

Abstract
Objective:
The cellular diversity of the inner ear has presented a technical challenge in obtaining molecular insight into its development and function. The application of technological advancements in cell type–specific expression enable clinicians and researchers to leap forward from classic genetics to obtaining mechanistic understanding of congenital and acquired hearing loss. This understanding is essential for development of therapeutics to prevent and reverse diseases of the inner ear, including hearing loss. The objective of this study is to describe and compare the available tools for cell type–specific analysis of the ear, as a means to support decision making in study design.
Study Design:
Three major approaches for cell type–specific analysis of the ear including fluorescence-activated cell sorting (FACS), ribosomal and RNA pulldown techniques, and single cell RNA-seq (scRNA-seq) are compared and contrasted using both published and original data.
Results:
We demonstrate the strength and weaknesses of these approaches leading to the inevitable conclusion that to maximize the utility of these approaches, it is important to match the experimental approach with the tissue of origin, cell type of interest, and the biological question. Often, a combined approach (eg, cell sorting and scRNA-seq or expression analysis using 2 separate approaches) is required. Finally, new tools for visualization and analysis of complex expression data, such as the gEAR platform (umgear.org), collate cell type–specific gene expression from the ear field and provide unprecedented access to both clinicians and researchers.
Keywords: Inner ear, transcriptome, RNA-seq, RiboTag, scRNA-seq
INTRODUCTION
The past 3 decades have been marked with a transformation in the tools available to study the molecular and genetic basis of disease, including auditory and vestibular disorders. With the advent of the polymerase chain reaction (PCR) and sequencing, individual genes could be amplified from small tissues such as mouse inner ears, affording researchers the ability to study their presence, structure and changes in expression levels during development or in response to injury.1,2 Additionally, the completion of sequencing the mouse and human genomes has provided a blueprint of the genetic code underlying hereditary disorders, opening the door to what is now known as the post-genomic era.3–5 This has resulted in a transformation of our knowledge of the molecular basis of hereditary hearing loss (HHL). While POU3F4, the first gene known to underlie human HHL was cloned in 1995,6 at the time this manuscript is written mutations in a total of 117 genes have been found to underlie hereditary nonsyndromic hearing loss (https://hereditaryhearingloss.org). Many of these genes have available mouse models, allowing patients and practitioners alike to obtain insight into the mechanism of HHL in affected individuals and serving as translational tools for developing treatments. Furthermore, the identification of the genetic basis of HHL has enabled the development of clinical diagnostic platforms7,8 and now serves as a basis for the active development of targeted therapeutics, for example, by gene editing or gene replacement.9 Thus, the advances in molecular biology and specifically in our knowledge of the genetic basis of HHL, have transformed how we approach our patients from a diagnostic perspective and are likely to also change our therapeutic interventions.
Yet, an in-depth understanding of the pathophysiology of disease does not end with the identification of the mutated gene or an overall response of a tissue to an insult such as noise, ototoxic drugs (eg, cisplatin, gentamycin) or aging. The function of any given cell is reflected by the compendium of genes that it expresses at any given moment (ie, the transcriptome). These represent only part of the genes that are encoded by the genome, which is identical across cells. In addition, the expressed structures of each gene (also known as isoforms) may change between cells, and even within one cell a gene may have several functionally significant isoforms. Thus, the key for the next step in understanding inner ear development, unraveling the pathways that are required for hair cell (HC) regeneration, or identifying signaling cascades that could be modified to ameliorate age- and noise-induced hearing loss is at least in part at the transcriptomic level. Importantly, unlike the identification of genetic mutations which can be found using DNA extracted from peripheral blood, the transcriptome is cell-specific. Thus, targeted analyses of individual cells/cell types are required.
The cochlea is divided into regions such as the lateral wall, organ of Corti, and neuronal compartment, and is comprised of epithelial, mesenchymal, neuronal, and vascular endothelial cell types, which differ in their basic biological properties. Within each subcategory of cell type, there exists of a variety of cells that differ in function, gene expression, response to injury, and regenerative capacity, to name a few variables (Fig. 1) (e.g., within the epithelial compartment inner and outer HCs [IHCs and OHCs], and the subtypes of supporting cells). Variability in gene expression has been observed even within a cell type based on the tonotopic position along the cochlear duct, age, and time of the day (also known as circadian changes in gene expression).10 Historically, only techniques based on examination of tissue sections or whole mounted inner ear tissue could provide cell type–specific information regarding gene/protein expression. These methods, while still useful and often used for validation, are both low throughput (examining a few genes at a time) and require advanced knowledge of the genes probed. The classic approach of dissecting regions within the ear for measuring gene expression suffers from signal averaging of all cell types, and signal dilution of the information from any individual cell. Fortunately, the past decade is marked with the development and application of a wide array of experimental approaches for cell type–specific analysis and their adaptation to the inner ear. Coupling these approaches with methods for high throughput recording of gene expression (whole transcriptome, ie, measuring changes in expression of all genes in the genome), is revolutionizing our understanding of inner ear function. In parallel, these new methods have resulted in the acquisition of terrific amounts of data that are accessible to anyone with advanced informatic skills, an expertise infrequently mastered by biological science researchers or clinicians—the natural end-users for the data. Consequent to the rapid development of tools and approaches, there is also considerable confusion as to how to best implement them for experiments, choose appropriate controls, and match a given approach to a research question. Here the current state of cell type–specific analysis of the inner ear is reviewed, alongside examples with original data and descriptions of tools that enable meaningful access of these data to a broad array of clinicians and scientists in the field. The goal is to provide a roadmap for clinicians and researchers planning to perform expression analyses in the ear or those interested in obtaining an in-depth understanding of available data and current techniques.
Fig. 1.
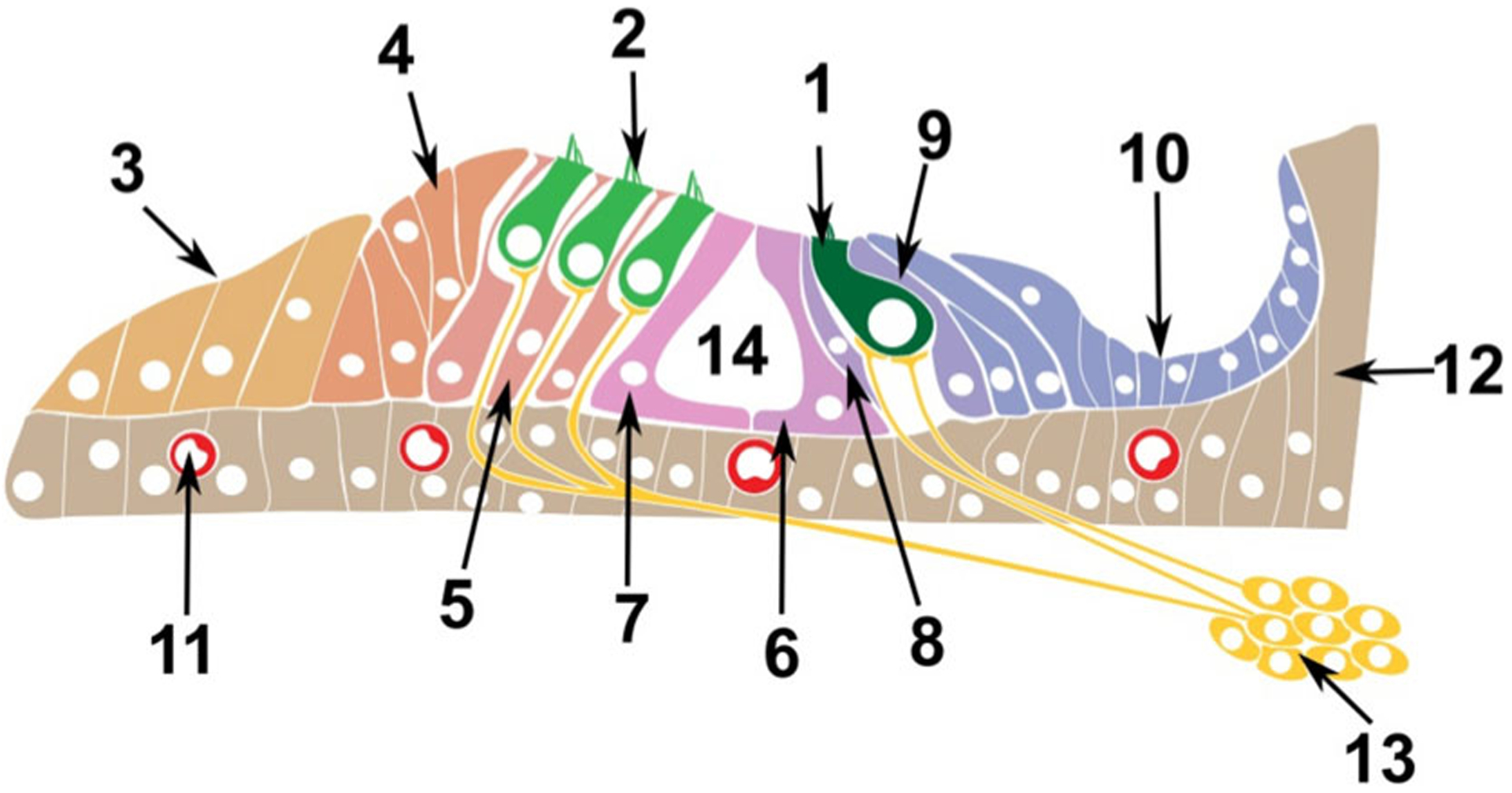
Schematic of the postnatal Organ of Corti. The Organ of Corti consists of multiple cell types. Epithelial cells comprising the sensory inner (1) and outer hair (2) cells, and the supporting cells of Claudius (3), Hensen (4), Deiters (5), as well as inner (6) and outer (7) pillar, inner phalangeal cells (8), border cells (9) and cells of the spiral limbus (10). Vascular Endothelial comprising the blood vessels (11), Mesenchymal cells (12) and Neuronal cells from the spiral ganglion (13). Many of these cell type can be further sub-divided to additional sub-populations. The tunnel of Corti is 14.
RESULTS
Cell Type–Specific Analysis Using Fluorescent-Activated Cell Sorting
Flow cytometry is a method used to count and pheno-type individual cells based on differences in size, granularity, or fluorescence—the latter achieved using endogenous markers, cellular uptake of fluorescent dyes or fluorescently conjugated antibodies to detect proteins expressed by the cells (ideally on the cell surface to avoid cellular permeabilization).11 Multiple fluorescent markers can be combined to enable complex phenotyping of cells (often 4–9 fluorophores are combined to define populations of interest using a binary serial “decision tree”). Importantly, as flow cytometry is broadly used in medicine (eg, for CD4 cell counts, immunophenotyping, HLA typing, or bone marrow biopsies), flow cytometers are readily available in nearly all clinical and medical research core facilities. Fluorescent-activated cell sorting (FACS) is the further use of flow cytometry to separate and collect individual cells based on their properties as identified by the cytometer. Thus, groups of cells that meet a set of criteria can be pooled into test tubes for further analyses (eg, RNA, DNA, or protein extraction), enabling their functional comparison to other cell types within the sample, or to the same cell type between samples of different conditions (eg, at different developmental time points or subject to treatment).
It is critical to understand tissue-specific considerations when applying FACS to study gene expression in the inner ear. For any cell type to be sorted by FACS, the tissue containing the cells of interest must first be dissociated into a single cell suspension. While trivial when analyzing whole blood, this is not the case for the inner ear. The inner ear consists of a complex epithelium with tight cell–cell junctions, and therefore a tissue dissociation step is necessary prior to further processing. Tissue dissociation is often performed by combining enzymatic and mechanical dissociation. Most of the enzymes used for tissue dissociation are effective at 37° C, and include Thermolysin (Sigma Aldrich #T7902, a thermostable extracellular metalloendopeptidase), Accutase (Sigma Aldrich #A6964, a cell detachment solution of proteolytic and collagenolytic enzymes), collagenase (Worthington Biochemical Corporation, endopeptidases that digest native collagen in the triple helix region), or papain (Worthington Biochemical Corporation #LK003150, a cysteine protease present in papaya).12,13 Additionally, DNase I can be added to reduce the viscosity of single cell suspensions secondary to free DNA from lysed cells. These enzymes are used in various combinations with mechanical trituration (using pipettes or syringes), followed by filtration of the dissociated tissue to obtain a single cell suspension (see methods).
Single cell suspensions are kept in solution, typically in a cold buffered media that helps maintain cell viability. After being loaded into the flow cytometer, a laser beam passes through each cell in order to measure forward scatter (the light scattered along the same plane as the laser, a proxy for cell diameter), side scatter (the light detected at 90 degrees from the laser, a proxy for cell granularity), as well as the emission of fluorescent markers.14 At the initial “gating” (marking of the cells to include in the analysis) it is important to exclude dead cells and debris. Typically, dead cells and debris have a much lower forward scatter and can be easily differentiated from the “primary population of cells” (also called population 1, or P1) (Fig. 2A). A second gating control consists of a doublet discrimination. This is particularly important when dissociating tissues with abundant tight junctions such as the inner ear. Despite the dissociation process, some cells may still be attached to other cells in doublets or triplets. A comparison of cell’s maximal signal (FSC-H) to signal width (FSC-W) allows users to gate on cells that are more most likely to be single cells for greatest accuracy (Fig. 2B).
Fig. 2.
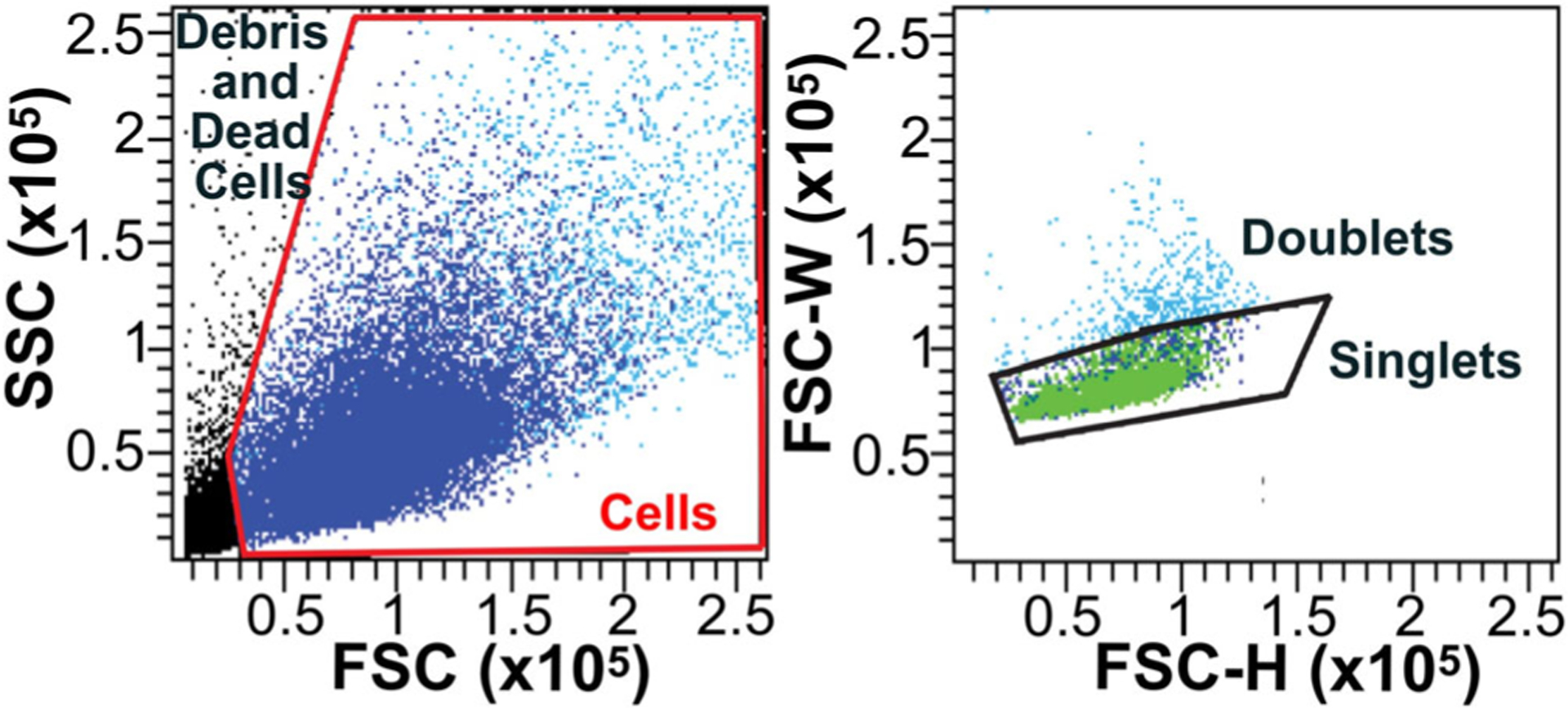
Gating methods to exclude debris, dead cells, and doublets. (A) Exclusion of debris/dead cells and gating of cells (red frame) to be included in subsequent steps of the analysis based on the forward scatter (FSC, related to size) and side scatter (SSC, related to granularity). (B) Gating of single cells (singlets) based on forward scatter width (FSC-W) and forward scatter height (FSC-H) and exclusion of cell clumps (doublets).
FACS inherently relies on the detection of a fluorescent emission to sort cell populations. Here, three different approaches to detect cellular populations based on fluorescence are discussed in the context of analyzing inner ear tissues. These approaches can be used independently, or in combination with each other. First, fluorescent labeling of cells can be performed by utilizing fluorescently conjugated antibodies that will recognize a cell surface marker, or by using antibodies for a cell surface marker followed by staining with a secondary antibody conjugated to a fluorescent molecule. Such antibodies will bind to and aggregate on the surface of cells that express the respective antigen, causing these cells, when excited by a laser, to fluoresce at variable wavelengths as dictated by the secondary antibody/conjugated fluorophore. Many of the cell surface markers that are used for cell sorting are cataloged as “CD” genes (clusters of differentiation), and over 60 of these genes are expressed in the newborn mouse inner ear.15 Commonly used markers to study the mouse inner ear include: CD326 (EpCAM), a marker for epithelial cells which include the sensory HCs, supporting cells and epithelial non-sensory cells (eg, epithelial cells of Reissner’s membrane and the stria vascularis); CD49f (integrin alpha 6) which in the newborn inner ear marks neurons, vascular endothelium and the sensory epithelial cells; and CD34, which marks vascular endothelial cells.12 The combination of these markers allows for the immunophenotyping of dissociated inner ear cells (both from the cochlea and the vestibular system) based on their combined expression (Fig. 3). Cells positive to CD326 are epithelial cells, cells negative to CD326 and CD34 but positive for CD49f are neurons, cells negative for CD326 and positive for CD34 and CD49f are vascular endothelial cells, and finally cells negative for CD326, CD49f, and CD34 (triple negative) can be further designated as mesenchymal cells12 (Fig. 3).
Fig. 3.
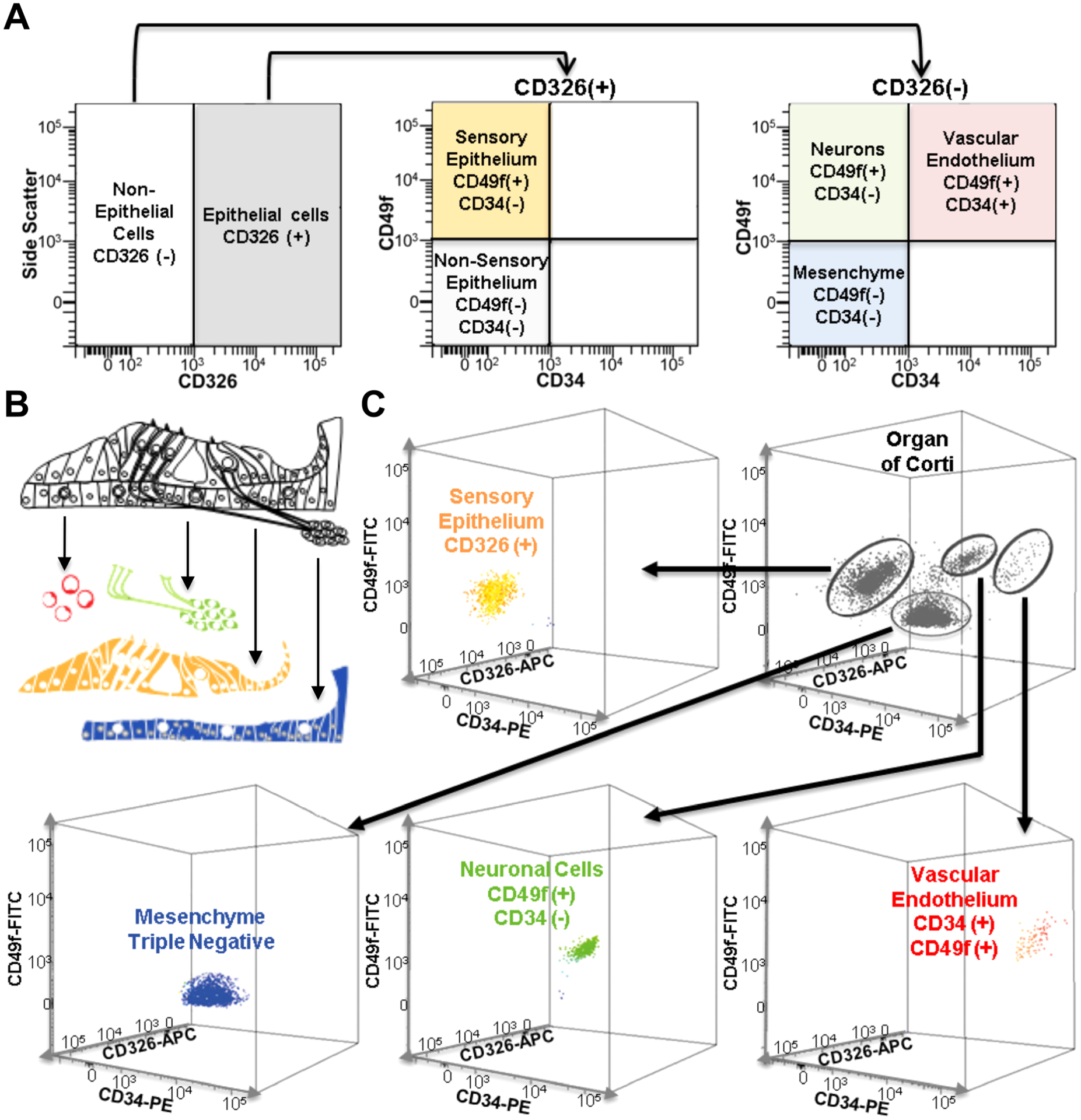
Cell type separation of newborn mouse cochleae using FACS. (A) Representation of expected flow cytometry immunophenotyping of cochlear major cell types when using antibodies for CD326, CD49f and CD34. (B) Schematic representing the organ of Corti and the four sorted cell populations. Red denotes vascular endothelium, green denote neurons, yellow denotes epithelial cells, and blue denotes mesenchyme. (C) 3-dimensional representation of flow cytometry analysis of a cochlear single cell suspension obtained from the organ of Corti using a combination of antibodies for CD326 (APC fluorescence), CD49f (FITC fluorescence), and CD34 (PE fluorescence) to separate the cells into epithelial, neuronal, vascular endothelial and mesenchymal cell populations.
Not all cell types have uniquely expressed cell surface proteins that also have validated antibodies to detect their extracellular domain. In these instances, one can either use 1) transgenic mouse lines that express a fluorescent protein in the cell type and time point of interest, or 2) mice induced to express a fluorescent protein in a cell type of interest via DNA recombination (Fig. 4A,B). The latter may provide more flexibility, relying on breeding mice that express the DNA modifying enzyme Cre-recombinase downstream of a cell type–specific promoter with a reporter mouse model to drive expression of a fluorescent marker after recombination. An example of this type of reporter mouse is the Ai14 mouse,16 which contains a construct to encode for the red fluorescent protein tdTomato within the Gt(ROSA)26Sor locus, along with a STOP cassette flanked by loxP sites. When crossed to a mouse model that expresses a cell type–specific Cre-recombinase, the Cre recombinase protein recognizes the loxP sites and cleaves them, removing the STOP codon, and allowing for tdTomato to be expressed (Fig. 4A).
Fig. 4.
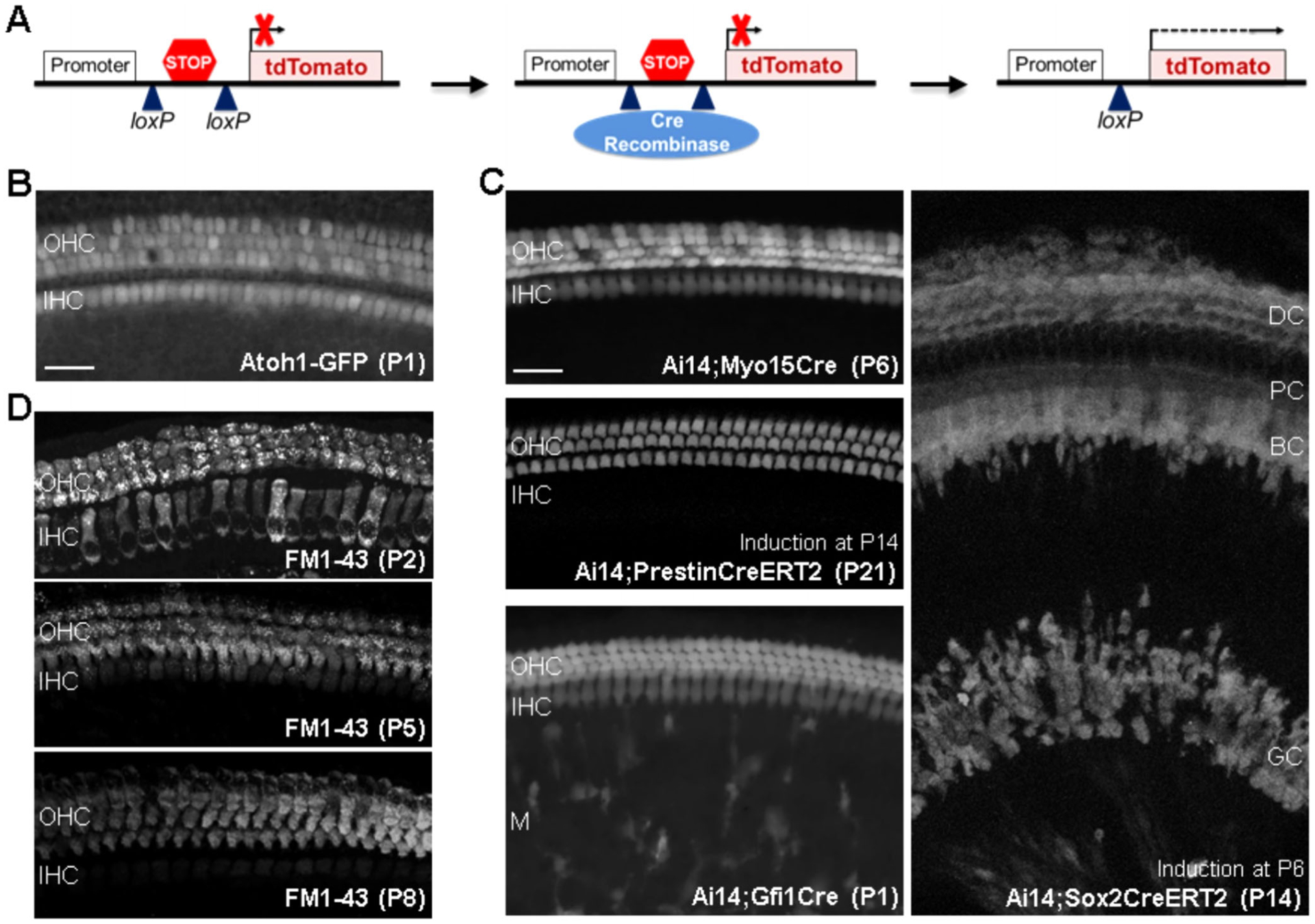
Examples of fluorescent markers used to sort cell-types in the cochlea. (A) Schematic representation of Cre-dependent expression of the reporter protein tdTomato in the Ai14 mouse model. (B) Expression of GFP in HCs in the transgenic Atoh1-GFP mouse expressing GFP under the control of the Atoh1 enhancer. (C) Specificity of different Cre mouse models assessed with a reporter mouse (Ai14). The Myo15 promoter is used to mark all HC starting at P4 (Myo15Cre). The Slc26a5 promoter (regulating the expression of the OHC protein prestin) has been utilized to drive inducible Cre recombinase expression specifically in OHCs (PrestinCreERT2, expression in all OHC starting at P7 with induction at P2). The promoter of Gfi1 (Gfi1Cre and Gfi1-P2A-GFP-CreERT2 (Gfi1-GCE)) is used to induce expression in all HCs starting at E16.5 but has been shown to induce recombination in Monocytes/Macropahges (M) as well. The Sox2 promoter has been used for recombination in embryonic prosensory cells, supporting cells (DC- Deiters Cells; PC- Pillar Cells; BC- Border Cells), and glial cells (GC) of the spiral ganglion, depending on the time point of induction (Sox2CreERT2). Scale bar is 20 μm. (D) Labeling of HCs with FM1–43. The dye in incorporated by OHC and IHC at P2 but mainly by OHC starting at P5.
Over the years, many cell type–specific Cre mouse models have been developed for inner ear research,17,18 providing researchers with flexibility to induce the expression of fluorescent markers in a variety of inner ear cell types (eg, all HCs, OHCs, supporting cells etc., Fig. 4C). Additionally, the fusion of Cre-recombinase with the estrogen receptor (CreER) in some of these models has allowed an additional level of temporal control of the DNA recombination, as recombination becomes dependent on the timing and dosage of a tamoxifen injection. The accuracy of the recombination and fluorescent marker expression also depends on the temporal and spatial specificity of the Cre-recombinase in the cell type of interest. Therefore, validation of the specificity by direct visualization in inner ear sections and/or whole mounted tissues with fluorescent microscopy is crucial (Fig. 4).19,20
Finally, a useful fluorescent marker in the ear is the vital dye FM1–43 (N-(3-Triethylammoniumpropyl)-4-(4-(Dibutylamino) Styryl) Pyridinium Dibromide), which can enter the inner ear HCs through the transduction channel. It can be used to mark HCs from freshly dissected mouse inner ear tissues from as early as postnatal day (P) 2 (Fig. 4D, see methods), and has been used in the past to sort HCs from chick epithelia.21 While wild type HCs with an intact mechanotransduction channel uptake the dye, the uptake is not specific only to HCs and may enter some other cell types. Future research may increase its utility for sorting HCs, possibly by combining with other cell surface markers.
Importantly, transgenic mice can also be utilized in combination with fluorescent markers to sort specific cell populations. Several previously reported combinations include using Ai14;Gfi1Cre mice with CD326 and CD45 to sort HCs, epithelial non-HCs and inner ear resident immune cells; Ai14;Myo15Cre mice and CD326 to specifically sort HCs; and Math1-GFP and CD326 to sort HCs, epithelial non-HCs and non-epithelial cells (here a combination of neurons, mesenchyme and vascular endothelium) (Fig. 5A).20,22,23 Finally, an important consideration in all flow cytometry-based experiments is the specificity of the sorting. While the user marks a population of interest on the screen for collection, it is imperative to include a control to ensure that the sought-after population is indeed the sorted population (Fig. 5B). Thus, a post sort analysis must be conducted, validating that the collected cells truly express the utilized markers and were sorted correctly by the flow cytometer. For this analysis, a small sample of each of the sorted groups is re-run through the flow cytometer to validate that the previously sorted cells fall into the correct gate (ie, double positive cells, single positive cells, or negative) (Fig. 5C).
Fig. 5.
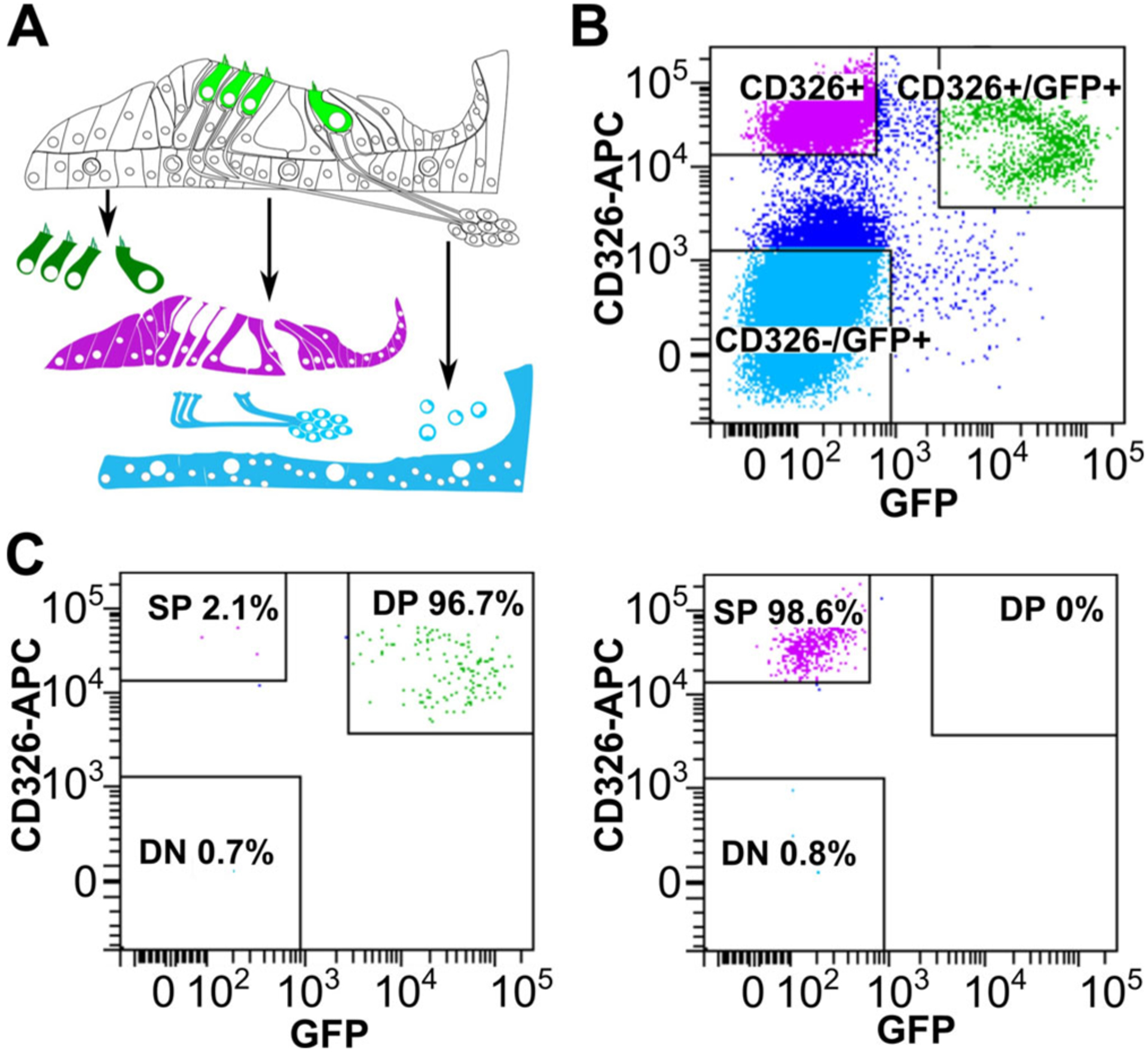
Cell sorting from a Math1-GFP mouse. (A) Schematic representing the organ of Corti and the three sorted cell populations. (B) Flow cytometry from cochlear tissue with HCs positive for CD326 and GFP (Double positive DP), supporting cells positive only for CD326 (Single Positive SP) and non-epithelial cells negative for both markers (Double negative DN). (C) Post-sort analyses of the cells sorted in (b) showing high purity for HCs (96.7%) and supporting cells (98.6%).
One caveat to FACS, as well as other methods that rely on tissue dissociation is that the process, in addition to taking the cells out of their normal tissue context and cell–cell signaling environment, leads to cell stress, causing changes in gene expression characterized by the activation and translation of heat shock and immediate early genes.24,25 These in turn can result in 1) a “false impression” of gene expression (ie, the transcriptome recorded from the dissociated cells is not representative of the native tissue), and 2) potential masking of relevant changes in gene expression (for example, when trying to assess gene expression in HCs following a noise exposure). A newly introduced method to decrease these potentially confounding changes in gene expression is the use of Cold Active Proteases (CAP), such as that obtained from Bacillus licheniformis, for tissue dissociation.26 Unlike other dissociation enzymes, CAP retain their function at colder temperatures, allowing the dissociation steps to be performed on ice (~4°C). Conducting dissociation steps at colder temperatures is beneficial, as the mammalian transcriptional machinery is much less active at these temperatures and stress induced gene changes will be less likely transcribed.26 CAP work just as effectively in dissociating cochlear cells as traditional 37°C enzymatic methods, such as Thermolysin and Accutase (Fig. 6A,B), and do not change expression of cell type–specific genes such as Atoh1 and Myo6 in HCs, Tubb3 in neurons, and Pou3f4 in mesenchymal cells (data not shown). Additionally, compared to non-dissociated control tissue, expression of apoptotic markers such as Casp3 and Bcl2 is not significantly increased in tissue dissociated using CAP or Thermolysin/Accutase (data not shown). However, unlike Thermolysin/Accutase dissociated cells, in which the expression of the immediate early genes Fosb and Junb is significantly increased due to the cellular stress of dissociation, CAP dissociated cells have much reduced levels of Fosb and Junb (Fig. 6C). These results suggest that CAP may be a preferable dissociation enzyme for inner ear tissues to avoid confounding changes in gene expression that result from the dissociation process. However, a recently identified pitfall for CAP is that it may completely or partially cleave cell surface markers, including the epithelial marker CD326 (Fig. 6D,E). Thus, for protocols that use CD326 as part of their workflow, CAP may not be a suitable approach.
Fig. 6.
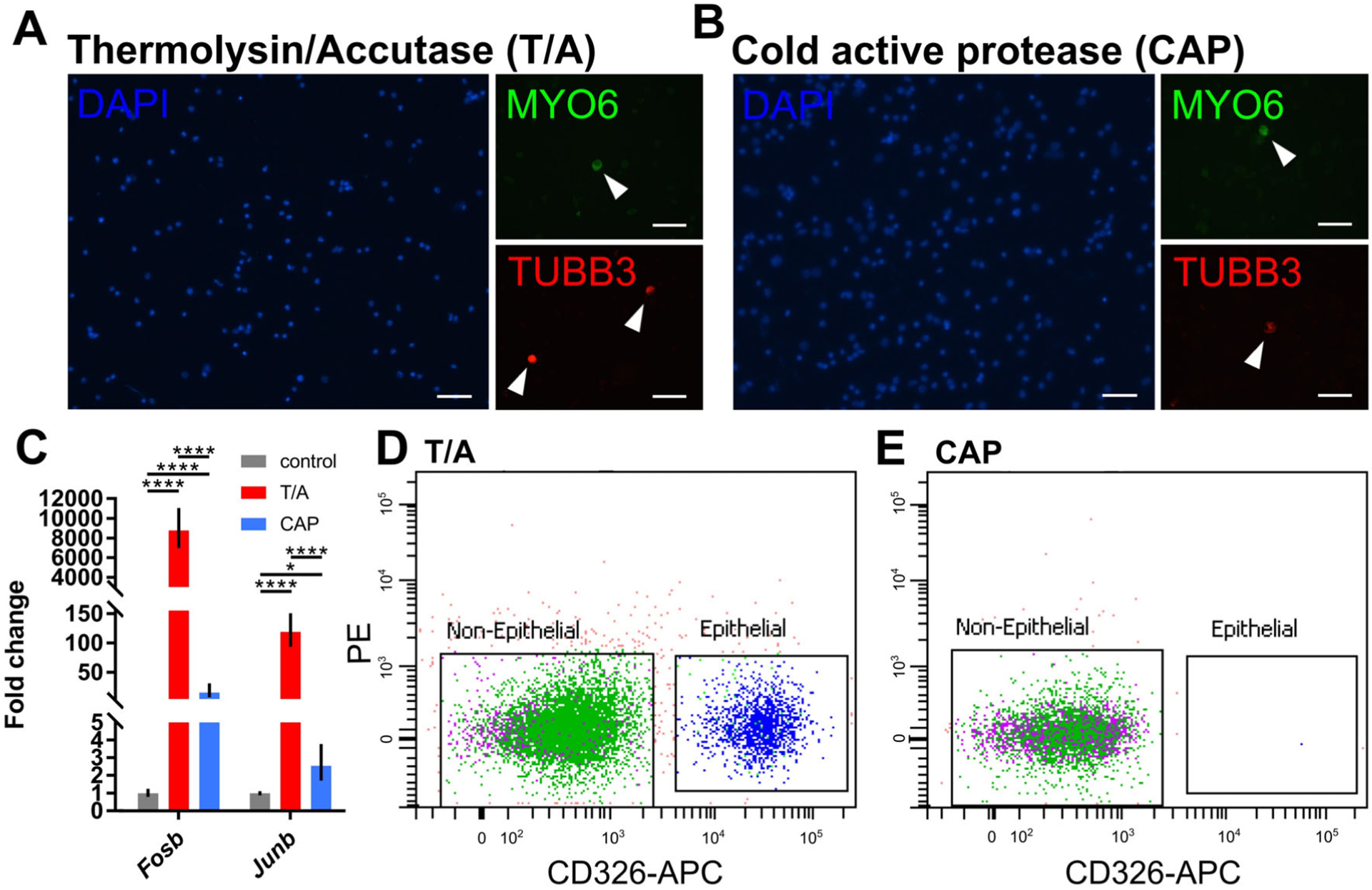
Validation of CAP in cochlear dissociations and FACS. (A-B) Immunofluorescence staining of cochlear cells dissociated with Thermolysin/Accutase (T/A) and cold active protease (CAP). Nuclei stained blue (DAPI), HCs stained green (MYO6) and neurons stained red (TUBB3). Scale bar is 50 μm. (C) Fold change difference in immediate early gene expression between non-, T/A- and CAP- dissociated cochlear cells as measured by RT-qPCR. Statistical significance was assessed by two-way analysis of variance with Tukey post-hoc test. (D-E) Flow cytometry of dissociated cochlear cells stained with CD326 to isolate cochlear epithelial cells. Dissociation with CAP results in a loss of immunoreactivity to CD326.
RNA Enrichment and Ribosomal Pulldown Techniques
While dissociation-based techniques can enable users to obtain near pure cell populations, not all tissues or cell types are amenable to dissociation. For example, the efficiency of dissociating tissues from the adult inner ear is significantly reduced due to increasing rigidity of the cytoskeleton and the tightening of cell–cell junctions as the tissue matures. Additionally, mature inner ear cells are more vulnerable to trauma, resulting in significantly lower yields of viable cells from FACS compared to their embryonic counterparts. Finally, the dissociation process itself induces changes in gene expression which may mask signaling pathways of relevance (Fig. 6C). To overcome these challenges, several animal models have been developed over the last decade to capture RNA from specific cell types without disrupting their native tissue context. These include models such as the BACarray, RiboTag and NuTRAP mice, which utilize ribosomal tagging to immunoprecipitate cell type–specific RNA, as well as a mouse model for immunoprecipitation of cell type–specific expressed thiouracil (TU) tagged RNA (Fig. 7A,B).27–30 In cells, mRNA that is actively translated to proteins is bound by ribosomes. For this reason, the BACarray, RiboTag, and NuTRAP models are considered methods for profiling the “translatome” (ie, the fraction of the mRNA in the cell that is actively being turned into protein at the time the experiment is done). All of the above mentioned models can be utilized for profiling cell type–specific gene expression beginning with “flash frozen” tissue and do not require cell dissociation, thereby providing an accurate representation of gene expression without dissociation artifacts. However, they too carry a separate set of challenges as described below.
Fig. 7.
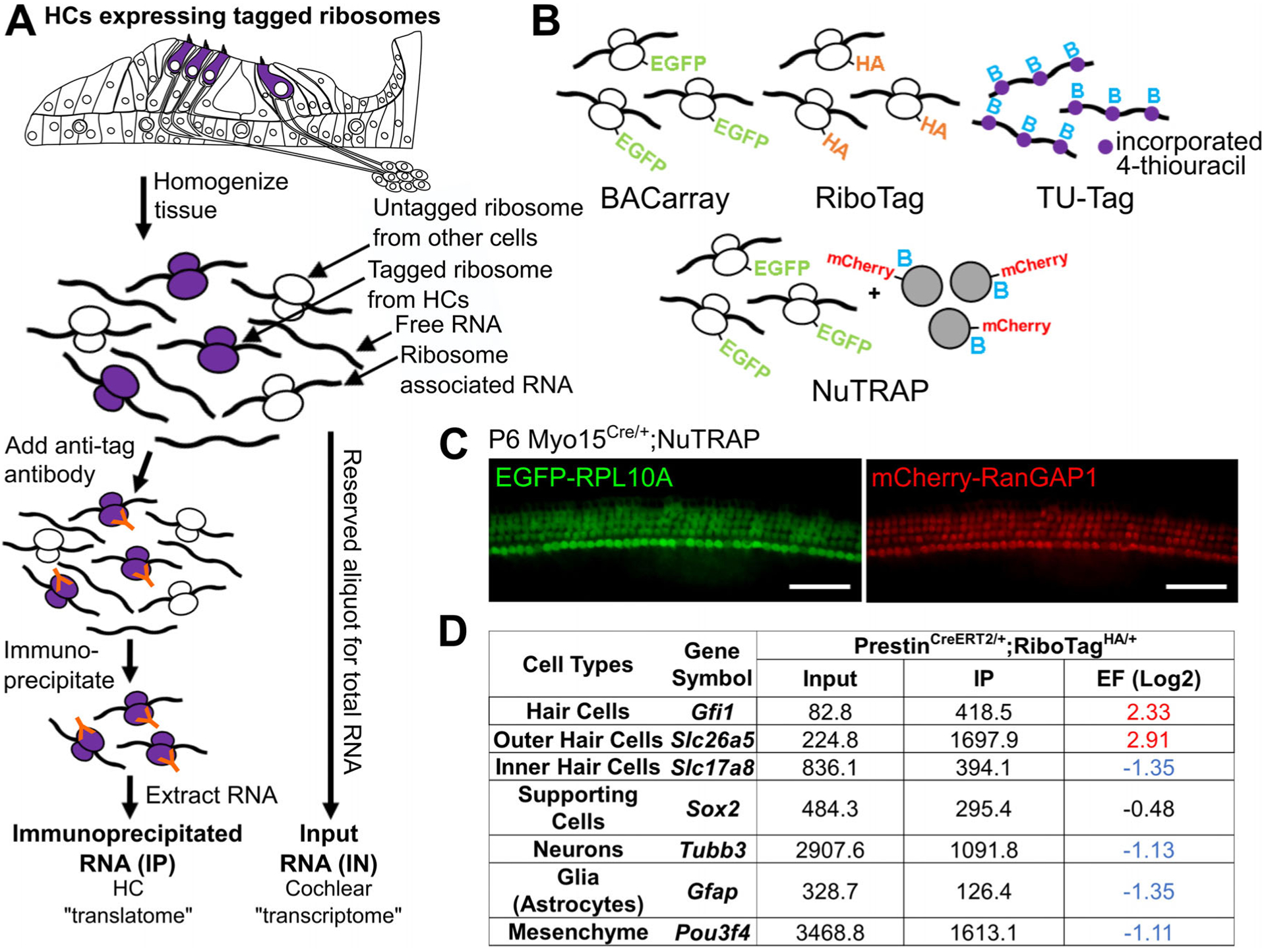
Models for RNA enrichment and ribosomal profiling. (A) Tagged ribosomes are expressed in a cell type–specific manner, either driven by BAC (BACarray) or Cre recombinase (RiboTag, NuTRAP). In this example, tagged ribosomes are expressed in cochlear HCs and immunoprecipitated using tag-specific antibodies from whole tissue lysates. RNA extracted from the immunoprecipitated ribosomes (IP) is enriched for the HC translatome, and can be compared to input RNA to identify HC-expressed genes. (B) Schematic representation of ribosome, RNA and nuclei tagging models used to perform cell type–specific analyses of whole complex tissues. EGFP = enhanced green fluorescent protein, HA = hemagglutinin, B = biotin. Purple circles represent 4-thiouracil (TU) incorporated into RNA, gray circles represent NuTRAP nuclei. (C) Cochlear whole mount preparation of a P6 NuTRAP mouse crossed to Myo15Cre showing HC-specific expression of GFP-RPL10A and nuclear mCherry-RanGAP1. Scale bar is 50 μm. (D) Sequencing results of a RiboTag immunoprecipitation experiment performed on 10 week old PrestinCreERT2/+;RiboTagHA/+ mice (OHC specific). Known cell-type specific transcripts are enriched (red) or depleted (blue) as expected according to the Cre driven RPL22-HA expressing cell-type. IP = immunoprecipitated, EF = enrichment factor (Log2 IP/input, corrected for transcript length).
The first ribosome tagging model introduced to the research community was the BACarray mouse model. This model results in overexpression of an RPL10A protein (a 60S ribosomal subunit) that is also tagged with an N-terminal enhanced green fluorescent protein (EGFP-RPL10A). Expression of the EGFP-tagged ribosomes is controlled via bacterial artificial chromosome (BAC), and expression is constitutive based on the utilized promoter (different BACs will drive expression in different cell types, but each model is generated separately). The RiboTag mouse also utilizes tagging of the 60S ribosomal subunit but offers greater spatiotemporal control of its expression. Specifically, the RiboTag mouse expresses a version of the 60S ribosomal subunit RPL22 that is tagged with a C-terminal hemagglutinin tag (RPL22-HA) in a Cre recombinase dependent manner. Therefore, the RiboTag mouse model can be crossed with any Cre recombinase model, either inducible or non-inducible, to control where and when RPL22-HA is expressed. For both models, cell type–specific expressed tagged ribosomes can be used to capture actively translated RNA from whole tissue lysates via immunoprecipitation utilizing an antibody for either the EGFP or the HA tag.
As an example, Figure 7A depicts a case in which these tagged ribosomes are specifically expressed within cochlear HCs. Upon dissection at any embryonic, postnatal, or adult time point, tissues can be flash frozen for storage, allowing for the pooling of tissues from different experiments or genotypes if necessary. Cochlear tissues from as little as two mice can be combined, homogenized to release all ribosomes into solution, and HC-expressed tagged ribosomes (and their associated RNAs) are then immunoprecipitated using an antibody for the ribosome tag. RNA extracted from the immunoprecipitated ribosomes (called the IP) represents actively translated RNA from HCs, or the HC “translatome.” As an important control, an aliquot of the whole tissue homogenate should also be saved for total RNA isolation (input control, see methods). This sample can be used to test the efficiency of the immunoprecipitation, as well as to assess cell type specificity of gene expression by calculation of an IP versus input enrichment factor (discussed further below).
In addition to the BACarray and RiboTag models, the recently introduced NuTRAP mouse model allows researchers to profile both gene expression of a cell type of interest using ribosomal immunoprecipitation, as well as perform cell type–specific epigenetic profiling by nuclear sorting or nuclear immunoprecipitation. At the Rosa26 locus, the NuTRAP mice harbor a construct to express 1) EGFP-RPL10A, 2) the biotinylation protein BirA, and 3) a biotin ligase recognition peptide (BLRP) and mCherry-fused RanGAP1 nuclear protein (Fig. 7C). Therefore, in the presence of Cre recombinase, EGFP-RPL10A is expressed for ribosomal pulldown, and the BLRP-mCherry-RanGAP1 is biotinylated by BirA and incorporated into the nuclear membrane for nuclear pulldown (using the biotin) or FACS (using the mCherry). For example, crossing the NuTRAP mice to the Myo15Cre model results in expression of both EGFP-RPL10A and mCherry-RanGAP1 in the cochlear HCs (Fig. 7C).
Actively transcribed mRNA represents only a fraction of the total RNA of a cell, as non-coding RNA, which plays critical roles in cell biology, is usually not bound by ribosomes. An additional model that has the advantage of not only profiling actively translated RNA, as with the ribosome immunoprecipitation models, but all newly synthesized RNA from the cell type of interest (ie, the transcriptome), is the thiouracil (TU) tagging mouse (Fig. 7B).29 For this model, a transgene encoding uracil phosphoribosyltransferase (UPRT) under control of the ubiquitous chicken β-actin/CMV (CAG) promoter is expressed in a Cre recombinase dependent manner. Therefore, upon injection of the animal with the uracil analog 4-thiouracil (4TU), UPRT expressing cells incorporate 4TU into all newly synthesized RNA in the cell type of interest. Total RNA from the whole tissue is then extracted, and cell type–specific 4TU-RNA is labeled with biotin before being purified. TU-tagging has been used successfully to study the HC transcriptome of the UPRT transgenic zebrafish (Tg[myo6b:UPRT]), identifying genes such as otofb, strc, and chrna9 as expressed in zebrafish HCs.31 However, compared with other methods, the overall efficiency of TU-tagging appears to be decreased, with a significantly smaller number of transcripts identified as enriched in the UPRT HCs than expected. One possible explanation for this decreased sensitivity is incorporation of 4TU into the RNA of “off target” cells not expressing UPRT, as was observed when wildtype zebrafish were exposed to 4TU.31 The sensitivity of this model may be improved by altering the time of exposure or concentration of 4TU, thereby decreasing the chances of 4TU incorporation into the RNA of non-hair cells.
Ribosomal immunoprecipitation models have been successfully utilized to study gene expression within the sensory HCs of mice and zebrafish.22,24 However, these and other studies highlight important considerations that should be taken into account when designing experiments and analyzing RiboTag data. Firstly, the interpretation of RiboTag data will rely on the specificity of the Cre recombinase model used for inducing HA-tagged ribosome expression, and it is recommended that the specificity of recombination be tested by crossing to a sensitive reporter model before use (for example: Ai14, utilized in Fig. 4C). Secondly, RiboTag immunoprecipitation experiments inherently rely on the enrichment, rather than the purification, of cell type–specific transcripts from a whole tissue. Similarly, RNA pulldown from TU tagging models also results in cell type–specific enrichment of transcripts. The immunoprecipitated RNA will also contain, to varying extents, RNA from other cell types regardless of the specificity of the Cre recombinase model or driving promoter utilized. To highlight this, in a RiboTag immunoprecipitation experiment performed on inner ear tissues from wild type 10-week-old PrestinCreERT2;RiboTag mice, the IP sample still contains a significant amount of non-OHC expressed transcripts, such as the neuronal marker gene Tubb3 and the mesenchymal marker gene Pou3f4, despite RPL22-HA being expressed only in the OHCs (Fig. 7D). However, when compared to the input sample (RNA extracted from the whole tissue lysate), these transcripts are considered depleted in the IP sample (ie, lower values in the IP compared to input). This introduces the concept of an enrichment factor (EF), or a log2 fold change between the IP RNA and input RNA, that can be used to assess the cell-type specificity of a given transcript. A general cutoff for transcript enrichment in the cell type of interest is EF ≥ 1 (ie, 2-fold higher transcript abundance in the IP compared to input), while a general cutoff for depletion is EF ≤ −1 (ie, 2-fold higher transcript abundance in the input compared to IP) (Fig. 7C). Transcripts that are enriched are more likely specifically expressed within the cell type of interest, whereas transcripts that are depleted are more likely to be expressed in other cell types contained within the tissue. In this same RiboTag immunoprecipitation experiment, transcripts for the HC and OHC expressed genes Gfi1 and Slc26a5 are enriched (EF ≥ 1) in the OHC IP compared to input, while transcripts for the IHC and non-HC expressed genes Slc17a8, Sox2, and Gfap are either not enriched or depleted (EF ≤ −1) (Fig. 7D).22 Of note, relying on an enrichment of transcripts between IP RNA and input allows only for the identification of significantly enriched and depleted transcripts; overlooking genes that may still be expressed, albeit at lower levels, in the cell type of interest, or also within other surrounding cell types. Another use for the RiboTag model is analysis of gene expression in a cell type of interest between different conditions (eg, drug exposure, developmental progression, mutation) through direct comparison of the IP RNA only. Analysis of the input RNA and calculation of an EF can then be used to further validate whether the changes in gene expression originate from the cell type of interest.
IP RNA from a tissue of interest can be utilized for a variety of techniques that measure gene expression, including RT-qPCR, NanoString analysis and RNA-seq. However, when proceeding to library preparation for sequencing, it is also important to consider how different library preparation methods will affect the results. A study by Song et al. demonstrated that different library preparation methods could affect the results of sequencing RiboTag immunoprecipitated RNA, and suggested that the TaKaRa SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing and the Illumina TruSeq RNA Library Prep Kit v2 provided better results, including more uniform transcript coverage and lesser representation of immature mRNAs.32 Indeed, successful and reproducible results could be obtained from as little as 250 pg of starting RNA using the TaKaRa SMART-Seq v4 kit, making it ideal for RiboTag analyses on rare cell populations or small amounts of tissue, such as that in the inner ear. Additionally, they noted a general increased efficiency for immunoprecipitation of longer transcripts, possibly as a result of a higher likelihood of polysome formation and therefore increased HA antibody binding on longer transcripts, which can be corrected for when calculating the EFs (as was done in Fig. 7D). This too was dependent on the kit used to generate the libraries, highlighting the need to validate and check every step of the protocols used for expression analysis.
Single Cell RNA Sequencing
When considering the cellular diversity of the inner ear, as well as the challenges and limitations presented with the methods described above, an ideal solution would be to perform RNA sequencing (RNA-seq) on all the cells of the inner ear, in situ. This would allow for comparison between cell types within the same sample, or of the entire tissue between developmental time points, health and disease, treatments or insults—all while knowing the physical source of the transcript. However, a single cell contains between 1 and 10 picograms of total RNA, and only less than a decade ago RNA-seq required at least 100 nanograms of total RNA per sample for successful analysis. A second challenge is the scale of the data—each cell expresses on average 12,000–15,000 transcripts. Visualizing/analyzing a gene expression matrix of 10,000 columns (representing cells) and 15,000 rows (representing genes) is not possible using simple spreadsheets. Finally, mapping back the data from the many individual cells to the tissue of origin or developing methods to perform these analyses on intact tissue sections presents yet another challenge. While merely a dream even a decade ago, and not completely possible yet, technological advances are progressing rapidly, with single cell RNA-sequencing (scRNA-seq) and Multiplexed Error-Robust Fluorescence In Situ Hybridization (MERFISH) at the leading edge.33,34
In recent years, scRNA-seq has been employed to study the transcriptome of thousands of individual cells simultaneously on a genomic scale.35 Unlike bulk RNA-seq, which represents an average of gene expression across thousands of cells, scRNA-seq indeed allows for the assessment of gene expression of cell populations at unprecedented resolution. Due to the cellular heterogeneity of inner ear structures, scRNA-seq is an ideal technique to study these different cell types, including rare populations such as sensory HCs (these represent only 3% of the cells in an intact inner ear sensory epithelium).36 Furthermore, scRNA-seq has a wide range of applications in the field of otolaryngology, from characterization of individual cells for developmental processes such as in studies of inner ear biology, to characterizing infiltrating cancer cells or the cellular heterogeneity of cancerous tumors. Additionally, in studies of neurodevelopment, it can be employed when cell surface markers or Cre mouse models are not available to isolate a specific cell type of interest by FACS for bulk RNA-seq. Additionally, scRNA-seq is particularly useful when studying complex mouse models where outcrossing the mice to yet another line to be able to isolate a specific cellular population may present a significant technical challenge.
scRNA-seq commercial platforms can be grossly divided into two types: 1) platforms that sequence a smaller number of cells with a greater depth of transcript coverage per cell (eg, the microfluidics Fluidigm platform), and 2) platforms that can sequence many more cells at a lower resolution and do not offer information about the full transcript length (eg, the droplet-based technique of 10x Genomics). Each technology has its own advantages and disadvantages when it comes to data usability, sequencing sensitivity, gene diversity and cost. Which platform is appropriate to use is based on the biological question. Fluidigm is a single cell capture microfluidic technology where each cell is captured into individual wells of a microchip to undergo automated single-cell lysis, RNA extraction, and cDNA synthesis for up to 800 cells at one time. The library can then be prepared and sent for sequencing, resulting in highly detailed transcriptomics of each cell and allowing for identification of lowly expressed genes. This approach is highly useful when sequencing an enriched population of cells or a highly prevalent cell type in an intact tissue, but can be less useful for analyzing a rarer cell type from a non-enriched population. For example, in a sample of 384 cells from a dissociated cochlea, one would expect less than 10 of the cells to be HCs, whereas over 100 would be otic mesenchymal cells. Given the high variability in gene expression when using single cell data (these data are normalized at a log10 scale), it is preferable to have a larger population of cells sequenced for reproducible results. These larger populations can be obtained with the 10× Genomics platform, as it allows for targeting of up to 10,000 cells, albeit with lower sequencing detail, by utilizing a micro-droplet technique. Here individual cells within a dissociated tissue are captured into microdroplets that contain the necessary reagents to proceed with library construction. The 10x Genomics platform has a lower sensitivity but a much higher throughput, giving the user the ability to gain a global view of the tissue of interest and in some cases obviate the need for cell type–specific enrichment. Ultimately, very similar results can be obtained using these two approaches. Recently two groups reported the cellular diversity of type I spiral ganglion neurons in the cochlea using scRNA-seq. Shrestha et al. used a low throughput high sequencing depth approach, whereas Sun et al., used the 10X Genomics platform.37,38 Both groups were able to identify the same three major subtypes of type I spiral ganglion neurons and their molecular composition; a result with important clinical significance as these neuronal subtypes differ in their spontaneous activity, threshold, and sensitivity to noise-induced trauma.37,38
Although the scRNA-seq platforms differ in how the single cells are captured before sequencing, they follow a conceptually similar methodological pipeline from tissue procurement to data analysis. First, tissue dissection provides an opportunity for crude selection of the cells of interest while limiting the number of cells that have little to no biological importance to the hypothesis. This can be seen in Figure 8, representing single cell datasets from P2 and P7 mice. Dissections were performed so that few spiral ganglion neurons were contained in the samples, increasing the chance to capture the most relevant cells for this study, which focused on HCs and supporting cells. Another key to a successful and robust dataset is an effective dissociation of the tissue to obtain a viable and healthy single cell suspension. This step is similar in approach and challenges to those described in the FACS section above. Tissue dissociation is done by using specific proteases that cleave the protein bonds between cells while keeping the cell integrity. Currently, many inner ear researchers have performed successful single-cell dissociations with different proteases, including those mentioned previously (Thermolysin, Papain, Accutase, and more recently, CAP).13 Alternatively, single nuclei preparations can be employed to avoid transcriptional artifacts from dissociation and can be used for transcriptomic profiling of difficult to dissociate time-points, such as adult inner ear tissues.39 This approach may be particularly useful for analysis of inner ear tissues from human, such as cochlear or vestibular sensory epithelia obtained during skull base surgery or procured from cadavers or organ donors. In these tissues, it may be nearly impossible to obtain healthy cellular populations at the single cell level. However, while to date sequencing results from nuclei are not similar in quality to whole cells, they are sufficient for cataloging cell types and obtaining an understanding of the cellular diversity within a tissue.39
Fig. 8.
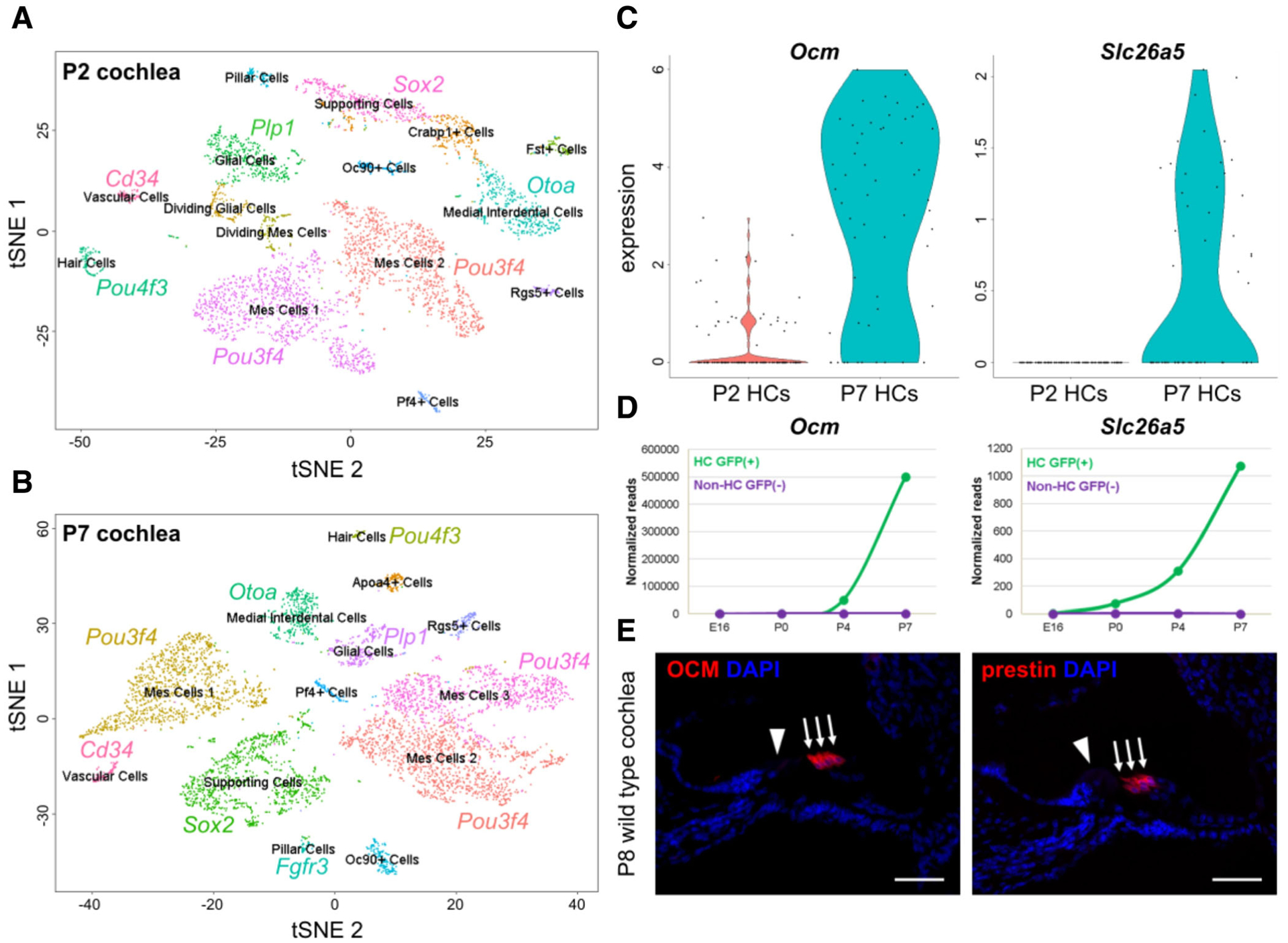
Initial analysis and gene identification in datasets of the P2 and P7 cochlea. Unbiased clustering of the P2 (A) and P7 (B) scRNA-seq datasets. Cluster identity is identified by known cell-type specific genes. (C) Expression of OCM and Slc26a5 in P2 and P7 HCs. (D) Validation of gene expression using the SHIELD database (adapted from the gEAR [umgear.org]). (E) Validation of protein expression using Immunostaining with antibodies to label OCM or prestin (red). Slides were counterstained with DAPI (blue). Scale bar is 50 μm.
Next, single cells are isolated (Fluidigm—each cell is in an individual well of a microchip; 10x Genomics—each cell is captured in a micro-droplet) and lysed, freeing the mRNA molecules from the cell. The mRNA is then captured and converted to cDNA via PCR using a reverse transcriptase and either poly[T] primers to target polyadenylated mRNAs or random hexamer primers that randomly prime regions of the mRNAs. Finally, the sequencing libraries are constructed by adding unique barcodes (short nucleotide sequences) to the mRNA from each individual cell, allowing for a reconstruction of which mRNAs came from each cell later in the analysis. Also, during this step, Illumina sequencing adapters are added to each end of the cDNA, mediating adhesion to the flow cell. Unique molecular identifiers (UMIs) are also added to address the technical issue of PCR duplicates, as scRNA-seq library preparation relies on amplification of small amounts of starting material. UMIs are random sequences that are added before PCR amplification that give each mRNA molecule a distinct identity, providing the ability during the analysis steps to correct for PCR amplification bias. Finally, the finished scRNA-seq library (cDNA that is amplified and tagged) from every cell is pooled and sequenced using an Illumina next generation sequencing platform.
After the scRNA-seq library has been sequenced, one can analyze the gene expression information of each single cell. First, the raw sequencing data are pre-processed into a count matrix, which includes aligning the reads to a reference genome and then assigning these reads to features (genes). This matrix represents read counts per genes per cell (each cell has a different barcode and all reads per cell per gene are summarized). Reads with identical UMIs are counted only once. Finally, the matrix is analyzed to identify cell types (based on similarities and differences in gene expression between individual cells) as well as additional downstream analyses (eg, trajectory analyses to identify patterns associated with changes in gene expression during development). Traditionally, these steps are handled by experienced bioinformaticians utilizing command line tools and programming languages to decipher the sequencing data. However, many new tools have become readily available that allow scientists, with minimal to no experience in bioinformatics, to tackle the initial analysis of scRNA-seq data. These programs include the 10x Genomics Loupe Cell Browser, Illumina’s cloud computing software (Basespace), Fluidigm’s SINGuLAR program and gEAR’s scRNA-seq workbench (Table I). However, a fundamental understanding of the analysis process is important for utilizing such tools. First, an important quality control step is necessary to remove unwanted captures (wells/droplets without cells and well/droplets with more than one cell) and filter by mitochondria content (high mitochondria content indicates cells undergoing apoptosis). After quality control has been completed, highly variable genes (HVGs) can then be identified. These genes contribute the most to cell-to-cell variation within the dataset, and are used to perform the Principal Component Analysis (PCA). PCA simplifies the complexity of the dataset while retaining trends and patterns, allowing for the visualization of the data structure.40 In essence, it identifies groups of genes whose shared expression pattern has the greatest effect on defining a subset of cells in the dataset. PC1 will have the strongest effect on the data, possibly dividing the dataset into two large groups of cells, and the following PCs with serial decrements. In the ear, for example, PC1 often separates a whole cochlear preparation to epithelial and non-epithelial cells, and PC2 the non epithelial cells to neuronal and non-neuronal. The t-Distributed Stochastic Neighbor Embedding (tSNE) technique is next implemented for dimensionality reduction and clustering of single cells based on gene expression. Of note, uniform manifold approximation and project (UMAP) has recently been used in place of tSNE for dimensionality reduction.41 These steps lay the groundwork to perform other basic analyses, such as finding marker genes for each cell cluster, comparing gene expression between cell clusters (ie, IHCs vs. OHCs) and computing differential gene expression (ie, control vs. mutant). However, when comparing two separate datasets, additional analyses need to be implemented to adjust for batch effects (differences in gene expression across samples processed on different days), technical variations due to dissociation protocols, library preparation techniques, and sequencing platforms. Overall, these initial analyses are sufficient to answer many biological questions at hand, but there are more advanced techniques which can expand the knowledge to be gained from a scRNA-seq dataset. This includes pseudo–time analysis to unravel cell fate decisions, as well as integrating epigenetic data to understand gene regulatory networks.42–44
TABLE I.
Non-Command Line–based Tools for Analyzing scRNA-seq Data.
| Analysis tool | Description | URL |
|---|---|---|
| Loupe Cell Browser (10x Genomics) | Allows for cell cluster analysis and finding significant genes of interest. To be used with the 10x genomics platform. | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| Basespace (Illumina) | Cloud computing to manage, analyze and share data; including scRNA-seq (cluster analysis and gene expression visualization). | https://www.fluidigm.com/softwarehttps://www.basespace.illumina.com/ |
| SINGuLAR (Fluidigm) | Identification of gene expression and cluster analysis. To be used with the Fluidigm platform. | https://www.fluidigm.com/software |
| scRNA-seq workbench (gEAR portal, RRID:SCR_017467) | Initial analysis of scRNA-seq datasets, including quality control, PCA, tSNE/UMAP, cluster analysis, comparing gene expression. | https://www.umgear.org |
After completion of the analyses, cell type–specific genes of interest that have been identified should be validated. This can be completed in a variety of ways with an array of commonly used molecular biology techniques. At the mRNA level, in situ hybridization of inner ear sections allow for visualization of mRNA expression, serving as a qualitative analysis of the location of the gene of interest (Fig. 9A). Additionally, RNAscope technology (a novel in situ hybridization assay) is a useful alternative to a standard in situ hybridization assay, as it can be quantitative, can label several genes on the same slide through the use of unique chromogenic or fluorescent probes for each gene, and limits background noise due to the unique probe design45 (Fig. 9B). For a quantitative analysis of expression, real-time quantitative reverse transcription PCR (RT-qPCR,) can be performed on either RNA from whole tissue, although this provides no information on location of expression or cell type, or FACS sorted tissue for the cell type of interest (Fig. 6C). At the protein level, immunostaining assays of inner ear sections or tissue whole mounts with antibodies recognizing proteins of interest will identify the localization of the gene product (Fig. 9C). Western blots can also be used for a quantitative analysis of protein expression, in either whole tissue or FACS sorted cell types. Finally, NanoString can be used for validation of numerous genes of interest at both the mRNA and protein levels.46
Fig. 9.
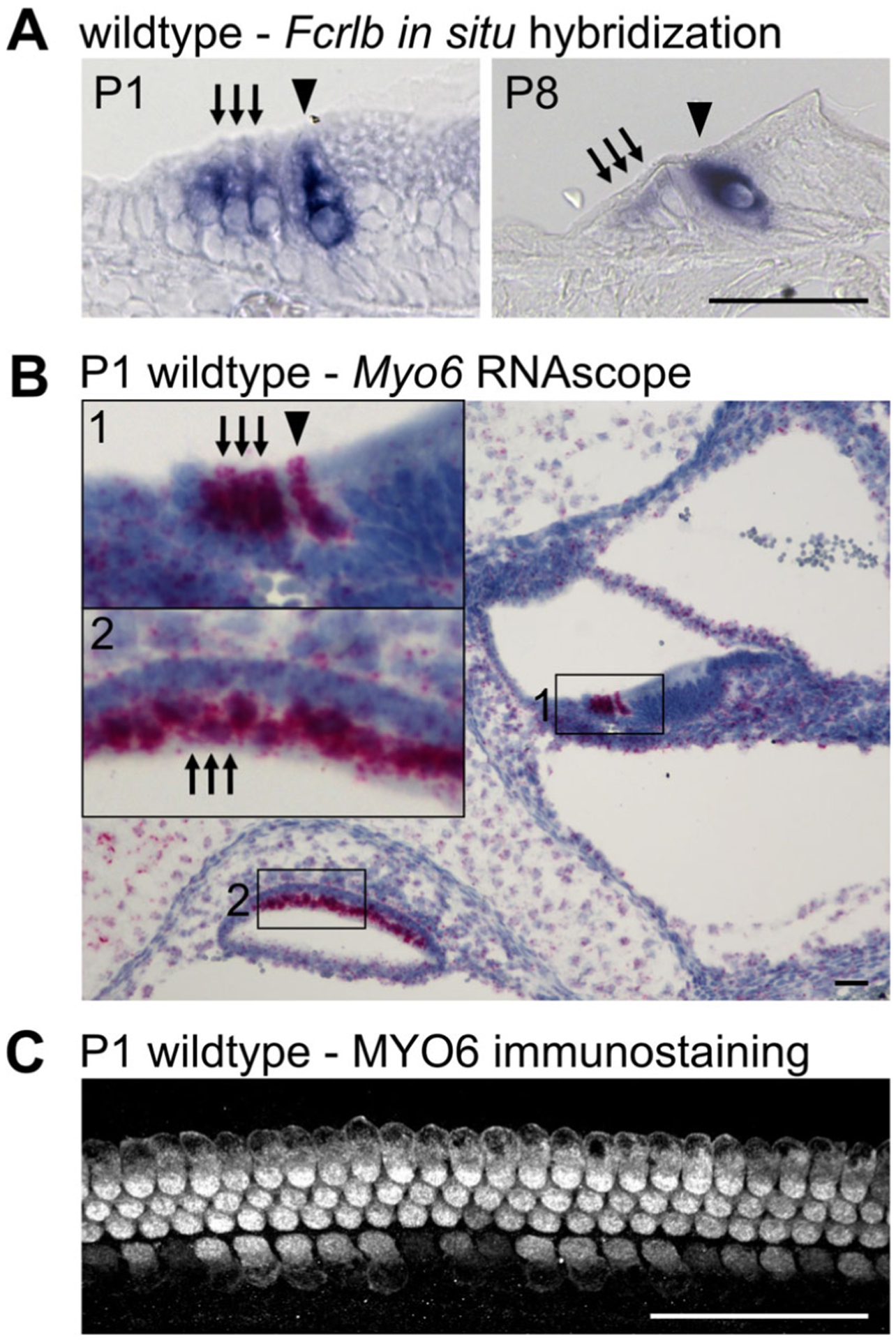
Validation of tissue specific expression. (A) In situ hybridization showing specific expression of Fcrlb in both IHCs and OHCs in the P1 cochlea. By P8, expression of Fcrlb is more restricted to IHCs. (B) Representative RNAscope for the HC marker Myo6 in P1 inner ear. Presence of mRNA is shown by distinct puncta, with one puncta representing one gene specific transcript. (C) Whole mount immunostaining showing HC-specific expression of the MYO6 protein in the P1 cochlea. Scale bar is 50 μm.
To illustrate the usefulness of scRNA-seq for inner ear development, a scRNA-seq was performed on P2 and P7 mouse cochleae (Fig. 8). The initial analysis was performed using the Seurat pipeline that follows the general methodology discussed above in this section. Furthermore, different cell clusters were identified and named based on published gene expression profiles for inner ear cell types (Fig. 8A,B). For example, the HC cluster within the tSNE was identified based on the expression of the known HC gene, Pou4f3. Next, the P2 and P7 datasets were aligned and integrated via canonical correlation analysis, which identified common sources of variation between the two datasets.47 This step is necessary to perform when comparing scRNA-seq datasets, as each dataset will have unique technical batch effects that need to be considered before comparing gene expression. This allows for gene expression differences to be observed between P2 and P7 cochlear HCs, providing information about the temporal dynamics of gene expression during postnatal HC maturation. Here, the OHC marker genes Ocm and Slc26a5 (encoding for the piezoelectric protein prestin) have a low expression at P2, but high expression at P7 (Fig. 8C). This pattern of expression was further validated by comparison to a well-known gene expression database for inner ear development, the SHIELD (Shared Harvard Inner-Ear Laboratory Database) (Fig. 8D),48 as well as by immunostaining with antibodies for both OCM and prestin at P8 to show OHC-specific expression (Fig. 8E). This simple experiment demonstrates how powerful scRNA-seq can be for the study of cell type–specific inner ear development. In fact, a simple scRNA-seq experiment that requires limited skill, just a couple of ears, and a very short timeline from tissue procurement to results can provide a magnitude of cell type–specific expression data that would previously have required decades of work, numerous assays and the efforts of many of laboratories.
The applications of scRNA-seq are endless, giving an unprecedented look into the transcriptome of the inner ear. There are currently a wealth of datasets that can be taken advantage of including the Human Atlas Project, which profiles the transcriptomes of a large variety of cell types, and inner ear datasets that are accessible through the gEAR portal (https://umgear.org/). Recent studies have also shown exciting data using a combination of other modalities and scRNA-seq. For example, Chessum et al. performed viral gene delivery to express Ikzf2, an essential transcription factor for OHC maturation and function, in the IHCs of a mouse that expressed a red fluorescent protein in all HCs (Ai14;Myo15Cre).22 This allowed them to sort the HCs, identify inner and outer HCs based on gene expression patterns, and discover using scRNA-seq that IKZF2 could induce expression of OHC genes and downregulate expression of IHC genes in the transduced IHCs.22 In other tissues, scRNA-seq is also being used along with CRISPR-based technologies to gain a comprehensive look at gene perturbations from knock-out mutations,49 and can be integrated with epigenetic data such as Assay for Transposase-Accessible Chromatin with high-throughput sequencing (ATAC-seq) to gain a full understanding of the gene regulatory networks for a gene of interest or developmental timepoint. Finally, scRNA-seq can be paired with multiplexed error-robust fluorescence In situ hybridization (MERFISH) which allows for thousands of RNA molecules to be identified, localized and counted in individual cells. At this time, this technique has not been applied to the inner ear, but would be an ideal application due to the inner ear distinct architecture and regional segregation of cell types.
DISCUSSION
The technological advancements of the past century have allowed clinicians and researchers to obtain an unprecedented understanding of cell type–specific gene expression of the inner ear. In this manuscript, three types of approaches for cell type–specific analysis of the ear have been reviewed—FACS, RNA/ribosomal pulldown techniques, and scRNA-seq. These methods differ in experimental design, resolution, depth and accuracy of gene expression, applicability to different scientific questions (eg, whether immediate early genes are activated), cost and ease of use, to name just a few parameters (see Table II for a comparative analysis). None of these presented methods are “perfect”; some allow for greater resolution and specificity but rely on tissue dissociation that inevitably induces changes in gene expression; whereas others that avoid dissociation are only methods of enrichment, and thereby lack the resolution of FACS or scRNA-seq–based approaches. While scRNA-seq provides the greatest resolution of gene expression, at present, it is limited by the need for tissue dissociation, the number of genes detected per cell, the stability of the results across samples (different cells), significant batch effects and often lack of full transcript representation. However, scRNA-seq, to date, is the only way one can obtain a representation of any and all cells of a sample without the need to use transgenic mouse lines, and therefore applicable also for the study of cell type–specific gene expression from human tissues.
TABLE II.
Comparative Analysis of Approached for Cell Type–Specific Analysis in the Ear.
| FACS | RNA/ribosomal pulldown | scRNA-seq | |
|---|---|---|---|
| Dissociation | Required | Not required | Required |
| Cell type–specific resolution | Antibody/marker dependent | Cre-recombinase/ transgene dependent | Maximal |
| Accuracy of cell type–specific resolution | Antibody/marker dependent | Moderate. Informs of enrichment or depletion | Maximal |
| Applicability to tissue without the use of transgenic models | Applicable but limited based on antibody availability | Not applicable | Applicable |
| Induction of dissociation-based molecular changes | + | − | + |
| Representation of gene expression | Low variability between samples, high accuracy, high flexibility in selection of library preparation kits | Higher variability between samples, lower accuracy as to source of signal | High variability between samples, incomplete representation of transcript length, partial representation of gene expression |
| Number of cell types analyzed per experiment | Up to four | One | Unlimited (can analyze all cell types in one experiment) |
| Translational impact (eg, for analysis of surgical inner ear specimens) | Low – adult tissues are difficult to dissociate | Low – human tissues do not have the necessary transgenes for pulldown | High—single nucleus RNA-seq is feasible and of likely high impact |
| Cost | $$ | $$ | $$$ |
The development of advanced approaches for cell type–specific isolation and analysis has resulted in even more applications for the data, catalyzing the development of further improved platforms. It is conceivable that within less than a decade, the methods used at this time will be considered archaic and analysis of gene expression will be performed in situ on intact tissue samples, obviating the need for dissociation and obtaining full expression data from all cells of a “captured region.” However, even now, the available tools provide great flexibility and insight into tissue development, response to injury, cell type diversity, and potential for regeneration. With the ability to obtain transcriptomic information from individual cells, even a single tissue provides sufficient material for robust representation of numerous cell types. The ability to measure gene expression using template-free approaches (ie, RNA-seq) allows us to perform cross-species comparisons of gene expression, comparing, for example, the response of inner ears with regenerative capacity from birds or fish to those of mammals which have only very limited regenerative capacity. As we continue to resolve the cellular complexity of the inner ear, the newly obtained data will provide the necessary path for the development of novel regenerative and protective interventions to treat and prevent hearing loss.
MATERIALS AND METHODS
Animals
All procedures involving animals were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals were approved by the Institutional Animal Care and Use Committee at the University of Maryland, Baltimore (protocol numbers 1112005, 1015003 and 0918005). Ai14 and Sox2-CreERT2 mice were purchased from the Jackson Laboratories (Stock # 007914, RRID:IMSR_JAX:007914 and # 017593, RRID: IMSR_JAX:017593 respectively), timed-pregnant CD-1 IGS mice were purchased from Charles River (Cat# CRL:022, RRID: IMSR_CRL:022). Many of the mouse models were generously provided by colleagues: RiboTag (RRID:IMSR_JAX:011029) by Dr. Mary Kay Lobo (University of Maryland Baltimore); prestin-CreERT2 by Dr. Jian Zuo (Creighton University); NuTRAP (RRID: IMSR_JAX:029899) by Dr. Evan Rosen (Beth Israel Deaconess Medical Center); Math1-GFP by Dr Jane Johnson (University of Texas Southwestern); and Myo15Cre by Drs. Christine Petit (Institut Pasteur) and Thomas Friedman (NIDCD).
Tissue Dissociation with Thermolysin and Accutase
Math1-GFP mice and CD-1 mice were euthanized at P1 and P2, respectively, and their temporal bone removed. Inner ear tissues were dissected and placed into 0.5 mg/ml Thermolysin (Sigma-Aldrich Cat# T7902) for 20 minutes at 37°C – 5% CO2. The Thermolysin was then replaced with Accutase (Sigma-Aldrich Cat# A6964) followed by three rounds of 3 minutes at 37°C and mechanical dissociation. The Accutase was inactivated with IMDM (Sigma-Aldrich, Cat# I6529) supplemented with 5% fetal bovine serum and the cell suspension was filtered through a 35-μm cell strainer to eliminate cell clumps. Dissociated cells were stained with CD326- APC (1:2,000; BioLegend Cat# 118213, RRID:AB_1134105), CD49falexa488 (1:100; BioLegend Cat# 313607, RRID:AB_493634), and CD34-PE (1:200; BioLegend Cat# 128609, RRID:AB_2074602) before FACS.
Tissue Dissociation with Cold Active Protease
Whole cochlear tissues were collected from P2 CD-1 mice and placed in cold PBS for cold active protease (CAP) dissociation, in six biological replicates (four ears from two mice per replicate). Additionally, tissues for six replicates of non-dissociated control were collected, snap frozen on dry ice and stored at −80°C until RNA extraction. CAP (Sigma-Aldrich Cat# P5380) was added to the cold PBS containing the tissue to a final concentration of 10 mg/ml, along with 5 mM CaCl2 and 125 U/ml DNAse (New England Biolabs Cat# M0303) and incubated for 15 minutes on ice. Tissues were then subjected to 10–15 rounds of mechanical dissociation using a pipette before filtering through a 35-μm cell strainer to eliminate cell clumps. Single cell suspensions were washed twice with 0.01% Bovine Serum Albumin in PBS (centrifugation 250 g at 4°C for 10 minutes) before proceeding with downstream analyses.
Flow Cytometry and FACS
Dissociation and flow cytometry using CD326-APC with T/A compared to CAP was performed on a BD LSR II (BD Biosciences) flow cytometer. FACS was performed on a BD FACSARIA Cell Sorter (BD Biosciences) at the University of Maryland Greenebaum Comprehensive Cancer Center Flow Cytometry Shared Service.
FM1-43 Dye Uptake
P2, P5, and P8 CD-1 mice were euthanized and their inner ears removed. The lateral wall and stria vascularis were removed from the cochlea to expose the organ of Corti. The tissue was incubated for 30 seconds in 3-μm FM1–43 (Thermo Fisher Scientific, Cat# T35356) in cold Hank’s Balanced Salt Solution (HBSS), followed by three washes in cold HBSS. Following the washes, the organ of Corti was dissected out of the cochlea and mounted on a slide with ProLong Gold antifade (Thermo Fisher Scientific Cat# P36934) to be imaged. Images were taken using an inverted Nikon W1 spinning disk (Nikon Instruments, Inc) at the University of Maryland School of Medicine Center for Innovative Biomedical Resources Confocal Microscopy Facility–Baltimore, Maryland.
Immunostaining of Dissociated Cells
Dissociated cells were fixed with 4% PFA for 10 minutes and attached to glass slides using a Cytospin 4 Cytocentrifuge (Thermo Fisher Scientific). Cells were permeabilized and the non-specific antigens blocked with a 30-minute incubation at room temperature in PBS-0.2% Tween20 supplemented with 5% normal goat serum. Cells were incubated at 4°C overnight with the following antibodies: rabbit anti-MYO6 (1:1000; Proteus Biosciences Cat# 25–6791, RRID:AB_10013626), mouse anti-TUBB3 (1:500; BioLegend Cat# 801202, RRID:AB_2313773). Following three washes in PBS-Tween, cells were incubated with a goat anti-rabbit IgG-Alexa Fluor 488 (1:1000; Thermo Fisher Scientific Cat# A-11034, RRID:AB_2576217), a Goat anti-Mouse-Alexa Fluor 546 (1:1000; Thermo Fisher Scientific Cat# A-11030, RRID: AB_2534089) and 4′,6-Diamidino-2-Phenylindole Dihydrochloride (DAPI to counterstain the nuclei; Thermo Fisher Scientific Cat# 62247). Cells were covered with a coverslip using ProLong Gold antifade (Thermo Fisher Scientific) and imaged using a Nikon Eclipse E600 coupled with an Infinity3 camera (Lumenera).
Immunostaining of Whole Mounted or Sectioned Tissue
Whole inner ears for section staining were fixed overnight in 4% PFA, decalcified using 0.5 M EDTA and mounted in Optimal Cutting Temperature embedding medium (OCT; Fisher Scientific Cat# 23-730-571) before cryosectioning to 10 μm. Immunostaining was performed as described for the dissociated cells (normal goat serum was replaced with 5% normal donkey serum when necessary) using the following primary antibodies: goat anti-prestin N-20 (1:200; Santa Cruz Biotechnology Cat# sc-22692, RRID:AB_2302038), goat anti-OCM N-19 (1:100; Santa Cruz Biotechnology Cat# sc-7446, RRID:AB_2267583), rabbit anti-MYO6 (1:1000; Proteus BioSciences). The secondary antibodies used were as followed: Donkey anti-Goat-Alexa Fluor 546 (1:1000; Thermo Fisher Scientific Cat# A-11056, RRID: AB_2534103) and Goat anti-Rabbit IgG-Alexa Fluor 488 (1:1000; Thermo Fisher Scientific). Nuclei were stained with DAPI. Images were taken using an inverted Nikon W1 spinning disk (Nikon Instruments Inc.) or a Nikon Eclipse E600 coupled with an Infinity3 camera (Lumenera). Confocal images were acquired at the University of Maryland School of Medicine Center for Innovative Biomedical Resources Confocal Microscopy Facility–Baltimore, Maryland.
RT-qPCR
RNA was extracted from non-dissociated and dissociated samples using TRIzol LS Reagent (ThermoFisher Scientific Cat# 10296010) and the Direct-zol RNA Mini-prep kit (Zymo Research Cat# R2050) following the manufacturer’s instructions. RNA was then reverse-transcribed using the Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific Cat# K1641), and RT-qPCR was performed using TaqMan Fast Advanced Master Mix (Applied Biosystems Cat# 44-445-57) and the following TaqMan assays: Mm00725448_s1 for Rplp0, Mm00500401_m1 for Fosb and Mm04243546_s1 for Junb. Relative gene expression was calculated using the comparative ΔΔCT method with Rplp0 as a control and statistical significance between samples was assessed by two-way analysis of variance with Tukey post hoc test.
RiboTag Immunoprecipitation
RiboTag immunoprecipitations were performed as described in Chessum et al., 2018. Briefly, cochlear ducts from eight mice were homogenized and centrifuged at 9400 g for 10 minutes at 4°C to pellet cell debris. Fifty microlitre of supernatant was stored at −80°C until RNA isolation (input sample) while 5 μg of haemagglutinin (HA) antibody (BioLegend Cat# 901502, RRID: AB_2565007) was added to the remaining supernatant and incubated at 4°C for 4 hours. Then the equivalent of 300 μl of rinsed Dynabeads Protein G magnetic beads (Thermo Fisher Scientific Cat# 10004D) were added to the sample and incubated overnight at 4°C. Following three washes in high-salt buffer, 350 μl of Buffer RLT (supplemented with β-mercaptoethanol) from the RNeasy Plus Micro kit (Qiagen Cat# 74034) was added to the beads and the input sample. RNA was extracted according to the manufacturer’s instructions. RNA integrity was assessed on a Bioanalyzer 2100 RNA pico chip (Agilent Technologies). The RNA was processed for RNA-seq using the Ovation RNA-Seq System V2 (NuGEN Cat# 7102-A01) to make the libraries and sequenced on a HiSeq 4000 system (Illumina) using a 75 bp paired end read configuration. Libraries and sequencing were performed at the Genomics Resource Center from the Institute for Genome Sciences at the University of Maryland.
Single Cell RNA-seq
Three P2 and 3 P7 CD1 mice were euthanized, the organs of Corti collected, and cells were dissociated by Thermolysin/Accutase as described above. Samples were then processed for single-cell RNA-seq using the Chromium Single Cell 3′ Reagent Kit and Chromium controller (10× Genomics Cat# PN-1000075) following the manufacturer’s instructions. The resulting samples were sequenced on a HiSeq 4000 system (Illumina) using a 75 bp paired end read configuration and one lane per sample. Library preparations and sequencing were performed at the Genomics Resource Center from the Institute for Genome Sciences at the University of Maryland. The scRNA-seq data are available via the gene Expression Analysis Resource (https://umgear.org/p?l=ed724158).
In situ Hybridization
Inner ear section in situ hybridization for Fcrlb was performed as described in Chessum et al., 2018. Sections obtained as described above were treated with 2 μg/ml Proteinase-K (New England Biolabs Cat# P8107S) for 10 minutes followed by a second fixation in 4% PFA and acetylation. Hybridization of the Fcrlb probe was performed at 65°C overnight. The detection of the probe was performed using a sheep-anti-digoxigenin antibody conjugated to alkaline phosphatase (1:100; Sigma-Aldrich Cat# 11093274910, RRID:AB_2734716) at 4°C overnight and incubation in BM purple AP substrate precipitating solution (Sigma-Aldrich Cat# 11442074001) at 28°C. Fcrlb probe primers are as follows: forward 5′-GTG GTG CTG CGC TGC GAG AC-3′; reverse 5′-CTA GCT GTC CAC TCG GCC CTC CA-3′. Images were acquired with a Nikon Eclipse E600 coupled with an Infinity3 camera (Lumenera).
RNAscope
RNAscope experiments were performed on paraffin-embedded inner ears of CD-1 mice at post-natal day 1 using the RNAscope 2.0 High Definition (HD)—Red Assay (Advanced Cell Diagnostics Cat# 320487) and following the manufacturer’s instructions with the following optimization: tissue was incubated with the Target Retrieval reagent for 10 minutes and incubated with the Protease Plus reagent for 15 minutes. The Myo6 probe was purchased from ACD (Cat# 313381). Images were acquired with a Nikon Eclipse E800 coupled with a Qimaging Retiga EXi Fast1394 camera.
Funding:
NIDCD/NIH R01DC013817 and R01DC03544 (R.H.), DOD CDMRP MR130240 (R.H.), Binational Science Foundation # 2017218 (R.H), NIDCD/NIH DC016595 (R.H.), The Hearing Restoration Project of the Hearing Health Foundation (R.H.), NIDCD/NIH T32DC00046 (M.S.M. and K.G.), F31DC016218 (M.S.M) and NIH/NHLBI T32HL007698-22A1S1 (K.R.).
We thank Amiel A. Dror, MD, PhD, for helping with the design of Figure 1. We thank Ferenc Livak, MD, for his help in generating Figure 3.
Footnotes
The authors have no conflicts of interest to declare.
Level of Evidence: N/A
BIBLIOGRAPHY
- 1.Mullis K, Faloona F, Scharf S, Saiki R, Horn G, Erlich H. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. 1986. Cold Spring Harb Symp Quant Biol 1986;51:263–273. [DOI] [PubMed] [Google Scholar]
- 2.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 1977;74:5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chinwalla AT, Cook LL, Delehaunty KD, et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002;420:520–562. [DOI] [PubMed] [Google Scholar]
- 4.Lander S, Linton LM, Birren B, et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001;409:860–921. [DOI] [PubMed] [Google Scholar]
- 5.Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science 2001;291:1304–1351. [DOI] [PubMed] [Google Scholar]
- 6.de Kok YJ, van der Maarel SM, Bitner-Glindzicz M, et al. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science 1995;267:685–688. [DOI] [PubMed] [Google Scholar]
- 7.Shearer AE, Smith RJH. Massively parallel sequencing for genetic diagnosis of hearing loss: the new standard of care. Otolaryngol Neck Surg 2015; 153:175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shearer AE, Shen J, Amr S, Morton CC, Smith RJ. A proposal for comprehensive newborn hearing screening to improve identification of deaf and hard-of-hearing children. Genet Med 2019;11:2614–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Omichi R, Shibata SB, Morton CC, Smith RJH. Gene therapy for hearing loss. Hum Mol Genet 2019;28(R1):R65–R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basinou V, Park J-S, Cederroth CR, Canlon B. Circadian regulation of auditory function. Hear Res 2017;347:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Betters DM. Use of flow cytometry in clinical practice. J Adv Pract Oncol 2015;6:435–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hertzano R, Elkon R, Kurima K, et al. Cell type – specific transcriptome analysis reveals a major role for Zeb1 and miR-200b in mouse inner ear morphogenesis. PLoS Genet 2011;7:e1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burns JC, Kelly MC, Hoa M, Morell RJ, Kelley MW. Single-cell RNA-Seq resolves cellular complexity in sensory organs from the neonatal inner ear. Nat Commun 2015;6:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bakke AC. The principles of flow cytometry. Lab Med 2001;32:207–211. [Google Scholar]
- 15.Hertzano R, Puligilla C, Chan S-L, et al. CD44 is a marker for the outer pillar cells in the early postnatal mouse inner ear. JARO 2010;11:407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Madisen L, Zwingman TA, Sunkin SM, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 2010;13:133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cox B, Liu Z, Mellado Lagarde MM, Zou J. Conditional gene expression in the mouse inner ear using Cre-loxP. JARO 2012;13:295–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stone JS, Wisner SR, Bucks SA, Mellado Lagarde MM, Cox BC. Characterization of adult vestibular organs in 11 CreER mouse lines. J Assoc Res Otolaryngol 2018;19:381–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walters BJ, Yamashita T, Zuo J. Sox2-CreER mice are useful for fate mapping of mature, but not neonatal, cochlear supporting cells in hair cell regeneration studies. Sci Rep 2015;5:11621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matern M, Vijayakumar S, Margulies Z, et al. Gfi1Cre mice have early onset progressive hearing loss and induce recombination in numerous inner ear non-hair cells. Sci Rep 2017;7:42079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herget M, Scheibinger M, Guo Z, et al. A simple method for purification of vestibular hair cells and non-sensory cells, and application for proteomic analysis. PLoS One. 2013;8:e66026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chessum L, Matern MS, Kelly MC, et al. Helios is a key transcriptional regulator of outer hair cell maturation. Nature 2018;563:696–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elkon R, Milon B, Morrison L, et al. RFX transcription factors are essential for hearing in mice. Nat Commun 2015;6:8549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matern MS, Beirl A, Ogawa Y, et al. Transcriptomic profiling of Zebrafish hair cells using RiboTag. Front Cell Dev Biol 2018;6:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van den Brink SC, Sage F, Vértesy A, et al. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat Methods 2017;14:395–396. [DOI] [PubMed] [Google Scholar]
- 26.Adam M, Potter AS, Potter SS. Psychrophilic proteases dramatically reduce single-cell RNA-seq artifacts: a molecular atlas of kidney development. Development 2017;144:3625–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heiman M, Schaefer A, Gong S, et al. Development of a BACarray translational profiling approach for the molecular characterization of CNS cell types. Cell Cell 2009;14:738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanz E, Yang L, Su T, Morris DR, Mcknight GS, Amieux PS. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. PNAS 2009;106:13939–13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gay L, Miller MR, Ventura PB, et al. Mouse TU tagging: a chemical/genetic intersectional method for purifying cell type-specific nascent RNA. Genes Dev 2013;27:98–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roh HC, Tsai LT-Y, Lyubetskaya A, Tenen D, Kumari M, Rosen ED. Simultaneous transcriptional and Epigenomic profiling from specific cell types within heterogeneous tissues in vivo. Cell Rep 2017;18: 1048–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Erickson T, Nicolson T. Identification of sensory hair-cell transcripts by thiouracil-tagging in zebrafish. BMC Genomics 2015;16:842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song Y, Milon B, Ott S, et al. A comparative analysis of library prep approaches for sequencing low input translatome samples. BMC Genomics 2018;19:696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015;348:aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol 2019;15:e8746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen X, Teichmann SA, Meyer KB. From tissues to cell types and Back: single-cell gene expression analysis of tissue architecture. Annu Rev Biomed Data Sci 2018;1:29–51. [Google Scholar]
- 36.Hertzano R, Elkon R. High throughput gene expression analysis of the inner ear. Hear Res 2012;288:77–88. [DOI] [PubMed] [Google Scholar]
- 37.Shrestha BR, Chia C, Wu L, Kujawa SG, Liberman MC, Goodrich LV. Sensory neuron diversity in the inner ear is shaped by activity. Cell. 2018; 174:1229–1246.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun S, Babola T, Pregernig G, et al. Hair cell Mechanotransduction regulates spontaneous activity and spiral ganglion subtype specification in the auditory system. Cell 2018;147(5):1247–1263. 10.1016/j.cell.2018.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bakken TE, Hodge RD, Miller JA, et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS One 2018; 13:e0209648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lever J, Krzywinski M, Altman N. Points of significance: principal component analysis. Nat Methods 2017;14:641–642. [Google Scholar]
- 41.Becht E, McInnes L, Healy J, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol 2019;37:38–44. [DOI] [PubMed] [Google Scholar]
- 42.Aibar S, González-Blas CB, Moerman T, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods 2017;14:1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ellwanger DC, Scheibinger M, Dumont RA, Barr-Gillespie PG, Heller S. Transcriptional dynamics of hair-bundle morphogenesis revealed with CellTrails. Cell Rep 2018;23:2901–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trapnell C, Cacchiarelli D, Grimsby J, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 2014;32:381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang F, Flanagan J, Su N, et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn 2012; 14:22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kulkarni MM. Digital multiplexed gene expression analysis using the NanoString nCounter system. Curr Protoc Mol Biol 2011;94:25B.10.1–25B.10.17. [DOI] [PubMed] [Google Scholar]
- 47.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018;36:411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen J, Scheffer DI, Kwan KY, Corey DP. SHIELD: an integrative gene expression database for inner ear research. Database 2015;2015:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Genga RMJ, Kernfeld EM, Parsi KM, Parsons TJ, Ziller MJ, Maehr R. Single-cell RNA-sequencing-based CRISPRi screening resolves molecular drivers of early human endoderm development. Cell Rep. 2019;27: 708–718.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]