

Summary
Fundamental aspects of DNA replication, such as the anatomy of replication stall sites, how replisomes are influenced by gene transcription and whether the progression of sister replisomes is coordinated are poorly understood. Available techniques do not allow the precise mapping of the positions of individual replisomes on chromatin. We have developed a method called Replicon-seq that entails the excision of full-length replicons by controlled nuclease cleavage at replication forks. Replicons are sequenced using Nanopore, which provides a single-molecule readout of long DNA. Using Replicon-seq, we found that sister replisomes function autonomously yet progress through chromatin with remarkable consistency. Replication forks that encounter obstacles pause for a short duration but rapidly resume synthesis. The helicase Rrm3 plays a critical role both in mitigating the effect of protein barriers and facilitating efficient termination. Replicon-seq provides a high-resolution means of defining how individual replisomes move across the genome.
Keywords: Replicon-seq, Nanopore sequencing, Sister replisome, DNA replication, Replisome pausing, Rrm3, rDNA, Replication termination
Graphic Abstract
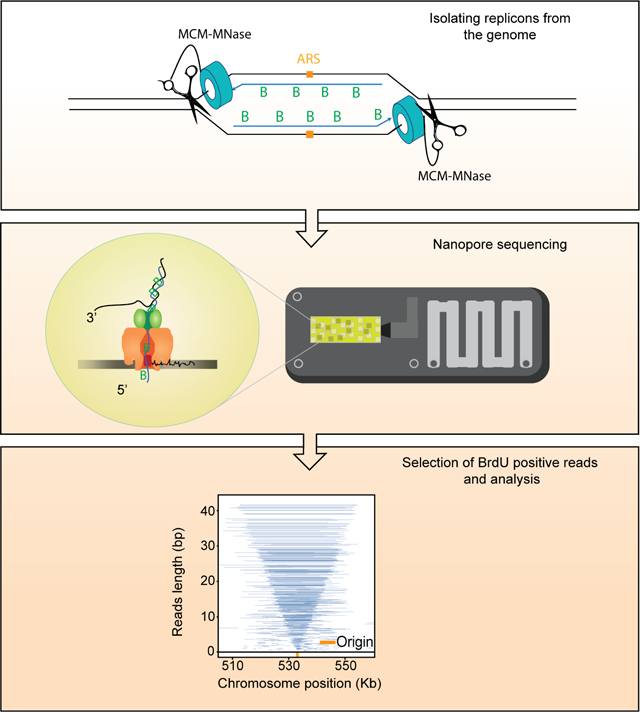
eTOC blurb (summary)
Claussin et al have developed a method that is able to precisely map the locations of DNA synthesis with single-molecule and single-nucleotide precision. “Replicon-seq” provides a new tool to study many aspects of DNA replication, including: initiation, elongation, termination and the various effects of replication impediments.
Introduction
In eukaryotes, DNA replication originates from multiple sites along chromosomes known as origins of replication. The replicative helicase – CMG – is coupled to several other replication factors and the replicative DNA polymerases to form the replisome, which collectively synthesizes the nascent genome (Burgers and Kunkel, 2017). Following initiation, two sister replisomes emanate from a replication origin and progress along the DNA template in opposite directions until they meet a DNA end, or a convergent replisome where a “termination” process results in CMG unloading and the ligation of two replicons (Dewar and Walter, 2017). During S-phase, sister replisomes can remain in proximity (Kitamura et al., 2006) and may be physically coupled (Yuan et al., 2019) which could allow some degree of coordination of their movement (Conti et al., 2007; Li et al., 2020; Yuan et al., 2019). However, in vitro evidence indicates that replisomes can function as monomers (Yardimci et al., 2010). Whether sister replisomes are functionally coupled in vivo – that is: a stall of one replisome is communicated to its sister – is poorly understood and has not been definitively addressed.
The study of DNA replication at nucleotide resolution with genomics has significantly lagged behind the transcription/RNA fields. This is principally because nascent DNA molecules are limited in copy number, differ in length by several orders of magnitude and are difficult to unambiguously separate from their template. Population based sequencing methods to study DNA replication are broadly insensitive to coincident or stochastic events affecting the replisome. Recent advances such as single-cell replication profiling (Chen et al., 2017; Dileep and Gilbert, 2018), high throughput fiber analysis (Wang et al., 2021), and nucleotide analogue detection with nanopore sequencing (Hennion et al., 2020; Muller et al., 2019) have begun to reveal DNA replication patterns at single molecule resolution. Yet, available methods have limited utility in precise mapping of replisome locations and are unsuitable for the fine scale analysis of replication defects.
We describe a new method which is able to overcome the many of the limitations of genome-wide approaches to study DNA replication. We utilize long-read Nanopore sequencing to identify and sequence nascent DNA molecules in their entirety. This approach maps the ends of nascent DNA molecules with nucleotide resolution and allows the precise localization of replisomes along the genome. We show that sister replisomes progress through tens of kilobases of chromatin at remarkably similar rates, we also find that a stall in one replisome is not communicated to its sister – indicating that replisomes function autonomously. Finally, we demonstrate that the DNA helicase Rrm3 performs a critical role in facilitating replication termination.
Design
We aimed to extract and sequence the DNA that has been synthesized by two replisomes that originated from the same origin of replication (replicon). We reasoned that creation of DNA strand breaks at functional replisomes would allow the position of any replisome to be identified by mapping the ends of nascent DNA by sequencing. The relative positions of sister replisomes could then be identified if nascent DNA chains could be sequenced in their entirety (Figure 1A) which is now possible using long-read Nanopore sequencing. Moreover, nascent DNA chains can be specifically identified using Nanopore, provided they are marked with nucleotide analogues such as BrdU (Hennion et al., 2020; Muller et al., 2019). Given that fusion of Micrococcal Nuclease (MNase) to candidate proteins allows the generation of specific DNA breaks (Schmid et al., 2004) including at replication origins (Foss et al., 2019), we chose to fuse MNase to the Mcm4 subunit of the replicative helicase, CMG, which encircles the leading strand template and is likely the most stable component of the replisome. Consistent with reports using MNase fusions (Schmid et al., 2004; Zentner et al., 2015), we found no growth defect of strains cultured at 25°C and 30°C in the absence of calcium, which is required by MNase for nuclease activity (Figure S1A, B). We tested whether the MNase fusion had the desired specificity by inducing DNA cleavage at origins in G1 (Figure S1C); surprisingly, we found that the C-terminal fusion give rise to far more DNA cleavage than the N-terminal fusion (Figure S1C). This difference likely reflects the N-N loading of two MCM2-7 hexamers at origins (Douglas et al., 2018; Georgescu et al., 2017) which may restrict access to the DNA of the N-terminal MNase fusion. Given that after helicase activation, the N-termini of a single MCM2-7 hexamer will be at the front of the replisome and no longer complexed with a second hexamer (Douglas et al., 2018; Georgescu et al., 2017), we reasoned that the use of the N-terminal fusion should provide a means of promoting parental DNA cleavage by MNase at “activated” helicases, rather than those that are loaded, but inactive.
Figure 1:
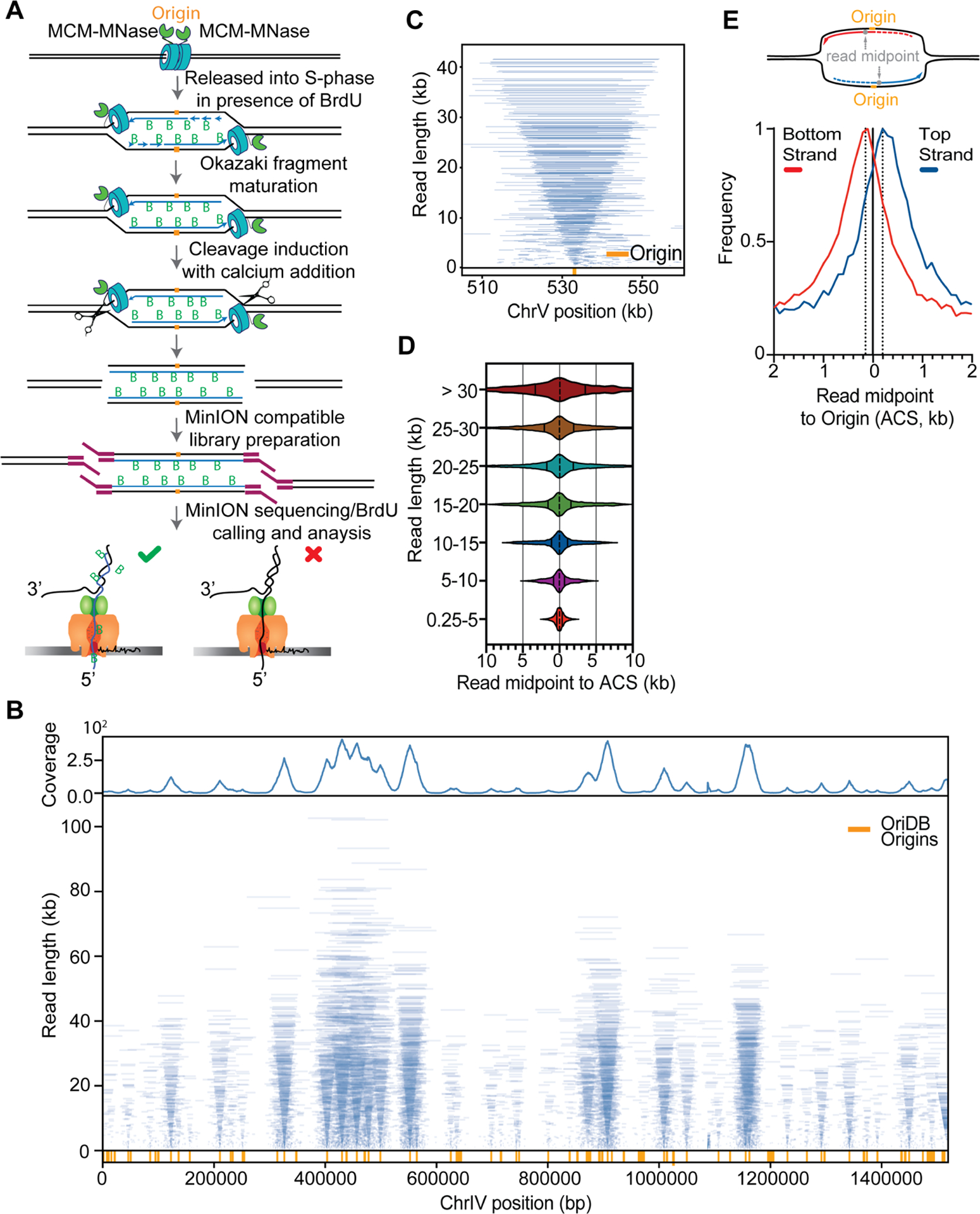
Replicon-seq a new method to study sister replication for movement. A. Schematic of Replicon-seq methodology. B. Tornado plot of Replicon-seq data. BrdU containing reads from combined WT runs in early S-Phase, each line represents a single read graphed by length and chromosome position, line transparency is scaled to reflect read abundance. Genome coverage of the displayed reads is shown on the upper track. Replication origins defined by OriDB are shown on the lower track. C. Zoom of representative origin (ARS520) showing symmetry of replicons, each blue line represents an individual read. D. Violin plot showing the positions of read midpoints relative to the ACS at replication origins for different length reads. E. Anchor plot showing the position of the midpoint of Top and Bottom-strand reads centered on the ACS. Illustration depicting the analysis of replicon midpoints to determine the relative lengths of the leading and lagging strands. Diagram shows two sister replication forks, which have emanated from a common replication origin (orange). The replicons produced during DNA synthesis are composed of the leading strand (solid line) and the lagging strand (dashed line). The position of the replicon midpoint (read midpoint, grey) relative to the position of the replication origin, indicates the relative lengths of the leading and lagging strands for each replicon. In this example, the leading strand is longer than the lagging strand. See Figures S1, S2, S3, S4 and S5 for more information of replicon-seq library generation, reproducibility and genome-wide replication patterns.
Results
Replicon-seq can map replisome movement by sequencing full-length replicons.
We performed experiments in which S. cerevisiae are arrested in G1 with alpha-factor and BrdU is added to allow labeling of nascent DNA upon release into S-phase. Cells were harvested in early S-phase, permeabilized and MNase activated with the addition of calcium for limited time, on ice (Methods). The DNA is directly sequenced using nanopore technology (Lu et al., 2016) with the output expected to be composed of nascent – BrdU containing molecules – as well as parental template and un-replicated DNA. BrdU containing reads were selected informatically (Boemo, 2021; Muller et al., 2019) (Methods).
Nanopore sequencing of replicons proceeds from the 5’ end of the lagging strand through to the 3’ end of the leading strand of the sister replisome (Figure 1A). Thus, the ends of replicons should provide a high-resolution, single-molecule readout of the relative position of sister replisomes along the genome. Plotting individual BrdU-containing reads according to genomic location and length reveals highly symmetric patterns which emanate from known replication origins (“tornado” Figure 1B; Figures S2, S3, S4). The median read-length of BrdU containing molecules is ~10kb but many molecules are greater than 50kb (Figure S1D). Small molecules are also present, and typically cluster around replication origins (Figure 1B, Figure S2, S3, S4), the genesis of this DNA is unclear, but most likely reflect sequencing of broken, or nicked replicons.
The symmetric pattern of replicons shown in Figure 1, B and C should only be apparent if two sister replisomes move at a similar rate. Indeed, provided that replisome progression is consistent, the midpoint of each replicon should overlap the origin from which it was derived. Using this logic, we can map replication origins (Figure S1E) and define the consistency of sister-replisome progression by comparing the distances each sister replisome traveled from their origin (Figure 1D) (Brewer and Fangman, 1987; Eaton et al., 2010; Nieduszynski et al., 2007). Although such measurements are potentially confounded by the variability in replication initiation sites, sequencing of truncated replicons, and replicon asymmetry brought about by termination; we find that the majority of sister replisomes progress at similar rates, regardless of distance traveled: the median deviation is ~1% and the distance traveled by 50% of all replisomes is within ~15% of their sister (Figure 1D). The remarkable consistency in replisome rate in WT cells does not prove that progression of sister replisomes is coordinated (Conti et al., 2007; Li et al., 2020), but does indicate that robust mechanisms exist to ensure consistent replisome progression through tens of kilobases of chromatin (Bellush and Whitehouse, 2017). Through further analysis, we also find that the synthesis of the contiguous lagging strand is typically delayed by ~350nt as compared to the leading strand (Figure 1E), a finding consistent with EM imaging (Lopes et al., 2006) and the expected size of un-ligated Okazaki fragments (Smith and Whitehouse, 2012). Our ability to detect a difference in position of leading and lagging strand ends indicates that MNase’s distinct preference for cleavage of single stranded DNA (Tucker et al., 1978), would likely result in preferential cleavage of the parental template - which is single-stranded at the replication fork. Such a cleavage pattern would likely occur under our tightly controlled reaction conditions (0°C, 10 seconds) and would preserve the integrity of the nascent DNA chains. Importantly, provided that the lagging strand template is cut, the 5’ ends of nascent DNA would be made accessible for sequencing which would allow us to assay both ends of nascent molecules (Figure S5).
Fob1-mediated replication fork pausing at rDNA
To test if Replicon-seq can identify sites of replication fork pausing, we analyzed the well-characterized (Replication Fork Block) RFB pausing site within the rDNA locus. The presence of Fob1 bound to the DNA results in a unidirectional pausing of replisomes to prevent convergence of RNA polymerase I and the replisome in rDNA repeats (Figure 2A) (Brewer and Fangman, 1988; Brewer et al., 1992; Kobayashi et al., 1992). As anticipated, we detected a pronounced accumulation of replicon ends directly adjacent to the annotated RFB, whereas progression of the sister replisome is apparently unimpeded (Figure 2B). We analyzed how DNA synthesis of the leading and lagging strands can proceed to the RFB. Although Fob1 can bind three closely spaced motifs: RFB1, RFB2 and RFB3, the highest preference is for RFB1 (Kobayashi, 2003).
Figure 2:
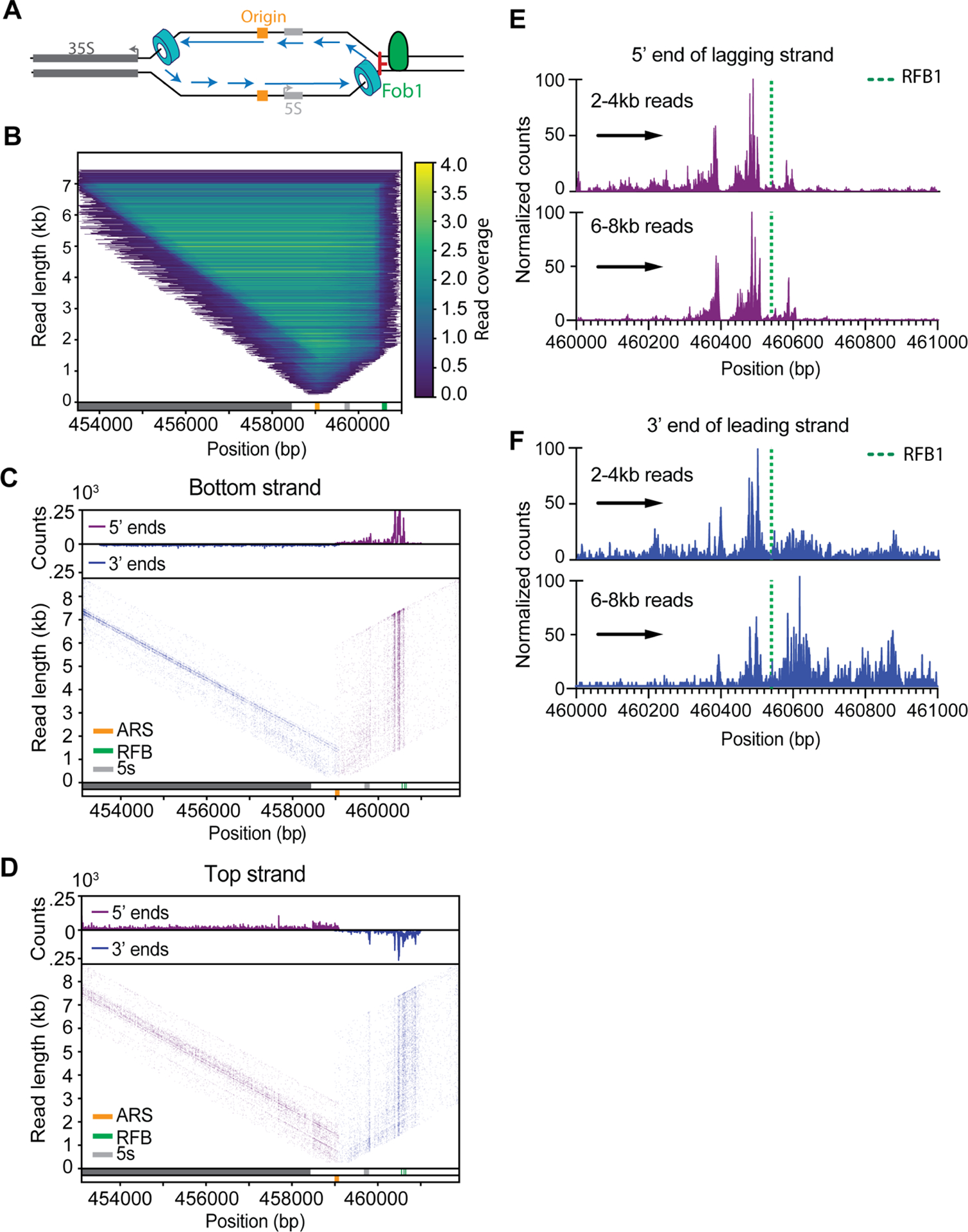
Replication fork block at the rDNA. A. Schematic of the rDNA locus showing the directional RFB mediated by Fob1. B. Heatmap of reads overlapping rDNA origin of replication (n = 5.58·104), each line represents an individual read, color is scaled to reflect density. C, D. 5’ and 3’ DNA ends are plotted for of either Bottom or Top-strand reads at the rDNA locus. Upper tracks show the genome coverage of the 3’ or 5’ ends of reads. Prominent rDNA features are displayed in the lower track. E, F. Counts of 5’ and 3’ends of the lagging or leading strands of replisomes approaching the RFB from the rightward moving fork (indicated by black arrow). Upper graphs show 2–4kb reads, lower show 6–8kb reads.
We found that the lagging strand 5’ ends primarily accumulate ~ 45–65nt upstream of RFB1 (Figure 2 C,E). This pattern may indicate that initiation of Okazaki fragments does not occur within ~40nt of the leading edge of the replication fork, similar to previous measurements (Duxin et al., 2014; Vrtis et al., 2021). The 3’ ends of the leading strand show a similar pattern to the 5’ ends of the lagging strand: a pronounced enrichment ~40–60nt upstream of RFB1, a distance which is likely defined by the footprint of CMG on the leading strand template (Figure 2 D,F) (Duxin et al., 2014). A significant fraction of the leading-strand 3’ ends extend beyond the RFB; it is noteworthy that extension through RFB becomes more prominent as replicons increase in size (Figure 2 D, F). Since rDNA repeats are replicated in clusters (Pasero et al., 2002), the majority of replisomes stalled at RFB soon encounter a converging replisome from an adjacent repeat. Thus, leading strand extension of longer replicons, through the RFB, likely occurs during termination; interestingly, bypass is rarely observed by the lagging strand, (Figure 2E) which may be indicative of the termination mechanism (Figure 5F). We also detect potential stall sites at the 3’ end of the 5S gene and ~150nt upstream of RFB1; these sites contain long poly dA-dT stretches of DNA, which may cause replisome/polymerase stall (Hile and Eckert, 2008; Tubbs et al., 2018) or may be aberrant cleavage by MNase (Tucker et al., 1978).
Figure 5:
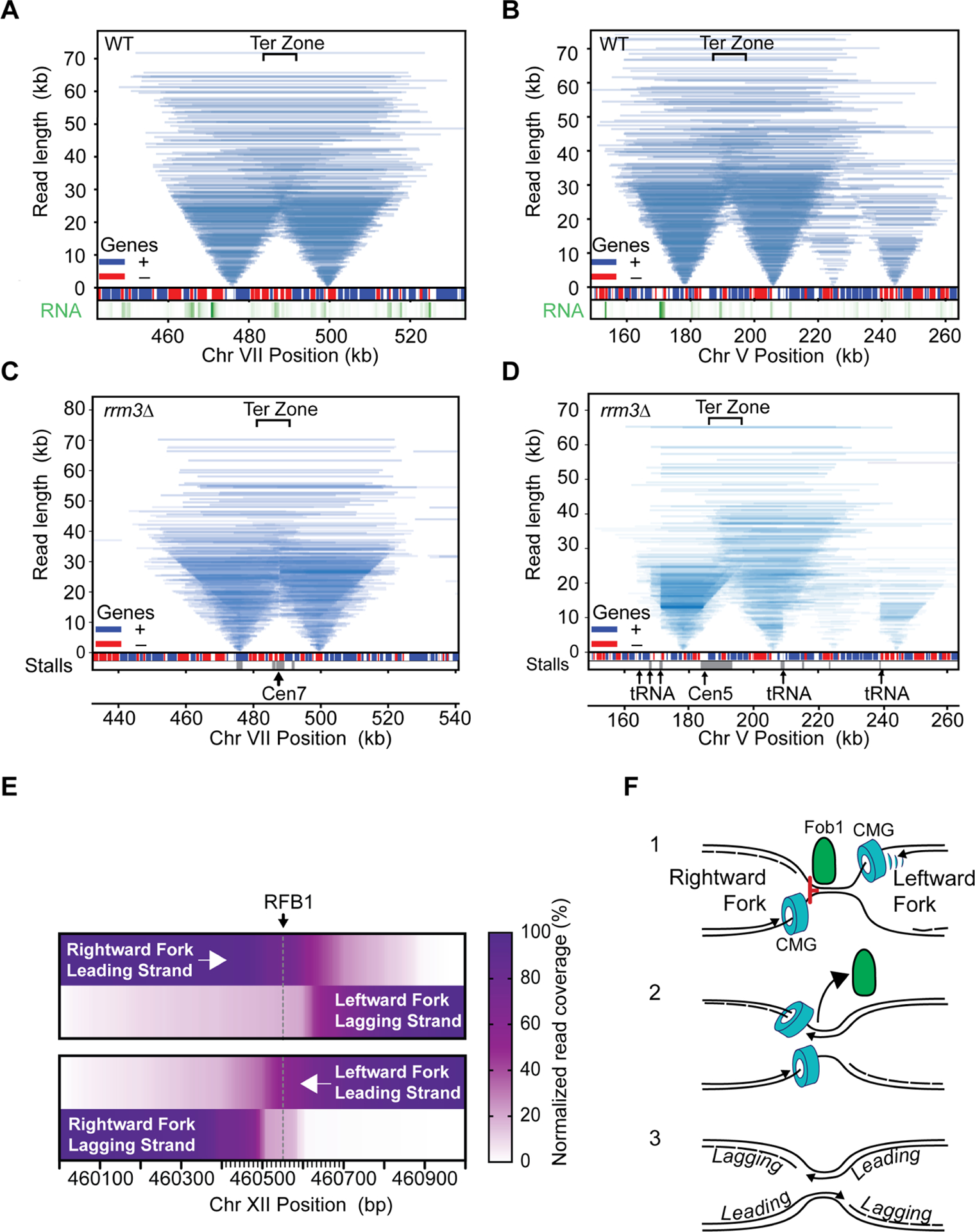
Tornado plot of replication termination zones. A, B. data from WT, C and D from rrm3Δ mutants. For clarity, only reads overlapping the replication origins are shown. Gene positions are shown a boxes (red=Bottom strand, blue=Top strand); nascent RNA levels are shown in the lower track (green); the positions of stall sites in rrm3Δ are shown at the bottom (grey). Prominent features are marked by arrows with text. E. Heatmap of read coverage of converging replication forks. Leading and lagging strands for left and right-ward moving forks are shown relative to RFB1. F. Model for replication termination at RFB site at the rDNA locus, see main text for details. Also see Figure S9.
Natural replication fork pausing sites in WT cells
Elements such as protein:DNA complexes, highly transcribed genes, R:loops and DNA structures can pose a significant challenge to the passage of the replisome and induce replisome stalling events (Shyian and Shore, 2021; Zeman and Cimprich, 2014). However, basic parameters such as the duration of the stall and the fate of the sister replisome remain poorly addressed. We therefore probed whether fork stalling in non-repetitive sequence could be detected by Replicon-seq. As shown in Figure 3, we find evidence of replisome stall in WT cells, which results in the accumulation of replicon ends at sites such as tRNA and centromeres (Deshpande and Newlon, 1996; Greenfeder and Newlon, 1992; Sekedat et al., 2010) (Figure 3A, B). Moreover, by following the progression of replicon ends past the impediment we can conclude that the replisome pauses – rather than permanently stalls at these sites. Importantly, progression of the sister replisome is not apparently affected by the stall, meaning that sister replisomes can move independently of each other, which confirms and extends previous findings (Doksani et al., 2009; Yardimci et al., 2010). We performed a meta-analysis of replicon-end accumulation across the genome to identify ~216 stall sites in early S-phase (Methods; Figure 3C). This analysis found tRNA, centromeres, origins and 208 genes transcribed by RNA polymerase II. We performed nascent RNA-seq in early S-phase to measure transcription but found that pause sites were not generally enriched for highly transcribed genes (Figure 3D). Closer inspection showed that many pause sites in genic regions are explained due to proximity to known impediments such as centromeres and tRNA. To directly assay the effect of RNA polymerase II transcription, we focused on the PDC1 gene whose transcription is >6 standard deviations above the S-phase mean (Figure 3D), which presumably increases the likelihood that the replisome will frequently encounter the transcription machinery. As shown in Figure 3, E and F, PDC1 shows a defect in replisome progression towards 5’ end of the body of the gene, nevertheless, even at PDC1 replisomes experience modest slowing and quickly resume synthesis at a similar rate to the sister (Figure 3F), illustrating the efficient mechanisms employed to mitigate transcription/replication conflicts in WT cells (Gomez-Gonzalez and Aguilera, 2019).
Figure 3:
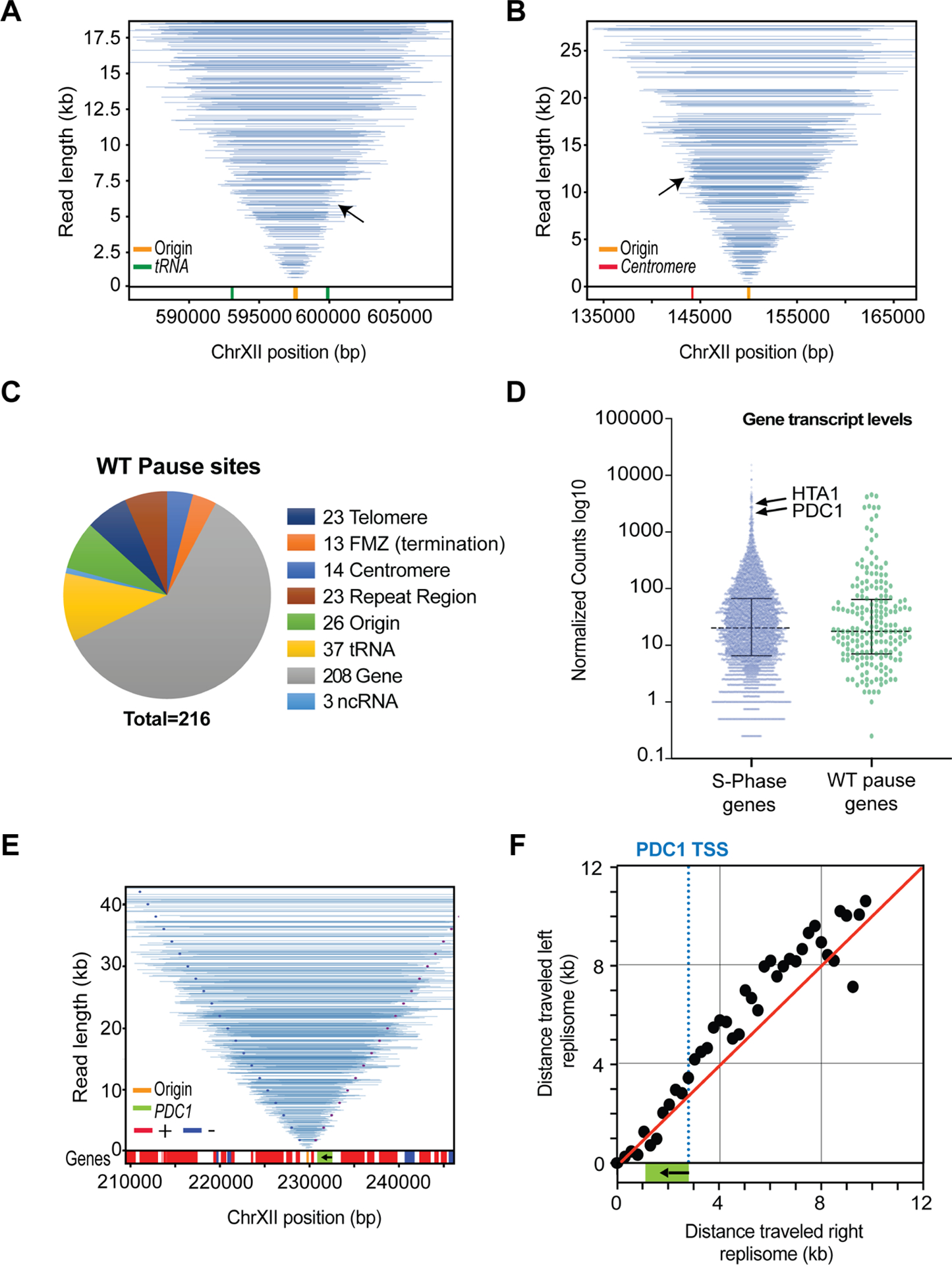
Replication fork movement in WT. Tornado plots show Replicon-seq reads and pausing events, for clarity, only reads overlapping the central origin are shown A. Pausing events at tRNA indicated by the black arrow, tRNA location is depicted in lower track in green. Also, see Figure S8A. B. Same as A, except the arrow in B is showing the Cen12 (lower track red), see Figure S8B for more information. C. Chart showing the relative proportions of genomic features found at stall sites in WT cells. D. Scatter dot-plot of nascent RNA seq transcription values for budding yeast genes in early S-Phase. E. Same as A&B but showing the PDC1 gene. The dotted lines represent expected positions of read ends if left and right replisomes progress at equal rates (left end in dark blue, right end in purple) from the origin. Gene position is depicted in lower track. PDC1 is green, arrow indicates transcription orientation. F. Median position of read ends in 250bp intervals was calculated for different length replicons emanating from the replication origin. The relative positions of the left and right replisomes are plotted at increasing distance from the origin. Dashed blue line represents the PDC1 TSS. Red line shows the expected trajectory if both replicons are moving at the same rate.
Rrm3 prevents replication fork pausing at t-RNAs and centromeres
The Pif1 family helicases – Rrm3 and Pif1 –promote replisome progression through a variety of elements, including protein DNA complexes, highly transcribed genes, tRNA and DNA structures such as G4 DNA (Muellner and Schmidt, 2020). We performed replicon-seq in rrm3Δ mutant strains and found that overall replication patterns appeared similar to WT, although with increased small or broken replicons (Figure 4A, Figure S6, S7). Closer inspection of the tornado plot reveals evidence that replisomes stall at tRNA, centromeres and other prominent sites, consistent with earlier reports (Figure 4B, Figure S8A, B and C) (Ivessa et al., 2003). Replisome stalls in rrm3Δ persist longer than WT as judged by accumulation of replisomes at the stall site and the extensive progression of the sister replisome. Despite extensive stalling, replication clearly resumes once the replisome has passed the impediment (Figure 4A, B) – a result that clearly demonstrates that forks do not terminally arrest at tRNA in rrm3Δ. We developed a protocol to measure replicon asymmetry induced by replisome stalling (Figure 4C), allowing us to plot the extent a replisome progresses whilst its sister is paused. Figure 4D shows a comparison of progression asymmetry of WT and rrm3Δ at tRNA in both head-on or co-directional orientations with respect to tRNA transcription. Assuming that replisomes travel ~2.0 kb per minute (Sekedat et al., 2010), we find that the pause duration in WT cells is typically less than 1 minute (Deshpande and Newlon, 1996), whereas the pause in rrm3Δ can extend past 4 minutes. We note that head-on conflicts take longer to resolve than co-directional conflicts – consistent with previous reports (Deshpande and Newlon, 1996; Ivessa et al., 2003; Osmundson et al., 2017; Tran et al., 2017). The abundance of stall events at tRNA genes in the rrm3Δ mutant allowed us to more closely investigate the specific replisome impediment. Figures 4E shows co-directional encounters result in a stall upstream of the tRNA with a prominent accumulation of 5’ nascent ends ~40–130nt upstream of TFIIIB binding site (Nagarajavel et al., 2013). Anti-directional encounters result in a similar accumulation of replicon ends downstream of the TSS/TFIIIB confirming that a primary impediment for replication through tRNA is likely TFIIIB (Yeung and Smith, 2020) (Figure S8D). Besides tRNA we identified prominent replisome stall events at all 16 centromeres, replication origins/dormant replication origins and replication termination zones (Azvolinsky et al., 2009; Fachinetti et al., 2010) (Figures 4F, Figure S8). However, only a minority of stalls occurred at genes transcribed at RNA polymerase II, leading us to conclude that Rrm3 plays a comparatively minor or infrequent role in mitigating transcription/replication conflicts (Osmundson et al., 2017). Notable exceptions are very highly transcribed genes such as PDC1 (Azvolinsky et al., 2009) where stalls also occur at adjacent dormant origins (Figure 4F). At PDC1, we find clear evidence of a distinct replisome stall at the TSS – indicating that Rrm3 is likely needed to help the replisome overcome the RNA polymerase II PIC.
Figure 4:
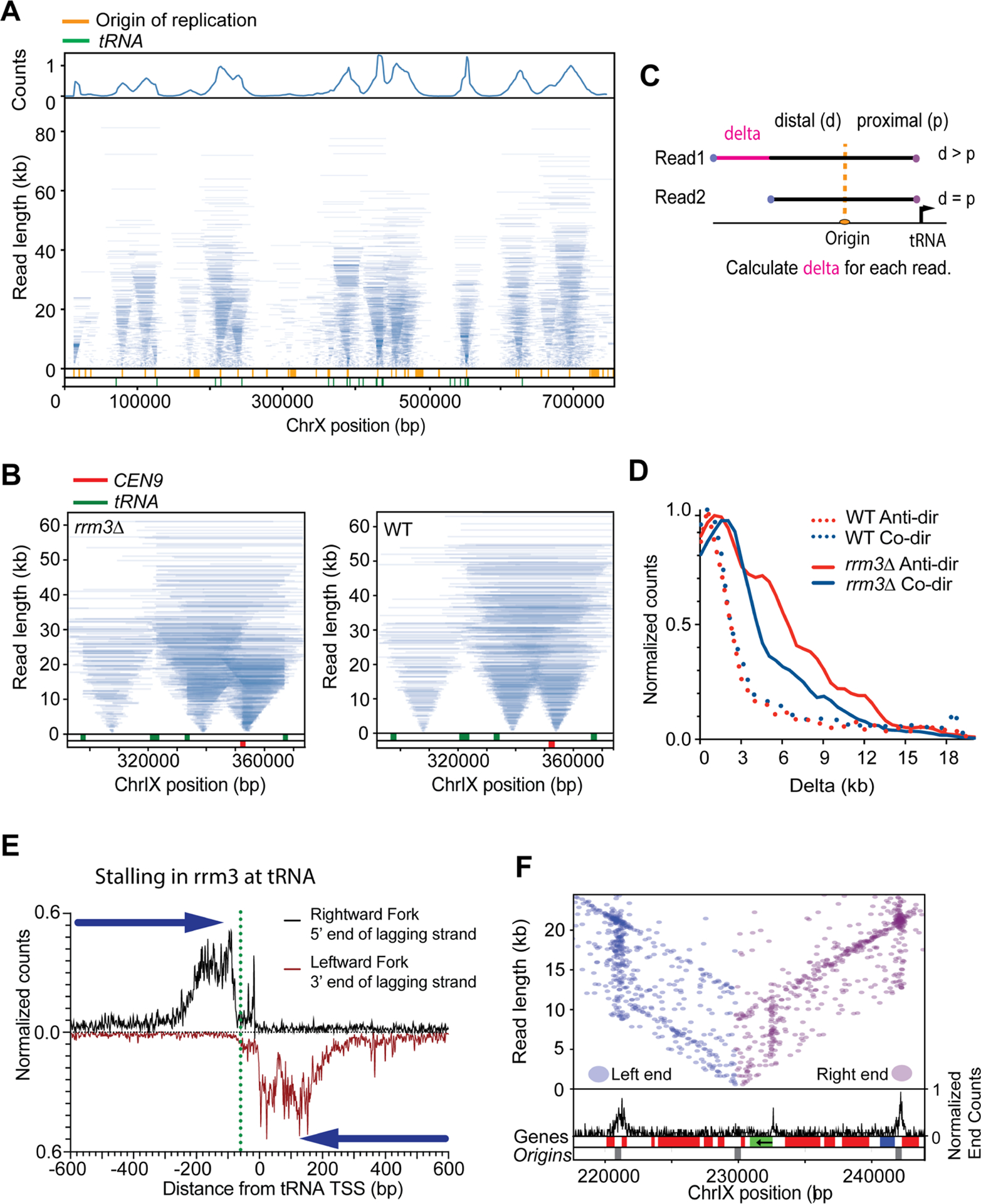
Replication fork movement in rrm3Δ. A. Tornado plot of rrm3Δ strain in early S-Phase, genome coverage is shown in the upper track, Origins of replication and tRNA locations are depicted in the lower track (orange and green, respectively). B. Enlargement of Tornado plot to show replication fork pausing at CEN9 (red) and tRNA (green) in rrm3Δ mutants. WT tornado plot is displayed for comparison at these sites. C. Schematic representing the calculation of replicon asymmetry at tRNA. D. Frequency plot showing extent of replicon asymmetry (delta) at tRNA genes for WT and rrm3Δ when tRNA are transcribed in a co-directional and anti-directional orientation with respect to the approaching replisome. E. Meta-plot showing the distance of the 5’ end of the lagging strand on the rightward moving fork (black) or the leftward moving fork (red) near the transcription site of 253 tRNA genes in rrm3Δ. Green dotted line shows the upstream edge of TFIIIB. F. Read end positions for replicons overlapping ARS1211 (left ends in blue, right ends in purple). Black graph shows read-end density across the highlighted region. Gene positions (red=Bottom strand, blue=Top strand) and replication origins (grey) are shown at the bottom; PDC1 gene is shown in green, arrow denotes transcription orientation. Note increased read-end density i.e. stall sites, at the 5’ end of PDC1 as well as the two origins flanking the central origin. See Figure S8 for more information.
Replication Termination
Replication termination zones are broadly dictated by the relative position and firing times of adjacent replication origins (Hawkins et al., 2013; McGuffee et al., 2013). In Replicon-seq, termination is evident as the fusion of two converging replication forks to produce a nascent molecule whose length is the sum of two joined replicons (Figure 5A and B). Converging replisomes do not appear to significantly slow (as compared with their sister replisomes, Figure S9) indicating that replisomes do not interfere with each other’s progress as they approach termination in vivo, as suggested in vitro (Dewar et al., 2015).
We noted that some stall sites detected in rrm3Δ overlap with replication termination zones (Fachinetti et al., 2010; McGuffee et al., 2013). Inspection of these sites provides evidence that adjacent replicons fail to terminate correctly in rrm3, resulting in an accumulation of replicon ends in the termination zone (Fig. 5C and D, Figure S6, S7). The delay in termination in rrm3Δ, appears to be impacted by two phenomena: first, tRNA, centromeres and dormant origins function as bi-directional barriers to replication fork progression – meaning that termination (and joining of nascent strands) cannot occur until the barrier is passed (Figure 5C), (Fachinetti et al., 2010). Second, even in the absence of known replication barriers, converging replication forks often fail to terminate in the termination zone (Figure 5D, Figure S6, S7), indicating that Rrm3 is required for a specific step in termination in vivo, consistent with in vitro studies and the detection of late replication intermediates on plasmids or at the rDNA (Deegan et al., 2019; Ivessa et al., 2000; Osmundson et al., 2017). The low coverage and diffuse nature of termination within the single copy genome prevented high-resolution analysis of termination; thus, we chose to investigate termination at the rDNA in rrm3Δ. Focusing on long replicons, which should be enriched for molecules in the process of termination, we calculated the read coverage on the leading and lagging strand, for the two converging forks at the RFB. Figure 5E shows that the 5’ end of the lagging strands of both converging forks stop some ~50nt from RFB1. Importantly, the 3’ end of both leading strands proceed through the RFB and stop directly adjacent to the lagging strand of the opposing fork (Figure 5E). Thus, leading strands of the two converging forks appear to pass one another and are extended to meet the 5’ ends of the lagging strands ahead of them. Termination at RFB is likely a multi-step process where the rightward moving fork is first arrested and waits for the leftward moving fork to displace Fob1 – which is a polar barrier (Brewer et al., 1992; Hizume and Araki, 2019; Kobayashi et al., 1992). Removal of Fob1 by the leftward moving fork would permit CMG progression, allowing both leading strands to be extended until they reach the nascent lagging strands ahead of them. Our high-resolution analysis provides the first in vivo evidence to show that leading strands pass one another during the termination process (Dewar et al., 2015). It remains unclear why Rrm3 is needed for termination; at RFB, Rrm3 may promote Fob1 removal (Mohanty et al., 2006), yet our data shows that pausing of forks at RFB in rrm3Δ mutants is not different to WT (compare Figure 2 and Figure S10). Figure 5E does show that the nascent ends of the converging forks are in close proximity to each other, thus it seems likely that small un-replicated gaps remain in rrm3Δ mutants (Deegan et al., 2019).
Discussion
The use of controlled nuclease digestion coupled with Nanopore sequencing provides a novel method for the analysis of coincident events on DNA. We have mapped the positions of sister replisomes in the process of DNA replication which has provided important new information on the relative rates of replisome progression, the sites of replisome stalls, and the mechanics of replication termination.
We find that sister replisomes move through many kilobases of chromatin with surprising consistency, allowing us to calculate that the leading strand is typically ~350nt longer than the lagging strand at advancing replisomes. This length is presumably dictated by size and rate of synthesis of Okazaki fragments, prior to their ligation into the nascent lagging strand. The high concordance of sister replisome progression also allows us to show that initiation of DNA synthesis occurs directly at the origin. Importantly, in both WT and rrm3Δ cells, arrest of one replisome is does not result in the arrest of the sister, demonstrating that the progression of sister replisomes is unlikely to be coordinated. Thus, despite their apparent colocalization (Kitamura et al., 2006), sister replisomes move independently but at a highly similar rate through a varying chromatin landscape. This finding illustrates that ancillary replisome factors, such as helicases and ATPases that promote fork progression through chromatin (Muellner and Schmidt, 2020; Shyian and Shore, 2021), are highly effective at mitigating the effect of potential impediments, including the transcription machinery (Gomez-Gonzalez and Aguilera, 2019). We confirm that Rrm3 is primarily required to overcome protein barriers, particularly TFIIIB at tRNA genes, centromeric nucleosomes and dormant origins (Azvolinsky et al., 2009), but we also find that Rrm3 is required for the replisome to overcome impediments in the promoter region of the PDC1 gene. Presumably, this indicates that the RNA polymerase II pre-initiation complex is, under certain conditions, an obstacle to the replisome. Nevertheless, we found that S-phase transcription rate is not a strong determinant of replisome stalling in WT or rrm3Δ cells.
When stalling at RFB, we find that the leading strand advances to within ~40nt of the impediment; while the precision of this measurement is lessened by our inability to define the exact position of the impediment, our measurements are consistent with previous reports and the finding that CMG prevents the leading strand from advancing to the fork junction (Duxin et al., 2014). Interestingly, despite the fact that the lagging strand template is not occluded by the helicase, the 5’ end of the lagging strand does not advance beyond the leading strand – indicating that unknown constraints prevent productive DNA synthesis on the lagging strand within ~50nt of the replication fork. We find a similar pattern of nascent DNA end accumulation at tRNA and centromere stalls in rrm3Δ cells; however, in these cases, DNA end positions are far more heterogeneous than at RFB. This pattern may indicate that replisomes significantly slow or stall in the ~200nt leading up to the DNA bound obstacle; or that a certain fraction of nascent DNA ends are processed following the prolonged stall.
Finally, we demonstrate that replicon-sequencing provides an unprecedented means to assay the process of replication termination. Our analysis shows that encroaching replisomes do not significantly slow as they approach each other, and that the sealing of converging replicons likely occurs without delay. Intriguingly, loss of Rrm3 activity results in a significant defect in replication termination at many sites across the genome. In part, this is explained by protein barriers acting as impediments to replisomes in termination zones (Fachinetti et al., 2010), however, even in the absence of protein barriers, converging replicons fail to efficiently join in rrm3Δ cells (Deegan et al., 2019). Such termination failure, though eventually resolved, results in a prolonged stall of the converging replisomes. The nature of the defect in rrm3Δ cells is presently unclear, but our data is consistent with biochemical data showing that Rrm3 and related helicases may be required to replicate through a small region separating two converging replisomes (Deegan et al., 2019).
Direct sequencing of DNA by Nanopore provides strand-specific information at nucleotide resolution, we anticipate that the roles of many replisome associated factors, key DNA processing events as well as chromatin assembly on nascent DNA can also be studied using this method.
Limitations of the study
DNA cleavage by MNase at a replication fork will generate three dsDNA ends: two from the replicated daughters, and one from the parental DNA ahead of the replication fork (Figure S5). Nanopore sequencing of DNA proceeds in a 5’–3’ direction, which means the nascent lagging strand, the leading strand template, and the “top” strand of the parental duplex ahead of the replisome will be passed into the nanopore. Thus, in ideal conditions, only 1/3 of sequenced molecules will be nascent. While each sequencing experiment currently generates a ~2–4 million sequencing reads, at present approximately 10% of reads are identified as BrdU-containing and of use for analysis. Such numbers of BrdU containing reads generate high genome coverage, but replisome stalls and progression are best measured by quantitation of ends of nascent molecules. Thus, with the present throughput, we estimate that we can detect consistent stalls lasting more than 1 minute. Use of larger flow-cells, and specific enrichment of BrdU or EdU molecules should improve throughput and allow the detection of less frequent or transient events. Another limitation of the study is our inability to precisely define how MNase cleavage influences the ends of nascent DNA molecules. While our data is consistent with previous studies that map ends of nascent chains at replisomes, it remains possible that MNase cleavage is influencing the readout in unanticipated ways. Finally, we have focused on the analysis of DNA replication in early S-phase; as a result, our study is not designed to generate a comprehensive list of all possible replisome impediments across the yeast genome.
Star methods
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Iestyn Whitehouse (whitehoi@mskcc.org)
Materials availability statement
All strains listed in this study will be provided upon request by the lead contact.
Data and code availability:
Sequencing data: fast5, fastq, and processed bed files are deposited on Gene Expression Omnibus (GEO) and publicly available as of the date of publication. Accession number is listed in the key resources table. Original southern blot images and flow cytometry data have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table.
All original codes have been included as supplementary files. All programs developed for the analysis of replicon-seq are available upon request as well.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Subject Details
Mnase-tag of MCM4
C-terminal fusion of codon optimized MNase was performed by transformation using pCC17 (MNase, yeast codon-optimized version of pGZ108); MNase is separated from MCM4 by an 8 amino acid linker. N-terminal fusion of codon optimized MNase was generated using CRISPR technology. Guide RNAs for CRISPR and donor sequence are listed in Key resources table and Figure S11, respectively.
Growth conditions
All strains used in this study are derived from W303 background (trp1-1, ade2-1 his3-11,15, can1-100) and are listed in Key resources table. Yeasts were grown in YPD at 25°C to OD600=0.35. Cells were arrested in G1 by addition of α-factor for 2h 45 minutes. Alpha factor was added each hour to a final concentration of 5ug/mL, 5ug/mL and 1ug/mL, respectively. 30min before the end of the arrest, the culture was supplemented with BrdU to 400ug/mL. Cells were washed 3 times with pre-warmed YPD and released in pre-warmed YPD supplemented with 400ug/mL of BrdU for 30 min at 25°C. 100mL of culture was pelleted at 4000rpm for 1 min, washed with ice-cold 20mM EDTA/1mM EGTA solution, pelleted at 4000rpm for 1min and flash frozen in liquid nitrogen.
Methods Details
Nascent RNA-sequencing
Cells were grown and prepared in the same conditions as the Replicon-seq assay except that BrdU was omitted and 4-thiouracil was added to the media 15 minutes after alpha factor release for 15 minutes. Nascent RNA was extracted as described in the protocol (Baptista and Devys, 2018) with the addition of a mRNA selection step using NEB mRNA selection kit (NEB cat # E7490S) after streptavidin enrichment of nascent RNAs. Library was prepared using Nanopore direct C-DNA protocol (SQK-DSC109) and sequenced on MinION FLO-MIN106 flow cells R9. We performed two biological replicates and found a strong correlation between replicates (Figure S12).
MNase cleavage and library preparation
MNase cleavage protocol was adapted from (Saleh et al., 2021). Briefly, cells were thawed on ice with 1mL of ice-cold 20mM EDTA/1mM EGTA solution for 10min, pelleted for 30sec at 10,000g. Cells were washed 3 times with 1mL of Solution A. (For 5mL of solution A: 15mM Tris-HCl pH 7.5, 80mM KCl, 0.1mM EGTA, ½ Roche mini-PIC EGTA-free, 0.5mM spermidine and 0.2mM spermine). Cells were resuspended into 570uL of Solution A, incubated 1min at 30°C. 30uL of 2% of digitonin was added to the cells and incubated 5min at 30°C. Cell were incubated on ice for 2min, before addition of 2mM CaCl2. MNase digestion was performed on ice for 10 seconds before addition of 610uL of 2X stop buffer (400mM NaCl, 20mM EDTA and 8mM EGTA). Cells were pelleted for 20 seconds at 10,000g. Cell walls were removed with the addition of 5mg of Zymolyase 100T (Nacalai Tesque) in a 250uL of Zymolyase digestion buffer (1M sorbitol, 1mM EGTA, 10mM B-mercaptoethanol, and 50mM Tris-HCl pH 7.5) for 3min at 30°C. Spheroplasts were washed 2 times with Zymolyase buffer, and one time with RNase buffer (50mM NaCl, 10mM DTT, 50mM Tris-Hcl pH8.5). Resuspended in 500uL of RNase buffer and supplemented with 10uL of RNAse cocktail (Invitrogen), and incubated at 37°C for 2h. 2% final concentration of SDS, 50mg of chelex 100 resin (Biorad) and 10uL of 20mg/mL proteinase K (Goldbio) was added to the spheroplasts and incubated for 1h at 55°C. DNA was extracted 2 times by phenol-chlorophorm extraction in phase lock tubes, the phenol was gently mixed by inversion, and spun at 13,000rpm for 5min. The aqueous phase was transferred by pouring into a new clean tube. DNA was ethanol precipitated and resuspended overnight in 40uL of nuclease-free water.
Replicon-seq libraries were performed using SQK-LSK109 and SQK-LSK110 library kit from Nanopore technologies. 1.2ug to 1.8ug of DNA of were used for library preparation. DNA repair and end-prep incubation time were changed to 10 minutes at 20°C. A-tailing was performed at room temperature for 30 minutes and elution from Ampure beads was performed at 37°C for 30min. 750ng to 1ug of library DNA was loaded on the MinION FLO-MIN106 flow cells R9, and sequenced for more than 48h, until pore exhaustion. We sequenced two biological replicates for WT and rrm3Δ mutant. WT run#1 we obtained 3.79.106 basecalled reads, with a total of 1.03.105 BrdU positive reads after BrdU calling by DNAscent, run #2 we obtained 4.87.106 basecalled reads, with a total of 1.25.105 BrdU positive reads after BrdU calling by DNAscent, rrm3Δ mutant run#1 we obtained 3.32.106 basecalled reads, with a total of 1.39.105 BrdU positive reads after BrdU calling by DNAscent, run #2 we obtained 4.31.106 basecalled reads, with a total of 1.61.105 BrdU positive reads after BrdU calling by DNAscent. Sequencing information on number of read sequenced, BrdU positive and read length N50 per runs are listed in Table 1.
Table 1:
Overview of sequencing runs and reads length N50
| Sample and run IDs | Total based call read (fast5) | Total mapped read/DNAscent reads (bed) | N50 total reads after DNAscent (Kb) | Total BrdU positive reads (≥0.5) in Kb | N50 BrdU positive reads (≥0.5) in Kb |
|---|---|---|---|---|---|
| WT run 1 | 3.79.106 | 1.45.106 | 7.091 | 1.03.105 | 7.439 |
| WT run2 | 4.87.106 | 1.81.106 | 7.639 | 1.25.105 | 7.787 |
| rrm3Δ run1 | 3.32.106 | 1.48.106 | 4.494 | 1.39.105 | 6.587 |
| rrm3Δ run2 | 4.31.106 | 2.03.106 | 4.290 | 1.61.105 | 7.369 |
Quantification and Statistical Analysis
All Fast5 files were converted into FastQ using Guppy (Oxford Nanopore Technologies). Sequencing reads were used to generate a custom reference genome using Canu (Koren et al., 2017), this allowed reliable mapping across transposable elements in the analyzed strain. All reads were mapped to the custom genome with Minimap2 (Li, 2018) using long read settings. BrdU containing reads were called using DNAscent v2 (Boemo, 2021). Using default settings, each read was split into 250bp intervals which were assigned as BrdU positive or negative. A BrdU score was then calculated for each read which represents the fraction of the BrdU positive intervals across the entire read. Only reads with a BrdU score ≥ 0.5 were used for analysis. These reads were mapped as Tornado plot using the Script S1. rDNA reads were plotted using the Script S2 as a heat map.
RNA seq data was converted to Fastq using Guppy and the mapped using Minimap2. The position and orientation of read primers were defined using LAST (Kielbasa et al., 2011) which allowed the orientation of the mRNA to be defined (Eccles, 2019). Median coverage for each gene was calculated using Bedtools (Quinlan and Hall, 2010).
Genome coordinates, Data plotting
With the exception of the rDNA, all genome coordinates reported correspond the custom genome assembly. Liftover for genome coordinates was performed using RATT (Otto et al., 2011). Tornado plots were generated using custom python scripts available on request. Figure 4 C & D: Only singleton tRNA genes (not within 10kb of another tRNA) and reads overlapping annotated ACS sequences (Eaton et al., 2010) were used for analysis. Delta (Figure 4C) was calculated by first selecting reads with one end within 2kb of an annotated tRNA TSS. For each read, the distance from the tRNA proximal end to the closest overlapping ACS was calculated. The proximal distance was then subtracted from the distal distance (i.e. the distance from the distal read end to the ACS) to generate a value for delta.
Stall sites and replication origins
Sites of replisome stalling were identified as regions with localized accumulation of read-ends. This was calculated by binning the genome into 200bp intervals and calculating the sum of ends for BrdU positive reads above 2kb in length in each interval. MACS2 (Zhang et al., 2008) was then used to call significant intervals using bdgpeakcall. Replication origins were identified by defining sites enriched for read midpoints. We mapped the midpoints of short reads (0–5kb) and long reads (5–20kb) to 200bp intervals along the genome. MACS2 was then used to call peaks using bdgpeakcall on short and long reads. We selected for peaks that overlap in both short reads and long reads as we found this reduces the bias introduced by the change in read midpoints during termination.
Supplementary Material
Highlights.
Replicon-seq provides a single-molecule readout of nascent DNA molecules.
DNA synthesis can be mapped with base-pair resolution.
Replisomes progress through chromatin at a consistent rate.
The Rrm3 helicase facilitates replication termination.
Acknowledgements:
We would like to thank Henry Cheng for work on analysis pipeline. M. Boemo, University of Cambridge, UK, for help with DNAscent. A. Viale and N. Mohibullah at the Sloan Kettering Integrated Genomics Operation for help with sequencing setup. D. Remus, X. Zhao, members of the Molecular Biology Program at SKI and members of the Whitehouse lab for comments and advice.
Funding:
This work was primarily supported by NIH grants R01RGM102253 and R01GM129058 awarded to IW. CC was also supported by an EMBO Long-term fellowship ALTF 246-2018.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: The authors declare no competing interests.
References
- Azvolinsky A, Giresi PG, Lieb JD, and Zakian VA (2009). Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol Cell 34, 722–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptista T, and Devys D (2018). Saccharomyces cerevisiae Metabolic Labeling with 4-thiouracil and the Quantification of Newly Synthesized mRNA As a Proxy for RNA Polymerase II Activity. J Vis Exp 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellush JM, and Whitehouse I (2017). DNA replication through a chromatin environment. Philos Trans R Soc Lond B Biol Sci 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boemo MA (2021). DNAscent v2: detecting replication forks in nanopore sequencing data with deep learning. BMC Genomics 22, 430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer BJ, and Fangman WL (1987). The localization of replication origins on ARS plasmids in S. cerevisiae. Cell 51, 463–471. [DOI] [PubMed] [Google Scholar]
- Brewer BJ, and Fangman WL (1988). A replication fork barrier at the 3’ end of yeast ribosomal RNA genes. Cell 55, 637–643. [DOI] [PubMed] [Google Scholar]
- Brewer BJ, Lockshon D, and Fangman WL (1992). The arrest of replication forks in the rDNA of yeast occurs independently of transcription. Cell 71, 267–276. [DOI] [PubMed] [Google Scholar]
- Burgers PMJ, and Kunkel TA (2017). Eukaryotic DNA Replication Fork. Annu Rev Biochem 86, 417–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Xing D, Tan L, Li H, Zhou G, Huang L, and Xie XS (2017). Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion (LIANTI). Science 356, 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti C, Sacca B, Herrick J, Lalou C, Pommier Y, and Bensimon A (2007). Replication fork velocities at adjacent replication origins are coordinately modified during DNA replication in human cells. Mol Biol Cell 18, 3059–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deegan TD, Baxter J, Ortiz Bazan MA, Yeeles JTP, and Labib KPM (2019). Pif1-Family Helicases Support Fork Convergence during DNA Replication Termination in Eukaryotes. Mol Cell 74, 231–244 e239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande AM, and Newlon CS (1996). DNA replication fork pause sites dependent on transcription. Science 272, 1030–1033. [DOI] [PubMed] [Google Scholar]
- Dewar JM, Budzowska M, and Walter JC (2015). The mechanism of DNA replication termination in vertebrates. Nature 525, 345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar JM, and Walter JC (2017). Mechanisms of DNA replication termination. Nat Rev Mol Cell Biol 18, 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dileep V, and Gilbert DM (2018). Single-cell replication profiling to measure stochastic variation in mammalian replication timing. Nat Commun 9, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doksani Y, Bermejo R, Fiorani S, Haber JE, and Foiani M (2009). Replicon dynamics, dormant origin firing, and terminal fork integrity after double-strand break formation. Cell 137, 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas ME, Ali FA, Costa A, and Diffley JFX (2018). The mechanism of eukaryotic CMG helicase activation. Nature 555, 265–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duxin JP, Dewar JM, Yardimci H, and Walter JC (2014). Repair of a DNA-protein crosslink by replication-coupled proteolysis. Cell 159, 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton ML, Galani K, Kang S, Bell SP, and MacAlpine DM (2010). Conserved nucleosome positioning defines replication origins. Genes Dev 24, 748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles D (2019). Stranded Mapping from Oriented Long Reads. In protocols io, p. 10.17504/protocols.io.17507vmhn17546. [DOI] [Google Scholar]
- Fachinetti D, Bermejo R, Cocito A, Minardi S, Katou Y, Kanoh Y, Shirahige K, Azvolinsky A, Zakian VA, and Foiani M (2010). Replication termination at eukaryotic chromosomes is mediated by Top2 and occurs at genomic loci containing pausing elements. Mol Cell 39, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foss EJ, Gatbonton-Schwager T, Thiesen AH, Taylor E, Soriano R, Lao U, MacAlpine DM, and Bedalov A (2019). Sir2 suppresses transcription-mediated displacement of MCM2-7 replicative helicases at the ribosomal DNA repeats. PLoS Genet 15, e1008138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu R, Yuan Z, Bai L, de Luna Almeida Santos R, Sun J, Zhang D, Yurieva O Li H, and O’Donnell ME (2017). Structure of eukaryotic CMG helicase at a replication fork and implications to replisome architecture and origin initiation. Proc Natl Acad Sci U S A 114, E697–E706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Gonzalez B, and Aguilera A (2019). Transcription-mediated replication hindrance: a major driver of genome instability. Genes Dev 33, 1008–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfeder SA, and Newlon CS (1992). Replication forks pause at yeast centromeres. Mol Cell Biol 12, 4056–4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins M, Retkute R, Muller CA, Saner N, Tanaka TU, de Moura AP, and Nieduszynski CA (2013). High-resolution replication profiles define the stochastic nature of genome replication initiation and termination. Cell Rep 5, 1132–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennion M, Arbona JM, Lacroix L, Cruaud C, Theulot B, Tallec BL, Proux F, Wu X, Novikova E, Engelen S, et al. (2020). FORK-seq: replication landscape of the Saccharomyces cerevisiae genome by nanopore sequencing. Genome Biol 21, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hile SE, and Eckert KA (2008). DNA polymerase kappa produces interrupted mutations and displays polar pausing within mononucleotide microsatellite sequences. Nucleic Acids Res 36, 688–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hizume K, and Araki H (2019). Replication fork pausing at protein barriers on chromosomes. FEBS Lett 593, 1449–1458. [DOI] [PubMed] [Google Scholar]
- Ivessa AS, Lenzmeier BA, Bessler JB, Goudsouzian LK, Schnakenberg SL, and Zakian VA (2003). The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol Cell 12, 1525–1536. [DOI] [PubMed] [Google Scholar]
- Ivessa AS, Zhou JQ, and Zakian VA (2000). The Saccharomyces Pif1p DNA helicase and the highly related Rrm3p have opposite effects on replication fork progression in ribosomal DNA. Cell 100, 479–489. [DOI] [PubMed] [Google Scholar]
- Kielbasa SM, Wan R, Sato K, Horton P, and Frith MC (2011). Adaptive seeds tame genomic sequence comparison. Genome Res 21, 487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura E, Blow JJ, and Tanaka TU (2006). Live-cell imaging reveals replication of individual replicons in eukaryotic replication factories. Cell 125, 1297–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T (2003). The replication fork barrier site forms a unique structure with Fob1p and inhibits the replication fork. Mol Cell Biol 23, 9178–9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Hidaka M, Nishizawa M, and Horiuchi T (1992). Identification of a site required for DNA replication fork blocking activity in the rRNA gene cluster in Saccharomyces cerevisiae. Mol Gen Genet 233, 355–362. [DOI] [PubMed] [Google Scholar]
- Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, and Phillippy AM (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Yao NY, and O’Donnell ME (2020). Anatomy of a twin DNA replication factory. Biochem Soc Trans 48, 2769–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes M, Foiani M, and Sogo JM (2006). Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell 21, 15–27. [DOI] [PubMed] [Google Scholar]
- Lu H, Giordano F, and Ning Z (2016). Oxford Nanopore MinION Sequencing and Genome Assembly. Genomics Proteomics Bioinformatics 14, 265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffee SR, Smith DJ, and Whitehouse I (2013). Quantitative, genome-wide analysis of eukaryotic replication initiation and termination. Mol Cell 50, 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty BK, Bairwa NK, and Bastia D (2006). The Tof1p-Csm3p protein complex counteracts the Rrm3p helicase to control replication termination of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 103, 897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muellner J, and Schmidt KH (2020). Yeast Genome Maintenance by the Multifunctional PIF1 DNA Helicase Family. Genes (Basel) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller CA, Boemo MA, Spingardi P, Kessler BM, Kriaucionis S, Simpson JT, and Nieduszynski CA (2019). Capturing the dynamics of genome replication on individual ultra-long nanopore sequence reads. Nat Methods 16, 429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajavel V, Iben JR, Howard BH, Maraia RJ, and Clark DJ (2013). Global ‘bootprinting’ reveals the elastic architecture of the yeast TFIIIB-TFIIIC transcription complex in vivo. Nucleic Acids Res 41, 8135–8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieduszynski CA, Hiraga S, Ak P, Benham CJ, and Donaldson AD (2007). OriDB: a DNA replication origin database. Nucleic Acids Res 35, D40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmundson JS, Kumar J, Yeung R, and Smith DJ (2017). Pif1-family helicases cooperatively suppress widespread replication-fork arrest at tRNA genes. Nat Struct Mol Biol 24, 162–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto TD, Dillon GP, Degrave WS, and Berriman M (2011). RATT: Rapid Annotation Transfer Tool. Nucleic Acids Res 39, e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasero P, Bensimon A, and Schwob E (2002). Single-molecule analysis reveals clustering and epigenetic regulation of replication origins at the yeast rDNA locus. Genes Dev 16, 2479–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh MM, Tourigny JP, and Zentner GE (2021). Genome-Wide Profiling of Protein-DNA Interactions with Chromatin Endogenous Cleavage and High-Throughput Sequencing (ChEC-Seq). Methods Mol Biol 2351, 289–303. [DOI] [PubMed] [Google Scholar]
- Schmid M, Durussel T, and Laemmli UK (2004). ChIC and ChEC; genomic mapping of chromatin proteins. Mol Cell 16, 147–157. [DOI] [PubMed] [Google Scholar]
- Sekedat MD, Fenyo D, Rogers RS, Tackett AJ, Aitchison JD, and Chait BT (2010). GINS motion reveals replication fork progression is remarkably uniform throughout the yeast genome. Mol Syst Biol 6, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyian M, and Shore D (2021). Approaching Protein Barriers: Emerging Mechanisms of Replication Pausing in Eukaryotes. Front Cell Dev Biol 9, 672510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DJ, and Whitehouse I (2012). Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature 483, 434–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PLT, Pohl TJ, Chen CF, Chan A, Pott S, and Zakian VA (2017). PIF1 family DNA helicases suppress R-loop mediated genome instability at tRNA genes. Nat Commun 8, 15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs A, Sridharan S, van Wietmarschen N, Maman Y, Callen E, Stanlie A, Wu W, Wu X, Day A, Wong N, et al. (2018). Dual Roles of Poly(dA:dT) Tracts in Replication Initiation and Fork Collapse. Cell 174, 1127–1142 e1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker PW, Hazen EE Jr., and Cotton FA (1978). Staphylococcal nuclease reviewed: a prototypic study in contemporary enzymology. I. Isolation; physical and enzymatic properties. Mol Cell Biochem 22, 67–77. [DOI] [PubMed] [Google Scholar]
- Vrtis KB, Dewar JM, Chistol G, Wu RA, Graham TGW, and Walter JC (2021). Single-strand DNA breaks cause replisome disassembly. Mol Cell 81, 1309–1318 e1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Klein KN, Proesmans K, Yang H, Marchal C, Zhu X, Borrman T, Hastie A, Weng Z, Bechhoefer J, et al. (2021). Genome-wide mapping of human DNA replication by optical replication mapping supports a stochastic model of eukaryotic replication. Mol Cell 81, 2975–2988 e2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardimci H, Loveland AB, Habuchi S, van Oijen AM, and Walter JC (2010). Uncoupling of sister replisomes during eukaryotic DNA replication. Mol Cell 40, 834–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung R, and Smith DJ (2020). Determinants of Replication-Fork Pausing at tRNA Genes in Saccharomyces cerevisiae. Genetics 214, 825–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z, Georgescu R, Santos RLA, Zhang D, Bai L, Yao NY, Zhao G, O’Donnell ME, and Li H (2019). Ctf4 organizes sister replisomes and Pol alpha into a replication factory. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, and Cimprich KA (2014). Causes and consequences of replication stress. Nat Cell Biol 16, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Kasinathan S, Xin B, Rohs R, and Henikoff S (2015). ChEC-seq kinetics discriminates transcription factor binding sites by DNA sequence and shape in vivo. Nat Commun 6, 8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data: fast5, fastq, and processed bed files are deposited on Gene Expression Omnibus (GEO) and publicly available as of the date of publication. Accession number is listed in the key resources table. Original southern blot images and flow cytometry data have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table.
All original codes have been included as supplementary files. All programs developed for the analysis of replicon-seq are available upon request as well.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.