

Abstract
Chronic liver injury results in cirrhosis and end-stage liver disease (ESLD) which represents a leading cause of death worldwide, affecting people in their most productive years of life. Medical therapy can extend life, but the only definitive treatment is liver transplantation (LT). However, LT remains limited by access to quality donor organs and suboptimal long-term outcomes. The degeneration from healthy-functioning livers to cirrhosis and ESLD involves a dynamic process of hepatocyte damage, diminished hepatic function, and adaptation. However, the mechanisms responsible for deterioration of hepatocyte function and ultimately hepatic failure in man are poorly understood. We review the current understanding of cirrhosis and ESLD as a dynamic process and outline the current mechanisms associated with the development of hepatic failure from the clinical manifestations to energy adaptations, regeneration, and regulation of nuclear transcription factors. A new generation of therapeutics could target stabilization of hepatocyte differentiation and function to avoid the need for transplantation in patients with cirrhosis and ESLD.
Keywords: cirrhosis, terminal liver failure, hepatocyte reprogramming
Chronic liver disease from cirrhosis is estimated to be present in around 5% of the general population and is the leading cause of liver-related mortality.1,2 Worldwide, complications of cirrhosis account for roughly 1 million deaths annually,3 and as of 2017, yearly deaths associated with chronic liver disease and cirrhosis in the United States exceeded 40,000.4,5 Regional variation in etiology of liver disease is notable, with alcoholic liver disease (ALD) and nonalcoholic fatty liver disease (NAFLD) representing the main causes of cirrhosis in Western and industrialized countries; while viral hepatitis (hepatitis B and C) is the primary cause in East Asian countries such as China and India.3 Importantly, new classes of direct-acting antiviral therapies targeting hepatitis C virus (HCV) protein products have demonstrated real-world cure rates exceeding 95%.6 As a result, the contribution of HCV to the burden of cirrhosis and end-stage liver disease (ESLD) is decreasing. As an example, in 2018, HCV was the primary diagnosis for 10.4% of liver transplant recipients in the U.S., compared with 24% in 2014.7 This success in HCV has at times been overshadowed by the emergence of NAFLD-related complications including nonalcoholic steatohepatitis (NASH). Currently, NASH is the most rapidly growing indication for liver transplantation (LT), combining with ALD as the two most common diagnoses in the U.S. among liver transplant recipients.7–9
LT has become the standard of care for those with an array of liver-based pathology. Once an experimental intervention with dismal outcomes, LT now provides durable life-saving therapy for many with otherwise devastating diseases. Still, challenges persist. Long-term outcomes of LT recipients have not improved significantly as recipients continue to succumb to complications of chronic immunosuppression such as infection, malignancy, and renal failure.10 Furthermore, chronic allograft injury and late graft failure remain significant contributors to morbidity and mortality in LT recipients.11 Even before a transplant occurs, barriers to access exist, most represented by the overwhelming disparity between the need for liver transplant and the shortage of donor organs.12 To address the “organ gap,” innovations have been established and continue to be explored to test the safety and recoverability of various donor organ sources. These include: the use of split-livers or living-related donation and the use of marginal donors, an ill-defined group comprised of donors over the age of 6013; donors with macrosteatosis9; donors with extended cold ischemia time; and nonheart-beating donors.14 However, even as the field continues to improve post-LT outcomes and pre-LT barriers to care, opportunities to avoid transplant all together, or enable extended survival with the native liver, are critical.
Ultimately, a greater understanding of the mechanistic underpinnings that drive development of ESLD is needed to optimize therapies. Historically, “cirrhosis” has at times been used interchangeably with “ESLD.” However, 1-year survival following the diagnosis of cirrhosis is highly variable15,16 suggesting an uncoupling of these terms is warranted and that the establishment of cirrhosis may not portend a definitive poor outcome. More recent efforts have looked to define “cirrhosis” in purely histopathologic terms while “ESLD” is used to describe a subgroup of patients with cirrhosis who have signs of decompensation that is generally irreversible. In those with cirrhosis but without signs of irreversible decompensation, more dynamic terms have been suggested (e.g., advance liver disease) to describe processes that can involve histological regression if the injurious agent is reduced or eliminated (e.g., hepatitis B virus [HBV], HCV, NAFLD).17 New findings at the hepatocellular, metabolic, and genetic level in livers with cirrhosis and terminal failure have changed our understanding of the biophysical environment in which the cells reside.18–21 Here, we review the current understanding of the dynamic degenerative changes that hepatocytes from cirrhotic livers with terminal failure undergo throughout the disease process. New evidence in dynamic changes of energy production, cell contacts, hepatocyte proliferation in response to injury, and inflammation indicates that hepatocytes experience intrinsic transcriptional alterations that result in the clinical features commonly observed in patients with ESLD. Novel concepts of ESLD such as transcriptional reprogramming will also be reviewed. The new paradigm in treating patients with cirrhosis and terminal liver failure will be to augment disease-specific therapy with the targeting of precise mechanisms in damaged hepatocytes to stabilize function and halt pathophysiologic progression, ultimately looking to avoid the need for LT.
Dynamic Evolution and Clinical Manifestations of End-Stage Liver Disease
The dynamic evolution of chronic liver disease that culminates in cirrhosis and ESLD comprises injury, inflammatory response, diffuse fibrosis through the activation of stellate cells, disruption of the normal lobular architecture of the liver with formation of regenerative nodules, and severe disruption of the vascular organization of the liver with loss of hepatocyte mass.16,22,23 Thus, there are myriad potential targets with which to intervene where benefit may be recognized. Phenotypic manifestations from decompensated liver disease reflect the broad metabolic functions of hepatocytes as well as the liver’s unique vascular anatomy, having inflow blood supply from both an arterial (hepatic artery) and venous (portal vein) sources. Symptoms include ascites, sepsis, variceal bleeding, hypoglycemia, coagulopathy, encephalopathy (with and without portosystemic shunting), and hyperbilirubinemia. Each of these clinical manifestations reflect dysfunction of hepatocyte-specific metabolic and synthetic capacities resulting from the fibrotic and inflamed microenvironment. However, most chronic liver disease that precedes ESLD is indolent and asymptomatic until complications from cirrhosis develop and/or hepatocyte dysfunction occurs.4,5 Therefore, prompt recognition and timely targeted intervention are most likely to optimize outcomes. Important initial changes are related to microvascular degeneration, characterized by remodeling in capillaries of the hepatic sinusoid (due to the extracellular matrix [ECM] deposition) and hepatic endothelial dysfunction. This endothelial dysfunction (characterized by insufficient release of vasodilators and increased production of vasoconstrictors)24 causes splanchnic vasodilatation and increases inflow of blood into the portal venous system resulting in an increased pressure.16 These effects concomitantly cause fluid and electrolyte disturbances, reduce the effective systemic blood volume, and can induce various extrahepatic complications such as hepatorenal syndrome (HRS) with deteriorating kidney function due to a reduction in kidney perfusion.25 Fluid balance remains an extremely challenging element of cirrhosis and ESLD. Ascites develops as a result of splanchnic vasodilation in combination with decreased arterial blood volume, which activates the renin-angiotensin-aldosterone system and prompts sodium retention.26 The presence of ascites increases 1-year mortality of liver diseases to 20%,15 and medically refractory ascites is often used to justify LT.27 However, prior to LT, surgical shunting via transjugular intrahepatic portosystemic shunt (TIPS), distal splenorenal shunt, or mesocaval shunt may be offered to increase transplant-free survival.28,29 Notably, these interventions do not affect the progressive nature of any particular underlying liver-based pathology, but merely act to decompress the portal vascular system in an effort to attenuate complications of portal hypertension.
Unpalliated portal hypertension from cirrhosis also increases the hepatic resistance to portal blood flow, driving the formation of portosystemic collaterals which enables abnormal shunting of portal blood directly to the systemic circulation, effectively bypassing the liver. The effects of this abnormal blood flow are wide ranging and can lead to various extrahepatic physical manifestations including hepatic encephalopathy (HE),16 hepatopulmonary syndrome,30 portopulmonary hypertension,31 and cirrhotic cardiomyopathy.32 A more common, and potentially devastating complication of cirrhosis and portal hypertension is the presence of esophageal varices which are well established to negatively impact clinical outcomes33 and increase the risk for decompensation and mortality.34
HE, in particular, is a devastating condition caused by ammonia accumulation in the systemic blood due to decreased urea synthesis and/or portal blood bypassing the liver (shunting). Regardless of etiology, when HE manifests in cirrhosis it portends a worrisome clinical course, with significant increases in 1-year mortality.35 Critically, HE has been generally considered a contraindication to decompressive surgical shunt procedures given the likelihood of exacerbation as more blood flow is subsequently rerouted to circumvent the liver.
Finally, patients with liver disease are known to be at increased risk for the development of systemic infections. This is in part related to the liver’s central role as an immunological organ with a high exposure to circulating antigens and endotoxins from the gut microbiota.36 Within the microenvironment of cirrhosis, key elements of immune responses are impaired, including antigen presentation capacity of monocytes and decreased phagocytic function of macrophages that are pivotal for antibacterial immune defense.37 As a result, the course of advanced cirrhosis, regardless of its etiology, is complicated by cirrhosis-associated immune dysfunction and this constitutes the pathophysiological hallmark of an increased susceptibility to bacterial infection, distinctive of the disease.37 Infections in liver cirrhosis have a poor prognosis with a 30% one-month mortality38 with the most common infections being bacterial peritonitis, urinary tract infections, pneumonia, and skin infections.39,40
Liver fibrosis has been defined as the pathological response to chronic injury to the liver that is formed by an excess of ECM. Cirrhosis, the final stage of liver fibrosis, is described histologically by the formation of parenchymal nodules and matrix cross-linking leading to vascular remodeling with portal hypertension and changes in hepatic metabolism.41 The space of Disse is filled with scar tissue and endothelial fenestrations are lost.42 Through liver fibrosis the total hepatocyte mass decreases, reducing the number of metabolically active hepatocytes. Liver fibrosis is potentially reversible if the inflammation stops43 as has been shown in patients with HBV, HCV, alcohol intake, and NAFLD. Histopathology has been the gold standard for diagnosis of liver fibrosis and cirrhosis. Depending on the underlying disease different histological scoring systems have been developed; the two most common being the METAVIR system and the Ishak score (►Table 1 ).44,45 Importantly, the histological analysis does not have a classification beyond cirrhosis, and to the extent that one looks to objectively assess ESLD; histology does not provide functional information.
Table 1.
Scoring systems for histologic fibrosis
| METAVIR score | F0 | F1 | F2 | F3 | F4 | ||
|---|---|---|---|---|---|---|---|
| No fibrosis | Fibrosis expansion of portal zones | Fibrosis expansion of most portal zones, occasional bridging | Marked bridging, occasional modules | Cirrhosis | |||
| Ishak Score | 0 | 1 | 2 | 3 | 4 | 5 | 6 |
| No fibrosis | Fibrosis expansion of some portal areas | Fibrous expansion of most portal areas | Portal to portal bridging | Portal to central bridging | Occasional nodules | Cirrhosis | |
| Representative histological photographs per scoring |
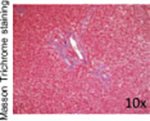
|
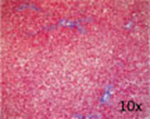
|
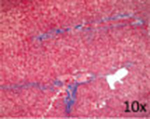
|
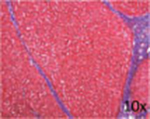
|
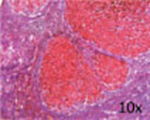
|
||
In order for clinicians to better gauge overall liver function and predict outcomes, numerous modeling tools have been developed using various objective data (i.e., biochemical parameters, portal pressure measurements) often in combination with clinical course assessments.46 Serum-based biomarkers are used to correlate directly with the ability of the hepatocyte to produce proteins or metabolize substrates. For instance, a damaged or dysfunctional hepatocyte will not be able to effectively produce various liver-specific proteins, including albumin and those critical to effective coagulation. Clinically, this will be reflected as hypoalbuminemia and ascites as well as prolonged bleeding times (elevated prothrombin time and international normalized ratio [INR]). Hyperammonemia with resultant HE, as previously noted, can occur with portosystemic shunting, but can also reflect decreased urea metabolism. Hypoglycemia and conjugated hyperbilirubinemia reflect additional metabolic and secretory mechanisms impacted by a damaged liver. These clinical parameters, and their physical manifestations, constitute many of the objective markers which comprise the clinical prediction modeling tools used in clinical practice. The two most commonly used tools include the Child-Turcotte-Pugh (CTP) classification47–50 (►Table 2) and the Model for End-stage Liver Disease (MELD) score51,52 (►Table 3). CTP was initially developed to predict perioperative risk of patients with liver disease and is now commonly used to categorize the severity of liver disease independently from the underlying cause.47 The CTP score gives points for bilirubin levels, albumin levels, INR, the presence of ascites, and the presence of encephalopathy. Notably, encephalopathy and ascites were found to be inherently subjective with categorizations such as “mild,” “moderate,” and “severe” used to identify severity and allocate points toward the overall CTP score. Points were additionally allocated based on the degree of measured dysfunction of the other markers with three “classes” (A, B, and C) ultimately being identified; class C having the highest perioperative mortality. In the early days of transplantation, it was the CTP classification, combined with time on the waitlist, that was used to allocate organs to recipients in need. However, the Department of Health and Human Services issued their “Final Rule” mandate in 2000 which tasked the transplant community to both eliminate waiting time and the subjective variables that were being used in organ allocation. Thus, the MELD was developed. Initially described to predict outcomes following TIPS procedures,53 the MELD score was adapted and found to be effective in predicting 3-month mortality of patients awaiting liver transplant. The subjective measures of ascites and HE were removed, replaced by a tool which included only the objective markers of bilirubin, INR, and serum creatinine (reflecting the negative impact that the HRS has on patients with ESLD). The MELD score ranges from 6 to 40 and is currently used to determine how urgently a patient may need LT. Later iterations have suggested adding sodium measurements (MELD-Na) or removing the 40-point max score as potential changes to improve the clinical capabilities; however, neither has yet to be fully accepted by the transplant community.54
Table 2.
Child-Turcotte-Pugh classification
| Child-Turcotte-Pugh score | Perioperative mortality | ||
|---|---|---|---|
| A | < 6 points | 10% | |
| B | 7–9 points | 30% | |
| C | > 9 points | 70–80% | |
| Measure | 1 point | 2 points | 3 points |
| Bilirubin (mg/dL) | < 2 | 2–3 | > 3 |
| Albumin (g/dL) | > 3.5 | 2.8–3.5 | < 2.8 |
| INR | < 1.7 | 1.71–2.30 | > 2.30 |
| Ascites | Mild | Moderate | Severe |
| Hepatic encephalopathy | Absent | Grade I-II | Grade III-IV |
Abbreviation: INR, international normalized ratio.
Table 3.
Model for End-stage Liver Disease (MELD) scoring system
| MELD score | 3-Month mortality |
|---|---|
| 6–9 | 1.9% |
| 10–19 | 6% |
| 20–29 | 19.6% |
| 30–39 | 52.6% |
| 40 | 71.3% |
| MELD score (6–40) | |
| (0.967*1oge(creatinine (mg/dL)) + 0.378 × loge(bilirubin (mg/dL)) + 1.120 × loge(INR) + 0.6431) × 10 | |
Abbreviation: INR, international normalized ratio.
Clinical manifestations in patients with ESLD are directly related to specific alternated metabolic pathways in failing hepatocytes and these pathways are regulated by specific genes and transcription factors. Current knowledge regarding control of cellular gene expression programs has had an important impact on our understanding of misregulation of gene expression in disease. For instance, it is well known that transcription factors are key to control cell status and they can function as reprogramming factors and control transcription initiation of the genes they regulate.55 Thus, genes and transcription factors can be targeted to treat the symptoms of patients with liver failure. Next, we will delineate the metabolic changes of hepatocytes with ESLD and introduce novel potentially therapeutical options.
Let’s Dig Down Deep into End-Stage Liver Disease
The process of how fibrosis evolves to cirrhosis and how ECM is produced in the liver through stellate cell activation has been well studied for decades as the cornerstone of ESLD.56 Numerous clinical trials testing antifibrotic molecules have been performed targeting different mechanisms of hepatic stellate activation with limited or controversial results.57,58 However, the mechanisms responsible for deterioration of hepatocyte function and ultimately hepatic failure are largely unknown. Several areas of investigation have been proposed to explain loss of hepatocyte function, resulting in the phenotype of ESLD. While the entirety of the picture remains underdeveloped, recent efforts have brought some clarity. Here, we review the cellular and molecular events that contribute to the development of ESLD in cirrhosis, as represented in ►Figs. 1 and 2.
Fig. 1.

Overview of end-stage liver disease (ESLD). (A) Several insults can lead to the loss of hepatic functions: There is a switch in energy metabolism from oxidative phosphorylation to glycolysis and a reduction in the mitochondrial content. The accumulation of collagen fibers in the extracellular matrix (ECM) leads to a loss of hepatocyte–hepatocyte contacts resulting in alterations in several pathways decreasing cell proliferation and liver regeneration. A reduced proliferative activity and liver regeneration can also be explained by the shortening of the telomere and a reduced activation of telomerase reverse transcriptase (TERT). The inflammatory milieu causes cell death and a loss in hepatic functions. An alteration in the transcription factor network, mainly caused by the downregulation and mislocalization of hepatocyte nuclear factor 4 alpha (HNF4α), contributes to hepatocyte dysfunction and clinical manifestations like reduced albumin production, reduced bilirubin and urea metabolism, and fewer coagulation factors. (B) With chronic injury the histopathologically healthy liver undergoes changes especially through the accumulation of extracellular matrix leading to fibrosis. As the disease progresses the liver parenchyma changes histologically into cirrhosis with nodule formation, the formation of scar tissue, and changes in hepatic blood flow leading to portal hypertension, hepatic encephalopathy, coagulopathy, hepatorenal syndrome, and hepatopulmonary syndrome causing the clinical symptoms/signs of ESLD. T2D, type 2 diabetes; NEA, nonessential amino acids.
Fig. 2.

Functional, histopathological, and genetic findings in end-stage liver disease (ESLD). Histologically, cirrhosis (METAVIR Score F4) is considered the terminal stage of liver disease. Histology does not provide functional information beyond the description of cirrhosis. The Child-Pugh score includes functional and symptomatic parameters to determine the mortality of end-stage liver disease. Genetically, ESLD leads to dedifferentiation of the hepatocyte and reduces the nuclear expression of hepatocyte nuclear factor 4 alpha (HNF4α), the “master regulator” of many hepatocellular functions (graph modified from refs. 113 and 114).
Hepatocytes Suffer Energy Changes
To orchestrate all of the biological, metabolic, and synthetic functions that the liver does, hepatocytes require efficient methods to produce energy. Through oxidative phosphorylation, hepatocytes produce adenosine triphosphate (ATP) as their main source of energy in the mitochondria, an organelle that comprises 13 to 20% of the liver volume.59–62 One of the first hypotheses that was reported as a cause of ESLD and hepatocyte functional decompensation was mitochondrial changes in hepatocytes. In 1977, Díaz Gil et al isolated the mitochondrial fraction from liver biopsies of patients with alcoholic cirrhosis, cryptogenic cirrhosis, and chronic hepatitis, and demonstrated a reduction in mitochondria which the authors postulated as the cause of the loss of hepatic function.63 Similarly, Möller and Dargel used an animal model of chronic liver injury and showed a decreased mitochondrial activity.64 In 1989, Krähenbühl et al reported a correlation between hepatocyte death and a reduction in oxygen uptake and mitochondrial enzyme activities.65
For a long time, the reduced number and function of mitochondria in hepatocytes were accepted as the major causes for the loss in hepatic function; however, with advances in analysis of mitochondrial function, the idea that mitochondrial function equates to the energy status of the cell has changed. Nishikawa et al, using a rat model of compensated and decompensated cirrhosis, measured the mitochondrial activity of hepatocytes derived from these animals, and reported that even in the early stages of the liver failure, there was a reduction of mitochondria content and function, but the energy status, measured by ATP production, was similar when compared with normal hepatocytes. This balance of energy was maintained by a switch in the source of ATP production, from oxidative phosphorylation to glycolysis.66 Glycolysis represents a less efficient compensatory mechanism to maintain energy homeostasis during early stages of liver injury, but leads to hepatocyte dysfunction during terminal stages of chronic liver disease because hepatocytes are unable to sustain high levels of energy production from glycolysis.66 Thus, it seems that mechanisms controlling energy homeostasis could be targeted to prolong or control energy production and supply in terminally diseased hepatocytes (►Fig. 1 ).
Hepatocytes are Losing Contacts in End-Stage Liver Disease
Hepatocytes, like many other epithelial cells, have two membrane domains: a luminal and a basolateral side, maintained by the expression of several junctional proteins including anchoring junctions, tight junctions, and gap junctions.67 This configuration is important to keep the hepatocyte’s functionality, since there is a localized expression of proteins related to specific processes in either the luminal or the basolateral membrane.67 For instance, bile formation requires the expression of transporters at the basolateral membrane for the uptake of products that will be converted into bile. Also, at the luminal membrane, known also as apical membrane, specific transporters are needed such as bile salt export pump and familial intrahepatic cholestasis type 1 for the effective secretion of bile salt and subsequent bile formation.68 Any perturbation in this system can lead to a disruption in the process of bile excretion and lead to intrahepatic cholestasis.68 Alteration in hepatocyte polarity is also related to diseases such as type 2 diabetes and NAFLD.69,70
In livers with ESLD, the diffuse presence of extra collagen fibers and the apoptotic process result in a loss of cell–cell contacts, leading to a decreased hepatocyte polarity.67,71 This event contributes to a loss of hepatic function. Some studies have reported a correlation between liver gap junction proteins, specifically the expression of connexins and liver injury.71 A total knockout of connexin 32 (cx32) in a mouse model increased inflammation, oxidative stress and liver injury after 8 weeks of choline-deficient high-fat diet.72 Interestingly, the expression cx43 showed a higher expression in the context of chronic liver disease and it was correlated with a propagation of a death signal mediated by caspase 3 through hepatocytes.73 How these paradoxical events in the connexin expression are correlated mechanistically with hepatocellular failure in cirrhotic livers remains unclear?
The perturbation of hepatic function by the loss of cell–cell contact can be explained partially by an alteration in the calcium (Ca2+) signaling. Several hepatic functions such as hepatocyte proliferation, apoptosis, gene transcription, lipid and glucose metabolism, and others have been reported to be under the control of Ca2+ signaling.74 Leite et al described that the liver has pacemaker cells, as reported in heart.75 The authors showed that the Ca2+ signaling starts in some cells and the signal travels through the lobule by the gap junctions, mediated mainly through the expression of cx32.75 The loss of hepatocellular interaction can alter the Ca2+ signal propagation leading to discoordination, causing dysfunction in hepatocytes on a functional level. The real contribution of the Ca2+ signaling in pathophysiology of liver failure needs to be investigated further.
Hepatocyte Regeneration is Over
The impairment of hepatocyte proliferation contributes to ESLD. It is well known that the liver has the capacity to regenerate and restore its functions after a partial hepatectomy.76 Massive hepatocyte death induced by different kinds of injuries induce a strong proliferation response. However, hepatocytes that reside in cirrhotic livers are largely senescent and cannot be induced to a proliferative state. This phenomenon is supported by the constant expression of markers of cell senescence (p16 and β-galactosidase).77 Moreover, Liu et al also reported that telomere length in hepatocytes derived from a decompensated cirrhotic liver are shorter when compared with healthy hepatocytes.18 This difference is supported by a downregulation of the enzyme telomerase reverse transcriptase (TERT) expression in addition to the decrease of its activity.18 To corroborate these findings and test the regenerative response of hepatocytes from cirrhotic livers with and without terminal liver failure, hepatocytes were transplanted into analbuminemic retrorsine-hepatectomy-preconditioned rats (a combination of interventions that allows a selective long-term survival and repopulation advantage to the engrafted donor hepatocytes). Healthy and cirrhotic hepatocytes without terminal liver failure repopulated the livers as expected in this kind of model. However, the engraftment and proliferation of transplanted cirrhotic hepatocytes with terminal liver failure was considerably lower, indicating the lack of proliferative capacity and intrinsic damage even in a regenerative microenvironment.18 However, eventually normalization of hepatocyte function occurs after a period of months, indicating that the intrinsic hepatocyte damage is reversible and can be influenced by the microenvironment (►Fig. 1).
Several reports have revealed a multifactorial contribution to the pathogenesis of ESLD involving lack of regeneration response by hepatocytes.78–80 Very recently, Paranjpe et al reported that downregulation of important growth factor receptors contributes to a reduction in hepatocyte proliferation.79,81 Mice with a systemic deletion of tyrosine-protein kinase Met and epidermal growth factor receptor, showed an impairment of liver regeneration due to a direct reduced hepatocyte proliferation.79 The authors also reported decrease of hepatocyte metabolism, protein synthesis, and cytochrome P450 activity and a switch from oxidative phosphorylation to glycolysis in hepatocytes.79 The downregulation of the growth factor receptors and the switch of energy source and decreased metabolism found in this animal model has also been confirmed in human livers.20,66,80
Transcriptional Reprogramming of Hepatocytes End-Stage Liver Disease
Liu et al reported the initial hypothesis that refers to the role of transcription factors specific to hepatocytes in the development of ESLD.18 Genome-wide analyses of hepatocytes derived from cirrhotic livers with terminal failure revealed that nuclear factor-kB was altered when compared with healthy hepatocytes. Such hepatic deprograming is evident in deterioration of other signals such as proliferation, regeneration, cell death, and apoptosis, as described previously (►Figs. 1 and 2). An important finding was related to the expression of transcription factors such as hepatocyte nuclear factor 4 alpha (HNF4α). This transcription factor was downregulated as cirrhosis progressed and terminal failure was identified.18
Liver-enriched transcription factors such as HNF4α,HNF1, FOXA, HNF6, and C/EBP and others, are responsible for maintaining the hepatocyte’s characteristics and functions.82,83 HNF4α, known as a master hepatic regulator, is encoded by a gene with two different promoters, which can generate, through alternative splicing, up to 12 isoforms of HNF4α.84 A global knockout of HNF4α is incompatible with life since it is important not only to the liver development, but also for the development of the pancreas and kidney.85,86 The concept of cellular reprogramming through overexpression of master transcription factors was conceived by Shinja Yamanaka and John B. Gurdon, who received the Nobel Prize in Physiology or Medicine in 2012. The overexpression of key transcription factors for stemness: Myc, Sox2, Klf4, and Oct4/Pou5f1, also known as Yamanka’s factors were sufficient to reprogram a mature fully differentiated cell into a pluripotent state, highlighting a new paradigm in cell biology: cellular reprogramming.87 This concept has also been applied in vivo, and such forced gene expression in specific cell types could be used to treat diseases. Heart failure in a mouse model has been treated by three transcription factors, Gata4, Mef2c, and Tbx5, that together are able to convert cardiac fibroblast into functional cardiomyocyte-like cells. After 4 weeks, the heart function was restored by the presence of these reprogrammed cells.88 A similar strategy has been pursued when mouse embryonic and fetal fibroblasts were converted into functional neurons, which were able to generate action potentials and synapses.89 In vivo reprogramming has been performed in the brain by Niu et al, delivering Sox2, a neural transcription factor, enabling astrocytes to transdifferentiate into neuroblasts.90 The forced expression of Ngn3, Pdx1, and Mafa in the pancreas of adult mice converted exocrine cells to insulin secreting functional β cells.91
Thus, the possibility of using hepatocyte-specific transcription factors as a therapy to treat ESLD is conceivable.92 HNF4α is important for the maintenance of hepatic homeostasis and functions18–20; Nishikawa et al forced the expression of HNF4α delivered by an adeno-associated virus (AAV) in a rat model of cirrhosis.92 An increase in nuclear HNF4α expression in hepatocytes was found to improve metabolic functions of hepatocytes, leading to an improvement in albumin secretion and lower serum total bilirubin levels, ascites, and ammonia levels. Notably, the transcriptional reprogramming using HNF4α-AAV did not alter the telomere length, suggesting that the reprogramming acted by phenotypically correcting diseased hepatocytes, rather than inducing hepatocyte growth or regeneration.92
Moreover, with an eye in the clinics, Guzman-Lepe et al showed a decreased expression of HNF4α in hepatocytes correlates with liver dysfunction, the stage of fibrosis, serum levels of total bilirubin, albumin, and prothrombin activity, revealing alterations in gene expression contribute to the development of ESLD in humans.19 Additionally, HNF4α must be expressed and translocated to the cell’s nucleus to be able to bind in the promoter regions of the target genes carrying out its function.84 Recently, Florentino et al showed that there is a correlation between the cellular localization of HNF4α and ESLD. We discovered that HNF4α protein expression is found in the cytoplasm of hepatocytes from explanted human cirrhotic livers with decompensated function. Moreover, we found that hepatic dysfunction correlated directly with a reduction in the nuclear acetylated HNF4α.20 Thus, posttranslational modifications are important for HNF4α localization in the nucleus. These results indicated that localization of HNF4α in the cytoplasm results from alterations of the molecular pathways, which maintain HNF4α in the nucleus during advanced stages of liver disease. Consequently, lack of HNF4α transcriptional activity may be responsible for deterioration of hepatocyte function in human cirrhotic livers with terminal failure (►Fig. 1) The next logical steps for these human studies are to induce the expression of HNF4α and possibly other molecules to re-establish nuclear localization and transcription of HNF4α and its target genes in hepatocytes from explanted human cirrhotic livers with decompensated function.
Several other reports have corroborated the initial observations18,92 of the crucial role of HNF4α and its complex roles controlling transcriptional networks related to hepatocyte metabolism and functions.21,93 Recently, Munroe et al using induced hepatocyte-like cells, showed that repression of HNF4α function leads to shortening in telomere length.94 Argemi et al found that livers from patients with alcoholic hepatitis showed downregulation of HNF4α and they proposed that HNF4α deregulation is in part transforming growth factor β (TGFβ1)-mediated. Moreover, Huang et al showed that upregulation of HNF4α in obese rats with fatty livers led to a significant improvement in serum lipids and glucose homeostasis. Thus, demonstrating the favorable metabolic rearrangement induced by HNF4α in fatty hepatocytes.93
The Inflammatory Microenvironment and Other Players in End-Stage Liver Disease
In ESLD, it is well known that the hepatic microenvironment leads to an activation of hepatic stellate cells (HSCs) which are initially located in the space of Disse.56,95,96 In liver injury, several factors can induce transformation of HSCs into myofibroblast which are defined by the loss of retinoid, gain of α-smooth muscle actin expression, production of TGF-β, platelet-derived growth factor, connective tissue growth factor, and other cytokines.56,96,97 HSC activation leads to liver fibrosis.22 Fibrosis is not only an increase in the amount and distribution of activated HSCs, but a diffuse ECM deposition, which increases the presence of collagens type I and III, mediated mainly by paracrine and autocrine TGFβ98–100 and cytokine signaling, such as interleukin (IL)-1β, tumor necrosis factor α (TNF-α), and IL-33.101,102
Perhaps the main contributor to the development of ESLD is inflammation. Activated by different pathways, the released cytokines, chemokines, and activated inflammatory cells induce a microenvironment that, in a vicious cycle, leads to hepatocyte death and long-term loss of hepatic function. The increase in gut permeability induced by infections, alcohol consumption, or any other stressors can lead to activation of Kupffer cells, a resident macrophage in the liver, through two main pathways: (1) activation of M1 phenotype Kupffer cells by lipopolysaccharide (LPS) and/or (2) IL-4 and IL-10 activation of M2 phenotype Kupffer cells.103 The binding of LPS to its receptor, Toll-like receptor 4, or the activation of complement receptors C3R and C4R of the complement system by pathogen-associated molecular patterns, activates M1 Kupffer cells.103,104 The activation of M1 phenotype cells trigger a cascade releasing TNF-α and IL-6, two proinflammatory cytokines, that have a dual function: stimulate hepatocyte proliferation and cause hepatocellular death.105 On the other hand, the activation of M2 Kupffer cells occurs through an alternative pathway. IL-4 and IL-10, produced by T helper type 2 cells (Th2), activate the M2 phenotype cells resulting in an anti-inflammatory cascade, mediated by TGF-β).103,106 TGF-β also plays a role in the activation of the HSCs and their transformation into myofibroblasts altering the hepatic parenchyma.107
Overall, the activation of Kupffer cells, causes an upregulation of cell adhesion molecules, such as intercellular adhesion molecule-1 and vascular cell adhesion molecule-1, and a downregulation of platelet endothelial cell adhesion molecule-1, allowing the migration of more inflammatory cells to the liver amplifying the inflammation cascade.108,109 A common finding in patients with liver failure is a high number of hepatic neutrophils, which often produce reactive oxygen species, leading to mitochondrial stress and cell death. Interestingly, the phagocytic activity of the neutrophils is compromised resulting in a higher rate of infections in these patients and exacerbating the severity of liver failure.110,111 All of these immune cellular responses are correlated with the survival rates of patients with ESLD due to their increased risk of sepsis and organ failure. This inflammatory milieu where hepatocytes from livers with end-stage disease reside contributes to a vicious cycle that affects the differentiation state and function of hepatocytes and involves highly complex signaling pathways and factors.111,112 However, the extent to which the inflammatory liver microenvironment affects hepatocyte differentiation and function is still unknown (►Fig. 1).
Conclusion
ESLD has often been defined by the finding of cirrhosis. However, cirrhosis is a histopathologic term that provides limited functional information of the liver. Some patients with liver cirrhosis progress into ESLD because of a reduced hepatocellular function, while other patients survive with 1-year mortality ranging from 1 to 57%.16 With an increasing prevalence of ESLD and limited treatment options, it is important to better understand the mechanisms behind ESLD and to investigate alternative therapies to orthotopic LT.
The changes impacting failing hepatocytes range from exposure to an inflammatory milieu in the cirrhotic liver, a loss of cell–cell contact caused by cell death and ECM deposition, and changes in energy metabolism and transcriptional deprogramming. These alterations in the hepatic microenvironment drive development of portal hypertension, esophageal varices, and ascites, which are indirect clinical manifestations of liver failure. The CTP classification and MELD score are helpful to evaluate the severity of hepatocellular failure because they include parameters that are related to metabolic and excretory functions of the failing hepatocyte (INR, bilirubin). Therefore, they are commonly used to determine the urgency for LT, which is the only available treatment for ESLD today.
The concept of cellular reprogramming using master transcription factors developed by Yamanaka and Gurdon, opens the door for a new concept in the treatment of diseases such as ESLD (►Fig. 2). HNF4α, a liver-enriched transcription factor, is a master regulator of liver genes and is responsible for regulating metabolic and excretory functions. In recent studies, we have found a downregulation of HNF4α expression and HNF4α localization outside of the nucleus in the failing hepatocytes. In an animal model of cirrhosis with ESLD, the forced re-expression of HNF4α showed promising results. Failing hepatocytes recovered and increased their metabolic and secretory activity after reprogramming with HNF4α in vivo. In the livers of patients with advanced cirrhosis, HNF4α ribonucleic acid expression levels decrease as hepatic function deteriorates and HNF4α protein expression is found largely in the cytoplasm.20 These findings could explain the impaired hepatic function in patients with degenerative liver disease. These findings have been corroborated in various scenarios of ESLD such as alcoholic cirrhosis, NASH,19,20 and severe alcoholic hepatitis.21 More studies will need to be performed to corroborate the efficacy of cellular reprogramming as a therapy for liver failure in humans (►Fig. 2).
Even now ESLD remains a devastating disease with a high mortality. But recent findings of the complex environment of hepatocellular failure are helping us to understand the disease better and might be the cornerstone for upcoming new approaches in the treatment of ESLD.
Main Concepts and Learning Points.
The degeneration from a healthy functioning liver to cirrhosis and end-stage liver disease involves a dynamic process of hepatocyte damage.
The environment in which the hepatocytes reside is characterized by an inflammatory milieu, extracellular matrix deposition, and a loss of cell–cell contacts.
Transcriptional deprogramming of the hepatocyte leads to a functional reduction of hepatocyte-specific functions.
Changes in the hepatocyte and the hepatic environment explain the symptoms in patients with end-stage liver disease.
Novel therapeutics targeting hepatocyte differentiation and function could avoid the need for transplantation in the future.
Financial Disclosure
This work was supported by grants from NIH, DK099257, DK117881, DK119973, DK096990, and TR002383 to A.S.-G. U.S. Department of Health and Human Services National Institute of Diabetes and Digestive and Kidney Diseases
Footnotes
Competing Interests Statement
A.S.-G. is a co-founder and have a financial interest in Von Baer Wolff, Inc., a company focused on biofabrication of autologous human hepatocytes from stem cells technology and programming liver failure and their interests are managed by the Conflict of Interest Office at the University of Pittsburgh in accordance with their policies.
Conflict of Interest
None declared.
References
- 1.Habka D, Mann D, Landes R, Soto-Gutierrez A. Future economics of liver transplantation: a 20-year cost modeling forecast and the prospect of bioengineering autologous liver grafts. PLoS One 2015;10(07):e0131764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.GBD 2017 Causes of Death Collaborators. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018;392 (10159):1736–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of liver diseases in the world. J Hepatol 2019;70(01):151–171 [DOI] [PubMed] [Google Scholar]
- 4.Lim Y-S, Kim WR. The global impact of hepatic fibrosis and end-stage liver disease. Clin Liver Dis 2008;12(04):733–746, vii [DOI] [PubMed] [Google Scholar]
- 5.Melato M, Sasso F, Zanconati F. Liver cirrhosis and liver cancer. A study of their relationship in 2563 autopsies. Zentralbl Pathol 1993;139(01 ):25–30 [PubMed] [Google Scholar]
- 6.Squires JE, Balistreri WF. Treatment of hepatitis C: a new paradigm toward viral eradication. J Pediatr 2020;221:12–22.e1, e11 [DOI] [PubMed] [Google Scholar]
- 7.Kwong A, Kim WR, Lake JR, et al. OPTN/SRTR 2018 annual data report: liver. Am J Transplant 2020;20(Suppl s1):193–299 [DOI] [PubMed] [Google Scholar]
- 8.Goldberg D, Ditah IC, Saeian K, et al. Changes in the prevalence of hepatitis C virus infection, nonalcoholic steatohepatitis, and alcoholic liver disease among patients with cirrhosis or liver failure on the waitlist for liver transplantation. Gastroenterology 2017;152(05):1090–1099.e1, e1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cholankeril G, Wong RJ, Hu M, et al. Liver transplantation for nonalcoholic steatohepatitis in the US: temporal trends and outcomes. Dig Dis Sci 2017;62(10):2915–2922 [DOI] [PubMed] [Google Scholar]
- 10.Jadlowiec CC, Taner T. Liver transplantation: current status and challenges. World J Gastroenterol 2016;22(18):4438–4445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomson AW, Vionnet J, Sanchez-Fueyo A. Understanding, predicting and achieving liver transplant tolerance: from bench to bedside. Nat Rev Gastroenterol Hepatol 2020;17:719–739. Doi: 10.1038/s41575-020-0334-4 [DOI] [PubMed] [Google Scholar]
- 12.Bodzin AS, BakerTB. Liver transplantation today: where we are now and where we are going. Liver Transpl 2018;24(10):1470–1475 [DOI] [PubMed] [Google Scholar]
- 13.Dasari BVM, Schlegel A, Mergental H, Perera MTPR. The use of old donors in liver transplantation. Best Pract Res Clin Gastroenterol 2017;31(02):211–217 [DOI] [PubMed] [Google Scholar]
- 14.Zarrinpar A, Busuttil RW. Liver transplantation: past, present and future. Nat Rev Gastroenterol Hepatol 2013;10(07):434–440 [DOI] [PubMed] [Google Scholar]
- 15.D’Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol 2006;44(01):217–231 [DOI] [PubMed] [Google Scholar]
- 16.Tsochatzis EA, Bosch J, Burroughs AK. Liver cirrhosis. Lancet 2014;383(9930):1749–1761 [DOI] [PubMed] [Google Scholar]
- 17.Marcellin P, Gane E, Buti M, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet 2013;381(9865):468–475 [DOI] [PubMed] [Google Scholar]
- 18.Liu L, Yannam GR, Nishikawa T, et al. The microenvironment in hepatocyte regeneration and function in rats with advanced cirrhosis. Hepatology 2012;55(05):1529–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guzman-Lepe J, Cervantes-Alvarez E, Collin de l’Hortet A, et al. Liver-enriched transcription factor expression relates to chronic hepatic failure in humans. Hepatol Commun 2018;2(05):582–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Florentino RM, Fraunhoffer NA, Morita K, et al. Cellular location of HNF4α is linked with terminal liver failure in humans. Hepatol Commun 2020;4(06):859–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Argemi J, Latasa MU, Atkinson SR, et al. Defective HNF4alpha-dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis. Nat Commun 2019;10(01):3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol 2014;14(03):181–194 [DOI] [PubMed] [Google Scholar]
- 23.Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut 2015;64(05):830–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fernández M, Semela D, Bruix J, Colle I, Pinzani M, Bosch J. Angiogenesis in liver disease. J Hepatol 2009;50(03):604–620 [DOI] [PubMed] [Google Scholar]
- 25.García-Pagán JC, Gracia-Sancho J, Bosch J. Functional aspects on the pathophysiology of portal hypertension in cirrhosis. J Hepatol 2012;57(02):458–461 [DOI] [PubMed] [Google Scholar]
- 26.Pose E, Cardenas A. Translating our current understanding of ascites management into new therapies for patients with cirrhosis and fluid retention. Dig Dis 2017;35(04):402–410 [DOI] [PubMed] [Google Scholar]
- 27.Runyon BA, Committee APGAASLD Practice Guidelines Committee. Management of adult patients with ascites due to cirrhosis: an update. Hepatology 2009;49(06):2087–2107 [DOI] [PubMed] [Google Scholar]
- 28.Salerno F, Cammà C, Enea M, Rössle M, Wong F. Transjugular intrahepatic portosystemic shunt for refractory ascites: a meta-analysis of individual patient data. Gastroenterology 2007;133(03):825–834 [DOI] [PubMed] [Google Scholar]
- 29.Fede G, D’Amico G, Arvaniti V, et al. Renal failure and cirrhosis: a systematic review of mortality and prognosis. J Hepatol 2012;56(04):810–818 [DOI] [PubMed] [Google Scholar]
- 30.Raevens S, Fallon MB. Potential clinical targets in hepatopulmonary syndrome: lessons from experimental models. Hepatology 2018;68(05):2016–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DuBrock HM, Krowka MJ. The myths and realities of portopulmonary hypertension. Hepatology 2020;•••;. Doi: 10.1002/hep.31415 [DOI] [PubMed] [Google Scholar]
- 32.Møller S, Lee SS. Cirrhotic cardiomyopathy. J Hepatol 2018;69(04):958–960 [DOI] [PubMed] [Google Scholar]
- 33.Burroughs AK, Thalheimer U. Hepatic venous pressure gradient in 2010: optimal measurement is key. Hepatology 2010;51(06): 1894–1896 [DOI] [PubMed] [Google Scholar]
- 34.Ripoll C, Groszmann R, Garcia-Tsao G, et al. ; Portal Hypertension Collaborative Group. Hepatic venous pressure gradient predicts clinical decompensation in patients with compensated cirrhosis. Gastroenterology 2007;133(02):481–488 [DOI] [PubMed] [Google Scholar]
- 35.Jepsen P, Ott P, Andersen PK, Sørensen HT, Vilstrup H. Clinical course of alcoholic liver cirrhosis: a Danish population-based cohort study. Hepatology 2010;51(05):1675–1682 [DOI] [PubMed] [Google Scholar]
- 36.Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol 2016;13(02):88–110 [DOI] [PubMed] [Google Scholar]
- 37.Albillos A, Lario M, Álvarez-Mon M. Cirrhosis-associated immune dysfunction: distinctive features and clinical relevance. J Hepatol 2014;61(06):1385–1396 [DOI] [PubMed] [Google Scholar]
- 38.Arvaniti V, D’Amico G, Fede G, et al. Infections in patients with cirrhosis increase mortality four-fold and should be used in determining prognosis. Gastroenterology 2010;139(04):1246–1256, 1256.e1–1256.e5 [DOI] [PubMed] [Google Scholar]
- 39.Borzio M, Salerno F, Piantoni L, et al. Bacterial infection in patients with advanced cirrhosis: a multicentre prospective study. Dig Liver Dis 2001;33(01):41–48 [DOI] [PubMed] [Google Scholar]
- 40.Fernández J, Navasa M, Gómez J, et al. Bacterial infections in cirrhosis: epidemiological changes with invasive procedures and norfloxacin prophylaxis. Hepatology 2002;35(01): 140–148 [DOI] [PubMed] [Google Scholar]
- 41.Anthony PP, Ishak KG, Nayak NC, Poulsen HE, Scheuer PJ, Sobin LH. The morphology of cirrhosis. Recommendations on definition, nomenclature, and classification by a working group sponsored by the World Health Organization. J Clin Pathol 1978;31(05):395–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schaffner F, Poper H. Capillarization of hepatic sinusoids in man. Gastroenterology 1963;44:239–242 [PubMed] [Google Scholar]
- 43.Pérez-Tamayo R Cirrhosis of the liver: a reversible disease? Pathol Annu 1979;14(Pt 2):183–213 [PubMed] [Google Scholar]
- 44.Ishak K, Baptista A, Bianchi L, et al. Histological grading and staging of chronic hepatitis. J Hepatol 1995;22(06):696–699 [DOI] [PubMed] [Google Scholar]
- 45.Bedossa P, Poynard TThe METAVIR Cooperative Study Group. An algorithm for the grading of activity in chronic hepatitis C. Hepatology 1996;24(02):289–293 [DOI] [PubMed] [Google Scholar]
- 46.Haj M, Rockey DC. Predictors of clinical outcomes in cirrhosis patients. Curr Opin Gastroenterol 2018;34(04):266–271 [DOI] [PubMed] [Google Scholar]
- 47.Pugh RNH, Murray-Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg 1973;60(08):646–649 [DOI] [PubMed] [Google Scholar]
- 48.Child CG, Turcotte JG. Surgery and portal hypertension. Major Probl Clin Surg 1964;1:1–85 [PubMed] [Google Scholar]
- 49.Mansour A, Watson W, Shayani V, Pickleman J. Abdominal operations in patients with cirrhosis: still a major surgical challenge. Surgery 1997;122(04):730–735, discussion 735–736 [DOI] [PubMed] [Google Scholar]
- 50.Garrison RN, Cryer HM, Howard DA, Polk HC Jr. Clarification of risk factors for abdominal operations in patients with hepatic cirrhosis. Ann Surg 1984;199(06):648–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kamath PS, Wiesner RH, Malinchoc M, et al. A model to predict survival in patients with end-stage liver disease. Hepatology 2001;33(02):464–470 [DOI] [PubMed] [Google Scholar]
- 52.Wiesner R, Edwards E, Freeman R, et al. ; United Network for Organ Sharing Liver Disease Severity Score Committee. Model for end-stage liver disease (MELD) and allocation of donor livers. Gastroenterology 2003;124(01 ):91–96 [DOI] [PubMed] [Google Scholar]
- 53.Malinchoc M, Kamath PS, Gordon FD, Peine CJ, Rank J, ter Borg PC. A model to predict poor survival in patients undergoing transjugular intrahepatic portosystemic shunts. Hepatology 2000;31 (04):864–871 [DOI] [PubMed] [Google Scholar]
- 54.Sacleux SC, Samuel D. A critical review of MELD as a reliable tool for transplant prioritization. Semin Liver Dis 2019;39(04):403–413 [DOI] [PubMed] [Google Scholar]
- 55.Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell 2013;152(06):1237–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 2017;14(07):397–411 [DOI] [PubMed] [Google Scholar]
- 57.Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest 2013;123(05):1887–1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoon YJ, Friedman SL, Lee YA. Antifibrotic therapies: where are we now? Semin Liver Dis 2016;36(01):87–98 [DOI] [PubMed] [Google Scholar]
- 59.Caneba CA, Bellance N, Yang L, Pabst L, Nagrath D. Pyruvate uptake is increased in highly invasive ovarian cancer cells under anoikis conditions for anaplerosis, mitochondrial function, and migration. Am J Physiol Endocrinol Metab 2012;303(08):E1036–E1052 [DOI] [PubMed] [Google Scholar]
- 60.Chen L-Y, Yang B, Zhou L, Ren F, Duan ZP, Ma YJ. Promotion of mitochondrial energy metabolism during hepatocyte apoptosis in a rat model of acute liver failure. Mol Med Rep 2015;12(04):5035–5041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Komurov K, Tseng JT, Muller M, et al. The glucose-deprivation network counteracts lapatinib-induced toxicity in resistant ErbB2-positive breast cancer cells. Mol Syst Biol 2012;8:596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nagrath D, Caneba C, Karedath T, et al. Metabolomics for mitochondrial and cancer studies. Biochimica et Biophysica Acta (BBA). Bioenergetics 2011;1807:650–663 [DOI] [PubMed] [Google Scholar]
- 63.Díaz Gil J, Rossi I, Escartín P, Segovia JM, Gosálvez M. Mitochondrial functions and content of microsomal and mitochondrial cytochromes in human cirrhosis. Clin Sci Mol Med 1977;52(06):599–606 [DOI] [PubMed] [Google Scholar]
- 64.Möller B, Dargel R. Structural and functional impairment of mitochondria from rat livers chronically injured by thioacetamide. Acta Pharmacol Toxicol (Copenh) 1984;55(02):126–132 [DOI] [PubMed] [Google Scholar]
- 65.Krähenbühl S, Stucki J, Reichen J. Mitochondrial function in carbon tetrachloride-induced cirrhosis in the rat. Qualitative and quantitative defects. Biochem Pharmacol 1989;38(10):1583–1588 [DOI] [PubMed] [Google Scholar]
- 66.Nishikawa T, Bellance N, Damm A, et al. A switch in the source of ATP production and a loss in capacity to perform glycolysis are hallmarks of hepatocyte failure in advance liver disease. J Hepatol 2014;60(06):1203–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Treyer A, Müsch A. Hepatocyte polarity. In: Terjung R, ed. Comprehensive Physiology. Hoboken, NJ: John Wiley & Sons, Inc.; 2013:c120009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Boyer JL. Bile formation and secretion. In: Terjung R, ed. Comprehensive Physiology. Hoboken, NJ: John Wiley & Sons, Inc.; 2013:c120027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luther J, Gala MK, Borren N, et al. Hepatic connexin 32 associates with nonalcoholic fatty liver disease severity. Hepatol Commun 2018;2(07):786–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wright JA, Richards T, Becker DL. Connexins and diabetes. Cardiol Res Pract 2012;2012:496904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hernández-Guerra M, Hadjihambi A, Jalan R. Gap junctions in liver disease: implications for pathogenesis and therapy. J Hepatol 2019;70(04):759–772 [DOI] [PubMed] [Google Scholar]
- 72.Tiburcio TC, Willebrords J, da Silva TC, et al. Connexin32 deficiency is associated with liver injury, inflammation and oxidative stress in experimental non-alcoholic steatohepatitis. Clin Exp Pharmacol Physiol 2017;44(02):197–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kameritsch P, Khandoga N, Pohl U, Pogoda K. Gap junctional communication promotes apoptosis in a connexin-type-dependent manner. Cell Death Dis 2013;4:e584–e584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Amaya MJ, Nathanson MH. Calcium signaling in the liver. In: Terjung R, ed. Comprehensive Physiology. Hoboken, NJ: John Wiley & Sons, Inc.; 2013:c120013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Leite MF, Hirata K, Pusl T, et al. Molecular basis for pacemaker cells in epithelia. J Biol Chem 2002;277(18):16313–16323 [DOI] [PubMed] [Google Scholar]
- 76.Michalopoulos GK. Hepatostat: liver regeneration and normal liver tissue maintenance. Hepatology 2017;65(04):1384–1392 [DOI] [PubMed] [Google Scholar]
- 77.Paradis V, Youssef N, Dargère D, et al. Replicative senescence in normal liver, chronic hepatitis C, and hepatocellular carcinomas. Hum Pathol 2001;32(03):327–332 [DOI] [PubMed] [Google Scholar]
- 78.Lu C, Xia J, Zhou Y, et al. Loss of Gsα impairs liver regeneration through a defect in the crosstalk between cAMP and growth factor signaling. J Hepatol 2016;64(02):342–351 [DOI] [PubMed] [Google Scholar]
- 79.Paranjpe S, Bowen WC, Mars WM, et al. Combined systemic elimination of MET and epidermal growth factor receptor signaling completely abolishes liver regeneration and leads to liver decompensation. Hepatology 2016;64(05):1711–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Michalopoulos GK, Bhushan B. Liver regeneration: biological and pathological mechanisms and implications. Nat Rev Gastroenterol Hepatol 2020;•••;. Doi: 10.1038/s41575-020-0342-4 [DOI] [PubMed] [Google Scholar]
- 81.Tsagianni A, Mars WM, Bhushan B, et al. Combined systemic disruption of MET and epidermal growth factor receptor signaling causes liver failure in normal mice. Am J Pathol 2018;188 (10):2223–2235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schrem H, Klempnauer J, Borlak J. Liver-enriched transcription factors in liver function and development. Part I: the hepatocyte nuclear factor network and liver-specific gene expression. Pharmacol Rev 2002;54(01):129–158 [DOI] [PubMed] [Google Scholar]
- 83.Schrem H, Klempnauer J, Borlak J. Liver-enriched transcription factors in liver function and development. Part II: the C/EBPs and D site-binding protein in cell cycle control, carcinogenesis, circadian gene regulation, liver regeneration, apoptosis, and liver-specific gene regulation. Pharmacol Rev 2004;56(02):291–330 [DOI] [PubMed] [Google Scholar]
- 84.Lau HH, Ng NHJ, Loo LSW, Jasmen JB, Teo AKK. The molecular functions of hepatocyte nuclear factors - in and beyond the liver. J Hepatol 2018;68(05):1033–1048 [DOI] [PubMed] [Google Scholar]
- 85.Chen WS, Manova K, Weinstein DC, et al. Disruption of the HNF-4 gene, expressed in visceral endoderm, leads to cell death in embryonic ectoderm and impaired gastrulation of mouse embryos. Genes Dev 1994;8(20):2466–2477 [DOI] [PubMed] [Google Scholar]
- 86.Kanazawa T, Konno A, Hashimoto Y, Kon Y. Hepatocyte nuclear factor4 alpha is related to survival of the condensed mesenchyme in the developing mouse kidney. Dev Dyn 2010;239(04):1145–1154 [DOI] [PubMed] [Google Scholar]
- 87.Okita K, Matsumura Y, Sato Y, et al. A more efficient method to generate integration-free human iPS cells. Nat Methods 2011;8 (05):409–412 [DOI] [PubMed] [Google Scholar]
- 88.Ieda M, Fu J-D, Delgado-Olguin P, et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010;142(03):375–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Südhof TC, Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010;463(7284):1035–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Niu W, Zang T, Smith DK, et al. SOX2 reprograms resident astrocytes into neural progenitors in the adult brain. Stem Cell Reports 2015;4(05):780–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to β-cells. Nature 2008;455(7213):627–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nishikawa T, Bell A, Brooks JM, et al. Resetting the transcription factor network reverses terminal chronic hepatic failure. J Clin Invest 2015;125(04):1533–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huang K-W, Reebye V, Czysz K, et al. Liver activation of hepatocellular nuclear factor-4α by small activating RNA rescues dyslipidemia and improves metabolic profile. Mol Ther Nucleic Acids 2020;19:361–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Munroe M, Niero EL, Fok WC, et al. Telomere dysfunction activates p53 and represses HNF4α expression leading to impaired human hepatocyte development and function. Hepatology 2020;•••;. Doi: 10.1002/hep.31414:hep.31414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jiang JX, Török NJ. Liver injury and the activation of the hepatic myofibroblasts. Curr Pathobiol Rep 2013;1(03):215–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yin C, Evason KJ, Asahina K, Stainier DY. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest 2013; 123(05):1902–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 2000;275 (04):2247–2250 [DOI] [PubMed] [Google Scholar]
- 98.Brown B, Lindberg K, Reing J, Stolz DB, Badylak SF. The basement membrane component of biologic scaffolds derived from extracellular matrix. Tissue Eng 2006;12(03):519–526 [DOI] [PubMed] [Google Scholar]
- 99.Dooley S, ten Dijke P. TGF-β in progression of liver disease. Cell Tissue Res 2012;347(01):245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wong L, Yamasaki G, Johnson RJ, Friedman SL. Induction of beta-platelet-derived growth factor receptor in rat hepatic lipocytes during cellular activation in vivo and in culture. J Clin Invest 1994;94(04):1563–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McHedlidze T, Waldner M, Zopf S, et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity 2013;39(02):357–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pradere J-P, Kluwe J, De Minicis S, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013;58(04):1461–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wan J, Benkdane M, Teixeira-Clerc F, et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014;59(01 ):130–142 [DOI] [PubMed] [Google Scholar]
- 104.Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology 2008;48(01):322–335 [DOI] [PubMed] [Google Scholar]
- 105.Desmots F, Rissel M, Gilot D, et al. Pro-inflammatory cytokines tumor necrosis factor a and interleukin-6 and survival factor epidermal growth factor positively regulate the murine GSTA4 enzyme in hepatocytes. J Biol Chem 2002;277(20): 17892–17900 [DOI] [PubMed] [Google Scholar]
- 106.Wasmuth HE, Kunz D, Yagmur E, et al. Patients with acute on chronic liver failure display “sepsis-like” immune paralysis. J Hepatol 2005;42(02):195–201 [DOI] [PubMed] [Google Scholar]
- 107.Rockey DC, Fouassier L, Chung JJ, et al. Cellular localization of endothelin-1 and increased production in liver injury in the rat: potential for autocrine and paracrine effects on stellate cells. Hepatology 1998;27(02):472–480 [DOI] [PubMed] [Google Scholar]
- 108.Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int 2006;26(10):1175–1186 [DOI] [PubMed] [Google Scholar]
- 109.Dixon LJ, Barnes M, Tang H, et al. Kupffer cells in the liver. In: Terjung R, ed. Comprehensive Physiology. Hoboken, NJ: John Wiley & Sons, Inc.; 2013:c120026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mookerjee RP, Stadlbauer V, Lidder S, et al. Neutrophil dysfunction in alcoholic hepatitis superimposed on cirrhosis is reversible and predicts the outcome. Hepatology 2007;46(03):831–840 [DOI] [PubMed] [Google Scholar]
- 111.Sarin SK, Choudhury A. Acute-on-chronic liver failure: terminology, mechanisms and management. Nat Rev Gastroenterol Hepatol 2016;13(03):131–149 [DOI] [PubMed] [Google Scholar]
- 112.Zhai S, Zhang L, Dang S, et al. The ratio of Th-17 to Treg cells is associated with survival of patients with acute-on-chronic hepatitis B liver failure. Viral Immunol 2011;24(04):303–310 [DOI] [PubMed] [Google Scholar]
- 113.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008;134(06):1655–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Garcia-Tsao G, Friedman S, Iredale J, Pinzani M. Now there are many (stages) where before there was one: In search of a pathophysiological classification of cirrhosis. Hepatology 2010;51(04):1445–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]