LA, 53BP17A and 53BP13LA7A mutants to support antibody class switch recombination in reconstituted Tp53bp1−/− mouse splenic B cells. We found that both 53BP13LA- and 53BP17A-expressing B cells could undergo class switching, albeit with lower efficiency than wild type 53BP1 (Figures 4B-C and S4B). In contrast, B cells expressing 53BP13LA7A had reduced switching to IgG+ cells comparable to those expressing the 53BP128A mutant or 53BP1D1521R, which inactivates the Tudor domain and disrupts 53BP1 localization to DNA damage sites (Botuyan et al., 2006). 53BP1-dependent end-joining during CSR therefore requires either the 53BP1-RIF1 interaction or the shieldin recruitment activity that is disabled in the 53BP17A mutant
Figure 4. Combination of 53BP1 3LA and 7A mutations impairs DSB repair.

(A) U2OS 53BP1-KO cells expressing the indicated 53BP1-FLAG constructs were transfected with the indicated siRNAs then irradiated (5 Gy) and processed for RAD51 immunofluorescence 4 h post-IR. Bar height corresponds to the mean ± s.d. N=3. Representative micrographs are shown in Figure S4A.
(B-C) Primary murine splenic B-cells were isolated from Tp53bp1−/− mice and infected with retroviral vectors encoding the indicated 53BP1-FLAG constructs and assayed for class switching to IgG1 by FACS (B). (C) Quantification of class switch recombination assay shown in (B). Bar height corresponds to mean ± s.d.; N=3. Expression of 53BP1 variants is shown in Figure S4B.
(D-E) Tp53bp1−/−Brca1Δ11/Δ11 MEFs were treated with olaparib (1 μM) for 16 h. Metaphase spreads were prepared and stained with DAPI and analyzed for radial chromosome formation. (D) Representative micrographs. Arrowheads indicate radial chromosomes. (E) Quantitation of the experiment shown in (D). Bar height corresponds to the mean ± s.d.; N=4 (WT and 28A), N=5 (3LA and 3LA7A), N=3 (7A). Expression of 53BP1 variants is shown in Figure S4C.
(F) Clonogenic survival of SUM149PT 53BP1-KO cells stably expressing the indicated 53BP1 variants treated with olaparib (9 d). Data is presented as the mean ± s.d.; N=3. Representative images and 53BP1 variant expression are shown in Expression of 53BP1 variants is shown in Figures S4D-E.
* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. ns, not significant as determined by unpaired two-tailed t-test.
Next, we assessed the ability of the various 53BP1 mutants to induce radial chromosome formation in PARP inhibitor-treated BRCA1-mutated cells, a hallmark of PARP inhibitor toxicity and indicative of inappropriate DNA end-joining (Bunting et al., 2010). We prepared metaphase spreads from olaparib-treated Tp53bp1−/−Brca1Δ11/Δ11 mouse embryonic fibroblasts (MEFs) expressing human 53BP1 variants. Consistent with the CSR assays, only the combination of the 3LA and 7A mutations (i.e. 3LA7A) resulted in a decrease of radial chromosome formation comparable to 53BP128A although we note that both the 3LA and 7A mutants showed a partial decrease in radial chromosome formation suggesting partial defects in 53BP1-dependent DSB repair (Figures 4D-E and S4C).
We also sought to test the impact of the 53BP1 mutations on PARP inhibitor sensitivity. However, clonogenic survival assays in MEFs proved inconclusive as we found that re-introduction of active 53BP1 impairs the growth of Tp53bp1−/−Brca1Δ11/Δ11 MEFs, presumably due to suppression of HR. We therefore assessed the impact of various 53BP1 mutants on the clonogenic survival of a BRCA1-mutated SUM149PT cell line in which 53BP1 was knocked-out (53BP1−/−) (Noordermeer et al., 2018). Re-introduction of 53BP1WT via retroviral expression restored PARP inhibitor sensitivity, as expected (Figures 4F and S4D-E). However, expression of 53BP13LA, 53BP17A, or 53BP13LA7A all failed to restore PARP inhibitor sensitivity, suggesting that they were unable to complement the loss of 53BP1 in this cell line. These results suggest that partially defective 53BP1 due to the 3LA or 7A mutations may provide enough HR activity to support viability in response to PARP inhibition, and it also highlights that the 3LA7A set of mutations abrogates 53BP1-dependent DNA repair in all assays tested.
Finally, since 53BP1 remains functional in the absence of the RIF1-recruiting phosphoepitopes as long as the shieldin-recruiting region is intact, we evaluated whether the converse was true. The 53BP1 [100-200]-FFR construct, which does not contain any of the residues mutated in 53BP17A, robustly recruits RIF1 to sites of DNA damage (Figure 1B). If focal accumulation of RIF1 alone is sufficient to mediate 53BP1 activity, this truncated construct should oppose HR. Indeed, 53BP1 [100-200]-FFR suppresses S-phase RAD51 focus formation dependent upon an intact RIF1-binding motif (Figure S4F-G). This result indicates that in the absence of a secondary mode of shieldin recruitment, 53BP1 depends on RIF1 binding for its activity.
RIF1 binds phosphorylated 53BP1 through basic surface residues
We next aimed to narrow down the RIF1 region involved in the recognition of phosphorylated 53BP1. Since large protein truncations in the RIF1 HEAT repeat region were not tolerated, we took advantage of the predicted structural conservation between human RIF1 and budding yeast Rif1. We substituted four segments of the human RIF1 NTD with their equivalent from ScRif1 and assayed the ability of the resulting chimeric protein to localize to FokI-induced DSBs (Figures 5A-B and S5A-B). While experiments with ScRif1-RIF1 chimeras containing substitutions within the N-terminal 632 residues were inconclusive due to loss of protein stability (Figure S5B) that impaired localization (Figure 5B), substitution of RIF1 residues 633-967 with its budding yeast counterpart produced a chimera with a clear ability to accumulate at DSB sites, indicating that the RIF1 633-967 residues are not essential for 53BP1 binding.
Figure 5. RIF1 binds phosphorylated 53BP1 through conserved basic surface residues.

(A-B) Segments of GFP-RIF1(1-967) were substituted with homologous ScRif1 sequences as indicated in (A) and evaluated for colocalization with FokI-induced DSBs by immunofluorescence in U2OS 2-6-3 cells in (B). Bar height corresponds to the mean ± s.d. N=3. Representative micrographs are shown in Figure S5A. § indicates poorly expressed variants (see Figure S5B).
(C) U2OS 2-6-3 cells expressing the indicated GFP-RIF1(1-967) variants were evaluated for colocalization with FokI-induced DSBs by immunofluorescence. Fluorescence intensity relative to nuclear signal is shown. Data from three biological replicates are presented as box plots. Whiskers indicate 10th and 90th percentiles, while the box indicate median and 25th and 75th percentiles. Statistical significance was determined using ANOVA with multiple comparisons to WT using Dunnett’s method. Representative micrographs are shown in Figure S5C.
(D) Colocalization of the indicated full-length RIF1 variants to FokI-induced DSBs were evaluated by immunofluorescence. Fluorescence intensity relative to nuclear signal is shown. Data from three biological replicates are presented as box plots. Whiskers indicate 10th and 90th percentiles, while the box indicate median, 25th and 75th percentiles. Representative micrographs are shown in Figure S5E.
(E-F) Peptide pulldown assays of nuclear extracts derived from cells expressing the indicated GFP-RIF1(1-967) proteins that were incubated with biotinylated 53BP1 peptides phosphorylated at residues S176 and S178. Bound proteins were analyzed by immunoblotting. Band intensities are quantitated in (F). Bar height corresponds to the mean ± s.d.; N=3. Significance was determined by unpaired two-tailed t-test.
(G) Graphical representation of crosslinking mass spectrometry results derived from the RIF1(1-980)-SHLD3-REV7-SUMO-SHLD2(1-65) complex purified from insect cells and treated with the crosslinker DSSO. Inter-protein crosslinks are shown as connecting lines.
(H) Fluorescence polarization assay of purified RIF1-SHLD3-REV7-SHLD2 incubated with Cy5-labeled pS176/pS178 (S) peptides. KD values ± 95% C.I. are shown, N=2.
(I) The predicted shieldin and 53BP1 binding sites on RIF1. Residues essential for DNA damage localization, K315/R316, and residues crosslinked to shieldin are indicated as spheres on an AlphaFold2-generated model of the RIF1 HEAT repeat domain.
* p < 0.05, ** p < 0.01, *** p < 0.001, **** p <0.0001. ns, not significant.
Since tandem phosphorylated residues are highly negatively charged, we hypothesized that the recognition of 53BP1 phosphopeptide may be mediated through positively charged surface residues on RIF1. Guided by the AlphaFold2-predicted structure of RIF1 HEAT repeats (Jumper et al., 2021), we generated 18 mutants in the context of RIF1 (1-967), substituting 20 conserved basic surface residues to alanine. The basic residues were all located within the N-terminal 632 residues of RIF1. We assayed the resulting mutant proteins for their ability to localize at FokI-induced DSBs (Figures 5C and S5C-D). 8 out of 18 mutants had impaired DSB localization, with the double K315A/R316A mutant displaying a near-complete loss of localization (Figures 5C and S5C-D). Introduction of the K315A/R316A mutations in the context of full-length RIF1 also produced a protein that failed to localize to FokI-induced DSBs (Figure 5D and Figure S5E-F. Analysis of the individual K315A or R316A mutants revealed that the K315 and R316 residues are both required for RIF1 localization, suggesting they may be part of a single phosphopeptide-binding interface (Figure S5G-H).
Finally, we performed pulldown assays with biotinylated pS176/pS178 peptides with nuclear extracts prepared from cells expressing various RIF1 variants. We found that the RIF1 R315A/K316A mutation impairs binding to the 53BP1-derived phosphopeptide (Figure 5E-F), suggesting that these residues may be directly involved in the recognition of phosphorylated 53BP1.
RIF1 binds shieldin and 53BP1 simultaneously
The critical role of RIF1 for shieldin recruitment to DSB sites suggests that they interact directly. The NTD of RIF1 (residues 1-980) is necessary and sufficient for shieldin recruitment to damaged chromatin (Noordermeer et al., 2018), indicating that this region mediates the RIF1-shieldin interaction. Indeed, RIF11-980, SHLD3, REV7, and SHLD21-65 form a stable complex when co-expressed in insect cells (Figure S5I). We employed disuccinimidyl sulfoxide (DSSO) crosslinking coupled to mass spectrometry to probe the topology of this RIF1-shieldin module (Figure 5G) and mapped the identified crosslinks on the structure of REV7-SHLD2-SHLD3 (PDB ID:6KTO) (Liang et al., 2020). We observed that 16/23 intra- and inter-links that could be mapped to the structure have Cα-Cα distances under 30 Å (Table S1), which is consistent with the lysine-linker-lysine chain length compounded with the flexibility of protein quaternary structure (Merkley et al., 2014). There were extensive crosslinks between the N-terminal 200 residues of RIF1 and both SHLD3 and REV7 (Table S2), suggesting that the RIF1-shieldin binding interface lies within this region. Additionally, the RIF1-SHLD3-REV7-SHLD21-65 complex interacts with the phosphorylated 53BP1(166-182) peptide with a similar affinity to that of RIF1 alone (Figure 5H), indicating that RIF1 can simultaneously bind shieldin and the phosphorylated 53BP1 motifs found in this study (Figure 5I).
Discussion
Our study identifies RIF1 as a phosphopeptide-binding protein that recognizes di-phosphorylated linear motifs located in the N-terminal region of 53BP1 and establishes that RIF1 is recruited to DNA damage sites via this phosphopeptide-binding activity. Interestingly, RIF1 does not contain any commonly known phosphopeptide-binding modules such as BRCT, FHA or WD40 domains, and this activity is instead embedded within the HEAT repeats of RIF1 as supported by our identification of two adjacent and conserved basic residues in the HEAT repeats, K315 and R316, that may directly participate in phosophopeptide recognition by RIF1. However, elucidation of the exact mechanism of phosphopeptide recognition by the RIF1 HEAT repeats must await structural determination of RIF1-phosphopeptide complexes.
In addition to DNA repair, RIF1 regulates other processes such as DNA replication origin firing and resolution of ultrafine bridges through its interaction with PP1 where the RIF1-PP1 complex promotes dephosphorylation of key factors such as MCM4 in the regulation of DNA replication (Hiraga et al., 2014; Hiraga et al., 2017). The fact that RIF1 is likely to be able to simultaneously bind to phosphorylated peptides and a protein phosphatase indicates that RIF1 action is intimately linked to reversible protein phosphorylation. This is analogous to the PP2A phosphatase, which can also be targeted to phosphorylated substrates via the HEAT repeats of its B56 subunit (Hertz et al., 2016). It is therefore attractive to consider that other RIF1-binding proteins, such as those involved in the initiation of DNA replication or ultrafine bridge resolution, may contain RIF1-binding phosphorylated epitopes. However, an initial search for such proteins containing 53BP1-like motifs did not yield any obvious candidate. If RIF1 indeed binds other phosphorylated proteins, these results suggest that it may not be limited to the consensus motif we deduced from the three 53BP1-derived phosphopeptides, or that phosphopeptide-binding may have evolved specifically for its metazoan DNA repair function, since this activity is not apparent in the RIF1 homolog in budding yeast.
While the phosphopeptide-binding activity of RIF1 is clearly linked to its accumulation at DNA damage sites, our work also reveals that RIF1 localization into IR-induced foci does not account, on its own, for all of its DNA repair activity. Indeed, cells expressing the RIF1-binding deficient 53BP13LA mutant retain noticeable end joining-promoting activity while being severely impaired in RIF1 recruitment to damaged chromatin. Since the genetic inactivation of RIF1 phenocopies 53BP1-null phenotypes (Chapman et al., 2013; Escribano-Diaz et al., 2013; Zimmermann et al., 2013), these observations suggest that RIF1 also promotes 53BP1-dependent DNA repair independently of these binding sites. Given that 53BP128A is completely defective in DNA repair, we searched for other ATM phosphorylation sites that contribute to RIF1-dependent action. A crucial hint for identifying these alternative sites came from studies in mouse cells, where work from the Nussenzweig and Di Virgilio groups converged on a set of overlapping phosphorylation sites that they assigned as being important for murine RIF1 accumulation to DNA damage sites (Callen et al., 2013; Sundaravinayagam et al., 2019). While these sites play only a minor role in RIF1 recruitment to DSBs in human cells (Figure 2H for 53BP17A derived from Callen et al., 2013; Figure S5J for 53BP1ΔRIF1 derived from Sundaravinayagam et al., 2019), they nevertheless play an important role in mediating RIF1-dependent DNA repair redundantly with the three RIF1-binding phosphoepitopes identified in this study. Our results therefore show that shieldin and RIF1 accumulation are not perfectly correlated, arguing against a linear 53BP1-RIF1-shieldin sequence of recruitment. While the function of the sites mutated in 53BP17A is linked to the recruitment of shieldin to DNA lesions, the details of this activity are still unclear and will represent a focus of future work.
Limitations of the Study
Our results show that RIF1 interacts with three phosphorylated motifs within the 53BP1 N-terminus that mediate its recruitment to DSB sites. Many of our conclusions are based on the observation that mutation of these three motifs on 53BP1 (i.e. the 3LA or 6STA mutations) abrogate RIF1 binding and DSB recruitment. However, we cannot completely rule out the existence of a residual 53BP1-RIF1 interaction, which could explain why this mutant maintains robust shieldin recruitment to DNA damage sites. Assessing RIF1 localization at DSB sites by chromatin immunoprecipitation may help address this issue. Secondly, some of our assays, such as RAD51 IR-induced foci in BRCA1-depleted cells, have a relatively small dynamic range, making the interpretation of some of the results challenging. Finally, while we evaluated the impact of disabling the RIF1-53BP1 interaction in several widely used assays, we did not assess other activities that are influenced by 53BP1 such as DNA repair synthesis, end-protection, DSB mobility or dysfunctional telomere fusions. It is possible that the loss of the 53BP1-RIF1 interaction has a strong impact on these processes.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and reagent request should be directed to Daniel Durocher (durocher@lunenfeld.ca).
Materials availability
Plasmids and cell lines generated in this study will be available upon request from the lead contact.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. Source data is available at doi:10.17632/ngnjxzy59n.1
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
RPE1 (Female), 293T (Female), and Phoenix-AMPHO (Female) cell lines were maintained in Dulbecco’s Modified Eagle Medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; Wisent), 50 IU penicillin and 50 μg/mL streptomycin (Wisent), 1x GlutaMax (Gibco), 1x MEM non-essential Amino Acids (NEAA; Gibco). U2OS (Female) cell lines were maintained in McCoy’s 5A (Modified) Medium (Gibco) supplemented with 10% FBS, 50 IU penicillin and 50 μg/mL streptomycin. Mouse embryonic fibroblasts were maintained in DMEM supplemented with 10% FBS, 50 IU penicillin, 50 μg/ml streptomycin, 1x GlutaMax, 1x NEAA, 1 mM sodium pyruvate (Thermo Scientific), and 60 μM β-mercaptoethanol. SUM149PT (Female) cell lines were maintained in DMEM/F-12 (Gibco) supplemented with 5% FBS, 50 IU penicillin and 50 μg/mL streptomycin (Wisent), 1 μg/mL hydrocortisone (Sigma-Aldrich) and 5 μg/mL insulin (Sigma-Aldrich). Sf9 cells were maintained in suspension in EX-CELL 420 serum-free medium (Sigma-Aldrich) and High Five cells were maintained in suspension in Sf-900 II SFM (Gibco). No cell line authentication service was utilized in this study aside from verification of knockout phenotypes by Western blot. U2OS 53BP1-KO (Orthwein et al., 2015) and U2OS 2-6-3 mCherry-LacR-FokI 53BP1-KO (Batenburg et al., 2017) were generated in previous studies.
Mice
Trp53bp1−/− mice were described previously (Ward et al., 2003). Both male and female mice between 20-25 weeks of age were used. Mice were maintained in a specific pathogen-free (SPF) facility, and all procedures followed protocol (20-042) approved by the National Institutes of Health Institutional Animal Care and Use Committee.
METHOD DETAILS
Plasmids
53BP1 fragment-expressing plasmids were generated by fusion PCR from pcDNA5-FRT/TO-eGFP-53BP1 (Full length 1-1972) (Escribano-Diaz et al., 2013) and Gateway-mediated cloning into the pDEST-FRT/TO-eGFP backbone. Amino acid substitutions and deletions were introduced by site-directed mutagenesis. RIF1 fragments and mutants were similarly generated from the pDEST-FRT/TO-eGFP-RIF1 vector (Escribano-Diaz et al., 2013). pMX-53BP1-ΔRif1 were a kind gift from Michela di Virgilio (Sundaravinayagam et al., 2019). Chimeric human- Saccharomyces cervisiae RIF1 mutants were generated by Gibson assembly using the pDEST-FRT/TO-eGFP-RIF1(1-967) backbone. Chimera (1-217) substituted HsRIF1(1-217) with ScRif1(177-456), chimera (218-452) substituted HsRIF1(218-462) with ScRif1(457-710), chimera (463-632) substituted HsRIF1(463-632) with ScRif1(711-857) and chimera (633-967) substituted HsRIF1(633-967) with ScRif1(734-1031). 53BP1 constructs used in the mCherry-LacR-FokI assays were generated by site-directed mutagenesis of the pMX-53BP1(1-1711)-HA-FLAG vector (Bothmer et al., 2011).
The insect cell expression construct of RIF1(1-980) was generated by restriction endonuclease cloning of a codon-optimized ORF sequence (Integrated DNA Technologies, Coralville) into the pFastBac-Strep-TEV backbone. SHLD3, REV7, and SHLD2 constructs were similarly generated using pAC8-derived Strep-, FLAG-, 6xHis-, or Strep-SUMO- transfer vectors (Abdulrahman et al., 2015).
PEI transfection
10-cm dishes of confluent 293T or 293T-derived cells were used for PEI transfection. 10 μg of plasmid DNA was incubated with 100 μg/mL polyethyleneimine (Polysciences Inc; linear 25 kDa) in 500 μL DMEM without serum or antibiotic for 20 minutes at room temperature. After incubation, 3 mL of complete DMEM was added to the transfection mixture. Cell media was then replaced with the transfection mixture, and the cells were subsequently incubated for 2-4 hours. After incubation, 10 mL of complete DMEM was added to the plates.
siRNA knockdown
Cells were either forward or reverse transfected with siRNA. For forward transfection, 100,000 U2OS cells were plated in 6-well plates. The next day, the cells were transfected with Lipofectamine RNAiMAX (Thermo Scientific) as per manufacturer recommendations. Briefly, 25 pmol of siRNA and 3 μL of RNAiMAX transfection reagent was incubated in 500 μL Opti-MEM (Thermo Scientific) and then added to cells in 2 ml media without antibiotics. For reverse transfections, 200,000 U2OS cells were plated in 6-well plates and the same transfection mix was added during plating. Cells were harvested for downstream applications 2-3 days after siRNA transfection.
Retrovirus production and infection
53BP1-expressing retroviruses were generated in two ways. First, a 10-cm dish of Phoenix-AMPHO helper-free retrovirus packaging cells (Kinsella and Nolan, 1996; Swift et al., 2001) was transfected with 10 μg of pMX-53BP1(1-1711)-HA-FLAG vectors using PEI. 24 hours after transfection, culture media was replaced with 6 mL complete DMEM. 48 hours after transfection, the virus-containing culture media was harvested and filtered using a 0.45 μm syringe filter. Retrovirus was used immediately after harvest. Alternatively, concentrated VSV-G pseudotyped retrovirus was generated by the University of Michigan Retroviral Core and concentrated 10X by ultracentrifugation. VSV-G retrovirus was snap frozen in liquid nitrogen and stored in −80°C until use.
Retroviral infection was conducted in 6-well plates using either 1 mL of unconcentrated virus or 200 μL of concentrated virus in 20 mM HEPES pH 7.4 and 8 μg/mL polybrene (Sigma-Aldrich). If selection was desired, puromycin was added either 24 or 48 hours post-infection (10 μg/mL for RPE1 or 2 μg/mL for MEFs, U2OS, and SUM149PT) for at least 48 hours. Transduction efficiency was determined by both immunoblotting and immunofluorescence analysis.
Immunofluorescence
Cells cultured on glass coverslips were harvested and rinsed with PBS. The coverslips were then fixed by incubation in 4% PFA in PBS for 10 minutes. After washing three times with PBS, the coverslips were permeabilized in 0.3% Triton X-100 in PBS for 30 minutes. For nuclear pre-extraction treatment, the coverslips are first incubated for 10 minutes on ice in nuclear pre-extraction buffer (20 mM HEPES pH 7.4, 20 mM NaCl, 5 mM MgCl2, 0.5% NP-40, 1 mM DTT, and 1x cOmplete EDTA-free protease inhibitor cocktail [Roche]), washed once with PBS, and fixed for 10 minutes at room temperature. After these treatments the coverslips were washed three times with PBS, then placed in a humidified chamber and incubated with blocking solution (either PBS + 1% BSA or PBS + 0.2% cold water fish gelatin + 0.5% BSA) for 30 minutes. The coverslips were then incubated with the primary antibody diluted in blocking solution for 1-2 hours at room temperature, followed by washing three times with PBS. The coverslips were then incubated with the secondary antibody diluted in blocking solution for 1h at room temperature and washed three times with PBS. Coverslips were then mounted on glass slides using ProLong Gold Antifade mounting media with DAPI (Invitrogen).
For EdU staining to enrich for S-phase cells, 10 μM EdU was added to cells 30 minutes prior to harvesting. The coverslips were treated as above, except that following secondary antibody incubation and washing, they were further fixed using 4% PFA for 5 minutes at room temperature, washed twice with PBS, and incubated for 30 minutes with EdU staining solution (100 mM Tris-HCl pH 8.5, 1 mM CuSO4, 100 mM ascorbic acid, 10 μM Alexa Fluor azide (either 647 or 555; Thermo-Fisher). The coverslips were then washed three times with PBS and mounted as above.
Complementation of RIF1 IRIFs
25,000 U2OS cells were plated in 24-well plates containing glass coverslips. After 24 hours, 53BP1 expression was knocked down by transfection of 10 nM 53BP1 siRNA#1 (ggacaagtctctcagctat; Dharmacon) with 1 μL RNAiMAX (total volume of 600 μL per well). 24 hours after siRNA transfection, 0.8 μg of 53BP1-expression plasmids were transfected using Lipofectamine 2000 (Thermo Scientific) according to the manufacturer guidelines. 24 hours after DNA transfection, cells were treated with 10 Gy of ionizing radiation using a Faxitron X-ray cabinet (Faxitron, Tucson AZ). After irradiation, cells were incubated for 1 hour, then fixed with 4% PFA for further immunofluorescence analysis.
Peptide pulldown with recombinant RIF1
5 μg of phosphorylated 53BP1 peptides (BioBasic, Markham; New England Peptide, Boston; Sigma-Aldrich) resuspended in manufacturer-recommended solvents were coupled to 10 μL Dynabeads M-280 streptavidin beads (Invitrogen) in 250 μl peptide pulldown buffer 1 (PPB1; 20 mM HEPES pH 7.8, 100 mM KCl, 0.2 mM EDTA, 1% BSA, 0.5 mM DTT, 0.2 mM PMSF, 1 mM β-glycerophosphate, 1 mM sodium orthovanadate) at 4°C. The peptides are defined in the Key Resources Table. Beads were collected after 30 minutes and washed twice with PPB1. Beads were resuspended in 250 μL PPB1 containing purified recombinant RIF1 (0.25 to 1 μg) and incubated by rotating at 4°C. Beads were collected and washed twice with 1 mL PPB1, and twice with 1 mL PPB2 (20 mM HEPES pH 7.8, 20 mM KCl, 0.2 mM EDTA, 0.5 mM DTT, 1 mM β-glycerophosphate, 1 mM sodium orthovanadate). The beads were then boiled in 25 μl SDS sample buffer (100 mM Tris-HCl pH 6.8, 4% SDS, 20% glycerol, 2% β-mercaptoethanol, 25 mM EDTA, 0.04% bromophenol blue). After magnetic removal of the beads, the supernatant was analyzed by SDS-PAGE and immunoblotting.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat polyclonal anti RIF1 | Santa Cruz | Cat #: sc-55979; RRID: AB_2126818 |
| Mouse monoclonal anti RIF1 | Santa Cruz | Cat #: sc-515573 |
| Rabbit polyclonal anti RIF1 | Bethyl Laboratories | Cat#: A300-569A; RRID: AB_669804 |
| Mouse monoclonal anti 53BP1 | BD Biosciences | Cat #: 612523; RRID: AB_399824 |
| Rabbit polyclonal anti 53BP1 | Bethyl Laboratories | Cat #: A300-273A; RRID: AB_185521 |
| Rabbit polyclonal anti 53BP1 | Novus Biologicals | Cat#: NB100-304; RRID:AB_1000303 |
| anti 53BP1 phospho S176/178 | Gift from J. Rouse (Jowsey et al. 2017) | N/A |
| anti 53BP1 phospho S166 | Gift from J. Rouse (Jowsey et al. 2017) | N/A |
| Rabbit polyclonal anti 53BP1 phospho S25 | Bethyl Laboratories | Cat #: A300-652A; RRID: AB_519340 |
| Rat monoclonal anti FLAG | BioLegend | Cat#: 637319; RRID: AB_2749907 |
| HRP-Mouse monoclonal anti FLAG | Sigma-Aldrich | Cat#: A8592; RRID: AB_439702 |
| Mouse monoclonal anti BRCA1 | Calbiochem | Cat#: OP92; RRID: AB_2750876 |
| Mouse monoclonal anti BRCA1 | Calbiochem | Cat#: OP93; RRID: AB_213440 |
| Rabbit polyclonal anti BRCA1 | (Escribano-Diaz et al. 2013) | N/A |
| Rabbit monoclonal anti REV7/MAD2L2 | Abcam | Cat#: ab180579 |
| Rabbit anti RAD51 serum | BioAcademia | Cat#: 70-001; RRID: AB_2177110 |
| Mouse monoclonal anti α-tubulin | EMD Millipore | Cat#: CP06; RRID: AB_2617116 |
| Rabbit polyclonal anti GFP | Gift from L. Pelletier | N/A |
| Rabbit polyclonal anti PTIP | Abcam | Cat#: ab2614; RRID: AB_303209 |
| Rabbit polyclonal anti Kap1 | Bethyl Laboratories | Cat#: A300-274A; RRID: AB_185559 |
| HRP-Sheep monoclonal anti mouse IgG | Cytiva | Cat#: 45-000-692 |
| HRP-Goat polyclonal anti rabbit IgG | Jackson Immunoresearch Labs | Cat#: 111-035-144; RRID: AB_2307391 |
| HRP-Bovine polyclonal anti goat IgG | Jackson Immunoresearch Labs | Cat#: 805-035-180; RRID:AB_2340874 |
| IRDye 680RD-Donkey polyclonal anti mouse IgG | LI-COR Biosciences | Cat#: 926-68072; RRID: AB_10953628 |
| IRDye 800CW-Donkey polyclonal anti goat IgG | LI-COR Biosciences | Cat#: 926-32214; RRID: AB_621846 |
| IRDye 800CW-Donkey polyclonal anti rabbit IgG | LI-COR Biosciences | Cat#: 926-32213; RRID: AB_621848 |
| IRDye 680RD-Goat polyclonal anti rabbit IgG | LI-COR Biosciences | Cat#: 926-68071; RRID: AB_10956166 |
| IRDye 800CW-Goat polyclonal anti mouse IgG | LI-COR Biosciences | Cat#: 926-32210; RRID: AB_621842 |
| Alexa Fluor 488-Donkey polyclonal anti rat IgG | Thermo Fisher | Cat#: A-21208; RRID: AB_141709 |
| Alexa Fluor 488-Donkey polyclonal anti mouse IgG | Thermo Fisher | Cat#: A-21202; RRID: AB_141607 |
| Alexa Fluor 555-Donkey polyclonal anti rabbit IgG | Thermo Fisher | Cat#: A-31570; RRID: AB_2536180 |
| Alexa Fluor 647-Donkey polyclonal anti mouse IgG | Thermo Fisher | Cat#: A-31571; RRID: AB_162542 |
| Alexa Fluor 647-Donkey polyclonal anti rabbit IgG | Thermo Fisher | Cat#: A-31573; RRID: AB_2536183 |
| Bacterial and virus strains | ||
| Bacteria: XL1-Blue competent cells | Agilent | Cat#: 200249 |
| Bacteria: DH10Bac competent cells | Thermo Fisher | Cat#: 10361012 |
| Retrovirus: pMX-53BP1-HA-FLAG and other mutants | (Bothmer et al. 2011), University of Michigan Retrovirus Core and this study. | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Biotin-53BP1(145-182); S176/S178 (L) | New England Peptide | N/A |
| Biotin-53BP1(145-182) pS176; pS176 (L) | New England Peptide | N/A |
| Biotin-53BP1(145-182) pS178; pS178 (L) | New England Peptide | N/A |
| Biotin-53BP1(145-182) pS176/pS178; pS176/pS178 (L) | New England Peptide | N/A |
| Biotin-53BP1(166-182); S176/S178 (S) | Sigma-Aldrich | N/A |
| Biotin-53BP1(166-182) pS176/pS178; pS176/pS178 (S) | Sigma-Aldrich | N/A |
| Biotin-53BP1(511-527); S518/S520 | New England Peptide | N/A |
| Biotin-53BP1(511-527) pS523; pS523 | New England Peptide | N/A |
| Biotin-53BP1(511-527) pS518/pS520; pS518/pS520 | BioBasic | N/A |
| Biotin-53BP1(686-702); T696/S698 | New England Peptide | N/A |
| Biotin-53BP1(686-702) pT696/pS698; pT696/pS698 | New England Peptide | N/A |
| Cy5-53BP1(166-182); S176/S178 (S) | New England Peptide | N/A |
| Cy5-53BP1(166-182) pS176/pS178; pS176/pS178 (S) | New England Peptide | N/A |
| Cy5-53BP1(166-182) S176E/pS178; S176E/pS178 (S) | New England Peptide | N/A |
| Cy5-53BP1(166-182) S176E/S178E; S176E/S178E (S) | New England Peptide | N/A |
| Cy5-53BP1(166-182) pS176/pS178 L173A; pS176/pS178 (S) L173A | New England Peptide | N/A |
| Cy5-53BP1(166-182) pS176/pS178 E174A; pS176/pS178 (S) E174A | New England Peptide | N/A |
| Cy5-53BP1(166-182) pS176/pS178 L175A; pS176/pS178 (S) L175A | New England Peptide | N/A |
| Cy5-53BP1(511-527) pS518/pS520; pS518/pS520 | New England Peptide | N/A |
| Cy5-53BP1(686-702) pT696/pS698; pT696/pS698 | New England Peptide | N/A |
| HeLa Nuclear Extracts | Accurate Chemical | Cat#: CC012050 |
| EdU (5-ethynyl-2’-deoxyuridine) | Thermo Fisher | Cat#: E10187 |
| Lipofectamine 2000 | Thermo Fisher | Cat#: 11668030 |
| Lipofectamine RNAiMAX | Thermo Fisher | Cat#: 13778100 |
| Puromycin | InvivoGen | Cat#: ant-pr |
| Blasticidin | InvivoGen | Cat#: ant-bl |
| Polybrene | Sigma-Aldrich | Cat#: TR-1003 |
| Polyethyleneimine, linear 25 kDa | Polysciences Inc | Cat #: 23966 |
| Penicillin/streptomycin | Wisent | Cat#: 450-200-EL |
| Fetal bovine serum | Wisent | Cat#: 080-150 |
| Insulin solution human | Sigma-Aldrich | Cat#: I9278 |
| Hydrocortisone | Sigma-Aldrich | Cat#: H0888 |
| Phosphate buffered saline | Gibco | Cat#: 10010023 |
| Dulbecco’s Modified Eagle Medium | Gibco | Cat#: C11965500BT |
| McCoy’s 5A (Modified) Medium | Gibco | Cat#: 16600082 |
| DMEM/F-12 | Gibco | Cat#: 11320033 |
| EX-CELL 420 Serum-free Medium | Sigma-Aldrich | Cat#: 14420C |
| Sf-900 II SFM | Gibco | Cat#: 10902088 |
| cOmplete Mini EDTA-free Protease Inhibitor Cocktail | Roche | Cat#: 11836170001 |
| cOmplete Mini Protease Inhibitor Cocktail | Roche | Cat#: 11836153001 |
| DAPI (4,6-diamidino-2-phenylindole) | Sigma-Aldrich | Cat# D9542 |
| Olaparib | Selleckchem | Cat#: AZD2281 |
| Lipopolysaccharide (LPS) from E. coli O111:B4 | Sigma-Aldrich | Cat#: L2630 |
| Interleukin 4 (IL-4) from mouse | Sigma-Aldrich | Cat#: I1020 |
| CD43 microbeads (Ly-48) | Miltenyi Biotec | Cat#: 130-049-801 |
| KaryoMAX Colcemid | Gibco | Cat#: 15210040 |
| StrepTactin Sepharose | IBA Lifesciences | Cat#: 2-1201-010 |
| FLAG M2 Magnetic Beads | EMD-Millipore | Cat#: M8823-5ML |
| Disuccinimidyl sulfoxide | Thermo Fisher | Cat#: A33545 |
| ProLong Gold Antifade Mountant with DAPI | Invitrogen | Cat#: P36931 |
| PfuTurbo DNA Polymerase | Agilent | Cat#: 600250 |
| T5 exonuclease | New England Biolabs | Cat#: M0363S |
| Phusion DNA polymerase | New England Biolabs | Cat#: M0530S |
| Taq DNA ligase | New England Biolabs | Cat#: M0208S |
| NAD | Sigma-Aldrich | Cat#: 10127965001 |
| PEG8000 | Sigma-Aldrich | Cat#: 81268 |
| Experimental models: Cell lines | ||
| Human: U-2-OS (U2OS) | ATCC | Cat#: HTB-96 |
| Human: U2OS 53BP1-KO | (Orthwein et al. 2015) | N/A |
| Human: U2OS 2-6-3 mCherry-LacR-FokI | Gift from R. Greenberg (Shanbhag et al. 2010) | N/A |
| Human: U2OS 2-6-3 mCherry-LacR-FokI 53BP1-KO | (Batenburg et al. 2017) | N/A |
| Human: RPE1-hTERT p53−/− FLAG-Cas9 | (Zimmermann et al. 2018) | N/A |
| Human: RPE1-hTERT p53−/− FLAG-Cas9 53BP1-KO | (Noordermeer et al. 2018)) | N/A |
| Human: SUM149PT | BioIVT | N/A |
| Human: SUM149PT 53BP1-KO | Gift from C.J. Lord (Noordermeer et al. 2018) | N/A |
| Human: 293T | ATCC | Cat#: CRL-3216 |
| Human: Phoenix-AMPHO | (Kinsella et al. 1996) | N/A |
| Human: BOSC23 | ATCC | Cat#: CRL-11270 |
| MEF: Tp53bp1−/− | (Bunting et al. 2010) | N/A |
| MEF: Tp53b91−/−BRCA1Δ11/Δ11 | (Bunting et al. 2010) | N/A |
| Insect: Spodoptera frugiperda Sf9 | Thermo Fisher | Cat#: B82501 |
| Insect: Trichoplusia ni High Five | Expression Systems | Cat#: 94-002F |
| Experimental models: Organisms/strains | ||
| Mouse: Tp53bp1−/−. B6/129 | (Ward et al. 2003) | N/A |
| Oligonucleotides | ||
| ON TARGETplus siRNA 53BP1 | Horizon Discovery | Cat#: J-003548-06-0005 |
| ON TARGETplus siRNA BRCA1 SMARTpool | Horizon Discovery | Cat#: L-003461-00-0005 |
| ON TARGETplus siRNA RIF1 | Horizon Discovery | Cat#: J-027983-10-0002 |
| Recombinant DNA | ||
| Plasmid: pMX-53BP1(1-1711)-HA-FLAG | (Bothmer et al. 2011) | N/A |
| Plasmid: pDEST-FRT/TO-eGFP-53BP1 | (Escribano-Diaz et al. 2013) | N/A |
| Plasmid: pMX-53BP1(100-200-FFR)-HA-FLAG | This study | N/A |
| Plasmid: pMX-53BP1(400-550-FFR)-HA-FLAG | This study | N/A |
| Plasmid: pMX-53BP1(650-800-FFR)-HA-FLAG | This study | N/A |
| Plasmid: pMX-53BP1(1000-1220-FFR)-HA-FLAG | This study | N/A |
| Plasmid: pMX-53BP1(750-1050-FFR)-HA-FLAG | This study | N/A |
| Plasmid: pMX-53BP1(500-650-FFR)-HA-FLAG | This study | N/A |
| Plasmid: pMX-53BP1(250-400-FFR)-HA-FLAG | This study | N/A |
| Plasmid: pMX-53BP1(1-300-FFR)-HA-FLAG | This study | N/A |
| Plasmid: pMX-53BP1(145-182-FFR)-HA-FLAG | This study | N/A |
| Plasmid: pMX-53BP1-ΔRif1 | (Sundaraviyanagam et al. 2019) | N/A |
| Plasmid: pDEST-FRT/TO-eGFP-RIF1(1-967) | (Escribano-Diaz et al. 2013) | N/A |
| Plasmid: pDEST-FRT/TO-eGFP-RIF1 | (Escribano-Diaz et al. 2013) | N/A |
| pDEST-FRT/TO-eGFP-RIF1(1-967) Sc(1-217) | This study | N/A |
| pDEST-FRT/TO-eGFP-RIF1(1-967) Sc(218-462) | This study | N/A |
| Plasmid: pDEST-FRT/TO-eGFP-RIF1(1-967) Sc(463-632) | This study | N/A |
| Plasmid: pDEST-FRT/TO-eGFP-RIF1(1-967) Sc(633-967) | This study | N/A |
| Plasmid: pFastBac-Strep-TEV-RIF1(1-980) | This study | N/A |
| Plasmid: pAC8-Strep-TEV-SHLD3 | This study | N/A |
| Plasmid: pAC8-6xHis-TEV-REV7 | This study | N/A |
| Plasmid: pAC8-Strep-SUMO-TEV-SHLD2(1-65) | This study | N/A |
| Plasmid: pCL-ECO | Addgene | Cat#: 12371 |
| Software and algorithms | ||
| ImageJ Fiji | (Schneider et al. 2012) | https://imagej.net/Fiji |
| CellProfiler 4.1.3 | (McQuin et al. 2018) | https://cellprofiler.org/ |
| PHERAstar FS | BMG Labtech | https://www.bmglabtech.com/software-updates/ |
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Chimera 1.13.1 | (Pettersen et al. 2004) | https://www.cgl.ucsf.edu/chimera/ |
| Proteome Discoverer 2.2 with XlinkX algorithm | Thermo Fisher, (Liu et al. 2017) | https://www.thermofisher.com/store/products/OPTON-30945#/OPTON-30945 |
| FlowJo 10.1 | FlowJo LLC | https://www.flowjo.com/ |
| LI-COR Image Studio 5.2 | LI-COR Biosciences | https://www.licor.com/bio/image-studio/ |
Peptide pulldown of RIF1 from nuclear extract
53BP1 peptides were coupled to streptavidin Dynabeads as above. Beads were resuspended in 450 μL PPB1 and 50 μL of 10 mg/mL HeLa nuclear extracts (Accurate Chemical, Carle Place NY) and incubated by rotating at 4°C. Beads were washed four times with 1 mL PPB1. Bound proteins were harvested by boiling in 25 μL SDS sample buffer and analyzed by SDS-PAGE and immunoblotting. For peptide pulldown of exogenously expressed RIF1 mutants, nuclear extracts were prepared from 293T PEI transfected with GFP-RIF1(1-967) plasmids using a modified Dignam protocol (Lee et al., 1988). One 15 cm dish of confluent 293T cells were harvested by scraping into PBS and pelleted by centrifugation at 200 xg for 5 minutes. Cell pellets were resuspended in one pellet volume of buffer A (10 mM HEPES-KOH pH 7.9, 1.5 mM MgCl2, 10 mM KCl, and 0.5 mM DTT) and incubated on ice for 15 minutes. Cells were then lysed by rapid ejection through a 26-gauge needle repeated five times. Lysates were centrifuged at 12,000 xg for 20 seconds to pellet nuclei. The nuclei were then resuspended in 2/3 of the original cell pellet volume in buffer C (20mM HEPES-KOH pH 7.9, 25% glycerol, 0.42M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5mM PMSF, and 0.5 mM DTT) and incubated end over end at 4°C for 30 minutes. The nuclear extract was then centrifuged at 21,000 xg for 5 minutes at 4°C and the supernatant was collected, then dialyzed in buffer D (20 mM HEPES-KOH pH 7.9, 20% glycerol, 0.1 mM KCl, 0.2 mM EDTA, 0.5 mM PMSF, and 0.5 mM DTT) for 2 hours at 4°C using 3.5 kDa MWCO Slide-A-Lyzer Dialysis Cassettes (Thermo Scientific). Dialyzed nuclear extracts were quantified by Bradford assay, and equal concentrations were used for peptide pulldown analysis. Pulldown of proteins were detected by Western blotting visualized using the LI-COR Odyssey CLx (LI-COR Biosciences) and quantified using LI-COR Image Studio software.
Co-immunoprecipitation of RIF1 and 53BP1
One confluent 10-cm dish of 293T cells transfected with 10 μg of pMX-53BP1(1-1711)-HA-FLAG was used for each co-immunoprecipitation experiment. Prior to harvesting, cells were treated with 10 Gy of X-ray irradiation using a Faxitron cabinet (Faxitron). 1h post irradiation, the cells were harvested by scraping into PBS and pelleted by centrifugation for 5 minutes at 1000xg at 4°C. The cells were then lysed by addition of lysis buffer (50 mM Tris-HCl pH 8, 100 mM NaCl, 2 mM EDTA, 10 mM NaF, 0.5% NP-40, 10 mM MgCl2, 1x cOmplete EDTA-free protease inhibitor tablet, 1x phosphatase inhibitor cocktail 3 [Sigma-Aldrich] and 5 U/mL of benzonase [Sigma-Aldrich]) and incubated on ice for 30 minutes. Lysates were then pre-cleared by centrifugation at 21,000xg at 4°C, followed by incubation with 10 μL bed volumes of anti-FLAG M2 magnetic beads for 1 hour at 4°C. The beads were then washed using 500 μL PPB1 buffer (see above), incubated with 450 μL PPB1 and 50 μL of 10 mg/ml HeLa nuclear extracts (Accurate Chemical) for 1 hour at 4°C. Beads were washed twice with 500 μL PPB1, then twice with 500 μL PPB1 without BSA. Bound protein was harvested by boiling beads in 50 μL SDS sample buffer and analyzed by SDS-PAGE and immunoblotting.
U2OS-2-6-3 FokI focus recruitment assay
200,000 U2OS-2-6-3 cells harboring 256 LacO arrays and an inducible mCherry-LacR-FokI (Shanbhag et al., 2010) were plated in a 6-well plate containing glass coverslips. The next day, cells were transfected using 1-2 μg of pMX-GFP or pMX-53BP1 vectors and 6 μL Lipofectamine 2000 (Thermo Scientific). The media was changed after 3 hours of incubation. 48 hours after transfection, mCherry-LacR-FokI expression was induced by addition of 10 μg/mL 4-hydroxytamoxifen (Sigma) and 1 μM of Shield-1 peptide (Clontech, Mountain View CA) for 4-6 hours. Cells were then fixed in 4% PFA for further immunofluorescence analysis. Images were quantified using ImageJ (Schneider et al., 2012), defining a ratio of ≥1.5 between the average fluorescence intensities colocalizing to mCherry-LacR signal and the nuclear signal as containing a focus. For RIF1 mutant localization experiments, the same procedure was used as above with the addition of RIF1 siRNA #2 (Dharmacon) transfection with RNAiMAX (1 μL in 24-well or 3 μL in 6-well format) during plating.
Laser microirradiation
Cells were first grown on 25 mm glass coverslips until the desired confluence. The cells were then sensitized to laser microirradiation by incubation with 2 μg/mL Hoechst 33342 (Invitrogen) for 10 minutes. DNA damage was induced by a 40 mW 355 nm laser (Coherent) via a 40x Plan-Apochromat 40x objective lens using an LSM780 laser scanning confocal microscope (Zeiss) with the following laser setting: 100% power, 128 x 128 frame size, line step 7, 25.21 μs pixel dwell time. After irradiation, cells were incubated for 60-90 minutes, processed for nuclear pre-extraction, and fixed with 4% PFA for immunofluorescence analysis. Images were quantified using ImageJ, defining a ratio of ≥2 between the average fluorescence intensities colocalizing with the 53BP1 stripe and the nucleus as containing a stripe.
RAD51 IR-induced focus formation
U2OS cells were reverse transfected with siRNA against BRCA1 using Lipofectamine RNAiMAX (Thermo Fisher) and plated in 6-well plates according to manufacturer’s instructions. 48 hours after transfection, cells were exposed to 5 Gy of X-ray irradiation using a Faxitron X-ray cabinet (Faxitron). After irradiation, cells were incubated for 4 hours, processed for nuclear pre-extraction, and fixed with 4% PFA for immunofluorescence analysis. 53BP1, BRCA1, and RAD51 foci were quantified using CellProfiler (McQuin et al., 2018). Cells containing 5 or more RAD51 foci were classified as RAD51-positive.
Metaphase spread analysis
MEF cells derived from Tp53bp1−/− Brca1Δ11/Δ11 mice (Bunting et al., 2010) were infected with 53BP1-expressing retroviruses. At approximately 70% confluence, cells were treated with DMSO or 1 μM of the PARPi olaparib (SelleckChem) for 24 hours. After this treatment, MEFs were arrested in mitosis by incubation with 0.2 μg/mL KaryoMax colcemid (Thermo Scientific) for 1 hour. Cells were then harvested by trypsinization, pelleted by centrifugation, and incubated with 75 mM KCl at 37°C for 30 minutes. After KCl incubation, cells were pelleted and fixed by dropwise addition of 500 μL Carnoy’s fixative (3:1 methanol and acetic acid). The mixture was further resuspended in 10 mL of fixative and stored at 4°C until preparation of slides. For metaphase analysis, fixed cells were pelleted by centrifugation and resuspended in 200-500 μL of fixative. Resuspended cells were dropped on glass coverslips, dried in ambient conditions, and coverslips were mounted using ProLong Gold Antifade mounting media with DAPI (Thermo Scientific).
Immunoglobulin class switch assay
Resting primary B lymphocytes (wild-type and Tp53bp1−/−) were purified from mouse spleens using anti-CD43 microbeads (Miltenyi Biotec). One million cells were stimulated to proliferate with a cytokine cocktail containing 25 μg/mL lipopolysaccharide (LPS, Sigma-Aldrich), 5 ng/mL interleukin-4 (IL-4, Sigma-Aldrich) and 0.5 μg/mL anti-CD180 (BD PharMingen). Infectious pMXs-based retroviruses encoding various 53BP1 proteins were assembled in BOSC23 packaging cells co-transfected with the pCL-Eco helper virus. Retroviral supernatant was collected 40 h later, passed through a sterile 0.45 μm syringe filter (VWR) and used to transduce activated B cells in the presence of 10 μg/mL polybrene (Sigma-Aldrich). Viral transduction was facilitated by centrifugation (2500 rpm, 1.5 h at 20°C, Sorvall Legend XTR, Thermo Scientific), after which cells were incubated in polybrene-containing media for an additional 6 h before being returned to regular B cell activation media. A second round of viral transduction was performed on the following day. Class switching to IgG1 was detected on day 4 by flow cytometry (FACSCantoTM II, BD Biosciences) using biotinylated anti-IgG1 followed by PE-conjugated streptavidin (BD Biosciences). Anti-B220 was used to confirm the purity of B cell samples.
Expression of endogenous and exogenous 53BP1 was confirmed by immunoblotting. Briefly, B cells were lysed on day 4 in a buffer containing 50 mM Tris-HCl (pH 7.5), 200 mM NaCl, 5% Tween-20, 2% Igepal CA-630, 2 mM PMSF, 50 mM β-glycerophosphate (all from Sigma-Aldrich) and protease inhibitor cocktail tablet (cOmplete Mini, Roche Diagnostics). Equal amounts of lysates were resolved by SDS-PAGE. Incubation with primary (polyclonal rabbit anti-53BP1, NB100-304, Novus Biologicals) and secondary (IRDye 680RD Goat anti-Rabbit IgG (H+L), LI-COR Biosciences) antibodies were performed according to standard procedures. Visualization of protein bands was achieved by fluorescence imaging (LI-COR Biosciences).
Olaparib clonogenic survival assay
500 cells of SUM149PT and SUM149PT 53BP1-KO stably expressing exogenous 53BP1 were plated in 6-well plates. Olaparib (SelleckChem) was added to the indicated concentrations, as well as 0.5 μg/ml puromycin to maintain selection for 53BP1-expressing cells. After 9 days, the plates were rinsed with PBS, stained for 30 minutes with crystal violet solution (0.5% w/v crystal violet [Sigma-Aldrich] in 20% methanol) and rinsed three times in water. Number of colonies was determined through manual counting.
Purification of recombinant proteins
Baculoviruses were generated in Spodoptera frugiperda Sf9 cells (Thermo Fisher) by cotransfecting linearized viral DNA with the transfer plasmid for the pAC8-derived vectors of Bac-to-Bac method for the pFastBac-derived vectors (Abdulrahman et al., 2015). For recombinant protein expression of Strep-TEV-RIF1(1-980) and Strep-TEV-RIF1(1-980)-Strep-TEV-SHLD3-His-TEV-REV7-Strep-SUMO-TEV-SHLD2(1-65), Trichoplusia ni High Five cells (Expression Systems) were infected (or coinfected) with baculoviruses encoding the desired proteins.
For purification of complexes, cells were harvested by centrifugation 36 h after infection, resuspended in lysis buffer (50 mM HEPES pH 7.4, 300 mM NaCl, 0.1% (v/v) Triton X-100, 1 mM TCEP, 5 mM MgCl2, 1 mM KCl, DNase (40 μg per litre of culture) supplemented with 1 × SigmaFast protease inhibitor cocktail (Sigma-Aldrich) and disrupted by sonication. Following high speed centrifugation, the supernatant was filtered through Miracloth (EMD Millipore) and subsequently applied to a 20 mL Strep-Tactin sepharose column (IBA Lifesciences). The affinity resin was washed (wash buffer: 50 mM HEPES pH 7.4, 300 mM NaCl, 1 mM TCEP) and the bound complex was eluted in 50 mM HEPES pH 7.4, 300 mM NaCl, 1 mM TCEP, 2.5 mM desthiobiotin. Fractions containing protein complexes were concentrated (Amicon Ultra-15, 10 kDa molecular weight cutoff) and purified by size exclusion chromatography on a HiLoad 16/60 Superdex 200 column (GE Healthcare), which was pre-equilibrated with buffer containing 50 mM HEPES pH 7.4, 300 mM NaCl, 5% glycerol and 0.5 mM TCEP. Strep-TEV-RIF1(1-980)-Strep-SHLD3-6xHis-REV7 and Strep-TEV-RIF1(1-980)-Strep-SHLD3-6xHis-REV7-Strep-SUMO-SHLD2 was overexpressed in High Five insect cells and purified as Strep tag fusion protein as described for RIF1(1-980). Purified samples were concentrated, quantified using a Bradford assay with bovine serum albumin standard and UV absorption on a Nanodrop spectrophotometer (ThermoFisher Scientific), flash frozen in liquid nitrogen and stored at −80°C.
Fluorescence polarization
N-terminally Cy5-labeled peptides (New England Peptide) dissolved in DMSO were diluted in binding buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 2.5 mM MgCl2, 0.25 mM TCEP, 50 μg/ml BSA (Sigma), and 0.05% Tween-20 to a concentration of 200 nM. Purified RIF1(1-980) or RIF1(1-980)-SHLD3-REV7 was diluted in binding buffer to generate a 20 μM stock and serially diluted to the desired concentration. 5 μL each of purified protein and peptide were mixed in a 384-well small volume microplate (Hibase, Greiner), and fluorescent polarization was measured with 200 flashes per second over a period of one hour using a PHERAstar FS microplate reader (BMG Labtech, Ortenberg). Peptide sequences can be found in the Key Resources Table.
Crosslinking coupled to mass spectrometry
Purified RIF1-SHLD3-REV7-Strep-SUMO-SHLD2(1-65) at a concentration of 0.5 mg/ml in buffer (50 mM HEPES pH 7.4, 300 mM NaCl, 0.5 mM TCEP) was incubated with 0.5, 1, or 2 mM disuccinimidyl sulfoxide (DSSO) for 60 minutes with shaking at room temperature. The reaction was quenched by the addition of 50 mM Tris-HCl pH 6.8. The crosslinked samples were then subjected to ultrafiltration using an Amicon Ultra 0.5 ml centrifugal filter (EMD-Millipore) to remove crosslinking reagent and non-crosslinked proteins. The crosslinked complex was resuspended in 400 μL 8M urea in 50 mM HEPES pH 8.5 to denature and wash the protein, then concentrated again for a total of two urea washes, and concentrated to 50 μL. The sample was reduced and alkylated by addition of reduction/alkylation solution (5 mM TCEP, 10 mM 2-chloroacetamide) for 30 minutes in the dark with shaking. The sample was then washed using a centrifugal spin filter with 8M urea in 50 mM HEPES pH 8.5 for a total of two washes and concentrated to 23.5 μl. The sample was digested by Lys-C addition (1:100 enzyme to protein ratio) for 1.5 hours at room temperature while shaking. The Lys-C digested samples were diluted to a final concentration of 50 mM HEPES pH 8.4 and 2M urea, followed by the addition of trypsin (1:100 enzyme to protein ratio) and digested overnight with shaking. An additional 1:100 ratio of trypsin was added, followed by acetonitrile to a final concentration of 5%. The sample was then incubated for 4 hours with shaking. The digested sample was acidified using 1% trifluoroacetic acid, sonicated, and centrifuged at 20,000 xg for 5 minutes yielding a final sample volume of 85 μl. 10 μl of the sample was subjected to single shot analysis, while the remaining 75 μl was subjected to strong cation exchange (SCX) fractionation. LC-MS analysis was performed using a 2 cm trapping column and a 15 cm analytical column coupled to an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher) using a 240 minute (single shot) or a 120 minute (SCX fractions) gradient and the MS2 MS3 method (Liu et al., 2017). Crosslinks were identified from a library constructed out of the primary amino acid sequences of the purified proteins used in the analysis with the XlinkX search node (Klykov et al., 2018) in Proteome Discoverer (version 2.2, Thermo Fisher) using the default cutoff score of 20. Identified crosslinks were validated by comparison to the SHLD3(1-64)-REV7-SHLD2(1-54) crystal structure (PDB:6KTO) (Liang et al., 2020), with atom-atom distances measured in UCSF Chimera (Pettersen et al., 2004).
QUANTIFICATION AND STATISTICAL ANALYSIS
All data presented are biological replicates unless otherwise stated. The statistical tests used, number of replicates, definition of error bars and center definitions are all defined within each figure or figure legend. Statistical tests performed in this study (Student’s unpaired t-test and ANOVA test with multiple comparisons using Dunnett’s method) as well as nonlinear fitting of ligand binding curves (single site model) were performed using Graphpad Prism 8. ns = p > 0.05, * = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.00001. No specific methods were used to determine whether the data met the assumptions of the statistical approach.
Supplementary Material
Table S2. Crosslinking mass spectrometry analysis of RIF1(1-980)-SHLD3-REV7-SUMO-SHLD2(1-65), related to Figure 4G
HIGHLIGHTS.
RIF1 is a phosphopeptide-binding protein
RIF1 recognizes three related diphosphorylated epitopes on 53BP1
RIF1-binding sites on 53BP1 are essential for RIF1 accumulation at DNA break sites
RIF1-dependent phosphopeptide recognition contributes to DNA repair
Acknowledgments
We thank R. Szilard for critical reading of the manuscript. We also thank members of the Durocher lab for helpful discussions. We thank J. Rouse for the phospho-53BP1 antibodies, R. Greenberg for the U2OS 2-6-3 cell line, C. Lord for the SUM149PT-53BP1-KO cell line, M. Di Virgilio for pMX-53BP1-ΔRif1 and J. Nilsson for pointing us to B56 as a HEAT-repeat phosphoprotein binding protein. DS held a postdoctoral fellowship from the Canadian Institutes for Health Research (CIHR) for most of this work. JKR was supported by a Boehringer Ingelheim Fonds PhD fellowship. DD is a Canada Research Chair (Tier I) and work in the DD lab was supported by grants from the CIHR (FDN143343) with additional support from OICR (Ovarian TRI) and the Krembil Foundation.
Footnotes
Declaration of interests
DD is a shareholder of and advisor to Repare Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdulrahman W, Radu L, Garzoni F, Kolesnikova O, Gupta K, Osz-Papai J, Berger I, and Poterszman A (2015). The production of multiprotein complexes in insect cells using the baculovirus expression system. Methods Mol Biol 1261, 91–114. [DOI] [PubMed] [Google Scholar]
- Anderson L, Henderson C, and Adachi Y (2001). Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol Cell Biol 21, 1719–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batenburg NL, Walker JR, Noordermeer SM, Moatti N, Durocher D, and Zhu XD (2017). ATM and CDK2 control chromatin remodeler CSB to inhibit RIF1 in DSB repair pathway choice. Nat Commun 8, 1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollen M, Peti W, Ragusa MJ, and Beullens M (2010). The extended PP1 toolkit: designed to create specificity. Trends Biochem Sci 35, 450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothmer A, Robbiani DF, Di Virgilio M, Bunting SF, Klein IA, Feldhahn N, Barlow J, Chen HT, Bosque D, Callen E, et al. (2011). Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol Cell 42, 319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, and Mer G (2006). Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 127, 1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. (2010). 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonomo SB, Wu Y, Ferguson D, and de Lange T (2009). Mammalian Rif1 contributes to replication stress survival and homology-directed repair. J Cell Biol 187, 385–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callen E, Di Virgilio M, Kruhlak MJ, Nieto-Soler M, Wong N, Chen HT, Faryabi RB, Polato F, Santos M, Starnes LM, et al. (2013). 53BP1 Mediates Productive and Mutagenic DNA Repair through Distinct Phosphoprotein Interactions. Cell 153, 1266–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callen E, Zong D, Wu W, Wong N, Stanlie A, Ishikawa M, Pavani R, Dumitrache LC, Byrum AK, Mendez-Dorantes C, et al. (2020). 53BP1 Enforces Distinct Pre- and Post-resection Blocks on Homologous Recombination. Mol Cell 77, 26–38 e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, and Boulton SJ (2013). RIF1 Is Essential for 53BP1-Dependent Nonhomologous End Joining and Suppression of DNA Double-Strand Break Resection. Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuella-Martin R, Oliveira C, Lockstone HE, Snellenberg S, Grolmusova N, and Chapman JR (2016). 53BP1 Integrates DNA Repair and p53-Dependent Cell Fate Decisions via Distinct Mechanisms. Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dev H, Chiang TW, Lescale C, de Krijger I, Martin AG, Pilger D, Coates J, Sczaniecka-Clift M, Wei W, Ostermaier M, et al. (2018). Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol 20, 954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT, et al. (2013). Rif1 Prevents Resection of DNA Breaks and Promotes Immunoglobulin Class Switching. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N, Chen YC, Spector DL, and de Lange T (2008). 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 456, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. (2013). A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol Cell 49, 872–883. [DOI] [PubMed] [Google Scholar]
- Findlay S, Heath J, Luo VM, Malina A, Morin T, Coulombe Y, Djerir B, Li Z, Samiei A, Simo-Cheyou E, et al. (2018). SHLD2/FAM35A co-operates with REV7 to coordinate DNA double-strand break repair pathway choice. EMBO J 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana GA, Reinert JK, Thoma NH, and Rass U (2018). Shepherding DNA ends: Rif1 protects telomeres and chromosome breaks. Microb Cell 5, 327–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradet-Turcotte A, Canny MD, Escribano-Diaz C, Orthwein A, Leung CC, Huang H, Landry MC, Kitevski-Leblanc J, Noordermeer SM, Sicheri F, et al. (2013). 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499, 50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Feng S, Ning S, Liu J, Zhao H, Xu Y, Shang J, Li K, Li Q, Guo R, et al. (2018). An OB-fold complex controls the repair pathways for DNA double-strand breaks. Nat Commun 9, 3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezraoui H, Oliveira C, Becker JR, Bilham K, Moralli D, Anzilotti C, Fischer R, Deobagkar-Lele M, Sanchiz-Calvo M, Fueyo-Marcos E, et al. (2018). 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 560, 122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Somyajit K, Narita T, Maskey E, Stanlie A, Kremer M, Typas D, Lammers M, Mailand N, Nussenzweig A, et al. (2018). DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 173, 972–988 e923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengeveld RC, de Boer HR, Schoonen PM, de Vries EG, Lens SM, and van Vugt MA (2015). Rif1 Is Required for Resolution of Ultrafine DNA Bridges in Anaphase to Ensure Genomic Stability. Dev Cell 34, 466–474. [DOI] [PubMed] [Google Scholar]
- Hertz EPT, Kruse T, Davey NE, Lopez-Mendez B, Sigurethsson JO, Montoya G, Olsen JV, and Nilsson J (2016). A Conserved Motif Provides Binding Specificity to the PP2A-B56 Phosphatase. Mol Cell 63, 686–695. [DOI] [PubMed] [Google Scholar]
- Hiraga S, Alvino GM, Chang F, Lian HY, Sridhar A, Kubota T, Brewer BJ, Weinreich M, Raghuraman MK, and Donaldson AD (2014). Rif1 controls DNA replication by directing Protein Phosphatase 1 to reverse Cdc7-mediated phosphorylation of the MCM complex. Genes Dev 28, 372–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraga SI, Ly T, Garzon J, Horejsi Z, Ohkubo YN, Endo A, Obuse C, Boulton SJ, Lamond AI, and Donaldson AD (2017). Human RIF1 and protein phosphatase 1 stimulate DNA replication origin licensing but suppress origin activation. EMBO Rep 18, 403–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, Latham V, and Sullivan M (2012). PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res 40, D261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jowsey P, Morrice NA, Hastie CJ, McLauchlan H, Toth R, and Rouse J (2007). Characterisation of the sites of DNA damage-induced 53BP1 phosphorylation catalysed by ATM and ATR. DNA Repair (Amst) 6, 1536–1544. [DOI] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsella TM, and Nolan GP (1996). Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Hum Gene Ther 7, 1405–1413. [DOI] [PubMed] [Google Scholar]
- Klykov O, Steigenberger B, Pektas S, Fasci D, Heck AJR, and Scheltema RA (2018). Efficient and robust proteome-wide approaches for cross-linking mass spectrometry. Nat Protoc 13, 2964–2990. [DOI] [PubMed] [Google Scholar]
- Lee KA, Bindereif A, and Green MR (1988). A small-scale procedure for preparation of nuclear extracts that support efficient transcription and pre-mRNA splicing. Gene Anal Tech 5, 22–31. [DOI] [PubMed] [Google Scholar]
- Liang L, Feng J, Zuo P, Yang J, Lu Y, and Yin Y (2020). Molecular basis for assembly of the shieldin complex and its implications for NHEJ. Nat Commun 11, 1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Lossl P, Scheltema R, Viner R, and Heck AJR (2017). Optimized fragmentation schemes and data analysis strategies for proteome-wide cross-link identification. Nat Commun 8, 15473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, and Carpenter PB (2004). 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat Immunol 5, 481–487. [DOI] [PubMed] [Google Scholar]
- Manke IA, Lowery DM, Nguyen A, and Yaffe MB (2003). BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 302, 636–639. [DOI] [PubMed] [Google Scholar]
- Mattarocci S, Reinert JK, Bunker RD, Fontana GA, Shi T, Klein D, Cavadini S, Faty M, Shyian M, Hafner L, et al. (2017). Rif1 maintains telomeres and mediates DNA repair by encasing DNA ends. Nat Struct Mol Biol 24, 588–595. [DOI] [PubMed] [Google Scholar]
- McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, et al. (2018). CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol 16, e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkley ED, Rysavy S, Kahraman A, Hafen RP, Daggett V, and Adkins JN (2014). Distance restraints from crosslinking mass spectrometry: mining a molecular dynamics simulation database to evaluate lysine-lysine distances. Protein Sci 23, 747–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirman Z, and de Lange T (2020). 53BP1: a DSB escort. Genes Dev 34, 7–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirman Z, Lottersberger F, Takai H, Kibe T, Gong Y, Takai K, Bianchi A, Zimmermann M, Durocher D, and de Lange T (2018). 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature 560, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz IM, Jowsey PA, Toth R, and Rouse J (2007). Phospho-epitope binding by the BRCT domains of hPTIP controls multiple aspects of the cellular response to DNA damage. Nucleic Acids Res 35, 5312–5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacson J, Krais JJ, Bernhardy AJ, Clausen E, Feng W, Wang Y, Nicolas E, Cai KQ, Tricarico R, Hua X, et al. (2018). BRCA1 Mutation-Specific Responses to 53BP1 Loss-Induced Homologous Recombination and PARP Inhibitor Resistance. Cell Rep 24, 3513–3527 e3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, Olivieri M, Alvarez-Quilon A, Moatti N, Zimmermann M, et al. (2018). The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560, 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochs F, Karemore G, Miron E, Brown J, Sedlackova H, Rask MB, Lampe M, Buckle V, Schermelleh L, Lukas J, et al. (2019). Stabilization of chromatin topology safeguards genome integrity. Nature 574, 571–574. [DOI] [PubMed] [Google Scholar]
- Orthwein A, Noordermeer SM, Wilson MD, Landry S, Enchev RI, Sherker A, Munro M, Pinder J, Salsman J, Dellaire G, et al. (2015). A mechanism for the suppression of homologous recombination in G1 cells. Nature 528, 422–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panier S, and Boulton SJ (2014). Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 15, 7–18. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Rappold I, Iwabuchi K, Date T, and Chen J (2001). Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J Cell Biol 153, 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setiaputra D, and Durocher D (2019). Shieldin - the protector of DNA ends. EMBO Rep 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, and Greenberg RA (2010). ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 141, 970–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, D'Souza RC, Tyanova S, Schaab C, Wisniewski JR, Cox J, and Mann M (2014). Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep 8, 1583–1594. [DOI] [PubMed] [Google Scholar]
- Sundaravinayagam D, Rahjouei A, Andreani M, Tupina D, Balasubramanian S, Saha T, Delgado-Benito V, Coralluzzo V, Daumke O, and Di Virgilio M (2019). 53BP1 Supports Immunoglobulin Class Switch Recombination Independently of Its DNA Double-Strand Break End Protection Function. Cell Rep 28, 1389–1399 e1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift S, Lorens J, Achacoso P, and Nolan GP (2001). Rapid production of retroviruses for efficient gene delivery to mammalian cells using 293T cell-based systems. Curr Protoc Immunol Chapter 10, Unit 10 17C. [DOI] [PubMed] [Google Scholar]
- Xu D, Muniandy P, Leo E, Yin J, Thangavel S, Shen X, Ii M, Agama K, Guo R, Fox D 3rd, et al. (2010). Rif1 provides a new DNA-binding interface for the Bloom syndrome complex to maintain normal replication. The EMBO journal 29, 3140–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, and Blackburn EH (2004). Human Rif1 protein binds aberrant telomeres and aligns along anaphase midzone microtubules. J Cell Biol 167, 819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Chini CC, He M, Mer G, and Chen J (2003). The BRCT domain is a phosphoprotein binding domain. Science 302, 639–642. [DOI] [PubMed] [Google Scholar]
- Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, and de Lange T (2013). 53BP1 Regulates DSB Repair Using Rif1 to Control 5' End Resection. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Crosslinking mass spectrometry analysis of RIF1(1-980)-SHLD3-REV7-SUMO-SHLD2(1-65), related to Figure 4G
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. Source data is available at doi:10.17632/ngnjxzy59n.1
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request