

Abstract
Background
People with cystic fibrosis are at increased risk of pulmonary nontuberculous mycobacteria (NTM) disease. Cystic fibrosis transmembrane conductance regulator (CFTR) modulators are associated with reduced lung infection with pathogens like Pseudomonas aeruginosa and Staphylococcus aureus. This association has not been studied with NTM.
Methods
Using encounter-level data from the US Cystic Fibrosis Foundation Patient Registry from 2011 to 2018, we identified individuals aged >12 years with one or more NTM-negative sputum culture and information on receipt of ivacaftor therapy. We used a Cox proportional hazards model to assess the relationship between CFTR modulator usage (any and monotherapy versus combination therapy) and NTM sputum culture positivity, controlling for sex, least severe class of CFTR mutation, receipt of chronic macrolides, age, body mass index and percentage predicted forced expiratory volume.
Results
Out of 25 987 unique individuals, 17 403 individuals met inclusion criteria. During follow-up, 42% of individuals received CFTR modulator therapy, and 23% had incident NTM. The median (interquartile range) time to event was 6.1 (4.0–7.3) years for those ever receiving CFTR modulators compared to 4.0 (1.6–6.5) years in those never receiving CFTR modulators. CFTR modulator use was associated with a significantly reduced hazard of NTM culture positivity (hazard ratio (HR) 0.88, 95% CI 0.79–0.97); there was no significant difference in the hazard between those receiving ivacaftor monotherapy versus combination therapy (combination HR 1.01, 95% CI 0.79–1.23).
Conclusions
CFTR modulator therapy is associated with a decreased risk of NTM positivity in individuals with cystic fibrosis.
Short abstract
Therapeutic use of cystic fibrosis transmembrane conductance regulator (CFTR) modulators is significantly associated with a decreased risk of NTM positivity in individuals with cystic fibrosis https://bit.ly/3GZC74b
Background
Cystic fibrosis is a genetic disease defined by a progressive decrease in lung function which is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene [1]. The CFTR protein acts as a chloride channel whose normal function in the lung allows appropriate mucus development and clearance. When defects in CFTR are present this process cannot occur or is inefficient, leading to increased mucus in the lungs. CFTR mutations can result in reduced or no protein production, incorrect protein processing and trafficking and/or insufficient gating and conduction [2]. There are >1800 known deleterious CFTR mutations [3].
In 2009, a CFTR potentiator was identified that increased the channel opening probability in individuals with G551D class III gating mutations [4], and in 2012, the drug ivacaftor was approved by the United States Food and Drug Administration for the treatment of individuals aged ≥6 years with the specific G551D mutation and in subsequent years for additional mutations such as R117H and for use in younger children. Monotherapy with ivacaftor has no effect in individuals with homozygous F508del CFTR mutations, the most common CFTR mutation (∼70% of individuals), as class II mutations cause misfolding of the CFTR protein and therefore no trafficking of CFTR to the surface of the cell [5]. However, when ivacaftor is given in combination therapy with a CFTR corrector drug, such as lumacaftor or tezacaftor, that assists with protein folding and trafficking, improvement in lung function is observed [6, 7]. These drugs have collectively been referred to as CFTR modulators.
Because of decreased mucus clearing from the lungs and other host defence alterations of defective CFTR function, people with cystic fibrosis are at increased risk of acquiring acute and chronic lung infections with pathogens such as Pseudomonas aeruginosa, Staphylococcus aureus and nontuberculous mycobacteria (NTM). Modulator therapy with ivacaftor has been associated with reduced lung infection with certain pathogens [8, 9]; however, this association has not been studied with NTM. The aim of this study was to assess the risk of developing an NTM-positive culture in individuals receiving CFTR modulator therapy versus those not receiving modulators, controlling for specific CFTR mutations and mono- versus combination therapy.
Methods
We conducted a retrospective cohort study using encounter-level data from the US Cystic Fibrosis Foundation Patient Registry (CFFPR) from 2011 to 2018. Individuals aged <12 years, those who received a lung transplant, died, were positive for Mycobacterium tuberculosis, were never tested for NTM, only had one encounter or had missing CFTR modulator treatment data for all years were excluded from analysis. Receipt of CFTR modulators between 2011 and 2018 was recorded, including whether individuals received ivacaftor monotherapy or combination therapy with lumacaftor or tezacaftor.
Survival analysis was conducted to assess the risk of NTM culture positivity comparing those who received CFTR modulator therapy to those who did not. A subanalysis was done assessing NTM culture-positivity risk by CFTR modulator type (ivacaftor monotherapy versus combination therapy with lumacaftor or tezacaftor), excluding individuals who had received both monotherapy and combination therapy (i.e. switched from one to the other). Species-specific analyses could not be run, due to small numbers in most strata. The first negative NTM culture for an individual was used as the entry date for this study. Time to event was calculated as the number of days from a negative NTM culture to the first instance of a positive culture. CFTR modulator treatment was included in the analysis as a time-varying covariate. Kaplan–Meier plots were created, and a Cox proportional-hazards model was used to assess the risk of having an NTM-positive lung culture using the “survival” package in R [10, 11]. Baseline covariates included in the model were an individual's sex and their least severe CFTR mutation class (class 1/2, class 3 or class 4/5). Encounter-level covariates included receipt of chronic macrolides, whether a mycobacterial culture was done, the number of sputum cultures an individual had up until that encounter, age (12–17 versus ≥18 years), body mass index (BMI) and percentage predicted Global Lung Function Initiative forced expiratory volume in 1 s (FEV1%). The last nonmissing observation for FEV1% was carried forward until the next nonmissing value, then categorised (<40% “severe”; ≥40% to <70% “moderate”; ≥70% to <90% “mild”; ≥90% “normal”) [12]. BMI was also carried forward and categorised, using BMI percentile for individuals aged 12–18 years (<5th “underweight”; ≥5th to <85th “normal”; ≥85th to <95th “overweight”; ≥95th “obese”) and BMI value for individuals aged ≥18 years (<18.5 kg·m−2 “underweight”; ≥18.5 to <25 kg·m−2 “normal”; ≥25 to <30 kg·m−2 “overweight”; ≥30 kg·m−2 “obese”). Logistic regression was used to assess the relationship between years of CFTR modulator usage and NTM positivity, controlling for sex, least severe mutation class, total number of sputum samples, age at end of follow-up, FEV1%, BMI, number of sputum cultures and years on chronic macrolides. Sensitivity analyses were conducted among people who had three or more positive NTM cultures (more likely to have NTM disease), people who provided at least one mycobacterial sample every other year (to control for detection bias) and without controlling for the cumulative number of sputum samples (in the event that sputum samples acted as a mediator for NTM positivity).
Data analysis was conducted using R version 3.6.2 [13]. The study was determined to be not human subject research by the National Institutes of Health Office of Human Subjects Research Protections, and was approved by the Cystic Fibrosis Foundation Patient Registry committee.
Results
From 2011 to 2018 there were 25 987 unique individuals aged ≥12 years with encounters in the CFFPR; 17 403 individuals met inclusion criteria (figure 1). This population was 52% male, and 67% received macrolides at least once during follow-up. At the time of positive NTM culture or end of follow-up, 79% were aged ≥18 years; 54% of individuals were in the normal or mild FEV1% categories; and 24% were overweight or obese. Class 1 mutations were found in 23% of individuals, class 2 in 86% (including 45% who were homozygous for the F508del mutation), class 3 in 5.8%, class 4 in 6.1% and class 5 in 6.4% (table 1). During follow-up, 42% of individuals received modulator therapy (of these 7384 people, 26% received ivacaftor monotherapy during the study and 79% received combination therapy). 23% of individuals had incident NTM (of these, 30% of positive cultures grew Mycobacterium abscessus only, 51% Mycobacterium avium complex only, 18% other only and 1.6% were mixed species). Median number of sputum samples was three for NTM-positive individuals and five for NTM-negative individuals. NTM was cultured from 10% of people who received CFTR modulator therapy and 33% of people who did not. Median (interquartile range (IQR)) follow-up time was 5.0 (2.4–7.0) years from the first negative culture.
FIGURE 1.
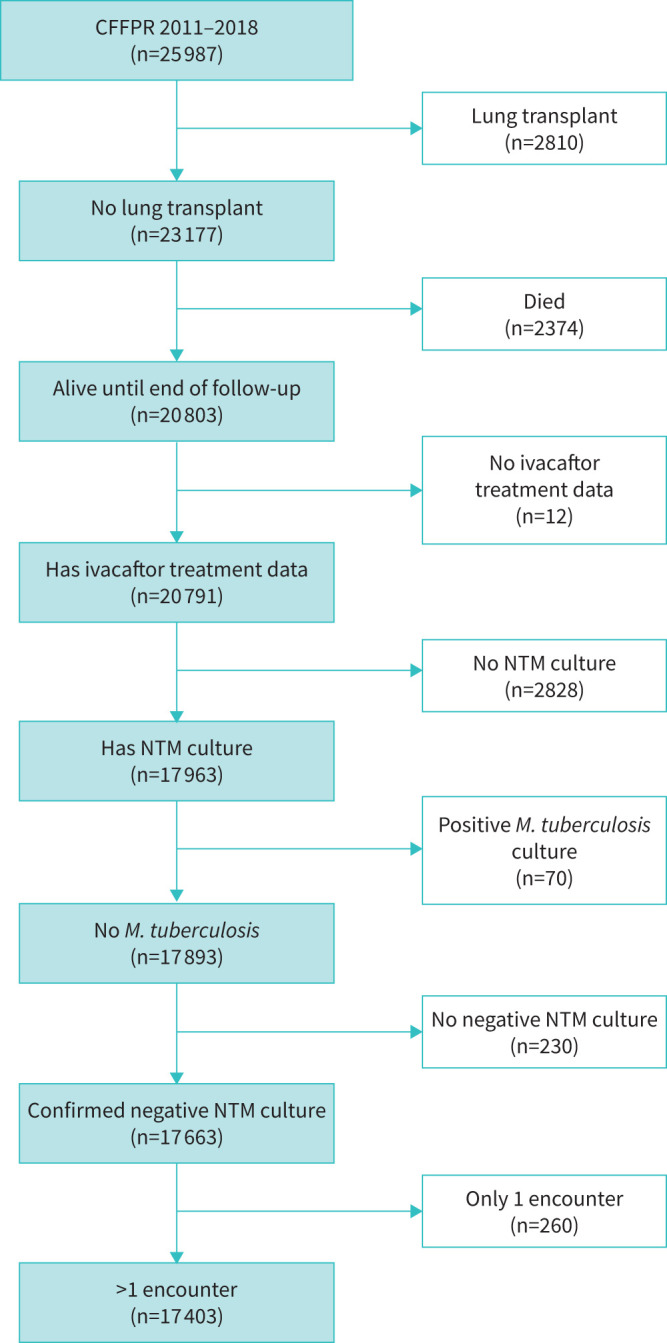
Flowchart depicting inclusion criteria for cystic fibrosis transmembrane conductance regulator (CFTR) modulator analysis from the Cystic Fibrosis Foundation Patient Registry (CFFPR), 2011–2018. These data included only individuals aged ≥12 years. NTM: nontuberculous mycobacteria; M. tuberculosis: Mycobacterium tuberculosis.
TABLE 1.
Individual and encounter-level characteristics at baseline and censoring time
| Baseline | Censored # | |
| Gender | ||
| Male | 9137 (52.5) | |
| Female | 8266 (47.5) | |
| Mutation category ¶ | ||
| Class 1 | 4062 (23.3) | |
| Class 2 | 15043 (86.4) | |
| Class 3 | 1007 (5.8) | |
| Class 4 | 1068 (6.1) | |
| Class 5 | 1120 (6.4) | |
| Unknown | 668 (3.8) | |
| F508del only | 7827 (45.0) | |
| Least severe mutation | ||
| Class 1/2 | 13614 (78.2) | |
| Class 3 | 950 (5.5) | |
| Class 4/5 | 2171 (12.5) | |
| Unknown | 668 (3.8) | |
| Age | ||
| 12 to <18 years | 6554 (37.7) | 3694 (21.2) |
| ≥18 years | 10849 (62.3) | 13709 (78.8) |
| FEV1pp | ||
| Normal | 3923 (22.5) | 4313 (24.8) |
| Mild | 4840 (27.8) | 5125 (29.4) |
| Moderate | 4959 (28.5) | 5597 (32.2) |
| Severe | 1210 (6.9) | 2256 (13.0) |
| Unknown | 2471 (14.2) | 112 (0.6) |
| BMI | ||
| Underweight | 1135 (6.5) | 1282 (7.4) |
| Normal | 11517 (66.2) | 10940 (62.9) |
| Overweight | 2345 (13.5) | 2852 (16.4) |
| Obese | 839 (4.8) | 1045 (6.0) |
| Unknown | 1567 (9.0) | 1284 (7.4) |
| Chronic macrolides | 7620 (43.8) | 8192 (47.1) |
| NTM positive | 4077 (23.4) | |
| Culture positive for MAB only | 1203 (6.9) | |
| Culture positive for MAC only | 2066 (11.9) | |
| Culture positive for other species only | 741 (4.3) | |
| Culture positive for multiple species | 67 (0.4) | |
| Any modulator therapy | 7384 (42.4) | |
| Ever received ivacaftor monotherapy | 1893 (10.9) | |
| Ever received combination therapy | 5869 (33.7) | |
| Neither | 10019 (57.6) |
Data are presented as n (%). FEV1pp: percentage predicted forced expiratory volume in 1 s; BMI: body mass index; NTM: nontuberculous mycobacteria; MAB: Mycobacterium abscessus; MAC: Mycobacterium avium complex. #: censor: NTM-positive culture or patient's last time point in dataset; ¶: ≥1.
The Kaplan–Meier plot shows a higher probability of incident NTM for the group not receiving CFTR modulators compared to those receiving modulator therapy (figure 2a). There was no difference in the probability of incident NTM between individuals receiving ivacaftor monotherapy versus combination therapy (figure 2b). The median (IQR) time to event was 6.1 (4.0–7.3) years for those ever receiving CFTR modulatory therapy compared to 4.0 (1.6–6.5) years in those never receiving modulators. Using an adjusted Cox proportional hazards model, CFTR modulator therapy was associated with a significantly reduced hazard of NTM culture positivity (hazard ratio (HR) 0.88, 95% CI 0.79–0.97). Receipt of combination modulator therapy compared to no therapy was associated with reduced hazard (HR 0.88, 95% CI 0.78–0.98); monotherapy alone was not associated with decreased risk of NTM (HR 0.89, 95% CI 0.73–1.08); nor was there a significant difference in the hazard between those receiving ivacaftor monotherapy versus combination therapy (combination HR 0.99, 95% CI 0.79–1.23). Compared to those whose least severe mutation was class 1/2, those whose least severe mutation was class 4/5 had significantly increased risk of NTM (HR 1.11, 95% CI 1.01–1.23), while those whose mutation class was unknown had a significantly reduced risk of NTM compared to individuals with class 1/2 (HR 0.79, 95% CI 0.65–0.97). The risk of NTM in class 3 was not significantly different from class 1/2 (HR 0.96, 95% CI 0.80–1.15). For every additional year on CFTR modulator therapy, the odds of NTM positivity decreased by 37% (OR 0.60, 95% CI 0.58–0.62), which is a larger effect size than the 16% decrease in the odds of NTM positivity for every additional year on chronic macrolides (OR 0.84, 95% CI 0.82–0.85). These results did not substantively differ in the sensitivity analyses (supplementary table).
FIGURE 2.

Kaplan–Meier plots and 95% confidence intervals evaluating the event probability of patients having incident nontuberculous mycobacteria (NTM) cultures. a) Probability of incident NTM comparing receipt of any cystic fibrosis transmembrane conductance regulator (CFTR) modulator therapy to nonreceipt of CFTR modulator therapy. b) Probability of incident NTM comparing receipt of ivacaftor monotherapy, combination therapy and no modulator therapy.
Discussion
Since the introduction of CFTR modulator therapy, there have been reports of decreased incidence of respiratory pathogens in people with cystic fibrosis who are on this treatment, including P. aeruginosa, S. aureus and Aspergillus spp. [8, 9]. However, trials assessing efficacy of CFTR modulators do not use acquisition of NTM, another important pathogen in the cystic fibrosis community, as an end-point [14], even considering it exclusionary in some trials [15, 16]. This study presents the first evidence that CFTR modulator therapy reduces the risk of infection with pulmonary NTM using data from the CFFPR to assess the incidence and time to first NTM-positive respiratory culture in individuals receiving modulator therapy compared to those who did not.
People with cystic fibrosis are at increased risk of developing pulmonary NTM due to their decreased ability to clear mycobacteria from the lungs, and NTM sputum positivity ranged from 14% to 19% across all ages in the United States in 2019 [12, 17]. In this study, we found that 23% of patients aged >12 years had at least one NTM positive culture during the study period, which is in the range of prevalence estimates found previously among teens and adults with cystic fibrosis in the United States [18, 19]. Because pulmonary NTM leads to progressive lung damage and decreasing pulmonary function, and increases the risk of death [20, 21], reducing the risk of NTM infection is of high priority. One of the frontline treatments for pulmonary NTM is macrolide therapy, which has been associated with improved outcomes in infected patients and reduced risk of infection the longer chronic macrolide therapy is received [18, 20]. However, macrolide resistance is an ever-present concern due to the use of chronic macrolide monotherapy for general disease management in people with cystic fibrosis and the possibility of acquired resistance in NTM [21]. Therefore, prevention of infection with a therapy that is unlikely to result in drug-resistance would be ideal.
Because CFTR modulator use results in increased mucus clearance and pulmonary function, it seems logical that this therapy would reduce the risk of pulmonary NTM; indeed, our results support this hypothesis. Overall, fewer people ever receiving CFTR modulator therapy had an NTM-positive sputum culture compared to those never receiving therapy (10% versus 33%), and time to culture positivity was significantly delayed among those receiving modulator therapy, a difference of 2.1 years. In adjusted analysis, the risk of having an NTM-positive culture decreased by 14%, regardless of whether CFTR monotherapy or combination therapy was received. It is possible that no difference was observed between monotherapy and combination therapy because, while combination therapy has been observed to have a greater therapeutic effect than monotherapy in residual function CFTR heterozygotes [7], it may only take a mild improvement in CFTR function to decrease the risk of NTM culture positivity. Residual function (milder) genotypes were at increased risk of incident NTM in this study. Previous studies showed that both NTM infections and milder CFTR genotypes were significantly more common in patients diagnosed with cystic fibrosis at a later age, likely due in part to greater longevity leading to longer NTM exposure time [18, 19].
One limitation of this study is that while a negative sputum culture was a requirement for inclusion in the analysis, we did not consider whether an individual had been NTM-positive previously, nor did we look at whether ivacaftor is associated with the risk of recurrence or reinfection after the first positive culture. It is possible that because recurrence is at least partially related to structural risk factors such as bronchiectasis, which itself is improved by restoration of CFTR function through modulator therapy, the risk of recurrence and reinfection would also be decreased [22]. Another potential limitation is that we included anyone meeting the study criteria, regardless of their formal eligibility for receipt of CFTR modulator therapy; however, excluding individuals who are presently ineligible to receive any form of modulator therapy would probably strengthen the associations seen in this study.
Conclusions
CFTR modulator therapy is associated with decreased risk of and longer time to NTM culture positivity. It is possible that this decreased risk, along with that seen in other respiratory pathogens could result in reduced treatment burden in people with cystic fibrosis over time.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00724-2021.SUPPLEMENT (16.7KB, xlsx)
Acknowledgements
The authors would like to thank the Cystic Fibrosis Foundation for the use of CF Foundation Patient Registry data to conduct this study. Additionally, we would like to thank the patients, care providers and clinic coordinators at CF centres throughout the United States for their contributions to the CF Foundation Patient Registry.
Provenance: Submitted article, peer reviewed.
Conflict of interest: K.N. Olivier declares a cooperative research and development agreement with Beyond Air, Inc, outside the current work. All other authors declare no competing interests.
Support statement: This research was supported by the Divisions of Intramural Research at the National Institute of Allergy and Infectious Diseases and the National Heart, Lung, and Blood Institute. Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.Riordan JR, Rommens JM, Kerem BS, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989; 245: 1066–1073. doi: 10.1126/science.2475911 [DOI] [PubMed] [Google Scholar]
- 2.Marson FAL, Bertuzzo CS, Ribeiro JD. Classification of CFTR mutation classes. Lancet Respir Med 2016; 4: e37–e38. doi: 10.1016/S2213-2600(16)30188-6 [DOI] [PubMed] [Google Scholar]
- 3.Yu H, Burton B, Huang CJ, et al. Ivacaftor potentiation of multiple CFTR channels with gating mutations. J Cyst Fibros 2012; 11: 237–245. doi: 10.1016/j.jcf.2011.12.005 [DOI] [PubMed] [Google Scholar]
- 4.Van Goor F, Hadida S, Grootenhuis PDJ, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA 2009; 106: 18825–18830. doi: 10.1073/pnas.0904709106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flume PA, Liou TG, Borowitz DS, et al. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest 2012; 142: 718–724. doi: 10.1378/chest.11-2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N Engl J Med 2017; 377: 2013–2023. doi: 10.1056/NEJMoa1709846 [DOI] [PubMed] [Google Scholar]
- 7.Rowe SM, Daines C, Ringshausen FC, et al. Tezacaftor–ivacaftor in residual-function heterozygotes with cystic fibrosis. N Engl J Med 2017; 377: 2024–2035. doi: 10.1056/NEJMoa1709847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heltshe SL, Mayer-Hamblett N, Burns JL, et al. Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor. Clin Infect Dis 2015; 60: 703–712. doi: 10.1093/cid/ciu944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frost FJ, Nazareth DS, Charman SC, et al. Ivacaftor is associated with reduced lung infection by key cystic fibrosis pathogens. A cohort study using national registry data. Ann Am Thorac Soc 2019; 16: 1375–1382. doi: 10.1513/AnnalsATS.201902-122OC [DOI] [PubMed] [Google Scholar]
- 10.Therneau T. A Package for Survival Analysis in R. R package version 3.2-7. 2020. Available from: https://CRAN.R-project.org/package=survival
- 11.Therneau TM, Grambsch PM. Modeling Survival Data: Extending the Cox Model. New York, Springer, 2000. [Google Scholar]
- 12.Cystic Fibrosis Foundation . 2019 Patient Registry Annual Data Report. 2019. Available from: www.cff.org/medical-professionals/patient-registry/
- 13.R Core Team . R: A Language and Environment for Statistical Computing. Vienna, R Foundation for Statistical Computing, 2020. Available from: www.r-project.org/ [Google Scholar]
- 14.Skilton M, Krishan A, Patel S, et al. Potentiators (specific therapies for class III and IV mutations) for cystic fibrosis. Cochrane Database Syst Rev 2019; 1: CD009841. doi: 10.1002/14651858.CD009841.pub3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies JC, Moskowitz SM, Brown C, et al. VX-659–tezacaftor–ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med 2018; 379: 1599–1611. doi: 10.1056/NEJMoa1807119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keating D, Marigowda G, Burr L, et al. VX-445–tezacaftor–ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med 2018; 379: 1612–1620. doi: 10.1056/NEJMoa1807120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martiniano SL, Nick JA, Daley CL. Nontuberculous mycobacterial infections in cystic fibrosis. Thorac Surg Clin 2019; 29: 95–108. doi: 10.1016/j.thorsurg.2018.09.008 [DOI] [PubMed] [Google Scholar]
- 18.Adjemian J, Olivier KN, Prevots DR. Epidemiology of pulmonary nontuberculous mycobacterial sputum positivity in patients with cystic fibrosis in the United States, 2010–2014. Ann Am Thorac Soc 2018; 15: 817–825. doi: 10.1513/AnnalsATS.201709-727OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodman DM, Polis JM, Heltshe SL, et al. Late diagnosis defines a unique population of long-term survivors of cystic fibrosis. Am J Respir Crit Care Med 2005; 171: 621–626. doi: 10.1164/rccm.200403-404OC [DOI] [PubMed] [Google Scholar]
- 20.Binder AM, Adjemian J, Olivier KN, et al. Epidemiology of nontuberculous mycobacterial infections and associated chronic macrolide use among persons with cystic fibrosis. Am J Respir Crit Care Med 2013; 188: 807–812. doi: 10.1164/rccm.201307-1200OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Floto RA, Olivier KN, Saiman L, et al. US Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus recommendations for the management of non-tuberculous mycobacteria in individuals with cystic fibrosis. Thorax 2016; 71: Suppl. 1, i1–i22. doi: 10.1136/thoraxjnl-2015-207360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheikh SI, Long FR, McCoy KS, et al. Computed tomography correlates with improvement with ivacaftor in cystic fibrosis patients with G551D mutation. J Cyst Fibros 2015; 14: 84–89. doi: 10.1016/j.jcf.2014.06.011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00724-2021.SUPPLEMENT (16.7KB, xlsx)