

SUMMARY
The sense of smell helps us navigate the environment, but its molecular architecture and underlying logic remain understudied. The spatial location of odorant receptor genes (Olfrs) in the nose is thought to be independent of the structural diversity of the odorants they detect. Using spatial transcriptomics, we create a genome-wide 3D atlas of the mouse olfactory mucosa (OM). Topographic maps of genes differentially expressed in space reveal that both Olfrs and non-Olfrs are distributed in a continuous and overlapping fashion over at least five broad zones in the OM. The spatial locations of Olfrs correlate with the mucus solubility of the odorants they recognize, providing direct evidence for the chromatographic theory of olfaction. This resource resolves the molecular architecture of the mouse OM and will inform future studies on mechanisms underlying Olfr gene choice, axonal pathfinding, patterning of the nervous system, and basic logic for the peripheral representation of smell.
Graphical Abstract
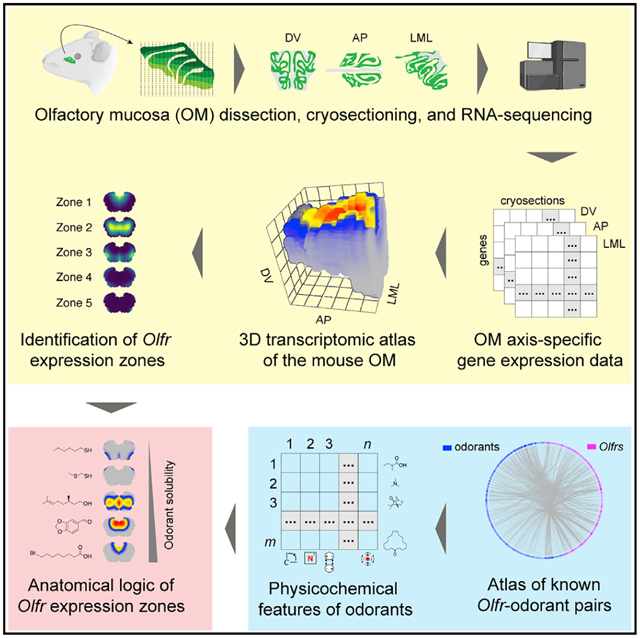
In brief
Ruiz Tejada Segura et al. employ a spatial transcriptomics approach to create a 3D map of gene expression of the mouse nose and combine it with single-cell RNA-seq, machine learning, and chemoinformatics to resolve its molecular architecture and shed light into the anatomical logic of smell.
INTRODUCTION
The functional logic underlying the topographic organization of primary receptor neurons and their receptive fields is well known for all sensory systems but olfaction (Kandel et al., 2013). The mammalian nose is constantly flooded with odorant cocktails. Powered by a sniff, air enters the nasal cavity until it reaches the olfactory mucosa (OM). There, myriad odorants activate odorant receptors (Olfrs) present in the cilia of olfactory sensory neurons (OSNs), triggering a cascade of events that culminate in the brain and result in odor perception (Buck and Axel, 1991; Kandel et al., 2013). Most mouse mature OSNs express a single allele of one out of ~1,100 Olfr genes (Olfrs) (Chess et al., 1994; Hanchate et al., 2015; Malnic et al., 1999; Saraiva et al., 2015b). Olfrs employ a combinatorial strategy to detect odorants, which maximizes their detection capacity (Malnic et al., 1999; Nara et al., 2011). OSNs expressing the same Olfr share similar odorant response profiles (Malnic et al., 1999; Nara et al., 2011) and drive their axons to the same glomeruli in the olfactory bulb (Mombaerts et al., 1996; Ressler et al., 1994; Vassar et al., 1994). Thus, Olfrs define functional units in the olfactory system and function as genetic markers to discriminate between different mature OSN subtypes (Ibarra-Soria et al., 2017; Saraiva et al., 2015b).
Another remarkable feature of the OSN subtypes is their spatial distribution in the OM. Early studies postulated that OSNs expressing different Olfrs are spatially segregated into four broad areas within the OM, called “zones,” and which define hemicylindrical rings with different radii (Ressler et al., 1993; Vassar et al., 1993). Subsequent studies identified Olfrs expressed across multiple zones, making clear that a division in four discrete zones might not accurately reflect the system, and a continuous numerical index representing the pattern of expression of each Olfr along the zones was implemented (Miyamichi et al., 2005; Strotmann et al., 1992). More recently, a study reconstructed Olfr expression patterns in three dimensions (3D) and qualitatively classified the expression areas of 68 Olfrs in nine overlapping zones (Zapiec and Mombaerts, 2020). However, all these studies sampled a fraction (~10%) of the total intact olfactory receptor gene repertoire and, most importantly, lack a quantitative and unbiased definition of zones or indices. We do not currently understand the full complexity of the OM and lack an unbiased and quantitative definition of zones. In effect, the exact number of zones, their anatomical boundaries, molecular identity, and functional relevance are yet to be determined.
One hypothesis is that the topographic distribution of Olfr and OSN subtypes evolved because it plays a key role in the process of Olfr choice in mature OSNs and/or in OSN axon guidance (Bashkirova et al., 2020; Coppola et al., 2013). An alternative hypothesis is that the spatial organization of Olfr/OSN subtypes is tuned to maximize the detection and discrimination of odorants in the peripheral olfactory system (Ressler et al., 1993). Interestingly, the receptive fields of mouse OSNs vary with their spatial location (Ma and Shepherd, 2000), which in some cases correlates with the patterns of odorant sorption in the mouse OM—this association was proposed as the “chromatographic hypothesis” decades before the discovery of the Olfrs (Mozell, 1966) and later rebranded as the “sorption hypothesis” in olfaction (Schoenfeld and Cleland, 2006; Scott et al., 2014). While some studies lend support to these hypotheses (reviewed in Secundo et al. 2014), others question their validity (Abaffy and Defazio, 2011; Coppola et al., 2019). Thus, the logic underlying the representation of smell in the peripheral olfactory system still remains unknown, and it is subject of great controversy (Kurian et al., 2021; Secundo et al., 2014).
Spatial transcriptomics, which combines spatial information with high-throughput gene expression profiling, expanded our knowledge of complex tissues, organs, or even entire organisms (Achim et al., 2015; Asp et al., 2019, 2020; Junker et al., 2014; Peng et al., 2016). In this study, we employed a spatial transcriptomics approach to create a 3D map of gene expression of the mouse nose, and we combined it with single-cell RNA sequencing (RNA-seq), machine learning, and chemoinformatics to resolve its molecular architecture and shed light onto the anatomical logic of smell.
RESULTS
A high-resolution spatial transcriptomic map of the mouse olfactory mucosa
We adapted the RNA-seq tomography (Tomo-seq) method (Junker et al., 2014) to create a spatially resolved genome-wide transcriptional atlas of the mouse nose. We obtained cryosections (35 μm) collected along the dorsal-ventral (DV), anterior-posterior (AP), and lateral-medial-lateral (LML) axes (n = 3 per axis) of the OM (Figure 1A) and performed RNA-seq on individual cryosections (see STAR Methods). After quality control (Figures S1A–S1D; Table S1; STAR Methods), we computationally refined the alignment of the cryosection along each axis, and we observed a high correlation between biological replicates (Figure 1B). Hence, we combined the three replicates into a single series of spatial data, including 54, 60, and 56 positions along the DV, AP, and LML axis, respectively (Figure 1C; STAR Methods). On average, we detected >18,000 genes per axis, representing a total of 19,249 genes for all axes combined (Figure 1D). Molecular markers for all canonical cell types known to populate the mouse OM were detected in all axes (Figure 1E) and expressed at the expected levels (Saraiva et al., 2015b).
Figure 1. Application of TOMO-seq to mouse OM.
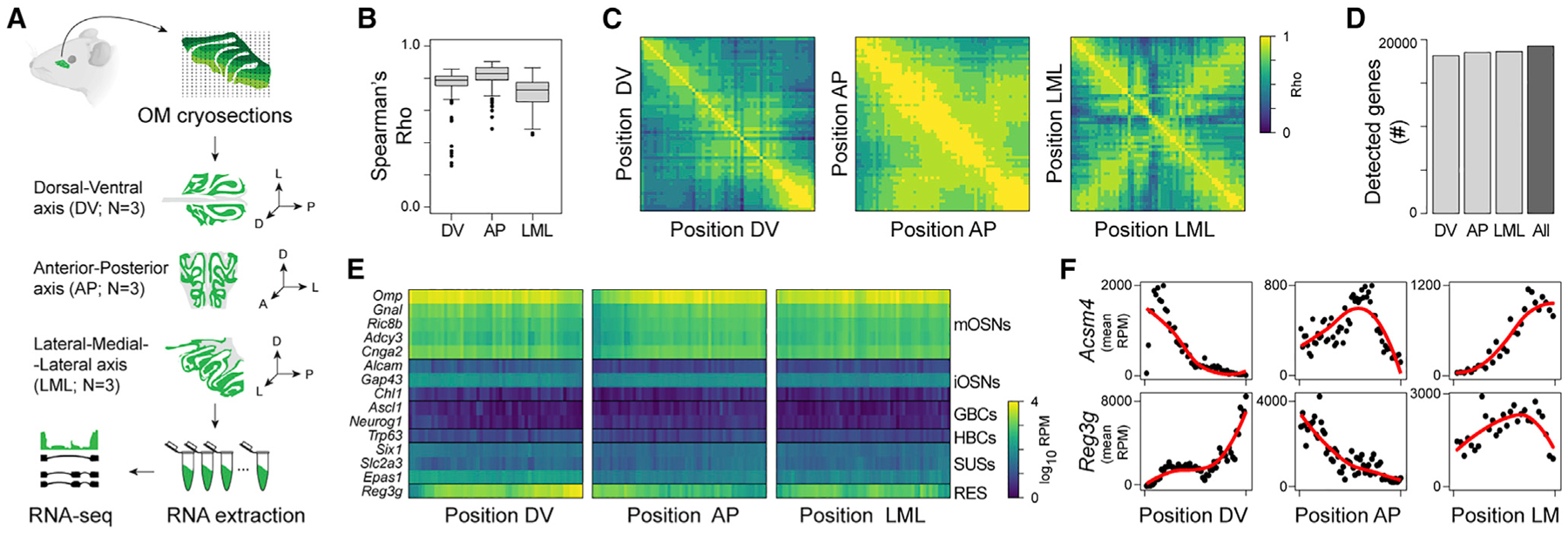
(A) Experimental design. TOMO-seq was performed on nine tissue samples, from which three were sliced along the dorsal-ventral axis (DV), three along the anterior-posterior axis (AP), and three along the lateral-medial-lateral axis (LML).
(B) Boxplots showing the distributions of Spearman’s correlation coefficients (rho) between replicates in each axis.
(C) Heatmaps showing Spearman’s correlation between gene expression patterns at different positions along the three axes.
(D) Number of detected genes along each axis separately or across the whole dataset. Genes were considered as detected when they had at least one normalized count in at least 10% of the samples from one axis.
(E) Heatmaps of log10 normalized expression (after combining the three replicates per axis) of OM canonical markers along the three axes (GBCs, globose basal cells; HBCs, horizontal basal cells; iOSNs, immature olfactory sensory neurons; mOSNs, mature olfactory sensory neurons; RESs, respiratory epithelium cells; RPM, reads per million; SUSs, sustentacular cells).
(F) Normalized expression of canonical OM spatial marker genes along the three axes. Red line shows fits with local polynomial models.
Next, we verified the presence of a spatial signal with the Moran’s I (Schmal et al., 2017; Figure S1E), whose value is significantly higher than 0 for the data along all axes (p < 2 × 10−16 for all axes), indicating that nearby sections have more similar patterns of gene expression than expected by chance. Given the left/right symmetry along the LML axis (Figure 1C), the data were centered and averaged on the two sides—henceforth, the LML axis will be presented and referred to as the lateral-medial (LM) axis (see STAR Methods). We could reproduce the expression patterns for known OM spatial markers, including the dorsomedial markers Acsm4 and Nqo1 (Gussing and Bohm, 2004; Oka et al., 2003) and the ventrolateral markers Ncam2 and Reg3g (Alenius and Bohm, 1997; Yu et al., 2005; Figures 1F and S1F).
Together, these results show that RNA tomography is a sensitive and reliable method to examine gene expression patterns in the mouse OM.
Spatial differential gene expression analysis identifies cell-type-specific expression patterns and functional hotspots in the OM
In the last 3 decades, multiple genes with spatially segregated expression patterns across the OM have been identified. Most of these genes are expressed in mature OSNs and encode chemosensory receptors, transcription factors, adhesion molecules, and many molecules involved in the downstream signaling cascade of receptor activation (Cho et al., 2007; Cloutier et al., 2002; Fulle et al., 1995; Greer et al., 2016; Gussing and Bohm, 2004; Juilfs et al., 1997; Liberles and Buck, 2006; Miyamichi et al., 2005; Norlin et al., 2001; Oka et al., 2003; Pacifico et al., 2012; Saraiva et al., 2015b; Tietjen et al., 2003, 2005; Vassar et al., 1993; Wang et al., 2004; Yoshihara et al., 1997; Yu et al., 2005; Zapiec and Mombaerts, 2020). A smaller number of zonally expressed genes (e.g., metabolizing enzymes, chemokines, and transcription factors) were found to be expressed in sustentacular cells, globose basal cells, olfactory ensheathing cells, Bowman’s gland cells, and respiratory epithelial cells (Cloutier et al., 2002; Duggan et al., 2008; Heron et al., 2013; Juilfs et al., 1997; Miyawaki et al., 1996; Norlin et al., 2001; Peluso et al., 2012; Whitby-Logan et al., 2004; Yu et al., 2005). Despite this progress, our knowledge on what genes display true zonal expression patterns and what cell types they are primarily expressed in is still very limited.
To identify axis-specific differentially expressed genes (DEGs) (hereafter referred to as spatial DEGs), we first filtered out lowly expressed genes, then binarized the expression levels at each position according to whether they were higher or lower than their median expression, and applied the Ljung-Box test to the autocorrelation function calculated on the binarized expression values (Figure S2A; STAR Methods). After correcting for multiple testing, we obtained a total of 12,303 spatial DEGs for the three axes combined (false discovery rate [FDR] < 0.01; Figure 2A)—the AP axis showed the highest number of spatial DEGs (10,855), followed by the DV axis (3,658) and the LM (1,318).
Figure 2. Genes with non-random spatial patterns across different cell types in the OM.
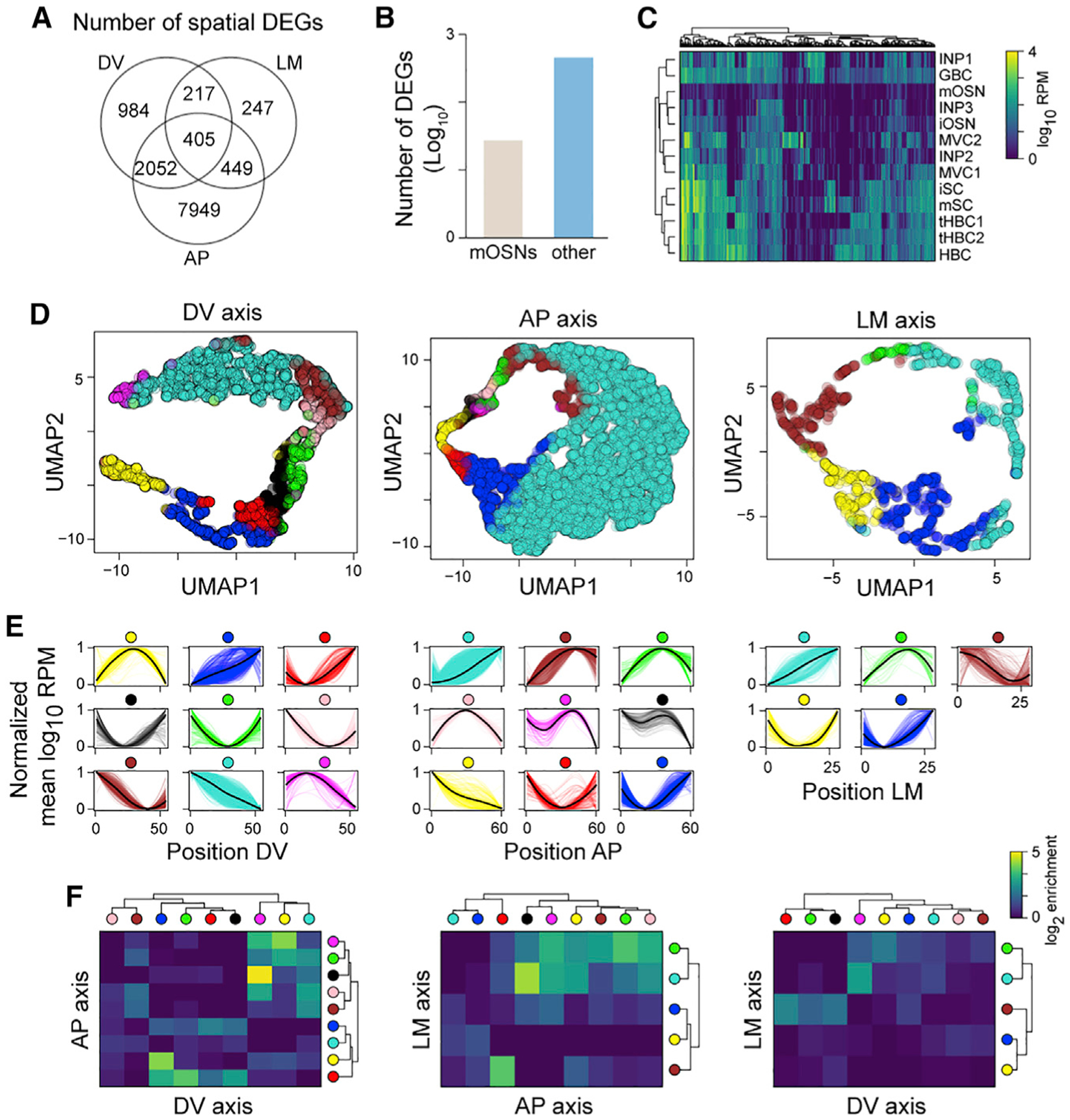
(A) Venn diagram showing the numbers of spatial differentially expressed genes (DEGs) along each axis.
(B) Bar plot showing the log10 number of spatial DEGs that are mOSN specific (“mOSNs”) or that are detected only in cell types other than mOSNs (“other”).
(C) Heatmap of log10 mean expression per cell type of genes that are not expressed in mOSNs but only in other OM cell types (INPs, immediate neuronal precursors; iSCs, immature sustentacular cells; mSCs, mature sustentacular cells; MVCs, microvillous cells; mSCs, mature sustentacular cells).
(D) UMAP plots of spatial DEGs along the three axes (n = 3 per axis). Each gene is colored according to the cluster it belongs to.
(E) Normalized average expression patterns of spatial DEGs clusters along the three axes.
(F) Heatmap showing the log2 enrichment over the random case for the intersection between lists of genes belonging to different clusters (indicated by colored circles) across pairs of axes.
Next, we added cell-type resolution to the spatial axes by combining our data with a single-cell RNA-seq (scRNA-seq) dataset from 13 cell types present in the mouse OM (Fletcher et al., 2017). We cataloged spatial DEGs based on their expression in mature OSNs (mOSNs) versus the 12 other cell types (non-mOSNs; Figures 2B and 2C; Table S2). This led to the identification of 456 spatial DEGs expressed exclusively in non-mOSNs, which are associated with gene ontology (GO) terms, such as transcription factors, norepinephrine metabolism, toxin metabolism, bone development, regulation of cell migration, T cell activation, and others (Table S2). Some genes are expressed across many cell types, but others are specific to one cell type (Figure 2C; Table S2). As expected, some of these genes are cell-specific markers with known spatial expression patterns, such as the sustentacular cells and Bowman’s glands markers Cyp2g1 and Gstm2 (Yu et al., 2005), the neural progenitor cell markers Eya2 and Hes6 (Tietjen et al., 2003), and the basal lamina and olfactory ensheathing cell markers Aldh1a7 and Aldh3a1 (Norlin et al., 2001; Table S2). We also identified spatial DEGs along a single axis or multiple axes and specific to one or few cell types (Figures S2B and S2C). For example, the ribosomal protein Rps21 plays a key role in ribosome biogenesis, cell growth, and death (Wang et al., 2020) and is primarily expressed in horizontal basal cells (HBCs), consistent with their role in the maintenance and regeneration of the OM (Leung et al., 2007). Another example is the extracellular proteinase inhibitor Wfdc18, which induces the immune system and apoptosis (Jung et al., 2004) and is expressed in microvillous cells type 1 (MVC1s), consistent with their role in immune responses to viral infection (Baxter et al., 2020). Two more examples are the fibroblast growth factor Fgf20 in immature sustentacular cells (iSCs) and the adapter protein Dab2 in mature sustentacular cells (mSCs) (Figures S2B and S2C). Fgf20 is expressed in several cell types, regulates the horizontal growth of the olfactory turbinates, and is preferentially expressed in the lateral OM (Yang et al., 2018), consistent with our data. Dab2 regulates mechanisms of tissue formation, modulates immune responses, and participates in the absorption of proteins (Finkielstein and Capelluto, 2016; Park et al., 2019), consistent with the known maintenance and support roles of mSCs in the OM (Brann et al., 2020).
A GO enrichment analysis on the axis-specific DEGs for non-mOSNs genes revealed a very wide variety of biological processes and molecular functions. Some of the notable terms identified were water and fluid transport (e.g., Ctfr and Aqp3), transcription factors (e.g., Hes1 and Dlx5), oxidation-reduction processes (e.g., Scd2 and Cyp2f2), microtubule cytoskeleton organization involved in mitosis (e.g., Stil and Aurkb), cell cycle (e.g., Mcm3 and Mcm4), cell division (e.g., Kif11 and Cdca3), negative regulation of apoptosis (e.g., Dab2 and Scg2), sensory perception of chemical stimulus (e.g., Olfr870 and Gnas), and cellular processes (e.g., Mal and Pthlh), among many others (Table S2).
The identification of thousands of spatial DEGs prompted us to examine their distribution patterns along each axis and the putative functions associated with such spatial clusters of gene expression. We started by using uniform manifold approximation and projection (UMAP) (Becht et al., 2018) and hierarchical clustering to visualize and cluster all spatial DEGs along the three axes. This analysis uncovered nine patterns of expression in the DV and AP axes each and five patterns in the LM axis (Figures 2D and 2E). These patterns include variations of four major shapes: monotonically increasing (/), monotonically decreasing (\), U-shape (W), and inverted U-shape (X) (Figure 2E). The latter two patterns present clear maximum and minimum at different positions along the axis—for example, the brown, green, pink, magenta, and black AP clusters show a similar inverted U-shape pattern, but their maximum moves along the axis (Figure 2E). As expected, the dorsomedial markers Acsm4 and Nqo1 belong to the turquoise clusters in both the DV and LM axes, while the ventrolateral marker Reg3g belongs to the blue cluster from the DV axis (Figure 1F; Table S3), mimicking their respective expression pattern in the mouse OM.
The total number of genes per cluster had a median value of 236 but varied greatly between clusters—ranging from 57 in the green LM cluster to 8,551 in the turquoise AP cluster (Figure 2D; Table S3). GO enrichment analyses on the spatial DEGs yielded enriched terms for 14 of the 23 spatial clusters (Table S3). For example, the turquoise AP cluster displaying a monotonically increasing pattern (Figure 2E) yielded GO terms associated with the molecular machinery of mOSNs—such as axonal transportation, RNA processing, ribosomal regulation, and regulation of histone deacetylation (Table S3). Interestingly, the brown DV cluster, which displays a monotonically decreasing expression pattern (Figure 2E), had similar GO term enrichment (Table S3). In agreement with these results, we found that most known OSN activity-dependent markers (Wang et al., 2017) are spatial DEGs belonging to the AP turquoise and brown clusters, which contain genes with expression peaks in the posterior region (Figure S2E; Table S3). We also observed a similar trend in the DV axis, with many of these markers being more highly expressed in the dorsal region (Figure S2E).
The results above show that OSN activity is enriched in the dorsoposterior region of the OM, which could be due to an enrichment of OSNs in that region. To test this hypothesis, we estimated the abundance and spatial variability of OSNs and five additional major cell types (HBCs, globose basal cells [GBCs], SCs, MVCs, and immediate neuronal precursors [INPs]) in each section through a cell deconvolution analysis (see STAR Methods). We observed statistically significant changes in the abundance of OSNs, which is predicted to be higher in the dorsoposterior region of the OM, as previously suggested (Nickell et al., 2012; Vedin et al., 2009). Conversely, other cell types like HBCs are predicted to have an opposite pattern, as they tend to be more abundant in the anteroventral region (Figure S2F; Table S2).
Next, we extended our GO analysis to the remaining spatial clusters and found additional terms enriched or shared between several clusters among the three axes. For example, GO terms enriched in the dorsomedial region (turquoise DV, pink AP, and LM green clusters) include detoxification of several metabolites and multiple metabolic and catabolic processes, suggesting that this region is involved in the OM detoxification (Table S3). Another example is the enrichment in terms related to the immune system—such as defense response and humoral immune response—in the anteromedial section along the AP axis (yellow, black, and magenta AP and turquoise LM clusters), which strongly hints at a role of this area in defending OM from pathogenic invaders (Table S3). The anteroventral and posteroventral regions (blue DV and blue AP clusters) are enriched in terms related to the cellular and anatomical organization (e.g., extracellular matrix organization and regulation of cell communication) and bone and cartilage development (e.g., ossification and biomineral tissue development), suggesting these locations are hotspots for the development and regulation of the OM structure. Finally, the ventral portion of the DV (red DV cluster) is associated with terms related to cilia movement and function (e.g., regulation of cilium movement and microtubule-based movement), consistent with both the location and functions of the respiratory epithelium (Yu et al., 2005).
Next, we further explored the relationships between the genes populating each cluster. We found that ventral genes (blue DV cluster) tend to reach a peak in expression in the anterior area of the OM (yellow AP cluster) more often than expected by chance (Figure 2F). We also observed that medial genes (turquoise LM cluster) are more highly expressed in the dorsal (magenta DV cluster) and anterior regions (black, yellow, and magenta AP cluster), while genes peaking in the lateral region (brown LM cluster) tend to be ventral (red DV cluster; Figure 2F). These conclusions hold, even when we exclude Olfrs from the analysis (Figure S2D).
These associations between the clusters of spatial DEGs along different axes suggest that the presence of complex 3D expression patterns in OM is not restricted to either Olfrs or OSNs. Moreover, our results show that our experimental approach can uncover spatially restricted functional hotspots within the OM.
A 3D transcriptomic atlas of the mouse OM
Since the OR discovery 3 decades ago (Buck and Axel, 1991), in situ hybridization (ISH) has been the method of choice to study spatial gene expression patterns across the OM. This method is technically challenging and inherently a very low-throughput experimental approach.
As we showed above, our Tomo-seq data enable a systematic and quantitative estimation of gene expression levels along the three axes of the OM. Here, we take this analysis one step further and generate a fully browsable tridimensional (3D) gene expression atlas of the mouse OM. First, we reconstructed the 3D shape of OM based on publicly available images of OM sections (STAR Methods). We then fed the shape information combined with the gene expression data along the three axes into the iterative proportional fitting (IPF) algorithm (Fienberg, 1970; Junker et al., 2014; Figure 3A). The 3D atlas of the OM faithfully reproduced the known 3D pattern of the dorsomedial marker Acsm4 (Oka et al., 2003; Figure 3B). To further validate our 3D gene expression atlas of the OM, we compared the 3D reconstructed patterns with conventional ISH patterns for five spatial DEGs identified in this study. The first gene validated was Cytl1, which we confirmed to be expressed along the septum throughout the OM (Figures 3C and 3D), consistent with the role Cytl1 plays in osteogenesis, chondrogenesis, and bone and cartilage homeostasis (Shin et al., 2019; Zhu et al., 2019). The four additional genes (Olfr309, Olfr618, Olfr727, and Moxd2) validated via ISH are presented elsewhere in this manuscript (Figures 4, 5, and S4).
Figure 3. The 3D reconstruction of the OM.
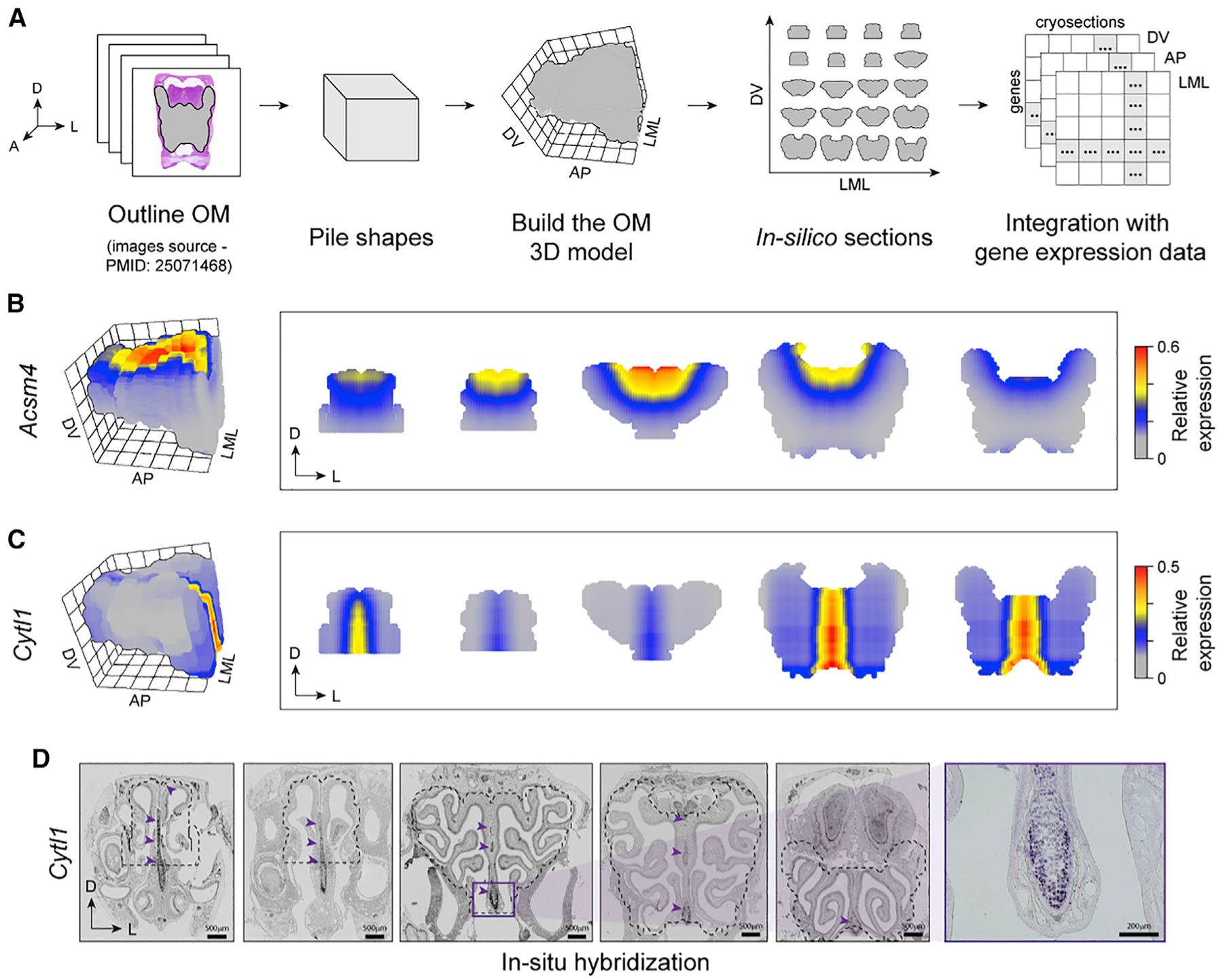
(A) Schematic of 3D shape reconstruction strategy. Images of 2D slices along the AP axis of the OM were piled together to build an in silico 3D model of OM, which can also be used to visualize in silico sections. This 3D model, together with the gene expression data along each axis, was the input of the iterative proportional fitting algorithm, which allowed us to estimate a 3D expression pattern for any gene.
(B and C) Reconstruction of the 3D expression patterns of the Acsm4 (B) and Cytl1 (C) in the OM, visualized in 3D and in OM coronal sections taken along the AP axis.
(D) ISH experiment validating Cytl1 spatial expression pattern reconstructed in (C); note that Cytl1 is expressed in the septal region all along the OM. Purple arrowheads indicate the location of labeled cells. The dotted outline marks the borders of the OM dissected and used in the RNA-seq experiments and for the construction of the 3D model.
Figure 4. Zonal organization of Olfrs in the OM.
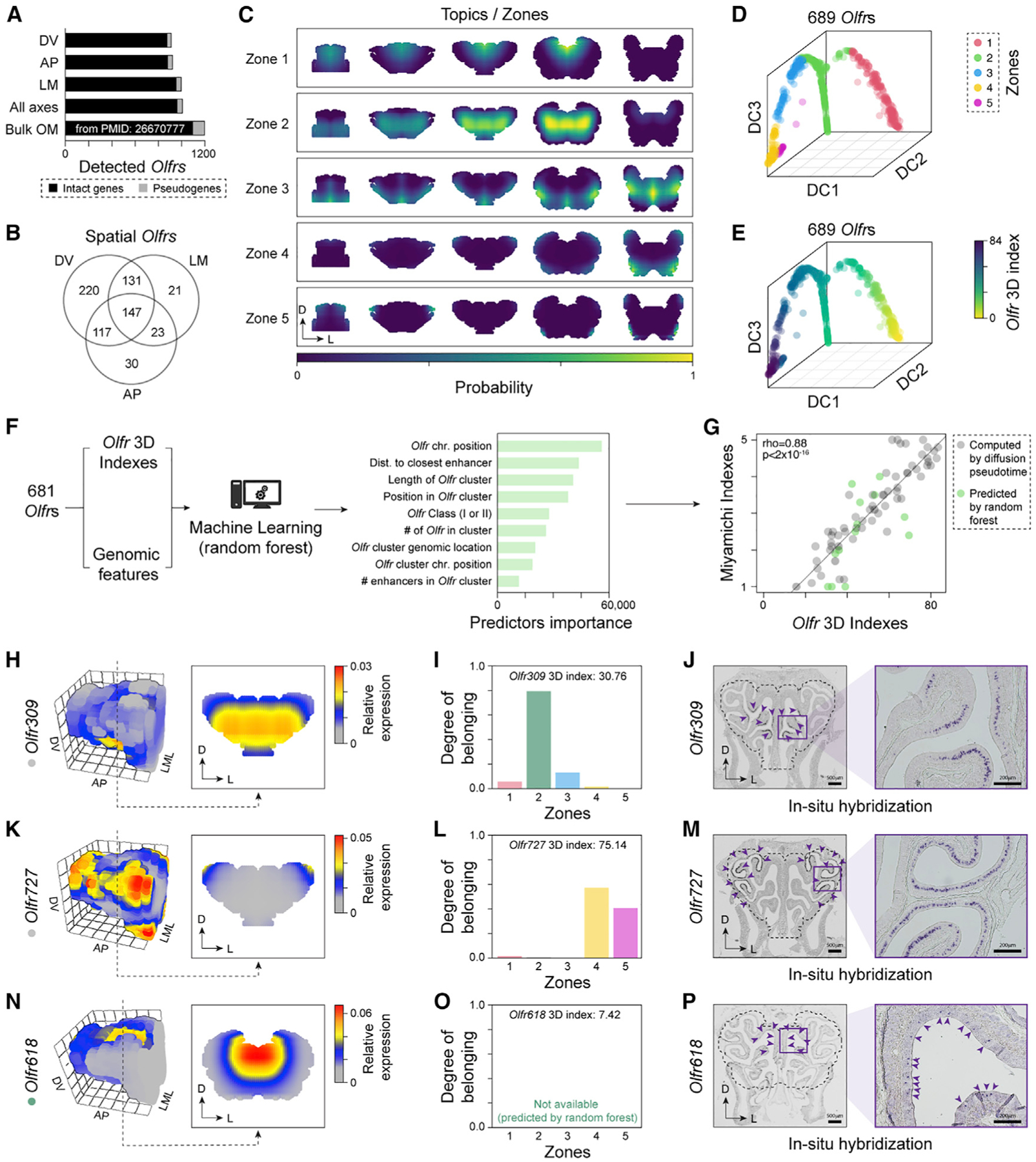
(A) Number of Olfrs detected in our data and in an OM bulk RNA-seq data (Saraiva et al., 2015b).
(B) Venn diagram of spatially differentially expressed Olfrs per axis (n = 3 per axis).
(C) Visualization of the five zones across the OM (coronal sections) estimated with a latent Dirichlet allocation algorithm. The colors indicate the probability (scaled by its maximum value) that a position belongs to a given zone.
(D) Diffusion map of Olfrs. Genes are colored based on the zone they fit in the most. DC, diffusion component.
(E) Same as (D), with Olfrs colored by their 3D index.
(F) We fitted a random forest algorithm to the 3D indices of 681 spatial Olfrs using nine genomic features as predictors. After training, the random forest was used to predict the 3D indices of 697 Olfrs that have too low levels in our data.
(G) 3D indices versus the indices of 80 Olfrs estimated in Miyamichi et al. (2005) from ISH data. Black circles indicate Olfrs detected in our dataset; green circles are Olfrs whose indices were predicted with random forest. The correlation coefficients computed separately on these two sets of Olfrs are, respectively, rho = 0.92 (p < 2 × 10−16) and rho = 0.69 (p = 0.009).
(H–P) Predicted expression patterns (H, K, and N), degrees of belonging (I, L, and O), and ISH (J, M, and P) for Olfr309, Olfr727, and Olfr618, respectively. Purple arrowheads indicate the location of labeled cells. The dotted outline marks the borders of the OM dissected and used in the RNA-seq experiments and for the construction of the 3D model.
Figure 5. Zonal organization of non-Olfr genes in the OM.
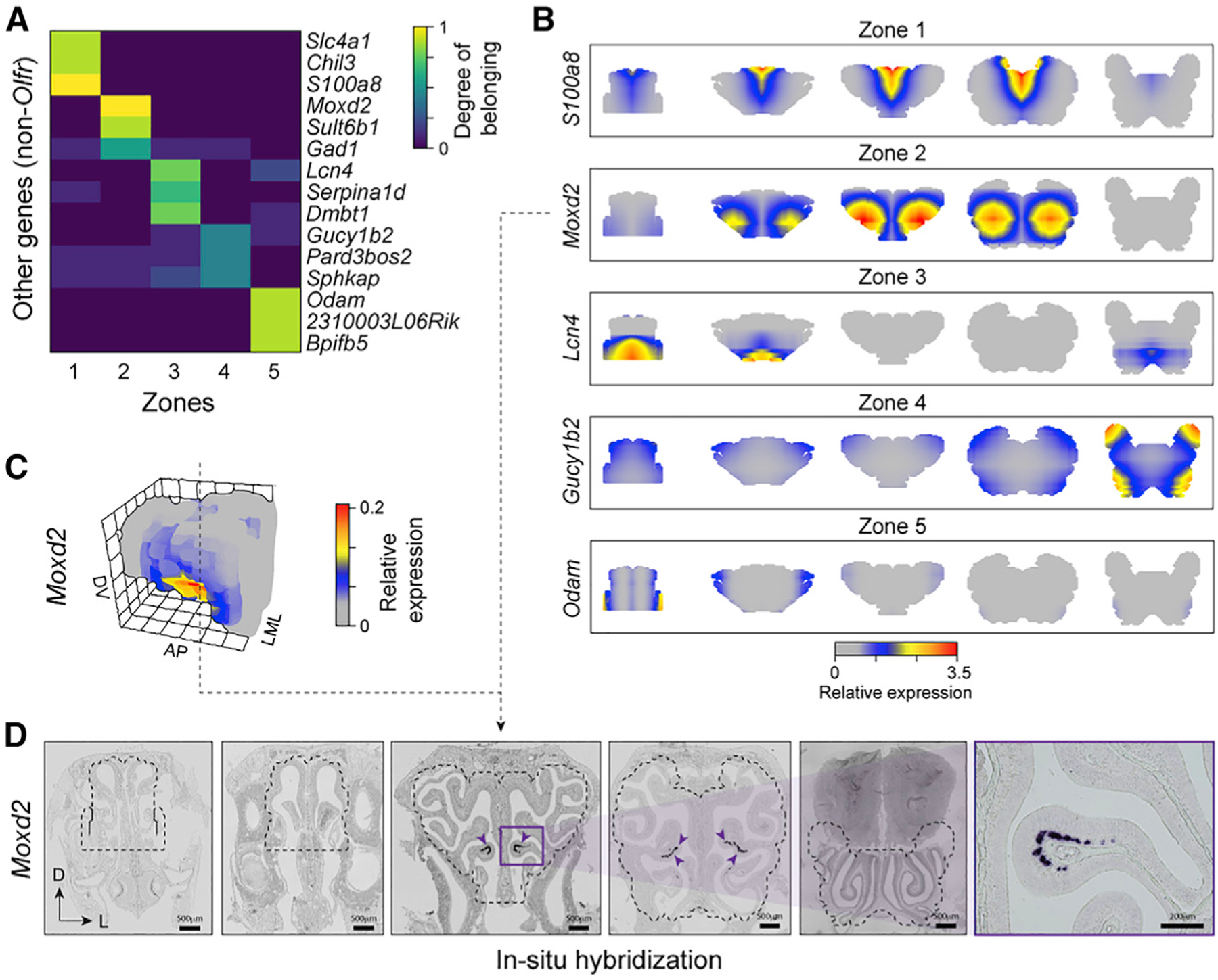
(A) Heatmap of degrees of belonging of most zone-specific non-Olfr genes.
(B) 3D gene expression pattern (coronal sections) of most topic-specific non-Olfrs for each topic along the AP axis.
(C) Reconstruction of the 3D expression pattern of the gene Moxd2 in the OM.
(D) ISH experiment validating Moxd2 spatial expression pattern reconstructed in (B) and (C). Purple arrowheads indicate the location of ISH-labeled cells. The dotted outline marks the borders of the OM dissected and used in the RNA-seq experiments and for the construction of the 3D model.
To make this 3D gene expression atlas of the mouse OM available to the scientific community, we created a web portal (available at http://atlas3dnose.helmholtz-muenchen.de:3838/atlas3Dnose) providing access to the spatial transcriptomic data described here in a browsable and user-friendly format. This portal contains search functionalities allowing the users to perform pattern search by gene, which returns (1) the normalized counts along each of the three axes, (2) the predicted expression pattern in 3D with a zoom function, (3) visualization of the expression patterns in virtual cryosections along the OM by selecting any possible pairwise intersection between two given axes (i.e., DVxAP, DVxLM, and APxLM), (4) the degrees of belonging for each “zone” (see results section below), and (5) single-cell expression data across 14 different cell types.
In sum, here, we generated and made publicly available a highly detailed and fully browsable 3D gene expression atlas of the mouse OM, which allows the exploration of the expression patterns for ~20,000 genes.
Topographical expression patterns of Olfrs
In our combined dataset, we detected a total of 959 Olfrs (Figure 4A), of which we confidently reconstructed the spatial expression patterns for 689 differentially expressed in space (FDR < 0.01; Figure 4B)—a number six times larger than the combined 112 Olfrs characterized by previous ISH studies (Miyamichi et al., 2005; Ressler et al., 1993; Vassar et al., 1994; Zapiec and Mombaerts, 2020). To define Olfr expression in 3D space in a rigorous, unbiased, and quantitative way, we ran a latent Dirichlet allocation (LDA) algorithm (STAR Methods; Liu et al., 2016) on the 689 spatially differentially expressed Olfrs. LDA is a generative statistical model that can infer the topics of a collection of documents based on the variability and frequency of specific words. In the context of this study, if the spatial expression data of Olfrs are considered equivalent to “documents,” the inferred topics correspond to “zones” (STAR Methods). We ran LDA for different numbers of zones, and the trend of the log likelihood function suggested that the minimal number of topics required to represent the diversity of patterns is five (Figure S3A; STAR Methods). Next, we visualized the spatial distribution of these five zones in our 3D OM model, with colors representing the probability that a given spatial position belongs to each zone. These five zones extend from the dorsomedial-posterior to the lateroventral-anterior region (Figure 4C), consistent with the previously described zones (Miyamichi et al., 2005; Ressler et al., 1993; Vassar et al., 1993).
The majority of Olfrs with known spatial patterns are restricted to a single zone, but a small number of Olfrs are expressed across multiple zones in a continuous or non-continuous fashion (Miyamichi et al., 2005; Strotmann et al., 1992; Zapiec and Mombaerts, 2020). Under this logic, each Olfr has a different probability of belonging to the five topics and zones we identified. To test this assumption, we used the same mathematical framework as above to compute the probabilities that the expression pattern of each Olfr belongs to a given zone, i.e., the “degree of belonging” (DOB) (Table S4). The DOBs represent a decomposition of the expression patterns in terms of the five zones (Figure 4C) and quantitatively describe the changes in patterns of genes with overlapping areas of expression (e.g., see Figure S3B). The width of the distribution of DOBs across the five zones, which can be measured with entropy, can distinguish genes whose patterns mostly fit in a single zone from those spanning multiple zones (Figure S3C; STAR Methods).
To visualize the global distribution of the 689 Olfrs, we applied the diffusion map algorithm (Haghverdi et al., 2015) to their DOBs. This showed that the genes are approximately distributed along a continuous line spanning the five zones and without clear borders between zones (Figure 4D), consistent with previous studies (Miyamichi et al., 2005; Strotmann et al., 1992; Zapiec and Mombaerts, 2020). With the diffusion pseudo-time algorithm (Haghverdi et al., 2016), we calculated an index (hereafter referred to as “3D index”) that tracks the position of each Olfr gene along the 1D curve in the diffusion map and represents its expression pattern (Figure 4E).
While our approach yielded an index for the 689 spatially differentially expressed Olfr genes used to build the diffusion map, there were 697 Olfrs that could not be analyzed, either because they were too lowly expressed or not detected at all in our dataset (Figure 4A). Since the spatial expression patterns for some Olfrs are partly associated with their chromosomal and genomic coordinates (Sullivan et al., 1996; Tan and Xie, 2018; Zhang et al., 2004), we hypothesized that we could use a machine-learning algorithm to predict the 3D indices for the 697 Olfrs missing from our dataset. Thus, we trained a random forest algorithm on the 3D indices of the spatially differentially expressed Olfrs in our dataset using nine genomic features as predictors, such as the chromosomal position, number of Olfrs in cluster, and distance to nearest known enhancer (Figure 4F; STAR Methods). The algorithm performance was confirmed by over 100 cross-validation iterations, which revealed a low root-mean-square error (≲10%) on the mean 3D index (Figure S4A; STAR Methods). The five most important predictors were features associated with chromosomal position, distance to the closest Olfr enhancer (Monahan et al., 2017), length of the Olfr cluster, position in the Olfr cluster, and phylogenetic Olfr class (Figure 4F). Using this machine-learning algorithm, we predicted the 3D indices for the 697 Olfrs missing reliable expression estimates in our dataset (Table S4).
Overall, through multiple unsupervised and supervised computational methods, we have quantitatively defined five spatial expression domains in the OM (called zones) and have provided accurate 3D spatial indices for 1,386 Olfrs, which represents ~98% of the annotated Olfrs.
Importantly, we found strong correlations between the “Miyamichi indices” inferred using ISH in Miyamichi et al. (2005) and our 3D indices (rho = 0.88; p < 2 × 10−16; Figure 4G). This correlation remains significant when we separately analyze the 3D indices computed by diffusion pseudo-time (rho = 0.92; p < 2 × 10−16) or predicted by random forest (rho = 0.69; p = 0.009). In addition, our indices also correlated with the “Zolfr indices” (Zapiec and Mombaerts, 2020; rho = 0.88; p < 2 × 10−16; Figure S4B), and with the “Tan indices” (Tan and Xie, 2018; rho = 0.89; p < 2 × 10−16; Figure S4C), inferred by ISH and RNA-seq, respectively.
To confirm our predictions, we performed ISH for three Olfrs that have not been characterized before—two detected in our dataset and for which the 3D index was calculated via diffusion pseudo-time (DPT) (Olfr309 and Olfr727) and one not detected in our dataset and for which the 3D index was predicted with the random forest algorithm (Olfr 618). Notably, all three Olfrs were expressed primarily within the zones they were predicted to be expressed in: zone 2 for Olfr309 (3D index = 30.76), zones 4 and 5 for Olfr727 (3D index = 75.14), and zone 1 for Olfr618 (3D index = 7.42; Figures 4H–4P and S4D–S4F; Table S4).
Topographical expression patterns for non-Olfr genes
A recent study performed RNA-seq in 12 randomly dissected OM pieces along the DV axis and identified ~700 non-Olfr genes with putatively spatial patterns (Tan and Xie, 2018), including many genes with zonal expression patterns identified previously (Duggan et al., 2008; Gussing and Bohm, 2004; Liberles and Buck, 2006; Ling et al., 2004; Norlin et al., 2001; Oka et al., 2003; Tietjen et al., 2003; Whitby-Logan et al., 2004; Yoshihara et al., 1997). By identifying 11,538 non-Olfr spatial DEGs (Figures 2 and 3; Table S5), we increased the list of non-Olfr genes with spatial zonation in the OM by 16-fold.
Using the mathematical framework based on topic modeling described above, we decomposed the expression patterns of non-Olfr genes onto the five zones we identified. This allowed us to identify genes showing zone specificity by calculating the entropy of the DOBs distributions. Interestingly, we found 28 genes highly specific for each of the five zones (i.e., with entropy <1; STAR Methods; Figure 5A; Table S5). For example, S100a8 (zone 1) codes for a calcium-binding protein involved in calcium signaling and inflammation (Yoshikawa et al., 2018), Moxd2 (zone 2) is a mono-oxygenase dopamine hydroxylase-like protein possibly involved in olfaction (Kim et al., 2014), Lcn4 (zone 3) is a lipocalin involved in transporting odorants and pheromones (Charkoftaki et al., 2019; Miyawaki et al., 1994), Gucy1b2 (zone 4) is a soluble guanylyl cyclase oxygen and nitric oxide (Bleymehl et al., 2016; Koglin et al., 2001), and Odam (zone 5) is a secretory calcium-binding phosphoprotein involved in cellular differentiation and matrix protein production and with antimicrobial functions of the junctional epithelium (Lee et al., 2012; Springer et al., 2019; Figure 5B). The high zone specificity of the expression pattern of these genes gives clues into possible biological processes taking place in the zones. Indeed, Gucy1b2 is a known genetic marker for a small OSN subpopulation localized in cul-de-sac regions in the lateral OM, consistent with our reconstruction (Figure 5B), and it regulates the sensing of environmental oxygen levels through the nose (Omura and Mombaerts, 2015; Saraiva et al., 2015b). In addition, our ISH experiments revealed that Moxd2 is expressed in a small ventrolateral patch of the OM (Figure 5D), validating its predicted 3D spatial pattern (Figures 5B and 5C) and highlighting a potential highly localized role of this protein in neurotransmitter metabolism (Goh et al., 2016) in the mouse OM.
A recent study showed that the transcription factors Nfia, Nfib, and Nfix regulate the zonal expression of Olfrs (Bashkirova et al., 2020). To get some insights into the signaling pathways involved in this process, we mined our dataset for genes encoding ligands and receptors (Efremova et al., 2020) correlated with the expression patterns of the Nfis (STAR Methods). This analysis returned 476 genes involved in biological processes associated with the regulation of neurogenesis, regulation of cell development, anatomical structure development, cellular component organization or biogenesis, and regulation of neuron differentiation (Table S5). As expected, some of these genes have known functions in the OM, such as segregating different cell lineages for Notch1–3 (Carson et al., 2006), genes associated with the development of the nervous system (e.g., Erbb2 and Lrp2; Britsch et al., 1998; Spuch et al., 2012), and many others associated with the semaphorin-plexin, ephrin-Eph, and Slit-Robo signaling complexes—which regulate OSN axon guidance and spatial patterning of the OM (Cloutier et al., 2002; Cutforth et al., 2003; Huber et al., 2003; Kania and Klein, 2016). Excitingly, the majority of these 476 genes still have unknown functions in the OM, thus highlighting the potential of our approach to discover genes and pathways involved in the regulation of zonal expression in the OM.
The anatomical logic of smell
For most sensory systems, the functional logic underlying the topographic organization of primary receptor neurons and their receptive fields is well known (Kandel et al., 2013). In contrast, the anatomic logic of smell still remains unknown, and it is subject of great controversy and debate (Kurian et al., 2021; Secundo et al., 2014).
To explore the underlying logic linked to the zonal distribution of Olfrs, we investigated possible biases between their expression patterns and the physicochemical properties of their cognate ligands. First, we compiled a list of known 738 Olfr-ligand pairs, representing 153 Olfrs and 221 odorants (Figure 6A; Table S6). Interestingly, Olfr pairs sharing at least one common ligand have more similar expression patterns (i.e., more similar 3D indices) than Olfrs detecting different sets of odorants (Wilcoxon rank-sum test; p < 2 × 10−16; Figure 6B). This observation is consistent with the hypothesis that the Olfr zonal distribution depends, at least partially, on the properties of the odorants they bind to.
Figure 6. Physiological role of the zones.
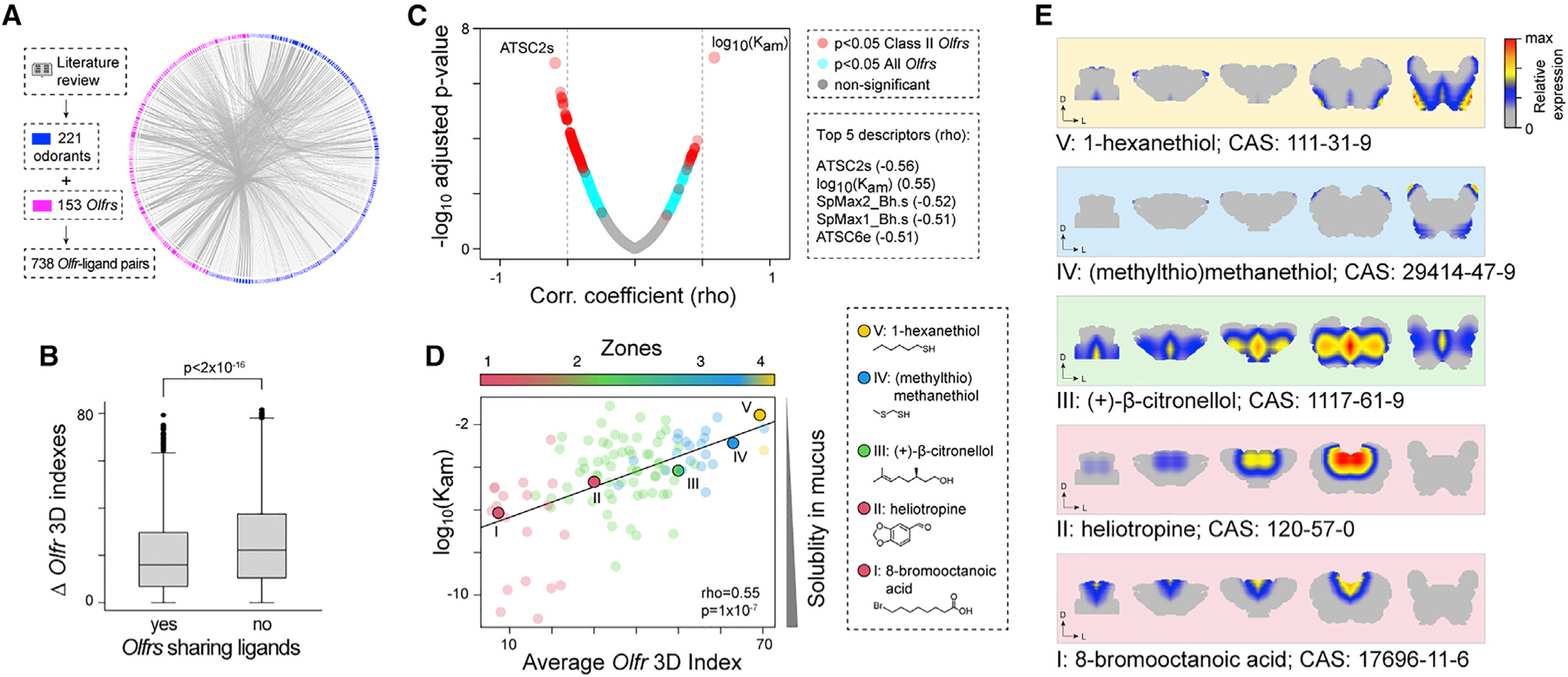
(A) Circular network illustrating the pairs of Olfrs and ligands that we found in the literature.
(B) Boxplots showing the distributions of the absolute value of 3D index differences between pairs of Olfrs sharing at least one ligand versus pairs of Olfrs without cognate ligands in common. The difference between the two distributions is statistically significant (p < 2 × 10−16; Wilcoxon rank-sum test).
(C) Scatterplot showing the Spearman correlation coefficients between the ligands’ mean 3D indices and molecular descriptors and the corresponding −log10(adjusted p value). Turquoise circles indicate the descriptors having a significant correlation only when both class I and II Olfrs are considered; red circles mark the descriptors with a significant correlation also when class I Olfrs are removed.
(D) Scatterplot illustrating the correlation between air/mucus partition coefficients of the odorants and the average 3D indices of their cognate Olfrs. Only odorants for which we know at least two cognate Olfrs (110) were used here. Odorants are colored according to the zone they belong to (defined as the zone with the highest average degree of belonging computed over all cognate receptors). The five odorants highlighted in the plot by larger circles are indicated on the right-hand side, along with their molecular structure and common name.
(E) Average expression pattern of the cognate Olfrs recognizing each of the five odorants highlighted in (D), including their respective CAS numbers.
Next, we considered a set of 1,210 physicochemical descriptors, including the molecular weight, the number of atoms, aromaticity index, lipophilicity, and the air/mucus partition coefficient (Kam), which quantifies the mucus solubility of each ligand (Rygg et al., 2017; Scott et al., 2014; STAR Methods). We then computed the Spearman’s correlation of each of these descriptors of the ligands with the average 3D indices of the Olfrs detecting them. We found a statistically significant correlation for 744 descriptors (FDR < 0.05; Figure 6C; Table S6). The top five highest correlations were with the air/mucus partition coefficient Kam (rho = 0.55; adjusted p = 1 × 10−7), ATSC2S (rho = −0.56; adjusted p = 2 × 10−7), SPmax2_Bh.s (rho = −0.52; adjusted p = 2 × 10−6), SPmax1_Bh.s (rho = −0.51; adjusted p = 3 × 10−6), and ATSC6e (rho = −0.51; adjusted p = 3 × 10−6; Figure 6C; Table S6). Interestingly, ATSC2S, SPmax1_Bh.s, and SPmax2_Bh.s are also related to solubility (Consonni and Todeschini, 2008; Devillers and Domine, 1997; Hollas, 2003). Notably, the association between Kam and the mean 3D indices does not depend on the number of zones defined with LDA (STAR Methods). Furthermore, it remains robust to changes in the set of ligands and/or Olfrs used for the analysis, namely, when we excluded Olfrs for which the 3D indices were predicted with the random forest model (rho = 0.48; p = 2 × 10−6; Figure S5B) or when only 3D indices from class II Olfrs were included in the analysis (rho = 0.5; p = 1 × 10−7; Figure S5C).
In particular, the positive correlation of the 3D indices with Kam (Figure 6D) indicates that the most soluble odorants (lower) preferentially activate Olfrs located in the most antero-dorsomedial region (zone 1) of the OM, while the least soluble odorants (higher Kam) activate Olfrs in the postero-ventrolateral region (zones 4 to 5). In other words, gradients of odorants sorption (as defined by their Kam) correlate with the gradients of Olfr expression from zone 1 to zone 5, consistent with the chromatographic/sorption hypothesis in olfaction (Mozell, 1966; Scott et al., 2014). This is exemplified by the plots in Figure 6E, illustrating the predicted average expression levels across OM sections of the Olfrs binding to five odorants with different values of Kam. These results show a direct association between Olfr spatial patterns and the calculated sorption patterns of their cognate ligands in the OM, providing a potential explanation for the physiological function of the zones in the OM.
DISCUSSION
Past studies yielded inconclusive and sometimes contradictory views on the basic logic underlying the peripheral representation of smell, partly because the topographic distribution of OSN subtypes and their receptive fields still remained vastly uncharted, data on Olfr-ligand pairs were scarce, and there were pitfalls associated with electro-olfactogram recordings used to study spatial patterns of odor recognition in the nose (Kurian et al., 2021; Scott and Scott-Johnson, 2002; Secundo et al., 2015). Here, we combined RNA-seq and computational approaches that utilize unsupervised and supervised machine learning methods to discover and quantitatively characterize spatial expression patterns in the OM. We created a 3D transcriptional map of the mouse OM, which allowed us to spatially characterize 17,628 genes, including ~98% of the annotated Olfrs. We identified and validated by ISH several spatial marker genes, and a clustering analysis pinpointed the OM locations where specific functions related to, e.g., the immune response might be carried out. We also mathematically defined Olfr expression zones in the OM with an unsupervised machine-learning method based on topic modeling. We estimated that the OM includes at least five zones, which can be used to decompose the expression patterns of all genes. However, our analysis showed that there is a continuous distribution of Olfrs patterns in the OM. Thus, while a discrete number of zones might be convenient to provide a first classification of Olfrs, these might obscure the complexity of the OM spatial patterns. To account for this, we adopted a mathematical framework that can rigorously define zones while capturing finer structures in the data, via the degrees of belonging and the 3D index, which are more suitable to describe Olfrs patterns crossing multiple zones.
The global transcriptomic landscape of the vertebrate OM is similar between individuals and broadly conserved among different vertebrate species, ranging from zebrafish to human (Bear et al., 2016; Saraiva et al., 2015a, 2019). Similarly, the spatial segregation of Olfrs into partially overlapping rings of expression, centered around the midline structure of the OM, is also conserved among vertebrates (Freitag et al., 1995; Horowitz et al., 2014; Marchand et al., 2004; Miyamichi et al., 2005; Octura et al., 2018; Ressler et al., 1993; Strotmann et al., 1992; Vassar et al., 1993; Weth et al., 1996). While the number of Olfr zones in zebrafish, frog, and salamander still remain unknown (Freitag et al., 1995; Marchand et al., 2004; Weth et al., 1996), ISH studies suggested that the total number of Olfr expression zones can vary between mammals—ranging from two in macaque (Horowitz et al., 2014) to four in rat (Vassar et al., 1993) and goat (Octura et al., 2018), and between four and nine in mouse (Miyamichi et al., 2005; Ressler et al., 1993; Zapiec and Mombaerts, 2020). While the exact number of Olfr expression zones in OM still remains under debate, our results are consistent with both another recent RNA-seq study (Tan and Xie, 2018) and the largest Olfr ISH study in the mouse OM (Miyamichi et al., 2005), thus supporting the existence of at least five overlapping Olfr expression zones in the mouse nose.
Taking into account how conserved the molecular organization of the OM is in vertebrates, the 2-fold reduction in the number of Olfr expression zones in macaque compared with rodents and goat (an ungulate) is puzzling. While we cannot exclude the presence of confounding factors (e.g., limited Olfr sampling and/or inconsistent definitions of “zones”), it is interesting that the 2-fold reduction in number of zones is associated with a 2-fold reduction in the number of annotated intact Olfrs in macaque (and other higher primates, including human) compared with other rodents and ungulates (Horowitz et al., 2014; Niimura et al., 2014; Saraiva et al., 2019). Since the accelerated loss of Olfr genes during primate evolution has been linked to the acquisition of trichromatic acute vision and dietary changes (Niimura et al., 2018), it is plausible that these evolutionary pressures also helped shape the spatial distribution of Olfrs in macaques and other primates, including human.
The quantitative framework we built for this dataset will facilitate the interrogation of gene expression patterns via an online tool we provide and help answer important questions on the physiology of the nose. Our approach could be easily applied to spatial transcriptomic data collected from other tissues to perform comparisons across tissues from different species or the same tissue across multiple developmental stages. Moreover, the results from this study serve as a template to start answering other important questions about olfaction, such as whether Olfr spatial expression maps can encode maps of odor perception. Because the general molecular mechanisms of olfaction, zonal organization of Olfrs, conservation of ligands among Olfr orthologs, and components of olfactory perception are conserved in mammals (Adipietro et al., 2012; Bear et al., 2016; Freitag et al., 1995; Horowitz et al., 2014; Kurian et al., 2021; Manoel et al., 2021; Octura et al., 2018; Saraiva et al., 2019; Weth et al., 1996), the association we uncovered here between Olfr zones and the solubility of odorants they detect can likely be extrapolated to other mammals, including humans. Finally, the functional logic underlying the mammalian topographic organization of primary receptor neurons and their receptive fields in smell is now starting to be exposed.
Limitations of the study
This study enabled us to answer fundamental and long-standing questions about the rationale behind the spatial organization of the peripheral olfactory system. Specifically, we provide evidence to the hypothesis that the spatial zones increase the discriminatory power of the olfactory system by distributing Olfrs in the areas of the OM more likely to be reached by their cognate ligands, based on their solubility in mucus. A caveat of this approach is that the Olfr-ligand list we compiled from the literature includes odorant libraries of different size and composition and tested using different experimental approaches. Moreover, highly abundant Olfrs have a higher probability of being deorphanized than lowly abundant Olfrs, and ecologically relevant odorants are more likely to activate Olfrs when compared with other odorants (Dunkel et al., 2014; Saraiva et al., 2019; Trimmer and Mainland, 2017). Despite having compiled and performed our analysis on the largest set of Olfr-ligand pairs assembled to date and carrying out multiple robustness checks, we cannot rule out that ascertainment bias might contribute to the associations we found between the Olfr spatial location and the properties of their respective ligands. Future studies investigating the activation profiles for all mouse Olfrs and/or mapping the in vivo activation patterns of mouse Olfrs in the olfactory mucosa will be key to stress test the conclusions of our study.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and data should be directed to and will be fulfilled by the Lead Contact Luis R. Saraiva (saraivalmr@gmail.com).
Materials availability
This study did not generate new unique reagents.
Data and code availability
RNA-seq raw data have been deposited and are publicly available as of the date of publication at ArrayExpress: E-MTAB-10211. All original code and scripts for the 3D nose atlas shiny app has been deposited at Github and can be found at the Github Repository: https://doi.org/10.5281/zenodo.6036047https://zenodo.org/badge/DOI/10.5281/zenodo.6045897.svg. The 3D nose atlas processed data can be browsed and visualized here: http://atlas3dnose.helmholtz-muenchen.de:3838/atlas3Dnose.
Any additional information required to reanalyze the data reported in this paper is available from the lead contacts upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
The animals used in this study were adult male C57Bl/6J mice (aged 8–14 weeks, The Jackson Laboratory, Stock # 00664) maintained in group-housed conditions on a 12:12 h light:dark schedule (lights on at 0700 hours). Each mouse was randomly assigned for cryosectioning along one of the three cartesian axes.
The use and care of animals used in this study was approved by the Internal Animal Care and Use Committee (IACUC) of Monell Chemical Senses Center, by the IACUC of the University of São Paulo, and by the Wellcome Trust Sanger Institute Animal Welfare and Ethics Review Board in accordance with UK Home Office regulations, the UK Animals (Scientific Procedures) Act of 1986.
METHOD DETAILS
Dissection of the olfactory mucosa, cryosections, and RNA-sequencing
The olfactory mucosa (OM) of 9 mice was carefully dissected, and all the surrounding tissue (including glands and bone) removed – this was necessary to ensure that the transcripts present in the surrounding tissue do not contaminate the RNA isolated from the OM. The OMs were then embedded in OCT (Tissue Tek), immediately frozen in dry-ice and kept at −80°C. Each OM was then cryosectioned along each of the 3 cartesian axes: dorsal-ventral (DV, n = 3), anterior-posterior (AP, n = 3), or lateral-medial-lateral (LML, N = 3). Every second cryosections (35 μm thick) was collected into 1.5 mL eppendorf tubes containing 350 μL RLT Plus Buffer (Qiagen) supplemented with 1% 2-mercaptoethanol, immediately frozen in dry-ice and kept at −80°C until extraction. RNA was extracted using the RNeasy Plus Micro Kit (Qiagen), together with a genomic DNA eliminator column and a 30-minute incubation with DNAse I (Qiagen). Reverse transcription and cDNA pre-amplification were performed using the SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing (Clontech/Takara). cDNA was harvested and quantified with the Bioanalyzer DNA High-Sensitivity kit (Agilent Technologies). Libraries were prepared using the Nextera XT DNA Sample Preparation Kit and the Nextera Index Kit (Illumina). Multiplexed libraries were pooled and paired-end 150-bp sequencing was performed on the Illumina HiSeq 4000 platform at Sidra Medicine, except for one library (DV-I) for which 125-bp paired-end sequencing was performed on the Illumina HiSeq 2500 platform at the Wellcome Sanger Institute. The raw data are available through ArrayExpress under accession number E-MTAB-10211.
RNA-seq data mapping and gene counting
Reads were aligned to the mm10 mouse genome (release 99). The sequences of the genes “Xntrpc” and “Capn5” were removed from the genome files as in Saraiva et al. (2015b). The alignment was performed with the software STAR version 2.7.3a (Dobin et al., 2013). Genome indexes were generated using STAR –runMode genomeGenerate with default parameters. Then, alignment of reads was performed with the following options: –runThreadN 48 –outSAMunmapped Within –outFilterMultimapNmax 1000 –outFilterMismatchNmax 4 –outFilterMatchNmin 100 –alignIntronMax 50000 –alignMatesGapMax 50500 –outSAMstrandField intronMotif –outFilterType BySJout. The resulting SAM files were converted to bam format and sorted using samtools (version 0.1.19–44428cd) (Li et al., 2009). The multi mapping reads were eliminated using the same software (samtools view -q 255). Finally, the reads for each gene were counted using htseq-count (version 0.11.2) with the options -m intersection-nonempty -s no -i gene_name -r pos (Anders et al., 2014).
Quality control
We excluded all the samples that fulfilled any of these criteria: they had less than 50% mapped reads, less than 4,000 detected genes, more than 20% mitochondrial reads, less than 10,000 total number of reads, or did not express any of the 3 canonical OSN markers Omp, Cnga and Gnal. This resulted in ~51 good-quality sections along the DV axis (~84% out of the collected sections), ~76 (~91% of total) along the AP axis and ~59 (~93% of total) along the LML axis, as averaged across the three replicates per axis.
Data normalization
Gene expression counts were normalized by reads-per-million (RPM), then genes detected in only one replicate and genes that were detected in less than 10% of all samples along one axis were eliminated. To check the similarity between replicates, we calculated Spearman correlations between the transcriptional profiles of sections along each axis (using the top 1000 Highly Variable Genes per axis). Close positions had the most similar transcriptional profiles (Figure 1C). Afterward, the 3 replicates for each axis were aligned as follows: the top 3,000 highly variable genes (HVGs) from each replicate were identified using the method implemented in the scran library in R (Lun et al., 2016) and the intersection of these 3 groups was used in the next steps. For the replicates’ alignment, we took as reference the replicate with the smallest number of slices. We used a sliding window approach that identified the range of consecutive positions on each replicate along which the average value of the Spearman’s correlation coefficient computed with the reference replicate over the HVG was maximum (mean Spearman’s Rho = 0.80, p < 0.05). To mitigate batch effects, the level of every gene was scaled in such a way that their average value in each replicate was equal to the average calculated across all replicates. After this scaling transformation, the data was then averaged between replicates. Once the 3 biological replicates were combined, we had 54 sections along the DV axis, 60 along the AP and 56 along the LML. Along the LML axis a symmetric pattern of expression is expected around the central position, where the septal bone is located. To confirm this in our data, first we identified the central position by analyzing the expression pattern of neuronal markers like Cnga2, Omp and Gnal, whose expression is lowest in the area around the septal bone. Indeed, all three marker genes reach a minimum at the same position along the LML axis (slice 28), which we considered to be the center. The expression patterns of ~90% of genes on either side of the central position show a positive correlation, and ~70% reach statistical significance (Spearman’s correlation computed on the highly variable genes having more than 50 normalized counts in at least 3 slices), further supporting the hypothesis of the bilateral symmetry. Hence, after replicates were averaged, LML axis was made symmetric averaging positions 1:28 and 56:29. Moreover, Olfrs were normalized by the geometric mean of neuronal markers Omp, Gnal and Cnga2, as done previously (Ibarra-Soria et al., 2017).
To verify the presence of a spatial signal, we calculated the Moran’s I and the associated p-values for the top 100 Highly Variable genes along each axis using the “Moran.I″ function from the “ape” library in R with default parameters (Paradis et al., 2004). The p-values of the genes along each axis were combined with the Simes’ method (Simes, 1986) using the function combinePValues from the scran R library (Figure S1E).
Identification of differentially expressed genes and gene clustering
Before testing for differential expression along a given axis, we filtered out genes whose expression levels had low variability. To this aim, for each gene we estimated their highest and lowest expression by taking the average of its three highest and three lowest values respectively. Then, we considered for downstream analyses only the genes that meet either of these two criteria: the highest expression value is greater than or equal to 5 normalized counts and the fold-change between the highest and lowest value is greater than 2; or the difference between the highest and the lowest value is greater than or equal to 4 normalized counts. The expression levels of the genes were binarized according to whether their value was higher or lower than their median expression along the axis. Finally, we used the “ts” function in R to transform the binarized expression values into time series objects, and we applied on them the Ljung-Box test (Box.test function in R with lag = (axis length)-10) which identifies genes with statistically significant autocorrelations, i.e., with non-random expression patterns along an axis. The resulting p-values were adjusted using the FDR method and genes with an FDR <0.01 were considered as differentially expressed. For the next steps, the log10 normalized expression of differentially expressed genes along each axis was fitted with a local regression using the locfit function in the R library locfit (Loader, 2007). Smoothing was defined in the local polynomial model term of the locfit model using the function “lp” from the same library with the following parameters: nn = 1 (Nearest neighbor component of the smoothing parameter) and deg = 2 (degree of polynomial). The fitted expression values of these genes along each axis were normalized between 0 and 1. Clustering was performed separately for each axis on the fitted and normalized patterns of the differentially expressed genes. We used the R function “hclust” to perform hierarchical clustering on the gene expression patterns, with a Spearman’s correlation-based distance (defined as ) and the “average” aggregation method. The number of clusters were defined with the cutreeDynamic function from the dynamicTreeCut R library, with the parameters minClusterSize = 50, method = “hybrid” and deepSplit = 0. To visualize the data in two dimensions, we applied the UMAP dimensionality reduction algorithm (umap function in the R library umap with default options; see Figure 2D) (Becht et al., 2018; McInnes et al., 2018). To analyze the relationship between the expression patterns of genes along different axes, we computed the intersections of the gene clusters between any pair of axes. The expected number of elements in each intersection under the assumption of independent sets is given by:
where A and B indicate the sets of genes in two clusters identified along two different axes and |•| indicates the cardinality of a set (i.e., the number of its elements). The ratio between the observed and the expected number of elements in the intersection |A∩B|obs / |A∩B|exp quantifies the enrichment/depletion of genes having a given pair of patterns across two axes with respect to the random case. The log2 values of (1 + |A∩B|obs / |A∩B|exp) are shown in Figure 2F.
Combining Tomo-seq with single-cell RNA-seq data
The TPM (transcripts per million)-normalized single cell RNA-seq (scRNA-seq) data collected from mouse olfactory epithelium available from Fletcher et al. (2017) was used to identify cell-type specific genes. To this aim, we computed the average expression level for each cell type in the scRNA-seq dataset for all the differentially expressed genes that we identified in our TOMO-seq data. The genes with an average expression above 100 TPM in mOSNs and below 10 TPM in all other cell types were considered mOSN-specific. Conversely, genes with an average expression above 100 TPM in any of the non-mOSN cell types and below 10 in mOSNs were considered to be specific for non-mOSN cells.
Gene ontology (GO) enrichment analysis
GO Enrichment analyses were performed using the GOrilla online tool (http://cbl-gorilla.cs.technion.ac.il) with the option “Two unranked lists of genes (target and background lists)”. For each axis, we used as background list the list of the genes we tested.
Cell type deconvolution analysis
To perform cell type deconvolution analysis, we used a previously published single-cell RNA-seq (scRNA-seq) data from the mouse OM (Fletcher et al., 2017). First, the cells included in unclassified clusters were removed and the data was rescaled using the function “pp.log1p” from the scanpy library (Wolf et al., 2018). Then, we obtained 2000 highly variable genes using the function “pp.highly_variable_genes” (scanpy library). In the following analysis, we merged clusters of similar cell populations and considered the following 6 cell types: 1-HBC = HBC1+HBC2+HBC3; 2-INP = INP1+INP2+INP3; 3-GBC = GBC, 4-SC = mSC + iSC, 5-OSN = iOSN + mOSN, 6-MVC = MVC1+MVC2.
This scRNA-seq data was used as input for the AutoGeneS algorithm (Aliee and Theis, 2021). The cell type assignment as well as the list of highly variable genes were passed as input to the function “ag.init” from AutogeneS, and then we estimated the optimal subset of genes to perform cell type deconvolution with the function “ag.optimize” (with parameters: “ngen” = 5000, “nfeatures” = 400 and “mode” = “fixed”). Finally, we deconvolved the Tomo-seq data along the three axes with the function “ag.deconvolve” using Nu Support Vector regression models (“model” = “nusvr”). The results were normalized such that the sums of cell type proportions per slice is equal to 1 (Figure S2F). To identify the cell types with non-random spatial distribution along the axes, we applied the Ljung-Box test as explained above (section “identification of differentially expressed genes and gene clustering“); the p values are reported in Table S2.
Identification of ligands and receptors associated with the NfiA, NfiB or NfiX transcription factors
The genes in the CellphoneDB ligands and receptor database (Efremova et al., 2020) that were among our spatially differentially expressed genes were selected and Spearman correlation tests between their 1D expression patterns and the 1D patterns for the Nfi transcription factors were performed. Correlation coefficients from the three axes were averaged and FDRs from the 3 axes were combined with the Simes’ method (Simes, 1986) using the function combinePValues from the scran R library. Combined FDR values <0.01 were taken as significant.
3D spatial reconstruction
The olfactory mucosa shape was obtained from publicly available images of the mouse nasal cavity along the posterior to the anterior axis published in Barrios et al. (2014). The area of the slices corresponding to the OM was manually selected and images of their silhouettes were made. Those images were then transformed into binary matrices having 1’s in the area occupied by the OM and 0’s in the remaining regions. The binary matrices were rescaled to match the spatial resolution in our dataset, which is composed of 54 voxels along the DV axis, 56 along the LML axis and 60 along the AP axis. Finally, matrices were piled in a 3D array in R to obtain an in-silico representation of the 3D shape of the OM, which, in total, was composed of 77,410 voxels. To perform the 3D reconstruction of the expression pattern for a given gene, first we normalized its expression levels by the volume of the slice at each corresponding position along the three axes, which was estimated using our 3D in silico representation of the OM. Then, we rescaled the data in such a way that the sum of the expression levels along each axis was equal to the average expression computed across the whole dataset. This rescaled dataset together with the binary matrix representing the 3D OM shape was used as input of the Iterative Proportional Fitting algorithm, which produced an estimation of the expression level of a gene in each voxel (Junker et al., 2014). Iterations stopped when the differences between the true and the reconstructed 1D values summed across the three axes was smaller than 1.
Definition of zones by topic modelling
In order to identify zones, we fitted a Latent Dirichlet Allocation (LDA) (Blei et al., 2003) algorithm to the 3D gene expression patterns (in log10 scale) of the differentially expressed Olfrs (689 Olfrs × 77,410 voxels).
The LDA algorithm was originally employed for document classification: based on the words included in each document, LDA can identify “topics”, in which the documents can then be classified. Using this linguistic analogy, in our application of LDA, we considered the genes as “documents”, and the spatial locations as “words”, with the matrix of gene expression levels being the analogous of the “bag-of-word” matrix (Liu et al., 2016). In this representation, the zones are the equivalent of “topics”, and they are automatically identified by LDA. We used the LDA implementation included in the R package “Countclust” (Dey et al., 2017), developed based on the “maptpx” library (Taddy, 2012), which performs a maximum a posteriori estimation to for model fitting. LDA was run for all possible numbers of topics K ∈ [2,9]. The following parameters were chosen: convergence tolerance = 0.1; max time optimization step = 180 seconds; n_init = 3. For each number of topics k, three independent runs were performed with different starting points, in order to avoid biases due to the choice of the initial condition. We estimated the number of topics by computing the log likelihood for each value of K ∈ [2,9]. As seen in Figure S3A, while the log-likelihood is a monotonically increasing function of the number of topic (as expected), for a number of topics around ~5 it shows a “knee” and starts to increase more slowly. This suggests that ~5 is the minimal number of topics needed to describe the complexity of the data without overfitting. Hence, we fix a number of topics equal to 5; however, we also verified that all our conclusions remain substantially unaffected if a different number of topics is chosen.
After running LDA with K = 5, we retrieved the model output, which consists of two probability distributions: the first is P(position| k) with k ∈ [1,5], which is the conditional probability distribution defining the topic k; the second probability distribution is P(k | gene), namely the probability distribution that quantifies the degrees of belonging of a given gene to the topics k∈[1,5]. With these probability distributions, we can identify the spatial positions that form each topic and how the different topics can be combined to generate the spatial expression pattern of each gene.
Being a generative model, once trained, LDA can also decompose into topics the spatial expression patterns of genes that were not used during the training procedure. We exploited this feature of LDA to estimate the degrees of belonging of non-olfactory receptor genes. To this aim, we utilized an algorithm based on the python gensim library Lda.Model.inference function (Rehurek and Sojka, 2010), using as input the estimated probability distribution P(position | k) with k ∈ [1,5]. The model fitting was performed using the Open Computing Cluster for Advanced data Manipulation (OCCAM), the High-Performance Computer designed and managed in collaboration between the University of Torino and the Torino division of the Istituto Nazionale di Fisica Nucleare (Aldinucci et al., 2017).
Definition of Olfr 3D indexes via diffusion pseudo-time
As explained in the section above, we can describe the spatial expression pattern of each gene through a set of five numbers, which represent the degrees of belonging to the five topics identified by LDA. We applied a diffusion map (Haghverdi et al., 2015) to the degrees of belonging of the Olfrs to visualize them in two dimensions by using the “DiffusionMap” function from the “destiny” R package (Angerer et al., 2016) (with distance = “rankcor” and default parameters). In this two-dimensional map, the Olfrs are approximately distributed along a curve that joins the most dorsal/medial genes (those in zones 1–2) with those that are more ventral/lateral (zones 3–5). To track the position of the genes along this curve, we computed a diffusion pseudo-time (DPT) coordinate (Haghverdi et al., 2016) with the “DPT” function from the “destiny” R package (taking as starting point the gene with the smallest first diffusion component among the genes suggested by the function find_tips from the same package). In order to make the indexes go from Dorsal to Ventral, as in previous studies (Miyamichi et al., 2005), we reversed the order of the DPT coordinates by substracting the maximum coordinate from all coordinates and multiplying them by (−1). By doing this, we obtained for each Olfr an index, which we called 3D index, representing its spatial expression pattern in the 3D space: more dorsal/medial genes (zones 1–2) have smaller 3D indexes than Olfrs expressed in the ventral/lateral regions (zones 3–5).
Prediction of zone index for undetected Olfrs with Random Forest
We fitted a Random Forest model to the 3D indexes of 681 of the 689 Olfrs we characterized with our dataset (i.e., those that are located in genomic clusters). The following nine features of each Olfr were used as predictors: genomic position (i.e., gene starting position divided by chromosome length); genomic cluster; genomic cluster length; number of Olfrs in the genomic cluster; number of enhancers in the genomic cluster; cluster position (i.e., starting position of the cluster divided by the chromosome length); distance to the closest enhancer; gene position within the cluster (i.e., the distance of the gene starting position from the end of the cluster divided by the cluster length); and phylogenetic class. These features were computed using the mm10 mouse genome in Biomart (Kinsella et al., 2011), while the list of enhancers and the genomic clusters assigned to each Olfr were taken from Monahan et al. (2017). The Random Forest model was fitted with the function “randomForest” (R library “randomForest” (Liaw and Wiener, 2002), with option “na.action = na.omit”). Afterward, we performed a cross-validation test with the function “rf.crossValidation” from the “rfUtilities” package (Rather et al., 2020) with default parameters. Over 100 cross-validation iterations, the root mean square error (RMSE) was ≲10% of the mean 3D index. The feature importance was computed with the “importance” function from the randomForest library with default parameters. Finally, the Random Forest model trained on the 681 Olfrs was used to predict the 3D indexes of 697 Olfrs that were too lowly expressed or were undetected in our dataset. Overall, we were able to compute or predict with Random Forest a 3D index for all the Olfrs annotated in the mouse genome, except for 28 of them that do not have any genomic cluster assigned. To quantify the consistency between our Olfr 3D indexes and indexes defined previously, we calculated the Spearman’s correlation coefficients between our indexes and those defined in three previous studies (Miyamichi et al., 2005; Tan and Xie, 2018; Zapiec and Mombaerts, 2020) (see Figures 4G, S4B, and S4C).
Odorant information and Olfr-ligand pairs
All odorant structures and associated CAS numbers were retrieved from either Sigma-Aldrich (www.sigmaaldrich.com) or PubChem (https://pubchem.ncbi.nlm.nih.gov). A comprehensive catalog of the cognate mouse Olfr-ligand pairs was collected (last update: March 2021) by combining data from the ODORactor database (Liu et al., 2011) and additional literature sources (Abaffy et al., 2006; Araneda et al., 2004; Bozza et al., 2002; Dunkel et al., 2014; Floriano et al., 2000; Gaillard et al., 2002; Godfrey et al., 2004; Grosmaitre et al., 2006, 2009; Jiang et al., 2015; Jones et al., 2019; Kajiya et al., 2001; Malnic et al., 1999, 2004; Nara et al., 2011; Nguyen et al., 2007; Oka et al., 2004, 2006, 2009; Pfister et al., 2020; Repicky and Luetje, 2009; Saito et al., 2004, 2017; Saraiva et al., 2019; Shirasu et al., 2014; Shirokova et al., 2005; von der Weid et al., 2015; Yoshikawa et al., 2013; Yoshikawa and Touhara, 2009; Yu et al., 2015; Zhuang and Matsunami, 2007).
This catalog includes 738 Olfr-ligand interactions for a total of 153 Olfrs and 221 odorants. These 153 Olfrs include 100 spatial Olfrs in our dataset and for which we have 3D indexes, and 49 additional Olfrs with predicted 3D indexes (see above). Next, we checked whether Olfrs pairs sharing at least one cognate ligand have more similar spatial expression patterns than pairs not sharing ligands. To do this, we computed the absolute values of the differences between the 3D indexes (Δ) of 1706 pairs of ORs sharing at least one odorant and 9,922 pairs of ORs that are known to bind to different odorants (Figure 6B). The two sets of Δ values were significantly different (Mann-Whitney U test, p value < 2 × 10−16). This test remained significant when excluding Olfrs for which 3D indexes were estimated by the Random Forests model (p value < 2 × 10−16), and also when excluding class I Olfrs (p value < 2 × 10−16).
Correlation analysis of physico-chemical descriptors with 3D index
Physicochemical descriptors for ligands were obtained from the Dragon 6.0 software (http://www.talete.mi.it/). After removing the descriptors showing 0 variance, a table of 1911 descriptors for 205 ligands was obtained. In addition to these, we estimated the air/mucus partition coefficients (Kam) of the odorants as done previously (Rygg et al., 2017; Scott et al., 2014). Briefly, we calculated the air/water partition coefficients (Kaw) for each odorant from the Henry’s Law constants obtained using the HENRYWIN model in the US EPA Estimation Program Interface (EPI) Suite (version 4.11; www.epa.gov/oppt/exposure/pubs/episuite.htm). Then, we computed the air/mucus partition coefficients (Kam) according to the formula:
where Kow indicates the octanol/water partition coefficient, which were obtained using the KOWWIN model in the EPI Suite.
To increase the robustness of our correlation analysis, we removed the descriptors with 20 or more identical values across our set of ligands, and we initially considered only the ligands having 2 or more known cognate receptors; these filters gave us 1,210 descriptors (including Kam) for 101 ligands.
We performed Spearman’s correlation tests between the physicochemical descriptors and mean 3D index of the cognate Olfrs, and we considered as statistically significant those correlations with FDR <0.05 (see Table S6). The descriptors with the largest correlation coefficients were Kam (rho = 0.55, p = 1 × 10−7) and ATSC2s (Centred Broto-Moreau Autocorrelation of lag 2 weighted by I-state, rho = −0.56, p = 2 × 10−7). We obtained statistically significant correlations between Kam and the mean 3D indexes also when excluding Olfrs with 3D indexes predicted by Random Forest (Rho = 0.48, p value = 2 × 10−6, based on 87 ligands; Figure S5B) or excluding class I Olfrs (Rho = 0.5, p value = 1 × 10−7, based on 101 ligands; Figure S5C).
In-situ hybridization
In-situ hybridization was basically performed as previously described (Ibarra-Soria et al., 2017). Adult 12-week-old male C57BL/6J mice anesthetized, and then perfused with 4% paraformaldehyde. The snouts containing the OM were dissected out, decalcified in RNase-free 0.45M EDTA solution (in 1× PBS) for two weeks – the bone and tissue encapsulating the OM are necessary to preserve the OM tissue integrity during the ISH. Next, the decalcified snouts were cryoprotected in RNase-free 30% sucrose solution (1× PBS), dried, embedded in OCT embedding medium, and frozen at −80°C. Sequential 16 μm sections were prepared with a cryostat and the sections were hybridized to digoxigenin-labeled cRNA probes prepared from the different genes using the following oligonucleotides: Cytl (5′-AAAGACACTACCTCTGTTGCTGCTG-3′ and 5′-GTAAGCAGAGACCAGAAAGAAGAGTG-3′), Moxd2 (5′-TGTACCTTTCTCCCACTCCCTATTGTC-3′ and 5′-CCCATGCAACTGGAGATGTTAATTCTG-3′), Olfr309 (5′-TACAATGGCCTATGACCGCTATGTG-3′ and 5′-TCCTGACTGCATCTCTTTGTTCCTG-3′), Olfr727 (5′-CGCTATGTTGCAATATGCAAGCCTC-3′ and 5′-GCTTTGACATTGCTGCTTTCACCTC-3′), and Olfr618 (5′-CATGAACCAATGTACCTTTTCCTCTC-3′ and 5′-AAACCTGTCTTGAATTTGCTTTGTC-3′). The PCR products were cloned into pGEM-T Easy vector and the probes were obtained by in vitro transcription of the plasmids, using SP6 or T7 RNA Polymerases (Roche) and DIG RNA Labeling mix (Roche).
QUANTIFICATION AND STATISTICAL ANALYSIS
Information on gene expression thresholds for spatial differential expression analysis is described in the method details section. The presence of a spatial signal along the 3 axes was verified via the Moran’s I statistic (see relevant section above). The presence of spatial non-random patterns was tested using the Ljung-Box test and the resulting p values were adjusted using the FDR method (see relevant section above). Consistency between different datasets and replicates, as well as association between independent data were tested using Spearman correlation tests. Mann-Whitney U tests were employed to test the statistical significance of differences between two distributions. Finally, a cross validation test was used to quantify the accuracy of our Random Forests model through the root mean square error (RMSE). Statistical tests were performed using R (version 4.1.2). Statistical details are reported in the Main text, Figures and Figure legends, the STAR Methods section and supplementary tables. N represents the number of biological replicates (animals) we analyzed. Boxplots are centered at the median of the distribution, the bottom and top of the box represent the 1st and 3rd quartiles respectively, and the whiskers extend for an additional 1.5 times the interquartile range.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Digoxigenin-AP, Fab fragments | Merck (Roche) | Cat# 11093274910, RRID:AB_514497 |
| Biological samples | ||
| Olfactory mucosae from C57Bl/6J mice (adult males) | The Jackson Laboratory | Stock # 00664 |
| Chemicals, peptides, and recombinant proteins | ||
| 30% Hydrogen Proxyde Solution | Merck (Sigma-Aldrich) | Cat. # H1009 |
| Triethanolamine | Merck (Sigma-Aldrich) | Cat. # T58300 |
| Acetic anhydride | Merck (Sigma-Aldrich) | Cat. #320102 |
| Deoinized formamide | Merck (Sigma-Aldrich) | Cat. # F9037 |
| Yeast tRNA | Merck (Roche) | Cat. # 10109495001 |
| Denhardt’s solution (50×) | Merck (Sigma-Aldrich) | Cat. # D9905 |
| Dextran sulfate solution (50%) | Merck (Chemicon) | Cat. # S4030 |
| 20× SSC | Merck (Calbiochem) | Cat. # 8310-OP |
| Tween-20 | Merck (Sigma-Aldrich) | Cat. #822184 |
| TSA Blocking Reagent | Perkin-Elmer | Cat. # FP1020 |
| NBT/BCIP Stock Solution | Merck (Roche) | Cat. # 11681451001 |
| Critical commercial assays | ||
| SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing | Clontech (Takara Bio) | Cat. # 634892 |
| Bioanalyzer DNA High-Sensitivity kit | Agilent Technologies | Cat. # 5067–4626 |
| Nextera XT DNA Library Preparation Kit (96 samples) | Illumina | Cat. # FC-131–1096 |
| Nextera XT Index Kit v2 Set A (96 indexes, 384 samples) | Illumina | Cat. # FC-131–2001 |
| pGEM®-T Easy Vector Systems | Promega | Cat. # A1360 |
| DIG RNA Labeling Kit (SP6/T7) | Merck (Roche) | Cat. # 11175025910 |
| ProbeQuant G-50 Micro Columns | Cytiva Biosciences | Cat. # 28903408 |
| Deposited data | ||
| TOMO-seq Olfactory Mucosa dataset | This study | https://www.ebi.ac.uk/arrayexpress/E-MTAB-10211 |
| Single cell RNA-seq data from the Olfactory Mucosa | Fletcher et al., 2017 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE95601 |
| Dragon database of molecular descriptors | Talete S.R.L. | http://www.talete.mi.it |
| CellphoneDB ligands and receptors database | Efremova et al., 2020 | https://github.com/ventolab/CellphoneDB |
| Experimental models: Organisms/strains | ||
| Adult male C57Bl/6J mice | The Jackson Laboratory | Stock # 00664 |
| Oligonucleotides | ||
| See “method details” section for oligonucleotides | This study | N/A |
| Software and algorithms | ||
| samtools version 0.1.19–44428cd | Li et al., 2009 | http://samtools.sourceforge.net/ |
| htseq-count version 0.11.2 | Anders et al., 2014 | https://github.com/htseq/htseq/ |
| R 4.1.2 | The R Foundation | https://www.r-project.org/ |
| Python 3.9.6 | Python Software Foundation | https://www.python.org/ |
| Scripts for TOMO-seq data analysis | This study |
https://doi.org/10.5281/zenodo.6036047
https://zenodo.org/badge/DOI/10.5281/zenodo.6045897.svg |
Highlights.
We generate a browsable 3D transcriptomic atlas of the mouse olfactory mucosa (OM)
We identify potential functional hotspots in the mouse OM
Odorant receptor genes (Olfrs) are continuously distributed over at least five zones
Spatial locations of Olfrs correlate with the mucus solubility of their ligands
ACKNOWLEDGMENTS
We would like to thank Ximena Ibarra-Soria for help with the initial analysis and Maria-Elena Torres-Padilla, Bob Datta, Giorgia Greco, and the members of the Saraiva and Scialdone Labs for the constructive feedback, insightful discussions, and helpful comments. This work was supported by the Helmholtz Association (A.S.), the Deutsche Forschungsgemeinschaft (SC280/2-1 to A.S.), the National Institutes of Health (R01DC016647 to J.R.), and Sidra Medicine (SDR400040 to L.R.S.)—a member of Qatar Foundation.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110547.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abaffy T, and Defazio AR (2011). The location of olfactory receptors within olfactory epithelium is independent of odorant volatility and solubility. BMC Res. Notes 4, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abaffy T, Matsunami H, and Luetje CW (2006). Functional analysis of a mammalian odorant receptor subfamily. J. Neurochem 97, 1506–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achim K, Pettit J-B, Saraiva LR, Gavriouchkina D, Larsson T, Arendt D, and Marioni JC (2015). High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin. Nat. Biotechnol 33, 503–509. [DOI] [PubMed] [Google Scholar]
- Adipietro KA, Mainland JD, and Matsunami H (2012). Functional evolution of mammalian odorant receptors. PLoS Genet. 8, e1002821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldinucci M, Bagnasco S, Lusso S, Pasteris P, Rabellino S, and Vallero S (2017). OCCAM: a flexible, multi-purpose and extendable HPC cluster. J. Phys. Conf. Ser 898, 082039. [Google Scholar]
- Alenius M, and Bohm S (1997). Identification of a novel neural cell adhesion molecule-related gene with a potential role in selective axonal projection. J. Biol. Chem 272, 26083–26086. [DOI] [PubMed] [Google Scholar]
- Aliee H, and Theis FJ (2021). AutoGeneS: automatic gene selection using multi-objective optimization for RNA-seq deconvolution. Cell Syst. 12, 706–715.e4. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2014). HTSeq - a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angerer P, Haghverdi L, Buttner M, Theis FJ, Marr C, and Buettner F (2016). destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics 32, 1241–1243. [DOI] [PubMed] [Google Scholar]
- Araneda RC, Peterlin Z, Zhang X, Chesler A, and Firestein S (2004). A pharmacological profile of the aldehyde receptor repertoire in rat olfactory epithelium. J. Physiol 555, 743–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asp M, Giacomello S, Larsson L, Wu C, Furth D, Qian X, Wardell E, Custodio J, Reimegard J, Salmen F, et al. (2019). A spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. Cell 179, 1647–1660.e19. [DOI] [PubMed] [Google Scholar]
- Asp M, Bergenstrahle J, and Lundeberg J (2020). Spatially resolved transcriptomes-next generation tools for tissue exploration. Bioessays 42, e1900221. [DOI] [PubMed] [Google Scholar]
- Barrios AW, Nunez G, Sanchez Quinteiro P, and Salazar I (2014). Anatomy, histochemistry, and immunohistochemistry of the olfactory subsystems in mice. Front. Neuroanat 8, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashkirova E, Monahan K, Campbell CE, Osinski JM, Tan L, Schieren I, Barnea G, Xie XS, Gronostajski RM, and Lomvardas S (2020). Homeotic regulation of olfactory receptor choice via NFI-dependent heterochromatic silencing and genomic compartmentalization. Preprint at bioRxiv. 10.1101/2020.08.30.274035. [DOI] [Google Scholar]
- Baxter BD, Larson ED, Merle L, Feinstein P, Polese AG, Bubak AN, Niemeyer CS, Hassell J, Shepherd D, Ramakrishnan VR, et al. (2020). Transcriptional profiling reveals potential involvement of microvillous TRPM5-expressing cells in viral infection of the olfactory epithelium. Preprint at bioRxiv. 10.1101/2020.05.14.096016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear DM, Lassance J-M, Hoekstra HE, and Datta SR (2016). The evolving neural and genetic architecture of vertebrate olfaction. Curr. Biol 26, R1039–R1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2018). Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol 37, 38–44. [DOI] [PubMed] [Google Scholar]
- Blei DM, Ng AY, and Jordan MI (2003). Latent dirichlet allocation. J. Mach. Learn. Res 3, 993–1022. [Google Scholar]
- Bleymehl K, Perez-Gomez A, Omura M, Moreno-Perez A, Macias D, Bai Z, Johnson RS, Leinders-Zufall T, Zufall F, and Mombaerts P (2016). A sensor for low environmental oxygen in the mouse main olfactory epithelium. Neuron 92, 1196–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozza T, Feinstein P, Zheng C, and Mombaerts P (2002). Odorant receptor expression defines functional units in the mouse olfactory system. J. Neurosci 22, 3033–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brann DH, Tsukahara T, Weinreb C, Lipovsek M, Van den Berge K, Gong B, Chance R, Macaulay IC, Chou HJ, Fletcher RB, et al. (2020). Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Sci. Adv 6, eabc5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britsch S, Li L, Kirchhoff S, Theuring F, Brinkmann V, Birchmeier C, and Riethmacher D (1998). The ErbB2 and ErbB3 receptors and their ligand, neuregulin-1, are essential for development of the sympathetic nervous system. Genes Dev. 12, 1825–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck L, and Axel R (1991). A novel multigene family may encode odorant receptors: a molecular basis for odor recognition. Cell 65, 175–187. [DOI] [PubMed] [Google Scholar]
- Carson C, Murdoch B, and Roskams AJ (2006). Notch 2 and Notch 1/3 segregate to neuronal and glial lineages of the developing olfactory epithelium. Dev. Dyn 235, 1678–1688. [DOI] [PubMed] [Google Scholar]
- Charkoftaki G, Wang Y, McAndrews M, Bruford EA, Thompson DC, Vasiliou V, and Nebert DW (2019). Update on the human and mouse lipocalin (LCN) gene family, including evidence the mouse Mup cluster is result of an “evolutionary bloom”. Hum. Genomics 13, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chess A, Simon I, Cedar H, and Axel R (1994). Allelic inactivation regulates olfactory receptor gene expression. Cell 78, 823–834. [DOI] [PubMed] [Google Scholar]
- Cho JH, Lepine M, Andrews W, Parnavelas J, and Cloutier JF (2007). Requirement for Slit-1 and Robo-2 in zonal segregation of olfactory sensory neuron axons in the main olfactory bulb. J. Neurosci 27, 9094–9104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloutier JF, Giger RJ, Koentges G, Dulac C, Kolodkin AL, and Ginty DD (2002). Neuropilin-2 mediates axonal fasciculation, zonal segregation, but not axonal convergence, of primary accessory olfactory neurons. Neuron 33, 877–892. [DOI] [PubMed] [Google Scholar]
- Consonni, and Todeschini R. (2008). New spectral indices for molecule description. MATCH Commun. Math. Comput. Sci 60, 3–14. [Google Scholar]
- Coppola DM, Fitzwater E, Rygg AD, and Craven BA (2019). Tests of the chromatographic theory of olfaction with highly soluble odors: a combined electro-olfactogram and computational fluid dynamics study in the mouse. Biol. Open 8, bio047217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola DM, Waggener CT, Radwani SM, and Brooks DA (2013). An electroolfactogram study of odor response patterns from the mouse olfactory epithelium with reference to receptor zones and odor sorptiveness. J. Neurophysiol 109, 2179–2191. [DOI] [PubMed] [Google Scholar]
- Cutforth T, Moring L, Mendelsohn M, Nemes A, Shah NM, Kim MM, Frisen J, and Axel R (2003). Axonal ephrin-As and odorant receptors: coordinate determination of the olfactory sensory map. Cell 114, 311–322. [DOI] [PubMed] [Google Scholar]
- Devillers J, and Domine D (1997). Comparison of reliability of log P values calculated from a group contribution approach and from the autocorrelation method. SAR QSAR Environ. Res. 7, 195–232. [Google Scholar]
- Dey KK, Hsiao CJ, and Stephens M (2017). Visualizing the structure of RNA-seq expression data using grade of membership models. PLoS Genet. 13, e1006599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggan CD, DeMaria S, Baudhuin A, Stafford D, and Ngai J (2008). Foxg1 is required for development of the vertebrate olfactory system. J. Neurosci 28, 5229–5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkel A, Steinhaus M, Kotthoff M, Nowak B, Krautwurst D, Schieberle P, and Hofmann T (2014). Nature’s chemical signatures in human olfaction: a foodborne perspective for future biotechnology. Angew. Chem. Int. Ed. Engl 53, 7124–7143. [DOI] [PubMed] [Google Scholar]
- Efremova M, Vento-Tormo M, Teichmann SA, and Vento-Tormo R (2020). CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc 15, 1484–1506. [DOI] [PubMed] [Google Scholar]
- Fienberg SE (1970). An iterative procedure for estimation in contingency tables. Ann. Math. Stat 41, 907–917. [Google Scholar]
- Finkielstein CV, and Capelluto DG (2016). Disabled-2: a modular scaffold protein with multifaceted functions in signaling. Bioessays 38, S45–S55. [DOI] [PubMed] [Google Scholar]
- Fletcher RB, Das D, Gadye L, Street KN, Baudhuin A, Wagner A, Cole MB, Flores Q, Choi YG, Yosef N, et al. (2017). Deconstructing olfactory stem cell trajectories at single-cell resolution. Cell Stem Cell 20, 817–830.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floriano WB, Vaidehi N, Goddard WA 3rd, Singer MS, and Shepherd GM (2000). Molecular mechanisms underlying differential odor responses of a mouse olfactory receptor. Proc. Natl. Acad. Sci. U S A 97, 10712–10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitag J, Krieger J, Strotmann J, and Breer H (1995). Two classes of olfactory receptors in Xenopus laevis. Neuron 15, 1383–1392. [DOI] [PubMed] [Google Scholar]
- Fulle HJ, Vassar R, Foster DC, Yang RB, Axel R, and Garbers DL (1995). A receptor guanylyl cyclase expressed specifically in olfactory sensory neurons. Proc. Natl. Acad. Sci. U S A 92, 3571–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard I, Rouquier S, Pin JP, Mollard P, Richard S, Barnabe C, Demaille J, and Giorgi D (2002). A single olfactory receptor specifically binds a set of odorant molecules. Eur. J. Neurosci 15, 409–418. [DOI] [PubMed] [Google Scholar]
- Godfrey PA, Malnic B, and Buck LB (2004). The mouse olfactory receptor gene family. Proc. Natl. Acad. Sci. U S A 101, 2156–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh CJ, Choi D, Park DB, Kim H, and Hahn Y (2016). MOXD2, a gene possibly associated with olfaction, is frequently inactivated in birds. PLoS One 11, e0152431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer PL, Bear DM, Lassance JM, Bloom ML, Tsukahara T, Pashkovski SL, Masuda FK, Nowlan AC, Kirchner R, Hoekstra HE, et al. (2016). A Family of non-GPCR chemosensors defines an alternative logic for mammalian olfaction. Cell 165, 1734–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosmaitre X, Vassalli A, Mombaerts P, Shepherd GM, and Ma M (2006). Odorant responses of olfactory sensory neurons expressing the odorant receptor MOR23: a patch clamp analysis in gene-targeted mice. Proc. Natl. Acad. Sci. U S A 103, 1970–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosmaitre X, Fuss SH, Lee AC, Adipietro KA, Matsunami H, Mombaerts P, and Ma M (2009). SR1, a mouse odorant receptor with an unusually broad response profile. J. Neurosci 29, 14545–14552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gussing F, and Bohm S (2004). NQO1 activity in the main and the accessory olfactory systems correlates with the zonal topography of projection maps. Eur. J. Neurosci 19, 2511–2518. [DOI] [PubMed] [Google Scholar]
- Haghverdi L, Buettner F, and Theis FJ (2015). Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics 31, 2989–2998. [DOI] [PubMed] [Google Scholar]
- Haghverdi L, Buttner M, Wolf FA, Buettner F, and Theis FJ (2016). Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 13, 845–848. [DOI] [PubMed] [Google Scholar]
- Hanchate NK, Kondoh K, Lu Z, Kuang D, Ye X, Qiu X, Pachter L, Trapnell C, and Buck LB (2015). Single-cell transcriptomics reveals receptor transformations during olfactory neurogenesis. Science 350, 1251–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heron PM, Stromberg AJ, Breheny P, and McClintock TS (2013). Molecular events in the cell types of the olfactory epithelium during adult neurogenesis. Mol. Brain 6, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollas B (2003). An analysis of the autocorrelation descriptor for molecules. J. Math. Chem 33, 91–101. [Google Scholar]
- Horowitz LF, Saraiva LR, Kuang D, Yoon KH, and Buck LB (2014). Olfactory receptor patterning in a higher primate. J. Neurosci 34, 12241–12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber AB, Kolodkin AL, Ginty DD, and Cloutier JF (2003). Signaling at the growth cone: ligand-receptor complexes and the control of axon growth and guidance. Annu. Rev. Neurosci 26, 509–563. [DOI] [PubMed] [Google Scholar]
- Ibarra-Soria X, Nakahara TS, Lilue J, Jiang Y, Trimmer C, Souza MA, Netto PH, Ikegami K, Murphy NR, Kusma M, et al. (2017). Variation in olfactory neuron repertoires is genetically controlled and environmentally modulated. Elife 6, e21476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Gong NN, Hu XS, Ni MJ, Pasi R, and Matsunami H (2015). Molecular profiling of activated olfactory neurons identifies odorant receptors for odors in vivo. Nat. Neurosci 18, 1446–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EM, Jajoo R, Cancilla D, Lubock NB, Wang J, Satyadi M, Cheung R, de March C, Bloom JS, Matsunami H, et al. (2019). A scalable, multiplexed assay for decoding GPCR-ligand interactions with RNA sequencing. Cell Syst. 8, 254–260.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juilfs DM, Fulle HJ, Zhao AZ, Houslay MD, Garbers DL, and Beavo JA (1997). A subset of olfactory neurons that selectively express cGMP-stimulated phosphodiesterase (PDE2) and guanylyl cyclase-D define a unique olfactory signal transduction pathway. Proc. Natl. Acad. Sci. U S A 94, 3388–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung DJ, Bong JJ, and Baik M (2004). Extracellular proteinase inhibitor-accelerated apoptosis is associated with B cell activating factor in mammary epithelial cells. Exp. Cell Res 292, 115–122. [DOI] [PubMed] [Google Scholar]
- Junker JP, Noel ES, Guryev V, Peterson KA, Shah G, Huisken J, McMahon AP, Berezikov E, Bakkers J, and van Oudenaarden A (2014). Genome-wide RNA tomography in the zebrafish embryo. Cell 159, 662–675. [DOI] [PubMed] [Google Scholar]
- Kajiya K, Inaki K, Tanaka M, Haga T, Kataoka H, and Touhara K (2001). Molecular bases of odor discrimination: reconstitution of olfactory receptors that recognize overlapping sets of odorants. J. Neurosci 21, 6018–6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel E, Schwartz J, Jessell T, Siegelbaum S, and Hudspeth A (2013). In Principles of Neural Science, Fifth edition (McGraw-Hill: ). [Google Scholar]
- Kania A, and Klein R (2016). Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat. Rev. Mol. Cell. Biol 17, 240–256. [DOI] [PubMed] [Google Scholar]
- Kim DS, Wang Y, Oh HJ, Lee K, and Hahn Y (2014). Frequent loss and alteration of the MOXD2 gene in catarrhines and whales: a possible connection with the evolution of olfaction. PLoS One 9, e104085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsella RJ, Kahari A, Haider S, Zamora J, Proctor G, Spudich G, Almeida-King J, Staines D, Derwent P, Kerhornou A, et al. (2011). Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database 2011, bar030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koglin M, Vehse K, Budaeus L, Scholz H, and Behrends S (2001). Nitric oxide activates the beta 2 subunit of soluble guanylyl cyclase in the absence of a second subunit. J. Biol. Chem 276, 30737–30743. [DOI] [PubMed] [Google Scholar]
- Kurian SM, Naressi RG, Manoel D, Barwich AS, Malnic B, and Saraiva LR (2021). Odor coding in the mammalian olfactory epithelium. Cell Tissue Res. 383, 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Park SJ, Oh HJ, Kim JW, Bae HS, and Park JC (2012). Expression pattern, subcellular localization, and functional implications of ODAM in ameloblasts, odontoblasts, osteoblasts, and various cancer cells. Gene Expr. Patterns 12, 102–108. [DOI] [PubMed] [Google Scholar]
- Leung CT, Coulombe PA, and Reed RR (2007). Contribution of olfactory neural stem cells to tissue maintenance and regeneration. Nat. Neurosci 10, 720–726. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw A, and Wiener M (2002). Classification and regression by randomForest. R News 2, 18–22. [Google Scholar]
- Liberles SD, and Buck LB (2006). A second class of chemosensory receptors in the olfactory epithelium. Nature 442, 645–650. [DOI] [PubMed] [Google Scholar]
- Ling G, Gu J, Genter MB, Zhuo X, and Ding X (2004). Regulation of cytochrome P450 gene expression in the olfactory mucosa. Chem. Biol. Interact 147, 247–258. [DOI] [PubMed] [Google Scholar]
- Liu L, Tang L, Dong W, Yao S, and Zhou W (2016). An overview of topic modeling and its current applications in bioinformatics. Springerplus 5, 1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Su X, Wang F, Huang Z, Wang Q, Li Z, Zhang R, Wu L, Pan Y, Chen Y, et al. (2011). ODORactor: a web server for deciphering olfactory coding. Bioinformatics 27, 2302–2303. [DOI] [PubMed] [Google Scholar]
- Loader C (2007). R Package “locfit”: Local Regression, Likelihood and Density Estimation (Version 1).
- Lun AT, McCarthy DJ, and Marioni JC (2016). A step-by-step workflow for low-level analysis of single-cell RNA-seq data with bioconductor. F1000Res. 5, 2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M, and Shepherd GM (2000). Functional mosaic organization of mouse olfactory receptor neurons. Proc. Natl. Acad. Sci. U S A 97, 12869–12874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malnic B, Godfrey PA, and Buck LB (2004). The human olfactory receptor gene family. Proc. Natl. Acad. Sci. U S A 101, 2584–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malnic B, Hirono J, Sato T, and Buck LB (1999). Combinatorial receptor codes for odors. Cell 96, 713–723. [DOI] [PubMed] [Google Scholar]
- Manoel D, Makhlouf M, Arayata CJ, Sathappan A, Da’as S, Abdelrahman D, Selvaraj S, Hasnah R, Mainland JD, Gerkin RC, et al. (2021). Deconstructing the mouse olfactory percept through an ethological atlas. Curr. Biol 31, 2809–2818.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand JE, Yang X, Chikaraishi D, Krieger J, Breer H, and Kauer JS (2004). Olfactory receptor gene expression in tiger salamander olfactory epithelium. J. Comp. Neurol 474, 453–467. [DOI] [PubMed] [Google Scholar]
- McInnes L, Healy J, and Melville J (2018). UMAP: uniform manifold approximation and projection for dimension reduction. Preprint at arXiv. 10.48550/arXiv.1802.03426. [DOI] [Google Scholar]
- Miyamichi K, Serizawa S, Kimura HM, and Sakano H (2005). Continuous and overlapping expression domains of odorant receptor genes in the olfactory epithelium determine the dorsal/ventral positioning of glomeruli in the olfactory bulb. J. Neurosci 25, 3586–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki A, Homma H, Tamura H, Matsui M, and Mikoshiba K (1996). Zonal distribution of sulfotransferase for phenol in olfactory sustentacular cells. EMBO J. 15, 2050–2055. [PMC free article] [PubMed] [Google Scholar]
- Miyawaki A, Matsushita F, Ryo Y, and Mikoshiba K (1994). Possible pheromone-carrier function of two lipocali proteins in the vomeronasal organ. EMBO J. 13, 5835–5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts P, Wang F, Dulac C, Chao SK, Nemes A, Mendelsohn M, Edmondson J, and Axel R (1996). Visualizing an olfactory sensory map. Cell 87, 675–686. [DOI] [PubMed] [Google Scholar]
- Monahan K, Schieren I, Cheung J, Mumbey-Wafula A, Monuki ES, and Lomvardas S (2017). Cooperative interactions enable singular olfactory receptor expression in mouse olfactory neurons. Elife 6, e28620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozell MM (1966). The spatiotemporal analysis of odorants at the level of the olfactory receptor sheet. J. Gen. Physiol 50, 25–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nara K, Saraiva LR, Ye X, and Buck LB (2011). A large-scale analysis of odor coding in the olfactory epithelium. J. Neurosci 31, 9179–9191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MQ, Zhou Z, Marks CA, Ryba NJ, and Belluscio L (2007). Prominent roles for odorant receptor coding sequences in allelic exclusion. Cell 131, 1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickell MD, Breheny P, Stromberg AJ, and McClintock TS (2012). Genomics of mature and immature olfactory sensory neurons. J. Comp. Neurol 520, 2608–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimura Y, Matsui A, and Touhara K (2014). Extreme expansion of the olfactory receptor gene repertoire in African elephants and evolutionary dynamics of orthologous gene groups in 13 placental mammals. Genome Res. 24, 1485–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimura Y, Matsui A, and Touhara K (2018). Acceleration of olfactory receptor gene loss in primate evolution: possible link to anatomical change in sensory systems and dietary transition. Mol. Biol. Evol 35, 1437–1450. [DOI] [PubMed] [Google Scholar]
- Norlin EM, Alenius M, Gussing F, Hagglund M, Vedin V, and Bohm S (2001). Evidence for gradients of gene expression correlating with zonal topography of the olfactory sensory map. Mol. Cell. Neurosci 18, 283–295. [DOI] [PubMed] [Google Scholar]
- Octura JER, Maeda KI, and Wakabayashi Y (2018). Structure and zonal expression of olfactory receptors in the olfactory epithelium of the goat, Capra hircus. J. Vet. Med. Sci 80, 913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka Y, Katada S, Omura M, Suwa M, Yoshihara Y, and Touhara K (2006). Odorant receptor map in the mouse olfactory bulb: in vivo sensitivity and specificity of receptor-defined glomeruli. Neuron 52, 857–869. [DOI] [PubMed] [Google Scholar]
- Oka Y, Kobayakawa K, Nishizumi H, Miyamichi K, Hirose S, Tsuboi A, and Sakano H (2003). O-MACS, a novel member of the medium-chain acyl-CoA synthetase family, specifically expressed in the olfactory epithelium in a zone-specific manner. Eur. J. Biochem 270, 1995–2004. [DOI] [PubMed] [Google Scholar]
- Oka Y, Omura M, Kataoka H, and Touhara K (2004). Olfactory receptor antagonism between odorants. EMBO J. 23, 120–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka Y, Takai Y, and Touhara K (2009). Nasal airflow rate affects the sensitivity and pattern of glomerular odorant responses in the mouse olfactory bulb. J. Neurosci 29, 12070–12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura M, and Mombaerts P (2015). Trpc2-expressing sensory neurons in the mouse main olfactory epithelium of type B express the soluble guanylate cyclase Gucy1b2. Mol. Cell. Neurosci 65, 114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacifico R, Dewan A, Cawley D, Guo C, and Bozza T (2012). An olfactory subsystem that mediates high-sensitivity detection of volatile amines. Cell Rep. 2, 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, Claude J, and Strimmer K (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. [DOI] [PubMed] [Google Scholar]
- Park J, Levic DS, Sumigray KD, Bagwell J, Eroglu O, Block CL, Eroglu C, Barry R, Lickwar CR, Rawls JF, et al. (2019). Lysosome-rich enterocytes mediate protein absorption in the vertebrate gut. Dev. Cell 51, 7–20.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peluso CE, Jang W, Drager UC, and Schwob JE (2012). Differential expression of components of the retinoic acid signaling pathway in the adult mouse olfactory epithelium. J. Comp. Neurol 520, 3707–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng G, Suo S, Chen J, Chen W, Liu C, Yu F, Wang R, Chen S, Sun N, Cui G, et al. (2016). Spatial transcriptome for the molecular annotation of lineage fates and cell identity in mid-gastrula mouse embryo. Dev. Cell 36, 681–697. [DOI] [PubMed] [Google Scholar]
- Pfister P, Smith BC, Evans BJ, Brann JH, Trimmer C, Sheikh M, Arroyave R, Reddy G, Jeong HY, Raps DA, et al. (2020). Odorant receptor inhibition is fundamental to odor encoding. Curr. Biol 30, 2574–2587.e6. [DOI] [PubMed] [Google Scholar]
- Rather TA, Kumar S, and Khan JA (2020). Multi-scale habitat modelling and predicting change in the distribution of tiger and leopard using random forest algorithm. Sci. Rep 10, 11473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehurek R, and Sojka P (2010). Software framework for topic modelling with large corpora. In Proceedings of the LREC 2010 Workshop on New Challenges for NLP Frameworks (ELRA), pp. 45–50. [Google Scholar]
- Repicky SE, and Luetje CW (2009). Molecular receptive range variation among mouse odorant receptors for aliphatic carboxylic acids. J. Neurochem 109, 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressler KJ, Sullivan SL, and Buck LB (1993). A zonal organization of odorant receptor gene expression in the olfactory epithelium. Cell 73, 597–609. [DOI] [PubMed] [Google Scholar]
- Ressler KJ, Sullivan SL, and Buck LB (1994). Information coding in the olfactory system: evidence for a stereotyped and highly organized epitope map in the olfactory bulb. Cell 79, 1245–1255. [DOI] [PubMed] [Google Scholar]
- Rygg AD, Van Valkenburgh B, and Craven BA (2017). The influence of sniffing on airflow and odorant deposition in the canine nasal cavity. Chem. Senses 42, 683–698. [DOI] [PubMed] [Google Scholar]
- Saito H, Kubota M, Roberts RW, Chi Q, and Matsunami H (2004). RTP family members induce functional expression of mammalian odorant receptors. Cell 119, 679–691. [DOI] [PubMed] [Google Scholar]
- Saito H, Nishizumi H, Suzuki S, Matsumoto H, Ieki N, Abe T, Kiyonari H, Morita M, Yokota H, Hirayama N, et al. (2017). Immobility responses are induced by photoactivation of single glomerular species responsive to fox odour TMT. Nat. Commun 8, 16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraiva LR, Ahuja G, Ivandic I, Syed AS, Marioni JC, Korsching SI, and Logan DW (2015a). Molecular and neuronal homology between the olfactory systems of zebrafish and mouse. Sci. Rep 5, 11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraiva LR, Ibarra-Soria X, Khan M, Omura M, Scialdone A, Mombaerts P, Marioni JC, and Logan DW (2015b). Hierarchical deconstruction of mouse olfactory sensory neurons: from whole mucosa to single-cell RNA-seq. Sci. Rep 5, 18178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraiva LR, Riveros-McKay F, Mezzavilla M, Abou-Moussa EH, Arayata CJ, Makhlouf M, Trimmer C, Ibarra-Soria X, Khan M, Van Gerven L, et al. (2019). A transcriptomic atlas of mammalian olfactory mucosae reveals an evolutionary influence on food odor detection in humans. Sci. Adv 5, eaax0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmal C, Myung J, Herzel H, and Bordyugov G (2017). Moran’s I quantifies spatio-temporal pattern formation in neural imaging data. Bioinformatics 33, 3072–3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfeld TA, and Cleland TA (2006). Anatomical contributions to odorant sampling and representation in rodents: zoning in on sniffing behavior. Chem. Senses 31, 131–144. [DOI] [PubMed] [Google Scholar]
- Scott JW, and Scott-Johnson PE (2002). The electroolfactogram: a review of its history and uses. Microsc. Res. Tech 58, 152–160. [DOI] [PubMed] [Google Scholar]
- Scott JW, Sherrill L, Jiang J, and Zhao K (2014). Tuning to odor solubility and sorption pattern in olfactory epithelial responses. J. Neurosci 34, 2025–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secundo L, Snitz K, and Sobel N (2014). The perceptual logic of smell. Curr. Opin. Neurobiol 25, 107–115. [DOI] [PubMed] [Google Scholar]
- Secundo L, Snitz K, Weissler K, Pinchover L, Shoenfeld Y, Loewenthal R, Agmon-Levin N, Frumin I, Bar-Zvi D, Shushan S, et al. (2015). Individual olfactory perception reveals meaningful nonolfactory genetic information. Proc. Natl. Acad. Sci. U S A 112, 8750–8755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y, Won Y, Yang JI, and Chun JS (2019). CYTL1 regulates bone homeostasis in mice by modulating osteogenesis of mesenchymal stem cells and osteoclastogenesis of bone marrow-derived macrophages. Cell Death Dis. 10, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasu M, Yoshikawa K, Takai Y, Nakashima A, Takeuchi H, Sakano H, and Touhara K (2014). Olfactory receptor and neural pathway responsible for highly selective sensing of musk odors. Neuron 81, 165–178. [DOI] [PubMed] [Google Scholar]
- Shirokova E, Schmiedeberg K, Bedner P, Niessen H, Willecke K, Raguse JD, Meyerhof W, and Krautwurst D (2005). Identification of specific ligands for orphan olfactory receptors. G protein-dependent agonism and antagonism of odorants. J. Biol. Chem 280, 11807–11815. [DOI] [PubMed] [Google Scholar]
- Simes RJ (1986). An improved Bonferroni procedure for multiple tests of significance. Biometrika 73, 751–754. [Google Scholar]
- Springer MS, Emerling CA, Gatesy J, Randall J, Collin MA, Hecker N, Hiller M, and Delsuc F (2019). Odontogenic ameloblast-associated (ODAM) is inactivated in toothless/enamelless placental mammals and toothed whales. BMC Evol. Biol 19, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spuch C, Ortolano S, and Navarro C (2012). LRP-1 and LRP-2 receptors function in the membrane neuron. Trafficking mechanisms and proteolytic processing in Alzheimer’s disease. Front. Physiol 3, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strotmann J, Wanner I, Krieger J, Raming K, and Breer H (1992). Expression of odorant receptors in spatially restricted subsets of chemosensory neurones. Neuroreport 3, 1053–1056. [DOI] [PubMed] [Google Scholar]
- Sullivan SL, Adamson MC, Ressler KJ, Kozak CA, and Buck LB (1996). The chromosomal distribution of mouse odorant receptor genes. Proc. Natl. Acad. Sci. U S A 93, 884–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddy M (2012). On estimation and selection for topic models. In Proceedings of the Fifteenth International Conference on Artificial Intelligence and Statistics, Neil DL and Mark G, eds., pp. 1184–1193, Proceedings of Machine Learning Research: PMLR. [Google Scholar]
- Tan L, and Xie XS (2018). A near-complete spatial map of olfactory receptors in the mouse main olfactory epithelium. Chem. Senses 43, 427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tietjen I, Rihel J, and Dulac CG (2005). Single-cell transcriptional profiles and spatial patterning of the mammalian olfactory epithelium. Int. J. Dev. Biol 49, 201–207. [DOI] [PubMed] [Google Scholar]
- Tietjen I, Rihel JM, Cao Y, Koentges G, Zakhary L, and Dulac C (2003). Single-cell transcriptional analysis of neuronal progenitors. Neuron 38, 161–175. [DOI] [PubMed] [Google Scholar]
- Trimmer C, and Mainland JD (2017). Simplifying the odor landscape. Chem. Senses 42, 177–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Chao SK, Sitcheran R, Nunez JM, Vosshall LB, and Axel R (1994). Topographic organization of sensory projections to the olfactory bulb. Cell 79, 981–991. [DOI] [PubMed] [Google Scholar]
- Vassar R, Ngai J, and Axel R (1993). Spatial segregation of odorant receptor expression in the mammalian olfactory epithelium. Cell 74, 309–318. [DOI] [PubMed] [Google Scholar]
- Vedin V, Molander M, Bohm S, and Berghard A (2009). Regional differences in olfactory epithelial homeostasis in the adult mouse. J. Comp. Neurol 513, 375–384. [DOI] [PubMed] [Google Scholar]
- von der Weid B, Rossier D, Lindup M, Tuberosa J, Widmer A, Col JD, Kan C, Carleton A, and Rodriguez I (2015). Large-scale transcriptional profiling of chemosensory neurons identifies receptor-ligand pairs in vivo. Nat. Neurosci 18, 1455–1463. [DOI] [PubMed] [Google Scholar]
- Wang Q, Titlow WB, McClintock DA, Stromberg AJ, and McClintock TS (2017). Activity-Dependent gene expression in the mammalian olfactory epithelium. Chem. Senses 42, 611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SS, Lewcock JW, Feinstein P, Mombaerts P, and Reed RR (2004). Genetic disruptions of O/E2 and O/E3 genes reveal involvement in olfactory receptor neuron projection. Development 131, 1377–1388. [DOI] [PubMed] [Google Scholar]
- Wang T, Wang ZY, Zeng LY, Gao YZ, Yan YX, and Zhang Q (2020). Down-regulation of ribosomal protein rps21 inhibits invasive behavior of osteosarcoma cells through the inactivation of MAPK pathway. Cancer Manag. Res 12, 4949–4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weth F, Nadler W, and Korsching S (1996). Nested expression domains for odorant receptors in zebrafish olfactory epithelium. Proc. Natl. Acad. Sci. U S A 93, 13321–13326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitby-Logan GK, Weech M, and Walters E (2004). Zonal expression and activity of glutathione S-transferase enzymes in the mouse olfactory mucosa. Brain Res. 995, 151–157. [DOI] [PubMed] [Google Scholar]
- Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LM, Huh SH, and Ornitz DM (2018). FGF20-expressing, wnt-responsive olfactory epithelial progenitors regulate underlying turbinate growth to optimize surface area. Dev. Cell 46, 564–580.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara Y, Kawasaki M, Tamada A, Fujita H, Hayashi H, Kagamiyama H, and Mori K (1997). OCAM: a new member of the neural cell adhesion molecule family related to zone-to-zone projection of olfactory and vomeronasal axons. J. Neurosci 17, 5830–5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa K, Nakagawa H, Mori N, Watanabe H, and Touhara K (2013). An unsaturated aliphatic alcohol as a natural ligand for a mouse odorant receptor. Nat. Chem. Biol 9, 160–162. [DOI] [PubMed] [Google Scholar]
- Yoshikawa K, and Touhara K (2009). Myr-Ric-8A enhances G(alpha15)-mediated Ca2+ response of vertebrate olfactory receptors. Chem. Senses 34, 15–23. [DOI] [PubMed] [Google Scholar]
- Yoshikawa K, Wang H, Jaen C, Haneoka M, Saito N, Nakamura J, Adappa ND, Cohen NA, and Dalton P (2018). The human olfactory cleft mucus proteome and its age-related changes. Sci. Rep 8, 17170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu TT, McIntyre JC, Bose SC, Hardin D, Owen MC, and McClintock TS (2005). Differentially expressed transcripts from phenotypically identified olfactory sensory neurons. J. Comp. Neurol 483, 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, de March CA, Ni MJ, Adipietro KA, Golebiowski J, Matsunami H, and Ma M (2015). Responsiveness of G protein-coupled odorant receptors is partially attributed to the activation mechanism. Proc. Natl. Acad. Sci. U S A 112, 14966–14971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapiec B, and Mombaerts P (2020). The zonal organization of odorant receptor gene choice in the main olfactory epithelium of the mouse. Cell Rep. 30, 4220–4234.e5. [DOI] [PubMed] [Google Scholar]
- Zhang X, Rogers M, Tian H, Zhang X, Zou DJ, Liu J, Ma M, Shepherd GM, and Firestein SJ (2004). High-throughput microarray detection of olfactory receptor gene expression in the mouse. Proc. Natl. Acad. Sci. U S A 101, 14168–14173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Kuek V, Bennett S, Xu H, Rosen V, and Xu J (2019). Protein Cytl1: its role in chondrogenesis, cartilage homeostasis, and disease. Cell Mol. Life Sci 76, 3515–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang H, and Matsunami H (2007). Synergism of accessory factors in functional expression of mammalian odorant receptors. J. Biol. Chem 282, 15284–15293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq raw data have been deposited and are publicly available as of the date of publication at ArrayExpress: E-MTAB-10211. All original code and scripts for the 3D nose atlas shiny app has been deposited at Github and can be found at the Github Repository: https://doi.org/10.5281/zenodo.6036047https://zenodo.org/badge/DOI/10.5281/zenodo.6045897.svg. The 3D nose atlas processed data can be browsed and visualized here: http://atlas3dnose.helmholtz-muenchen.de:3838/atlas3Dnose.
Any additional information required to reanalyze the data reported in this paper is available from the lead contacts upon request.