

Summary
Hepatitis E virus (HEV) genotype (gt) 3 infection is food-borne causing sporadic infections in older individuals and gt1 infection is waterborne, often causing epidemics affecting primarily young adults. Although HEV infection causes self-limited disease, gt3 induces chronic infection in immunocompromised individuals. Hepatic host gene expression against gt3 infection remains unknown. Host gene expression profiles for HEV gt1 (n = 3) and gt3 (n = 7) infections were analysed in the livers of experimentally infected rhesus macaques. HEV RNA was detected from 2 to 24 days after inoculation (DAI) in stool and serum, elevated alanine aminotransferase (ALT) activity was detected from 7 to 31 DAI, and anti-HEV antibody became detectable between 12 and 42 DAI. All 10 animals cleared the infection between 34 and 68 DAI. We found that 24%, 48% and 41% of hepatic immune response genes against gt3 infection were upregulated during the early, peak and decline phases of HEV RNA replication. For gt1 infection, 25% of hepatic immune response-related genes were downregulated during early viremia, but 6%, 34% and 37% of genes were upregulated at the early, peak and during decline of HEV RNA replication, respectively. Our study demonstrated distinct differences in the expression profiles of host immune response-related genes of HEV gt3 and gt1 infections in experimentally infected rhesus macaques.
Keywords: acute infection, genotype 1, genotype 3, hepatitis E virus, host immune response-related genes, rhesus macaques
1 |. INTRODUCTION
Hepatitis E virus (HEV) infection presents a significant public health problem in Asia and Africa where HEV can cause large waterborne epidemics of acute jaundice.1 Clinical attack rates are more common among young adults than other age groups during epidemics. HEV infection is one of the most frequent causes of acute hepatitis, with an estimated 20 million incident infections worldwide and 3.3 million cases of acute hepatitis per year, and a 20% probability of death in infected pregnant women.1
HEV is classified into 8 genotypes (gt), five of these, genotypes 1, 2, 3, 4 and 7 are known to infect humans.2,3 HEV gt1 tends to be more common in Southern and Central Asian countries and gt3 infection occurs worldwide, including the Americas, Europe, China and Japan.4 Disease caused by HEV gt1 is often self-resolving but can lead to fulminant liver failure, particularly in pregnant women; and gt3 infection is associated with individual cases and sporadic outbreaks linked to exposure to pigs and consumption of raw or undercooked meat from infected animals.5 In addition, chronic HEV infection and cirrhosis has been reported in immunosuppressed individuals, patients co-infected with HIV and haematological patients.6 To date, all chronic HEV infection cases are associated with HEV gt3, gt4 and gt7.7–9 No chronic HEV infections related to HEV gt1 or 2 have been reported.
Macaques and chimpanzees are the only animal models for studying pathogenesis of viral hepatitis E and are susceptible to infections with HEV genotypes 1–4.10,11 In addition, human liver chimeric mice were also shown to be susceptible to HEV gt1 and gt3 infection.12–14 Host response to HEV gt1 was reported in the livers of experimentally infected chimpanzees and chimeric mice.12,15 A large number of innate and adaptive immune response-related genes were upregulated in HEV gt1-infected chimpanzees. HEV-specific T-cell responses in peripheral blood of patients with HEV gt3 infection have been reported.16,17 However, the hepatic host response to HEV gt3 infection has not been studied.
In this study, host gene expression profiles were analysed in liver biopsy tissue specimens from experimentally infected rhesus macaques. Hepatic immune response-related host gene expression in HEV gt1 and gt3 infections was analysed in experimentally infected rhesus macaques.
2 |. MATERIALS AND METHODS
2.1 |. Animal care
The Comparative Medicine Branch at the Centers for Disease Control and Prevention (CDC) provided care and husbandry for the animals in accordance with the Guide for the Care and Use of Laboratory Animals and the use of nonhuman primates in research.18 All animal protocols and procedures were reviewed and approved by the CDC Institutional Animal Care and Use Committee (IACUC).
2.2 |. Inoculum
The HEV inoculum used to infect each animal is shown in Table 1. Six rhesus macaques (RH623, RH633, RH620, RH625, RH635 and RH634) were inoculated intravenously with stool specimen of a rhesus macaque that had been originally inoculated with the human HEV gt1 Sar-55 strain.14 RH633, RH635 and RH634 were excluded from the study because of co-infection with SIV. Seven animals (RH654, RH641, RH644, RH639, RH645, RH642 and RH650) were inoculated intravenously with stool specimens of rhesus macaques that were originally inoculated with material from a kidney transplant patient infected with HEV gt3 strain (gene bank access number, JN837481).19
TABLE 1.
Clinical characteristics of each animal at the time of liver biopsy
| RHID | HEV | Inoculum (Log10 WHO IU/ml) | HEV RNA (Log10 WHO IU/mLa) |
ALT activity/cut-off value (U/L) | Anti-HEV AB |
Viremia stage | |||
|---|---|---|---|---|---|---|---|---|---|
| Liver | Stoo | Serum | IgM | IgG | |||||
| RA1073 | naive | n/a | n/a | n/a | n/a | 24 | NEG | NEG | n/a |
| RA1092 | naive | n/a | n/a | n/a | n/a | 16 | NEG | NEG | n/a |
| RH623 | gt1 | 6.4 | 8.8 | 7.2 | 5.3 | 34/54 | NEG | NEG | early |
| RH620 | gt1 | 3.5 | 9.7 | 9.1 | 5.1 | 46/53 | POS | POS | peak |
| RH625 | gt1 | 4.7 | 4 | 4.0 | NEG | 113/40 | POS | POS | decline |
| RH654 | gt3 | 4 | 6.3 | 4.0 | NEG | 35/44 | NEG | NEG | early |
| RH641 | gt3 | 2.5 | 5.2 | 5.0 | 1.9 | 35/43 | NEG | NEG | early |
| RH644 | gt3 | 9.5 | 8.8 | 8.9 | 3.7 | 231/71 | NEG | NEG | peak |
| RH639 | gt3 | 7.8 | 7.7 | 9.3 | 3.5 | 65/36 | POS | NEG | peak |
| RH645 | gt3 | 9.5 | 7 | 7.9 | 4 | 110/36 | POS | POS | peak |
| RH642 | gt3 | 7.8 | 3.7 | 5.8 | 1.8 | 38/29 | POS | POS | peak |
| RH650 | gt3 | 7.8 | 4.1 | 7.3 | 1.7 | 308/58 | POS | POS | decline |
n/a, not available.
Detection limit of HEV RNA by real-time PCR is 20 WHO IU/mL. HEV RNA was negative in the second liver biopsied tissues obtained from gt1 and gt3 infected animal.
2.3 |. Determination of HEV RNA titre by real-time PCR
Total RNA extraction was either performed using the RiboPure RNA purification Kit (Life Technology, Carlsbad, CA) or the Total Nucleic Acid Isolation Kit (Roche Applied Science, Indianapolis, IN) on the MagNA Pure LC 2.0 instrument (Roche Applied Science) according to the manufacturer’s recommended procedure. The HEV RNA titre in liver, stool and serum specimens was determined as an RNA value in IU/mL by comparison with a standard curve of serial log10 dilutions of HEV with known potency in WHO international units.20
2.4 |. Viremia stages during acute HEV infection
Based on HEV RNA titre in daily stool samples during the study period (115 days), viremia stages were characterized as early, peak, decline and clearance of the infection. Early viremia was the initial phase of infection before the peak of viremia, and decline of viremia was defined the phase after the peak viremia.
2.5 |. Collection of liver tissues
Time of biopsy samples from each animal was organized based on the stage of viremia (Table 1). Two anti-HEV antibody negative animals were used as baseline controls for determination of differential gene expression. Liver needle biopsy samples were frozen in liquid nitrogen immediately after the biopsy and subsequently stored at −80°C before RNA isolation. Total RNA from the frozen liver specimens was extracted using the RiboPure RNA purification Kit (Life Technology, Carlsbad, CA). The quality and quantity of RNA were analysed using a 2100 Bioanalyzer (Agilent, Santa Clara, CA).
2.6 |. ALT activity and detection of anti-HEV antibodies
Multiple pre-inoculation, weekly post-inoculation sera collected from the monkeys were tested for alanine aminotransferase (ALT) activity using a VetScan VS2 (ABAXIS, Union City, CA) and anti-HEV antibody response using commercially available enzyme immunoassays (DSI, Milan, Italy) performed in accordance with the manufacturer’s instructions, and recorded as signal-to-cut-off ratios.
2.7 |. Rhesus macaque-specific immune gene array
Each cDNA was synthesized with 500 ng of total RNA using the RT2 First Strand kit (Qiagen, Carlsbad, CA) and used to estimate rhesus macaque-specific innate and adaptive immune responses (PAQQ-052Z), NF-κB signalling pathway (PAQQ-025Z), Th1 and Th2 responses (PAQQ-034Z), and Type I interferon response (PAQQ-016Z) RT2 profiler PCR array (Qiagen) according to the manufacturer’s recommended procedures. Briefly, cDNA template was combined with an instrument-specific and ready-to-use RT2 SYBR Green qPCR Master Mix (Qiagen). Equal aliquots of this mixture (25 μL) were added to each well of the same PCR array plate containing the pre-dispensed gene-specific primer sets. PCR was conducted on a ViiA7 Real-Time PCR System (Life Technology, Carlsbad, CA). Fold changes in gene expression were calculated using the Web-Based PCR Array Data Analysis from Sabiociences (Valencia, CA, USA). Five genes, ACTB, B2M, GAPDH, HGPRT and RPL13A, in each PCR array were used as housekeeping control genes (Data S1).
3 |. RESULTS
3.1 |. RNA and ALT activity in HEV-infected rhesus macaques
The six HEV gt1-infected and the seven HEV gt3-infected rhesus macaques exhibited the typical course of acute HEV infection. However, 3 of 6 HEVgt-1 infected animals were found to have been exposed to simian immunodeficiency virus. The results from these 3 animals are excluded from the rest of data analysis. HEV RNA was detected in stools from 2 to 9 days after inoculation (DAI) and from 3 to 24 DAI in serum and cleared between 34 and 68 DAI in all of the animals (Figure 1). Serum ALT activity was elevated from 7 to 31 DAI in all of the gt1- and gt3-infected animals. In gt1-infected animals, anti-HEV antibody became detectable from 35 to 42 DAI and gt3-infected animals became anti-HEV antibody positive from 12 to 24 DAI.
FIGURE 1.
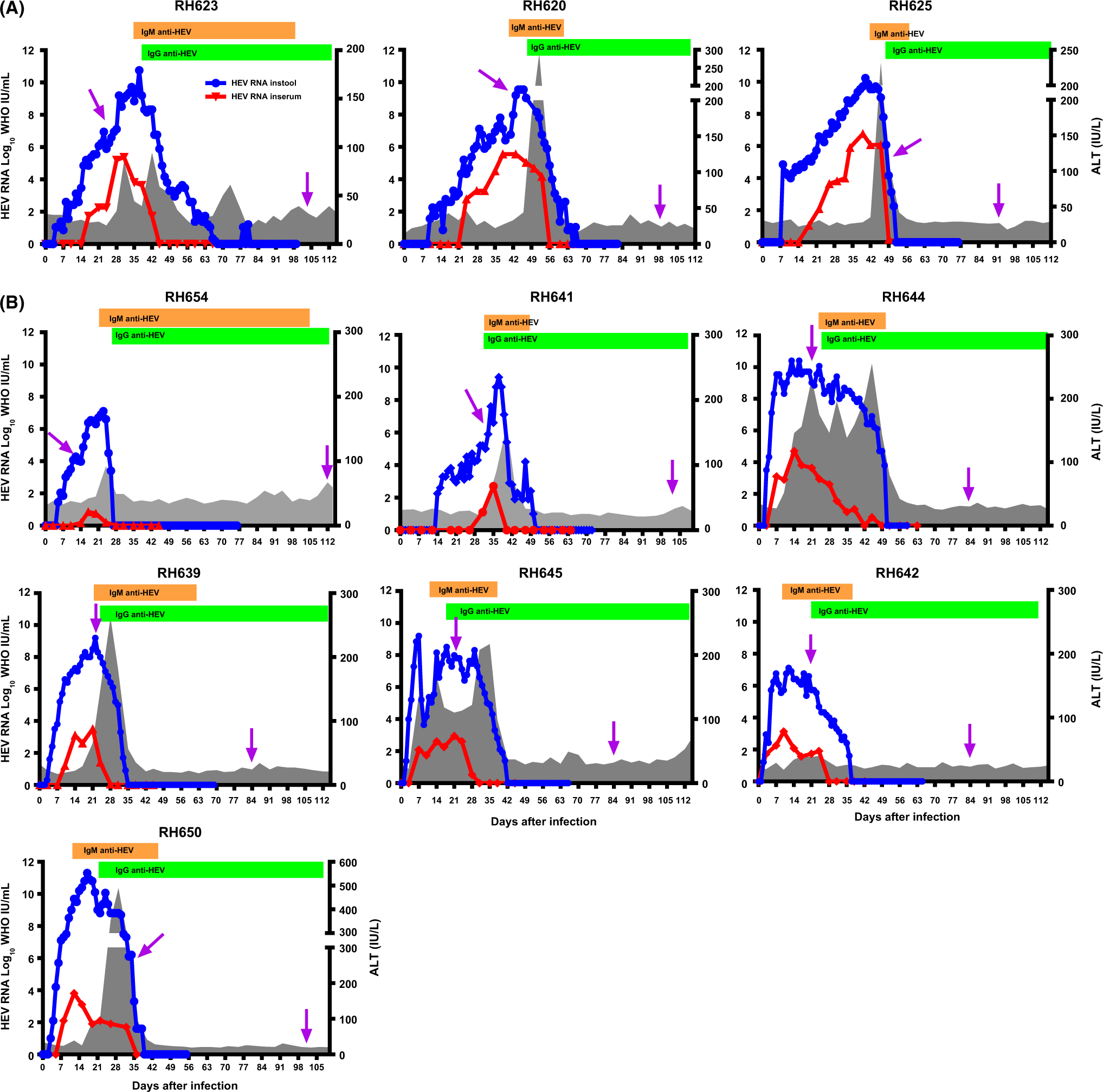
Course of HEV infection. A, HEV genotype 1 (gt) infection and B, HEV gt3 infection were observed for 115 days after infection. Levels of alanine aminotransferase (ALT) activities, antibodies and HEV RNA in daily stool and weekly serum were analysed. PCR arrays were performed on the liver biopsied samples (purple arrow). The blue line shows levels of HEV RNA in stool and the red line shows RNA levels in serum samples. Bar graphs represent anti-HEV antibody responses and ALT activities are shown as gray area graphs. Dates of liver biopsies from each animal are as follows: days 28 and 104 in RH623, days 46 and 99 in RH620, days 49 and 92 in RH625; days14 and 114 in RH654; 32 and 103 in RH641; 22 and 84 in RH644, RH639, RH645, RH642; days 32 and 103 in RH650
3.2 |. Liver tissues from the rhesus macaques with HEV gt1 and gt3 infection
Two liver biopsies were obtained from each HEV-infected animal. Characteristics of the first liver biopsy are shown in Table 1. For the gt1-infected animals, the first liver biopsy was obtained during early viremia from RH623, peak viremia from RH620 and decline of viremia from RH625 (Figure 1A).
For the gt3-infected animals, the first liver biopsy specimens were obtained during early viremia from RH654 and RH641, peak viremia from RH644, RH639, RH642 and RH645, and decline of viremia from RH650 (Figure 1B and Table 1). A second liver biopsy sample was obtained from all of gt1- and gt3-infected rhesus macaques when HEV RNA cleared in liver, stool and serum. Gene expression profiles in the second liver biopsy were analysed separately, and the average fold change for differentially regulated genes from each gene array was calculated and labelled as HEV clearance in Figure 2.
FIGURE 2.
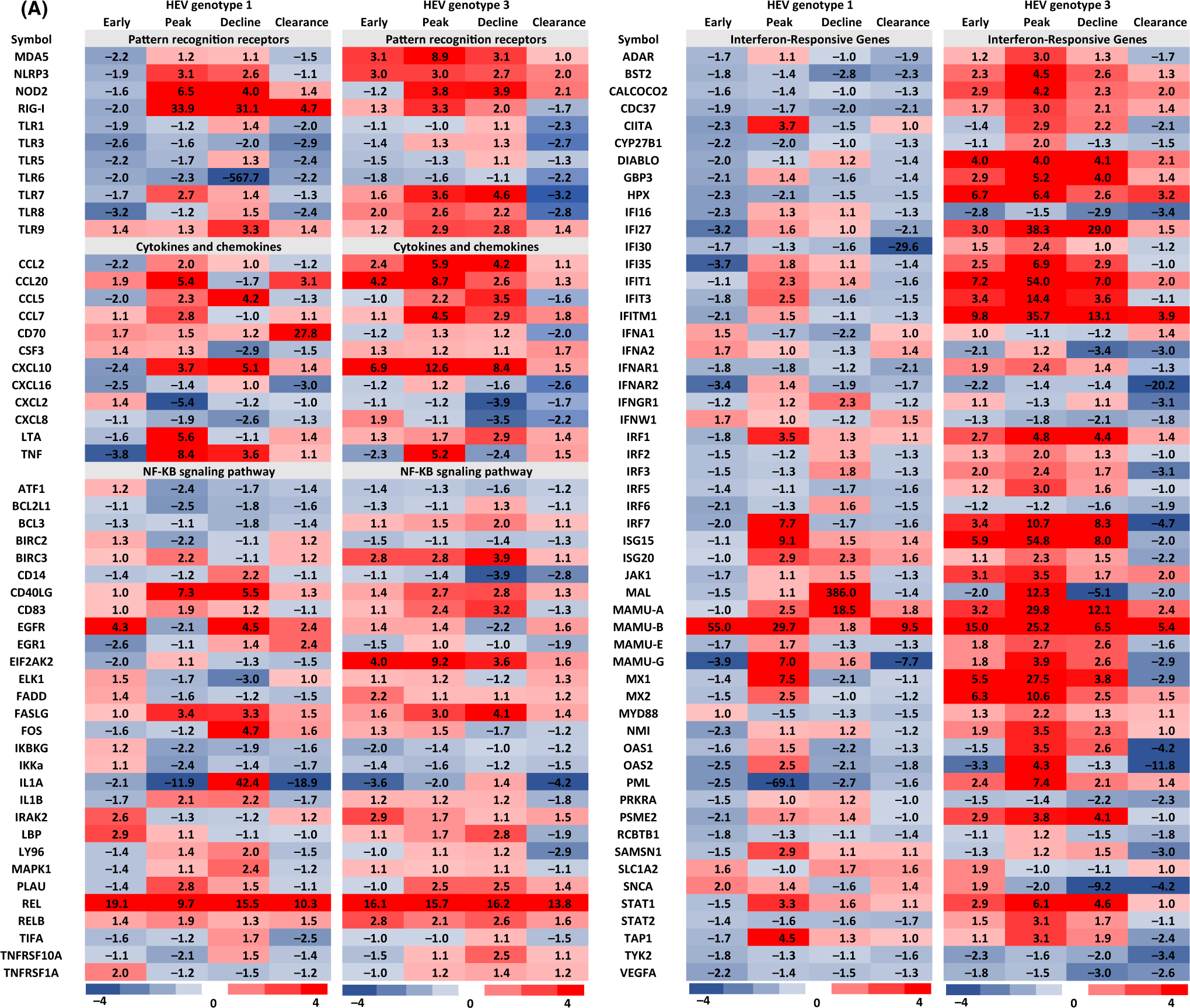
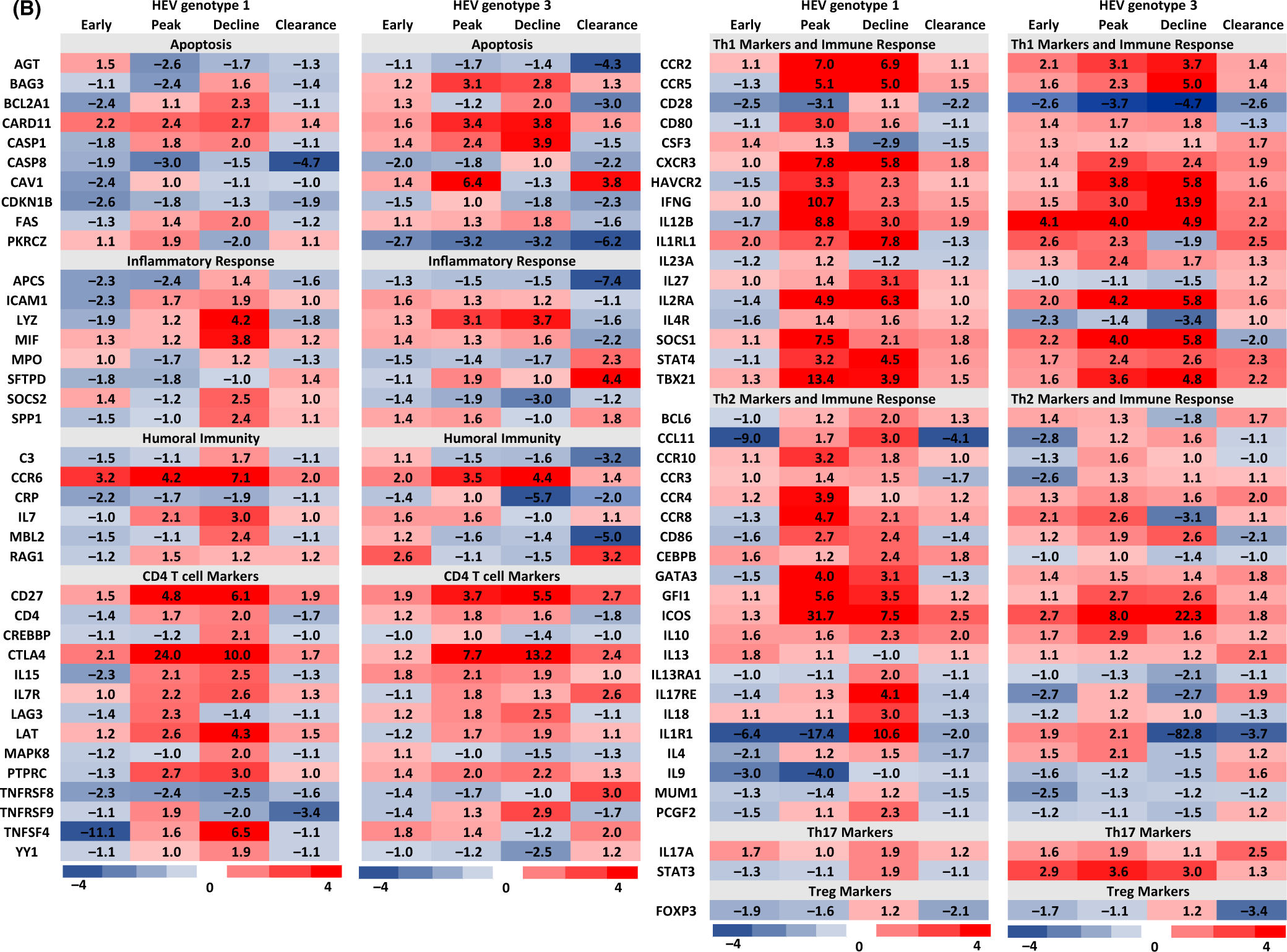
Differentially regulated hepatic host genes in HEV gt1 and gt3 infections in experimentally infected rhesus macaques. Heat map gene expression profiles were created using data from rhesus macaque-specific real-time PCR arrays. Gene expression changes in gt1 and gt3 infected animals were compared against two naive animals and expression levels >2-fold were considered significant. Gene expression profiles of clearance of HEV gt1 and gt3 infections as well as early and peak viremia of HEV gt3 were pooled and calculated the average fold change for differentially regulated genes from each animal. Red color represents upregulated genes and blue color represents downregulated genes.
3.3 |. Immune response gene arrays
Genes showing twofold and more changes in expression compared to baseline were considered to be significantly altered by HEV gt1 or gt3 infection, and this generated a list of 185 genes (Figure 2). In the gt1 infection, 25% (46 genes) of hepatic immune response genes were downregulated, while 6% (11 genes) were upregulated in early viremia. During the peak and decline of gt1 viremia, 34% (62 genes) and 37% (68 genes) of immune response genes were upregulated, respectively. In the gt3 infection, however, significantly higher numbers of immune response genes [24%, (44 genes)] were upregulated in early viremia, the peak [49%, (90 genes)] or decline of viremia [41%, (75 genes)]. Venn diagrams were used to identify genes exhibiting up- or downregulation in each viremia group in HEV gt1 and gt3 infections (Figure 3). In the gt1 infection, 3 downregulated and 5 upregulated genes were common among the infected animals and 37 upregulated genes were found to be common during gt3 viremia.
FIGURE 3.
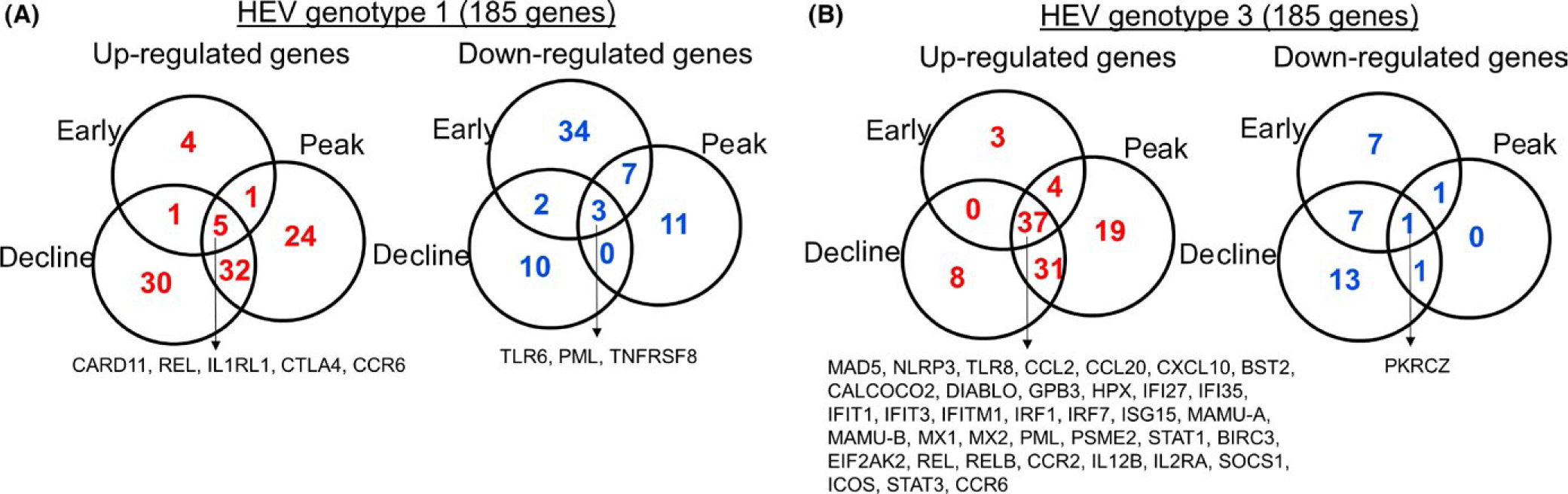
Expression of hepatic immune response genes against HEV gt1 and gt3 infection. Venn diagrams showing the numbers of up- and downregulated genes in the gt1 infection (A) and gt3 infection (B) during early, at the peak and decline of viremia
Most notable differences were associated with the timing of upregulated gene expression (Figure 4). In early viremia, hepatic immune response-related genes were mostly downregulated in gt1 infected animals; however, they were upregulated in gt3 infected animals. During the peak of viremia, when gt1 infected animals became anti-HEV antibody positive, hepatic immune response genes were upregulated and then expression levels were significantly reduced or normalized when the infection cleared. For gt3 infected animals, levels of hepatic immune response gene expression were upregulated during early and peak of viremia and most of the immune response-related gene expression were downregulated or normalized when the infection was cleared in gt3 infected animals, but a large number of interferon responsive genes were downregulated (Figure 4).
FIGURE 4.
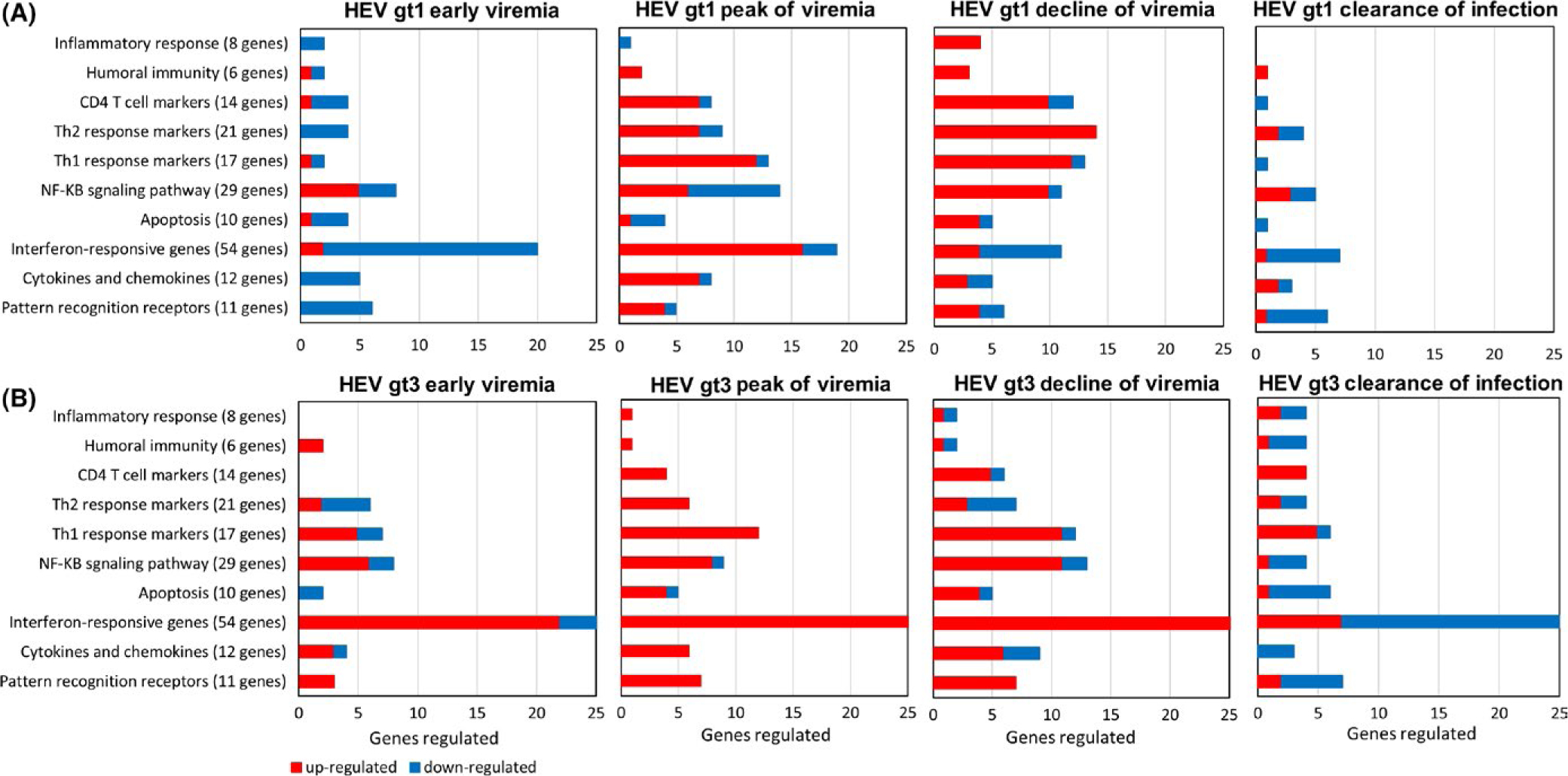
Classification of the differentially regulated genes in HEV gt1 and gt3 infection. Up- and downregulated hepatic immune response genes are organized according to biological process in early, at the peak, decline and clearance of the gt1 (A), and gt3 infections (B). Red bar represents upregulated genes and blue bar represents downregulated genes
3.4 |. Pattern recognition receptor (PRR)
Six of 11 PRR genes, MDA5, RIG-I, TLR3, TLR5, TLR6 and TLR8 were downregulated in early gt1 infection. MDA5, NLRP3 and TLR8 were upregulated in early gt3 infection (Figure 2). NLRP3, NOD2 and RIG-I were upregulated at the peak and decline of both gt1 and gt3 infections. MDA5 and TLR8 were upregulated at the peak and decline of gt3 infection. TLR7 was upregulated at the peak of both gt1 and gt3 infections (Figure 2). TLR9 was upregulated during decline of both gt1 and gt3 infection as well as at peak of gt3 infection. Higher levels of RIG-I gene expression were detected in the gt1 infection compared to the gt3 infection. High levels of MDA5 and TLR8 expression were detected in the gt3 infection, but not in the HEV gt1 infection. After the clearance of infection, TLR1, TLR3, TLR6 and TLR8 were downregulated both in the gt1 and gt3 infection.
3.5 |. Chemokine and cytokines
Expression of CCL2, CCL5, CXCL10, CXCL16 and TNF was downregulated in early viremia, but they were upregulated in the peak of the gt1 infection except CXCL16. High levels of CCL2, CCL20 and CXCL10 expression were detected in early viremia and continued to be upregulated until the decline of gt3 infection (Figure 2). At the peak and decline of gt1 and gt3 infection, CCL2, CCL20, CCL5, CCL7,CXCL10, LTA and TNF gene expression was upregulated, and CXCL2 and CXCL8 were downregulated in both gt1 and gt3 infections. Upregulation of CD70 gene expression was detected after clearance of gt1 infection.
3.6 |. Type 1 interferon response
In early viremia, 18 genes of 54 IFN response-related genes were downregulated and 2 genes were upregulated in the gt1 infection, whereas 22 genes were upregulated and 6 genes were downregulated in the gt3 infection (Figures 2 and 4). Forty and 26 genes were upregulated at the peak and decline in the gt3 virus replication; 16 and 4 genes were upregulated in the peak and decline of gt1 viremia, respectively. IFNGR1, SAMSN1 and SNCA were upregulated in the gt1 infection, and ADAR, BST2, CALCOCO2, CDC37, DIABLO, GBP3, HPX, IFI27, IFI30, IFI35, IFITM1, IFNAR1, IRF2, IRF3, IRF5, JAK1, MAMU-E, MYD88, NMI, OAS1, PML, PSME2 and STAT2 were upregulated in the gt3 infection. CIITA, IFIT1, IFIT3, IRF1, IRF7, ISG15, ISG20, MAL, MAMU-A, MAMU-B, MAMU-G, MX1, MX2, OAS2, STAT1 and TAP1 were upregulated in both infections.
3.7 |. Apoptosis
Upregulation of CARD11 was detected during gt1 replication. In early viremia, downregulation of BCL2A1, CAV1, CDKN1 and CASP8 and PKRCZ was observed in gt1 and gt3 infections, respectively. BAG3, CARD11, CASP1 and CAV1 were upregulated at the peak of the gt3 infection. Upregulation of BCL2A1, CARD11 and CASP1 was detected during decline of both infections. During decline of infection, FAS was upregulated in gt1 infection and BAG3, BCL2A1, CARD11 and CASP1 were upregulated in the gt3 infection. After clearance of infection, downregulation of CASP8 was detected in both infections, and AGT, BCL2A1, CDKN1B and PKRCZ were downregulated, while CAV1 was upregulated in the gt3 infection.
3.8 |. NF-κB signalling pathways
In early infection, EGFR, LBP and TNFRSF1A were upregulated in the gt1 infection and BIRC3, EIF2AK2, FADD, RELB were upregulated in the gt3 infection. IRAK2 and REL were upregulated in both infections during early viremia. At the peak of infection, IL1B was detected in the gt1 infection, and expression of CD83, EIF2AK2, FASLG and RELB was elevated in the gt3 infection. BIRC3, CD40LG, PLAU and REL were upregulated in both infections. During decline of infection, CD14, EGFR, FOS, IL1A, IL1B, LY96, MAPK1 were upregulated in the gt1 infection and BCL3, BIRC3, CD83, EIF2AK2, LBP, PLAU, RELB and TNFRSF10A were upregulated in the gt3 infection. CD40LG, FASLG and REL were upregulated in both infections. After the clearance of infection, EGFR and EGR1 were upregulated in the gt1 infection and CD14 and LY96 were downregulated in the gt3 infection. REL was upregulated, and IL1A was downregulated in both infections.
3.9 |. Th1 and Th2 responses
Similar levels of Th1 response genes were induced by both infections except during early viremia. During early infection, upregulation of CCR2, IL12B, IL2RA and SOCS1 was detected in the gt3 infection and IL1RL1 in both infections. At the peak and decline of infection, CCR2, CCR5, CXCR3, HAVCR2 (Tim-3), IFN-γ, IL12B, IL2RA, SOCS1, STAT4 and TBX21 were upregulated in both infections. Upregulation of CD80 was seen in the gt1 infection and IL23A in the gt3 infection, with IL1RL1 in both infections detected at the peak of infection. ILRL1 and Il-27 were upregulated in the decline of gt1 infection. Upregulation of IFN-γ, IL12B, IL1RL1, STAT4 and TBX21 was detected after the clearance of the gt3 infection. Higher numbers of Th2 cytokines were upregulated in the gt1 infection than the gt3 infection (Figure 2). IL1R1 and IL9 were downregulated in early gt1 infection. CCR8 and ICOS were upregulated, and CCR3 and IL17RE were downregulated in early gt3 infection. At the peak of infection, CCR10, CCR4, CD86 and GATA3 were upregulated in the gt1 and IL10, IL1R1 and IL4 were upregulated in the gt3 infection. CCR8, GFI1 and ICOS were upregulated in both infections. During the decline infection, BCL6, CCL11, CCR8, CEBPB, GATA3, IL10, IL13RA1, IL17RE, IL18 and ILR1 were upregulated in the gt1 infection and upregulation of CD86, GFI, and ICOS was induced by both infections. After clearance of infection, upregulation of ICOS and IL10 in gt1, CCR4, and IL14 in the gt3 were detected, and downregulation of ILR1 was detected in both infections. STAT3 (Th17 maker gene) was upregulated during the gt3 infection, and IL17A was upregulated after clearance of gt3 infection. CD4 T-cell response gene, CTLA4 was upregulated in early gt1 infection. CD27, CTLA4 and PTPRC were upregulated at the peak and decline of both infections. During decline of infection CD4, CREBBP, IL-15, IL17R, LAT, MAPK8 and TNFSF4 were upregulated with gt1, and LAG3 and TNFRSF9 were upregulated with gt3.
3.10 |. Inflammatory and humoral immune responses
APCS and ICAM1 were downregulated in early gt1 infection. Upregulation of LYZ was detected at the peak of gt3 infection and during decline of both infections. MIF, SOCS2 and SPP1 were upregulated during decline of gt1 infection. MPO and SFTPD were upregulated after clearance of the gt3 infection. Humoral immune response gene, CCR6 was upregulated during replication of both viruses and after clearance of gt1 infection. IL7 and MBL2 were upregulated at the peak and decline of the gt1 infection, and RAG1 was upregulated in early and after clearance of gt3 infection.
4 |. DISCUSSION
Hepatic expression of host immune response-related genes in the gt3 infection was investigated and compared to gt1 infection using experimentally infected rhesus macaques. Profiles of host gene expression were analysed in the livers of experimentally infected rhesus macaques. Although two liver biopsy samples from each infected rhesus macaque were obtained during acute HEV infection, the liver tissues represented different clinical characteristics at time of biopsy (Table 1). We found that during the early phase of HEV RNA replication, the hepatic immune response-related genes were downregulated in the gt1 infection, but were upregulated in the gt3 infection (Figure 2). Upregulation of hepatic immune response-related genes was detected at the peak and decline of HEV RNA replication and after the anti-HEV antibody response in the gt1 infection (Figures 2 and 4). In the gt3 infection, IFN responsive genes were upregulated during the infection, while they were downregulated after the clearance of the infection. Our study suggests that HEV gt1 and gt3 infections may induce different host pathogenic mechanisms to control viral infection.
In a previous microarray study conducted in experimentally infected chimpanzees, most of the host response genes in HEV gt1 infection were upregulated during the first week of viremia before the anti-HEV antibody response.15 Although the timing of upregulated gene expression was different, chemokines (CXCL10) and a large number of ISG genes (IFIT1, IFIT3, ISG15, ISG20, OAS2, MX1, IRF7, and STAT1, MANU-A, MANU-B and TAP-1) were upregulated in gt1 infected rhesus macaques, which were also upregulated in gt1-infected chimpanzees. In addition, hepatic innate immune response genes were upregulated in humanized mice during gt1 infection.12 The humanized mice had more pronounced innate immune response gene expression after 9 weeks compared to 4 weeks after infection. This is similar to our observation where large numbers of immune response genes were upregulated during peak or decline of viremia in the gt1 infection. In addition, our data showed that hepatic immune response gene expression was downregulated during early viremia in the gt1 infection (Figure 2). Differences between our study and previous studies remain unclear. These differences could be pathobiological differences between rhesus and chimpanzee hepatocytes against HEV infection. Additionally, rhesus macaque-specific arrays were used with 500 ng of total RNA extracted from liver tissue to synthesize cDNA without amplification cycles in our study, whereas in the gt1 chimpanzee infection study a human microarray was used in which 6 ng of total RNA was amplified twice to generate cRNA, which may have induced selection bias. In our study, gene expression profiles for each macaque were analysed separately in which different animals with similar clinical markers had similar patterns of differentially regulated genes. These results indicated the reproducibility of hepatic immune gene expression profile identified in the gt1 and gt3 infected rhesus macaques (Table 1 and Figure 2). In addition, we found that when the infection was cleared, a large number of IFN responsive genes (18 of 54 genes) were downregulated in the gt3 infection (Figure 4). Even after HEV infection was cleared, proliferation and functional HEV-specific T-cell responses were detected in patients with acute gt3 infection.17 Previously, a significantly reduced induction of IFN-sensitive genes in activated T cells against vesicular stomatitis virus infection was reported.21 In addition, transcription factor forkhead box protein O3 (FOXO3), a negative regulator of IRF7 transcription, was found to shut down type I IFN response during the resolution phase in mice with VSV infection.22 These observations suggest that inhibition of the IFN response after clearance of the gt3 infection may be one way to balance host defence with inflammatory damage.
HEV gt1 and gt3 infections have distinct epidemiologic characteristics. Gt1 infects humans as a waterborne disease and is transmitted faecal orally. Gt3 is transmitted zoonotically, primarily through the consumption of raw or undercooked meat.5 Gt3 has been shown to grow in cell lines,23 but gt1 cell culture is inefficient and limited.24 Our expression profiles of immune response-related genes may provide additional evidence for the differences between gt1 and gt3 infections. Even though high levels of gt1 viral RNA were detected in the liver, stool and serum before the anti-HEV response; most immune response genes, especially IFN response and PRR related genes, were found to be downregulated in the gt1 infection, but not in the gt3 infection (Figures 2 and 4). The lack of IFN response gene expression in gt1 infected animals may be attributed to HEV gt1 virus’s ability to block PRR expression or interferon-α/β signalling via the ORF1 polyprotein,25 or the absence of exogenous interferon during the early phase of HEV RNA replication. On the other hand, high levels of IFN response genes (IFI27, IFI35, IFNA2 and OAS1) in the gt3 infection may be caused by enhancement of IFN induction by the ORF3 protein.26 However, in vitro studies have reported conflicting observations. Nan et al26 found that enhancement of IFN induction in both gt1 and gt3, where gt1 and gt3 ORF3 produced higher IFN-β response than was seen with gt2 or gt4, and Dong et al,19 showed suppression of IFN-α signalling in gt3-infected A549 cells. In addition, expression of the MDA5 gene was observed only in gt3 infection in our study. Higher levels of RIG-I expression were detected in the gt1 infection (Figure 2). RIG-I and MDA5 have similar signalling features and structure homology27; however, these two helicases are found to discriminate among different ligands to trigger the innate immune response to RNA viruses.28 A recent study found that RIG-I exerts antiviral activity through IFN-independent mechanisms against HEV infection.29 These observations provide evidence on different antiviral mechanisms induced against HEV gt1 and gt3 infection.
Large numbers of CD4 T-cell and Th1/Th2-related immune response genes were upregulated in gt1 infection, and IFN-response genes (IFI27, IFI35, IFNA2, IFITM1 and OAS1) were upregulated in gt3 infection. Gt1 and gt3 infections induced upregulation of different ligands and receptors (IL1A and IL1B in the gt1 infection and CD83 in the gt3 infection), and kinase [MAPK1 in the gt1 infection and EIF2AK2 in the gt3 infection] genes in the NF-κB signalling pathway responses (Figure 2). Additionally, in early viremia and before anti-HEV antibody response, 5 and 6 of 24 NF-kB signalling pathway genes were upregulated in the gt1 and gt3 infections, respectively, indicating early induction of the NF-κB signalling pathway during HEV replication. These results further support that early innate immune responses in HEV gt1 and gt3 infections may induce different pathological mechanisms to control virus infection.
In summary, we identified expression profiles of hepatic immune response genes in HEV gt1 and gt3 infection. Hepatic immune response-related genes were found to be substantially different during HEV gt1 and gt3 virus replication, as well as the genes associated with viral clearance in experimentally infected rhesus macaques. Gene expression profiles induced by the gt3 infection indicate high levels of innate immune responses expressed at the early viremic stage of infection. For the gt1 infection, early host immune responses are inhibited until the activation of adaptive/humoral immune response. It may also be possible that host gene expression profiles identified in our study could be specific to a strain of HEV gt1 and gt3. Testing additional stains of HEV gt1 and gt3 viruses is warranted for future study.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Michael A. Purdy (Division of Viral Hepatitis, CDC) for critical reviews of the manuscript and helpful discussions, and animal caretakers (Comparative Medicine Branch, CDC) for husbanding the animals. This information has not been formally disseminated by the Centers for Disease Control and Prevention/Agency for Toxic Substances and Disease Registry. It does not represent and should not be construed to represent any agency determination or policy. Use of trade names is for identification only and does not imply endorsement by the U.S. Department of Health and Human Services, the Public Health Service, or the Centers for Disease Control and Prevention. The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention.
Funding information
National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention, CDC.
Abbreviations:
- ALT
alanine aminotransferase
- ALT
alanine aminotransferase
- CDC
Centers for Disease Control and Prevention
- CTL
cytotoxic T lymphocyte
- DAI
days after inoculation
- gt
genotype
- HEV
hepatitis E virus
- IACUC
Institutional Animal Care and Use Committee
- NF-kB
Nuclear factor k enhancer binding protein
- ORF
open reading frames
- PAMP
pathogen-associated molecular patterns
- PRR
pattern recognition receptor
- RH
rhesus macaques
Footnotes
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article.
REFERENCES
- 1.Rein DB, Stevens GA, Theaker J, Wittenborn JS, Wiersma ST. The global burden of hepatitis E virus genotypes 1 and 2 in 2005. Hepatology. 2012;55:988–997. [DOI] [PubMed] [Google Scholar]
- 2.Smith DB, Simmonds P, Izopet J, et al. Proposed reference sequences for hepatitis E virus subtypes. J Gen Virol. 2016;97:537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woo PC, Lau SK, Teng JL, et al. New hepatitis E virus genotype in Bactrian Camels, Xinjiang, China, 2013. Emerg Infect Dis. 2016;22:2219–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kamar N, Dalton HR, Abravanel F, Izopet J. Hepatitis E virus infection. Clin Microbiol Rev. 2014;27:116–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meng XJ. Zoonotic and foodborne transmission of hepatitis E virus. Semin Liver Dis. 2013;33:41–49. [DOI] [PubMed] [Google Scholar]
- 6.Sayed IM, Vercouter AS, Abdelwahab SF, Vercauteren K, Meuleman P. Is hepatitis E virus an emerging problem in industrialized countries? Hepatology. 2015;62:1883–1892. [DOI] [PubMed] [Google Scholar]
- 7.Lee GH, Tan BH, Chi-Yuan Teo E, et al. Chronic infection with camelid hepatitis E virus in a liver transplant recipient who regularly consumes camel meat and milk. Gastroenterology. 2016;150:355–357e3. [DOI] [PubMed] [Google Scholar]
- 8.Geng Y, Zhang H, Huang W, et al. Persistent hepatitis e virus genotype 4 infection in a child with acute lymphoblastic leukemia. Hepat Mon. 2014;14:e15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kamar N, Weclawiak H, Guilbeau-Frugier C, et al. Hepatitis E virus and the kidney in solid-organ transplant patients. Transplantation. 2012;93:617–623. [DOI] [PubMed] [Google Scholar]
- 10.Emerson SU, Zhang M, Meng XJ, et al. Recombinant hepatitis E virus genomes infectious for primates: importance of capping and discovery of a cis-reactive element. Proc Natl Acad Sci USA. 2001;98:15270–15275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Purcell RH, Engle RE, Govindarajan S, et al. Pathobiology of hepatitis E: lessons learned from primate models. Emerg Microbes Infect. 2013;2:e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sayed IM, Verhoye L, Cocquerel L, et al. Study of hepatitis E virus infection of genotype 1 and 3 in mice with humanised liver. Gut. 2016;66:920–929. [DOI] [PubMed] [Google Scholar]
- 13.Allweiss L, Gass S, Giersch K, et al. Human liver chimeric mice as a new model of chronic hepatitis E virus infection and preclinical drug evaluation. J Hepatol. 2016;64:1033–1040. [DOI] [PubMed] [Google Scholar]
- 14.van de Garde MDB, Pas SD, van Oord GW, et al. Interferon-alpha treatment rapidly clears Hepatitis E virus infection in humanized mice. Sci Rep. 2017;7:8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu C, Boon D, McDonald SL, et al. Pathogenesis of hepatitis E virus and hepatitis C virus in chimpanzees: similarities and differences. J Virol. 2010;84:11264–11278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suneetha PV, Pischke S, Schlaphoff V, et al. Hepatitis E virus (HEV)-specific T-cell responses are associated with control of HEV infection. Hepatology. 2012;55:695–708. [DOI] [PubMed] [Google Scholar]
- 17.Gisa A, Suneetha PV, Behrendt P, et al. Cross-genotype-specific T-cell responses in acute hepatitis E virus (HEV) infection. J Viral Hepat. 2016;23:305–315. [DOI] [PubMed] [Google Scholar]
- 18.Weatherall D The use of non-Human Primates in Research: A Working Group Report Chaired by Sir David Weatherall FRS FMedSci. London, UK: The Academy of Medical Sciences, Medical Research Council, The Royal Society, Wellcome Trust; 2006. [Google Scholar]
- 19.Dong C, Zafrullah M, Mixson-Hayden T, et al. Suppression of interferon-alpha signaling by hepatitis E virus. Hepatology. 2012;55:1324–1332. [DOI] [PubMed] [Google Scholar]
- 20.Baylis SA, Blumel J, Mizusawa S, et al. World Health Organization International Standard to harmonize assays for detection of hepatitis E virus RNA. Emerg Infect Dis. 2013;19:729–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dondi E, Rogge L, Lutfalla G, Uze G, Pellegrini S. Down-modulation of responses to type I IFN upon T cell activation. J Immunol. 2003;170:749–756. [DOI] [PubMed] [Google Scholar]
- 22.Litvak V, Ratushny AV, Lampano AE, et al. A FOXO3-IRF7 gene regulatory circuit limits inflammatory sequelae of antiviral responses. Nature. 2012;490:421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okamoto H Culture systems for hepatitis E virus. J Gastroenterol. 2013;48:147–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen HT, Shukla P, Torian U, Faulk K, Emerson SU. Hepatitis E virus genotype 1 infection of swine kidney cells in vitro is inhibited at multiple levels. J Virol. 2014;88:868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nan Y, Yu Y, Ma Z, Khattar SK, Fredericksen B, Zhang YJ. Hepatitis E virus inhibits type I interferon induction by ORF1 products. J Virol. 2014;88:11924–11932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nan Y, Ma Z, Wang R, et al. Enhancement of interferon induction by ORF3 product of hepatitis E virus. J Virol. 2014;88:8696–8705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoneyama M, Kikuchi M, Matsumoto K, et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175:2851–2858. [DOI] [PubMed] [Google Scholar]
- 28.Loo YM, Fornek J, Crochet N, et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82:335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu L, Wang W, Li Y, et al. RIG-I is a key antiviral interferon-stimulated gene against hepatitis E virus regardless of interferon production. Hepatology. 2017;65:1823–1839. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.