

Abstract
The development and continuous optimization of newborn screening (NBS) programs remains an important and challenging task due to the low prevalence of screened diseases and high sensitivity requirements for screening methods. Recently, different machine learning (ML) methods have been applied to support NBS. However, most studies only focus on single diseases or specific ML techniques making it difficult to draw conclusions on which methods are best to implement. Therefore, we performed a systematic literature review of peer‐reviewed publications on ML‐based NBS methods. Overall, 125 related papers, published in the past two decades, were collected for the study, and 17 met the inclusion criteria. We analyzed the opportunities and challenges of ML methods for NBS including data preprocessing, classification models and pattern recognition methods based on their underlying approaches, data requirements, interpretability on a modular level, and performance. In general, ML methods have the potential to reduce the false positive rate and identify so far unknown metabolic patterns within NBS data. Our analysis revealed, that, among the presented, logistic regression analysis and support vector machines seem to be valuable candidates for NBS. However, due to the variety of diseases and methods, a general recommendation for a single method in NBS is not possible. Instead, these methods should be further investigated and compared to other approaches in comprehensive studies as they show promising results in NBS applications.
Keywords: data mining, data preprocessing, data science, deep learning, machine learning, modeling, neonatal screening, pattern recognition
SYNOPSIS.
Machine learning can help to further improve newborn screening programs by reducing the false positive rate and hereby increasing specificity and the positive predictive value as well as identifying so far unknown metabolic patterns within the data.
1. INTRODUCTION
For more than 50 years, newborn screening (NBS) programs aim at early, ideally presymptomatic, identification of treatable rare diseases with significant health burden to reduce morbidity and mortality. With the introduction of tandem mass spectrometry (MS/MS) 1 , 2 and recently genetic methods, NBS panels expanded worldwide 3 , 4 and include many inherited metabolic diseases as well as endocrine, hematological, immune and neurological disorders, and cystic fibrosis. NBS programs refer to the screening principles of Wilson and Jungner 5 which demand a very high sensitivity (ideally 100%) to avoid false negatives and very high specificity (at least 99.5%) to keep the number of false positives low. This is especially challenging in NBS because birth prevalences of the target diseases are very low (1:10 000–<1:1 000 000). 6 Traditional cut‐off‐based approaches in NBS integrate only a fraction of the available information and focus on the primary variables of the metabolic pathway affected in a particular metabolic disease. Here, laboratory physicians are needed to evaluate these findings and workload directly depends on the number of false positives. Moreover, cut‐off‐based methods cannot deal with complex relationships among metabolites. 7 To improve the diagnostic specificity of NBS programs an increasing number of second and multiple tier strategies have been developed combining different biochemical 8 , 9 as well as biochemical and genetic methods. 10 , 11 In contrast to these analytical improvements, mathematical‐based methods are still rarely used to exploit the complete information of NBS test results to improve specificity and positive prediction of NBS. Thanks to advances in data mining and machine learning (ML) as well as the computing landscape in recent years, new opportunities have been created to examine large datasets with high dimensional feature spaces by implementing a ML pipeline for NBS (Figure 1). ML‐based NBS aims at building a classification model, which is part of the essential classification models module to predict the outcome of unknown test data and reduce the number of false positive classifications. The high data imbalance caused by the low birth prevalences of the target diseases makes this task very challenging. Thus, often data preprocessing methods such as data sampling, feature construction, and feature selection are applied before classification. 12 Furthermore, pattern recognition techniques help to detect hidden metabolic interactions within the data. 13 Hence, the goal of this systematic literature review is to present and evaluate current approaches of ML‐based NBS, to find an overall consensus on its applicability, and to provide future research directions.
FIGURE 1.
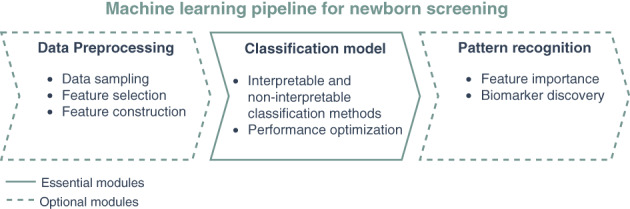
Illustration of machine learning pipeline in NBS. The classification model is the essential part of the ML pipeline in NBS including the interpretable and noninterpretable classification methods and their performance optimization. Data preprocessing is an optional module applied before the classification model. It can include data sampling, feature selection and feature construction methods. Pattern recognition is applied after the classification method evaluating feature importance for biomarker discovery. ML, machine learning; NBS, newborn screening
2. METHODS
This study was conducted and reported according to the preferred reporting items for systematic reviews and meta‐analyses (PRISMA) guidelines (www.prisma-statement.org).
2.1. Research questions
The primary outcome of this systematic literature review was to assess the applicability, advantages and limitations of ML‐based NBS. Therefore, we analyzed published studies according to the following questions:
• Which diseases were investigated in NBS?
• Which data preprocessing methods have been applied in NBS?
• Which ML classification algorithms have been applied in NBS and how did they perform?
• How were pattern recognition techniques implemented in NBS?
2.2. Search strategy
A two‐stage search procedure was conducted to compile relevant papers. In the initial phase, five electronic databases (ScienceDirect, IEEE, ACM, Sage, and PubMed) were searched in May 2021 and October 2021 to collect literature. The search keywords were Newborn Screening AND (Machine Learning OR Deep Learning OR Data Mining). In the second phase, cross‐references from eligible literature of the first phase were searched via Google Scholar and expert advice was added to compile the final literature collection.
2.3. Inclusion and exclusion criteria for study selection
All included studies applied an ML classification method in advanced NBS and were published between January 2000 and September 2021. Studies were excluded if they do not concern NBS, do not use data obtained from MS/MS, or do not apply ML algorithms for disease classification.
2.4. Study eligibility
Duplicates were removed before assessment. First, titles and abstracts were screened and studies not relating to the research question were excluded. Then, full‐text articles were reviewed for inclusion. In case of exclusion, the reason was reported.
2.5. Data extraction and synthesis
Data from all studies, including information on authors, data preprocessing, classification models, performance, and pattern recognition were extracted and summarized in Table 1. For the data analysis we consider key indicators based on their underlying approaches, data requirements, interpretability on a modular level, and performance. The classification performance was evaluated based on the sensitivity, specificity, and positive predictive value (PPV) as summarized in Table 2. These were compared to reference values and other ML methods in comparative studies. For studies lacking sensitivity or specificity values, we calculated these based on the published contingency tables. The studies were insufficient for a meta‐analysis, hence, the findings were synthesized into an overall narrative.
TABLE 1.
Summary of all reviewed studies on applied data imbalance, feature construction, feature selection and ML classification methods
| Author | Disease | Data imbalance | Feature construction | Feature selection | ML classification |
|---|---|---|---|---|---|
| Baumgartner et al. 13 | PKU | Random sampling | Information gain | DT, LRA | |
| Baumgartner et al. 14 | MCADD, PKU | Random sampling | Information gain, relief‐based | LDA, DT, KNN, LRA, NN, SVM | |
| Baumgartner et al. 15 | 3‐MCCD*, MCADD, PKU | Random sampling | Diagnostic flag | DT, LRA | |
| Baumgartner et al. 16 | 3‐MCCD*, PKU, GA1, MMA, PA, MCADD, LCHADD | Random sampling | Discriminatory threshold | KNN, LRA, Naive Bayes, NN, SVM | |
| Ho et al. 12 | MCADD | Informed sampling | Arithmetic ratio | χ 2 | Rule learner |
| Hsieh et al. 17 | MMA | Pearson coefficient | SVM | ||
| Hsieh et al. 18 | MMA | Random sampling | Pearson coefficient | SVM | |
| Van den Bulcke et al. 19 | MCADD | Oversampling | Arithmetic ratio | Variable set optimization | DT, LRA, Ridge‐LRA |
| Chen et al. 20 | PKU | Fisher score | SVM | ||
| Chen et al. 21 | 3‐MCCD*, PKU, MET | Arithmetic ratio | Fisher score, Variable set optimization | SVM | |
| Lin et al. 7 | CIT1, CIT2, CPT1D, GA1, IBDD, IVA, MADD, MET, MMA, MSUD, PA, PKU, PTPSD, SCADD*, VLCADD | Random sampling, oversampling, informed sampling | χ 2, ANOVA, mutual information, L1‐norm, tree‐based | Bagging, Boosting, DT, KNN, LDA, LRA, RF, SVM | |
| Peng et al. 22 | MMA | Oversampling | RF | ||
| Wang et al. 23 | SCADD*, MCADD, VLCADD | Arithmetic ratio | Discriminatory threshold | LRA | |
| Zarin Mousavi et al. 24 | CH | χ 2, information gain, expert consultation | Bagging, Boosting, DT, NN, SVM | ||
| Peng et al. 25 | GA1, MMA, OTCD, VLCADD | Second tier | RF | ||
| Zhu et al. 26 | PKU | Arithmetic ratio | Pearson coefficient, LVQ | LRA | |
| Lasarev et al. 27 | CAH | Informed sampling | PCA | DT |
Note: Diseases with * are biochemical variations nowadays known as nondiseases.
Abbreviations: CAH, congenital adrenal hyperplasia; CH, congenital hypothyroidism; CIT1, citrullinemia type I; CIT2, citrullinemia type II; CPT1D, carnitine palmitoyltransferase I deficiency; DT, decision tree; GA1, glutaric aciduria type I; IBDD, isobutyryl‐CoA dehydrogenase deficiency; IVA, isovaleric aciduria; KNN, K‐nearest neighbors; LCHADD, long‐chain hydroxyacyl‐CoA deficiency; LDA, linear discriminant analysis; LRA, logistic regression analysis; LVQ, learned vector quantization; MADD, multiple acyl‐CoA dehydrogenase deficiency; MCADD, medium‐chain acyl‐CoA dehydrogenase deficiency; 3‐MCCD, 3‐methylcrotonyl‐CoA carboxylase deficiency; MET, hypermethioninemia; MMA, methylmalonic aciduria; MSUD, maple syrup urine disease; NN, neural network; OTCD, ornithine transcarbamylase deficiency; PA, propionic aciduria; PCA, principal component analysis; PKU, phenylketonuria; PTPSD, 6‐pyruvoyl‐tetrahydrobiopterin synthetase deficiency; RF, random forest; Ridge‐LRA, logistic ridge regression; SCADD, short‐chain acyl‐CoA dehydrogenase deficiency; SVM, support vector machine; VLCADD, very long‐chain acyl‐CoA dehydrogenase deficiency.
TABLE 2.
Sensitivity, specificity and positive predictive value (PPV) of considered ML classification methods
| Disease | ML classification | Sensitivity (%) | Specificity (%) | PPV (%) | Author |
|---|---|---|---|---|---|
| (A) Comparative ML classification studies | |||||
| PKU | LRA | 100 | 99.793 | 17.41 | Baumgartner et al. 14 |
| LRA | 98.0 | 99.9 | – | Baumgartner et al. 13 | |
| LRA | 96.809 | 99.905 | 49.46 | Baumgartner et al. 15 | |
| MMA | NN | 98.0 | – | 98.0 | Baumgartner et al. 16 |
| MCADD | Ridge‐LRA | 100 | 99.987 | 33.90 | Van den Bulcke et al. 19 |
| LRA | 96.83 | 99.992 | 88.41 | Baumgartner et al. 14 | |
| LRA | 95.238 | 99.992 | 88.24 | Baumgartner et al. 15 | |
| 3‐MCCD* | LRA | 95.455 | 99.957 | 33.33 | Baumgartner et al. 15 |
| CH | Bagging‐SVM | 73.33 | 100 | – | Zarin Mousavi et al. 24 |
| CIT2, MET, MMA, PKU, SCADD* | SVM | 91.30 | 36.36 | 19.29 | Lin et al. 7 |
| (B) Single ML classification studies | |||||
| PKU | SVM | 100 | 99.997 (99.971) | – | Chen et al. 21 |
| SVM | 100 (100) | 99.98 (99.96) | – | Chen et al. 20 | |
| LRA | 97.66 | 31.61 | 24.59 | Zhu et al. 26 | |
| MMA | SVM | 100 (100) | 100 (99.79) | – | Hsieh et al. 18 |
| RF | 100 (100) | 89.678 (81.226) | 26.40 (16.40) | Peng et al. 25 | |
| RF | 96.117 (96.117) | 65.143 (28.286) | 28.9 (16.5) | Peng et al. 22 | |
| SVM | 95.9 (81.4) | 95.6 (76.2) | – | Hsieh et al. 17 | |
| MCADD | LRA | 100 (100) | 99.988 (99.924) | 18.2 (3.4) | Wang et al. 23 |
| RL | 100 (100) | 99.901 (98.463) | 93.75 (49.18) | Ho et al. 12 | |
| GA1 | RF | 100 (100) | 94.503 (50.751) | 22.30 (3.10) | Peng et al. 25 |
| 3‐MCCD* | SVM | 100 | 99.936 (99.711) | – | Chen et al. 21 |
| MET | SVM | 100 | 99.986 (99.958) | – | Chen et al. 21 |
| VLCADD | LRA | 100 (100) | 100 (100) | 100 (100) | Wang et al. 23 |
| RF | 100 (100) | 92.786 (92.639) | 23.40 (23.10) | Peng et al. 25 | |
| OTCD | RF | 100 (100) | 99.601 (81.983) | 62.10 (3.50) | Peng et al. 25 |
| SCADD* | LRA | 100 (100) | 99.997 (99.974) | 73.3 (22.0) | Wang et al. 23 |
| CAH | DT | 90.909 (100) | 100 (87.194) | 66.7 (20) | Lasarev et al. 27 |
Note: (A) Values of best performing ML classification methods with highest sensitivity and specificity in comparative studies. If presented in the study, these are the results from largest or unknown validation datasets. (B) All results of studies applying a single classification method. If sensitivity and specificity were not stated in the study, the results are calculated based on the published contingency table and given in italics. Results in brackets show comparison to traditional NBS, where given. Diseases with * are biochemical variations nowadays none as nondiseases. The results from Lin et al. 7 are presented in a separate row, since they only report average evaluation results for groups diseases. Most studies applied sampling algorithms, changing the sick‐to‐control ratio, and reduced datasets, such as only including false positive patients from traditional screening. Hence, the performance results and reference values of Table 2 have to be evaluated and compared carefully.
Abbreviations: CAH, congenital adrenal hyperplasia; CH, congenital hypothyroidism; CIT2, citrullinemia type II; DT, decision tree; GA1, glutaric aciduria type I; LRA, logistic regression analysis; MCADD, medium‐chain acyl‐CoA dehydrogenase deficiency; 3‐MCCD, 3‐methylcrotonyl‐CoA carboxylase deficiency; MET, hypermethioninemia; MMA, methylmalonic aciduria; NN, neural network; OTCD, ornithine transcarbamylase deficiency; PKU, phenylketonuria; RF, random forest; RL, rule learner; Ridge‐LRA, logistic ridge regression; SCADD, short‐chain acyl‐CoA dehydrogenase deficiency; SVM, support vector machine; VLCADD, very long‐chain acyl‐CoA dehydrogenase deficiency.
3. RESULTS
Detailed search results of the literature identification process based on the predefined inclusion and exclusion criteria are presented in the PRISMA flow diagram in Figure 2. From the 99 unique publications, we identified 14 as highly relevant. The main reasons for dismissing papers were that they did not apply ML classification methods, 28 investigated other diseases from NBS programs such as hearing disabilities 29 or did not use data obtained from MS/MS. 30 Publications from different screening centers in Europe, 12 , 13 , 14 , 15 , 16 , 19 Asia, 7 , 17 , 18 , 20 , 21 , 23 , 24 , 26 and North America 22 , 25 , 27 are reviewed in this work.
FIGURE 2.
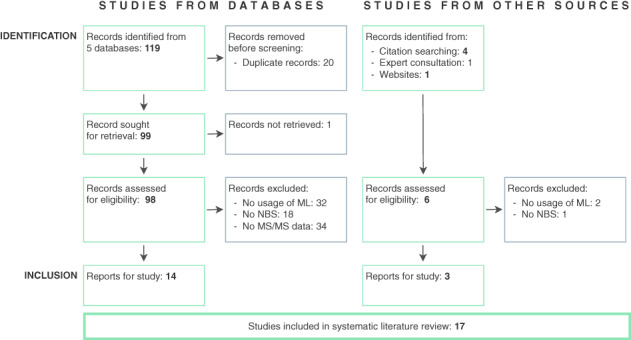
PRISMA flow diagram describing the two‐stage search procedure for studies identified, screened, included, and excluded for this review
3.1. Diversity of NBS disease panels
Studies included in the systematic literature review focused on NBS programs for early detection of inherited metabolic diseases and endocrine disorders in newborns, which endanger the physical and mental development of infected children to an extent. Some studies also included biochemical variations nowadays known as nondiseases benign conditions (i.e. short‐chain acyl‐CoA dehydrogenase deficiency, 3‐methylcrotonyl‐CoA carboxylase deficiency). As they were part of the disease panels, they were not excluded from the analysis, but marked as nondiseases in Table 1. The number of diseases included in NBS programs varies over time and depends on the screening center location. In total, 21 diseases were examined in the reviewed studies, whereby only nine were considered in more than one publication (Table 1). From these, phenylketonuria (PKU), methylmalonic aciduria, and medium‐chain acyl‐CoA dehydrogenase deficiency were the most frequently examined (Table 1).
3.2. Applied data preprocessing methods
Data preprocessing is usually the first step in the ML pipeline (Figure 1) and deals with preparing and transforming the data into a suitable form for classification algorithms. It includes data imbalance, feature construction, and feature selection methods in NBS (Table 1). All of the evaluated studies applied at least one preprocessing method.
3.2.1. Data imbalance
Common methods to overcome data imbalance are sampling methods, which either increase (oversampling) or decrease (undersampling) the data 31 (Figure 3). In NBS, informed sampling is applied to include special subsets of healthy patients. The inclusion is mainly based on clinical criteria such as healthy patients with elevated primary markers, 12 particularly removing samples close to the decision boundary, 7 one‐sided selection, 7 or healthy patients with varying birth weight and gestational age. 27 Other inclusion criteria are based on Tomek links and edited nearest neighbors. 7 Random sampling is applied to change the data imbalance to ratios, for instance, between 1:4 18 and 1:25 14 by randomly excluding data points. In contrast, oversampling methods are applied rarely and create synthetic data samples from the minority class by applying randomness or cluster‐based methods such as synthetic minority oversampling technique (SMOTE) 7 and Borderline‐SMOTE. 7 Furthermore, spiked blood samples which are designed to resemble sick blood samples are added to enrich the datasets 19 and mixed models such as SMOTE + ENN 7 were applied. For studies that applied ML in second tier analysis, the data was less imbalanced since it only contained false positive screening results from the first tier. 25
FIGURE 3.
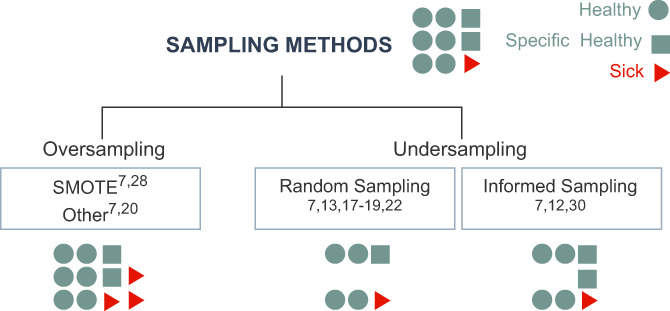
Applied sampling methods. Imbalanced datasets consist of healthy patients (), special subsets of healthy patients (), and sick patients (). Oversampling adds synthetically created sick patients to the dataset. Undersampling methods reduce the number of data points: random sampling randomly excludes healthy patients, informed sampling excludes only specific subsets of healthy patients. In each box the studies applying the respective method are given
3.2.2. Feature construction
Most feature construction methods combine existing numerical features to build new complex features. In NBS, mostly arithmetic operators on original features are used to construct features (Table 1). A new feature can be built from two features by calculating their ratio 19 , 21 , 23 , 26 :
or by combining several original features. 12 Here, can be all original features 12 or a subset of disease‐specific primary markers. 19 Other approaches applied principal component analysis, 27 which computes eigenvectors of the data's covariance matrix, the principal components, which are then used as features or applied self‐developed algorithms 21 to identify relevant features for feature construction.
3.2.3. Feature selection
Feature selection methods aim at identifying the most relevant features and reducing the dimensionality of the feature space. Either a fixed number 18 , 21 or adaptive approaches 12 are applied to decide how many features should be selected. They can be distinguished by their application procedure, before (pre) or after (post) a classification algorithm and are grouped in filter, wrapper, and embedded methods 32 (Figure 4). For NBS, a fourth category, informed methods, was added. Sometimes, several of these methods were applied sequentially 16 , 26 (Table 1).
FIGURE 4.
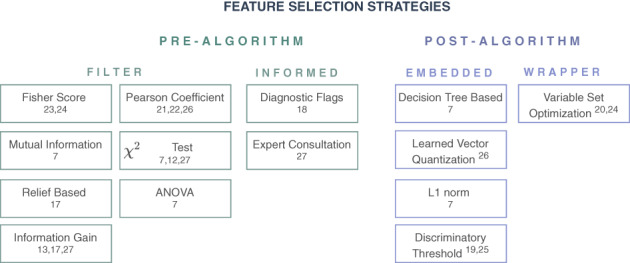
Applied feature selection strategies. Prealgorithm strategies (left) work independent of the ML classification method and are filter methods, using statistical properties, or informed methods, using clinical knowledge. Postalgorithm methods (right) are directly embedded within the classification method or wrapped around it via an iterative loop. In each box the studies applying the respective method are given. ML, machine learning
In NBS, filter methods are frequently applied. They select features based on statistical measures and properties such as analysis of variance (ANOVA), 7 χ 2 tests, 7 , 12 , 24 mutual information, 7 Pearson‐like formula, 17 , 18 , 26 Fisher score, 20 , 21 information gain, 13 , 14 , 24 and relief‐based methods. 14 Informed approaches apply prior knowledge obtained from experts or literature to select relevant features. In NBS, these are established diagnostic flags, which are developed by biochemical and medical experts 15 or important features based on results of consultation with a pediatric endocrinologist. 24
Embedded methods exploit the architecture of the classification method to understand the impact different features have on its performance. In NBS, decision tree splitting rules, 7 the discriminatory threshold from logistic regression analysis (LRA), 16 , 23 learned vector quantization, 26 and underlying cost functions, such as L1 norm 7 were analyzed for feature selection. Wrapper methods choose different subsets of all features 19 or subsets preselected by another method 21 and iterate through the algorithm to detect feature combinations which optimize the performance of the classification method.
3.3. Application and performance of ML classification algorithms
NBS data contain individuals with confirmed diagnosis, hence, only supervised classification methods are applied (Table 1). We grouped these methods according to their interpretability and functionality (Figure 5). There are various definitions of interpretability where we here apply interpretability on a modular level, referring to methods that can inherently explain how parts of the model affect predictions. 33
FIGURE 5.
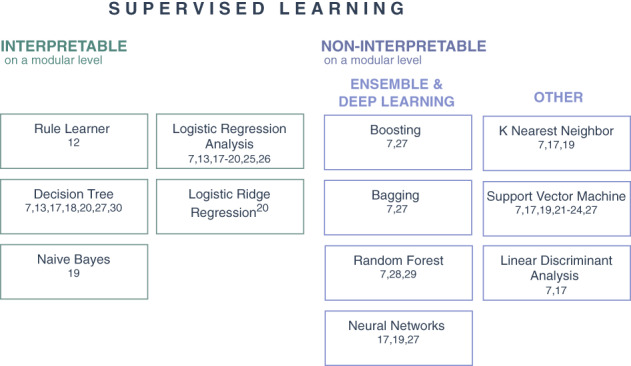
ML classification methods applied in NBS. The methods are distinguished according to their interpretability and functionality. Interpretable methods on a modular level (left) and noninterpretable methods on a modular level (right) which can be split into ensemble and deep learners or other methods. In each box the studies applying the respective method are given. ML, machine learning; NBS, newborn screening
3.3.1. Interpretable methods
LRA 7 , 13 , 14 , 15 , 16 , 19 , 23 , 26 is based on modeling the distribution of discrete dependent features. For instance, the LRA model from Baumgartner et al. 13 for PKU was stated as
where is the amount of the measured phenylalanine concentration and yields the probability of a patient suffering from PKU. Ridge logistic regression analysis (Ridge‐LRA) 19 extends this method by adding a penalty term to the logistic regression function and is applied when independent features are highly correlated. For both methods, the weights of the resulting regression coefficients are the interpretable part of the model. 33 Decision trees 7 , 13 , 14 , 15 , 19 , 24 , 27 are used to subsequently divide the dataset into subsets by applying an impurity index to minimize the impurity of the subsets. They can be interpreted using the splitting decisions and leaf node predictions. Rule learners 12 classify patients by finding interpretable decision rules which can be applied for classifying unseen datasets. Naive Bayes 16 is a probabilistic method based on applying Bayes' theorem with strong independence assumptions. It can be interpreted on a modular level by interpreting the conditional probability through estimating how much each feature contributes to a specific classification.
3.3.2. Noninterpretable methods
Ensemble methods combine several weak learners to obtain one strong classification algorithm and are noninterpretable even if the underlying weak learner is interpretable on a modular level. Random forest 7 , 22 , 25 combines several randomly initialized decision trees to obtain one powerful classifier. Boosting algorithms such as adaptive boosting (ADA), 7 , 24 extremely randomized trees, and gradient boosting are ensemble meta‐algorithms for primarily reducing bias and variance, where each weak learner tries to correct the model predictions of its predecessors. Furthermore, bagging methods such as Bagging‐SVM 7 , 24 were applied as meta‐estimators to train several base‐classifiers on randomly sampled subsets and aggregate the predictions. Neural networks 14 , 16 , 24 try to mimic the signaling processes in the human brain by leading information through multilayer perceptrons. Other methods such as K‐nearest neighbor 7 , 14 , 16 consider the distance between data points and identify clusters of healthy and sick patients within the data. Support vector machines (SVMs) 7 , 14 , 16 , 17 , 18 , 20 , 21 , 24 attempt to find the largest separating band between sick and healthy patients by transforming the features into a higher dimensional space with linear kernels, 7 , 16 radial basis function, 16 , 18 , 20 , 21 or polynomial (degree 2 or 3) 14 , 16 kernels. Linear discriminant analysis 7 , 14 is a linear classifier that aims to find a discriminant line (plane or hyperplane) by fitting weights of features that are optimal for maximizing the between‐class variance.
3.3.3. Performance results
For parameter optimization, grid search is commonly applied 17 , 19 and iterates through a set of parameter combinations returning the combination with the best performance. To test the robustness of the methods and estimate the performance on different subsets, cross‐validation 17 , 21 or stratified cross‐validation 19 and evaluation of receiver operating characteristic curves 25 is applied. The classification performance is evaluated using classification sensitivity, specificity, and PPV, which are calculated using the amount of true positive (TP), false positive (FP), true negative (TN), and false negative (FN) predicted patients,
Here, 100% sensitivity reflects finding all sick newborns and an increasing specificity implies fewer false positive patients. PPV expresses the probability that positively predicted patients are truly suffering from a disease. Table 2 gives an overview of all published results from (A) comparative studies, displaying only the results of the best from several applied classification methods and (B) single studies, applying only one classification method.
Hence, from all 12 considered ML classification methods only LRA, Ridge‐LRA, SVM, Bagging‐SVM, rule learner, neural network, random forest, and decision tree are included in the performance evaluation. From these, LRA and SVM were applied most frequently. In the comparative studies, LRA, Ridge‐LRA, SVM, Bagging‐SVM, and neural networks were the best performing algorithms for different diseases. Moreover, 10 studies reported reference values from traditional NBS on the same datasets. In all of these studies, the ML classification improved the PPV compared to the reference values. Furthermore, applying SVM, random forest, LRA, or rule learners maintained or improved the sensitivity, compared to the reference value in every study. While maintaining the high sensitivity, mostly 100%, all of these methods improved the specificity. Overall, for 9 of the 21 evaluated diseases, ML classification methods achieved 100% sensitivity. Lin et al. 7 presented the most comprehensive study, where SVM, LRA, and linear discriminant analysis showed the best average evaluation results on groups of 5 and 16 diseases. However, when they evaluated single diseases, they showed that also ADA is appropriate for NBS if the dataset includes sufficient patients suffering from a specific disease.
3.4. Implementation of pattern recognition techniques in NBS
Pattern recognition techniques are strongly related to feature selection since they aim to recognize patterns within the features' importances. Therefore, the results of the feature selection methods are often compared to established primary markers 7 and analyzed by clinical and biochemical experts. 15 Furthermore, for interpretable methods, built‐in decision functions 12 , 14 , 15 , 24 and discriminatory thresholds 14 , 16 can be used to identify the biomarkers on which the classifier based its classification. For noninterpretable ML methods, model agnostic approaches such as mean decrease accuracy are applied to identify the individual contribution of specific metabolites. 22 , 25
4. DISCUSSION
Further development and optimization of NBS for inherited metabolic diseases remains an important and challenging task. Based on a systematic review, we identified opportunities and challenges of the whole ML pipeline for NBS, including the application of data preprocessing, classification models and pattern recognition methods.
4.1. Opportunities
The evaluated studies showed that ML methods can be applied for classifying NBS data and presented high classification sensitivity and specificity on several diseases. They were able to decrease the number of false positives compared to reference values from traditional screening, 14 to find metabolic markers without prior assumptions which correspond to the established biochemical knowledge, 13 , 15 and to identify so far unknown metabolic patterns within the data. 14 From all included classification methods, LRA, Ridge‐LRA, SVM, Bagging‐SVM, and neural network achieved best results in comparative studies (Table 2A) indicating that these methods outperform others for NBS classification. Results from single method studies (Table 2B) are more difficult to evaluate as a comparison on the specific dataset is missing. We analyzed the performance of the classification methods based on their frequency of application, ability to achieve 100% sensitivity, a comparison with other classification methods, and reference values. From the reported results, LRA and SVM seem to be valuable candidate methods for NBS classification. Both algorithms are frequently applied in NBS, achieve 100% sensitivity for various diseases in several studies, are the best algorithms in most comparative studies, and can increase the sensitivity, specificity, and PPV compared to reference values from traditional screening (Table 2). Also, advanced versions of these methods such as Bagging‐SVM and Ridge‐LRA achieved the best results in two comparative studies. 19 , 24 Furthermore, we analyzed their interpretability on a modular level, referring to whether they can inherently explain how parts of the model affect the prediction. 33 LRA is interpretable, as the model and weights can be intuitively interpreted. The separating hyperplane of SVM is difficult to interpret on a modular level, particularly, when the original variables are embedded into a higher dimensional space with a kernel trick. 14 , 16 , 18 , 20 , 21
However, the classification methods should not be evaluated on their own, as they are usually part of a whole ML pipeline (Figure 1) including data preprocessing methods, which can further influence the classification performance. Sampling methods can be applied to decrease false positive classifications utilizing expert knowledge on primary markers. Feature construction methods enable to build complex features, which can account for nonlinear correlations spread over several metabolites and discover hidden interactions. 12 , 19 , 23 This can increase the accuracy of LRA and other classifiers such as rule learner methods, which do not perform well for problems requiring diagonal partitioning. 12 , 34
Feature selection techniques are employed to eliminate irrelevant and redundant information for the classification method to reduce dimensionality and overfitting and allow classification algorithms to operate faster and more efficiently. 14 They can reduce the number of positive NBS results and improve sensitivity and specificity in NBS classification. 13 , 20 , 26 Furthermore, pattern recognition showed great potential by confirming primary diagnostic markers and identifying markers without any other a priori assumptions or conditions. 15 Model agnostic pattern recognition can be applied for noninterpretable methods by discovering nonexplainable incidents such as a higher percentage of false positive newborns with Hispanic ethnicity. 22 , 35 This can be especially beneficial for varying prevalence between racial/ethnic groups and populations. 25 , 26 , 35 Furthermore, these methods can help to identify other risk factors such as gender, family disease history, and chronic diseases to identify infants with potential disease risk. 24
4.2. Limitations and future work
The heterogeneity of the 17 studies, including data from 10 screening centers, investigating 21 diseases, applying 12 classification methods and 14 feature selection strategies (Table 1) makes an evaluation of the results challenging, and requires a careful interpretation.
4.2.1. Preprocessing methods
Sampling methods are a promising approach to handle the data imbalance in NBS. However, oversampling methods could pose a problem since it cannot be verified whether the synthetically created samples correspond to a positive confirmation diagnosis. Moreover, sampling methods artificially change the sick‐to‐control ratio of a patient dataset, which could change the model's accuracy on a real population. 13 , 19 Hence, sampling methods should be chosen carefully and evaluated on real populations to verify performance measures in real settings.
Feature selection is applied to support the classification method by identifying relevant features. When deciding which method to choose, several criteria have to be taken into consideration. Prealgorithm methods are independent of the classification method and its respective computational costs. However, they do not take into account the biases of the classifiers which can be problematic when classification methods are highly sensitive to the feature selection procedure. 36 In contrast, postalgorithm methods depend on the specific biases and heuristics of the classification method. This can make them computationally more expensive, as wrapper methods for instance iterate through subsets of all features. Wrapper methods such as mean decrease in accuracy can also be used to rank the relative importance of individual features in a random forest model for pattern recognition. 22 , 25 Furthermore, the applicability for NBS has to be evaluated based on its specific data requirements. NBS has numerical input and categorical output data. However, χ 2 and mutual information expect a categorical input and Pearson's correlation coefficient expects numerical output values whereas ANOVA expects numerical input and categorical output values, which would be most appropriate for NBS. Informed methods allow to include expert knowledge into the feature selection process which can be beneficial for well‐studied diseases but lowers the chances of discovering new metabolic patterns.
4.2.2. ML classification methods
Most studies applied sampling algorithms, changing the sick‐to‐control ratio, and reduced datasets, such as only including false positive patients from traditional screening. Hence, the performance results and reference values in Table 2 have to be evaluated and compared carefully. Classification methods require a certain minimum amount of data to learn the underlying classification processes depending on the task and data. NBS suffers from data limitations due to the rare true positives which lead to diseases being excluded for ML‐based NBS. 14 However, first experimental studies showed methods trained with more than 20 positive patients achieve stable results. 7
Furthermore, in many medical ML applications, noninterpretable methods are state‐of‐the‐art. 37 In NBS, methods such as ensemble learners and neural networks are applied rarely but could surpass interpretable methods in comparative studies. 16 , 24 Reasons for this could be that they are not well‐suited for NBS, or their lack of interpretability makes them less applicable. These points could be addressed in a comparative study including different diseases, classification methods, and datasets, ideally, a benchmark dataset. The results of the study should be analyzed with respect to large variations in parameter settings to estimate the stability of the performance. 19 The lack of interpretability could be addressed by integrating explainable artificial intelligence methods such as SHAP 38 and LIME 39 to explain which metabolites contributed to the algorithm's classification results. Furthermore, the developed ML methods could be applied for future NBS conditions aiming at reducing false negative classification results. Here, feature importance and explainable artificial intelligence methods could be implemented for pattern recognition and could play a key role in identifying so far unknown metabolic patterns within the data. Nevertheless, the proposed biomarkers require further validation and evaluation regarding their biochemical role and underlying biological processes in health and disease. 13 , 16 Although ML methods showed great potential in classifying NBS conditions based on screening data, their reliability has to be proven by thorough validation studies to adhere to regulatory and quality requirements before they can be integrated into NBS programs. Here, explainable AI methods, can contribute to enhance reliability of ML methods for clinical integration.
4.3. Conclusion
Through technical advances, ML‐based NBS enables new opportunities in reducing false positive rates and identifying so far unknown metabolic patterns by relying on complex feature combinations instead of predefined cut‐off values. These mathematical strategies should be regarded as complementary to the combined use of biochemical and genetic tests aiming at improving the diagnostic specificity of NBS programs through second and multiple tier analysis. However, due to the variety of diseases and methods, a general recommendation for a single ML method in NBS is currently not possible. Instead, a thorough analysis of different methods is necessary for all applications. Among the presented, LRA and SVM seem to be valuable candidates for NBS classification since they are often applied, achieve high performance in general and in comparative studies, and handle multidimensional data. Comparing both methods, LRA is interpretable on a modular level, whereas SVM is not and therefore, LRA might be more applicable for NBS. Yet, with the rise of ensemble and deep learning methods, also noninterpretable extensions of these methods such as Ridge‐LRA and Bagging‐SVM showed promising results. 19 , 24 In combination with explainable artificial intelligence methods, these noninterpretable methods could be applied more frequently, which will be investigated in comprehensive future studies.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
All authors have participated in planning, conducting, reporting, and revision of the manuscript before submission.
ETHICS STATEMENT
This study did not require ethical approval or informed consent by patients.
Supporting information
Supplementary Material 1. PRISMA checklist for “Opportunities and Challenges in Machine Learning‐based Newborn Screening ‐ A systematic literature review”
ACKNOWLEDGMENTS
The authors acknowledge the support of the Informatics for Life project funded by the Klaus Tschira Foundation, the Heidelberg Institute for Theoretical Studies (HITS), as well as the Dietmar Hopp Foundation, St.Leon Rot, Germany (Grant numbers 2311221, 1DH2011117 and 1DH1911376). The present contribution is supported by the Helmholtz Association under the joint research school HIDSS4Health – Helmholtz Information and Data Science School for Health. The authors confirm independence from the sponsors; the content of the article has not been influenced by the sponsors.
Open access funding enabled and organized by Projekt DEAL.
Zaunseder E, Haupt S, Mütze U, Garbade SF, Kölker S, Heuveline V. Opportunities and challenges in machine learning‐based newborn screening—A systematic literature review. JIMD Reports. 2022;63(3):250‐261. doi: 10.1002/jmd2.12285
Communicating Editor: Jaak Jaeken
Funding information Dietmar Hopp Stiftung, Grant/Award Numbers: 2311221, 1DH2011117, 1DH1911376; Klaus Tschira Stiftung
DATA AVAILABILITY STATEMENT
This study has no associated data.
REFERENCES
- 1. Wilcken B, Wiley V, Hammond J, Carpenter K. Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N Engl J Med. 2003;348(23):2304‐2312. doi: 10.1056/nejmoa025225 [DOI] [PubMed] [Google Scholar]
- 2. Schulze A, Lindner M, Kohlmüller D, Olgemöller K, Mayatepek E, Hoffmann GF. Expanded newborn screening for inborn errors of metabolism by electrospray ionization‐tandem mass spectrometry: results, outcome, and implications. Pediatrics. 2003;111(6):1399‐1406. doi: 10.1542/peds.111.6.1399 [DOI] [PubMed] [Google Scholar]
- 3. Loeber JG, Platis D, Zetterström RH, et al. Neonatal screening in Europe revisited: an ISNS perspective on the current state and developments since 2010. Int J Neonatal Screen. 2021;7(1):15. doi: 10.3390/ijns7010015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Therrell BL, Padilla CD, Loeber JG, et al. Current status of newborn screening worldwide: 2015. Semin Perinatol. 2015;39(3):171‐187. doi: 10.1053/j.semperi.2015.03.002 [DOI] [PubMed] [Google Scholar]
- 5. Wilson JMG, Jungner G. Principles and practice of screening for disease. WHO Public Health Papers . 1968;34. Accessed February 2, 2022. https://apps.who.int/iris/handle/10665/37650 [Google Scholar]
- 6. Mütze U, Garbade SF, Gramer G, et al. Long‐term outcomes of individuals with metabolic diseases identified through newborn screening. Pediatrics. 2020;146(5):e20200444. doi: 10.1542/peds.2020-0444 [DOI] [PubMed] [Google Scholar]
- 7. Lin B, Yin J, Shu Q, et al. Integration of machine learning techniques as auxiliary diagnosis of inherited metabolic disorders: promising experience with newborn screening data. In: Wang X, Gao H, Iqbal M, Min G, eds. Collaborative Computing: Networking, Applications and Worksharing. Vol 292. Springer; 2019:334‐349. doi: 10.1007/978-3-030-30146-0_23 [DOI] [Google Scholar]
- 8. Gramer G, Fang‐Hoffmann J, Feyh P, et al. Newborn screening for vitamin B12 deficiency in Germany‐strategies, results, and public health implications. J Pediatr. 2019;216:165‐172.e4. doi: 10.1016/j.jpeds.2019.07.052 [DOI] [PubMed] [Google Scholar]
- 9. Monostori P, Klinke G, Richter S, et al. Simultaneous determination of 3‐hydroxypropionic acid, methylmalonic acid and methylcitric acid in dried blood spots: second‐tier LC‐MS/MS assay for newborn screening of propionic acidemia, methylmalonic acidemias and combined remethylation disorders. PLOS One. 2017;12(9):e0184897. doi: 10.1371/journal.pone.0184897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Luo X, Wang R, Fan Y, Gu X, Yu Y. Next‐generation sequencing as a second‐tier diagnostic test for newborn screening. J Pediatr Endocrinol Metab. 2018;31(8):927‐931. doi: 10.1515/jpem-2018-0088 [DOI] [PubMed] [Google Scholar]
- 11. Ruiz‐Schultz N, Sant D, Norcross S, et al. Methods and feasibility study for exome sequencing as a universal second‐tier test in newborn screening. Genet Med. 2021;23(4):767‐776. doi: 10.1038/s41436-020-01058-w [DOI] [PubMed] [Google Scholar]
- 12. Ho S, Lukacs Z, Hoffmann GF, Lindner M, Wetter T. Feature construction can improve diagnostic criteria for high‐dimensional metabolic data in newborn screening for medium‐chain acyl‐CoA dehydrogenase deficiency. Clin Chem. 2007;53(7):1330‐1337. doi: 10.1373/clinchem.2006.081802 [DOI] [PubMed] [Google Scholar]
- 13. Baumgartner C, Baumgartner D, Böhm C. Classification on high dimensional metabolic data: phenylketonuria as an example. Paper presented at: Proceedings of the IASTED International Conference on Biomedical Engineering. 2004. Accessed February 2, 2022. https://www.dbs.ifi.lmu.de/boehm/publications/pku.pdf
- 14. Baumgartner C, Böhm C, Baumgartner D, et al. Supervised machine learning techniques for the classification of metabolic disorders in newborns. Bioinformatics. 2004;20(17):2985‐2996. doi: 10.1093/bioinformatics/bth343 [DOI] [PubMed] [Google Scholar]
- 15. Baumgartner C, Böhm C, Baumgartner D. Modelling of classification rules on metabolic patterns including machine learning and expert knowledge. J Biomed Inform. 2005;38(2):89‐98. doi: 10.1016/j.jbi.2004.08.009 [DOI] [PubMed] [Google Scholar]
- 16. Baumgartner C, Baumgartner D. Biomarker discovery, disease classification, and similarity query processing on high‐throughput MS/MS data of inborn errors of metabolism. J Biomol Screen. 2006;11(1):90‐99. doi: 10.1177/1087057105280518 [DOI] [PubMed] [Google Scholar]
- 17. Hsieh SH, Hsieh SL, Chien YH, Wang Z, Weng YC & Lai F A newborn screening system based on service‐oriented architecture embedded support vector machine. Paper presented at: Proceedings of the 2008 IEEE International Symposium on Service‐Oriented System Engineering. 2008:196‐201. 10.1109/SOSE.2008.58
- 18. Hsieh SH, Chien YH, Shen CP, et al. Newborn screening system based on adaptive feature selection and support vector machines. Paper presented at: 2009 Ninth IEEE International Conference on Bioinformatics and BioEngineering. 2009:344‐347. 10.1109/BIBE.2009.72 [DOI]
- 19. Van den Bulcke T, Vanden Broucke P, Van Hoof V, et al. Data mining methods for classification of medium‐chain acyl‐CoA dehydrogenase deficiency (MCADD) using non‐derivatized tandem MS neonatal screening data. J Biomed Inform. 2011;44(2):319‐325. doi: 10.1016/j.jbi.2010.12.001 [DOI] [PubMed] [Google Scholar]
- 20. Chen WH, Chen HP, Tseng YJ, et al. Newborn screening for phenylketonuria: machine learning vs clinicians. Paper presented at: 2012 IEEE/ACM International Conference on Advances in Social Networks Analysis and Mining. 2012:798‐803. 10.1109/ASONAM.2012.145 [DOI]
- 21. Chen WH, Hsieh SL, Hsu KP, et al. Web‐based newborn screening system for metabolic diseases: machine learning versus clinicians. J Med Internet Res. 2013;15(5):e98. doi: 10.2196/jmir.2495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Peng G, Shen P, Gandotra N, et al. Combining newborn metabolic and DNA analysis for second‐tier testing of methylmalonic acidemia. Genet Med. 2019;21(4):896‐903. doi: 10.1038/s41436-018-0272-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang B, Zhang Q, Gao A, et al. New ratios for performance improvement for identifying acyl‐CoA dehydrogenase deficiencies in expanded newborn screening: a retrospective study. Front Genet. 2019;10:811. doi: 10.3389/fgene.2019.00811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zarin Mousavi SS, Mohammadi Zanjireh M, Oghbaie M. Applying computational classification methods to diagnose congenital hypothyroidism: a comparative study. Inform Med Unlocked. 2020;18:100281. doi: 10.1016/j.imu.2019.100281 [DOI] [Google Scholar]
- 25. Peng G, Tang Y, Cowan TM, Enns GM, Zhao H, Scharfe C. Reducing false‐positive results in newborn screening using machine learning. Int J Neonatal Screen. 2020;6(1):16. doi: 10.3390/ijns6010016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhu Z, Gu J, Genchev GZ, et al. Improving the diagnosis of phenylketonuria by using a machine learning–based screening model of neonatal MRM data. Front Mol Biosci. 2020;7:115. doi: 10.3389/fmolb.2020.00115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lasarev MR, Bialk ER, Allen DB, Held PK. Application of principal component analysis to newborn screening for congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2020;105(8):dgaa371. doi: 10.1210/clinem/dgaa371 [DOI] [PubMed] [Google Scholar]
- 28. Segundo U, Aldámiz‐Echevarría L, López‐Cuadrado J, et al. Improvement of newborn screening using a fuzzy inference system. Expert Syst Appl. 2017;78:301‐318. doi: 10.1016/j.eswa.2017.02.022 [DOI] [Google Scholar]
- 29. Paulraj MP, Subramaniam K, Yaccob SB, Adom AHB, Hema CR. A machine learning approach for distinguishing hearing perception level using auditory evoked potentials. Paper presented at: 2014 IEEE Conference on Biomedical Engineering and Sciences (IECBES). 2014:991‐996. 10.1109/IECBES.2014.7047661 [DOI]
- 30. Hajjar Y, El‐Sayed M, Al Hajjar AES, Daya B. Correlation analysis between EEG parameters to enhance the performance of intelligent predictive models for the neonatal newborn sick effects. Paper presented at: Proceedings of the 9th International Conference on Information Systems and Technologies. 2019:1‐5. 10.1145/3361570.3361615 [DOI]
- 31. Haixiang G, Li Y, Shang J, Mingyun G, Yuanyue H, Gong B. Learning from class‐imbalanced data: review of methods and applications. Expert Syst Appl. 2017;73:220‐239. doi: 10.1016/j.eswa.2016.12.035 [DOI] [Google Scholar]
- 32. Kumar V, Minz S. Feature selection: a literature review. Smart Computing Review. 2014;4(3):211‐229. Accessed February 2, 2022. https://faculty.cc.gatech.edu/hic/CS7616/Papers/Kumar-Minz-2014.pdf [Google Scholar]
- 33. Carvalho DV, Pereira EM, Cardoso JS. Machine learning interpretability: a survey on methods and metrics. Electronics. 2019;8(8):832. doi: 10.3390/electronics8080832 [DOI] [Google Scholar]
- 34. Kotsiantis SB. Machine learning: a review of classification and combining techniques. Artif Intell Rev. 2006;26:159‐190. doi: 10.1007/s10462-007-9052-3 [DOI] [Google Scholar]
- 35. Peng G, Tang Y, Gandotra N, et al. Ethnic variability in newborn metabolic screening markers associated with false‐positive outcomes. J Inherit Metab Dis. 2020;43(5):934‐943. doi: 10.1002/jimd.12236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guyon I, Elisseeff A. An introduction to variable and feature selection. J Mach Learn Res. 2003;3:1157‐1182. doi: 10.1162/153244303322753616 [DOI] [Google Scholar]
- 37. Cabitza F, Banfi G. Machine learning in laboratory medicine: waiting for the flood? Clin Chem Lab Med. 2018;56(4):516‐524. doi: 10.1515/cclm-2017-0287 [DOI] [PubMed] [Google Scholar]
- 38. Lundberg SM & Lee SI A unified approach to interpreting model predictions. Paper presented at: Proceedings of the 31st International Conference on Neural Information Processing Systems. 2017:4768‐4777. 10.5555/3295222.3295230
- 39. Ribeiro MT, Singh S, Guestrin C. “Why Should I Trust You?”: Explaining the Predictions of Any Classifier. Paper presented at: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. 2016:1135‐1144. 10.1145/2939672.2939778 [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material 1. PRISMA checklist for “Opportunities and Challenges in Machine Learning‐based Newborn Screening ‐ A systematic literature review”
Data Availability Statement
This study has no associated data.