

Abstract
Variants of uncertain significance (VUS) are commonly found following genomic sequencing, particularly in ethnically diverse populations that are underrepresented in large population databases. Functional characterization of VUS may assist in variant reclassification, however these studies are not readily available and often rely on research funding and good will. We present four individuals from three families at different stages of their diagnostic trajectory with recurrent acute liver failure (RALF) and biallelic NBAS variants, confirmed by either trio analysis or cDNA studies. Functional characterization was undertaken, measuring NBAS and p31 levels by Western blotting, demonstrating reduced NBAS levels in two of three families, and reduced p31 levels in all three families. These results provided functional characterization of the molecular impact of a missense VUS, allowing reclassification of the variant and molecular confirmation of NBAS‐associated RALF. Importantly, p31 was decreased in all individuals, including an individual with two missense variants where NBAS protein levels were preserved. These results highlight the importance of access to timely functional studies after identification of putative variants, and the importance of considering a range of assays to validate variants whose pathogenicity is uncertain. We suggest that funding models for genomic sequencing should consider incorporating capabilities for adjunct RNA, protein, biochemical, and other specialized tests to increase the diagnostic yield which will lead to improved medical care, increased equity, and access to molecular diagnoses for all patients.
Keywords: functional genomics, genome sequencing, pediatrics, rapid genomic sequencing, recurrent acute liver failure, variant classification
SYNOPSIS.
Access to rapid genomic and adjunct functional studies will improve clinical care of patients with NBAS‐associated recurrent acute liver failure and other monogenic conditions.
1. INTRODUCTION
Rapid genomic sequencing is revolutionizing management of seriously ill infants and children, with the potential to confirm a molecular diagnosis within a few days with a high degree of clinician‐reported clinical utility. 1 , 2 However, many patients receive uncertain results due to the lack of evidence supporting pathogenicity of rare or novel variants. Reclassification of variants of uncertain significance (VUS) in genes that are phenotypically concordant with the clinical presentation remains challenging in all clinical settings, but particularly when a rapid diagnosis is needed to guide clinical management. Functional characterization of VUS may be required to determine pathogenicity and allow a definitive molecular diagnosis, however these studies are often not readily available, are time consuming, and rely on research funding and good will. Improved access to supplementary adjunct studies allows reclassification of splicing VUS in a clinically meaningful timeframe, 3 a model that may be applied to functional genomics studies.
Biallelic NBAS variants were first associated with short stature, optic nerve atrophy, and Pelger‐Huet anomaly (SOPH, MIM 614800) in 2010, 4 followed by reports of an association between biallelic NBAS variants and recurrent acute liver failure (RALF) or Infantile liver failure syndrome 2 in 2015 (ILFS2, MIM 616483). 5 Since that time, over 100 individuals with biallelic NBAS variants and RALF have been reported, with a mutational spectrum including loss of function, missense, and deep intronic variants. 6 The neuroblastoma amplified sequence (NBAS) protein is a component of the endoplasmic reticulum tethering complex involved in retrograde Golgi to endoplasmic reticulum transport, interacting with a number of intracellular proteins including p31. 5 Disease‐causing pathogenic NBAS variants may result in decreased NBAS protein levels, however some variants, particularly biallelic missense variants, may be associated with normal NBAS protein levels despite altered NBAS function. 7 Previous studies have demonstrated that disease‐causing NBAS variants result in decreased p31 protein levels. 5 , 7
We present clinical, genomic and functional data from three families with biallelic variants in NBAS. Our functional data assisted reclassification of one VUS to likely pathogenic, and two likely pathogenic variants to pathogenic, thus confidently securing molecular diagnoses in these families.
2. METHODS
2.1. Case descriptions
2.1.1. Individual A
An 18‐month‐old female presented with RALF. At 8 months, she was admitted to hospital with vomiting and fever and was found to have alanine aminotransferase (ALT) activity of 739 IU/L (normal range 4–45), which resolved with supportive treatment. Coagulation studies were not performed. At age 11 months, she developed symptoms suspicious for sepsis and was found to have hypoglycemia, hypernatremia, lactic acidosis, coagulopathy, and liver derangement (ALT 9683 IU/L, AST 14760 IU/L, international normalized ratio (INR) 9.9 (normal range 0.8–1.2)). She was treated with intravenous fluids, antibiotics, antivirals, fresh frozen plasma, vitamin K and corticosteroids, and her symptoms resolved over a few days. Adenovirus was isolated from a stool sample. At 18 months, she developed symptoms of a viral respiratory infection and was found to have an ALT of 11 524 IU/L and lactate dehydrogenase (LDH) of 20 823 U/L (normal range 180–300) accompanied by a coagulopathy with an INR of 9.1 and unexplained hypoglycemia. There was no history of an ischemic insult or exposure to toxins. A liver biopsy was performed for diagnostic purposes, as investigations for metabolic and infective causes of liver failure were non diagnostic. Results were nonspecific with severe diffuse acute pauci‐inflammatory hepatocellular injury, with zonal hepatocyte necrosis. Extensive metabolic investigations were nondiagnostic. There was no history of an ischemic insult or exposure to toxins.
She has subsequently experienced three further episodes requiring hospital admission (at 2 years 6 months, 2 years 11 months, and 3 years 2 months), all in the context of viral precipitants. The peak INR at these presentations was 2.0, 1.0, and 2.7, respectively. All episodes resolved with supportive therapy. Apart from these six episodes, she remains otherwise well with normal intervening blood test results. She has normal growth and neurodevelopment with no craniofacial dysmorphism. There is no family history of liver disease.
2.1.2. Individuals B1 and B2
Individual B1 had an antenatal history of intrauterine growth restriction from 32 weeks of gestation. Physical examination showed rhizomelia and large fontanelles. Although reported to be irritable, his neurodevelopment was normal prior to the onset of viral illness with elevated liver enzymes at age 7 months. Hepatitis A and B serology and blood lactate and pyruvate at this time were normal. Subsequently, he had recurrent episodes characterized by initial signs of a viral illness and then deterioration with prostration, drowsiness, and markedly elevated liver enzymes. However, coagulopathy was not documented. Fasting studies showed a blood glucose level of 2.5 mmol/L with B‐hydroxybutyrate increased markedly, reaching 9.1 mmol/L. Succinyl‐CoA transferase and acetoacetyl‐CoA thiolase activities in cultured skin fibroblasts were normal. The episodes would resolve in a few days with liver enzyme levels returning to baseline. He had several admissions to hospital until he died at age of 14 months. Respiratory chain enzymology in skin fibroblasts and skeletal muscle were normal, while liver homogenate results were deficient for complex II in both a biopsy and perimortem sample (17% and 19% of control mean relative to protein and 23% and 16% of control mean when expressed relative to citrate synthase, respectively). His female sibling, individual B2, had a similar presentation with episodic severe RALF with intercurrent viral infections. Unlike her brother, she had no documented hypoglycemia, ketosis or metabolic acidosis. The episodes would resolve in a few days with the liver function enzymes returning to baseline. Fibroblast fatty acid oxidation screen, and specific assays for Long‐chain L‐3‐hydroxyacyl‐CoA dehydrogenase deficiency and short chain l‐3‐hydroxyacyl‐CoA dehydrogenase were normal. She died aged 2 years 9 months during an acute episode. Respiratory chain enzymology in liver was borderline low for complex III and normal for all other enzymes.
2.1.3. Individual C
Individual C has been previously described. 8 Briefly, a 13‐month old female presented with RALF, usually prompted by febrile infections. They were characterized by transaminases being in the tens of thousands, severe coagulopathy with INR up to 9.1 (RR 1.0–1.2), and lactic acidosis with or without hypoketotic hypoglycemia. Urgent liver transplantation was considered. However, with supportive dextrose parenteral infusions, the episodes resolved within 2 weeks and the frequency of RALF reduced with age, with her last episode being at 6 years. Between episodes, laboratory, and imaging findings were normal. Respiratory chain enzymology in liver was normal, while muscle results demonstrated a possible complex II + III deficiency (22% of normal relative to protein, 14% relative to citrate synthase, and 15% relative to complex II). She was treated with coenzyme Q and L‐carnitine. Despite these episodes, her growth and neurodevelopment were normal.
2.2. Genomic sequencing
Genomic sequencing was performed by clinically accredited laboratories at the Victorian Clinical Genetics Services (Melbourne, Australia) (individual A), The Center for Applied Genomics (The Children's Hospital of Philadelphia, USA) (individuals B1 and B2), and Kinghorn Centre for Clinical Genomics (Garvan Institute, Sydney, Australia) (individual C), using genome sequencing (GS) or exome sequencing (ES) techniques. For individual A, GS was performed using massively parallel sequencing (MPS) (Nextera DNA Flex Library Prep kit (Illumina Sequencers, San Diego, CA, USA) with a mean target coverage of 30×, and a minimum of 90% of bases sequenced to at least 10×. Data were processed, including read alignment to the reference genome (GRCh38) and variant calling, using Cpipe 9 or a functionally equivalent analysis with the Illumina Dragen System (Illumina). Variant analysis and interpretation within the selected target region (RefSeq genes ± 1 kb) was performed using Alissa Interpret (Agilent). Variants were annotated against all RefSeq gene transcripts and reported in accordance with HGVS nomenclature. 10 Copy number variants (CNV) were screened for using an internal CNV detection tool. 11 All reported CNVs were orthogonally validated unless otherwise stated. Individuals B1 and B2 had ES performed as previously described. 12 In brief, libraries were constructed and sequenced on an Illumina HiSeq 2000 Sequencing System (Illumina, San Diego, CA, USA). Reads were aligned to reference genome (UCSC hg19), using Burrows–Wheeler alignment (BWA). 13 Genomic variants were then called, including single nucleotide variants (SNVs) and small insertions/deletions (indels), by an integrated in‐house pipeline, using Genome Analysis Tool Kit (GATK, v1.4). 14 In individual C, trio GS was also performed as previously described, 8 , 15 and samples were sequenced on an Illumina HiSeq × Ten sequencer (Illumina, Software, v3.0.29.0). Reads were aligned to the b37d5 reference genome using BWA, 13 sorted using Novosort v.1.03.01 (Novocraft Technologies), then realigned and recalibrated using GATK v.3.3. Variants were identified using GATK HaplotypeCaller v.3.3. 16 Databases were imported into SEAVE, which was used to perform variant filtration and prioritization. 17
Curation of variants was phenotype‐driven with pre‐curated or custom gene lists used for variant prioritization. Variant classification was based on modified ACMG/AMP guidelines. 18
2.3. Cell culture and Western blotting
All fibroblast cell lines were tested for Mycoplasma. Skin fibroblasts were cultured at 37°C in 5% CO2 in Dulbecco's Modified Eagle Medium (DMEM) 3.7 g/L NaHCO3 (HyClone) supplemented with 10% Fetal Bovine Serum (FBS) and 1% penicillin/streptomycin. Protein was extracted from cultured fibroblasts by resuspending in radioimmunoprecipitation assay (RIPA) buffer with protease inhibitor cocktail (Roche), then sonicated and incubated on ice for 30 min before centrifugation at 18000g for 20 min at 4°C. Total protein concentration was determined with the Pierce Bicinchoninic acid (BCA) kit (ThermoFisher) following the manufacturer's protocol. Protein lysates containing 20 μg of protein samples were analyzed by SDS‐PAGE Western blot (BioRad system) using primary antibodies against NBAS (1:500, ThermoFisher, PA5‐103963), USE1 (p31) (1:250 Sigma Aldrich, HPA026851) and GAPDH (1:10 000; Sigma Aldrich, G9545), and detected by anti‐rabbit IgG secondary‐horseradish peroxidase (HRP) conjugated antibody (1:5000, Cell Signaling Technology, #7074S) using Enhanced chemiluminescence reagents (ECL) (GE Healthcare). Protein band intensities were quantified using ImageJ software and normalized to GAPDH.
2.4. Cycloheximide treatment and cDNA studies
To inhibit nonsense‐mediated decay, Case B1 and control fibroblasts at ~70% confluency were treated with 100 ng/μL cycloheximide (Sigma‐Aldrich, #C6255) for 24 h before harvesting. 19 RNA was extracted using the miRNeasy Mini kit (Qiagen) and cDNA was synthesized using the Superscript III kit (Thermo Fisher Scientific) following the manufacturer's protocol. cDNA was amplified by polymerase chain reaction (PCR) using primers designed to sequence NBAS exons 12–15 (5′ACTGAGCATCTGGGCGATTC3′, 5′AGTAATGGTTCGTGGGCGTT3′) and 25–30 (5′GCCTACCAGTGGATGGTTCC3′, 5′CTGGCAGCCAAAACCAAGTC 3′).
3. RESULTS
3.1. Genomic sequencing
3.1.1. Individual A
Ultra‐rapid trio GS, including analysis of the mitochondrial genome, was performed during an acute episode of RALF, identifying compound heterozygous missense variants in NBAS with results available in 71 h. The paternally inherited variant, NM_015909.3(NBAS):c.2951T>G; p.(Ile984Ser), was classified as likely pathogenic according to modified ACMG/AMP criteria. 18 The variant was present in a large population database at a frequency of <0.01% (3 heterozygotes, 0 homozygotes) (gnomAD v2.1.1, 20 https://gnomad.broadinstitute.org/ [accessed 15/11/2019]). It has been previously reported in individuals with RALF. 5 , 6 Computational evidence for pathogenicity is conflicting with uninformative conservation (100 vertebrates, UCSC, 21 https://genome.ucsc.edu/ [accessed 15/11/2019]). There is a large physicochemical difference between isoleucine and serine (Grantham Distance of 142). 22 The variant is located in a well‐established functional domain (Sec39). The maternally inherited variant, NM_015909.3(NBAS):c.406A>G; p.(Arg136Gly), was classified as a VUS according to modified ACMG/AMP criteria. 18 The variant was present in a large population database at a frequency of <0.01% (1 heterozygote, 0 homozygotes) (gnomAD v2.1.1, 20 https://gnomad.broadinstitute.org/ [accessed 15/11/2019]). It has not been previously reported in association with disease. Computational tools predict a deleterious effect of the variant on protein function, and the variant is highly conserved (100 vertebrates, UCSC, 21 https://genome.ucsc.edu/ [accessed 15/11/2019]). There is a large physicochemical difference between arginine and glycine (Grantham Distance of 125). 22 The variant is located in the β‐propeller domain. 23 The phenotype of individual A was considered to be a strong and specific match for the gene of interest (NBAS).
3.1.2. Individuals B1 and B2
Exome sequencing was undertaken decades after death from stored DNA of individual B1, identifying two heterozygous missense variants in NBAS: the NM_015909.3(NBAS):c.2951T>G; p.(Ile984Ser) likely pathogenic variant described in individual A, and a novel variant, NM_015909.3(NBAS):c.1213C>T; p.(Arg405*), classified as pathogenic according to modified ACMG/AMP criteria. The p.(Arg405*) variant was absent from a large population database (gnomAD v2.1.1 20 [accessed 07/10/2020]). It has not been previously reported in association with clinical disease, however many upstream and downstream variants also resulting in a premature termination codon have been reported. 6 cDNA studies confirmed the transcript to be subject to degradation by nonsense mediated decay (NMD) (Figure 1). The phenotype of individual B1 was considered to be a strong and specific match for the gene of interest. Sanger sequencing of his affected sibling B2 identified both variants. Parental DNA was not available for segregation, however cDNA studies in B1 confirmed compound heterozygosity of the variants (Figure 1).
FIGURE 1.
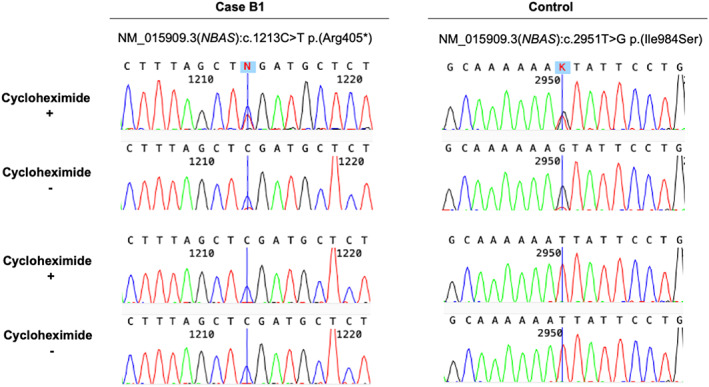
cDNA studies in Individual B1. The NM_015909.3(NBAS):c.2951T>G, p.(Ile984Ser) variant appeared heterozygous when amplifying the cDNA generated from cells with cycloheximide treatment and as homozygous when amplifying the cDNA generated from cells grown without cycloheximide, suggesting that the allele with the p.(Arg405*) is in trans and largely degraded by nonsense‐mediated decay (NMD)
3.1.3. Individual C
ES, GS and cDNA studies were performed, identifying biallelic variants in NBAS as previously described. 8 These variants were previously described in a case report of individual C. 8 Briefly, the maternally inherited variant NM_015909.4(NBAS):c.2617C>T p.(Arg873Trp) was classified as likely pathogenic according to modified ACMG/AMP criteria. The variant was present in a large population database at a frequency of <0.01% (2 heterozygotes, 0 homozygotes) (gnomAD v2.1.1, 20 [accessed 12/10/2020]). Computational evidence for pathogenicity is consistently predicted to be damaging and the variant is highly conserved. There is a large physicochemical difference between arginine and tryptophan (Grantham Distance of 101). 22 The variant is located in a well‐established functional domain (Sec39). The paternally inherited variant, NM_015909.4(NBAS):c.2423+404G>C, is a deep intronic variant and was classified as likely pathogenic. The variant was present in a large population database at a frequency of <0.01% (2 heterozygotes, 0 homozygotes) (gnomAD v2.1.1, 20 [accessed 12/10/2020]). cDNA studies confirmed a splicing defect leading to the inclusion of a pseudo‐exon and premature termination codon leading to nonsense‐mediated RNA decay (NMD). 8 Many upstream and downstream variants also resulting in NMD have been reported. 6 The phenotype of individual C was considered to be a strong and specific match for the gene of interest.
3.2. Patient fibroblasts display decreased levels of p31
Western blotting of fibroblast cell lysates showed decreased NBAS protein expression in individual B1 and individual C compared to control cells (50% and 60% relative reduction, respectively). However, the NBAS protein levels in individual A who carried biallelic missense variants, were similar to controls. There was an overall reduction in p31 levels in all cases compared to controls (Figure 2).
FIGURE 2.
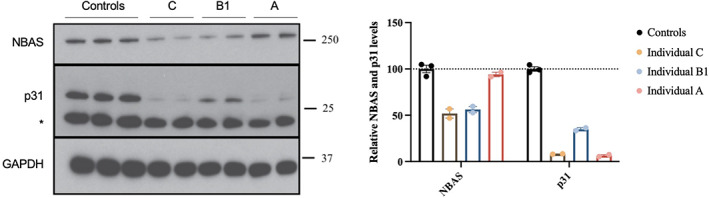
NBAS and p31 protein expression in fibroblasts. Representative Western blot and densitometry analysis suggest NBAS protein levels of 40%–50% of control mean in individual C and B1, and p31 levels of 5% in individual A, 35% in individual B1 and 10% in individual C relative to control mean, GAPDH was used as a protein loading control
3.3. Variant reclassification
The missense VUS (p.(Arg136Gly)) identified in individual A was reviewed by the diagnostic genomic laboratory following completion of the adjunct functional studies, and reclassified as Likely Pathogenic. The missense variant identified in individual A and B, previously classified as Likely Pathogenic (p.(Ile984Ser)), was upgraded to Pathogenic, based on the functional data. Likewise, the p.(Arg873Trp) and NM_015909.4(NBAS):c.2423+404G>C variants identified in individual C could be reclassified to Pathogenic.
4. DISCUSSION
This study describes three families who received a molecular diagnosis of NBAS‐associated RALF, including one individual where adjunct studies were used to reclassify a VUS, highlighting the importance of access to timely functional studies after identification of putative variants, and the importance of selecting appropriate orthogonal analyses to reclassify VUS.
This study raises several important aspects. We have confirmed that biallelic missense variants may lead to normal NBAS protein levels but markedly reduced p31 levels, indicating protein dysfunction, as previously described. 5 , 7 Individual A, with biallelic missense variants, demonstrated normal NBAS protein levels despite evidence of NBAS dysfunction as shown by very low p31 levels. In contrast, individuals B1 and C, who each had a heterozygous missense variant in trans with a nonsense variant and deep intronic splicing variant, respectively, had reduced NBAS levels and reduced p31 levels. These results highlight the importance of selecting the correct functional assay depending on the mutational mechanism of the gene and the variants found, as measurement of NBAS protein levels alone would have been insufficient to allow reclassification of the missense VUS in individual A.
Interestingly, though perhaps not unsurprisingly, each of the families described in this case series had at least one NBAS missense variant affecting the Sec39 domain (Table 1). Variants in the Sec39 domain are more likely to be seen in individuals with a RALF phenotype, while variants in the β‐propeller domain often present with a mixed phenotype. 6 None of the patients in our case series of individuals with NBAS‐related RALF had a variant in the C‐terminal domain, which is more commonly associated with short stature, optic atrophy, and Pelger‐Huët anomaly. 6
TABLE 1.
Clinical and molecular characteristics of individuals with NBAS variants
| Individual | A | B1 | B2 | C |
|---|---|---|---|---|
| Alive | Yes | No | No | Yes |
| Age at last follow‐up | 3 years 3 months | NA | NA | 9 years |
| Age at death | NA | 1 year 2 months | 2 years 9 months | NA |
| Previously published | No | No | No | Yes 8 |
| Facial features | No | NK | NK | No |
| Abnormality of the liver, HP:0001392 | ||||
| Acute liver failure | Yes | Yes | Yes | Yes |
| Elevated hepatic transaminase | Yes | Yes | Yes | Yes |
| Age at onset of first ALF / ELT | 8 months | 7 months | NK | 13 months |
| Growth abnormality, HP:0001507 | ||||
| Intrauterine growth retardation | No | Yes | NK | No |
| Short stature | No | Yes | NK | No |
| Abnormality of the nervous system, HP:0000707 | ||||
| Motor delay | No | No | No | No |
| Intellectual disability | No | No | No | No |
| Other | Irritable but well until 7 months | |||
| Skeletal system, HP:0000924 | ||||
| Reduced bone mineral density | No | NK | NK | No |
| Delayed closure of the anterior fontanelle | No | Yes | NK | NK |
| Abnormality of the vertebral column | No | NK | NK | NK |
| Other | Proximal limb shortening | |||
| Abnormality of the musculature, HP:0003011 | ||||
| Hypotonia | No | No | NK | No |
| Skeletal muscle atrophy | No | No | NK | No |
| Abnormality of the eye, HP:0000478 | ||||
| Optic atrophy | No | NK | NK | No |
| Abnormality of the skin, HP:0000951 | ||||
| Cutis laxa | No | NK | NK | No |
| Abnormality of the immune system, HP:0002715 | ||||
| Reduced IgG levels | No | NK | NK | No |
| Reduced NK cell count | NK | NK | NK | NK |
| Pelger‐Huët anomaly | Occasional | NK | NK | NK |
| Biopsies/RCE | A liver biopsy was nonspecific with severe diffuse acute pauci‐inflammatory hepatocellular injury, with zonal hepatocyte necrosis. | RCE in liver were deficient for CII and borderline low for all other enzymes. | RCE in liver was borderline low for CIII and normal for all other enzymes. | Liver biopsy during an acute episode showed microvesicular steatosis. RCE in muscle showed low levels of CII + III activities. |
| Genomic results | ||||
| Allele 1 |
c.2951T>G; p.(Ile984Ser) (Sec39 domain) |
c.2951T>G; p.(Ile984Ser) (Sec39 domain) |
c.2617C>T p.(Arg873Trp) (Sec39 domain) |
|
| Allele 2 |
c.406A>G; p.(Arg136Gly) (β‐propeller domain) |
c.1213C>T; p.(Arg405*) | c.2423+404G>C | |
Abbreviations: NK, not known; RCE, respiratory chain enzymology.
We note that individuals B1 and B2, and individual C, underwent extensive investigations for a possible mitochondrial disorder before genomic sequencing identified biallelic NBAS variants. Indeed, individual B1 had liver respiratory chain enzyme results suggesting complex II deficiency. We reported previously that complex II is more labile than complexes I, IV and citrate synthase in severe liver disease 24 so caution is needed in interpreting low complex II levels in this scenario, and we regard the low complex II activity in B1 as secondary to liver failure. This emphasizes that NBAS‐associated RALF should be considered as a possible differential diagnosis in individuals with a suspected mitochondrial disorder with prominent liver dysfunction.
The three families presented here represent a spectrum of the diagnostic experience, with the first patient (individual A) receiving ultra‐rapid genomic sequencing results which required functional characterization to confirm pathogenicity of a VUS, the fourth patient (individual C) receiving stepwise genetic results until a molecular diagnosis was achieved, and the second family (individuals B1 and B2) receiving molecular confirmation of a diagnosis over 30 years after death. Ideally, infants and children presenting with RALF and other conditions suspicious for a monogenic disorder should be able to access rapid genomic testing followed by timely adjunct studies (if required) to allow reclassification of VUS in a clinically relevant timeframe. A timely diagnosis of NBAS‐associated RALF can encourage aggressive management of acute presentations, potentially obviating the need for liver transplantation. Currently, these adjunct functional studies are not readily available, are usually performed as part of a research study, are time consuming, are not adequately funded, and rely on the good will of the researchers and clinicians involved. Further, VUS are more frequent in ethnically diverse populations that are underrepresented in large population databases, 25 which further increases inequity. We therefore recommend increasing the availability and funding for timely supportive studies, including RNA, protein, and biochemical studies, to provide functional evidence supporting pathogenicity for VUS detected by genomic sequencing. This will require developing standards and guidelines for integration of functional genomic data into curation pathways and the development of flexible funding models. 26
There are no published management guidelines for NBAS‐related RALF. Based on published case reports and series', NBAS‐related acute liver failure is usually self‐limiting and resolves with conservative and supportive management in most, 7 , 27 , 28 , 29 but not all 30 individuals. Invasive investigations such as liver biopsy are generally not required. Confirmatory diagnosis of NBAS‐related RALF may provide reassurance to treating clinicians and avoid invasive or expensive investigations.
In summary, adjunct functional studies have an important role in assisting with reclassification of VUS identified by genomic sequencing. Funding models for genomic sequencing services should consider incorporating capabilities for adjunct functional studies as part of their service.
CONFLICT OF INTEREST
All authors declare that they have no conflict of interest.
ETHICS APPROVAL AND PATIENT CONSENT
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation and with the Helsinki Declaration of 1975, as revised in 2000 Parents provided written informed consent for participation in the study. Human research ethics committee approvals were obtained from the Royal Children's Hospital Human Research Ethics Committee (HREC/16/RCHM/150, HREC36291A, HREC 2016.224 [Individual A]) and the Children's Hospital at Westmead (HREC 10/CHW/114 [Individuals B1 and B2] and #10/CHW/113 [Individual C]). Parents provided written informed consent for participation in the study.
Akesson LS, Rius R, Brown NJ, et al. Distinct diagnostic trajectories in NBAS‐associated acute liver failure highlights the need for timely functional studies. JIMD Reports. 2022;63(3):240‐249. doi: 10.1002/jmd2.12280
Lauren S. Akesson and Rocio Rius contributed equally to this manuscript and should be considered joint first authors.
Funding InformationThis research was funded by a grant from the Australian Genomics Health Alliance, which is funded by the National Health and Medical Research Council (NHMRC) 1113531 (J.C., D.R.T.) as well as other NHMRC grants and fellowships 1164479 (D.R.T., J.C.); 1155244 (D.R.T.), plus grants from the US Department of Defense Congressionally Directed Medical Research Programs PR170396 (D.R.T., J.C.), the Australian Mito Foundation (A.G.C., D.R.T., J.C.), the Vincent Chiodo Charitable Trust (D.R.T.) and the New South Wales Office of Health and Medical Research Council Sydney Genomics Collaborative grant (J.C.). The Chair in Genomic Medicine awarded to J.C. is generously supported by The Royal Children's Hospital Foundation. The Australian Genomics Health Alliance (Australian Genomics) Acute Care project was funded by grant GNT1113531 from the National Health and Medical Research Council Targeted Call for Research, grant 2017‐906 from the Royal Children's Hospital Foundation, and unspecified grants from the Sydney Children's Hospital Network and the Channel 7 Children's Research Foundation. The research conducted at the Murdoch Children's Research Institute was supported by the Victorian government's operational infrastructure support program. Whole exome sequencing and data analysis for individual B1 and B2 were done in the Center for Applied Genomics at the Children's Hospital of Philadelphia through research funding from Aevi Genomic Medicine Inc. We are grateful to the Crane and Perkins families for their generous financial support.
Communicating Editor: Peter Witters
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Australian Genomics Health Alliance Acute Care F , Lunke S, Eggers S, et al. Feasibility of ultra‐rapid exome sequencing in critically ill infants and children with suspected monogenic conditions in the Australian public health care system. Jama. 2020;323(24):2503‐2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dimmock D, Caylor S, Waldman B, et al. Project baby bear: rapid precision care incorporating rWGS in 5 California children's hospitals demonstrates improved clinical outcomes and reduced costs of care. Am J Hum Genet. 2021;108(7):1231‐1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Akesson LS, Bournazos A, Fennell A, et al. Rapid exome sequencing and adjunct RNA studies confirm the pathogenicity of a novel homozygous ASNS splicing variant in a critically ill neonate. Hum Mutat. 2020;41(11):1884‐1891. [DOI] [PubMed] [Google Scholar]
- 4. Maksimova N, Hara K, Nikolaeva I, et al. Neuroblastoma amplified sequence gene is associated with a novel short stature syndrome characterised by optic nerve atrophy and Pelger‐Huet anomaly. J Med Genet. 2010;47(8):538‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Haack TB, Staufner C, Kopke MG, et al. Biallelic mutations in NBAS cause recurrent acute liver failure with onset in infancy. Am J Hum Genet. 2015;97(1):163‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Staufner C, Peters B, Wagner M, et al. Defining clinical subgroups and genotype‐phenotype correlations in NBAS‐associated disease across 110 patients. Genet Med. 2020;22(3):610‐621. [DOI] [PubMed] [Google Scholar]
- 7. Ono S, Matsuda J, Watanabe E, et al. Novel neuroblastoma amplified sequence (NBAS) mutations in a Japanese boy with fever‐triggered recurrent acute liver failure. Hum Genome Var. 2019;6:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rius R, Riley LG, Guo Y, et al. Cryptic intronic NBAS variant reveals the genetic basis of recurrent liver failure in a child. Mol Genet Metab. 2019;126(1):77‐82. [DOI] [PubMed] [Google Scholar]
- 9. Sadedin SP, Dashnow H, James PA, et al. Cpipe: a shared variant detection pipeline designed for diagnostic settings. Genome Med. 2015;7(1):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. den Dunnen JT. Describing sequence variants using HGVS nomenclature. Methods Mol Biol. 2017;1492:243‐251. [DOI] [PubMed] [Google Scholar]
- 11. Sadedin SP, Ellis JA, Masters SL, Oshlack A. Ximmer: a system for improving accuracy and consistency of CNV calling from exome data. Gigascience. 2018;7(10):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guo Y, Menezes MJ, Menezes MP, et al. Delayed diagnosis of congenital myasthenia due to associated mitochondrial enzyme defect. Neuromuscul Disord. 2015;25(3):257‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li H, Durbin R. Fast and accurate long‐read alignment with burrows‐wheeler transform. Bioinformatics. 2010;26(5):589‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 2010;20(9):1297‐1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Riley LG, Cowley MJ, Gayevskiy V, et al. The diagnostic utility of genome sequencing in a pediatric cohort with suspected mitochondrial disease. Genet Med. 2020;22(7):1254‐1261. [DOI] [PubMed] [Google Scholar]
- 16. DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet. 2011;43(5):491‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gayevskiy V, Roscioli T, Dinger ME, Cowley MJ. Seave: a comprehensive web platform for storing and interrogating human genomic variation. Bioinformatics. 2019;35(1):122‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lamande SR, Bateman JF, Hutchison W, et al. Reduced collagen VI causes Bethlem myopathy: a heterozygous COL6A1 nonsense mutation results in mRNA decay and functional haploinsufficiency. Hum Mol Genet. 1998;7(6):981‐989. [DOI] [PubMed] [Google Scholar]
- 20. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res. 2002;12(6):996‐1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185(4154):862‐864. [DOI] [PubMed] [Google Scholar]
- 23. Carli D, Giorgio E, Pantaleoni F, et al. NBAS pathogenic variants: defining the associated clinical and facial phenotype and genotype‐phenotype correlations. Hum Mutat. 2019;40(6):721‐728. [DOI] [PubMed] [Google Scholar]
- 24. Thorburn DR, Chow CW, Kirby DM. Respiratory chain enzyme analysis in muscle and liver. Mitochondrion. 2004;4(5–6):363‐375. [DOI] [PubMed] [Google Scholar]
- 25. Ricks‐Santi L, McDonald JT, Gold B, et al. Next generation sequencing reveals high prevalence of BRCA1 and BRCA2 variants of unknown significance in early‐onset breast cancer in African American women. Ethn Dis. 2017;27(2):169‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brnich SE, Abou Tayoun AN, Couch FJ, et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 2019;12(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li W, Zhu Y, Guo Q, Wan C. Infantile fever‐triggered acute liver failure caused by novel neuroblastoma amplified sequence mutations: a case report. BMC Gastroenterol. 2020;20(1):308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jiang B, Xiao F, Li X, Xiao Y, Wang Y, Zhang T. Case report: pediatric recurrent acute liver failure caused by neuroblastoma amplified sequence (NBAS) gene mutations. Front Pediatr. 2020;8:607005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Staufner C, Haack TB, Kopke MG, et al. Recurrent acute liver failure due to NBAS deficiency: phenotypic spectrum, disease mechanisms, and therapeutic concepts. J Inherit Metab Dis. 2016;39(1):3‐16. [DOI] [PubMed] [Google Scholar]
- 30. Nazmi F, Ozdogan E, Mungan NO, Arikan C. Liver involvement in neuroblastoma amplified sequence gene deficiency is not limited to acute injury: fibrosis silently continues. Liver Int. 2021;41(10):2433‐2439. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.