

Abstract
Background:
Spina bifida (SB) is the second most common nonlethal congenital malformation. The existence of monogenic SB mouse models and human monogenic syndromes with SB features indicate that human SB may be caused by monogenic genes. We hypothesized that whole exome sequencing (WES) allows identification of potential candidate genes by i) generating a list of 136 candidate genes for SB, and ii) by unbiased exome-wide analysis.
Methods:
We generated a list of 136 potential candidate genes from three categories: and evaluated WES data of 50 unrelated SB cases for likely deleterious variants in 136 potential candidate genes, and for potential SB candidate genes exome-wide.
Results:
We identified 6 likely deleterious variants in 6 of the 136 potential SB candidate genes in 6 of the 50 SB cases, whereof 4 genes were derived from mouse models, 1 gene was derived from human non-syndromic SB, and 1 gene was derived from candidate genes known to cause human syndromic SB. In addition, by unbiased exome-wide analysis, we identified 12 genes as potential candidates for SB.
Conclusions:
Identification of these 18 potential candidate genes in larger SB cohorts will help decide which ones can be considered as novel monogenic causes of human SB.
Keywords: Birth defect, monogenic disease, molecular genetic diagnosis, spina bifida, whole exome sequencing
INTRODUCTION
Spina bifida (SB) constitutes the most common neural tube defect (NTD) compatible with life. With an incidence of around 1 per 1,000 births, it is one of the most common birth defects after congenital heart defects (incidence about 1%; (Reller, Strickland, Riehle-Colarusso, Mahle, & Correa, 2008)) and besides congenital anomalies of the kidneys and urinary tract (CAKUT; incidence ranging from 0.3 to 1.6 per 1,000 births) (Andrés-Jensen et al., 2016; Caiulo et al., 2012; Salih, Murshid, & Seidahmed, 2014). SB arises from incomplete closure of the distal end of the neural tube during embryonic development (Copp et al., 2015). The phenotypic severity is governed by the extent of the cleft and thus the tissues that are directly affected. This can include, from the body surface towards the interior, the skin, subcutaneous tissue, skeletal muscles, vertebrae, meninges, and the spinal cord (Mohd-Zin, Marwan, Abou Chaar, Ahmad-Annuar, & Abdul-Aziz, 2017). Clinically, two major entities of SB can be discerned: spina bifida occulta (SBO), the mildest and often ‘hidden’ type with unaffected meninges and spinal cord, and spina bifida aperta (SBA), the more severe form with a macroscopically overt cleft formation (Copp, et al., 2015). Among cases with SBA, myelomeningocele (MMC) is the most common and most severe phenotype. Individuals with MMC often exhibit multiple complications, such as bladder or intestinal motility disorders, hindbrain herniation (Chiari malformation type II), and associated hydrocephalus, which often requires shunting (Copp, et al., 2015). SBA leads to a limited life expectancy (Yi, Lindemann, Colligs, & Snowball, 2011).
Neural tube formation is a rapid, multi-step process, which is precisely regulated by genes that act in a wide range of molecular pathways forming a complex biological network (Wang, Marco, Capra, & Kibar, 2019). The pathogenesis of NTD is based on the disturbance of the normal neural tube closure and can be due to the disruption of many genes involved in this process (Copp & Greene, 2010). To date, the etiology of SB has not been well established. Previous studies have shown both, environmental (non-genetic) and genetic factors, to be involved in the pathogenesis of SB (Mohd-Zin, et al., 2017). Reduced maternal folic acid (FA) intake, by far, is the most extensively studied environmental factor to generate a risk for this particular birth defect. The prevalence of NTD was shown to be significantly reduced by up to 70–80% in response to maternal FA supplementation (Berry et al., 1999). However, a large proportion of NTD remains folate-resistant and cannot be prevented by FA supplementation (Copp, Stanier, & Greene, 2013; Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. MRC Vitamin Study Research Group, 1991).
Over 300 mouse models have been found to exhibit NTD, and, among them, over 40 have been extensively studied to better understand the underlying mechanisms of SB (reviewed in (Lee & Gleeson, 2020); Table S2). This includes genes that converge in developmental pathways, like the planar cell polarity (PCP) and non-canonical Wnt pathways (Juriloff & Harris, 2012). Furthermore, rare deleterious variants in PCP pathway genes derived from mouse models, including CELSR1, VANGL1, VANGL2, GRHL3, SCRIB, and LRP6, have been identified in human SB/NTD (Chen et al., 2018; Kibar et al., 2011).
We deemed it likely that human spina bifida may be caused by deleterious variants in monogenic genes, because of i) the congenital nature of spina bifida; ii) familial occurrence of spina bifida (Detrait et al., 2005); iii) the existence of monogenic mouse models with spina bifida (Table S2); iv) the existence of spina bifida as part of the phenotypic manifestation of known monogenic multi-organ syndromes (Table S4); and v) the knowledge that specific master genes govern neural tube morphogenesis (Beaumont et al., 2019; Kibar et al., 2007; Torban, Wang, Groulx, & Gros, 2004; Wang, et al., 2019). Several studies have been performed to explore the genetic nature of SB. However, most of them are case-control studies that focused on single candidate genes or pathways (Kibar, et al., 2007) (Kim et al., 2019; Lei et al., 2014; Ye et al., 2020). Previous studies, also from our group, showed that whole exome sequencing (WES) is a powerful approach to identify causative monogenic genes in pediatric diseases of the kidneys and urinary tract(Connaughton et al., 2020; Hildebrandt, Benzing, & Katsanis, 2011; Kopp et al., 2020; Sadowski et al., 2015). WES allowed the identification of 22 monogenic CAKUT-causing genes by our group (reviewed in (Kohl, Habbig, Weber, & Liebau, 2021)), and, intriguingly, many of them were derived from monogenic mouse models (reviewed in (van der Ven, Vivante, & Hildebrandt, 2018)). A major strength of WES is the potential of an unbiased evaluation; therefore WES can complement a candidate-gene based search, purely based on genetic and functional criteria of deleteriousness.
We therefore hypothesized that WES enables identification of potential monogenic candidate genes for SB in a dual approach, by i) generation of a list of 136 potential candidate genes derived from (A) mouse models, (B) potentially causing isolated human SB, and (C) syndromic human SB genes, as well as by ii) an exome-wide, unbiased query for likely deleterious variants in potential novel genes. We here report the results from our study utilizing this approach on 50 SB cases from 50 unrelated, international families and identifying 6 potential monogenic SB candidate genes from 136 candidate genes we generated, as well as 12 additional potential candidate genes from an unbiased evaluation. In summary, we detected a likely deleterious variant in one of 18 potential monogenic candidate genes in 18 of 50 (36%) SB cases. Detecting additional families with likely deleterious variants conducted in larger cohorts will be necessary to strengthen which of the 18 candidate genes will be confirmed as a novel monogenic cause of SB.
MATERIALS AND METHODS
Human Subjects
This study was approved by the Institutional Review Board (IRB) of Boston Children’s Hospital as well as the IRBs of the institutions from where we have recruited families. Following informed consent, we obtained clinical data using a standardized questionnaire and collected blood or saliva samples from individuals with spina bifida and their relatives.
A total of 89 individuals (50 affected members with SB and 39 reportedly unaffected family members) from 50 unrelated families, who met the inclusion criteria, were enrolled. All individuals with the clinical diagnosis of SB were referred to us by their health care provider. SB was defined as the demonstration of spina bifida occulta, meningocele, myelocele, diastematomyelia, myelomeningocele, lipomyelomeningocele, and/or tethered spinal cord. The patients were further classified into two clinical subtypes: isolated spina bifida and syndromic spina bifida. Isolated spina bifida was defined as SB with or without relatable findings or complications of SB, i.e., neurogenic bladder, bowel/bladder incontinence, Chiari malformation type II, and paraplegia. Syndromic spina bifida was defined as the presence of organ involvement beyond SB that was not secondary to SB.
For tailored WES evaluation based on Mendelian principles, we allotted the families to subgroups based on pedigree structure (Fig. 1): (1) singlet (17 of 50 families), only affected proband’s DNA available for analysis; (2) Duo or Multi-Duo (29 of 50 families), affected proband and one of the parent’s DNA available for analysis, and (3) Trio (4 of 50 families), affected proband and both parents’ DNA available for analysis (Fig. 1, Table S1).
Figure 1. Generation of a SB candidate gene list of 136 potential candidate genes in three categories from 4 sources.
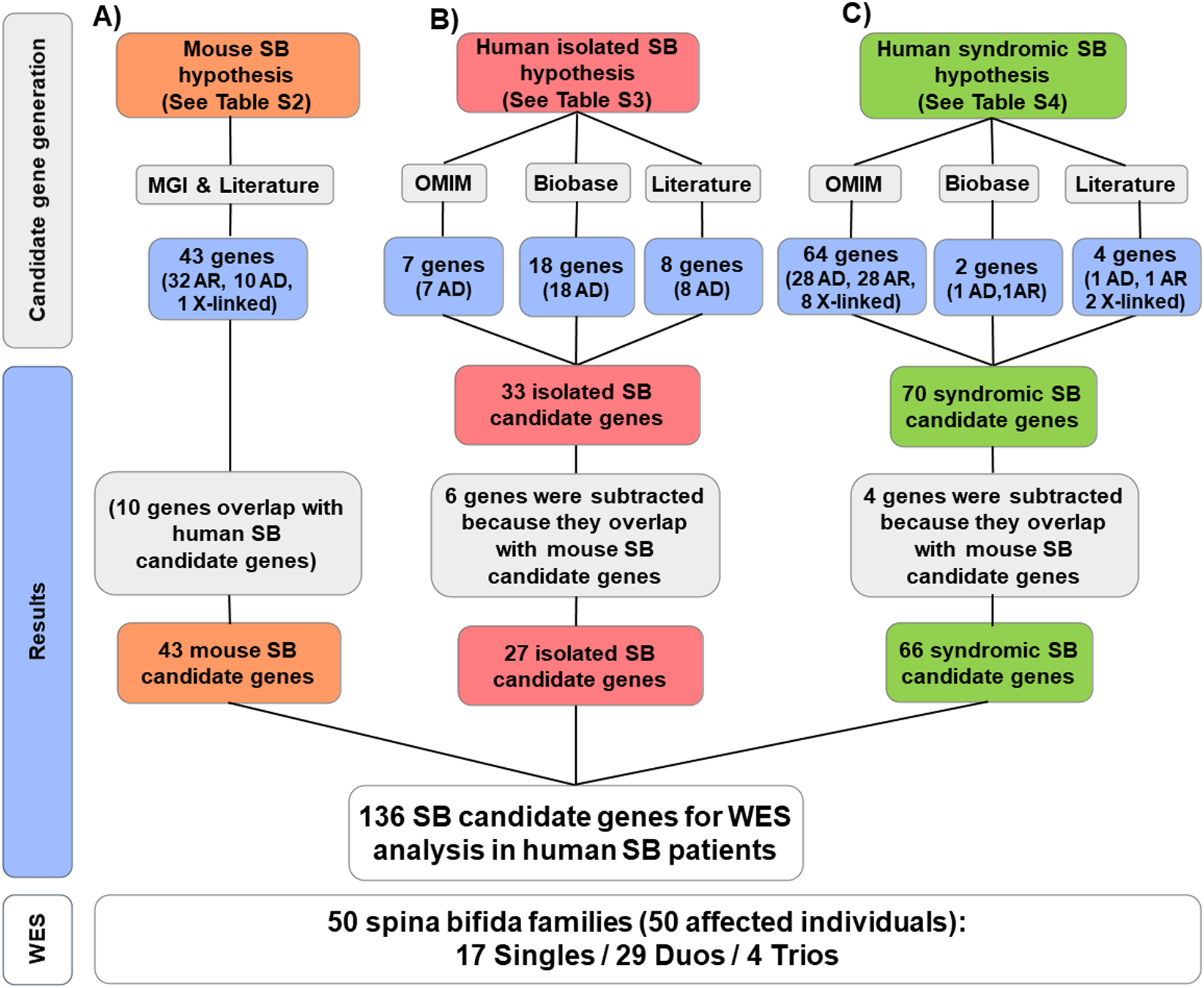
The following databases, MGI (Mouse Genome Informatics; http://www.informatics.jax.org/), Literature (See Methods), OMIM (Online Mendelian Inheritance in Man; https://www.omim.org/), and Biobase-HGMD (Human Gene Mutation Database; https://portal.biobase-international.com/hgmd), were searched to generate a set of four lists of SB candidate genes from four categories of candidate hypotheses: A) 43 candidate genes were derived from mouse models of SB (Table S2); B) 27 candidate genes were derived from existing evidence to potentially cause human non-syndromic SB (Table S3); and C) 66 candidate genes were derived from known causes of human clinical syndromes with facultative SB features (Table S4). Note that we consider candidate status derived from mouse SB as the strongest hypothesis, and since 10 genes in this category overlapped with category B) and C), 6 genes were subtracted from B (non-syndromic human SB candidate genes, red color), and 4) genes were subtracted from C (mouse SB candidate genes, green color). A total of 50 affected individuals with spina bifida from 50 unrelated families were subjected to WES, and their WES data were analyzed for variants in these 136 potential candidate genes (lower panel).
AD, autosomal dominant; AR, autosomal recessive; SB, Spina bifida.
Generation of a List of Candidate Genes for Spina Bifida
We generated a list of candidate genes for SB based on preexisting evidence and classified them into three categories (Fig. 1): A) candidate genes known to cause SB in mouse models; B) candidate genes known to potentially cause human isolated SB; and C) candidate genes known to cause human clinical syndromes with facultative SB features. Candidate genes for these three categories (A-C) were obtained from the databases OMIM, Biobase-HGMD, and Mouse Genome Informatics (See Website Resources; Fig. 1). We then reviewed the literature for additional SB candidate genes reported (Harris & Juriloff, 2007, 2010; Mohd-Zin, et al., 2017). By hypothetical strength, we consider that there is a decreasing level of evidence from categories A through C.
Establishment of Mode of Inheritance
For candidate genes known to cause SB in mouse models (A), we hypothesized a dominant and/or recessive inheritance mode if both heterozygous and homozygous mice were reported to present with a SB phenotype (Fig. S1). In addition, we considered a dominant and/or recessive mode of inheritance if the human ortholog of a gene with a recessive mouse model was known to cause neural tube defects (or susceptibility to those) in human or in the heterozygous state as reported in OMIM. We considered a recessive mode of inheritance if only the homozygous mice presented with SB phenotypes (Fig. S1).
For candidate genes known to potentially cause human isolated SB (category B; Table S3) or human syndromes with facultative SB features (category C; Table S4), we adopted the presumed mode of inheritance recessive or dominant from the reports in the primary literature (Tables S3–4).
Whole Exome Sequencing and Variant Calling
Research-based WES was performed as previously described (Braun et al., 2016). In brief, genomic DNA was isolated from blood lymphocytes or saliva samples and subjected to exome capture using Agilent SureSelect™ human exome capture arrays (Life Technologies™), followed by next generation sequencing on an Illumina HiSeq™ sequencing platform. Sequence reads were mapped to the human reference genome assembly (National Center for Biotechnology Information build 37/hg19) using CLC Genomics Workbench™ software (version 6.5.2, CLC bio, Aarhus, Denmark). Following alignment to the human reference genome, variants were filtered for most likely deleterious variants as previously described (Mann et al., 2019; Sadowski, et al., 2015). In brief, variant filtering based on population frequency was performed using population databases (Genome Aggregation Database and the 1,000 Genomes Project) to include only rare alleles (i.e., minor allele frequency <1%). Synonymous and intronic variants that were not located within splice site regions were excluded. Remaining variants included non-synonymous variants and splice site variants.
Mutation Calling in 136 Candidate Genes of Human or Mouse Spina Bifida
We evaluated WES data for likely deleterious variants within the list of 136 SB candidate genes that we generated (Fig. 1, Table S2–4). The variants were ranked on the basis of their probable effect on the function of the encoded protein considering evolutionary conservation among orthologues using ENSEMBL Genome Browser and assembled using Clustal Omega, as well as PolyPhen-2 (Adzhubei et al., 2010), SIFT (Kumar, Henikoff, & Ng, 2009), MutationTaster (Schwarz, Cooper, Schuelke, & Seelow, 2014), and CADD score (Rentzsch, Witten, Cooper, Shendure, & Kircher, 2019). Variants were designated as likely deleterious based on criteria given by Table S5, derived from our previous publications (van der Ven et al., 2018; Wu et al., 2020). Remaining variants were confirmed in original patient DNA by Sanger sequencing. A limited number of parental samples were available for segregation analysis. Where available, we sought to determine segregation of the allele in question.
Identification of Potential Novel Candidate Genes for SB by Familial Analysis
In parallel, for all SB families, a family-based unbiased evaluation was done based on pedigree structure (Fig. 1). Given a negative family history in all recruited families, we assumed a recessive mode of inheritance. For singlets, we only queried for homozygous variants; for Duos and Multi-Duos, we queried for homozygous and compound heterozygous variants, and for Trio families, we queried for homozygous, compound heterozygous, and de novo variants. Variants were designated as likely deleterious on the basis of criteria listed in Table S5.
Control Cohorts
All likely deleterious variants we identified were also tested for presence in an in-house negative control population. This negative control cohort consisted of 50 cases with steroid-resistant nephrotic syndrome (SRNS) in whom a definitive underlying monogenic cause had already been established.
WEB RESOURCES
Clustal Omega, http://www.ebi.ac.uk/Tools/msa/clustal
Combined Annotation Dependent Depletion (CADD), https://cadd.gs.washington.edu/
Ensembl Genome Browser, http://www.ensembl.org/
Exome Variant Server, http://evs.gs.washington.edu/EVS
HGMD Professional 2016.3, https://portal.biobase-international.com/hgmd
Genome Aggregation Database (gnomAD), http://gnomad.broadinstitute.org/
Mouse Genome Informatics, http://www.informatics.jax.org/
Mutation Taster, http://www.mutationtaster.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
OMIM, https://www.omim.org/
PhastCons/phyloP, http://compgen.cshl.edu/phast/
Polyphen2, http://genetics.bwh.harvard.edu/pph2
Sorting Intolerant From Tolerant (SIFT), http://sift.jcvi.org/
UCSC Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
Uniprot Consortium, http://www.uniprot.org/
1000 Genomes Browser, http://browser.1000genomes.org/
RESULTS
Generation of a List of 136 Potential Candidate Genes for Human Spina Bifida
In order to generate candidate gene lists for potential monogenic candidate genes of SB, we queried 4 different sources which generated a list of 136 genes with existing evidence to be potential candidate genes for human spina bifida (see Methods; Fig. 1). Based on the following 3 candidate gene hypotheses: A) 43 candidate genes known to cause SB in mouse models (Table S2), B) 27 genes known to potentially cause human isolated SB (Table S3), and C) 66 genes known to cause human syndromes with facultative SB features (Table S4). By hypothetical strength, we consider that there is a decreasing level of evidence from categories A through C.
Clinical Characteristics of 50 SB cases
We enrolled an international cohort of 50 individuals with SB from 50 unrelated families and subjected their DNA samples to WES (Fig. 1). These included 36 families from Boston Children’s Hospital (BCH) and 14 families from external hospitals (Fig. S1). Clinical characteristics of the 50 affected individuals are outlined in Table S1. Female-to-male ratio in individuals with SB was 27:23. The most frequent ethnicity was Caucasian (31/50; 62%). The majority of individuals were from non-consanguineous families (47/50; 94%). All cases were sporadic without any reported family history of SB (One individual was adopted, family history was unknown). 20/50 (40%) of individuals presented with syndromic SB features. 36/50 (72%) individuals presented with myelomeningocele, the most severe type of SB, with or without tethered spinal cord. 39/50 (78%) of SB individuals presented with at least one of the complications related to SB, i.e., neurogenic bladder, neurogenic bowel (see Methods for case definitions; Table S2).
Analysis of 136 Potential Candidate Genes Identifies Likely Deleterious Variants in Candidate genes in 6/50 (12%) of Cases with SB
We first evaluated WES data from the 50 families with SB for likely deleterious variants within the list of 136 potential SB candidate genes that we generated based on our three candidate gene hypotheses: A) mouse SB candidate genes, B) human isolated SB potential candidate genes, and C) human syndromes with facultative SB features (Fig. 1 and Table S2–4, and see METHODS).
We deemed the 43 genes derived from SB mouse models (group A) as the strongest candidates (Fig. 1A). We based the assumed mode of inheritance of these genes on the mouse models. Dominant inheritance was considered if both the heterozygous and homozygous mouse model showed an SB phenotype. Recessive inheritance was considered if only the homozygous mouse model showed an SB phenotype (Fig. 1). As shown in Fig. 2, by evaluating WES for these 136 candidate genes, we identified four likely deleterious variants in 4 unrelated individuals with SB in 4 of 43 mouse SB genes (Fig. 2, inner ring, Table 1; orange segment). These variants included one homozygous variant in the gene AMBRA1 and three heterozygous missense variants in the genes AXIN1, TBXT, and TULP3 (Table 1) respectively. The homozygous AMBRA1 variant was absent from the control database gnomAD and has not been previously reported in SB individuals. The heterozygous variants in AXIN1, TBXT, and TULP3 were either unreported or present heterozygously in <10 of 125,748 control individuals in the gnomAD database (see Table 1). All variants were predicted as deleterious by at least three of four algorithms (PolyPhen-2, Mutation Taster, SIFT, and CADD) (Table 1). The amino acid residues at the positions were evolutionarily conserved (Table 1).
Figure 2. WES in 50 cases with SB identifies 6 potential SB genes from 136 potential candidate genes and 12 potential novel SB genes from unbiased WES analysis.
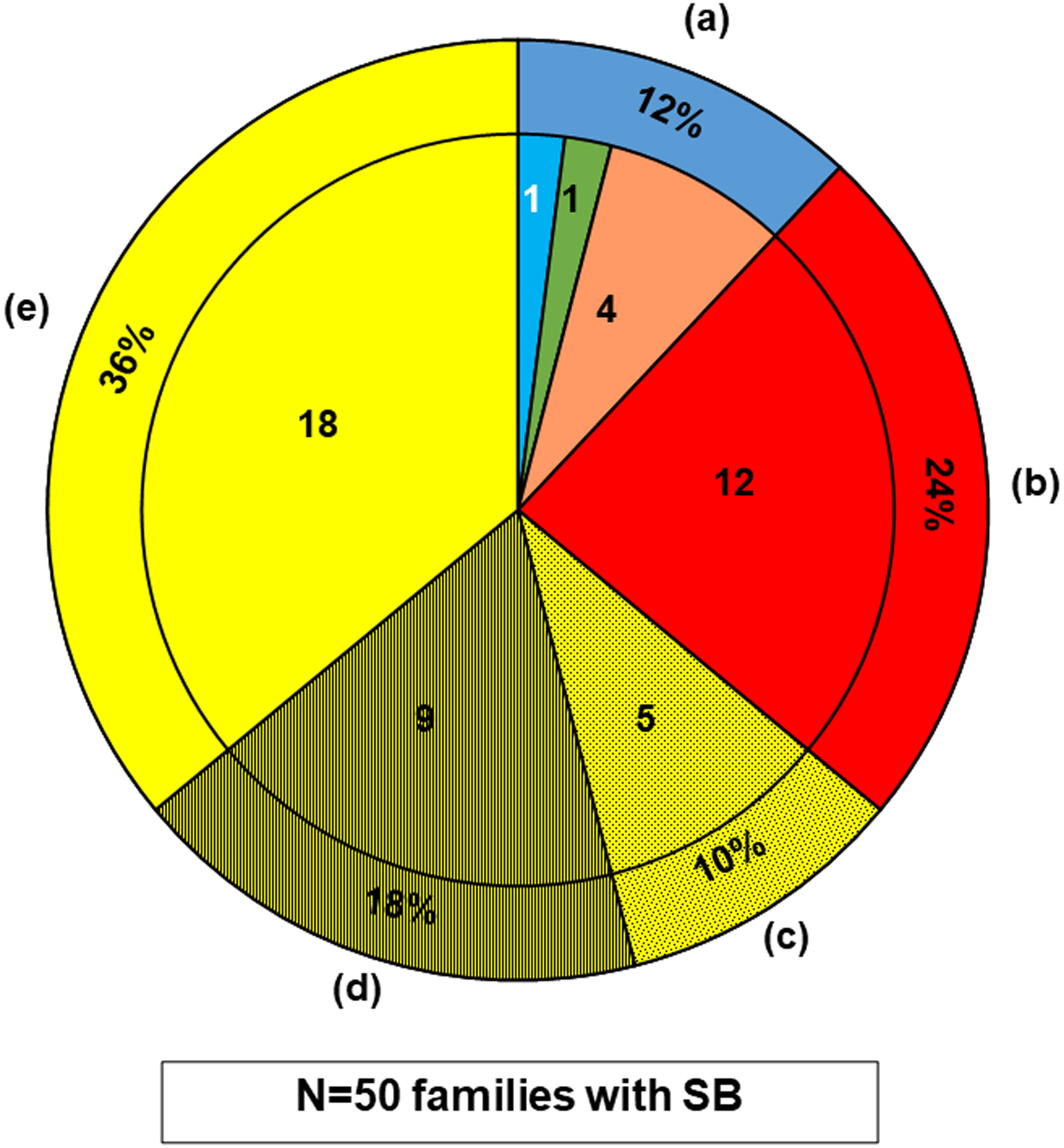
Whole exome sequencing (WES) data of 50 affected individuals with SB from 50 unrelated families was analyzed for likely deleterious variants in the candidate genes assumed to cause, if mutated.
The outer ring shows the percentage, and the inner segments shows the number of families for the 50 SB cases.
The pie chart summarizes the findings for all 50 SB cases, which is divided into the following subgroups:
(a)The outer ring dark blue segment denotes the 6/50 (12%) cases, in whom a likely deleterious variant in one of the potential SB candidate genes we generated was detected (Table 1). The inner sub-segments show a breakdown of these genes by their origin from the predefined candidate gene lists (Tables S2–S5), it shows that a likely deleterious variant was detected in an SB candidate gene (see Fig.1) in 6 cases (12%), i.e., 4 variants in mouse SB candidate genes (orange); and 1 variant in a candidate gene known to potentially cause human non-syndromic SB (light blue); and 1 variant in a potential candidate gene known to cause a human syndrome with facultative SB (green).
(b)The red segment denotes the 12/50 (24%) cases, in whom a likely deleterious variant in a potential novel gene was detected by WES (Table 2).
(c)The dotted yellow segment denotes the 5/50 (10%) cases, in whom likely deleterious variants in more than one gene were identified without a preferred candidate, leaving them inconclusive (Table S7).
(d)The hatched yellow segment denotes the 9/50 (18%) cases, in whom likely deleterious variants in a single candidate gene were detected, but negative reverse phenotyping, conflicting segregation analysis, or a comparable burden in our in-house control cohort (Table S8) rendered them unlikely deleterious.
(e)The clear yellow segment denotes the 18/50 (36%) cases, in whom no likely deleterious variant was detected
Table 1. Information on likely deleterious variants in 6 of 36 potential SB candidate genes known to cause SB in mice, humans with non-syndromic SB, or humans with clinical syndromes with facultative SB.
Families are listed by corresponding candidate gene category: A) mouse SB candidate gene (orange); B) candidate genes known to potentially cause human non-syndromic SB (blue); C) candidate genes known to cause human syndromes with facultative SB (green).
| Family ID | Gene | NM number | Nucleotide change | Amino acid change | State | Evolutionary conservationa phastCons / phyloP | PP2 SIFT MT | CADD Score | EVSb | gnom ADc | HGMDd OMIM# | Phenotypes In this patient | Segregation (M,P) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B3747 | AMBRA1 | NM_001267782.1 | c.2266C>T | p.Leu 756Phe |
hom |
D. rerio 1.00 / 5.88 |
0.99 Tol. D.C |
29.9 | NP | 0/31/251,478 | Gene | Myelomeningocele, neurogenic bladder, Chiari malformation type 2 | M: NA P: NA |
| B1337 | AXIN1 | NM_003502.3 | c.1003C>T | p.Leu 335Phe |
het |
D. rerio 1.00 / 3.13 |
0.99 Del. D.C |
23.4 | NP | 0/1/250,476 | Gene Disease (#603816)e |
spina bifida occulta, BL RHD, facial dysmorphism | M: NA P: NA |
| B4121 | TBXT | NM_001366285.2 | c.301C>T | p.Arg 101Cys |
het |
D. melanogaster 1.00 / 2.58 |
1.00 Del. D.C. |
28.9 | NP | NP | Gene Disease (#1615709, #182940)f |
Myelomeningocele, tethered spinal cord, neurogenic bladder, neurogenic bowel, Chiari malformation type 2, hydrocephalus, hip deformity, accommodative esotropia | M: WT P: NA |
| B4197 | TULP3 | NM_001160408.1 | c.703C>T | p.Leu 235Phe |
het |
D. rerio 1.00 / 1.75 |
1.00 Del. D.C |
26.0 | 0/2/4,298 | 0/8/282,550 | Gene | Myelomeningocele, neurogenic bladder, neurogenic bowel, Chiari malformation type 2, hydrocephalus | Adopted M: NA P: NA |
| B4126 | PRICKLE1 | NM_001144881.1 | c.311G>A | p.Arg 104Gln |
het |
D. rerio 1.00 / 4.45 |
1.00 Del. D.C |
28.8 | 0/1/2,202 | 0/5/282,818 | DM Disease (#608500)g |
Myelomeningocele, neurogenic bladder, hydrocephalus, gastroesophageal reflux disease | M: WT P: NA MGM: WT |
| B4072 | FOXC2 | NM_005251.3 | c.127G>C | p.Gly 43Arg |
het |
G. gallus 1.00 / 2.30 |
0.977 Del / |
25.6 | NP | NP | Gene Disease (#153400)h |
Myelomeningocele, hydronephrosis, BL VUR, growth retardation, skeletal deformity, club feet, hydrocephalus, divergent squint, edema, cellulitis | M: NA P: NA |
BL, bilateral; CADD, Combined Annotation Dependent Depletion (https://cadd.gs.washington.edu/); D.C., Disease Causing; Del., Deleterious; EVS, Exome Variant Server; gnomAD, Genome Aggregation Database; MGM, maternal grandmother; hom, homozygous; het, heterozygous; M, Maternal; MT, Mutation Taster (http://www.mutationtaster.org); NA, not available; NP, not present; P, Paternal; PP2, PolyPhen 2 (http://genetics.bwh.harvard.edu/pph2); RHD, renal/hypodysplasia; SIFT, Sorting intolerant from tolerant (http://sift.jcvi.org); Tol, tolerate; VUR, vesicoureteral reflux. WT, wildtype.
Evolutionary conservation was assessed across phylogeny: G. gallus, Gallus gallus; D. rerio, Danio rerio; D. melanogaster, Drosphilia melanogaster.
Variant frequencies listed for homozygous/ heterozygous/ total alleles detected in the gnomAD population.
HGMD, (https://portal.biobaseinternational.com/hgmd). If the exact variant has previously been reported and classified as a pathogenic mutation to be disease causing, the variant is denoted as “DM”. If the gene, but not the exact variant has been reported for the corresponding phenotype, “Gene” is indicated.
AXIN1 gene is known to cause caudal duplication anomaly in OMIM.
TBXT gene is known to cause sacral agenesis with vertebral anomalies, as well as known as risk factor gene in OMIM.
PRICKLE1 gene is known to cause epilepsy, progressive myoclonic 1B type in OMIM.
FOXC2 gene is known to cause lymphedema-distichiasis syndrome in OMIM.
By WES evaluation of the 27 genes with evidence of potential SB involvement in humans (Fig. 1B, Table S2), we identified one likely deleterious heterozygous variant in the candidate gene PRICKLE1 in an individual with non-syndromic SB features (Fig. 2, inner ring, Table 1; light blue segment). The Arg104 residue in PRICKLE1 occurred 5 times heterozygously in the gnomAD database and yielded predominantly deleterious prediction scores by all four algorithms (PolyPhen-2, MutationTaster, SIFT, and CADD; Table 1). The variant is well conserved across evolution from Homo sapiens to Danio rerio (Table 1). Segregation analysis showed that neither the healthy mother nor the maternal grandmother carried this variant, the father’s DNA was not available for segregation analysis.
Finally, by WES evaluation of the 66 genes known to cause human clinical syndromes with facultative SB features, likely deleterious variants can be detected in individuals with syndromic SB features (Fig. 1C, Table S4). We detected one likely deleterious heterozygous variant in the syndromic SB candidate gene FOXC2 in one individual with syndromic SB (Figure 2a, inner ring, Table 1; green segment). This variant is unreported in the gnomAD database and was predicted to be deleterious by three of four algorithms (PolyPhen-2, SIFT, and CADD) (Table 1). Heterozygous FOXC2 mutations are known to cause Lymphedema-distichiasis syndrome (LDS; OMIM#153400), and a milder SB phenotype (spinal extradural cysts) has been reported in human LDS individuals (Ogura et al., 2015). By reverse phenotyping, i.e., requesting the physician to reexamine the patient regarding related symptoms of LDS in OMIM, we found that our patient has additional symptoms of LDS (see Table 1), which strengthens this FOXC2 variant as likely deleterious in this family.
All 6 variants we identified in the 136 potential candidate genes in 50 SB cases were absent from 50 matched in-house negative individuals with the clinical diagnosis of steroid resistant nephrotic syndrome (SRNS), in whom a definitive underlying monogenic cause had already been established (Table S8). In addition, we performed WES evaluation in parallel in these 6 families and did not find any potential candidate genes. Altogether, this approach renders these six genes as the strongest potential candidate genes for human SB in these six families.
Unbiased Exome-wide Analysis Identifies 12 Potential Candidate Genes in 12 of 50 (24%) Cases with SB
Next, we searched for potential novel candidate genes for SB by an unbiased exome-wide analysis in the 50 SB cases. We therefore evaluated WES data for (1) recessive genes by evaluation of homozygous regions in consanguineous families (Hildebrandt et al., 2009) (3 of 50; 6%) or (2) recessive and/or dominant mutations by Singlet, Duo, and Trio analysis depending on pedigree structure (Fig. 1). Since the parents of these 50 individuals were all reported as unaffected individuals, we searched for biallellic variants of an autosomal recessive gene in these families or a de novo variant of an autosomal dominant gene in these families. After filtering variants based on a priori genetic criteria (see Methods, Table S5), we identified each a single potential candidate gene in 12 of the 50 cases (24%) with SB (Fig. 2b, Table 2; red segment).
Table 2. Information on potentially deleterious variants identified per family.
Families are listed in alphabetical order by gene symbol.
| Family ID | Gene | NM number | Nucleotide change | Amino acid change | State | Evolutionary conservationa phastCons / phyloP | PP2 SIFT MT | CADD Score | EVSb | gnom ADc | HGMDd | Phenotypes | Segregation (M,P) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B4212 | ATG2B | NM_018036.6 | c.2938G>A | p.Glu 980Lys |
het |
X. tropicalis 1.00 / 5.77 |
0.83 Del. / |
24.6 | NP | 0/4/280,668 | Gene | Lipomeningocele, tethered cord, neurogenic bladder, L VUR, neurogenic bowel, acrocyanosis | M: WT P: het |
| c.1246T>A | p.Ser 416Thr |
het |
X. tropicalis 1.00 / 4.92 |
0.98 Del. / |
23.7 | NP | NP | Gene | h: Het P: WT |
||||
| B4124 | EWSR1 | NM_013986.3 | c.13+4A>G | ess | Hom | / 0.99 / 2.15 | / | / | 0/1/2202 | 0/11/281,120 | Gene | Myelomeningocele, thoracolumbar, myelodysplasia, kyphoscoliosis and scoliosis, myelodysplastic syndrome, neurogenic bladder, neurogenic bowel, Chiari malformation type 2, hydrocephalus, intellectual disability, epilepsy, porencephaly, thrombocytopenia, | M: NA P: het |
| B4109 | GPR83 | NM_016540.3 | c.820G>C | p.Asp 274His |
het |
D. rerio 1.00 / 5.63 |
1.00 Del. D.C. |
24.2 | NP | NP | NP | Lipomyelomeningocele, neurogenic bladder BL VUR, neurogenic bowel, Chiari malformation type 2, hydrocephalus |
M: WT P: WT |
| B4271 | IGBP1 | NM_001551.2 | c.212T>C | p.Ile 71Thr |
hemi |
C. intestinalis 1.00 / 3.60 |
0.93 Del. D.C. |
26.4 | NP | NP | Gene | Myelomeningocele, neurogenic bladder, neurogenic bowel, Chiari malformation type 2, hydrocephalus, hip flexion contracture | M: het P: WT |
| B4104 | MAML1 | NM_014757.4 | c.202G>A | p.Ala 68Thr |
het |
D. rerio 1.00 / 4.56 |
0.99 Del. D.C. |
25 | NP | 0/5/101,280 | Gene | Myelomeningocele, neurogenic bladder, neurogenic bowel, Chiari malformation type 2, hydrocephalus, macrocephaly | M: het P: NA |
| c.1657G>A | p.Glu 553Lys |
het |
X. tropicalis 1.00 / 3.46 |
0.93 Del. D.C. |
24.4 | 0/16/ 4284 |
1/443/ 276,670 |
Gene | M: WT P: NA |
||||
| B4127 | MTMR8 | NM_017677.3 | c.866–2 A>G |
100% ESS |
hemi | / 1.00 / 1.84 | / | / | NP | 0/0/1/166,539 | Gene | Myelomeningocele, tethered spinal cord, neurogenic bladder PUV, BL hydronephrosis, neurogenic bowel, Chiari malformation type 2, corpus callosum agenesis, | M: het P: NA Sib: het |
| B4225 | MAGI3 | NM_0042782.1 | c.706C>G | p.Leu 236Val |
het |
X. tropicalis 1.00 / 4.03 |
0.13 Del. D.C. |
18.7 | 0/12/4,288 | 1/310/282,358 | Gene | Myelomeningocele, neurogenic bladder, Chiari malformation type 2, hydrocephalus | M: het P: NA |
| c.1786C>A | p.Pro 596Thr |
het |
D. rerio 1.00 / 5.74 |
0.99 Del. D.C. |
26.0 | NP | 0/1/251,442 | Gene | M: WT P: NA |
||||
| B4110 | NUP205 | NM_015135.2 | c.1218+4 delA |
ess | het | / 0.98 / 2.15 | / | / | 0/1/4,126 | 0/14/279,876 | Gene | Myelomeningocele, Neurogenic bladder, renal scar, neurogenic bowel, Chiari malformation type II, hydrocephalus, hypercholesterolemia | M: WT P: NA |
| c.5386C>T | p.Arg 1796Trp |
het |
X. tropicalis 0.99 / 0.24 |
0.99 Del. D.C. |
24.8 | NP | 0/25/282,834 | Gene | M: het P: NA |
||||
| B4144 | PIK3R4 | NM_014602.2 | c.1039C>T | p.Arg 347Trp |
hom |
D. rerio 1.00 / 2.75 |
0.63 Tol. D.C. |
27.6 | 0/14/4,286 | 0/369/282,816 | Gene | Myelomeningocele, scoliosis, neurogenic bladder, neurogenic bowel, Chiari malformation type 2, stenogyria, thinning of corpus callosum | M: het P: NA |
| B3893 | TSPEAR | NM_144991.2 | c.668C>T | p.Ser 223Leu |
hom |
D. rerio 1.00 / 5.47 |
0.52 Del. D.C. |
26.8 | 0/4/4,296 | 1/201/276,8820 | Gene | Myelomeningocele, neurogenic bladder, Chiari malformation type 2, hydrocephalus | M: NA P: NA |
| B4303 | TTC21A | NM_145755.2 | c.1416+1 G>A |
100% ESS |
het | / 1.00 / 5.55 | / | / | 0/13/4,140 | 0/100/279,732 | Gene | Myelomeningocele, scoliosis, neurogenic bladder, BL hydronephrosis, neurogenic bowel, Chiari malformation type 2, hydrocephalus, restrict lung disease | M: het P: NA |
| c.2910delA | p.Lys 970fs28 |
het | / 1.00 / 0.92 | / | / | 0/1/3,931 | 0/5/280,964 | Gene | M: WT P: NA |
||||
| B4194 | ZNF790 | NM_001242800.1 | c.964C>T | p.Arg 322* |
het | / 0.95 / 2.45 | / | / | 1/34/2,168 | 1/163/281,656 | Gene | Myelomeningocele, neurogenic bladder, neurogenic bowel, Chiari malformation type 2, coxa valga | M: het P: NA |
| c.904A>T | p.Arg 302* |
het | / 0.00 / −0.60 | / | / | NP | 0/1/314,06 | Gene | M: WT P: NA |
BL, bilateral; CADD, Combined Annotation Dependent Depletion (https://cadd.gs.washington.edu/); D.C., Disease Causing; Del., Deleterious; del; deletion, ESS, Essential Splice Site; ess, extended splice site; EVS, Exome Variant Server; fs, frameshift; gnomAD, Genome Aggregation Database; het, heterozygous; Hom, homozygous; hemi, hemizygous; L, left; M, Maternal; MT, Mutation Taster (http://www.mutationtaster.org); NA, not available; NP, not present; P, Paternal; PP2, PolyPhen 2 (http://genetics.bwh.harvard.edu/pph2); PUV, posterior urethral valve; RHD, renal/hypodysplasia; SIFT, Sorting intolerant from tolerant (http://sift.jcvi.org); Sib, sibling; Tol, tolerated; VUR, vesicoureteral reflux. WT, wildtype.
Evolutionary conservation was assessed across phylogeny: X. tropicalis, Xenopus tropicalis; D. rerio, Danio rerio; C. intestinalis, Ciona intestinalis.
Variant frequencies listed for homozygous/ hemizygous (if applicable)/ heterozygous/ total alleles detected in the population.
HGMD, (https://portal.biobaseinternational.com/hgmd). If the exact variant has previously been reported and classified as a pathogenic mutation to be disease causing, variant denoted as “DM”. If the gene but not the exact variant has been reported for the corresponding phenotype “Gene” is indicated.
Specifically, we identified three homozygous/hemizygous missense variants in three genes (IGBP1, PIK3R4, and TSPEAR) in three SB individuals, one hemizygous essential splice site variant in MTMR8 gene in one SB individual, one homozygous extended splice site mutation in EWSR1 gene in one SB individual, and six compound heterozygous variants in six genes (ATG2B, MAML1, MAGI3, NUP205, TTC21A, and ZNF790) (Table 2, Fig. 2) in six SB individuals respectively. We also identified one de novo heterozygous missense variant in GPR83 gene (Table 2). The variants in these novel genes were absent from our in-house SRNS negative control cohort of 50 individuals (Table S8). The detailed information about these novel genes is summarized in Table 2.
In order to include a larger cohort of SB cases, we screened our collaborator’s spina bifida WES cohort of 511 myelomeningocele individuals (Hebert et al., 2020) for the variants in the total of 18 potential candidate genes. However, no likely deleterious variants were identified in these 18 genes.
In additional 5 SB cases listed in Table S6, multiple candidate genes were detected. All variants identified in these 5 SB cases were predicted as deleterious and well conserved in mutant amino acid residues, as well as absent from 50 matched in-house control individuals with the clinical diagnosis of SRNS.
Discussion
In this study, we performed WES in an international cohort of 50 SB cases for a list of 136 potential candidate genes that were derived from: (A) 43 candidate genes derived from mouse models, (B) 27 potential candidate genes derived isolated human SB genes, and (C) 66 potential candidate genes derived from monogenic human syndromes with SB features. We detected a likely deleterious variant in 6 of the 136 potential candidate genes in 6 (6/50, 12%) SB cases. In addition, by unbiased exome-wide analysis, we identified 12 potential candidate genes for SB in 12 of 50 (24%) SB cases.
To our knowledge, our study is the first comprehensive study that combines candidate gene evaluation with an unbiased exome-wide sequencing evaluation to test the genetic architecture of SB. Several studies have been performed to test the genetic nature of neural tube defect or spina bifida. Beaumont et al. found that targeted panel sequencing enabled detection of candidate pathogenic variants in up to 36% for 52 NTDs/SB individuals (Beaumont, et al., 2019). However, they did not test for the presence or absence of these variants in a negative control population. They also included all the variants inherited from unaffected healthy parents, potentially explaining their high percentage of variant detection in known candidate genes. Hebert et al. evaluated the burden of rare deleterious variants in WNT signaling genes among 511 myelomeningocele patients and showed that ten WNT signaling genes were disrupted with a higher mutational burden among Mexican American myelomeningocele subjects compared to reference subjects (Hebert, et al., 2020). Lemay et al showed that, by applying WES in 43 trio NTDs families, loss-of-function (LOF) de novo mutations were identified in 14% (6 of 43) individuals. Importantly, 3 of 6 LOF variants in two genes (SHROOM3, PAX3) are known as monogenic causes of SB in mouse SB (Lemay et al., 2015).
Human SB Candidate Genes Can Be Derived from Murine SB Genes
Mouse SB models have been studied to understand the disease mechanism of SB, as they provide functional evidence for genes causing human SB (reviewed in (Lee & Gleeson, 2020)). Mouse models clearly replicate SB phenotypes (Table S2), so we considered that SB mouse models as generating the strongest hypothesis for a candidate gene list (Fig. 1A). Of note, this is only a subset of a large list of mouse models with a neural tube defect phenotype (Lee & Gleeson, 2020), as we used a primarily MGI-based approach to identify potential candidate genes for our analysis (Fig. 1A). We identified likely deleterious variants in 8% (4 of 50) of cases with SB in one recessive gene (AMBRA1) and three dominant genes (AXIN1, TBXT, and TULP3). The potential causative role of these variants is strengthened by the finding that we did not find any competing likely deleterious candidate genes by unbiased exome-wide analysis in these 4 cases. In addition, no deleterious variants in these four genes were found in 50 cases’ data of in-house negative control. However, the variant in TUPL3 has been reported to be present heterozygously in 8 individuals in gnomAD (Table S2). Although gnomAD aims at removing individuals with severe pediatric diseases(Karczewski et al., 2020), there might still be individuals with hypomorphic SB phenotypes (i.e., spina bifida occulta, which has potentially not been established as a clinical diagnosis) included in the dataset. Combining all evidence, we consider these 4 genes as the strongest potential candidates.
Our previous studies showed that WES is a powerful approach to identify causative monogenic genes in congenital anomalies of the kidney and urinary tract (van der Ven, Connaughton, et al., 2018) Many established CAKUT genes encode transcription factors and follow a dominant pattern of inheritance (reviewed in (van der Ven, Vivante, et al., 2018)). Given that, 1) SB is a developmental disorder, 2) the fact that SB and CAKUT share several pathogenic pathways, such as the PCP/Wnt pathway and sonic hedgehog signaling pathway, and 3) and that both SB and CAKUT can be caused by genes that encode transcription factors, like FOX genes (FOXC1, FOXC2), we hypothesized that similar to CAKUT, there may be a high percentage of SB with genetic causes. They might also exhibit features of incomplete penetrance and variable expressivity. As is the case in monogenic CAKUT genes, to answer this question will require more evidence from large pedigrees with affected individuals from multiple generations as well as functional studies.
Potential Candidate Genes Can Be Derived from WES
Using unbiased exome-wide evaluation in different family structures, we were able to identify likely deleterious variants in 12/50 cases (24%) with SB (Fig. 1b, Table 2). However, we did not identify a second SB family with a variant in these 12 genes in a larger cohort of 511 MMC individuals. The identification of additional families is difficult because of the rarity of these genes in rare diseases, phenotypic heterogeneity, and multiple genes causing the same phenotype. Additional families and further experimental evidence are needed to confirm the causality of these potential candidate genes. In 5/50 (10%) of 50 SB cases, after variant filtering, we were left with multiple potential causative genes (Table S6).
Limitation of the Study
In total, 18 of 50 cases with SB (36%) remained without any findings (Fig. 2e). Similar to the WES study on CAKUT, no variant was detected in 56% cases (van der Ven, Connaughton, et al., 2018), and in steroid-resistant nephrotic syndrome, no mutation was detected in 44% cases (Warejko et al., 2018). The reason for this is likely multifold. First, it has been demonstrated that loss-of-function de novo variants can be detected in 14% of individuals with NTDs [40], which can be difficult to find in our cohort since we only have four trios. Second, it has been shown that de novo copy number variants were detected in 19% of SB cases, which can be difficult to detect using WES (Bassuk et al., 2013; Wolujewicz et al., 2021). Third, recently, one study showed somatic mutations in PCP genes in neural tissue from human fetuses with NTDs, which cannot be detected through a genomic DNA extracted from blood sample (Tian et al., 2020).
Conclusion
For each of these potential candidate genes for monogenic SB, we need more cases with likely deleterious variants in the same gene to strengthen the causality, and loss-of-function studies should be conducted before it can be accepted as a novel SB gene.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to the families who contributed to this study. We thank Wanxia Wu for excellent technical assistance and Meredith Weaver, Alana Gerald, Leslie Spaneas, and Rebecca Sherlock for recruitment of patients at Boston Children’s Hospital.
F.H. is the William E. Harmon Professor of Pediatrics at Harvard Medical School. This research was supported by grants from the National Institutes of Health to F.H. (DK076683). Sequencing and data processing was performed by the Yale Centers for Mendelian Genomics funded by the National Human Genome Research Institute (U54 HG006504). S.Se. is supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation; 442070894). B.Z. is supported by the program of China Scholarships Council (No. 201908320472). C.-H.W.W. is supported by funding from the National Institutes of Health (grant T32-GM007748) and the American College of Medical Genetics and Genomics Foundation (ACMG/Takeda Next-Generation Biochemical Genetics Award). K.S.A. is supported by funding from the National Institutes of Health (R01HD073434). N.M. is supported by funding from the National Institutes of Health T32-DK007726 grant and by an NIHK08 funding (#DK127011). T.M.K. was supported by a Post-Doctoral Fellowship award from the KRESCENT Program, a national kidney research training partnership of the Kidney Foundation of Canada, the Canadian Society of Nephrology, and the Canadian Institutes of Health Research. D.M.C. is funded by Health Research Board, Ireland (HPF-206-674), the International Pediatric Research Foundation Early Investigators’ Exchange Program and the Amgen® Irish Nephrology Society Specialist Registrar Bursary. She is also funded by the Eugen Drewlo Chair for Kidney Research and Innovation at the Schulich School of Medicine & Dentistry at Western University, London, Ontario, Canada. C.E., is supported by Nanji Myelodysplasia Research Fund. F.H. and S.Sh. are supported by grants from the Begg Family Foundation and by the Isabella Forrest Julian Research Fund for Pediatric Post Kidney Transplant Research.
Footnotes
CONFLICT OF INTEREST STATEMENT
F.H. is a cofounder and S.A.C. member of Goldfinch-Bio. All other authors declare that they have no competing financial interests.
DATA AVAILABILITY STATEMENT
Depersonalized data that this study is based on is available from the corresponding author upon request.
REFERENCES
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, … Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nat Methods, 7(4), 248–249. doi: 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrés-Jensen L, Jørgensen FS, Thorup J, Flachs J, Madsen JL, Maroun LL, … Cortes D (2016). The outcome of antenatal ultrasound diagnosed anomalies of the kidney and urinary tract in a large Danish birth cohort. Arch Dis Child, 101(9), 819–824. doi: 10.1136/archdischild-2015-309784 [DOI] [PubMed] [Google Scholar]
- Bassuk AG, Muthuswamy LB, Boland R, Smith TL, Hulstrand AM, Northrup H, … Manak JR (2013). Copy number variation analysis implicates the cell polarity gene glypican 5 as a human spina bifida candidate gene. Hum Mol Genet, 22(6), 1097–1111. doi: 10.1093/hmg/dds515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont M, Akloul L, Carre W, Quelin C, Journel H, Pasquier L, … David V (2019). Targeted panel sequencing establishes the implication of planar cell polarity pathway and involves new candidate genes in neural tube defect disorders. Hum Genet, 138(4), 363–374. doi: 10.1007/s00439-019-01993-y [DOI] [PubMed] [Google Scholar]
- Berry RJ, Li Z, Erickson JD, Li S, Moore CA, Wang H, … Correa A (1999). Prevention of neural-tube defects with folic acid in China. China-U.S. Collaborative Project for Neural Tube Defect Prevention. N Engl J Med, 341(20), 1485–1490. doi: 10.1056/NEJM199911113412001 [DOI] [PubMed] [Google Scholar]
- Braun DA, Sadowski CE, Kohl S, Lovric S, Astrinidis SA, Pabst WL, … Hildebrandt F (2016). Mutations in nuclear pore genes NUP93, NUP205 and XPO5 cause steroid-resistant nephrotic syndrome. Nat Genet, 48(4), 457–465. doi: 10.1038/ng.3512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caiulo VA, Caiulo S, Gargasole C, Chiriacò G, Latini G, Cataldi L, & Mele G (2012). Ultrasound mass screening for congenital anomalies of the kidney and urinary tract. Pediatr Nephrol, 27(6), 949–953. doi: 10.1007/s00467-011-2098-0 [DOI] [PubMed] [Google Scholar]
- Chen Z, Lei Y, Cao X, Zheng Y, Wang F, Bao Y, … Wang H (2018). Genetic analysis of Wnt/PCP genes in neural tube defects. BMC Med Genomics, 11(1), 38. doi: 10.1186/s12920-018-0355-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connaughton DM, Dai R, Owen DJ, Marquez J, Mann N, Graham-Paquin AL, … Hildebrandt F (2020). Mutations of the Transcriptional Corepressor ZMYM2 Cause Syndromic Urinary Tract Malformations. Am J Hum Genet, 107(4), 727–742. doi: 10.1016/j.ajhg.2020.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp AJ, Adzick NS, Chitty LS, Fletcher JM, Holmbeck GN, & Shaw GM (2015). Spina bifida. Nat Rev Dis Primers, 1, 15007. doi: 10.1038/nrdp.2015.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp AJ, & Greene N (2010). Genetics and development of neural tube defects. The Journal of Pathology, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp AJ, Stanier P, & Greene ND (2013). Neural tube defects: recent advances, unsolved questions, and controversies. Lancet Neurol, 12(8), 799–810. doi: 10.1016/S1474-4422(13)70110-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detrait ER, George TM, Etchevers HC, Gilbert JR, Vekemans M, & Speer MC (2005). Human neural tube defects: developmental biology, epidemiology, and genetics. Neurotoxicol Teratol, 27(3), 515–524. doi: 10.1016/j.ntt.2004.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris MJ, & Juriloff DM (2007). Mouse mutants with neural tube closure defects and their role in understanding human neural tube defects. Birth Defects Res A Clin Mol Teratol, 79(3), 187–210. doi: 10.1002/bdra.20333 [DOI] [PubMed] [Google Scholar]
- Harris MJ, & Juriloff DM (2010). An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth Defects Res A Clin Mol Teratol, 88(8), 653–669. doi: 10.1002/bdra.20676 [DOI] [PubMed] [Google Scholar]
- Hebert L, Hillman P, Baker C, Brown M, Ashley-Koch A, Hixson JE, … Au KS (2020). Burden of rare deleterious variants in WNT signaling genes among 511 myelomeningocele patients. PLoS One, 15(9), e0239083. doi: 10.1371/journal.pone.0239083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt F, Benzing T, & Katsanis N (2011). Ciliopathies. N Engl J Med, 364(16), 1533–1543. doi: 10.1056/NEJMra1010172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt F, Heeringa SF, Ruschendorf F, Attanasio M, Nurnberg G, Becker C, … Otto EA (2009). A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS Genet, 5(1), e1000353. doi: 10.1371/journal.pgen.1000353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juriloff DM, & Harris MJ (2012). A consideration of the evidence that genetic defects in planar cell polarity contribute to the etiology of human neural tube defects. Birth Defects Res A Clin Mol Teratol, 94(10), 824–840. doi: 10.1002/bdra.23079 [DOI] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, … MacArthur DG (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), 434–443. doi: 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibar Z, Salem S, Bosoi CM, Pauwels E, De Marco P, Merello E, … Gros P (2011). Contribution of VANGL2 mutations to isolated neural tube defects. Clin Genet, 80(1), 76–82. doi: 10.1111/j.1399-0004.2010.01515.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibar Z, Torban E, McDearmid JR, Reynolds A, Berghout J, Mathieu M, … Gros P (2007). Mutations in VANGL1 associated with neural-tube defects. N Engl J Med, 356(14), 1432–1437. doi: 10.1056/NEJMoa060651 [DOI] [PubMed] [Google Scholar]
- Kim SE, Lei Y, Hwang SH, Wlodarczyk BJ, Mukhopadhyay S, Shaw GM, … Finnell RH (2019). Dominant negative GPR161 rare variants are risk factors of human spina bifida. Hum Mol Genet, 28(2), 200–208. doi: 10.1093/hmg/ddy339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl S, Habbig S, Weber LT, & Liebau MC (2021). Molecular causes of congenital anomalies of the kidney and urinary tract (CAKUT). Mol Cell Pediatr, 8(1), 2. doi: 10.1186/s40348-021-00112-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp JB, Anders HJ, Susztak K, Podesta MA, Remuzzi G, Hildebrandt F, & Romagnani P (2020). Podocytopathies. Nat Rev Dis Primers, 6(1), 68. doi: 10.1038/s41572-020-0196-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, & Ng PC (2009). Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc, 4(7), 1073–1081. doi: 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- Lee S, & Gleeson JG (2020). Closing in on Mechanisms of Open Neural Tube Defects. Trends Neurosci, 43(7), 519–532. doi: 10.1016/j.tins.2020.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Y, Zhu H, Yang W, Ross ME, Shaw GM, & Finnell RH (2014). Identification of novel CELSR1 mutations in spina bifida. PLoS One, 9(3), e92207. doi: 10.1371/journal.pone.0092207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemay P, Guyot MC, Tremblay E, Dionne-Laporte A, Spiegelman D, Henrion E, … Kibar Z (2015). Loss-of-function de novo mutations play an important role in severe human neural tube defects. J Med Genet, 52(7), 493–497. doi: 10.1136/jmedgenet-2015-103027 [DOI] [PubMed] [Google Scholar]
- Mann N, Braun DA, Amann K, Tan W, Shril S, Connaughton DM, … Hildebrandt F (2019). Whole-Exome Sequencing Enables a Precision Medicine Approach for Kidney Transplant Recipients. J Am Soc Nephrol, 30(2), 201–215. doi: 10.1681/ASN.2018060575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohd-Zin SW, Marwan AI, Abou Chaar MK, Ahmad-Annuar A, & Abdul-Aziz NM (2017). Spina Bifida: Pathogenesis, Mechanisms, and Genes in Mice and Humans. Scientifica (Cairo), 2017, 5364827. doi: 10.1155/2017/5364827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura Y, Fujibayashi S, Iida A, Kou I, Nakajima M, Okada E, … Ikegawa S (2015). A novel FOXC2 mutation in spinal extradural arachnoid cyst. Hum Genome Var, 2, 15032. doi: 10.1038/hgv.2015.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. MRC Vitamin Study Research Group. (1991). Lancet, 338(8760), 131–137. [PubMed] [Google Scholar]
- Reller MD, Strickland MJ, Riehle-Colarusso T, Mahle WT, & Correa A (2008). Prevalence of congenital heart defects in metropolitan Atlanta, 1998–2005. J Pediatr, 153(6), 807–813. doi: 10.1016/j.jpeds.2008.05.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzsch P, Witten D, Cooper GM, Shendure J, & Kircher M (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res, 47(D1), D886–D894. doi: 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, … Hildebrandt F (2015). A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol, 26(6), 1279–1289. doi: 10.1681/ASN.2014050489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salih MA, Murshid WR, & Seidahmed MZ (2014). Epidemiology, prenatal management, and prevention of neural tube defects. Saudi Med J, 35 Suppl 1, S15–28. [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Cooper DN, Schuelke M, & Seelow D (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods, 11(4), 361–362. doi: 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Tian T, Lei Y, Chen Y, Karki M, Jin L, Finnell RH, … Ren A (2020). Somatic mutations in planar cell polarity genes in neural tissue from human fetuses with neural tube defects. Hum Genet, 139(10), 1299–1314. doi: 10.1007/s00439-020-02172-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torban E, Wang HJ, Groulx N, & Gros P (2004). Independent mutations in mouse Vangl2 that cause neural tube defects in looptail mice impair interaction with members of the Dishevelled family. J Biol Chem, 279(50), 52703–52713. doi: 10.1074/jbc.M408675200 [DOI] [PubMed] [Google Scholar]
- van der Ven AT, Connaughton DM, Ityel H, Mann N, Nakayama M, Chen J, … Hildebrandt F (2018). Whole-Exome Sequencing Identifies Causative Mutations in Families with Congenital Anomalies of the Kidney and Urinary Tract. J Am Soc Nephrol, 29(9), 2348–2361. doi: 10.1681/ASN.2017121265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Ven AT, Vivante A, & Hildebrandt F (2018). Novel Insights into the Pathogenesis of Monogenic Congenital Anomalies of the Kidney and Urinary Tract. J Am Soc Nephrol, 29(1), 36–50. doi: 10.1681/ASN.2017050561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Marco P, Capra V, & Kibar Z (2019). Update on the Role of the Non-Canonical Wnt/Planar Cell Polarity Pathway in Neural Tube Defects. Cells, 8(10). doi: 10.3390/cells8101198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warejko JK, Tan W, Daga A, Schapiro D, Lawson JA, Shril S, … Hildebrandt F (2018). Whole Exome Sequencing of Patients with Steroid-Resistant Nephrotic Syndrome. Clin J Am Soc Nephrol, 13(1), 53–62. doi: 10.2215/CJN.04120417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolujewicz P, Aguiar-Pulido V, AbdelAleem A, Nair V, Thareja G, Suhre K, … Ross ME (2021). Genome-wide investigation identifies a rare copy-number variant burden associated with human spina bifida. Genet Med, 23(7), 1211–1218. doi: 10.1038/s41436-021-01126-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CW, Mann N, Nakayama M, Connaughton DM, Dai R, Kolvenbach CM, … Hildebrandt F (2020). Phenotype expansion of heterozygous FOXC1 pathogenic variants toward involvement of congenital anomalies of the kidneys and urinary tract (CAKUT). Genet Med, 22(10), 1673–1681. doi: 10.1038/s41436-020-0844-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Tong Y, Lv J, Peng R, Chen S, Kuang L, … Wang H (2020). Rare mutations in the autophagy-regulating gene AMBRA1 contribute to human neural tube defects. Hum Mutat, 41(8), 1383–1393. doi: 10.1002/humu.24028 [DOI] [PubMed] [Google Scholar]
- Yi Y, Lindemann M, Colligs A, & Snowball C (2011). Economic burden of neural tube defects and impact of prevention with folic acid: a literature review. Eur J Pediatr, 170(11), 1391–1400. doi: 10.1007/s00431-011-1492-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Depersonalized data that this study is based on is available from the corresponding author upon request.