

Abstract
Endometrial cancer (EC) is the most common gynecological cancer that affects the female reproductive organs. The standard therapy for EC for at least the past two decades has been chemotherapy and/or radiotherapy. PD98059 is a reversible MEK inhibitor that was found in these studies to increase the cytotoxicity of paclitaxel (PTX) against human endometrial cancer cells (Hec50co) in a synergistic and dose dependent manner. Additionally, while PD98059 arrested Hec50co cells at the G0/G1 phase, and PTX increased accumulation of cells at the G2/M phase, the combination treatment increased accumulation at both the G0/G1 and G2/M phases at low PTX concentrations. We recently developed poly(lactide-co-glycolide) (PLGA) nanoparticles (NPs) modified with polyethylene glycol (PEG) and coated with polyamidoamine (PAMAM) (referred to here as PGM NPs) which have favorable biodistribution profiles in mice, compared to non-PEGylated PLGA NPs, or PEGylated PLGA NPs with no PAMAM. Here, in order to enhance tissue distribution of PD98059, PD98059-loaded PGM NPs were prepared and characterized. The average size, zeta potential, and % encapsulation efficiency (%EE) of these NPs was approximately 184 nm, +18 mV, and 23%, respectively. The PD98059-loaded PGM NPs released ~25% of the total load within 3 days in vitro. In vivo murine studies revealed that the pharmacokinetics and biodistribution profile of intravenous (IV) injected PD98059 was improved when delivered as PD98059-loaded PGM NPs as opposed to soluble PD98059. Further investigation of the in vivo efficacy and safety of this formulation is expected to emphasize the potential of its clinical application in combination with commercial PTX formulations against different cancers.
Graphical Abstract
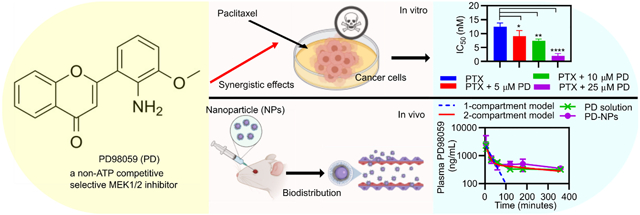
Introduction
The mitogen-activated protein kinase (MAPK) cascade is a protein kinase signaling pathway that plays a crucial role in multiple cellular processes including cell survival, proliferation, differentiation, cell development, apoptosis, metabolism, and migration [1-4]. The MAPK pathway is crucial for the development and proliferation of many cancer cell types [5-9] and is known to promote anti-apoptotic and survival signals [10, 11]. The use of MAPK inhibitors against cancer, including those that target mutations along the MAPK pathway such as BRAF mutations, either as single agents or in combination with other chemotherapeutics to overcome chemo-resistance has been widely described [12]. Recently, we investigated the use of different combinations designed to diminish chemoresistance of cancer cells to paclitaxel (PTX) in various tumor models such as endometrial cancer [13] and melanoma [14]. Several researchers confirmed the incidence of PTX-induced activation of MAPK pathway in some cancers, which triggers subsequent survival signals, and induces its own resistance [15, 16], which justifies the benefit of using MAPK inhibitors in combination with PTX [16, 17].
PD98059 (2-(2-Amino-3-methoxyphenyl)-4H-1-benzopyran-4-one) is a non-ATP competitive selective MEK1/2 inhibitor that specifically inhibits MEK-1-mediated activation of MAPK and does not directly inhibit ERK1/2 [18, 19]. Its antiproliferative effect through inducing G1 cell cycle arrest and apoptosis in tumor cells has been previously described [20]. Previous studies reported synergistic antiproliferative activity against cancer cells when PD98059 was used in combination with other agents such as resveratrol [21], docetaxel [22], curcumin [23], and PTX [15]. However, most of the reports utilizing PD98059 with chemotherapeutics either used it in vitro or administered it in vivo in non-clinically feasible formulations [15, 24], with certain exceptions, such as those reported by Basu et al. [25], Naguib et al. [26, 27], and Mekkawy, et al. [28]. This highlights the need to find a suitable nanocarrier for PD98059 to improve its clinical applicability while avoiding side effects due to generalized MEK1/2 inhibition.
Nanoparticles (NPs) with average diameters of 10-200 nm, are widely used in drug delivery systems to improve efficacy, prolong circulation time, and modify biodistribution of several therapeutic agents, including chemotherapeutics [29-33]. Polyethylene glycol (PEG) modified (PEGylated) NPs evade uptake by the cells of the mononuclear phagocyte system (MPS) that tend to clear circulation of the lipophilic circulating NPs after the latter are coated with opsonins [34-37]. Furthermore, cationic NPs have long been utilized to improve cellular uptake and tissue accumulation [38]. Kaps et al., used cationic nanohydrogel composites to improve the cellular uptake of siRNA by M2 macrophages in the liver [39]. Hybrid polyamidoamine (PAMAM)-modified poly(lactide-co-glycolide) (PLGA) hydrogel NPs achieved higher corneal cellular uptake and corneal tissue uptake compared to unmodified hydrogels or NPs [40].
In the current study, to overcome the short plasma half-life of PD98059 [26] and improve its tissue biodistribution profile, we loaded the compound into our recently developed PEG-PLGA/ PAMAM hybrid NP formulation (PGM). PAMAM operates as a surface charge modifier of NPs to increase uptake and can improve their transportation across the blood brain barrier [34, 41] and across other tissues in the body [40]. Many reports demonstrate that cationic, long-circulating nanocarriers were superior to their non-cationic counterparts for brain and tissue delivery [42-44].
The objective for this study was to develop a novel PGM formulation containing PD98059 to improve the drug’s pharmacokinetics and tissue distribution profile. To confirm the therapeutic potential of this formulation against cancer, we first tested it in combination with PTX using the loss-of-function (LOF) p53 human endometrial cancer cell line, Hec50co. We chose Hec50co as a model cell line because it is known to develop several chemoresistance mechanisms against PTX and we wanted to determine other possible PTX chemoresistance mechanisms beyond what we have previously investigated [13]. The purpose of developing this NP formulation was primarily to modify the drug’s biodistribution profile, and in particular, explore the potential of the NPs to improve drug accumulation in the brain.
Methods
Cell culture and cell viability assay
Hec50co cells were generously provided by Prof. Kimberly Leslie (University of Iowa) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Invitrogen, Madison, WI) supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) penicillin/streptomycin (100 U/mL, Invitrogen, Madison, WI). Cells were maintained at 37°C, 5% CO2 in a humidified incubator. The treatments were made from serial dilutions of PD98059 (Selleck Chemicals, Houston, TX) and PTX (LC Laboratories, Woburn, MA) solutions in DMSO and combinations of the two. One day before treatment, Hec50co cells were seeded in 96-well plates at a density of 1500 cells/well. Treatments were added in a volume of 50 μL per well and 150 μL of complete fresh media in case of single treatment or 100 μL of media in case of combination treatments (final volume: 200 μl). The untreated control cells were incubated with 200 μL of media. After three days, the media from the wells was discarded and replaced with 100 μL complete fresh media and 20 μL of the MTS reagent (CellTiter 96® AQueous One Solution Cell Proliferation Assay, Promega Corporation) and incubated at 37° C for 2 hours (hr). Absorbance was measured using a Spectra Max plus 384 Absorbance Microplate (Molecular Devices) at wavelength 490 nm. Cell viability was expressed as the percentage of the absorbance reading from the treated cell samples compared to the absorbance from the untreated cell samples. IC50 values for PTX and its combinations with PD98059 were calculated using GraphPad prism software for Windows version 8.3.0 (GraphPad Software Inc.). Additionally, synergy between PTX and PD98059 was assessed using CompuSyn software (CompuSyn Inc.) by establishing dose-response curves for PTX and its combinations with PD98059. Combination index (CI) values were calculated, and synergy was indicated when CI < 1.
Cell cycle analysis by flow cytometry
For cell cycle analysis, Hec50co cells were seeded in 6-well plates at a density of 5×103 cells/well in 2 mL complete DMEM media and incubated in a humidified incubator at 37°C, 5% CO2 for 24 hrs. Then, cells were treated with either different concentrations of PTX (5 nM and 10 nM), 25 μM PD98059 or a combination of these and incubated for another 24 hr. Subsequently, media was aspirated, and cells were washed twice with phosphate-buffered saline (PBS, Invitrogen, Madison, WI), trypsinized and quenched with fresh media. Cells were collected and fixed in ice chilled 70% ethanol (Sigma Aldrich, St. Louis, MO) and incubated for 30 minutes (min) at 4°C. Afterwards, cells were centrifuged at 230 xg for 5 min, resuspended in 250 μL PBS containing 1% NP40 (Sigma Aldrich) and 100 μg/mL RNAse solution (Thermo Fisher Scientific, Rockford, IL), and incubated at 37°C for 30 min. Then, 250 μL of PBS containing 1% NP40 and 50 μg/mL propidium iodide (Thermo Fisher Scientific) solution were added to each tube and samples were then analyzed using a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ). Data was further analyzed using ModFit LT software. Also, cell percentage in each cell cycle stage including G0/G1, S or G2/M was determined using GraphPad Prism software.
Synthesis of PLGA-b-PEG-COOH copolymer
250 mg of PLGA-COOH (Creative PEGWorks, Durham, NC) was dissolved in 2.0 mL of dichloromethane (DCM, Sigma Aldrich). Next, 3 mg of N-hydroxysuccinimide (NHS, Sigma Aldrich) and 4.8 mg of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC, Sigma Aldrich) were dissolved in 2.5 mL of DCM, and then added into the PLGA-COOH solution. This mixture was stirred at room temperature during which time the PLGA-COOH reacted with EDC/NHS to become PLGA-NHS (amine-reactive sulfo-NHS ester). A 20 mL mixture of ethyl ether and methanol (Sigma Aldrich) (50:50 %v/v, “washing solution”) was added to the previously prepared solution and centrifuged at 5000 xg for 10 min (Eppendorf 5810-R, Eppendorf®, Westbury, NY) twice to remove any residual EDC/NHS. The PLGA-NHS was then dried under rotary evaporator vacuum for 30 min. The PLGA-NHS was dissolved in 4.0 mL of DCM followed by the addition of 11 μL of N, N-diisopropylethylamine (DIPEA). To this, 30 mg of NH2-PEG-COOH (3.4kDa) was added and stirred at room temperature for 24 hr. Then the washing solution was added to the PLGA-b-PEG-COOH solution and centrifuged at 5000 xg for 10 min twice to remove any unreacted NH2-PEG-COOH. The PLGA-b-PEG-COOH copolymer was then dried under rotary evaporation.
The PLGA-b-PEG-COOH was examined using a Proton Nuclear Magnetic Resonance (1H NMR) spectrometer with samples dissolved in deuterated chloroform (CDCl3). Chemical shifts were reported in ppm (δ units).
Preparation of PD98059-loaded PLGA-b-PEG-PAMAM NPs
PD98059-loaded PLGA-b-PEG-PAMAM (PGM) NPs were prepared using a modification of a previously described nanoprecipitation method [45]. Briefly, 2 mg of PD98059 was dissolved by vortexing in 5 mL acetone (Sigma Aldrich). Next, 50 mg of the PLGA-b-PEG copolymer was added into the PD98059 solution, vortexed, and 125 μL of generation 5 (G5) PAMAM (Sigma Aldrich) solution (5 % wt. in methanol) was added to the mixture. This mixture was vortexed and transferred to a 5 mL syringe fitted with a G26 needle (0.45 mm x 13 mm) where the solution was added dropwise into 15 mL of 0.1% poly-(vinyl alcohol) (PVA, hydrolyzed 80% (Mw 9,000 - 10,000), Sigma Aldrich) aqueous solution while continuously stirring for 30 min. The NPs were collected, and the organic solvents were removed by rotary evaporation (Buchi, Flawil, Switzerland) under vacuum and reduced pressure for 4 hr. The suspension was then washed four times using Nanopure water and collected using Amicon ultra-15 centrifugal filter units (Mw cutoff = 100 kDa) (Millipore Sigma, St. Louis, MO) at 4000 xg for 20 min using centrifugation (Eppendorf 5810-R, Eppendorf®, Westbury, NY). The NPs were lyophilized using FreeZone (Freeze Dry Systems, −105°C, 4.5 Liter Benchtop Model; Labconco Co., Kansas City, MO) with cryoprotectant (10% w/v sucrose), and the NPs were frozen at −20 °C for 24 hr prior to the lyophilization cycle.
NP physicochemical characterization
The physicochemical properties of the PGM NPs such as size distribution and zeta (ζ) potential were measured by Dynamic Light Scattering (DLS) and by laser Doppler velocimetry (LDV), respectively. NPs were suspended in Nanopure water and measurements were performed using a Zetasizer Nano ZS particle analyzer (Malvern Instrument Ltd., Westborough, MA). The morphology of the NPs was determined using Scanning Electron Microscopy (SEM) (Hitachi S- 4800 SEM, Hitachi High-Tech America, Inc., Schaumburg, IL). A suspension of NPs was added onto a silicon wafer mounted on SEM stubs using double sided carbon tape. The NP suspension was dried at room temperature for 24 hr, and then coated with gold-palladium using an argon beam K550 sputter coater (Emitech Ltd., Kent, England).
Determination of PD98059 loading and percent encapsulation efficiency
The PD98059 loading and percent encapsulation efficiency (%EE) were determined using an HPLC analytical method that has been described in our previous publications [26-28]. High-performance liquid chromatography (HPLC) with an ultraviolet (UV) detector (Agilent 1100 series with VWD detector (Agilent, Santa Clara, CA)) was used. The NPs were dissolved in acetonitrile (ACN, Sigma Aldrich) and vortexed until the solution became clear. This mixture was diluted with methanol:water (Barnstead™ Nanopure, Thermo Fisher, Rockford, IL) (56:44 v/v), sonicated until the solution became clear, and centrifuged at 14000 xg for 2 min to separate undissolved NPs. The solution was injected into the HPLC and the PD98059 concentration was quantified by using a calibration curve of known sample concentrations.
Chromatographic separation of PD98059 was performed at room temperature. Reverse phase C-18 column (Waters, Symmetry® 5μm, 4.6 x 150 mm) was used in the assay and the mobile phase used consisted of methanol:water (70:30 v/v) with 0.1% trifluoroacetic acid (TFA) (Sigma Aldrich) pumped at a flow rate of 1 mL/min. The detection wavelength was 320 nm and the injection volume was 50 μL. Drug loading in the PGM NPs and EE were calculated using equations 1 and 2, respectively.
| (Equation 1) |
| (Equation 2) |
In vitro release study of NPs
The cumulative release of PD98059 from PGM NPs was studied using Float-A-Lyzer dialysis (50 KD MWCO, 1-mL capacity, regenerated cellulose membrane). Briefly, triplicate samples of the PD98059-loaded PGM NPs were suspended in 1 mL of release medium (PBS, pH7.4 + 0.4% Tween 80) and transferred into the Float-A-Lyzer which was placed in a 50 mL tube containing 12 mL release medium. These tubes were left shaking at 300 rpm and 37°C and samples withdrawn at predetermined time points. Release medium was removed at each time point and replaced with fresh medium to maintain sink conditions. The PD98059 concentration in each sample was measured using the previously described HPLC-UV method.
Biodistribution studies
BALB/CJ mice 8 weeks old with a weight range of 20 – 23 g were used and allowed uncontrolled access to food and water. Mice were obtained and experiments carried out following the animal protocol approved by the institutional animal care and use committee (IACUC).
The mice were injected IV with saline, 113 μg of PD98059 solution, or 3 mg of PD98059-loaded PLGA-b-PEG-PAMAM NPs (equivalent to 113 μg of PD98059) by retro-orbital injection. The PD98059 was extracted using a modified extraction technique [46]. Briefly, the collected organs were first carefully rinsed with 1X DPBS (Life Science, Waltham, MA), and then they were placed into cryotubes, and 250 μL of 1X PBS and disruption beads were added into the cryotube [26, 47]. To extract PD98059, the organs were homogenized using a homogenizer (Fisherbrand™ Bead Mill 4, Fisher scientific, Pittsburgh, PA) for approximately 2 min at speed 5 and transferred into 15-mL centrifuge tubes. Then 15 μL of the internal standard (10 μg/mL 7-hydroxyflavone) was added into each centrifuge tube and vortexed. Then, 2 mL of ethyl acetate (EA, Sigma Aldrich) was added to the tubes and centrifuged at 4° C, 5000 xg for 5 min for liquid-liquid extraction, where PD98059 was separated and dissolved into the EA layer. This step was repeated, and the top EA layer was transferred into a glass tube. The EA layer was evaporated under a stream of nitrogen (TurboVap® LV, Caliper life Sciences, Hopkinton, MA) for approximately 2 hr. The amount of PD98059 was calculated by adding 85 μL of methanol to the tubes, which were then vortexed and sonicated until resuspended. Ultrapure distilled water (Invitrogen, Madison, WI) was then added and mixed to obtain a final volume of 160 μL. Finally, the mixture was centrifuged at 4° C, 5000 xg for 20 min and the supernatant was injected into the HPLC and analyzed using the previously mentioned HPLC-UV method [26-28].
The anesthetized mice were euthanized 5, 30, 60, 120, 180 and 360 min after injection of drug or drug-loaded PGM NPs. For the biodistribution studies, mouse organs were subjected to the aforementioned extraction procedure, and HPLC-UV analysis was performed to determine the concentration of PD98059 in each tissue. Then, the relationship between PD98059 concentration and time was determined by plotting concentrations versus the area ratio between PD98059 and internal standard. Pharmacokinetic analysis was performed using the collected mouse serum at the previously mentioned time points. Briefly, the obtained blood was centrifuged for 5 min at 10,600 xg. Plasma was then separated from the top of the rest of blood components and subjected to an extraction procedure and analyzed for drug content using HPLC-UV. Using GraphPad Prism software (Version 8.3.0) and the PK Solver add-in program, the plot of PD98059 concentration versus time was prepared. Pharmacokinetic parameters were calculated in mouse plasma and other organs.
Statistical analysis
To compare data between two groups, the data was analyzed using Student’s two-tailed t-test (GraphPad Prism version 8.3.0 for Windows, GraphPad Software, La Jolla, CA). To compare data between more than two groups, one-way analysis of variance (ANOVA) test was used followed by Tukey’s post-hoc test. Analysis of the pharmacokinetic study was performed using the RStudio software (RStudio, Inc., Boston, MA) and PK-solver Excel® add-in. All plots were created using GraphPad Prism. A statistically significant difference was considered for p-values < 0.05.
Results and discussion
Cell viability assay and cell cycle analysis
The cell viability study demonstrated that the addition of PD98059 significantly increased the cytotoxicity of PTX in a dose dependent manner (Figure 1A). A 6.5-fold decrease in the IC50 of PTX on Hec50co cells was observed when treated concomitantly with 25 μM PD98059 (Figure 1B). It is worth mentioning that 5,10 and 25 μM PD98059 alone showed negligible cytotoxicity against the Hec50co cell line (3.8, 5.3 and 17 %, respectively). The combination index (CI) was calculated and showed a noticeable synergy between PTX and different concentrations of PD98059 where CI < 1 indicated synergy (Figure 1C).
Figure 1:

Cytotoxicity study of PTX +/− PD98059. (A) Cell viability study of Hec50co cells cultured with increasing concentrations of soluble PTX +/− indicated concentrations of PD98059 for 72 hr and measured using an MTS assay. (B) IC50 of PTX with various PD98059 concentrations. (C) Synergy between PTX and PD98059, where the CI was plotted versus fraction affected (Fa) where CI values < 1 indicate synergy. (D) Cell cycle profiles of Hec50co cells treated with 5 and 10 nM PTX, and 25 μM PD98059 or their combination for 24 hr and (E) representative histograms of the percentages of the cells that were in each phase. Results are reported as mean ± SD (n = 3). Statistical analysis was performed using one-way ANOVA; **** P < 0.0001, ** P < 0.01, * P < 0.05.
The effect of the combination treatment on cell cycle progression was investigated using 5 and 10 nM PTX on Hec50co cells. The cells were treated with soluble PTX and/or PD98059, and the cell cycle progression was analyzed by flow cytometry. PD98059 arrested Hec50co cells at the G0/G1 phase (63.04%), while 5 nM and 10 nM PTX increased accumulation of cells at the G2/M phase in a dose dependent manner, 37.2 % and 45.74 % respectively, compared to untreated cells (27.95 %) (Figure 1D and E). However, addition of 25 μM PD98059 to 5 nM PTX resulted in a slight increase of percentage of cells in the G2/M phase (42.15%); and this cell accumulation into the G2/M phase was markedly increased (74.99%) when a higher PTX concentration (10 nM) was used. PD98059 was reported to down-regulate the p-glycoprotein (p-gp) in p-gp positive tumor cells [48]. This can lead to an increase in the intracellular concentration of the co-administered PTX by preventing the efflux of PTX [13].
Previous studies showed that PD98059 induced a notable apoptosis of HeLa S3 (human cervix carcinoma) cells in a time and concentration-dependent manner [49]. The ability of PTX to activate the MAPK pathway can compromise its anticancer efficacy. The combination of MEK1/2 inhibitors and PTX has been shown to be beneficial in inhibiting PTX-induced ERK1/2 activation and enhancing the cell death of some cancer cell lines [16]. Moreover, the combination treatment may help achieve the same therapeutic efficacy as current high dose chemotherapy with lower doses of chemotherapy, thus lowering the risk of chemotherapy-related adverse events. Moreover, the p-gp inhibitory effect that was exerted by PD98059 could provide a means of reversing the p-gp mediated resistance to chemotherapy.
Synthesis of PLGA-b-PEG-COOH copolymer
The PLGA-b-PEG-COOH copolymers were successfully fabricated as determined by 1H NMR spectrometry (Supplementary Figure. 1A). The peaks appeared at δ 1.56 ppm, δ 3.66 ppm, δ 4.81 ppm and δ 5.21 ppm which corresponded with the 1H NMR spectrum for PLGA (at δ 1.63 ppm, δ 4.83 ppm and δ 5.18 ppm) and the 1H NMR spectrum for NH2-PEG-COOH (at δ 3.67 ppm) (Supplementary Figure. 1B and 1C, respectively). The prepared PLGA-b-PEG-COOH copolymers were used for the synthesis of the NPs in an attempt to modify the pharmacokinetics and biodistribution profile of the drug [50].
NP formulation and characterization
We used PLGA-b-PEG-COOH copolymers to develop our new delivery system, PD98059-loaded PLGA-PEG-PAMAM (PGM) NPs. Size, surface charge and morphology of the NPs were determined by DLS, LDV, and SEM, respectively and the values are summarized in Table 1. The average diameter and ζ-potential of the nanoparticles were 184.2 nm and 18.3 mV, respectively. The nanoprecipitation method yielded drug %EE of greater than 20%. SEM images showed spherical-shaped nanoparticles (Figure 2A). Cumulative release of PD98059 from the NPs in the first 72 h is shown in Figure 2B. The release study revealed that approximately 25% of the drug was released over 72 h, which is much slower that what we found previously for free PD98059, where more than 80% of it was dissolved in 24 h [27]. These results confirm that the prepared NPs are expected to prevent dose dumping of PD98059 in the circulation within the first few hours after IV injection.
Table 1.
Summary of the characterization of the NPs.
| Nanoparticles | Average diameter (nm) |
Morphology | PDI | ζ-potential (mV) |
Loading (μg/1mg NPs) |
EE% |
|---|---|---|---|---|---|---|
| PD98059-PGM | 184.2 ± 0.4 | Spherical | 0.076 | 18.3 ± 6.7 | 37.7 ± 3.4 | 22.6 ± 2.0 |
Figure 2.
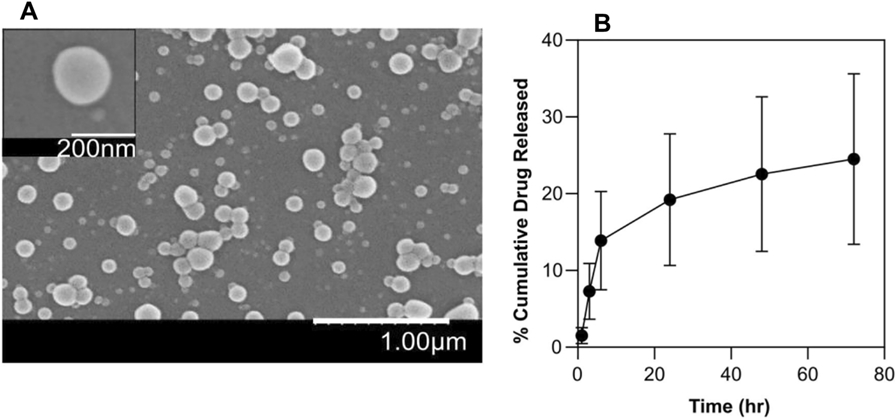
(A) SEM image of PD98059-loaded PGM NPs. (B) In vitro release profile of PD98059 from PGM NPs over 72 hr. Results are reported as mean ± SD (n = 3).
Pharmacokinetics and biodistribution of PD98059 in free form and in PGM NPs
The biodistribution-time profiles of PD98059 in different organs was quantified as mice were euthanized at each time point (n=3/time point). PD98059 accumulation data showed statistically significant differences between PD98059 solution and PD98059-PGM NPs in the pancreas, kidney, heart, and muscle, while other organs like the brain, lung, spleen, and liver did not show significant differences (Figure 3). However, there remarkably higher accumulation in other organs, especially the brain, in favor the PGM NPs compared to free PD98059 was also found, as will be discussed in the following section.
Figure 3.

Biodistribution of PD98059 for each organ 5, 30, 60, 120, 180 and 360 minutes after injection displaying the PD98059 accumulation/g of indicated organ. The amount of PD98059 for each organ was measured using HPLC-UV. Student’s T-test was used to compare accumulation of PD98059 (resulting from soluble PD98059 versus PD98059-PGM NP delivery) in a specific tissue at a specific time point. Where p values were between 0.05 – 0.1, the values are shown (indicating a possible trend) while p-values > 0.1 are not shown in the graph; * p < 0.05 and ** p < 0.01. Results are reported as mean ± S.D. (n = 3).
The PD98059 plasma concentration versus time curve for drug-loaded PGM NPs and free drug after IV injection in mice is shown in Figure 4. The pharmacokinetics of PD98059 were studied after data were fitted to a 1-compartment model (blue dotted line) or 2-compartment model (red line) using PK Solver add-in. The R2 values of 1-compartments al and 2-compartmental fitting were 0.9715 and 0.9964, respectively. Our previous studies in rats showed that PD98059 elimination follows a 2-compartment open model [26], with a distribution half-life of less than 10 minutes, and a relatively high volume of distribution, which indicates that the drug is widely distributed to organs. Hence, it was expected that soluble PD98059 would distribute to various organs within a few minutes after IV injection as shown in Figure 3. Even though we also found a short distribution half-life in mice comparable to that previously found in rats [26], the relatively longer elimination half-life in mice may indicate dose-dependent pharmacokinetics of PD98059 solution [51], which can be the subject of further studies.
Figure 4.

Plasma PD98059 concentration versus time subsequent to administration of mice with PD98059 solution and PD98059-PGM NPs. The pharmacokinetics of PD98059 were evaluated using PK Solver add-in. Results are reported as mean ± SD (n = 3). The 1- and 2-compartment models fitting was done for the free PD98059 plasma levels.
The pharmacokinetic parameters of PD98059 solution and PGM NPs were best fit with the 2-compartment model and first order elimination as shown in Figure 4 and Tables 2 and 3. Compared to PD98059 solution, NPs showed an increase in elimination half-life by ~1.7-fold compared to free drug solution, which may be attributed to PEGylation.
Table 2.
Pharmacokinetic parameters of PD98059 in plasma after IV administration of PD98059 solution to healthy mice.
| Non compartment model | One-compartment model | Two-compartment model | |||
|---|---|---|---|---|---|
| Parameters | PD98059 solution |
Parameters | PD98059 solution |
parameters | PD98059 solution |
| K (min −1) | 0.0022 | K (min −1) | 0.033 | A (μg/mL) | 2.65 |
| T1/2 (min) | 314.87 | T1/2 (min) | 20.91 | α (min −1) | 0.076 |
| C0 (μg/mL) | 2.91 | C0 (μg/mL) | 2.63 | B (μg/mL) | 0.49 |
| AUC0-t (μg x min/mL) |
175.72 | AUC0-t (μg x min/mL) |
79.61 | β (min −1) | 0.0016 |
| AUC0-inf (μg x min/mL) |
320.68 | AUC0-inf (μg x min/mL) |
79.61 | K10 (min −1) | 0.0092 |
| AUMC0-inf (μg x min2/mL) |
140,206.69 | AUMC0-inf (μg x min2/mL) |
2,402.29 | K12 (min −1) | 0.055 |
| MRT0-inf (min) | 437.20 | MRT0-inf (min) | 30.17 | K21 (min −1) | 0.013 |
| V (mL) | 141.65 | V (mL) | 37.90 | T1/2(α) (min) | 9.06 |
| Cl (mL/min) | 0.31 | Cl (mL/min) | 1.25 | T1/2(β) (min) | 427.63 |
| Vss (mL) | 136.33 | Vss | 37.90 | C0 (μg/mL) | 3.15 |
| R2 | 0.9715 | V (mL) | 31.72 | ||
| Cl (mL/min) | 0.29 | ||||
| AUC0-t (μg x min/mL) |
170.01 | ||||
| AUC0-inf (μg x min/mL) |
340.78 | ||||
| AUMC0-inf (μg x min2/mL) |
189,279.12 | ||||
| MRT0-inf (min) | 555.42 | ||||
| Vss (mL) | 162.98 | ||||
| R2 | 0.9964 | ||||
K: elimination rate constant, T1/2: plasma half-life, C0: plasma concentration at time 0, AUC: area under the plasma concentration-time curve, AUMC: area under the moment curve, MRT: mean residence time, V: volume of distribution, Cl: clearance and Vss: Volume of distribution at steady state
Table 3.
Pharmacokinetic parameters of PD98059 in plasma after IV administration of PD98059-PGM NPs to healthy mice.
| Non compartment model | One-compartment model | Two-compartment model | |||
|---|---|---|---|---|---|
| Parameters | PD98059-PGM NPs |
Parameters | PD98059-PGM NPs |
Parameters | PD98059-PGM NPs |
| K (min −1) | 0.0014 | K (min −1) | 0.038 | A (μg/mL) | 3.13 |
| T1/2 (min) | 466.75 | T1/2 (min) | 17.80 | α (min −1) | 0.087 |
| C0 (μg/mL) | 3.26 | C0 (μg/mL) | 3.04 | B (μg/mL) | 0.52 |
| AUC0-t (μg x min/mL) |
209.72 | AUC0-t (μg x min/mL) |
78.23 | β (min −1) | 0.00093 |
| AUC0-inf (μg x min/mL) |
445.54 | AUC0-inf (μg x min/mL) |
78.23 | K10 (min −1) | 0.0061 |
| AUMC0-inf (μg x min2/mL) |
271,559.48 | AUMC0-inf (μg x min2/mL) |
2,009.44 | K12 (min −1) | 0.068 |
| MRT0-inf (min) | 609.49 | MRT0-inf (min) | 25.68 | K21 (min −1) | 0.013 |
| V (mL) | 151.14 | V (mL) | 32.82 | T1/2(α) (min) | 7.95 |
| Cl (mL/min) | 0.22 | Cl (mL/min) | 1.27 | T1/2(β) (min) | 745.09 |
| Vss (mL) | 136.79 | Vss (mL) | 32.82 | C0 (μg/mL) | 3.66 |
| R2 | 0.9699 | V (mL) | 27.28 | ||
| Cl (mL/min) | 0.16 | ||||
| AUC0-t (μg x min/mL) |
198.05 | ||||
| AUC0-inf (μg x min/mL) |
605.49 | ||||
| AUMC0-inf (μg x min2/mL) |
612,620.73 | ||||
| MRT0-inf (min) | 1,011.76 | ||||
| Vss (mL) | 167.09 | ||||
| R2 | 0.9991 | ||||
K: elimination rate constant, T1/2: plasma half-life, C0: plasma concentration at time 0, AUC: area under the plasma concentration-time curve, AUMC: area under the moment curve, MRT: mean residence time, V: volume of distribution, Cl: clearance and Vss: Volume of distribution at steady state.
In addition, the area under the curve from zero to infinity (AUC0-inf), increased in the case of PGM NPs by ~1.8-fold compared to free drug solution (Tables 2 and 3), with a reduction in PD98059 clearance (Cl) from plasma when the PGM NP formulation was used (0.16 mL/min) compared to free drug solution (0.29 mL/min). Moreover, the mean residence time (MRT) of PD98059 in the PGM NPs exhibited an increase of ~1.8-fold compared to PD98059 solution. Even though the change in the volume of distribution (V) following the injection of PD98059-PGM NPs was not remarkably different compared to that obtained following the injection of the solution (27.28 mL and 31.72 mL, respectively), the PGM NPs changed the accumulation of PD98059 in certain organs as will be discussed later.
PD98059 distribution in mouse organs
Figure 3 shows the concentration versus time profile of PD98059 in mouse organs after IV administration. In the brain, plasma levels reached a maximum value of about 3 μg/g 5 minutes following the IV injection of the PD98059 solution, and this value increased about 6-fold when the PGM NPs were injected. Furthermore, the area under the curve from zero to 6 hr (AUC0-6) showed a more than 3-fold increase for the PGM NPs compared to free drug solution (1160.83 μg x min/mL compared to 334.25 μg x min/mL, respectively). This trend can be attributed to the cationic nature of the PGM NPs which allows them to massively accumulate at the vascular endothelium and increases their ability to cross the blood brain barrier [34]. The molecules of a positive charge surface modifier such as PAMAM may have deposited on the surface of the NPs rendering them cationic, which in turn could have improved brain uptake [34, 41]. Throughout the study period, the drug concentration from PGM NPs was higher than that from free drug solution in the brain at almost all time points. The absence of statistical significance in the brain accumulation when PGM NPs were used compared to free drug solution may be achieved if the number of animals per time point is increased, considering the dramatic increase in the AUC0-6 value when the PGM NPs were used, and the large variability between data points.
The heart levels of PD98059 following IV injection of PGM NPs were relatively higher compared to solution, especially at the earlier time points. After 60 minutes, the heart levels declined markedly and reached values very close to those obtained after IV injection of the PD98059 solution. This rapid disappearance from the cardiac tissues may be a promising sign that the PGM NPs do not accumulate in the heart or cause damage, however, safety of these NPs needs to be evaluated to confirm that there are no detectable heart-related adverse events.
The gradually declining trend in PD98059 concentration over time was evident for both free drug and drug loaded into PGM NPs (Figure 3). The liver concentration of PD98059 following the PD98059-PGM NP injection was ~5-fold higher after 5 min compared to PD98059 in solution. This may be attributed to the net positive surface charge of the PD98059-PGM NPs due to PAMAM and resultingly enhancing liver accumulation. Even though there was not a significant difference between both formulations, the AUC0-6 was increased by ~1.6-fold (197.93 μg x min/mL for PD98059-PGM NPs compared to 125.38 μg x min/mL for PD98059 in solution).
PD98059 levels in the kidneys tended to be higher for mice injected with the PGM NPs versus those injected with free drug solution. The relatively higher AUC0-6 following the PGM NPs injection compared to the solution injection ((555.96 and 301.47 μg x min/mL, respectively) may be attributed to the cationic nature of the NPs as well as the large particle size which makes them unsuitable candidates for glomerular filtration. PGM NPs showed higher accumulation level of PD98059 in pancreas, spleen and muscle when compared to drug solution. Table 4 shows that the AUC0-6 of PD98059 in organs was increased by approximately 1.3-fold, 1.2-fold and 1.1-fold in pancreas, spleen, and muscle, respectively when the PGM NPs were injected compared to free drug solution.
Table 4.
Pharmacokinetic parameter (AUC) of PD98059 in organs after IV administration of free drug solution and PD98059-PGM NPs to healthy mice.
| Organs | AUC0-6 (μg x min/mL) | |
|---|---|---|
| PD98059 solution | PD98059-PGM NPs | |
| Brain | 334.25 | 1,160.83 |
| Heart | 160.37 | 212.05 |
| Lung | 459.51 | 561.27 |
| Pancreas | 177.37 | 233.56 |
| Spleen | 129.72 | 160.68 |
| Kidney | 301.47 | 555.96 |
| Liver | 125.38 | 197.93 |
| Muscle | 129.86 | 153.86 |
Further investigation is required to determine if the drug encapsulated in PGM NPs can accumulate in tumors (especially in orthotopic tumor models) to a greater or longer extent than soluble drug. In addition, testing the antitumor efficacy of this proposed combination therapy will elucidate the potential benefit of PTX and PD98059 treatment strategy. Our future research will focus on testing this therapeutic strategy against endometrial cancer tumor models. Since the PGM NP formulation showed superior brain accumulation compared to free drug, we will also direct our future research to use this PGM NPs platform to treat brain cancer. Dendrimers and other PAMAM-based formulations are promising drug delivery systems that tend to maximize tissue accumulation of their loaded cargos due to their cationic nature [30, 33, 34]. However, their cationic nature can also lead to toxicity [52]; an issue which has been at least partially addressed through PEGylation. Our future studies will include deciphering the safety and toxicity of this PGM NP formulation in mice, which includes determination of the maximum tolerated dose, their short-term toxicity and their long-term toxicity.
Conclusion
This study demonstrated that PD98059 increased the cytotoxicity of PTX in a dose dependent manner and showed a 2.3-fold decrease in the IC50 of PTX against Hec50co cells when treated concomitantly with 25 μM PD98059. The CI demonstrated a noticeable synergistic therapeutic efficacy between PD98059 and PTX. In addition, cell cycle analysis showed that PD98059 arrested Hec50co cells at the G0/G1 phase, and PTX increased accumulation of cells at the G2/M phase, while their combination treatment increased both the G0/G1 and the G2/M phases at the lower PTX concentration. This information emphasized the significance of PD98059 as a suitable drug candidate to be used with chemotherapy and warrants further investigation into developing a delivery system that maximizes the accumulation of PD98059 in tissues of interest.
In this work, we developed PGM NPs composed of PLGA-PEG-PAMAM which modified the pharmacokinetics and biodistribution of PD98059 when encapsulated into these NPs compared to free drug solution. The PGM NPs enhanced PD98059 accumulation (compared to when soluble PD98059 was delivered) in the heart, muscle, pancreas and the kidney, albeit transiently. These findings emphasize the application of these PGM NPs as potential carriers of PD98059 to target certain cancers.
Supplementary Material
Acknowledgements
K.W. acknowledges support from the Government Pharmaceutical Organization (GPO) scholarship. J.C.Q. acknowledges support from the Alfred P. Sloan Foundation, the University of Iowa Graduate College, and the American Association for University Women. A.K.S acknowledges support from the Cancer Center support grant (P30 CA086862) and the Lyle and Sharon Bighley Chair of Pharmaceutical Sciences.
Funding
This work was supported by the Cancer Center support grant (P30 CA086862).
Footnotes
Ethics approval and consent to participate
Animal studies were carried out by Kanawat Wiwatchaitawee, Youssef W. Naguib and Kareem Ebeid for this article.
Animal husbandry and veterinary care were provided by the Office of Animal Resources at the University of Iowa. All animal protocols used in these studies were in agreement with and approved by Institutional Animal Care and Use Committee (IACUC).
Consent for publication
All authors have given approval to publication.
Availability of data and materials
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
Competing interest
The authors declare no competing interests.
References
- 1.Chang L and Karin M, Mammalian MAP kinase signalling cascades. Nature, 2001. 410(6824): p. 37–40. [DOI] [PubMed] [Google Scholar]
- 2.Pearson G, et al. , Mitogen-Activated Protein (MAP) Kinase Pathways: Regulation and Physiological Functions*. Endocrine Reviews, 2001. 22(2): p. 153–183. [DOI] [PubMed] [Google Scholar]
- 3.Zhang W and Liu HT, MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Research, 2002. 12(1): p. 9–18. [DOI] [PubMed] [Google Scholar]
- 4.Munshi A and Ramesh R, Mitogen-activated protein kinases and their role in radiation response. Genes Cancer, 2013. 4(9-10): p. 401–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li L, et al. , The Ras/Raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC (Review). Oncol Lett, 2016. 12(5): p. 3045–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costigan DC and Dong F, The extended spectrum of RAS-MAPK pathway mutations in colorectal cancer. Genes, Chromosomes and Cancer, 2020. 59(3): p. 152–159. [DOI] [PubMed] [Google Scholar]
- 7.Li S, Balmain A, and Counter CM, A model for RAS mutation patterns in cancers: finding the sweet spot. Nature Reviews Cancer, 2018. 18(12): p. 767–777. [DOI] [PubMed] [Google Scholar]
- 8.Satoh R, et al. , Discovery of new benzhydrol biscarbonate esters as potent and selective apoptosis inducers of human melanomas bearing the activated ERK pathway: SAR studies on an ERK MAPK signaling modulator, ACA-28. Bioorganic Chemistry, 2020. 103: p. 104137. [DOI] [PubMed] [Google Scholar]
- 9.Wang A-X and Qi X-Y, Targeting RAS/RAF/MEK/ERK signaling in metastatic melanoma. IUBMB Life, 2013. 65(9): p. 748–758. [DOI] [PubMed] [Google Scholar]
- 10.Montor WR, Salas AROSE, and Melo F.H.M.d., Receptor tyrosine kinases and downstream pathways as druggable targets for cancer treatment: the current arsenal of inhibitors. Molecular cancer, 2018. 17(1): p. 55–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng Y and Tian H, Current Development Status of MEK Inhibitors. Molecules (Basel, Switzerland), 2017. 22(10): p. 1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Menzies AM and Long GV, Dabrafenib and Trametinib, Alone and in Combination for BRAF-Mutant Metastatic Melanoma. Clinical Cancer Research, 2014. 20(8): p. 2035–2043. [DOI] [PubMed] [Google Scholar]
- 13.Ebeid K, et al. , Synthetically lethal nanoparticles for treatment of endometrial cancer. Nat Nanotechnol, 2018. 13(1): p. 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hodi FS, et al. , Phase II study of paclitaxel and carboplatin for malignant melanoma. Am J Clin Oncol, 2002. 25(3): p. 283–6. [DOI] [PubMed] [Google Scholar]
- 15.Xu R, et al. , Enhancement of paclitaxel-induced apoptosis by inhibition of mitogen-activated protein kinase pathway in colon cancer cells. Anticancer Res, 2009. 29(1): p. 261–70. [PubMed] [Google Scholar]
- 16.MacKeigan JP, Collins TS, and Ting JP, MEK inhibition enhances paclitaxel-induced tumor apoptosis. J Biol Chem, 2000. 275(50): p. 38953–6. [DOI] [PubMed] [Google Scholar]
- 17.McDaid HM and Horwitz SB, Selective potentiation of paclitaxel (taxol)-induced cell death by mitogen-activated protein kinase kinase inhibition in human cancer cell lines. Mol Pharmacol, 2001. 60(2): p. 290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dudley DT, et al. , A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proceedings of the National Academy of Sciences, 1995. 92(17): p. 7686–7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muslin AJ, MAPK signalling in cardiovascular health and disease: molecular mechanisms and therapeutic targets. Clinical science (London, England : 1979), 2008. 115(7): p. 203–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoshino R, et al. , Blockade of the Extracellular Signal-regulated Kinase Pathway Induces Marked G1 Cell Cycle Arrest and Apoptosis in Tumor Cells in Which the Pathway Is Constitutively Activated: UP-REGULATION OF p27Kip1. Journal of Biological Chemistry, 2001. 276(4): p. 2686–2692. [DOI] [PubMed] [Google Scholar]
- 21.Alkhalaf M and Jaffal S, Potent antiproliferative effects of resveratrol on human osteosarcoma SJSA1 cells: novel cellular mechanisms involving the ERKs/p53 cascade. Free Radical Biology and Medicine, 2006. 41(2): p. 318–325. [DOI] [PubMed] [Google Scholar]
- 22.Zelivianski S, et al. , ERK inhibitor PD98059 enhances docetaxel-induced apoptosis of androgen-independent human prostate cancer cells. Int J Cancer, 2003. 107(3): p. 478–85. [DOI] [PubMed] [Google Scholar]
- 23.Qiang Z, et al. , Curcumin regulates the miR-21/PTEN/Akt pathway and acts in synergy with PD98059 to induce apoptosis of human gastric cancer MGC-803 cells. Journal of International Medical Research, 2019. 47(3): p. 1288–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clemons AP, et al. , Cerulein-induced acute pancreatitis in the rat is significantly ameliorated by treatment with MEK1/2 inhibitors U0126 and PD98059. Pancreas, 2002. 25(3): p. 251–9. [DOI] [PubMed] [Google Scholar]
- 25.Basu S, et al. , Nanoparticle-mediated targeting of MAPK signaling predisposes tumor to chemotherapy. Proceedings of the National Academy of Sciences, 2009. 106(19): p. 7957–7961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naguib YW, et al. , An injectable microparticle formulation for the sustained release of the specific MEK inhibitor PD98059: in vitro evaluation and pharmacokinetics. Drug Delivery and Translational Research, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naguib YW, et al. , An Injectable Microparticle Formulation Provides Long-Term Inhibition of Hypothalamic ERK1/2 Activity and Sympathetic Excitation in Rats with Heart Failure. Molecular Pharmaceutics, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mekkawy AI, et al. , Paclitaxel anticancer activity is enhanced by the MEK 1/2 inhibitor PD98059 in vitro and by PD98059-loaded nanoparticles in BRAF(V600E) melanoma-bearing mice. Int J Pharm, 2021. 606: p. 120876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fang J, Nakamura H, and Maeda H, The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Advanced Drug Delivery Reviews, 2011. 63(3): p. 136–151. [DOI] [PubMed] [Google Scholar]
- 30.Saraiva C, et al. , Nanoparticle-mediated brain drug delivery: Overcoming blood–brain barrier to treat neurodegenerative diseases. Journal of Controlled Release, 2016. 235: p. 34–47. [DOI] [PubMed] [Google Scholar]
- 31.He C, et al. , Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials, 2010. 31(13): p. 3657–3666. [DOI] [PubMed] [Google Scholar]
- 32.Naguib YW, et al. , Solid lipid nanoparticle formulations of docetaxel prepared with high melting point triglycerides: in vitro and in vivo evaluation. Mol Pharm, 2014. 11(4): p. 1239–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X, Naguib YW, and Cui Z, In vivo distribution of zoledronic acid in a bisphosphonate-metal complex-based nanoparticle formulation synthesized by a reverse microemulsion method. International journal of pharmaceutics, 2017. 526(1-2): p. 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Santos SD, et al. , PAMAM dendrimers: blood-brain barrier transport and neuronal uptake after focal brain ischemia. J Control Release, 2018. 291: p. 65–79. [DOI] [PubMed] [Google Scholar]
- 35.Ernsting M, et al. , Factors Controlling the Pharmacokinetics, Biodistribution and Intratumoral Penetration of Nanoparticles. Journal of controlled release : official journal of the Controlled Release Society, 2013. 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh R and Lillard JW, Nanoparticle-based targeted drug delivery. Experimental and Molecular Pathology, 2009. 86(3): p. 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klibanov AL, et al. , Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett, 1990. 268(1): p. 235–7. [DOI] [PubMed] [Google Scholar]
- 38.Intracellular Release of 17-β Estradiol from Cationic Polyamidoamine Dendrimer Surface-Modified Poly (Lactic-co-Glycolic Acid) Microparticles Improves Osteogenic Differentiation of Human Mesenchymal Stromal Cells. Tissue Engineering Part C: Methods, 2011. 17(3): p. 319–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaps L, et al. , In Vivo siRNA Delivery to Immunosuppressive Liver Macrophages by α-Mannosyl-Functionalized Cationic Nanohydrogel Particles. Cells, 2020. 9(8): p. 1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang H, et al. , Hybrid Dendrimer Hydrogel/PLGA Nanoparticle Platform Sustains Drug Delivery for One Week and Antiglaucoma Effects for Four Days Following One-Time Topical Administration. ACS Nano, 2012. 6(9): p. 7595–7606. [DOI] [PubMed] [Google Scholar]
- 41.Jingyan L and Cristina S, PLA/PLGA nanoparticles for delivery of drugs across the blood-brain barrier. Nanotechnology Reviews, 2013. 2(3): p. 241–257. [Google Scholar]
- 42.Wiwatchaitawee K, et al. , Enhancement of Therapies for Glioblastoma (GBM) Using Nanoparticle-based Delivery Systems. AAPS PharmSciTech, 2021. 22(2): p. 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao J, et al. , CREKA peptide-conjugated dendrimer nanoparticles for glioblastoma multiforme delivery. Journal of Colloid and Interface Science, 2015. 450: p. 396–403. [DOI] [PubMed] [Google Scholar]
- 44.Jallouli Y, et al. , Influence of surface charge and inner composition of porous nanoparticles to cross blood-brain barrier in vitro. Int J Pharm, 2007. 344(1-2): p. 103–9. [DOI] [PubMed] [Google Scholar]
- 45.Bilati U, Allémann E, and Doelker E, Development of a nanoprecipitation method intended for the entrapment of hydrophilic drugs into nanoparticles. European Journal of Pharmaceutical Sciences, 2005. 24(1): p. 67–75. [DOI] [PubMed] [Google Scholar]
- 46.Annesley TM and Clayton L, Simple Extraction Protocol for Analysis of Immunosuppressant Drugs in Whole Blood. Clinical Chemistry, 2004. 50(10): p. 1845. [DOI] [PubMed] [Google Scholar]
- 47.Pan K, et al. , Efficacy, Pharmacokinetics, Biodistribution and Excretion of a Novel Acylated Long-Acting Insulin Analogue INS061 in Rats. Drug Des Devel Ther, 2021. 15: p. 3487–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Katayama K, et al. , Inhibition of the mitogen-activated protein kinase pathway results in the down-regulation of P-glycoprotein. Mol Cancer Ther, 2007. 6(7): p. 2092–102. [DOI] [PubMed] [Google Scholar]
- 49.Yang E-J and Chang J-H, PD98059 Induces the Apoptosis of Human Cervical Cancer Cells by Regulating the Expression of Bcl2 and ERK2. Biomedical Science Letters, 2011. 17(4): p. 291–295. [Google Scholar]
- 50.Rafiei P and Haddadi A, Docetaxel-loaded PLGA and PLGA-PEG nanoparticles for intravenous application: pharmacokinetics and biodistribution profile. International journal of nanomedicine, 2017. 12: p. 935–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee JS and Kim SH, Dose-Dependent Pharmacokinetics of Tofacitinib in Rats: Influence of Hepatic and Intestinal First-Pass Metabolism. Pharmaceutics, 2019. 11(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Janaszewska A, et al. , Cytotoxicity of Dendrimers. Biomolecules, 2019. 9(8): p. 330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.