

Abstract
Proteolysis-targeting chimeras (PROTACs) are engineered techniques for targeted protein degradation. A bifunctional PROTAC molecule with two covalently-linked ligands recruits target protein and E3 ubiquitin ligase together to trigger proteasomal degradation of target protein by the ubiquitin-proteasome system. PROTAC has emerged as a promising approach for targeted therapy in various diseases, particularly in cancers. In this review, we introduce the principle and development of PROTAC technology, as well as the advantages of PROTACs over traditional anti-cancer therapies. Moreover, we summarize the application of PROTACs in targeting critical oncoproteins, provide the guidelines for the molecular design of PROTACs and discuss the challenges in the targeted degradation by PROTACs.
Keywords: PROTAC, Targeted cancer therapy, Ubiquitin-proteasome system, Protein degradation
Introduction
Targeted cancer therapies aim to target cancer-associated biomolecules (such as oncoproteins) and interfere with their oncogenic cellular processes in cancer tissues. In the past several decades, targeted therapies have achieved remarkable advances in cancers and become a powerful treatment strategy for cancer patients. For example, small molecular inhibitors or monoclonal antibodies have been successfully developed to target overexpressed or overactivated proteins in cancer [1]. However, due to limited therapeutic benefit, drug resistance and off-target effect of these targeted therapies, researchers are still seeking more effective and specific strategy to target cancer-related oncoproteins.
Inspired by the fact that cells employ the ubiquitin-proteasome system (UPS) to maintain intracellular protein homeostasis, Deshaies laboratory designed and synthesized the functional molecule Protac-1 to induce the degradation of methionine aminopeptidase-2 (MetAP-2) via recruiting UPS in 2001. Protac-1 consists of three covalently-linked segments: a domain containing the IκBα phosphopeptide that is recognized by Skp1-Cullin-F-box complex (SCF, an E3 ligase to initiate protein ubiquitination and degradation by UPS), a domain having ovalicin (MetAP-2 inhibitor), and a linker connecting these two domains [2]. This work proposed the initial concept of proteolysis-targeting chimeras (PROTACs), an engineered technique that induces degradation of protein of interest (POI) via UPS in living cells. Subsequently, researchers developed different peptide-based PROTACs to eliminate the disease-promoting proteins, such as androgen receptor (AR), estrogen receptor (ER), FK506 binding protein (FKBP12) and aryl hydrocarbon receptor (AHR) [3–6]. Because the peptide backbones have low lipophilicity (unfavorable to cross cell membrane) and are easily hydrolyzed by digestive enzymes, these peptide-based PROTACs have poor cell permeability and low stability, limiting their application.
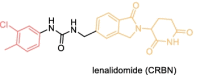
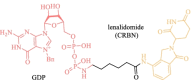
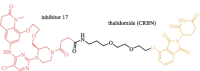
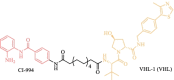
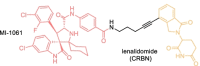
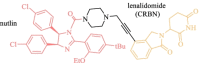
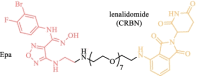
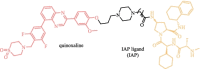
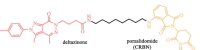
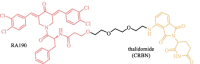
Given that some small chemical molecules exhibit stronger lipophilicity, Crews’ group developed the first small-molecule based PROTAC in 2008 to effectively degrade AR in cancer cells. This cell permeable PROTAC comprises the chemical nutlin (the E3 ligase MDM2 inhibitor) and a non-steroidal AR ligand (SARM), connected by a PEG-based linker [7]. In 2010, Itoh et al. utilized the chemical methyl bestatin to synthesize another PROTAC molecule, thus recruiting the E3 ligase inhibitor-of-apoptosis-protein (IAP) to degrade POI [8]. To increase potency and target selectivity, small molecules with high affinity and specificity, such as phthalimides recruiting the E3 ligase cereblon (CRBN) [9–14] or VHL-1 recognizing the E3 ligase Von Hippel-Lindau (VHL) [15–17], were introduced into PROTACs to downregulate numerous cancer targets, such as Ikaros family zinc finger protein 1/3 (IKZF1/3) and estrogen-related receptor alpha (ERRα). The breakthroughs of small molecule-based PROTACs pave the way for PROTACs as therapeutic anticancer strategies.
Recently, a series of novel PROTACs have been developed to expand their applications with more advantages, such as RNA-PROTAC for degrading undruggable RNA-binding proteins [18], PhotoPROTAC for optical control of protein degradation [19–25], and CLIPTAC for increasing bioavailability [26]. Importantly, PROTAC is a highly promising technology for clinical applications, given that Arvinas Therapeutics Company has initiated the first-in-human trial in 2019 (i.e., PROTAC ARV-110 targeting AR for the treatment of prostate cancer), and at least 15 targeted degraders are expected to enter clinical trials by the end of 2021 [27].
In this review, we introduce the principle and development of PROTAC technology and summarize the application of PROTACs in targeting crucial oncoproteins. Furthermore, we discuss the challenges in PROTAC realm and propose the guidelines to design excellent PROTACs for targeted cancer therapy.
Principle of PROTACs
PROTACs hijack the ubiquitin-proteasome system (UPS)
UPS is a highly conserved mechanism for degradation of both normal and misfolded proteins in eukaryotic cells, thus keeping intracellular protein homeostasis [28–30]. In UPS, proteins to be degraded are covalently tagged with ubiquitin (Ub, a 76-amino acid protein), and this tagging process is catalyzed by three enzymes known as Ub-activating enzyme (E1), Ub-conjugating enzyme (E2) and Ub-ligase (E3): free Ub is activated by E1 and then attached to the cysteine residue (Cys) of E1 to form a thioester bond via an ATP-dependent reaction; the Ub-tagged E1 transfers its Ub to the Cys of E2 through a trans-thioesterification reaction; E3 recruits Ub-tagged E2 and E3 substrate to label the ubiquitin at the lysine residue (Lys) of the substrate. Such repeated ubiquitination processes generate a poly-Ub chain (mainly linked through Lys48 of Ub) on the target protein, which guides the substrate to 26S proteasome for degradation [31, 32] (Fig. 1). In human proteome, there are two E1s, about forty E2s and more than 600 E3s. Among them, the E3 ligases are responsible for specifically recognizing substrates.
Fig. 1.

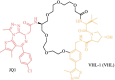
The mechanism of PROTACs based on the UPS. UPS consists of specific enzymes (E1, E2 and E3) modifying proteins with ubiquitin and the proteasome degrading the ubiquitin-tagging proteins. PROTAC contains a POI ligand, an E3 ligand and a linker. The E3-PROTAC-POI ternary complex induces the polyubiquitination and proteasome-mediated degradation of POIs. The presented PROTAC is BRD4 degrader MZ1 that is composed of POI ligand JQ1 (red) and E3 ligand VHL-1 (yellow)
Inspired by UPS, researchers designed PROTACs to hijack the UPS and degrade POI. PROTAC molecule consists of three covalently-bonded moieties: a ligand to bind POI (POI ligand), another ligand to recognize E3 ligase (E3 ligand) and a linker to conjugate the two ligands. PROTAC simultaneously recruits E3 ligase and POI, forming the “E3-PROTAC-POI” ternary complex. Gadd et al. solved the crystal structure of bromodomain-containing protein 4 (BRD4) PROTAC MZ1 in complex with human VHL and BRD4 bromodomain, supporting the formation of the ternary complex [33]. This complex potentiates the substrate recognition by E3 ligase and promotes the transfer of Ub to POI, accelerating the poly-ubiquitination and subsequent proteasome-mediated degradation of POI [34] (Fig. 1).
Hook effect of PROTAC
The bifunctional molecule (“B”) interacts with its two substrates (“A” and “C”), forming “A-B-C” ternary complex to exert its biological functions. When the concentration of “B” exceeds a certain range, “B” prefers to form “A-B” and “B-C” binary complexes, instead of “A-B-C” ternary complex, thus reducing the activity of “B”. This phenomenon is termed as the “hook effect” [35]. As a bifunctional molecule, high-dose of PROTAC tends to form “PROTAC-POI” and/or “PROTAC-E3” complexes rather than “POI-PROTAC-E3” ternary complex (required for POI degradation), thus reducing its degradation potency [36–39]. Hook effect exists in most known PROTACs, thereby this effect is available to check whether the synthesized PROTAC is bifunctional.
To avoid hook effect, a wide range of PROTAC concentrations should be tested in cellular activity assays to determine the maximal concentration without hook effect [40]. Unfortunately, the research on hook effect of PROTACs in vivo is lacking, so it’s hard to choose an appropriate concentration of PROTAC in in vivo application [41, 42]. Intriguingly, some PROTACs could trigger the positive cooperative assembly of ternary complexes by inducing “neocontacts” between E3 and POI (e.g., “neocontacts” between VHL and BRD4 caused by PROTAC MZ1). These “neocontacts” stabilize “POI-PROTAC-E3” ternary complex and increase the threshold for triggering the hook effect [33]. Therefore, optimizing the PROTAC structure to enhance this “neocontacts” is a potential method to avoid hook effect to some extent.
Advantages and disadvantages of PROTAC
Diverse therapeutic strategies, such as small-molecule inhibitor, monoclonal antibody, RNA interference and CRISPR/Cas9, have been developed to treat human cancers [43–46]. The unique chemical and biological features of PROTAC endow it with advantages and disadvantages in cancer therapy (Table 1).
Table 1.
Comparisons of different targeted cancer therapies
| PROTAC | CRISPR/Cas9 | RNA interfering | small-molecule inhibitor | monoclonal antibody | |
|---|---|---|---|---|---|
| Requirement of active sites | No | No | No | Yes | Yes |
| Elimination of pathogenic proteins | Yes | Yes | Yes | No | No |
| Undruggable targets | Yes | Yes | Yes | No | Yes |
| Tissue penetration | Moderate | Poor | Poor | Yes | Poor |
| Intracellular targets | Yes | Yes | Yes | Yes | No |
| Systemic delivery | Yes | Poor | No | Yes | Yes |
| Catalytic mechanism of action | Yes | Yes | Yes | No | No |
| Route of administration | PO/IV/SC | IV | IV/SC | PO/IV/SC | IV/SC |
Note: IV intravenous injection, PO peros, SC Subcutaneous injection
Advantages of PROTAC
Event-driven mechanism
The activity of small molecule drugs, especially the FDA-approved inhibitors, is usually driven by occupancy of target (called “occupancy-driven mechanism”), while PROTACs act as catalysts to initiate degradation event of target protein in a repeatable manner (called “event-driven mechanism”) [36]. Thus, one equivalent of PROTAC could degrade multiple equivalents of POI, allowing the dosage, administration frequency and toxicity of PROTACs lower than those of small-molecule drugs. Additionally, due to the catalytic behavior of PROTAC, transient or low-abundance ternary complexes are sufficient to achieve target degradation. Therefore, ligands with lower POI/E3 affinity and high selectivity are favorable for PROTAC activity, enabling the rapid assembly/disassembly of functional ternary complex. Moreover, PROTACs eliminate the whole functions of targets, overcoming the therapeutic challenges (commonly occurred in the treatment by small-molecule inhibitors) caused by the non-catalytic functions or gain/loss-of-function mutations of POIs [47].
Degrading “undruggable” targets
Many proteins, such as DNA-binding proteins (DBPs, e.g. transcriptional factor c-myc) and RNA-binding proteins (RBPs, e.g. IGF2BPs), play important roles in cancer initiation and progression and are regarded as high-value therapeutic targets. But these proteins generally lack targetable pockets (orthosteric or allosteric sites), so they are deemed “undruggable” by small-molecule inhibitors. PROTACs could use the low-affinity small-molecule ligands (transiently associated with the possible binding site of POI) or the oligonucleotides as protein decoys to release the dependence on the well-defined targetable pockets, providing opportunities to degrade “undruggable” proteins [48].
Avoiding compensatory protein expression
Targeted therapies, such as small-molecule inhibitors, may trigger compensatory protein expression after administration, which decreases drug efficacy and increases side effects [49]. For instance, treatment with statin, HMG-CoA reductase (HMGCR) inhibitor, increased HMGCR level by enhancing gene transcription and retarding protein degradation, thus attenuating statin’s activity to treat cardiovascular diseases [50]. PROTAC can potently downregulate POI protein level through accelerating UPS-mediated degradation, thus offering a pathway to prevent compensatory protein expression of POI. Moreover, genetical interference with short hairpin RNA might induce a secondary cellular response (e.g., by triggering the compensatory mechanism) to maintain cell homeostasis, so it is difficult to disclose the bona fide function of proteins. Because of acute and reversible depletion of protein, PROTAC could be a molecular tool to dissect protein function [40].
Disadvantages of PROTAC
PROTAC needs to enter cells to mobilize intracellular UPS, so its membrane permeability is the key to PROTAC’s function. Currently, the penetration mechanism of PROTAC has not yet been elucidated. Most known PROTACs have the molecular weights (M.W.) of 1000–2000 Da [51, 52], so they penetrate cell membrane mainly through passive diffusion and active transport. Nevertheless, large M.W. and large exposed polar surface area of PROTACs makes their cell/tissue permeability worse than small molecules. Various strategies have been employed to improve permeability of PROTACs. The common ways are to limit its M.W. below 1000 Da [53] or to split the molecule into two smaller precursors and generate mature PROTAC in cells (CLIPTAC) [26]. Additionally, the cell permeability of PROTAC could be increased by introducing long flexible linkers to form intramolecular hydrogen bonds that partially reduce polarity [54], or attaching cell-permeable peptides (such as poly-D-arginine sequence) to E3 ligands [3]. Except for modifying PROTAC itself, application of nanoparticles such as liposomes to deliver PROTAC also significantly enhanced the cellular uptake of PROTACs [55].
Currently, the design of PROTACs needs known POI/E3 ligands as protein decoys, so PROTAC development largely depends on the discovery and optimization of these ligands. Moreover, some known POI/E3 ligands exhibit low specificity, making such PROTACs have off-target effects [34]. Therefore, identifying highly specific POI/E3 ligands is critical for developing good PROTACs.
Development of PROTAC technology
Classification of PROTAC
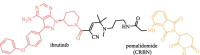
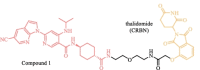
According to the chemical structure of POI ligands, PROTACs could be divided into peptide-based, small molecule-based and nucleotide-based ones. Peptide-based PROTACs contain peptidic POI ligands mimicking the sequences of native POI-binding proteins. For example, Signal transducer and activator of transcription 3 (STAT3) is a critical transcription factor and its hyperactivation is tightly associated with cancer initiation and progression [56]. SI-109, a peptide stemmed from the STAT3-binding motif of the protein gp130, was utilized to develop a peptide-based PROTAC termed as SD-36, which achieved potent STAT3 degradation and inhibited leukemia and lymphoma in vitro and in vivo [57]. Peptide-based PROTACs have advantages in binding affinity, target specificity and chemical synthesis, while they suffer from limited membrane permeability and digestive intolerance [58]. Thus, peptide-based PROTAC is usually intravenously injected and especially suitable for the membrane proteins or the treatment of hematological diseases.
Small molecules, especially FDA-approved anticancer inhibitors, could be used as the POI ligands to build small molecule-based PROTACs. For instance, BRAFV600E (a mutant of RAF kinase) prevalently occurs in melanoma and colorectal cancer, driving oncogenic ERK signaling even in the absence of activated RAS [59]. Posternak et al. introduced the small-molecule BRAFV600E inhibitor BI882370 as POI ligand and pomalidomide as the E3 ligand for CRBN. The obtained PROTAC P4B exhibited effective BRAF degradation to inhibit melanoma and colon cancer harboring BRAF mutation [60]. Compared with peptide-based PROTAC, small molecule-based one displays improved cell permeability and resistance to digestion, thus allowing more manners of administration and expanded target scope. Notably, small-molecule ligands usually have poor target specificity [58], so more concerns should be given to this point when selecting or optimizing small-molecule POI ligands.
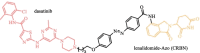
Recently, nucleotide-based PROTACs have been developed, which use oligonucleotides as POI ligands. Numerous RBPs (e.g. Lin28, IGF2BPs and Musashi-1/2) and DBPs (e.g. NF-κB, c-myc and STAT3) are overexpressed and/or overactivated in human cancers, promoting tumorigenesis and cancer development. But discovery of RBP/DPB-targeted drugs is challenging due to the lack of targetable binding pockets within RBPs/DBPs. For instance, Lin28 is a highly conserved RBP that promotes tumorigenesis by interacting with let-7 precursor (pre-let-7) to inhibit the biogenesis of let-7, a tumor suppressive microRNA [61]. But Lin28 doesn’t have well-defined targetable pockets for small-molecule intervention. Ghidini et al. utilized the Lin28-binding oligoribonucleotides derived from pre-let-7 as POI ligand to synthesize a nucleotide-based PROTAC termed RNA-PROTAC. This PROTAC accomplished remarkable Lin28 degradation in leukemia cells with high selectivity and negligible toxicity [18]. In addition, Samarasinghe et al. developed DNA-based PROTAC (termed TRAFTAC), which used DNA sequence as POI ligand to recognize the transcription factor NF-κB for targeted degradation [62]. These nucleotide-based PROTACs expand the concept of PROTACs and provide a novel strategy for cancer treatment.
New concepts of PROTAC technologies
PhotoPROTAC
Through optically-controlled generation or release of active small-molecule modulators, light with high spatiotemporal resolution has been widely used in biomedical research and disease treatment [63]. Some moieties (e.g. azobenzene) within molecules could be reversibly or irreversibly changed under light stimulation, altering the spatial configuration and the physical/chemical/biological properties of molecules. This concept inspired the development of PhotoPROTAC, which utilized the photoswitches [64] or the photocages [65] to realize the spatiotemporal control of PROTAC function (Fig. 2a).
Fig. 2.

The emerging new concepts of PROTAC technologies. a photoswitchable PROTAC (upper) achieves reversible optical control of protein degradation by interconverting between inactive and active conformers, and photocaged PROTAC (lower) irreversibly achieves light-induced protein degradation by removing photocaging group. b CLIPTAC can be formed intracellularly through click combination of two tagged precursors. c HaloPROTAC and dTAG system utilize tag fusion POI and ligand that bind to tag protein
Photoswitchable PROTACs optically control protein degradation in a reversible manner by using a photoswitchable moiety (e.g. azobenzene) on linker or E3 ligand. Without light irradiation, PROTAC maintains the inactive conformation that is unable to form a stable ternary complex. Upon light exposure at the designed wavelength, PROTAC switches to the active conformation, forming a functional ternary complex to degrade target [19–21]. For example, Reynders et al. designed PHOTACs involving azobenzene moiety on the linker to degrade BET family proteins and suppress acute lymphoblastic leukemia (ALL) cells in the presence of 390 nm UV light [21]. Notably, with appropriate light exposure (e.g. 500 nm for azobenzene-containing PROTAC), target degradation could be halted by converting the PROTAC into an inactive conformation [19–21].
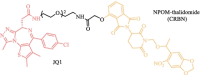
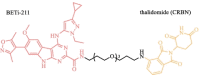
Photocaged PROTACs irreversibly accomplish protein degradation by incorporating photolabile blocking groups (e.g. nitroveratryloxycarbonyl group, NVOC). Without light stimulation, the photocaging group labeling to the E3 ligand impairs the binding between PROTAC and E3 ligase. Upon light exposure, the photocaging group is released from PROTAC, benefiting the formation of POI-PROTAC-E3 ternary complex [22–25]. For instance, Liu et al. used NVOC on CRBN ligand to synthesize photocaged PROTACs. These obtained opto-PROTACs were able to degrade IKZF1/3, BRDs or ALK fusion protein (using corresponding POI ligands) upon 365 nm UV irradiation, inhibiting cancer cell proliferation in an optical-controlled manner [22].
CLIPTAC
PROTACs usually have large M.W., limiting their solubility, pharmacokinetics and bioavailability, thus how to reduce the M.W. of PROTACs is critical. Click chemistry, coined by Sharpless group, is used to describe chemical reactions with the advantages of benign reaction condition, high yielding, high selectivity as well as wide scope [66]. To date, click chemistry has developed as a fundamental technology to covalently modify biomolecules under physiological conditions, which is particularly suitable for building conjugated skeletons from two small precursors in cells [67, 68]. Inspired by the concept of click chemistry, Lebraud et al. prepared a tetrazine-tagged E3 ligand and a trans-cyclooctene-tagged POI ligand as the precursors. Via the click reaction between tetrazine and trans-cyclooctene, generating a covalent six-membered ring moiety, these two precursors formed integrated PROTACs (termed as CLIPTAC) in cells (Fig. 2b) that successfully degraded oncogenic BRD4 or ERK1/2 [26]. Therefore, the CLIPTAC has become an attractive solution for reducing the M.W. of PROTACs.
Tag-based PROTAC
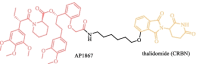
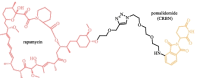
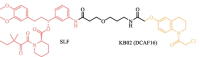
The ab initio development of PROTAC is a time-consuming and multistep process, including molecular design, chemical synthesis and cell−/animal-based evaluation [51]. The selection of appropriate E3 ligase/E3 ligand system is crucial for the progress of PROTAC research. However, there are more than 600 E3 ligases in human proteome and the atlas for POI-E3 ligase interactions is far from clear. Thus, researchers have established the tag-based PROTAC systems, in which the tag-POI fusion protein was expressed in cells and the universal PROTAC molecule was administrated to recruit the candidate E3 ligase and the tag of tag-POI protein. Measuring the abundance of tag-POI protein was able to verify whether the candidate E3 ligase could initiate POI degradation. The most widely-used tag-based PROTACs are HaloPROTAC and dTAG (Fig. 2c) [69–72]. These tag-based PROTACs suggest promising molecular tools to check whether a candidate E3 ligase/E3 ligand system is suitable for PROTAC, but they could not be used as the therapeutics for disease treatment.
PROTACs in targeted cancer therapy
Cancer initiation and progression is a complex process characterized by sustaining proliferative signaling, evading growth suppressors, resisting cell death, inducing angiogenesis, activating invasion and metastasis [73]. Compelling evidence has demonstrated that some overexpressed and/or overactivated proteins play crucial roles in tumorigenesis and act as potential therapeutic targets. Here we summarize the applications of PROTACs in targeted cancer therapy.
Targeting cancer cell proliferation
The growth-promoting signals, including RAS-RAF-MEK-ERK pathway, are frequently hyperactivated in tumors, eliciting cell cycle progression to induce the uncontrolled cell proliferation [74, 75] (Fig. 3). PROTAC technology has been applied to target the overexpressed, overactivated or mutated proteins involved in cell cycle regulation (Table 2).
Fig. 3.

PROTACs targeting cancer proliferation. In cell cycle regulation, some proteins (e.g. c-Myc, p21) act as accelerator or inhibitor to regulate CDK expression. CDKs and their chaperones phosphorylate retinoblastoma protein (Rb), thus releasing transcription factor E2F and promoting DNA replication. The RAS-RAF-MEK-ERK pathway also plays a central role in growth-promoting signaling and elicits cell cycle progression. These key elements in cancer proliferation can be targeted by diverse PROTACs (red arrow). Tumor-suppressor proteins are indicated in blue and oncogenic proteins are indicated in red. In the presented pathways, PROTACs have been developed targeting BRD4 [12, 76], CDK4/6 [77, 78], EGFR [79, 80], AURORA-A [81], Raf [60], BRD7/9 [82, 83], CDK2/5 [84], ERK1/2 [26], HER2 [79], MEK1/2 [85, 86], Ras [87] and Wee1 [88]
Table 2.
The structures of PROTAC molecules targeting cell proliferation in cancers. (red: POI ligand; yellow: E3 ligand)
| Target | PROTAC | PROTAC structure | Cancer | Ref | Target | PROTAC | PROTAC structure | Cancer | Ref |
|---|---|---|---|---|---|---|---|---|---|
| BRD4 | ARV-825 |
|
BL | [39] | BRD4 | SNIPER(BRD4)-1 |
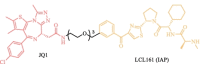
|
BC | [89] |
| BRD4 | dBET1 |
|
AML | [12] | BRD4 | A1874 |
|
CRC, M, BL, et al. | [90] |
| BRD4 | MZ1 |
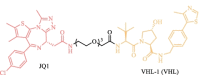
|
CC | [33, 91] | BRD4 | CLIPTAC-BRD4 |
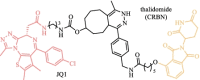
|
CC, M, CRC | [26] |
| BRD4 | dBET6 |
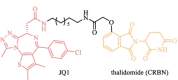
|
T-ALL | [92] | BRD4 |
opto-dBE T1 |
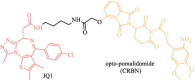
|
MM, PC, ALCL, et al. | [22] |
| BRD4 | ZXH-3-26 |
|
MM | [93] | BRD4 | PROTAC4 |
|
BC, PC | [23] |
| BRD4 | ARV-771 |
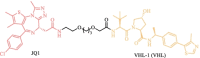
|
PC | [76] | BRD4 | pc-PROTAC1 |
|
BL, HCC | [25] |
| BRD4 | macroPROTAC-1 |
|
PC, AML | [94] | BRD4 | PHOTAC-I-3 |
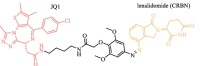
|
ALL | [21] |
| BRD4 | BETd-246 |
|
TNBC | [95] | BRD4 | photoPROTAC-1 |
|
BL | [20] |
| BRD4 | BETd-260 |
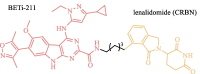
|
AML, ALL | [96] | BRD4 | PROTAC3 |
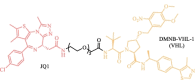
|
CC | [24] |
| BRD4 | KB02-JQ1 |
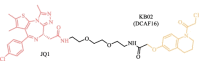
|
/ | [97] | EGFR | compound 1 |
|
OC, CC | [79] |
| BRD4 | XH2 |
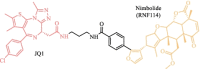
|
BC | [98] | EGFR | compound 14o |
|
NSCLC | [99] |
| BRD4 | DP1 |
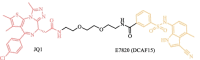
|
DLBCL | [100] | EGFR | compound P3 |
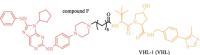
|
LC | [101] |
| CDK4/6 | BSJ-03-123 |
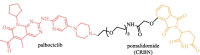
|
AML | [102] | EGFR | MS154 |
|
LC | [103] |
| CDK4/6 | pal-pom |

|
TNBC | [104] | EGFR | DDC-01-163 |
|
LC | [80] |
| CDK4/6 | BSJ-02-162 |
|
MCL | [77] | AURORA-A | JB170 |
|
AML, OS, NB, HCC | [81] |
| CDK4/6 | BSJ-04-132 |
|
T-ALL | [77] | Raf | P4B |
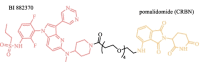
|
M, CRC | [60] |
| CDK4/6 | YX-2-107 |
|
ALL | [78] | AR | PROTAC 14 |

|
CC | [7] |
| CDK4/6 | compound 11 |
|
M | [105] | AR | ARCC-4 |
|
PC | [106] |
| CDK4/6 | PROTAC 34 |
|
ALL, AML, MM, TNBC | [107] | AR | TD-802 |
|
PC | [108] |
| AR | MTX-23 |
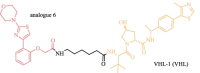
|
PC | [109] | BRD7/9 | ACBI1 |
|
AML, M, NSCLC | [83] |
| AR | ARD-69 |
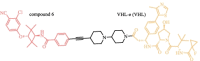
|
PC | [110] | CDK2/5 | TMX-2172 |
|
OC | [84] |
| AR | SNIPER(AR)-51 |
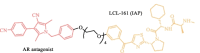
|
PC | [111] | CDK8 | JH-XI-10-02 |
|
T-ALL | [112] |
| ALK | MS4077 |
|
NSCLC, ALCL | [113] | CDK9 | PROTAC 3 |
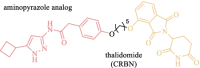
|
CRC | [114] |
| ALK | TL13–12 |
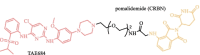
|
ALCL, NB, NSCLC | [52] | CDK9 | THAL-SNS-032 |
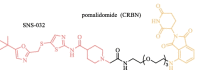
|
T-ALL | [38] |
| ALK | opto-dALK |
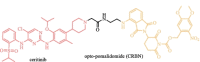
|
NSCLC, ALCL | [22] | CDK9 | B03 |
|
AML | [115] |
| ALK | SIAIS117 |
|
NSCLC, ALCL | [116] | CDK9 | compound F3 |
|
PC | [117] |
| BLK | PROTAC 7 |
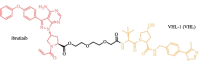
|
CML | [118] | Cdc20 | CP5V |
|
BC | [119] |
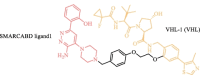
| BRD7/9 | dBRD9 |
|
AML | [82] | c-Met | PROTAC 7 |
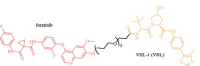
|
TNBC, GC | [79] |
| BRD7/9 | VZ185 |
|
MRT | [120] | CREPT | PRTC |
|
PaC | [121] |
| CYP1B1 | compound 6C |
|
PC | [122] | MEK1/2 | MS432 |
|
CRC, M | [86] |
| DHODH | probe 10 |
|
PaC | [123] | Ras | LC-2 |
|
NSCLC, PaC | [87] |
| ER | ERD-308 |

|
BC | [124] | GSPT1 | CC-885 |
|
AML | [125] |
| ER | compound I-6 |
|
BC | [126] | PLK1 | HBL-4 |
|
AML | [127] |
| ER | TD-PROTAC |
|
BC | [128] | SLC9A1 | d9A-2 |
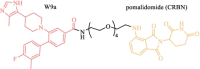
|
CML | [129] |
| ER | SNIPER(ER)-87 |
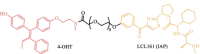
|
BC | [89] | TACC3 | SNIPER(TACC3)-1 |
|
FS, OS, CRC | [130] |
| ERK1/2 | ERK-CLIPTAC |
|
CC, M, CRC | [26] | TRIM 24 | dTRIM24 |
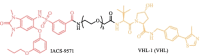
|
AML | [131] |
| FLT-3 | FLT-3 PROTAC |
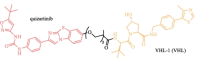
|
AML | [132] | TRKA/C | CG416 |
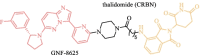
|
CRC, AML | [133] |
| HER2 | PROTAC 1 |
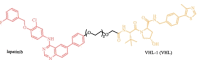
|
OC, BC | [79] | Wee1 | ZNL-02-096 |
|
OC, T-ALL | [88] |
| MEK1/2 | compound 3 |
|
M | [85] | α1A-AR | α1A-AR |
|
PC | [134] |
BRD4
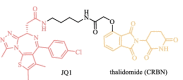
BRD4, a member of the bromodomains and extraterminal (BET) family, is an epigenetic reader of histone acetylation, triggering the transcription of pro-proliferative genes, such as c-myc [135]. Small-molecule BRD4 inhibitors including JQ1 and BETi-211 can downregulate c-myc level and induce potent anti-proliferative response [136]. However, high dose of BRD4 inhibitors is required to ensure sufficient BRD4 inhibition [39] and their antitumor efficacy might be unsatisfactory only by disrupting the bromodomain of BRD4 [12].
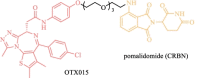
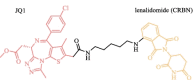
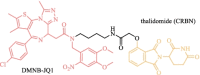
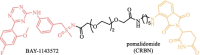
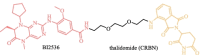
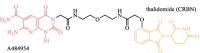
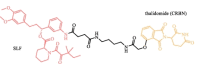
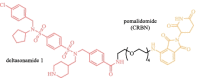
In 2015, Bradner’s group used JQ1 and thalidomide (a ligand for CRBN E3 ligase) to develop the BRD4-targeting PROTAC dBET1 with a DC50 value of 430 nM, attenuating tumor progression in vitro and in vivo by reducing the expression of BRD4 and c-myc [12]. To improve the degradation potency, Hines et al. synthesized the nutlin-base PROTAC A1874 to recruit MDM2 E3 ligase to effectively degrade BRD4 with DC50 of 32 nM [90]. Moreover, by utilizing VHL ligand and replacing the “(CH2CH2O)3” moiety of A1874 linker with “CH2CH2OCH2CH2CH2O”, the new PROTAC ARV-771 exhibited rapid BRD4 degradation (DC50 value < 1 nM) and potent antitumor effects in castration-resistant prostate cancer [76]. JQ1’s optimized analogue OTX015 was also applied to PROTAC ARV-825 to obtain a DC50 value of < 1 nM, leading to prolonged c-myc loss and enhanced anti-proliferative effects in Burkitt’s lymphoma cells [39].
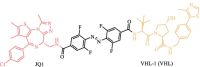
As JQ1 and OTX015 were non-selective BRD4 inhibitors, dBET1 and ARV-825 also caused the degradation of both BRD2 and BRD3. Intriguingly, Zengerle et al. described a JQ1-based PROTAC MZ1, choosing VH032 as VHL ligand, exhibiting preferential degradation of BRD4 over BRD2/3 in cervical cancer cells [91]. This evidence indicated that PROTAC might gain selectivity, even starting with non-selective ligands. Recently, Gadd et al. resolved the crystal structure of BRD4-MZ1-VHL ternary complex, which suggested a BRD4-VHL “neocontacts” resulted from the MZ1-induced cooperative recognition [33]. Nowak et al. demonstrated that such “neocontacts” were plastic and generated several distinct BRD4-VHL conformations. Suitable length of PROTAC linker could reinforce the cooperative interaction between BRD4 and VHL, thereby conferring PROTAC the selectivity toward BRD4. This finding guided the development of BRD4-selective PROTAC ZXH-3-26 by adjusting the length and modification site of linker to generate a favorable BRD4-CRBN binding conformation [93]. Besides, a number of other BRD4-based PROTACs have also been developed for cancer therapy [89, 92, 94–98, 100, 137–140].
CDK4/CDK6
Cyclin-dependent kinases (CDKs) control cell cycle progression in response to extracellular pro-proliferative signals. Among them, CDK4/6 phosphorylate retinoblastoma protein (Rb) and activate the transcription factor E2F to promote gene transcription, mediating the G1 to S phase transition [141]. In cancer cells, CDK4/6 are usually overactivated by their upstream oncogenes (e.g. c-myc) and serve as potential targets for cancer therapies [142, 143].
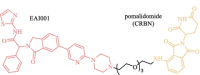
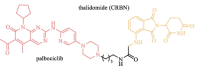
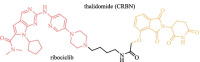
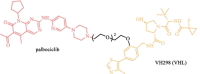
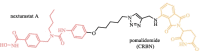
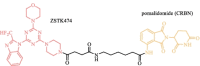
In 2019, Zhao et al. exploited the CRBN ligand and palbociclib (a CDK4/6 inhibitor) to synthesis the PROTAC Pal-pom that degraded CDK4/6 with DC50 values of 20–50 nM, thus preventing Rb phosphorylation and inducing cell cycle arrest in triple negative breast cancer (TNBC) cells [104]. Subsequently, Jiang et al. obtained the new PROTAC BSJ-02-162 based on Pal-pom by introducing a shorter alkyl chain and removing the 1,2,3-triazole moiety, which degraded both CDK4/6 and IKZF1/3 to exhibit increased anti-proliferative function in mantle cell lymphoma cells [77]. Another CDK inhibitor ribociclib was used into the first orally bioavailable prodrug of PROTAC, which degraded CDK 2/4/6 in vivo [105].
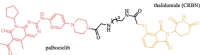
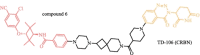
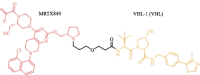
The high sequence similarity of CDK4 and CDK6 near their active sites makes them difficult to be distinguished by current CDK inhibitors. Interestingly, increasing evidence demonstrates that PROTAC exhibits good substrate selectivity after its optimization or molecule modifications. For example, Gray’s group optimized linkers to successfully develop the CDK6-selective degrader BSJ-03-123 based on palbociclib [102] and the CDK4-selective degrader BSJ-04-132 based on ribociclib [77]. This selectivity might be caused by the cooperative CDK-CRBN interactions as described in the “neocontacts” of BRD4-based PROTACs [33, 93]. Additionally, through adding oxygen or nitrogen atom to the linker of BSJ-02-162, PROTACs CP-10 and YX-2-107 can selectively degrade CDK6 [78, 144]. Notably, CDK6 exerts its functions in both kinase-dependent and -independent manners, and only its kinase-independent function is required for the growth of Philadelphia-positive acute lymphoblastic leukemia (Ph+-ALL). PROTAC YX-2-107 was demonstrated to inhibit CDK6’s kinase-independent function, thus more efficiently suppressing Ph+-ALL cells compared to palbociclib (inactive to CDK6’s kinase-independent function) [78]. Except for CRBN, Steinebach et al. found that VHL also had the potential to selectively degrade CDK6 in leukemia, myeloma and breast cancer cells [107].
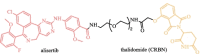
AURORA-a
Aurora kinase A (AURORA-A) drives centrosome separation to induce cell cycle progression from G2 to M phase. Overexpressed AURORA-A could transform normal epithelial cells to cancer cells in mouse models, highlighting AURORA-A as a prior cancer target [145]. The potent AURORA-A inhibitor alisertib is in multiple clinical trials. Besides the catalytic activity, AURORA-A has additional non-catalytic functions that are difficult to target by conventional small molecules, which may explain why some trials exhibit low therapeutic efficacy [146, 147]. To overcome this problem, Adhikari et al. developed a potent AURORA-A degrader JB170 by connecting alisertib to VHL ligand, which induced rapid, durable and highly-specific degradation of AURORA-A in leukemia and neuroblastoma cells [81]. Moreover, AURORA-A degradation by JB170 arrested S-phase progression and this effect was not observed upon kinase inhibition, further supporting the important non-catalytic function of AURORA-A during DNA replication [81].
EGFR
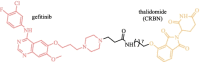
Epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase (RTK) that activates several oncogenic signals, promoting cell proliferation and differentiation. Overactivation or gain-of-function mutation of EGFR are prevalent in a variety of epithelial cancers (e.g. breast and lung cancers) [75, 148]. EGFR inhibitors, such as gefitinib, lapatinib and afatinib, have been approved to treat cancers, but severe drug resistance of EGFR inhibitors leads to low clinical response, which may be caused by drug-induced EGFR mutations (e.g. EGFRL858R, EGFRT790M or EGFRC797S) [149].
Based on lapatinib, gefitinib and afatinib, PROTACs compound 1/3/4 were respectively developed by linking VHL ligands, exhibiting anti-proliferative activity against breast cancer and lung cancer cells [79]. These PROTACs were selective for different EGFRs: compound 1 degraded wild-type or exon-20 insertion EGFRs; compound 2 preferred exon-19 deletion or L858R EGFRs; compound 3 degraded L858R/T790M dual mutant EGFR [79]. Novel EGFRL858R/T790M selective inhibitors XTF-262 and EGFRT790M/C797S selective inhibitors EAI001 were also utilized to synthesis PROTAC 14o and DDC-01-163, respectively, which exhibited anti-proliferative activities in lung cancer cells with corresponding EGFR-mutations [80, 99]. Overall, the selectivity of EGFR inhibitor-based PROTACs was consistent with that of their parental inhibitors [79, 80, 99, 101, 103, 150], so it’s necessary to conduct molecular typing of EGFR before PROTAC treatment.
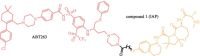
BRAF
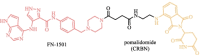
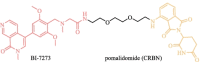
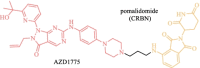
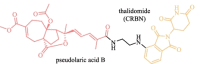
The RAF family kinases are key regulators of RAS-RAF-MEK-ERK pathway, transmitting oncogenic signals to promote cell proliferation [75]. Gain-of-function mutations in RAF (e.g. BRAFV600E) act as potent drivers of human cancers [151]. BRAFV600E inhibitors have shown great efficacy in cancer therapy, but long-term effectiveness is limited by RTKs and/or RAS activation or by secondary BRAF mutations [152, 153]. PROTAC provides an alternative strategy to therapeutically constrain oncogenic BRAF [60, 154]. Posterna et al. conjugated BRAF inhibitor BI-882370 and CRBN ligand to synthesize the PROTAC P4B, which specifically suppressed melanoma and colorectal cancer cells harboring BRAFV600E or other BRAF mutations [60].
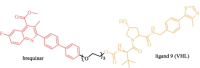
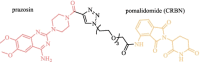
Except for these targets, the following proteins related to cancer cell proliferation could also be targeted by PROATCs: AR [7, 106, 108–111, 155, 156], ALK [22, 52, 113, 116, 157], BLK [118], BRD7/9 [82, 83, 120, 158], CDK2/5 [84], CDK8 [112], CDK9 [38, 114, 115, 117, 159], Cdc20 [119], c-Met [79], CREPT [121], CYP1B1 [122], DHODH [123], ER [89, 124, 126, 128, 160], ERK1/2 [26], FLT-3 [132, 161], HER2 [79], MEK1/2 [85, 86, 162], KRASG12C [87, 163], GSPT1 [125], PLK1 [127], SLC9A1 [129], TACC3 [130], TRIM24 [131], TRKA/C [133, 164], Wee1 [88], α1A-AR [134].
Targeting cancer apoptosis
Apoptosis (or programmed cell death) is an evolutionarily conserved process that maintains tissue homeostasis upon the simulation by cellular stress, DNA damage and immune surveillance. However, cancer cells upregulate anti-apoptotic proteins (e.g., Bcl-2 and Bcl-xL) or downregulate pro-apoptotic factors (e.g., Puma, Bax) to evade apoptosis, supporting their abnormal survival, therapeutic resistance and cancer recurrence [165, 166]. Therefore, targeting apoptosis could initiate programmed cell death of cancer cells and improve their response to anticancer drugs (Fig. 4) (Table 3).
Fig. 4.

PROTACs targeting apoptosis and angiogenesis. Apoptosis is accomplished by downregulation of antiapoptotic proteins (e.g. Bcl-2, Bcl-6, Bcl-xL, Mcl-1, etc) or upregulation of proapoptotic factors (e.g. Puma, Bax). This process can be regulated by various proteins (e.g. p53) and signaling pathways (e.g. PI3K/AKT). PI3K/AKT activation also promotes VEGF expression and angiogenesis. These key elements involved in cancer apoptosis and angiogenesis can be targeted by PROTACs (red arrow). Tumor-suppressor proteins are indicated in blue and oncogenic proteins are indicated in red. In the presented pathways, PROTACs have been developed targeting Bcl-xL [167, 168], PARP1 [169, 170], BCR-ABL [171, 172], AKT [173], Bcl-2 [174], c-IAP [175], eIF4E [176], Mcl-1 [174, 177], MDM2 [178, 179], PI3K [180], CBP/p300 [181], SirT2 [182] and VEGFR2 [183]
Table 3.
The structures of PROTAC molecules targeting apoptosis or angiogenesis in cancers. (red: POI ligand; yellow: E3 ligand)
| Target | PROTAC | PROTAC structure | Cancer | Ref | Target | PROTAC | PROTAC structure | Cancer | Ref |
|---|---|---|---|---|---|---|---|---|---|
| Bcl-xL | DT2216 |
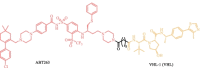
|
T-ALL | [167] | BCR-ABL | DAS-6-2-2-6-CRBN |

|
CML | [171] |
| Bcl-xL | XZ739 |
|
T-ALL | [184] | BCR-ABL | Azo-PROTAC-4C |
|
CML | [19] |
| Bcl-xL | compound 8a |
|
TL | [185] | BCR-ABL | SNIPER(ABL)-38 |
|
CML | [89] |
| Bcl-xL | XZ424 |
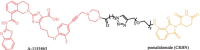
|
T-ALL | [168] | BCR-ABL | compound 19 |
|
CML | [186] |
| Bcl-xL | PROTAC 6 |
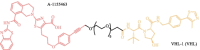
|
AML | [187] | BCR-ABL | GMB-475 |
|
CML | [172] |
| PARP1 | compound 2 |
|
CRC | [188] | BCR-ABL | GMB-805 |
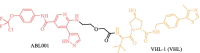
|
CML | [189] |
| PARP1 | iRucaparib-AP5 |
|
CC, RCC, BC, PC | [170] | BCR-ABL | SIAIS178 |
|
CML | [190] |
| PARP1 | compound 3 |
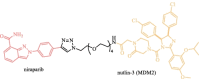
|
TNBC | [191] | BCR-ABL | BT1 |
|
CML | [192] |
| PARP1 | SK-575 |
|
BC, CRC, PC, PaC | [169] | AKT | INY-03-041 |
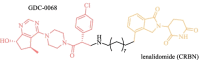
|
BC | [173] |
| Bcl-2 | C5 |
|
CC, CML, NSCLC | [174] | eIF4E | PROTAC 23a |
|
TNBC, CML | [176] |
| Bcl-6 | PROTAC 15 |
|
DLBCL | [193] | HDAC1/2/3 | PROTAC 4 |
|
CRC | [194] |
| c-IAP | compound 6 |
|
FS | [175] | HDAC1/2/3 | XZ9002 |
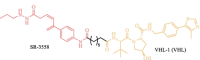
|
TNBC | [195] |
| CK2 | PROTAC 2 |
|
TNBC, NSCLC | [196] | HDAC6 | PROTAC 4 |
|
OSCC, GB | [197] |
| CRABP I/II | compound 4b |
|
NB | [8] | HDAC6 | degrader 12d |
|
MM | [198] |
| CRABP I/II | compound 6 |
|
FS | [175] | HDAC6 | NP8 |

|
MM, CC | [199] |
| CRABP I/II | β-NF-ATRA |
|
BC, NB | [140] | HDAC6 | NH2 |
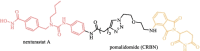
|
MM, CC | [200] |
| eEF2K | compound 11l |
|
BC | [201] | HDAC6 | P1 |
|
MM | [202] |
| HDAC6 | compound 3j |
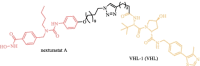
|
MM | [203] |
CBP/ p300 |
dCBP-1 |
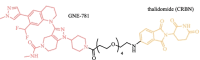
|
MM | [181] |
| Mcl-1 | dMCL1-2 |
|
MM | [177] | RIPK2 | PROTAC_RIPK2 |
|
BC, AML | [17] |
| Mcl-1 | C3 |
|
CC, CML, NSCLC | [174] | SGK3 | SGK3-PROTAC1 |
|
BC | [204] |
| MDM2 | MD-224 |
|
ALL, AML | [179] | SirT2 | PROTAC 12 |
|
CC | [182] |
| MDM2 | degrader 32 |
|
ALL | [178] | VEGFR2 | PROTAC-5 |
|
/ | [183] |
| PI3K | compound D |
|
HCC | [180] |
Bcl-xL
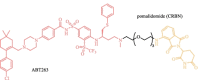
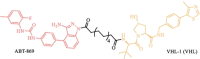
Bcl-xL inactivates the intrinsic apoptotic pathway to promote cell survival. Overexpression of Bcl-xL occurs in many tumor cells and is highly correlated with the resistance to cancer therapy, so Bcl-xL is a well-validated cancer target [165]. However, the low target engagement and dose-limiting thrombocytopenia limits the use of Bcl-xL inhibitors (e.g. ABT263 and A-1155463) as safe and effective anticancer agents [205].
Zhou and his coworkers linked ABT263 to VHL ligand to develop the PROTAC DT2216, which effectively degraded Bcl-xL and suppressed Bcl-xL-dependent leukemia cells in vitro and in vivo, without causing thrombocytopenia due to the poor expression of VHL in platelets [167]. Since CRBN is poorly expressed in platelets, they designed another PROTAC XZ739 containing CRBN ligand and ABT263, treating T cell acute lymphoblastic leukemia (T-ALL) with less toxicity to platelets [184]. Because VHL and CRBN expressions are extremely low in cutaneous T-cell lymphoma (CTCL) cells, the activity of VHL- and CRBN-based Bcl-xL PROTACs against CTCL were unfavorable. Zhou group further designed PROTAC 8a involving the ligand of IAP E3 ligase (with high level in CTCL) to efficiently degrade Bcl-xL in CTCL cells [185]. The selective Bcl-xL inhibitor A-1155463 was also utilized to develop XZ424 and PROTAC 6, showing increased selectivity in Bcl-xL-dependent T-ALL cells [168, 187].
PARP1
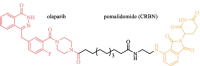
Poly(ADP-ribose) polymerase 1 (PARP1) participates in DNA damage repair to maintain genomic stability, and is overexpressed in human cancers to evade apoptosis [166]. Small-molecule PARP1 inhibitors, such as niraparib, rucaparib and olaparib, have been developed to treat cancers [206]. However, these inhibitors prevent PARP1 from dissociating DNA lesions to block DNA replication, leading to high cytotoxicity to normal cells [206].
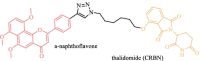
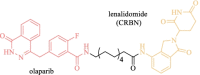
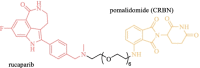
In 2019, by connecting niraparib and the MDM2 ligand nutlin-3, Zhao et al. synthesized the PARP1-targeting PROTAC compound 3 to induce significant apoptosis of TNBC cells without cytotoxicity against normal cells [191]. Olaparib was also used to design CRBN-recruiting PROTACs to trigger apoptosis in multiple cancers [169, 188]. To improve selectivity, Wang et al. utilized rucaparib (a selective PARP1 inhibitor) and CRBN ligand to develop PARP1 degrader iRucaparib-AP5, which exerted highly specific PARP1 degradation in cervical, breast, renal and prostate cancer cells [170].
BCR-ABL
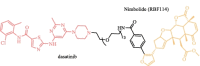
The oncogenic fusion kinase BCR-ABL activates the anti-apoptotic protein Bcl-2 to protect mitochondria from DNA-damaged signals and prevent apoptosis in chronic myelogenous leukemia (CML) [207, 208]. BCR-ABL inhibitors (e.g. dasatinib, ponatinib and imatinib) have successfully treated CML patients. But lifelong drug administration is required due to the persistent CML stem cells that rely on BCR-ABL’s kinase-independent function for survival [171, 209]. Moreover, BCR-ABL mutations can also cause drug resistance [210].
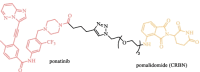
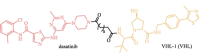
Lai et al. synthesized the BCR-ABL degrader DAS-6-2-2-6-CRBN containing dasatinib and CRBN ligand, which showed potent degradation of BCR-ABL and growth inhibition in CML cells [171]. Other E3 ligases including IAP, VHL and RNF114 were also recruited by dasatinib-based PROTACs that achieved effective BCR-ABL degradation to suppress CML cells [89, 190, 192]. Ponatinib and two novel BCR-ABL inhibitors (GNF5 and ABL001) were utilized into PROTACs development, and the obtained degraders showed increased degradation ability and better selectivity with less adverse effects [172, 186, 189].
Targeting cancer angiogenesis
Tumors require neovasculature, generated by angiogenesis, to supply nutrients and oxygen as well as to evacuate metabolic wastes and carbon dioxide [73]. Angiogenesis is triggered by hypoxia that activates the expression of multiple growth factors, such as vascular endothelial growth factor (VEGF), a pivotal growth factor that specifically recognizes vascular endothelial growth factor receptor (VEGFR) to induce the formation of neovasculature (Fig. 4) [211]. The blockade of VEGF/VEGFR signaling to suppress angiogenesis has been developed for cancer therapy (Fig. 4, Table 3).
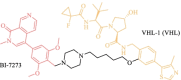
VEGFR-2 is the main VEGFRs to mediate proliferation and angiogenesis of vascular endothelial cells, and targeting VEGFR2 is a promising strategy for cancer treatment. Based on the VEGFR-2 inhibitor S7, Shan et al. developed PROTAC-2 and PROTAC-5 to exhibit potent VEGFR-2 elimination and anti-proliferative activity in human umbilical vein endothelial cells. Moreover, these PROTACs had low cytotoxicity to HEK-293 cells (human embryonic kidney cells, VEGFR-2 negative), displaying excellent safety to VEGFR-2 negative cells [183].
Targeting cancer immunity and inflammation
To sustain cell survival, cancer cells induce inflammation and immune evasion by reprogramming tumor microenvironment that involves regulatory cells (e.g., regulatory T cells), B-cell receptor (BCR) signaling and T-cell receptor (TCR) signaling [212–214] (Fig. 5).
Fig. 5.

PROTACs targeting cancer immune evasion and inflammation. Cancer cells promote immune evasion and inflammation by reprogramming tumor microenvironment that involves BCR, TCR and JAK-STAT pathways. These components in cancer immune evasion and inflammation can be targeted by PROTACs (red arrow). Tumor-suppressor proteins are indicated in blue and oncogenic proteins are indicated in red. In the presented pathways, PROTACs have been developed targeting PD-L1 [215], BTK [216–219], STAT3 [57], HPK1 [220], IDO1 [221], ITK [161], JAK [222] and SHP2 [223]
Immunotherapies by the immune-checkpoint inhibitors are new therapeutics that relieve immunosuppression and enable immune-mediated tumor clearance [224]. However, some patients have innate or acquired resistance to immunotherapies. To overcome these problems, PROTACs targeting immunity and inflammation have been developed (Table 4).
Table 4.
The structures of PROTAC molecules targeting cancer immune evasion or inflammation. (red: POI ligand; yellow: E3 ligand)
| Target | PROTAC | PROTAC structure | Cancer | Ref | Target | PROTAC | PROTAC structure | Cancer | Ref |
|---|---|---|---|---|---|---|---|---|---|
| PD-L1 | P22 |
|
NSCLC TNBC, M | [215] | FKBP12 | dFKBP-1 |
|
AML | [12] |
| BTK | DD-04-015 |
|
DLBCL | [161] | FKBP12 | dTAG-13 |
|
AML | [225] |
| BTK | P13I |
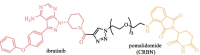
|
BL, DLBCL, MCL | [216] | FKBP12 | RC32 |
|
T-ALL, BL, PC, BC, CC | [226] |
| BTK | RC-3 |
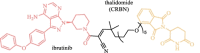
|
BL, MCL, CML | [217] | FKBP12 | KB02-SLF |
|
/ | [97] |
| BTK | RC-1 |
|
AML, MCL | [227] | HPK1 | SS44 |
|
BL, MM, CML, et al |
[220] |
| BTK | BC5P |
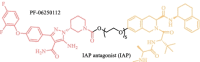
|
AML | [219] | IDO1 | degrader 2c |
|
CC | [221] |
| BTK | DD-03-171 |
|
MCL, DLBCL | [218] | IKZF1/3 | DD-03-171 |
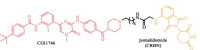
|
MCL, DLBCL | [218] |
| BTK | compound 10 |
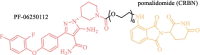
|
BL, AML | [228] | IRAK4 | degrader-5 |
|
DLBCL | [229] |
| BTK | PROTAC 7 |
|
BL, CML | [118] | ITK | TL12-186 |

|
AML, T-ALL | [161] |
| STAT3 | SD-36 |
|
AML, ALCL | [57] | JAK | JP-6 |
|
AML | [222] |
| CD147 | compound 6a |
|
M | [230] | Lin28 | ORN3P1 |
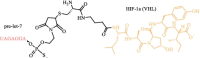
|
CML | [18] |
| PDE4 | SNIPER(PDE4)-9 |
|
FS | [89] | PRC2 | PROTAC 1 |
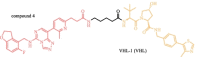
|
DLCBL | [231] |
| PDEδ | compound 17f |
|
CRC | [232] | PRMT5 | MS4322 |
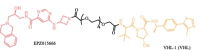
|
BC | [233] |
| PDEδ | PROTAC 3 |
|
T-ALL, PaC, CC | [234] | Rpn13 | WL-40 |
|
MM | [235] |
| Pirin | CCT367766 |
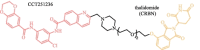
|
OC | [236] | SHP2 | SHP2-D26 |
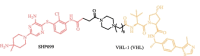
|
EC, AML | [223] |
| PRC2 | UNC6852 |
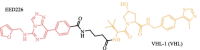
|
CC, DLCBL | [237] | TBK1 | PROTAC 3i |
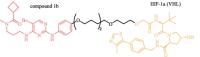
|
NSCLC | [238] |
PD-L1
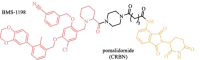
Programmed death-ligand 1 (PD-L1) is frequently overexpressed in cancer cells. The binding of PD-L1 on cancer cells to its receptor programmed death 1 (PD-1) on T cells counteracts T cell-activating signals, inhibiting anti-tumor immunity and promoting immune escape [239]. Chen group used BMS-1198 (a small-molecule PD-L1 inhibitor) and pomalidomide (a CRBN ligand) to synthesize PROTAC P22, which moderately degraded PD-L1 in lung and breast cancer cells [215]. Thus, it’s possible to develop PD-L1-targeting PROTAC based on small-molecule PD-L1 inhibitors. However, due to the hydrophobic and flat binding pocket of PD-L1, there are few known small-molecule PD-L1 inhibitors, so it’s challenging to develop effective PD-L1 PROTAC currently.
BTK
Bruton’s tyrosine kinase (BTK) is a non-receptor tyrosine kinase, playing pivotal roles in B-cell development and immune responses. BTK inhibitors (e.g. ibrutinib) have been developed to treat chronic lymphocytic leukemia (CLL) and mantle cell lymphoma (MCL) by blocking BCR signaling and regulating innate/adaptive immunity [240]. But many patients exhibit drug resistance due to BTK mutations in the ibrutinib binding site (BTKC481S).
Based on ibrutinib and CRBN ligand, Rao group developed PROTACs P13I and L18I, two irreversible covalent PROTACs, to degrade wide-type and C481S-mutant BTKs and suppress diffuse large B cell lymphoma (DLBCL) and MCL cells [216, 241]. As P13I and L18I formed irreversible covalent bonds with BTK, so they did not follow “event-driven mechanism”, even though they used the PROTAC-like structure. To solve this problem, another two groups designed PROTACs RC-1 and RC-3 using cyano-acrylamide moiety to shape up reversible covalent bonds with BTK, exhibiting enhanced selectivity and efficacy over irreversible PROTACs [217, 227]. Additionally, a new generation of non-covalent BTK inhibitors (e.g., RN486 and CGI1746) was utilized to develop PROTACs (e.g., DD-04-015 and DD-03-171) that efficiently degraded BTKs and inhibited cancer cell growth [161, 218]. Intriguingly, Calabrese group found that alleviation of steric clashes between BTK and CRBN by adjusting PROTAC linker length allowed potent BTK degradation in the absence of thermodynamic cooperativity [228], indicating increased BTK-PROTAC-IAP ternary complex stability was not always related to increased degradation efficiency [219]. However, its underlying mechanism remains obscure.
Targeting cancer metastasis
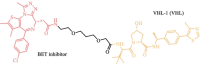
Tumor cells extravasate, disseminate and successfully colonize distant organs from the primary foci via circulatory systems to achieve metastasis, causing ~ 90% of cancer deaths worldwide [242, 243]. Epithelial-to-mesenchymal transition (EMT) is a key step during metastasis and can be activated by several upstream cellular signaling pathways including Integrin/FAK/PI3K/AKT axis (Fig. 6) [243–245]. In the past decades, PROTACs targeting EMT-related proteins have been developed to manage cancer metastasis (Table 5).
Fig. 6.

PROTACs targeting cancer metastasis. Activation of Integrin/FAK/PI3K/AKT, TGF-β/SMAD and Wnt/β-catenin pathways significantly increase the expression of pro-EMT transcription factors (e.g. ZEB, SNAIL and TWIST), leading to the downregulation of E-cadherin (E-Cad) that maintains epithelial integrity, and the upregulation of N-cadherin (N-Cad) and vimentin (VIM) that implicate in motility and invasion. These key elements involved in metastasis can be targeted by PROTACs (red arrow). Tumor-suppressor proteins are indicated in blue and oncogenic proteins are indicated in red. In the presented pathways, PROTACs have been developed targeting FAK [246, 247], IGF-1R [248], p38 [249, 250], Smad3 [251], Src [248, 252], TCF [253], TGF-β1 [254] and β-catenin [255]
Table 5.
The structures of PROTAC molecules targeting cancer metastasis. (red: POI ligand; yellow: E3 ligand)
| Target | PROTAC | PROTAC structure | Cancer | Ref | Target | PROTAC | PROTAC structure | Cancer | Ref |
|---|---|---|---|---|---|---|---|---|---|
| FAK (PTK2) | PROTAC-3 |
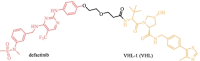
|
TNBC, PC | [256] | p38 | NR-7h |
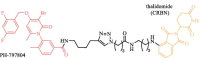
|
BC, CRC, CC | [257] |
| FAK (PTK2) | BI-0319 |
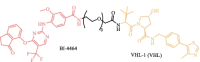
|
HCC | [246] | Smad3 | PROTAC_Smad3 |
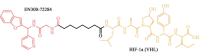
|
RCC | [251] |
| FAK (PTK2) | BI-3663 |

|
HCC | [246] | Src | ND1-YL2 |
|
TNBC | [252] |
| FAK (PTK2) | FC-11 |
|
OC, BC, PC, BL | [247] | Src | 12b |
|
BC, NSCLC | [248] |
| IGF-1R | 12b |
|
BC, NSCLC | [248] | TCF | ARV771 |
|
DLBCL | [253] |
| p38 | SJFα |
|
BC, CC | [249] | TGF-β1 | DT-6 |
|
HCC, BC, NSCLC | [254] |
| p38 | PROTAC 1 |
|
BC, CC | [250] | β-catenin | xStAx-VHLL |
|
CRC | [255] |
FAK
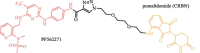
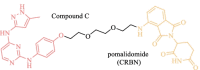
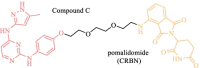
Focal adhesion kinase (FAK) is one of the most prominent effectors of integrin signaling. Overexpressed FAK, correlating with poor clinical outcome, drives cancer invasion and migration through exerting both kinase-dependent and independent functions [258–260]. Several FAK kinase inhibitors have been developed, such as defactinib, BI-4464 and PF-562271 [261]. Nevertheless, the critical kinase-independent scaffolding function of FAK is beyond the ability of current inhibitors [262].
Cromm et al. used the clinical candidate defactinib as the FAK ligand and (S,R,S)-AHPC as the VHL ligand to prepare a selective FAK degrader PROTAC-3. PROTAC-3 dramatically suppressed FAK signaling as well as FAK-mediated cell migration and invasion in TNBC and prostate cancer cells [256]. Based on small-molecule FAK inhibitor BI-4464 and CRBN ligand pomalidomide, Popow et al. presented a highly selective PROTAC BI-3663 to hijack UPS for FAK degradation, showing a DC50 of 30 nM in a panel of hepatocellular carcinoma cell lines [246]. In addition, FAK inhibitor PF562271 was also included in PROTAC study, leading to the establishment of PROTAC FC-11 that exhibited rapid FAK degradation with picomolar DC50 in several cancer cells [247].
Guidelines for PROTAC design
Developing anticancer PROTAC aims to improve the effectiveness and precision of targeted cancer therapy. In the molecular design of PROTACs, many critical issues about POI ligand, E3 ligand and linker should be comprehensively considered, such as specificity, solubility, stability, drug safety and bioavailability.
For POI/E3 ligands, the known ligands as well as the newly designed ligands based on 3D structure of POI/E3 could be used in PROTAC design. Notably, the ligand with high target affinity is not favorable, because this makes the ligand difficult to dissociate from target protein and is more likely to exert “occupancy-driven mechanism” instead of “event-driven mechanism” [250, 263]. A transient or low-abundance “POI-PROTAC-E3” ternary complex is enough to achieve adequate degradation, thus the ligand with low target affinity without affecting the assembly of ternary complex is acceptable. Moreover, selective inhibitors could be utilized to increase the precision of PROTACs, while the multitargeted inhibitors are also useful to develop PROTAC degraders that exert anticancer activities by simultaneously degrading multiple proteins [161]. Besides, there are ~ 600 E3 ligases in human and their expressions usually exhibit tissue−/tumor-specific, so selecting appropriate E3 ligase/E3 ligand system should consider the cellular context of tumors to increase the efficacy and reduce the toxicity [27, 36].
For the linker, the first issue is to define the ligand site that binds to the linker. The protein- or cell-based biological assays should be performed to test the activity of ligands with chemical modifications at different sites, aiming to find the promising sites that maintain the ligand’s function. Secondly, since the physical and chemical properties of linkers affect PROTAC’s selectivity and efficiency via adjusting the POI-E3 interface, a series of linkers with different lengths and chemical compositions should be designed, simulated (e.g., by structural modelling or molecular simulation), synthesized and biologically evaluated. Importantly, for the convenience in preparation and purification of PROTACs, the hydrophilicity/liposolubility of linker needs to match the properties of POI ligand and E3 ligand.
In addition, since PROTACs with high M.W. may influence their bioavailability, the idea of CLIPTACs that design a pair of smaller precursors is feasible to increase cell permeability [26]. It’s also recommended to use computer-aided drug design (CADD) software (e.g. Discovery Studio or Schrodinger Suites) to in silico predict the solubility and ADMET (Absorption, Distribution, Metabolism, Excretion and Toxicity) properties of molecules before PROTAC design.
Conclusions and prospects
From the establishment of the PROTAC concept in 2001, extensive efforts have been devoted to improving the efficacy, expanding the target scope and overcoming the disadvantages of PROTAC. Therefore, PROTAC has become an attractive technique for cancer treatment. Until now, many PROTACs have been developed to control cancer progression, exhibiting clinical potential in cancer therapy. However, there is still a demand to accelerate the development of PROTACs.
Expanding the POI spectrum is urgent for cancer therapy. Currently, although inhibitors of some proteins (e.g. kinases) have been successfully developed, there are many oncogenic proteins (e.g. RBPs and DBPs) that can’t be targeted by small molecules. Interestingly, taking advantage of the fact that RBPs and DBPs bind to specific nucleotide sequences, researchers utilized oligonucleotides as POI ligands to develop the RNA-PROTAC and TF-PROTAC that induced the degradation of RBPs and DBPs [18, 55]. Therefore, these techniques open up a new direction for targeting undruggable pathogenic proteins. In future, the design and optimization of these oligonucleotide-based PROTACs, targeting oncogenic RBPs (e.g. IGF2BPs and YBX1) and DBPs (e.g. c-myc and STAT3), should be extensively investigated for targeted cancer therapy.
Less than 10 of ~ 600 E3 ligases have been utilized in PROTAC so far, other E3 ligases could be considered to develop new PROTACs. For example, Cotton et al. established antibody-based PROTACs (AbTACs) that the recombinant bispecific antibodies recruit the membrane-bound E3 ligase RNF43 for the degradation of the cell-surface protein PD-L1 [264]. Moreover, the proteasome-independent protein degradation systems (including endosome, lysosome or autophagosome systems) have been harnessed to develop novel targeted degradation techniques, such as lysosome-targeting chimera (LYTAC), autophagy-targeting chimera (AUTAC) and autophagosome-tethering compound (ATTEC) [265–267], providing different strategies for targeted cancer therapy. Additionally, the ribonuclease targeting chimera (RIBOTAC) used RNA-targeting small molecules and RNase L to accomplish the degradation of intracellular RNAs [268, 269], suggesting a new idea for the degradation of oncogenic RNAs for cancer therapy. Therefore, PROTAC and related degradation techniques are powerful tools for specifically degrading oncogenic proteins or RNA molecules and will be used clinically for cancer therapy.
Acknowledgements
We’d like to thank Dr. Jiao Li for her discussion during manuscript preparation.
Abbreviations
- ALCL
Anaplastic large cell lymphoma
- ALL
Acute lymphoblastic leukemia
- AML
Acute myeloid leukemia
- BC
Breast cancer
- BL
Burkitt’s lymphoma
- BRD4
Bromodomain-containing protein 4
- BTK
Bruton’s tyrosine kinase
- CC
Cervical cancer
- CDK
Cyclin-dependent kinase
- CML
Chronic myelogenous leukemia
- CRBN
Cereblon
- CRC
Colorectal cancer
- DLBCL
Diffuse large B cell lymphoma
- EC
Esophageal cancer
- EGFR
Epidermal growth factor receptor
- FAK
Focal adhesion kinase
- FS
Fibrosarcoma
- GB
Glioblastoma
- GC
Gastric cancer
- HCC
Hepatocellular cancer
- IAP
Inhibitor-of-apoptosis-protein
- LC
Lung cancer
- M
Melanoma
- MCL
Mantle cell lymphoma
- MM
Multiple myeloma
- MRT
Malignant rhabdoid tumor
- NB
Neuroblastoma
- NSCLC
Non-small cell lung cancer
- OC
Ovarian cancer
- OS
Osteosarcoma
- OSCC
Oral squamous cell carcinoma
- PaC
Pancreatic cancer
- PARP1
Poly(ADP-ribose) polymerase 1
- PC
Prostate cancer
- PD-L1
Programmed death-ligand 1
- POI
Protein of interest
- PROTAC
Proteolysis-targeting chimeras
- RCC
Renal cell cancer
- SS
Synovial sarcoma
- T-ALL
T cell acute lymphoblastic leukemia
- TL
T-cell lymphoma
- TNBC
Triple negative breast cancer
- Ub
Ubiquitin
- UPS
Ubiquitin-proteasome system
- VEGFR
Vascular endothelial growth factor receptor
- VHL
Von Hippel-Lindau
Authors’ contributions
YP and SC conceived the structure of manuscript and revised the manuscript; XL, WP, QZ and MA drafted the initial manuscript and revised it. All authors read and approved the final manuscript.
Funding
This work was supported by National Natural Science Foundation of China (81772960, 81821002 and 82073319), Science and Technology Foundation of Sichuan Province, China (2022YFS0046), the 1.3.5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (ZYJC18030 and ZYGD20008).
Availability of data and materials
Not applicable.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Xinyi Li and Wenchen Pu contributed equally to this work.
Contributor Information
Song Chen, Email: songchen882002@hotmail.com.
Yong Peng, Email: yongpeng@scu.edu.cn.
References
- 1.Gharwan H, Groninger H. Kinase inhibitors and monoclonal antibodies in oncology: clinical implications. Nat Rev Clin Oncol. 2016;13(4):209–227. doi: 10.1038/nrclinonc.2015.213. [DOI] [PubMed] [Google Scholar]
- 2.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98(15):8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schneekloth JS, Fonseca FN, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K, et al. Chemical genetic control of protein levels: selective in vivo targeted degradation. J Am Chem Soc. 2004;126(12):3748–3754. doi: 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez-Gonzalez A, Cyrus K, Salcius M, Kim K, Crews C, Deshaies R, et al. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene. 2008;27(57):7201–7211. doi: 10.1038/onc.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee H, Puppala D, Choi EY, Swanson H, Kim KB. Targeted degradation of the aryl hydrocarbon receptor by the PROTAC approach: a useful chemical genetic tool. Chembiochem. 2007;8(17):2058–2062. doi: 10.1002/cbic.200700438. [DOI] [PubMed] [Google Scholar]
- 6.Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, Crews CM, et al. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol Cell Proteomics. 2003;2(12):1350–1358. doi: 10.1074/mcp.T300009-MCP200. [DOI] [PubMed] [Google Scholar]
- 7.Schneekloth AR, Pucheault M, Tae HS, Crews CM. Targeted intracellular protein degradation induced by a small molecule: en route to chemical proteomics. Bioorg Med Chem Lett. 2008;18(22):5904–5908. doi: 10.1016/j.bmcl.2008.07.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Itoh Y, Ishikawa M, Naito M, Hashimoto Y. Protein knockdown using methyl bestatin-ligand hybrid molecules: design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J Am Chem Soc. 2010;132(16):5820–5826. doi: 10.1021/ja100691p. [DOI] [PubMed] [Google Scholar]
- 9.Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327(5971):1345–1350. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- 10.Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343(6168):301–305. doi: 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343(6168):305–309. doi: 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, et al. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348(6241):1376–1381. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sievers QL, Petzold G, Bunker RD, Renneville A, Słabicki M, Liddicoat BJ, et al. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science. 2018;362(6414):eaat0572. doi: 10.1126/science.aat0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jan M, Sperling AS, Ebert BL. Cancer therapies based on targeted protein degradation—lessons learned with lenalidomide. Nat Rev Clin Oncol. 2021. 10.1038/s41571-021-00479-z. [DOI] [PMC free article] [PubMed]
- 15.Buckley DL, Van Molle I, Gareiss PC, Tae HS, Michel J, Noblin DJ, et al. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1α interaction. J Am Chem Soc. 2012;134(10):4465–4468. doi: 10.1021/ja209924v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buckley DL, Gustafson JL, Van Molle I, Roth AG, Tae HS, Gareiss PC, et al. Small-molecule inhibitors of the interaction between the E3 ligase VHL and HIF1α. Angew Chem Int Ed Engl. 2012;51(46):11463–11467. doi: 10.1002/anie.201206231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bondeson DP, Mares A, Smith IED, Ko E, Crews CM. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. 2015;11(8):611–617. doi: 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghidini A, Cléry A, Halloy F, Allain FH, Hall J. RNA-PROTACs: degraders of RNA-binding proteins. Angew Chem Int Ed Engl. 2021;60(6):3163–3169. doi: 10.1002/anie.202012330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin YH, Lu MC, Wang Y, Shan WX, Wang XY, You QD, et al. Azo-PROTAC: novel light-controlled small-molecule tool for protein knockdown. J Med Chem. 2020;63(9):4644–4654. doi: 10.1021/acs.jmedchem.9b02058. [DOI] [PubMed] [Google Scholar]
- 20.Pfaff P, Samarasinghe KT, Crews CM, Carreira EM. Reversible spatiotemporal control of induced protein degradation by bistable photoPROTACs. ACS Cent Sci. 2019;5(10):1682–1690. doi: 10.1021/acscentsci.9b00713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reynders M, Matsuura BS, Bérouti M, Simoneschi D, Marzio A, Pagano M, et al. PHOTACs enable optical control of protein degradation. Sci Adv. 2020;6(8):eaay5064. doi: 10.1126/sciadv.aay5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu J, Chen H, Ma L, He Z, Wang D, Liu Y, et al. Light-induced control of protein destruction by opto-PROTAC. Sci Adv. 2020;6(8):eaay5154. doi: 10.1126/sciadv.aay5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naro Y, Darrah K, Deiters A. Optical control of small molecule-induced protein degradation. J Am Chem Soc. 2020;142(5):2193–2197. doi: 10.1021/jacs.9b12718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kounde CS, Shchepinova MM, Saunders CN, Muelbaier M, Rackham MD, Harling JD, et al. A caged E3 ligase ligand for PROTAC-mediated protein degradation with light. Chem Commun. 2020;56(41):5532–5535. doi: 10.1039/d0cc00523a. [DOI] [PubMed] [Google Scholar]
- 25.Xue G, Wang K, Zhou D, Zhong H, Pan Z. Light-induced protein degradation with photocaged PROTACs. J Am Chem Soc. 2019;141(46):18370–18374. doi: 10.1021/jacs.9b06422. [DOI] [PubMed] [Google Scholar]
- 26.Lebraud H, Wright DJ, Johnson CN, Heightman TD. Protein degradation by in-cell self-assembly of proteolysis targeting chimeras. ACS Cent Sci. 2016;2(12):927–934. doi: 10.1021/acscentsci.6b00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mullard A. Targeted protein degraders crowd into the clinic. Nat Rev Drug Discov. 2021;20(4):247–250. doi: 10.1038/d41573-021-00052-4. [DOI] [PubMed] [Google Scholar]
- 28.Bard JA, Goodall EA, Greene ER, Jonsson E, Dong KC, Martin A. Structure and function of the 26S proteasome. Annu Rev Biochem. 2018;87:697–724. doi: 10.1146/annurev-biochem-062917-011931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kleiger G, Mayor T. Perilous journey: a tour of the ubiquitin–proteasome system. Trends Cell Biol. 2014;24(6):352–359. doi: 10.1016/j.tcb.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hipp MS, Kasturi P, Hartl FU. The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol. 2019;20(7):421–435. doi: 10.1038/s41580-019-0101-y. [DOI] [PubMed] [Google Scholar]
- 31.Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 32.Yau R, Rape M. The increasing complexity of the ubiquitin code. Nat Cell Biol. 2016;18(6):579–586. doi: 10.1038/ncb3358. [DOI] [PubMed] [Google Scholar]
- 33.Gadd MS, Testa A, Lucas X, Chan KH, Chen W, Lamont DJ, et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol. 2017;13(5):514–521. doi: 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017;16(2):101–114. doi: 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miles LE, Lipschitz DA, Bieber CP, Cook JD. Measurement of serum ferritin by a 2-site immunoradiometric assay. Anal Biochem. 1974;61(1):209–224. doi: 10.1016/0003-2697(74)90347-9. [DOI] [PubMed] [Google Scholar]
- 36.Schapira M, Calabrese MF, Bullock AN, Crews CM. Targeted protein degradation: expanding the toolbox. Nat Rev Drug Discov. 2019:1–15. [DOI] [PubMed]
- 37.Verma R, Mohl D, Deshaies RJ. Harnessing the power of proteolysis for targeted protein inactivation. Mol Cell. 2020;77(3):446–460. doi: 10.1016/j.molcel.2020.01.010. [DOI] [PubMed] [Google Scholar]
- 38.Olson CM, Jiang B, Erb MA, Liang Y, Doctor ZM, Zhang Z, et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat Chem Biol. 2018;14(2):163–170. doi: 10.1038/nchembio.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem Biol. 2015;22(6):755–763. doi: 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burslem GM, Crews CM. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell. 2020;181(1):102–114. doi: 10.1016/j.cell.2019.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chamberlain PP, Hamann LG. Development of targeted protein degradation therapeutics. Nat Chem Biol. 2019;15(10):937–944. doi: 10.1038/s41589-019-0362-y. [DOI] [PubMed] [Google Scholar]
- 42.Edmondson SD, Yang B, Fallan C. Proteolysis targeting chimeras (PROTACs) in ‘beyond rule-of-five’chemical space: recent progress and future challenges. Bioorg Med Chem Lett. 2019;29(13):1555–1564. doi: 10.1016/j.bmcl.2019.04.030. [DOI] [PubMed] [Google Scholar]
- 43.Bedard PL, Hyman DM, Davids MS, Siu LL. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet. 2020;395(10229):1078–1088. doi: 10.1016/S0140-6736(20)30164-1. [DOI] [PubMed] [Google Scholar]
- 44.Mayes PA, Hance KW, Hoos A. The promise and challenges of immune agonist antibody development in cancer. Nat Rev Drug Discov. 2018;17(7):509–527. doi: 10.1038/nrd.2018.75. [DOI] [PubMed] [Google Scholar]
- 45.Zuckerman JE, Davis ME. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat Rev Drug Discov. 2015;14(12):843–856. doi: 10.1038/nrd4685. [DOI] [PubMed] [Google Scholar]
- 46.Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367(6481):eaba7365. doi: 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paiva SL, Crews CM. Targeted protein degradation: elements of PROTAC design. Curr Opin Chem Biol. 2019;50:111–119. doi: 10.1016/j.cbpa.2019.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nalawansha DA, Crews CM. PROTACs: an emerging therapeutic modality in precision medicine. Cell Chem Biol. 2020;27(8):998–1014. doi: 10.1016/j.chembiol.2020.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu H, Xue Y, Yu GK, Arias C, Lin J, Fong S, et al. Compensatory induction of MYC expression by sustained CDK9 inhibition via a BRD4-dependent mechanism. Elife. 2015;4:e06535. doi: 10.7554/eLife.06535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang S, Li H, Tang J, Wang J, Luo J, Liu B, et al. Discovery of a potent HMG-CoA reductase degrader that eliminates statin-induced reductase accumulation and lowers cholesterol. Nat Commun. 2018;9:5138. doi: 10.1038/s41467-018-07590-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cecchini C, Pannilunghi S, Tardy S, Scapozza L. From conception to development: investigating PROTACs features for improved cell permeability and successful protein degradation. Front Chem. 2021;9:672267. doi: 10.3389/fchem.2021.672267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Powell C, Gao Y, Tan L, Donovan K, Nowak R, Loehr A, et al. Chemically induced degradation of anaplastic lymphoma kinase (ALK) J Med Chem. 2018;61(9):4249–4255. doi: 10.1021/acs.jmedchem.7b01655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klein V, Townsend C, Testa A, Zengerle M, Maniaci C, Hughes S, et al. Understanding and improving the membrane permeability of VH032-based PROTACs. ACS Med Chem Lett. 2020;11(9):1732–1738. doi: 10.1021/acsmedchemlett.0c00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsson P, Kihlberg J. How big is too big for cell permeability? J Med Chem. 2017;60(5):1662–1664. doi: 10.1021/acs.jmedchem.7b00237. [DOI] [PubMed] [Google Scholar]
- 55.Liu J, Chen H, Kaniskan H, Xie L, Chen X, Jin J, et al. TF-PROTACs enable targeted degradation of transcription factors. J Am Chem Soc. 2021;143(23):8902–8910. doi: 10.1021/jacs.1c03852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johnson DE, O'Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signaling axis in cancer. Nat Rev Clin Oncol. 2018;15(4):234. doi: 10.1038/nrclinonc.2018.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bai L, Zhou H, Xu R, Zhao Y, Chinnaswamy K, McEachern D, et al. A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell. 2019;36(5):498–511.e17. doi: 10.1016/j.ccell.2019.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jin J, Wu Y, Chen J, Shen Y, Zhang L, Zhang H, et al. The peptide PROTAC modality: a novel strategy for targeted protein ubiquitination. Theranostics. 2020;10(22):10141–10153. doi: 10.7150/thno.46985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Poulikakos PI, Rosen N. Mutant BRAF melanomas-dependence and resistance. Cancer Cell. 2011;19(1):11–15. doi: 10.1016/j.ccr.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 60.Posternak G, Tang X, Maisonneuve P, Jin T, Lavoie H, Daou S, et al. Functional characterization of a PROTAC directed against BRAF mutant V600E. Nat Chem Biol. 2020;16(11):1170–1178. doi: 10.1038/s41589-020-0609-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet. 2009;41(7):843–848. doi: 10.1038/ng.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Samarasinghe KT, Jaime-Figueroa S, Burgess M, Nalawansha DA, Dai K, Hu Z, et al. Targeted degradation of transcription factors by TRAFTACs: transcription factor targeting chimeras. Cell Chem Biol. 2021;28(5):648–661. doi: 10.1016/j.chembiol.2021.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Szymański W, Beierle JM, Kistemaker HA, Velema WA, Feringa BL. Reversible photocontrol of biological systems by the incorporation of molecular photoswitches. Chem Rev. 2013;113(8):6114–6178. doi: 10.1021/cr300179f. [DOI] [PubMed] [Google Scholar]
- 64.Hüll K, Morstein J, Trauner D. In vivo photopharmacology. Chem Rev. 2018;118(21):10710–10747. doi: 10.1021/acs.chemrev.8b00037. [DOI] [PubMed] [Google Scholar]
- 65.Silva JM, Silva E, Reis RL. Light-triggered release of photocaged therapeutics - where are we now? J Control Release. 2019;298:154–176. doi: 10.1016/j.jconrel.2019.02.006. [DOI] [PubMed] [Google Scholar]
- 66.Kolb HC, Finn MG, Sharpless KB. Click chemistry: diverse chemical function from a few good reactions. Angew Chem Int Ed Engl. 2001;40(11):2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 67.Ji X, Pan Z, Yu B, De La Cruz LK, Zheng Y, Ke B, et al. Click and release: bioorthogonal approaches to “on-demand” activation of prodrugs. Chem Soc Rev. 2019;48(4):1077–1094. doi: 10.1039/c8cs00395e. [DOI] [PubMed] [Google Scholar]
- 68.Smeenk ML, Agramunt J, Bonger KM. Recent developments in bioorthogonal chemistry and the orthogonality within. Curr Opin Chem Biol. 2021;60:79–88. doi: 10.1016/j.cbpa.2020.09.002. [DOI] [PubMed] [Google Scholar]
- 69.Buckley DL, Raina K, Darricarrere N, Hines J, Gustafson JL, Smith IE, et al. HaloPROTACS: use of small molecule PROTACs to induce degradation of HaloTag fusion proteins. ACS Chem Biol. 2015;10(8):1831–1837. doi: 10.1021/acschembio.5b00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tomoshige S, Naito M, Hashimoto Y, Ishikawa M. Degradation of HaloTag-fused nuclear proteins using bestatin-HaloTag ligand hybrid molecules. Org Biomol Chem. 2015;13(38):9746–9750. doi: 10.1039/c5ob01395j. [DOI] [PubMed] [Google Scholar]
- 71.England CG, Luo H, Cai W. HaloTag technology: a versatile platform for biomedical applications. Bioconjug Chem. 2015;26(6):97586. doi: 10.1021/acs.bioconjchem.5b00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nabet B, Roberts JM, Buckley DL, Paulk J, Dastjerdi S, Yang A, et al. The dTAG system for immediate and targetspecific protein degradation. Nat Chem Biol. 2018;14(5):43141. doi: 10.1038/s41589-018-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 74.Martin GS. Cell signaling and cancer. Cancer Cell. 2003;4(3):167–174. doi: 10.1016/s1535-6108(03)00216-2. [DOI] [PubMed] [Google Scholar]
- 75.Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13(12):928–942. doi: 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- 76.Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AMK, et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci U S A. 2016;113(26):7124–7129. doi: 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jiang B, Wang ES, Donovan KA, Liang Y, Fischer ES, Zhang T, et al. Development of dual and selective degraders of cyclin-dependent kinases 4 and 6. Angew Chem Int Ed Engl. 2019;58(19):6321–6326. doi: 10.1002/anie.201901336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.De Dominici M, Porazzi P, Xiao Y, Chao A, Tang HY, Kumar G, et al. Selective inhibition of Ph-positive ALL cell growth through kinase-dependent and-independent effects by CDK6-specific PROTACs. Blood. 2020;135(18):1560–1573. doi: 10.1182/blood.2019003604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Burslem GM, Smith BE, Lai AC, Jaime-Figueroa S, McQuaid DC, Bondeson DP, et al. The advantages of targeted protein degradation over inhibition: an RTK case study. Cell Chem Biol. 2018;25(1):67–77. e3. doi: 10.1016/j.chembiol.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jang J, To C. De Clercq DJ, Park E, Ponthier CM, Shin BH, et al. Mutant-selective allosteric EGFR degraders are effective against a broad range of drug-resistant mutations. Angew Chem Int Ed Engl. 2020;132(34):14589–14597. doi: 10.1002/anie.202003500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Adhikari B, Bozilovic J, Diebold M, Schwarz JD, Hofstetter J, Schröder M, et al. PROTAC-mediated degradation reveals a non-catalytic function of AURORA-A kinase. Nat Chem Biol. 2020;16(11):1179–1188. doi: 10.1038/s41589-020-00652-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Remillard D, Buckley DL, Paulk J, Brien GL, Sonnett M, Seo HS, et al. Degradation of the BAF complex factor BRD9 by heterobifunctional ligands. Angew Chem Int Ed Engl. 2017;56(21):5738–5743. doi: 10.1002/anie.201611281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Farnaby W, Koegl M, Roy MJ, Whitworth C, Diers E, Trainor N, et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat Chem Biol. 2019;15(7):672–680. doi: 10.1038/s41589-019-0294-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Teng M, Jiang J, He Z, Kwiatkowski NP, Donovan KA, Mills CE, et al. Development of CDK2 and CDK5 dual degrader TMX-2172. Angew Chem Int Ed Engl. 2020;59(33):13865–13870. doi: 10.1002/anie.202004087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vollmer S, Cunoosamy D, Lv H, Feng H, Li X, Nan Z, et al. Design, synthesis, and biological evaluation of MEK PROTACs. J Med Chem. 2019;63(1):157–162. doi: 10.1021/acs.jmedchem.9b00810. [DOI] [PubMed] [Google Scholar]
- 86.Wei J, Hu J, Wang L, Xie L, Jin MS, Chen X, et al. Discovery of a first-in-class mitogen-activated protein kinase kinase 1/2 degrader. J Med Chem. 2019;62(23):10897–10911. doi: 10.1021/acs.jmedchem.9b01528. [DOI] [PubMed] [Google Scholar]
- 87.Bond MJ, Chu L, Nalawansha DA, Li K, Crews CM. Targeted degradation of oncogenic KRAS(G12C) by VHL-recruiting PROTACs. ACS Cent Sci. 2020;6(8):1367–1375. doi: 10.1021/acscentsci.0c00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li Z, Pinch BJ, Olson CM, Donovan KA, Nowak RP, Mills CE, et al. Development and characterization of a Wee1 kinase degrader. Cell Chem Biol. 2020;27(1):57–65. e9. doi: 10.1016/j.chembiol.2019.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ohoka N, Okuhira K, Ito M, Nagai K, Shibata N, Hattori T, et al. In vivo knockdown of pathogenic proteins via specific and nongenetic inhibitor of apoptosis protein (IAP)-dependent protein erasers (SNIPERs) J Biol Chem. 2017;292(11):4556–4570. doi: 10.1074/jbc.M116.768853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hines J, Lartigue S, Dong H, Qian Y, Crews CM. MDM2-recruiting PROTAC offers superior, synergistic antiproliferative activity via simultaneous degradation of BRD4 and stabilization of p53. Cancer Res. 2019;79(1):251–262. doi: 10.1158/0008-5472.CAN-18-2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zengerle M, Chan KH, Ciulli A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem Biol. 2015;10(8):1770–1777. doi: 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Winter GE, Mayer A, Buckley DL, Erb MA, Roderick JE, Vittori S, et al. BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol Cell. 2017;67(1):5–18. e9. doi: 10.1016/j.molcel.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nowak RP, DeAngelo SL, Buckley D, He Z, Donovan KA, An J, et al. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat Chem Biol. 2018;14(7):706–714. doi: 10.1038/s41589-018-0055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Testa A, Hughes SJ, Lucas X, Wright JE, Ciulli A. Structure-based design of a macrocyclic PROTAC. Angew Chem Int Ed Engl. 2020;59(4):1727–1734. doi: 10.1002/anie.201914396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bai L, Zhou B, Yang CY, Ji J, McEachern D, Przybranowski S, et al. Targeted degradation of BET proteins in triple-negative breast cancer. Cancer Res. 2017;77(9):2476–2487. doi: 10.1158/0008-5472.CAN-16-2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhou B, Hu J, Xu F, Chen Z, Bai L, Fernandez-Salas E, et al. Discovery of a small-molecule degrader of bromodomain and extra-terminal (BET) proteins with picomolar cellular potencies and capable of achieving tumor regression. J Med Chem. 2018;61(2):462–481. doi: 10.1021/acs.jmedchem.6b01816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang X, Crowley VM, Wucherpfennig TG, Dix MM, Cravatt BF. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat Chem Biol. 2019;15(7):737–746. doi: 10.1038/s41589-019-0279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Spradlin JN, Hu X, Ward CC, Brittain SM, Jones MD, Ou L, et al. Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat Chem Biol. 2019;15(7):747–755. doi: 10.1038/s41589-019-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang X, Xu F, Tong L, Zhang T, Xie H, Lu X, et al. Design and synthesis of selective degraders of EGFRL858R/T790M mutant. Eur J Med Chem. 2020;192:112199. doi: 10.1016/j.ejmech.2020.112199. [DOI] [PubMed] [Google Scholar]
- 100.Li L, Mi D, Pei H, Duan Q, Wang X, Zhou W, et al. In vivo target protein degradation induced by PROTACs based on E3 ligase DCAF15. Signal Transduct Target Ther. 2020;5(1):1–3. doi: 10.1038/s41392-020-00245-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhao HY, Yang XY, Lei H, Xi XX, Lu SM, Zhang JJ, et al. Discovery of potent small molecule PROTACs targeting mutant EGFR. Eur J Med Chem. 2020;208:112781. doi: 10.1016/j.ejmech.2020.112781. [DOI] [PubMed] [Google Scholar]
- 102.Brand M, Jiang B, Bauer S, Donovan KA, Liang Y, Wang ES, et al. Homolog-selective degradation as a strategy to probe the function of CDK6 in AML. Cell Chem Biol. 2019;26(2):300–6. e9. doi: 10.1016/j.chembiol.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cheng M, Yu X, Lu K, Xie L, Wang L, Meng F, et al. Discovery of potent and selective epidermal growth factor receptor (EGFR) bifunctional small-molecule degraders. J Med Chem. 2020;63(3):1216–1232. doi: 10.1021/acs.jmedchem.9b01566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhao B, Burgess K. PROTACs suppression of CDK4/6, crucial kinases for cell cycle regulation in cancer. Chem Commun. 2019;55(18):2704–2707. doi: 10.1039/c9cc00163h. [DOI] [PubMed] [Google Scholar]
- 105.Wei M, Zhao R, Cao Y, Wei Y, Li M, Dong Z, et al. First orally bioavailable prodrug of proteolysis targeting chimera (PROTAC) degrades cyclin-dependent kinases 2/4/6 in vivo. Eur J Med Chem. 2021;209:112903. doi: 10.1016/j.ejmech.2020.112903. [DOI] [PubMed] [Google Scholar]
- 106.Salami J, Alabi S, Willard RR, Vitale NJ, Wang J, Dong H, et al. Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun Biol. 2018;1(1):1–9. doi: 10.1038/s42003-018-0105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Steinebach C, Ng YLD, Sosič I, Lee C-S, Chen S, Lindner S, et al. Systematic exploration of different E3 ubiquitin ligases: an approach towards potent and selective CDK6 degraders. Chem Sci. 2020;11(13):3474–3486. doi: 10.1039/d0sc00167h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Takwale AD, Jo SH, Jeon YU, Kim HS, Shin CH, Lee HK, et al. Design and characterization of cereblon-mediated androgen receptor proteolysis-targeting chimeras. Eur J Med Chem. 2020;208:112769. doi: 10.1016/j.ejmech.2020.112769. [DOI] [PubMed] [Google Scholar]
- 109.Lee GT, Nagaya N, Desantis J, Madura K, Sabaawy HE, Kim WJ, et al. Effects of MTX-23, a novel PROTAC of androgen receptor splice variant-7 and androgen receptor, on CRPC resistant to second-line antiandrogen therapy. Mol Cancer Ther. 2020;20(3):490–499. doi: 10.1158/1535-7163.MCT-20-0417. [DOI] [PubMed] [Google Scholar]
- 110.Han X, Wang C, Qin C, Xiang W, Fernandez-Salas E, Yang CY, et al. Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J Med Chem. 2019;62(2):941–964. doi: 10.1021/acs.jmedchem.8b01631. [DOI] [PubMed] [Google Scholar]
- 111.Shibata N, Nagai K, Morita Y, Ujikawa O, Ohoka N, Hattori T, et al. Development of protein degradation inducers of androgen receptor by conjugation of androgen receptor ligands and inhibitor of apoptosis protein ligands. J Med Chem. 2018;61(2):543–575. doi: 10.1021/acs.jmedchem.7b00168. [DOI] [PubMed] [Google Scholar]
- 112.Hatcher JM, Wang ES, Johannessen L, Kwiatkowski N, Sim T, Gray NS. Development of highly potent and selective steroidal inhibitors and degraders of CDK8. ACS Med Chem Lett. 2018;9(6):540–545. doi: 10.1021/acsmedchemlett.8b00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang C, Han XR, Yang X, Jiang B, Liu J, Xiong Y, et al. Proteolysis targeting chimeras (PROTACs) of anaplastic lymphoma kinase (ALK) Eur J Med Chem. 2018;151:304–314. doi: 10.1016/j.ejmech.2018.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Robb CM, Contreras JI, Kour S, Taylor MA, Abid M, Sonawane YA, et al. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC) Chem Commun. 2017;53(54):7577–7580. doi: 10.1039/c7cc03879h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Qiu X, Li Y, Yu B, Ren J, Huang H, Wang M, et al. Discovery of selective CDK9 degraders with enhancing antiproliferative activity through PROTAC conversion. Eur J Med Chem. 2021;211:113091. doi: 10.1016/j.ejmech.2020.113091. [DOI] [PubMed] [Google Scholar]
- 116.Sun N, Ren C, Kong Y, Zhong H, Chen J, Li Y, et al. Development of a Brigatinib degrader (SIAIS117) as a potential treatment for ALK positive cancer resistance. Eur J Med Chem. 2020;193:112190. doi: 10.1016/j.ejmech.2020.112190. [DOI] [PubMed] [Google Scholar]
- 117.Zhou F, Chen L, Cao C, Yu J, Luo X, Zhou P, et al. Development of selective mono or dual PROTAC degrader probe of CDK isoforms. Eur J Med Chem. 2020;187:111952. doi: 10.1016/j.ejmech.2019.111952. [DOI] [PubMed] [Google Scholar]
- 118.Xue G, Chen J, Liu L, Zhou D, Zuo Y, Fu T, et al. Protein degradation through covalent inhibitor-based PROTACs. Chem Commun. 2020;56(10):1521–1524. doi: 10.1039/c9cc08238g. [DOI] [PubMed] [Google Scholar]
- 119.Chi JJ, Li H, Zhou Z, Izquierdo-Ferrer J, Xue Y, Wavelet CM, et al. A novel strategy to block mitotic progression for targeted therapy. EBioMedicine. 2019;49:40–54. doi: 10.1016/j.ebiom.2019.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zoppi V, Hughes SJ, Maniaci C, Testa A, Gmaschitz T, Wieshofer C, et al. Iterative design and optimization of initially inactive proteolysis targeting chimeras (PROTACs) identify VZ185 as a potent, fast, and selective von Hippel-Lindau (VHL) based dual degrader probe of BRD9 and BRD7. J Med Chem. 2018;62(2):699–726. doi: 10.1021/acs.jmedchem.8b01413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ma D, Zou Y, Chu Y, Liu Z, Liu G, Chu J, et al. A cell-permeable peptide-based PROTAC against the oncoprotein CREPT proficiently inhibits pancreatic cancer. Theranostics. 2020;10(8):3708. doi: 10.7150/thno.41677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhou L, Chen W, Cao C, Shi Y, Ye W, Hu J, et al. Design and synthesis of α-naphthoflavone chimera derivatives able to eliminate cytochrome P450 (CYP) 1B1-mediated drug resistance via targeted CYP1B1 degradation. Eur J Med Chem. 2020;189:112028. doi: 10.1016/j.ejmech.2019.112028. [DOI] [PubMed] [Google Scholar]
- 123.Madak JT, Cuthbertson CR, Chen W, Showalter HD, Neamati N. Design, synthesis, and characterization of brequinar conjugates as probes to study DHODH inhibition. Chemistry (Easton) 2017;23(56):13875–13878. doi: 10.1002/chem.201702999. [DOI] [PubMed] [Google Scholar]
- 124.Hu J, Hu B, Wang M, Xu F, Miao B, Yang CY, et al. Discovery of ERD-308 as a highly potent proteolysis targeting chimera (PROTAC) degrader of estrogen receptor (ER) J Med Chem. 2019;62(3):1420–1442. doi: 10.1021/acs.jmedchem.8b01572. [DOI] [PubMed] [Google Scholar]
- 125.Matyskiela ME, Lu G, Ito T, Pagarigan B, Lu CC, Miller K, et al. A novel cereblon modulator recruits GSPT1 to the CRL4 CRBN ubiquitin ligase. Nature. 2016;535(7611):252–257. doi: 10.1038/nature18611. [DOI] [PubMed] [Google Scholar]
- 126.Dai Y, Yue N, Gong J, Liu C, Li Q, Zhou J, et al. Development of cell-permeable peptide-based PROTACs targeting estrogen receptor α. Eur J Med Chem. 2020;187:111967. doi: 10.1016/j.ejmech.2019.111967. [DOI] [PubMed] [Google Scholar]
- 127.Mu X, Bai L, Xu Y, Wang J, Lu H. Protein targeting chimeric molecules specific for dual bromodomain 4 (BRD4) and polo-like kinase 1 (PLK1) proteins in acute myeloid leukemia cells. Biochem Biophys Res Commun. 2020;521(4):833–839. doi: 10.1016/j.bbrc.2019.11.007. [DOI] [PubMed] [Google Scholar]
- 128.Jiang Y, Deng Q, Zhao H, Xie M, Chen L, Yin F, et al. Development of stabilized peptide-based PROTACs against estrogen receptor α. ACS Chem Biol. 2017;13(3):628–635. doi: 10.1021/acschembio.7b00985. [DOI] [PubMed] [Google Scholar]
- 129.Bensimon A, Pizzagalli MD, Kartnig F, Dvorak V, Essletzbichler P, Winter GE, et al. Targeted degradation of SLC transporters reveals amenability of multi-pass transmembrane proteins to ligand-induced proteolysis. Cell Chem Biol. 2020;27(6):728–39.e9. doi: 10.1016/j.chembiol.2020.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ohoka N, Nagai K, Hattori T, Okuhira K, Shibata N, Cho N, et al. Cancer cell death induced by novel small molecules degrading the TACC3 protein via the ubiquitin–proteasome pathway. Cell Death Dis. 2014;5(11):e1513. doi: 10.1038/cddis.2014.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gechijian LN, Buckley DL, Lawlor MA, Reyes JM, Paulk J, Ott CJ, et al. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat Chem Biol. 2018;14(4):405–412. doi: 10.1038/s41589-018-0010-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Burslem GM, Song J, Chen X, Hines J, Crews CM. Enhancing antiproliferative activity and selectivity of a FLT-3 inhibitor by proteolysis targeting chimera conversion. J Am Chem Soc. 2018;140(48):16428–16432. doi: 10.1021/jacs.8b10320. [DOI] [PubMed] [Google Scholar]
- 133.Chen L, Chen Y, Zhang C, Jiao B, Liang S, Tan Q, et al. Discovery of first-in-class potent and selective tropomyosin receptor kinase degraders. J Med Chem. 2020;63(23):14562–14575. doi: 10.1021/acs.jmedchem.0c01342. [DOI] [PubMed] [Google Scholar]
- 134.Li Z, Lin Y, Song H, Qin X, Yu Z, Zhang Z, et al. First small-molecule PROTACs for G protein-coupled receptors: inducing α (1A)-adrenergic receptor degradation. Acta Pharm Sin B. 2020;10(9):1669–1679. doi: 10.1016/j.apsb.2020.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer. 2012;12(7):465–477. doi: 10.1038/nrc3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Qin C, Hu Y, Zhou B, Fernandez-Salas E, Yang CY, Liu L, et al. Discovery of QCA570 as an exceptionally potent and efficacious proteolysis targeting chimera (PROTAC) degrader of the bromodomain and extra-terminal (BET) proteins capable of inducing complete and durable tumor regression. J Med Chem. 2018;61(15):6685–6704. doi: 10.1021/acs.jmedchem.8b00506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhang J, Chen P, Zhu P, Zheng P, Wang T, Wang L, et al. Development of small-molecule BRD4 degraders based on pyrrolopyridone derivative. Bioorg Chem. 2020;99:103817. doi: 10.1016/j.bioorg.2020.103817. [DOI] [PubMed] [Google Scholar]
- 139.Ward CC, Kleinman JI, Brittain SM, Lee PS, Chung CYS, Kim K, et al. Covalent ligand screening uncovers a RNF4 E3 ligase recruiter for targeted protein degradation applications. ACS Chem Biol. 2019;14(11):2430–2440. doi: 10.1021/acschembio.8b01083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ohoka N, Tsuji G, Shoda T, Fujisato T, Kurihara M, Demizu Y, et al. Development of small molecule chimeras that recruit AhR E3 ligase to target proteins. ACS Chem Biol. 2019;14(12):2822–2832. doi: 10.1021/acschembio.9b00704. [DOI] [PubMed] [Google Scholar]
- 141.O'leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13(7):417–430. doi: 10.1038/nrclinonc.2016.26. [DOI] [PubMed] [Google Scholar]
- 142.Goel S, DeCristo MJ, McAllister SS, Zhao JJ. CDK4/6 inhibition in cancer: beyond cell cycle arrest. Trends Cell Biol. 2018;28(11):911–925. doi: 10.1016/j.tcb.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Álvarez-Fernández M, Malumbres M. Mechanisms of sensitivity and resistance to CDK4/6 inhibition. Cancer Cell. 2020;37(4):514–529. doi: 10.1016/j.ccell.2020.03.010. [DOI] [PubMed] [Google Scholar]
- 144.Su S, Yang Z, Gao H, Yang H, Zhu S, An Z, et al. Potent and preferential degradation of CDK6 via proteolysis targeting chimera degraders. J Med Chem. 2019;62(16):7575–7582. doi: 10.1021/acs.jmedchem.9b00871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. 2001;2(1):21–32. doi: 10.1038/35048096. [DOI] [PubMed] [Google Scholar]
- 146.O’Connor OA, Özcan M, Jacobsen ED, Roncero JM, Trotman J, Demeter J, et al. Randomized phase III study of alisertib or investigator’s choice (selected single agent) in patients with relapsed or refractory peripheral T-cell lymphoma. J Clin Oncol. 2019;37(8):613–623. doi: 10.1200/JCO.18.00899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Beltran H, Oromendia C, Danila DC, Montgomery B, Hoimes C, Szmulewitz RZ, et al. A phase II trial of the aurora kinase a inhibitor alisertib for patients with castration-resistant and neuroendocrine prostate cancer: efficacy and biomarkers. Clin Cancer Res. 2019;25(1):43–51. doi: 10.1158/1078-0432.CCR-18-1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358(11):1160–1174. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- 149.Chong CR, Jänne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19(11):1389–1400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhang H, Zhao HY, Xi XX, Liu YJ, Xin M, Mao S, et al. Discovery of potent epidermal growth factor receptor (EGFR) degraders by proteolysis targeting chimera (PROTAC) Eur J Med Chem. 2020;189:112061. doi: 10.1016/j.ejmech.2020.112061. [DOI] [PubMed] [Google Scholar]
- 151.Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol. 2015;16(5):281–298. doi: 10.1038/nrm3979. [DOI] [PubMed] [Google Scholar]
- 152.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF (V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF (V600E) Nature. 2011;480(7377):387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chen H, Chen F, Pei S, Gou S. Pomalidomide hybrids act as proteolysis targeting chimeras: synthesis, anticancer activity and B-Raf degradation. Bioorg Chem. 2019;87:191–199. doi: 10.1016/j.bioorg.2019.03.035. [DOI] [PubMed] [Google Scholar]
- 155.Han X, Zhao L, Xiang W, Qin C, Miao B, Xu T, et al. Discovery of highly potent and efficient PROTAC degraders of androgen receptor (AR) by employing weak binding affinity VHL E3 ligase ligands. J Med Chem. 2019;62(24):11218–11231. doi: 10.1021/acs.jmedchem.9b01393. [DOI] [PubMed] [Google Scholar]
- 156.Kregel S, Wang C, Han X, Xiao L, Fernandez-Salas E, Bawa P, et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia. 2020;22(2):111–119. doi: 10.1016/j.neo.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yan G, Zhong X, Yue L, Pu C, Shan H, Lan S, et al. Discovery of a PROTAC targeting ALK with in vivo activity. Eur J Med Chem. 2021;212:113150. doi: 10.1016/j.ejmech.2020.113150. [DOI] [PubMed] [Google Scholar]
- 158.Brien GL, Remillard D, Shi J, Hemming ML, Chabon J, Wynne K, et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. Elife. 2018;7:e41305. doi: 10.7554/eLife.41305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bian J, Ren J, Li Y, Wang J, Xu X, Feng Y, et al. Discovery of Wogonin-based PROTACs against CDK9 and capable of achieving antitumor activity. Bioorg Chem. 2018;81:373–381. doi: 10.1016/j.bioorg.2018.08.028. [DOI] [PubMed] [Google Scholar]
- 160.Ohoka N, Morita Y, Nagai K, Shimokawa K, Ujikawa O, Fujimori I, et al. Derivatization of inhibitor of apoptosis protein (IAP) ligands yields improved inducers of estrogen receptor α degradation. J Biol Chem. 2018;293(18):6776–6790. doi: 10.1074/jbc.RA117.001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Huang HT, Dobrovolsky D, Paulk J, Yang G, Weisberg EL, Doctor ZM, et al. A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chem Biol. 2018;25(1):88–99. e6. doi: 10.1016/j.chembiol.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hu J, Wei J, Yim H, Wang L, Xie L, Jin MS, et al. Potent and selective mitogen-activated protein kinase kinase 1/2 (MEK1/2) heterobifunctional small-molecule degraders. J Med Chem. 2020;63(24):15883–15905. doi: 10.1021/acs.jmedchem.0c01609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zeng M, Xiong Y, Safaee N, Nowak RP, Donovan KA, Yuan CJ, et al. Exploring targeted degradation strategy for oncogenic KRASG12C. Cell Chem Biol. 2020;27(1):19–31. e6. doi: 10.1016/j.chembiol.2019.12.006. [DOI] [PubMed] [Google Scholar]
- 164.Zhao B, Burgess K. TrkC-targeted kinase inhibitors and PROTACs. Mol Pharm. 2019;16(10):4313–4318. doi: 10.1021/acs.molpharmaceut.9b00673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17(7):395–417. doi: 10.1038/s41571-020-0341-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Mohammad RM, Muqbil I, Lowe L, Yedjou C, Hsu HY, Lin LT, et al. Broad targeting of resistance to apoptosis in cancer. Semin Cancer Biol. 2015;35:S78–S103. doi: 10.1016/j.semcancer.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Khan S, Zhang X, Lv D, Zhang Q, He Y, Zhang P, et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat Med. 2019;25(12):1938–1947. doi: 10.1038/s41591-019-0668-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zhang X, Thummuri D, He Y, Liu X, Zhang P, Zhou D, et al. Utilizing PROTAC technology to address the on-target platelet toxicity associated with inhibition of BCL-XL. Chem Commun. 2019;55(98):14765–14768. doi: 10.1039/c9cc07217a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Cao C, Yang J, Chen Y, Zhou P, Wang Y, Du W, et al. Discovery of SK-575 as a highly potent and efficacious proteolysis-targeting chimera degrader of PARP1 for treating cancers. J Med Chem. 2020;63(19):11012–11033. doi: 10.1021/acs.jmedchem.0c00821. [DOI] [PubMed] [Google Scholar]
- 170.Wang S, Han L, Han J, Li P, Ding Q, Zhang QJ, et al. Uncoupling of PARP1 trapping and inhibition using selective PARP1 degradation. Nat Chem Biol. 2019;15(12):1223–1231. doi: 10.1038/s41589-019-0379-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lai AC, Toure M, Hellerschmied D, Salami J, Jaime-Figueroa S, Ko E, et al. Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew Chem Int Ed Engl. 2016;55(2):807–810. doi: 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Burslem GM, Schultz AR, Bondeson DP, Eide CA, Stevens SLS, Druker BJ, et al. Targeting BCR-ABL1 in chronic myeloid leukemia by PROTAC-mediated targeted protein degradation. Cancer Res. 2019;79(18):4744–4753. doi: 10.1158/0008-5472.CAN-19-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.You I, Erickson EC, Donovan KA, Eleuteri NA, Fischer ES, Gray NS, et al. Discovery of an AKT degrader with prolonged inhibition of downstream signaling. Cell Chem Biol. 2020;27(1):66–73. e7. doi: 10.1016/j.chembiol.2019.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wang Z, He N, Guo Z, Niu C, Song T, Guo Y, et al. Proteolysis targeting chimeras for the selective degradation of mcl-1/Bcl-2 derived from nonselective target binding ligands. J Med Chem. 2019;62(17):8152–8163. doi: 10.1021/acs.jmedchem.9b00919. [DOI] [PubMed] [Google Scholar]
- 175.Itoh Y, Ishikawa M, Kitaguchi R, Okuhira K, Naito M, Hashimoto Y. Double protein knockdown of cIAP1 and CRABP-II using a hybrid molecule consisting of ATRA and IAPs antagonist. Bioorg Med Chem Lett. 2012;22(13):4453–4457. doi: 10.1016/j.bmcl.2012.04.134. [DOI] [PubMed] [Google Scholar]
- 176.Kaur T, Menon A, Garner AL. Synthesis of 7-benzylguanosine cap-analogue conjugates for eIF4E targeted degradation. Eur J Med Chem. 2019;166:339–350. doi: 10.1016/j.ejmech.2019.01.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Papatzimas JW, Gorobets E, Maity R, Muniyat MI, MacCallum JL, Neri P, et al. From inhibition to degradation: targeting the antiapoptotic protein myeloid cell leukemia 1 (MCL1) J Med Chem. 2019;62(11):5522–5540. doi: 10.1021/acs.jmedchem.9b00455. [DOI] [PubMed] [Google Scholar]
- 178.Wang B, Wu S, Liu J, Yang K, Xie H, Tang W. Development of selective small molecule MDM2 degraders based on nutlin. Eur J Med Chem. 2019;176:476–491. doi: 10.1016/j.ejmech.2019.05.046. [DOI] [PubMed] [Google Scholar]
- 179.Li Y, Yang J, Aguilar A, McEachern D, Przybranowski S, Liu L, et al. Discovery of MD-224 as a first-in-class, highly potent, and efficacious proteolysis targeting chimera murine double minute 2 degrader capable of achieving complete and durable tumor regression. J Med Chem. 2018;62(2):448–466. doi: 10.1021/acs.jmedchem.8b00909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Li W, Gao C, Zhao L, Yuan Z, Chen Y, Jiang Y. Phthalimide conjugations for the degradation of oncogenic PI3K. Eur J Med Chem. 2018;151:237–247. doi: 10.1016/j.ejmech.2018.03.066. [DOI] [PubMed] [Google Scholar]
- 181.Vannam R, Sayilgan J, Ojeda S, Karakyriakou B, Hu E, Kreuzer J, et al. Targeted degradation of the enhancer lysine acetyltransferases CBP and p300. Cell Chem Biol. 2021;28(4):503–14.e12. doi: 10.1016/j.chembiol.2020.12.004. [DOI] [PubMed] [Google Scholar]
- 182.Schiedel M, Herp D, Hammelmann SR, Swyter SR, Lehotzky A, Robaa D, et al. Chemically induced degradation of sirtuin 2 (Sirt2) by a proteolysis targeting chimera (PROTAC) based on sirtuin rearranging ligands (SirReals) J Med Chem. 2018;61(2):482–491. doi: 10.1021/acs.jmedchem.6b01872. [DOI] [PubMed] [Google Scholar]
- 183.Shan Y, Si R, Wang J, Zhang Q, Li J, Ma Y, et al. Discovery of novel anti-angiogenesis agents. Part 11: Development of PROTACs based on active molecules with potency of promoting vascular normalization. Eur J Med Chem. 2020;205:112654. doi: 10.1016/j.ejmech.2020.112654. [DOI] [PubMed] [Google Scholar]
- 184.Zhang X, Thummuri D, Liu X, Hu W, Zhang P, Khan S, et al. Discovery of PROTAC BCL-XL degraders as potent anticancer agents with low on-target platelet toxicity. Eur J Med Chem. 2020;192:112186. doi: 10.1016/j.ejmech.2020.112186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhang X, He Y, Zhang P, Budamagunta V, Lv D, Thummuri D, et al. Discovery of IAP-recruiting BCL-XL PROTACs as potent degraders across multiple cancer cell lines. Eur J Med Chem. 2020;199:112397. doi: 10.1016/j.ejmech.2020.112397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Yang Y, Gao H, Sun X, Sun Y, Qiu Y, Weng Q, et al. Global PROTAC toolbox for degrading BCR-ABL overcomes drug-resistant mutants and adverse effects. J Med Chem. 2020;63(15):8567–8583. doi: 10.1021/acs.jmedchem.0c00967. [DOI] [PubMed] [Google Scholar]
- 187.Chung CW, Dai H, Fernandez E, Tinworth CP, Churcher I, Cryan J, et al. Structural insights into PROTAC-mediated degradation of Bcl-xL. ACS Chem Biol. 2020;15(9):2316–2323. doi: 10.1021/acschembio.0c00266. [DOI] [PubMed] [Google Scholar]
- 188.Zhang Z, Chang X, Zhang C, Zeng S, Liang M, Ma Z, et al. Identification of probe-quality degraders for poly (ADP-ribose) polymerase-1 (PARP-1) J Enzyme Inhib Med Chem. 2020;35(1):1606–1615. doi: 10.1080/14756366.2020.1804382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Burslem GM, Bondeson DP, Crews CM. Scaffold hopping enables direct access to more potent PROTACs with in vivo activity. Chem Commun. 2020;56(50):6890–6892. doi: 10.1039/d0cc02201b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zhao Q, Ren C, Liu L, Chen J, Shao Y, Sun N, et al. Discovery of SIAIS178 as an effective BCR-ABL degrader by recruiting von Hippel-Lindau (VHL) E3 ubiquitin ligase. J Med Chem. 2019;62(20):9281–9298. doi: 10.1021/acs.jmedchem.9b01264. [DOI] [PubMed] [Google Scholar]
- 191.Zhao Q, Lan T, Su S, Rao Y. Induction of apoptosis in MDA-MB-231 breast cancer cells by a PARP1-targeting PROTAC small molecule. Chem Commun. 2019;55(3):369–372. doi: 10.1039/c8cc07813k. [DOI] [PubMed] [Google Scholar]
- 192.Tong B, Spradlin JN, Novaes LF, Zhang E, Hu X, Moeller M, et al. A nimbolide-based kinase degrader preferentially degrades oncogenic BCR-ABL. ACS Chem Biol. 2020;15(7):1788–1794. doi: 10.1021/acschembio.0c00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.McCoull W, Cheung T, Anderson E, Barton P, Burgess J, Byth K, et al. Development of a novel B-cell lymphoma 6 (BCL6) PROTAC to provide insight into small molecule targeting of BCL6. ACS Chem Biol. 2018;13(11):3131–3141. doi: 10.1021/acschembio.8b00698. [DOI] [PubMed] [Google Scholar]
- 194.James K, John W, MichaeláCowley S. PROTAC-mediated degradation of class I histone deacetylase enzymes in corepressor complexes. Chem Commun. 2020;56(32):4476–4479. doi: 10.1039/d0cc01485k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Xiao Y, Wang J, Zhao LY, Chen X, Zheng G, Zhang X, et al. Discovery of histone deacetylase 3 (HDAC3)-specific PROTACs. Chem Commun. 2020;56(68):9866–9869. doi: 10.1039/d0cc03243c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Chen H, Chen F, Liu N, Wang X, Gou S. Chemically induced degradation of CK2 by proteolysis targeting chimeras based on a ubiquitin-proteasome pathway. Bioorg Chem. 2018;81:536–544. doi: 10.1016/j.bioorg.2018.09.005. [DOI] [PubMed] [Google Scholar]
- 197.Sinatra L, Bandolik JJ, Roatsch M, Sönnichsen M, Schoeder CT, Hamacher A, et al. Hydroxamic acids immobilized on resins (HAIRs): synthesis of dual-targeting HDAC inhibitors and HDAC degraders (PROTACs) Angew Chem Int Ed Engl. 2020;59(50):22494–22499. doi: 10.1002/anie.202006725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Wu H, Yang K, Zhang Z, Leisten ED, Li Z, Xie H, et al. Development of multifunctional histone deacetylase 6 degraders with potent antimyeloma activity. J Med Chem. 2019;62(15):7042–7057. doi: 10.1021/acs.jmedchem.9b00516. [DOI] [PubMed] [Google Scholar]
- 199.An Z, Lv W, Su S, Wu W, Rao Y. Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell. 2019;10(8):606–609. doi: 10.1007/s13238-018-0602-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Yang H, Lv W, He M, Deng H, Li H, Wu W, et al. Plasticity in designing PROTACs for selective and potent degradation of HDAC6. Chem Commun. 2019;55(98):14848–14851. doi: 10.1039/c9cc08509b. [DOI] [PubMed] [Google Scholar]
- 201.Liu Y, Zhen Y, Wang G, Yang G, Fu L, Liu B, et al. Designing an eEF2K-targeting PROTAC small molecule that induces apoptosis in MDA-MB-231 cells. Eur J Med Chem. 2020;204:112505. doi: 10.1016/j.ejmech.2020.112505. [DOI] [PubMed] [Google Scholar]
- 202.Cao J, Zhao W, Zhao C, Liu Q, Li S, Zhang G, et al. Development of a bestatin-SAHA hybrid with dual inhibitory activity against APN and HDAC. Molecules. 2020;25(21):4991. doi: 10.3390/molecules25214991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Yang K, Wu H, Zhang Z, Leisten ED, Nie X, Liu B, et al. Development of selective histone deacetylase 6 (HDAC6) degraders recruiting Von Hippel-Lindau (VHL) E3 ubiquitin ligase. ACS Med Chem Lett. 2020;11(4):575–581. doi: 10.1021/acsmedchemlett.0c00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Tovell H, Testa A, Zhou H, Shpiro N, Crafter C, Ciulli A, et al. Design and characterization of SGK3-PROTAC1, an isoform specific SGK3 kinase PROTAC degrader. ACS Chem Biol. 2019;14(9):2024–2034. doi: 10.1021/acschembio.9b00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Schoenwaelder SM, Jarman KE, Gardiner EE, Hua M, Qiao J, White MJ, et al. Bcl-xL-inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood. 2011;118(6):1663–1674. doi: 10.1182/blood-2011-04-347849. [DOI] [PubMed] [Google Scholar]
- 206.Curtin NJ, Szabo C. Poly (ADP-ribose) polymerase inhibition: past, present and future. Nat Rev Drug Discov. 2020;19(10):711–736. doi: 10.1038/s41573-020-0076-6. [DOI] [PubMed] [Google Scholar]
- 207.Skorski T. BCR/ABL regulates response to DNA damage: the role in resistance to genotoxic treatment and in genomic instability. Oncogene. 2002;21(56):8591–8604. doi: 10.1038/sj.onc.1206087. [DOI] [PubMed] [Google Scholar]
- 208.Quintás-Cardama A, Cortes J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood. 2009;113(8):1619–1630. doi: 10.1182/blood-2008-03-144790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest. 2011;121(1):396–409. doi: 10.1172/JCI35721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Soverini S, Hochhaus A, Nicolini FE, Gruber F, Lange T, Saglio G, et al. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood. 2011;118(5):1208–1215. doi: 10.1182/blood-2010-12-326405. [DOI] [PubMed] [Google Scholar]
- 211.Apte RS, Chen DS, Ferrara N. VEGF in signaling and disease: beyond discovery and development. Cell. 2019;176(6):1248–1264. doi: 10.1016/j.cell.2019.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Garner H, de Visser KE. Immune crosstalk in cancer progression and metastatic spread: a complex conversation. Nat Rev Immunol. 2020;20(8):483–497. doi: 10.1038/s41577-019-0271-z. [DOI] [PubMed] [Google Scholar]
- 213.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 214.Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015;35:S185–SS98. doi: 10.1016/j.semcancer.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 215.Cheng B, Ren Y, Cao H, Chen J. Discovery of novel resorcinol diphenyl ether-based PROTAC-like molecules as dual inhibitors and degraders of PD-L1. Eur J Med Chem. 2020;199:112377. doi: 10.1016/j.ejmech.2020.112377. [DOI] [PubMed] [Google Scholar]
- 216.Sun Y, Zhao X, Ding N, Gao H, Wu Y, Yang Y, et al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 2018;28(7):779–781. doi: 10.1038/s41422-018-0055-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Gabizon R, Shraga A, Gehrtz P, Livnah E, Shorer Y, Gurwicz N, et al. Efficient targeted degradation via reversible and irreversible covalent PROTACs. J Am Chem Soc. 2020;142(27):11734–11742. doi: 10.1021/jacs.9b13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Dobrovolsky D, Wang ES, Morrow S, Leahy C, Faust T, Nowak RP, et al. Bruton tyrosine kinase degradation as a therapeutic strategy for cancer. Blood. 2019;133(9):952–961. doi: 10.1182/blood-2018-07-862953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Schiemer J, Horst R, Meng Y, Montgomery JI, Xu Y, Feng X, et al. Snapshots and ensembles of BTK and cIAP1 protein degrader ternary complexes. Nat Chem Biol. 2020;17(2):152–160. doi: 10.1038/s41589-020-00686-2. [DOI] [PubMed] [Google Scholar]
- 220.Si J, Shi X, Sun S, Zou B, Li Y, An D, et al. Hematopoietic progenitor kinase1 (HPK1) mediates T cell dysfunction and is a druggable target for T cell-based immunotherapies. Cancer Cell. 2020;38(4):551–66. e11. doi: 10.1016/j.ccell.2020.08.001. [DOI] [PubMed] [Google Scholar]
- 221.Hu M, Zhou W, Wang Y, Yao D, Ye T, Yao Y, et al. Discovery of the first potent proteolysis targeting chimera (PROTAC) degrader of indoleamine 2, 3-dioxygenase 1. Acta Pharm Sin B. 2020;10(10):1943–1953. doi: 10.1016/j.apsb.2020.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Shah RR, Redmond JM, Mihut A, Menon M, Evans JP, Murphy JA, et al. Hi-JAK-ing the ubiquitin system: the design and physicochemical optimisation of JAK PROTACs. Bioorg Med Chem. 2020;28(5):115326. doi: 10.1016/j.bmc.2020.115326. [DOI] [PubMed] [Google Scholar]
- 223.Wang M, Lu J, Wang M, Yang CY, Wang S. Discovery of SHP2-D26 as a first, potent, and effective PROTAC degrader of SHP2 protein. J Med Chem. 2020;63(14):7510–7528. doi: 10.1021/acs.jmedchem.0c00471. [DOI] [PubMed] [Google Scholar]
- 224.O’Donnell JS, Teng MW, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16(3):151–167. doi: 10.1038/s41571-018-0142-8. [DOI] [PubMed] [Google Scholar]
- 225.Erb MA, Scott TG, Li BE, Xie H, Paulk J, Seo HS, et al. Transcription control by the ENL YEATS domain in acute leukaemia. Nature. 2017;543(7644):270–274. doi: 10.1038/nature21688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Sun X, Wang J, Yao X, Zheng W, Mao Y, Lan T, et al. A chemical approach for global protein knockdown from mice to non-human primates. Cell Discov. 2019;5(1):1–13. doi: 10.1038/s41421-018-0079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Guo WH, Qi X, Yu X, Liu Y, Chung CI, Bai F, et al. Enhancing intracellular accumulation and target engagement of PROTACs with reversible covalent chemistry. Nat Commun. 2020;11(1):1–16. doi: 10.1038/s41467-020-17997-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Zorba A, Nguyen C, Xu Y, Starr J, Borzilleri K, Smith J, et al. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc Natl Acad Sci U S A. 2018;115(31):E7285–E7E92. doi: 10.1073/pnas.1803662115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Zhang J, Fu L, Shen B, Liu Y, Wang W, Cai X, et al. Assessing IRAK4 functions in ABC DLBCL by IRAK4 kinase inhibition and protein degradation. Cell Chem Biol. 2020;27(12):1500–9. e13. doi: 10.1016/j.chembiol.2020.08.010. [DOI] [PubMed] [Google Scholar]
- 230.Zhou Z, Long J, Wang Y, Li Y, Zhang X, Tang L, et al. Targeted degradation of CD147 proteins in melanoma. Bioorg Chem. 2020;105:104453. doi: 10.1016/j.bioorg.2020.104453. [DOI] [PubMed] [Google Scholar]
- 231.Hsu JHR, Rasmusson T, Robinson J, Pachl F, Read J, Kawatkar S, et al. EED-targeted PROTACs degrade EED, EZH2, and SUZ12 in the PRC2 complex. Cell Chem Biol. 2020;27(1):41–6. e17. doi: 10.1016/j.chembiol.2019.11.004. [DOI] [PubMed] [Google Scholar]
- 232.Cheng J, Li Y, Wang X, Dong G, Sheng C. Discovery of novel PDEδ degraders for the treatment of KRAS mutant colorectal cancer. J Med Chem. 2020;63(14):7892–7905. doi: 10.1021/acs.jmedchem.0c00929. [DOI] [PubMed] [Google Scholar]
- 233.Shen Y, Gao G, Yu X, Kim H, Wang L, Xie L, et al. Discovery of first-in-class protein arginine methyltransferase 5 (PRMT5) degraders. J Med Chem. 2020;63(17):9977–9989. doi: 10.1021/acs.jmedchem.0c01111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Winzker M, Friese A, Koch U, Janning P, Ziegler S, Waldmann H. Development of a PDEδ-targeting PROTACs that impair lipid metabolism. Angew Chem Int Ed Engl. 2020;59(14):5595–5601. doi: 10.1002/anie.201913904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Song Y, Park PM, Wu L, Ray A, Picaud S, Li D, et al. Development and preclinical validation of a novel covalent ubiquitin receptor Rpn13 degrader in multiple myeloma. Leukemia. 2019;33(11):2685–2694. doi: 10.1038/s41375-019-0467-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Chessum NE, Sharp SY, Caldwell JJ, Pasqua AE, Wilding B, Colombano G, et al. Demonstrating in-cell target engagement using a pirin protein degradation probe (CCT367766) J Med Chem. 2018;61(3):918–933. doi: 10.1021/acs.jmedchem.7b01406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Potjewyd F, Turner AMW, Beri J, Rectenwald JM, Norris-Drouin JL, Cholensky SH, et al. Degradation of polycomb repressive complex 2 with an EED-targeted bivalent chemical degrader. Cell Chem Biol. 2020;27(1):47–56. e15. doi: 10.1016/j.chembiol.2019.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Crew AP, Raina K, Dong H, Qian Y, Wang J, Vigil D, et al. Identification and characterization of Von Hippel-Lindau-recruiting proteolysis targeting chimeras (PROTACs) of TANK-binding kinase 1. J Med Chem. 2018;61(2):583–598. doi: 10.1021/acs.jmedchem.7b00635. [DOI] [PubMed] [Google Scholar]
- 239.Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. 2018;48(3):434–452. doi: 10.1016/j.immuni.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Burger JA, Wiestner A. Targeting B cell receptor signalling in cancer: preclinical and clinical advances. Nat Rev Cancer. 2018;18(3):148. doi: 10.1038/nrc.2017.121. [DOI] [PubMed] [Google Scholar]
- 241.Sun Y, Ding N, Song Y, Yang Z, Liu W, Zhu J, et al. Degradation of Bruton’s tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-Hodgkin lymphomas. Leukemia. 2019;33(8):2105–2110. doi: 10.1038/s41375-019-0440-x. [DOI] [PubMed] [Google Scholar]
- 242.Vanharanta S, Massagué J. Origins of metastatic traits. Cancer Cell. 2013;24(4):410–421. doi: 10.1016/j.ccr.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Reymond N, d'Água BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. 2013;13(12):858–870. doi: 10.1038/nrc3628. [DOI] [PubMed] [Google Scholar]
- 244.Hamidi H, Ivaska J. Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer. 2018;18(9):533–548. doi: 10.1038/s41568-018-0038-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Singh M, Yelle N, Venugopal C, Singh SK. EMT: mechanisms and therapeutic implications. Pharmacol Ther. 2018;182:80–94. doi: 10.1016/j.pharmthera.2017.08.009. [DOI] [PubMed] [Google Scholar]
- 246.Popow J, Arnhof H, Bader G, Berger H, Ciulli A, Covini D, et al. Highly selective PTK2 proteolysis targeting chimeras to probe focal adhesion kinase scaffolding functions. J Med Chem. 2019;62(5):2508–2520. doi: 10.1021/acs.jmedchem.8b01826. [DOI] [PubMed] [Google Scholar]
- 247.Gao H, Wu Y, Sun Y, Yang Y, Zhou G, Rao Y. Design, synthesis, and evaluation of highly potent FAK-targeting PROTACs. ACS Med Chem Lett. 2019;11(10):1855–1862. doi: 10.1021/acsmedchemlett.9b00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Manda S, Lee NK, Oh DC, Lee J. Design, synthesis, and biological evaluation of proteolysis targeting chimeras (PROTACs) for the dual degradation of IGF-1R and Src. Molecules. 2020;25(8):1948. doi: 10.3390/molecules25081948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Smith BE, Wang SL, Jaime-Figueroa S, Harbin A, Wang J, Hamman BD, et al. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat Commun. 2019;10(1):1–13. doi: 10.1038/s41467-018-08027-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Bondeson DP, Smith BE, Burslem GM, Buhimschi AD, Hines J, Jaime-Figueroa S, et al. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem Biol. 2018;25(1):78–87. e5. doi: 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Wang X, Feng S, Fan J, Li X, Wen Q, Luo N. New strategy for renal fibrosis: targeting Smad3 proteins for ubiquitination and degradation. Biochem Pharmacol. 2016;116:200–209. doi: 10.1016/j.bcp.2016.07.017. [DOI] [PubMed] [Google Scholar]
- 252.Lee Y, Heo J, Jeong H, Hong KT, Kwon DH, Shin MH, et al. Targeted degradation of transcription coactivator SRC-1 through the N-degron pathway. Angew Chem Int Ed Engl. 2020;132(40):17701–17708. doi: 10.1002/anie.202005004. [DOI] [PubMed] [Google Scholar]
- 253.Jain N, Hartert K, Tadros S, Fiskus W, Havranek O, Ma MCJ, et al. Targetable genetic alterations of TCF4 (E2–2) drive immunoglobulin expression in diffuse large B cell lymphoma. Sci Transl Med. 2019;11(497):eaav5599. doi: 10.1126/scitranslmed.aav5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Feng Y, Su H, Li Y, Luo C, Xu H, Wang Y, et al. Degradation of intracellular TGF-β1 by PROTACs efficiently reverses M2 macrophage induced malignant pathological events. Chem Commun. 2020;56(19):2881–2884. doi: 10.1039/c9cc08391j. [DOI] [PubMed] [Google Scholar]
- 255.Liao H, Li X, Zhao L, Wang Y, Wang X, Wu Y, et al. A PROTAC peptide induces durable β-catenin degradation and suppresses Wnt-dependent intestinal cancer. Cell Discov. 2020;6(1):1–12. doi: 10.1038/s41421-020-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Cromm PM, Samarasinghe KT, Hines J, Crews CM. Addressing kinase-independent functions of Fak via PROTAC-mediated degradation. J Am Chem Soc. 2018;140(49):17019–17026. doi: 10.1021/jacs.8b08008. [DOI] [PubMed] [Google Scholar]
- 257.Donoghue C, Cubillos-Rojas M, Gutierrez-Prat N, Sanchez-Zarzalejo C, Verdaguer X, Riera A, et al. Optimal linker length for small molecule PROTACs that selectively target p38α and p38β for degradation. Eur J Med Chem. 2020;201:112451. doi: 10.1016/j.ejmech.2020.112451. [DOI] [PubMed] [Google Scholar]
- 258.Cooper J, Giancotti FG. Integrin signaling in cancer: mechanotransduction, stemness, epithelial plasticity, and therapeutic resistance. Cancer Cell. 2019;35(3):347–367. doi: 10.1016/j.ccell.2019.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014;14(9):598–610. doi: 10.1038/nrc3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Fujii T, Koshikawa K, Nomoto S, Okochi O, Kaneko T, Inoue S, et al. Focal adhesion kinase is overexpressed in hepatocellular carcinoma and can be served as an independent prognostic factor. J Hepatol. 2004;41(1):104–111. doi: 10.1016/j.jhep.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 261.Lu Y, Sun H. Progress in the development of small molecular inhibitors of focal adhesion kinase (FAK) J Med Chem. 2020;63(23):14382–14403. doi: 10.1021/acs.jmedchem.0c01248. [DOI] [PubMed] [Google Scholar]
- 262.Cance WG, Kurenova E, Marlowe T, Golubovskaya V. Disrupting the scaffold to improve focal adhesion kinase–targeted cancer therapeutics. Sci Signal. 2013;6(268):pe10. [DOI] [PMC free article] [PubMed]
- 263.Donovan KA, Ferguson FM, Bushman JW, Eleuteri NA, Bhunia D, Ryu S, et al. Mapping the degradable kinome provides a resource for expedited degrader development. Cell. 2020;183(6):1714–31. e10. doi: 10.1016/j.cell.2020.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Cotton AD, Nguyen DP, Gramespacher JA, Seiple IB, Wells JA. Development of antibody-based PROTACs for the degradation of the cell-surface immune checkpoint protein PD-L1. J Am Chem Soc. 2021;143(2):593–598. doi: 10.1021/jacs.0c10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Banik S, Pedram K, Wisnovsky S, Riley N, Bertozzi C. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature. 2020;584(7820):291–297. doi: 10.1038/s41586-020-2545-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Takahashi D, Moriyama J, Nakamura T, Miki E, Takahashi E, Sato A, et al. AUTACs: cargo-specific degraders using selective autophagy. Mol Cell. 2019;76(5):797–810. e10. doi: 10.1016/j.molcel.2019.09.009. [DOI] [PubMed] [Google Scholar]
- 267.Li Z, Wang C, Wang Z, Zhu C, Li J, Sha T, et al. Allele-selective lowering of mutant HTT protein by HTT-LC3 linker compounds. Nature. 2019;575(7781):203–209. doi: 10.1038/s41586-019-1722-1. [DOI] [PubMed] [Google Scholar]
- 268.Liu X, Haniff HS, Childs-Disney JL, Shuster A, Aikawa H, Adibekian A, et al. Targeted degradation of the oncogenic microRNA 17-92 cluster by structure-targeting ligands. J Am Chem Soc. 2020;142(15):6970–6982. doi: 10.1021/jacs.9b13159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Haniff HS, Tong Y, Liu X, Chen JL, Suresh BM, Andrews RJ, et al. Targeting the SARS-CoV-2 RNA genome with small molecule binders and ribonuclease targeting chimera (RIBOTAC) degraders. ACS Cent Sci. 2020;6(10):1713–1721. doi: 10.1021/acscentsci.0c00984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.