

Abstract
Background
Advanced renal cell carcinoma has been resistant to drug therapy of different types and new types of drug therapy are needed. Targeted agents inhibit known molecular pathways and have been tested in renal cancer for just over a decade.
Objectives
1) To provide a systematic and regularly updated review of randomized studies testing targeted agents in advanced renal cell cancer. 2) To identify the type and degree of clinical benefit of targeted agents over the prevailing standard of care.
Search methods
Period of search: January 2000 to June 2010. 1) Electronic search of CENTRAL, MEDLINE and EMBASE databases. 2) Hand search of international cancer meeting abstracts.
Selection criteria
Randomized, controlled studies, including a targeted agent in patients with advanced renal cell cancer reporting any pre‐specified cancer outcome by allocation.
Data collection and analysis
The majority of the standardized search and data extraction was conducted independently by two investigators with subsequent resolution of differences. Handsearching, quality of life and toxicity data extraction, most of the initial analysis, and risk of bias assessment, was carried out by one investigator and verified by additional authors as required. Twenty‐five fully eligible studies tested thirteen different targeted agents in a total of 7484 patients with mostly Stage IV disease; 61% had not received prior systemic treatment. The majority of patients were good performance status (ECOG (Eastern Cooperative Oncology Group) 0 to 1). Most comparisons were each examined in only a single study. Risk of bias was considered low for studies that were placebo‐controlled, had a primary outcome of overall survival, or that evaluated progression by independent radiologic reviewers unaware of the intervention allocation.
Main results
Most progress has been made in patients with advanced renal cancer of the clear cell subtype, a condition with a clearly defined molecular pathology promoting angiogenesis. In systemically untreated patients, two approaches to angiogenesis inhibition have demonstrated benefit. Compared with interferon‐alfa monotherapy, oral sunitinib improved multiple outcomes including overall survival (18% risk reduction for death; median survival improved from 21.8 to 26.4 months, P = 0.049) without correction for crossover) in patients with mostly good or moderate prognosis. In the same setting, two studies have shown that the addition of biweekly intravenous bevacizumab to interferon‐alfa also improved the chance of major remission and prolonged progression‐free survival. These two bevacizumab plus interferon studies each observed improved overall survival approaching statistical significance (each study observed a 14% risk reduction for death). Additional anti‐angiogenesis agents, such as pazopanib and tivozanib, are in earlier stages of evaluation. After progression of clear cell disease on prior cytokine therapy, oral sorafenib results in a better quality of life than placebo. In patients with clear cell disease with progression on or within 6 months of first‐line targeted therapy with sunitinib or sorafenib, the targeted oral mTOR (mammalian target of rapamycin) inhibitor everolimus resulted in prolonged disease‐free survival without detriment to quality of life. Remissions were very infrequent and no improvement in overall survival was observed in this study where the majority of placebo‐assigned patients received everolimus at disease progression. In untreated patients with unselected renal cancer histology and poor prognostic features, weekly intravenous temsirolimus, an mTOR inhibitor, improved outcomes compared with interferon‐alfa (median overall survival improved from 7.3 to 10.9 months, P = 0.008). Of particular interest, an exploratory analysis observed a marked reduction in hazard for death in the non‐clear cell subgroup. Combinations of targeted agents are being evaluated, but toxicity is problematic.
Authors' conclusions
Several agents with specified molecular targets have demonstrated clinically useful benefits over interferon‐alfa, and also after either prior cytokine or initial anti‐angiogenesis therapy. More research is required to fully establish the role of targeted agents in this condition.
Plain language summary
Targeted drug therapy for advanced kidney cancer
Background
Cancer of the kidney is an important health problem with over 15,000 deaths in North America annually. Kidney cancer in adults that has spread or is too advanced for surgery is incurable and is resistant to conventional chemotherapy drugs. Drugs that affect the body's immune system have been standard care in the past two decades but have been associated with unpleasant side effects and poor results in most patients. Recent advances in understanding the molecular biology of kidney cancer have resulted in the development of drugs that target known molecular pathways (targeted therapy). This review critically examines reports of clinical trials that have directly compared the new targeted "designer" drugs with previous standard therapies for this condition, to see if these drugs could be considered an advance in care. Studies identified A systematic survey of reports published in electronically available medical journals and cancer meeting reports since 2000 identified 25 studies that looked at 13 different new drugs in a total of over 7000 patients. Patients were generally representative of those with advanced kidney cancer, with the exception of being fully ambulatory, and with no evidence of spread to the brain. Most studies were restricted to patients with renal carcinoma of the clear‐cell subtype. Over 60% of patients had not received any prior drug treatment. All patients consented to be randomly assigned to receive the test program or standard care, often with the opportunity to receive the test drug later if beneficial (this ethical approach may have reduced any differences in survival between groups). Results of studies demonstrating important benefits A. Untreated patients with advanced kidney cancers of the clear‐cell subtype 1. In patients with no prior drug therapy and most with a predicted survival of over 12 months, a drug called sunitinib was given daily by mouth for 4 weeks out of 6. Sunitinib caused more frequent major remissions (at least 50% shrinkage of cancer) than standard interferon‐alfa given by injection under the skin three times per week (major remissions in 39% of treated patients versus 8% respectively). This benefit was associated with improved average sense of well‐being and other measures of quality of life, though patient's responses may have been influenced because they knew whether they were getting the new treatment or not. Interferon caused more fatigue, whereas sunitinib caused more diarrhea, high blood pressure, and skin problems. On average, sunitinib was associated with an extra 6 months delay in the time before the cancer grew on X‐rays, and an extra 4.6 months of survival. Sunitinib has been approved for use in North America, the European Union, and elsewhere. 2. Two studies also in untreated patients, one in Europe and one in North America, observed greater anti‐cancer effect by adding bevacizumab by vein on alternate weeks to interferon‐alfa. The magnitude of the benefits was similar to those seen with sunitinib, and the combination is in use in Europe. However, this regimen is less convenient than oral sunitinib and has side‐effects associated with both interferon and bevacizumab. 3. In untreated patients with poor predicted survival, weekly intravenous temsirolimus was associated with longer survival (10.9 versus 7.3 months) and better quality of life than interferon alfa. However, remissions were uncommon. B. Patients previously treated with drug therapy
1. Following initial interferon therapy, sorafenib improved quality of life and delayed disease growth compared to placebo.
2. Following initial targeted therapy with sunitinib or sorafenib, daily oral everolimus delayed cancer growth compared to placebo but did not result in remissions or improve quality of life. Survival was similar but most placebo‐assigned patients received everolimus later, making survival interpretation difficult. C. Patients with advanced kidney cancers of the non clear‐cell subtypes
These cancers lack the primary target for sunitinib or sorafenib, consequently patients with non clear‐cell kidney cancers have been excluded from comparative studies of those and similar drugs. Temsirolimus may help some patients in this group, according to one analysis. Implications for care
About three‐quarters of patients with advanced kidney cancer have the clear‐cell subtype and the new targeted drugs can modestly improve the quantity and quality of life in this setting. Most oncologists in North America consider oral sunitinib to be the current standard of initial drug care in appropriately selected patients. Additional after‐market studies have extended these results to patients who are older or only partially ambulatory. The expense of these drugs limits their availability in some regions, an aspect beyond the scope of this review. Complete disappearance of advanced kidney cancer remains very uncommon and will be the main objective for further research.
Summary of findings
Background
*Additional reference names are marked with an asterisk to distinguish them from study names with no asterisk.
Advanced renal cell cancer has been one of the most drug‐resistant malignancies. Hormonal and cytotoxic chemotherapy agents have not been demonstrated to improve overall survival for this condition, and remissions occur at a frequency similar to that seen with no therapy (Oliver 1989*) or with placebo (Gleave 1998*). For most of the past 20 years immunotherapy has been the main focus of the search for an effective drug therapy for renal cancer, and is the subject of a companion Cochrane systematic review (Coppin 2006*). In summary, immunotherapy is associated with very modest survival benefit at best. Pretreatment prognostic factors that predict survival are useful for stratification purposes and have been determined in both the first‐line setting (Leibovich 2005*) and in previously treated patients (Motzer 2004*). There is evidence that the distribution of risk categories in clinical trials is changing to a more favourable profile (Patil 2010*). Until now, interferon‐alfa has been considered the standard comparator for first‐line therapy of advanced kidney cancer (Mickisch 2003*; Motzer 2002*), and placebo‐controlled trials have been appropriate in the second‐line setting.
There is clearly high interest in finding more effective treatment for advanced renal cell cancer. The search for specific targets for therapy goes back at least to Paul Ehrlich's "magic bullet" over a century ago. This concept has recently received an enormous boost with the knowledge explosion of molecular targets and the potential for associated therapies that are target specific and therefore might have greater efficacy with less toxicity (Sawyers 2004*). Clinical proof of concept came with the remarkable success of single agent imatinib for chronic myeloid leukemia (Deiniger 2005*). In advanced solid tumours, bevacizumab has been demonstrated, if used in combination with chemotherapy, to improve survival in colorectal cancer (Hicklin 2005*).
Molecular analysis of renal cell carcinoma has shown that kidney cancer is not a homogeneous condition (Linehan 2005*; Hacker 2010*). A high proportion of sporadic clear cell renal cell cancers have biallelic abnormalities of the VHL (Von Hippel‐Lindau) tumour‐suppressor gene (Young 2009*), whereas other subtypes do not. Absence of the active VHL gene product results in unregulated activation of the hypoxia‐inducible system and accumulation of growth factors such as vascular endothelial growth factor (VEGF). In future, it will be necessary to distinguish the impact of therapy on different molecularly defined tumour types but the necessary technology has not yet reached routine use. The molecular complexities of both the disease (renal cell cancer) and the treatment (targeted therapy) are resulting in a rapidly evolving and exciting phase in the history of the treatment of advanced kidney cancer. According to one reviewer (Uzzo 2003*), "an understanding of the basic biology of renal cell carcinoma is more advanced than that of any other solid malignancy". Further molecular subclassification within clear cell renal cancer may well become feasible (Kaelin 2008*).
Molecular pathways with multiple targets that are of particular interest in renal cell cancer currently fall into two major groups including angiogenesis (Rini 2005*), and intracellular signal transduction pathways (Adjei 2005*). The presence of a target may or may not translate into benefit from a targeted agent (Bergsland 2006*). Some agents have activity against multiple targets. Classic immunotherapies such as interferon‐alfa may have anti angiogenic activity but are considered as a separate class of agent (Coppin 2006*). Suitably large randomized controlled trials have a high financial and resource cost so that selection of agents for Phase III testing requires strategic decision making (Roberts 2003*). Since the new agents are not necessarily cytotoxic, it is possible that tumour shrinkage may not be a reliable indicator of drug activity (Stadler 2006*); for example, stabilization of previously progressive disease might result in extension of overall survival. Additionally, objective response of renal cell carcinoma has not hitherto been associated with subsequent regulatory approval of an agent (Goffin 2005*). Our review examines the primary and secondary endpoints used by investigators, including progression‐free survival. Additional background will be provided in the 'Results' section for individual agents tested in the studies identified by this review.
Objectives
The primary objectives of this review are to provide a systematic survey of randomized trials of targeted agents in advanced renal cancer, and to assess whether any of these agents provide better clinically relevant outcomes than other therapies for advanced renal cell cancer. An additional objective is to provide a continuously updated reference resource in this rapidly developing field.
Methods
Criteria for considering studies for this review
Types of studies
Randomized controlled trials, except that randomized Phase I trials were excluded.
Types of participants
Adult patients with metastatic or locally inoperable renal cell carcinoma, histologically verified at presentation or relapse. Some patients with "operable" tumours but who had serious co morbidities were enrolled in these studies. Studies of mixed solid tumours were eligible only if patients with renal cell carcinoma were stratified and reported separately from other tumour types. Patients may or may not have received prior systemic therapy.
Types of interventions
Agents with known or presumed molecular targets and known or presumed anti‐angiogenesis agents must have been part of the therapeutic regimen of at least one study arm. Classic immunotherapy agents, including recombinant cytokines and their predecessors, were excluded from this definition of targeted therapy, but may have been included as part of the regimen in any study arm. Studies in which maintenance therapy by a targeted agent was the randomized variable were eligible.
Types of outcome measures
Studies reported at least one efficacy outcome by allocation arm to be eligible for inclusion. Eligible efficacy outcomes were categorical or time‐dependent. Categorical efficacy outcomes included achievement of tumour shrinkage or disease stabilization according to commonly recognized criteria. Time‐dependent outcomes included overall survival or progression‐free survival from date of randomization. Quality‐of‐life outcomes were examined where available. Adverse events were examined in studies reporting superior efficacy or decreased toxicity for the investigational arm. Studies that reported only adverse events were not eligible.
Search methods for identification of studies
See also the Cochrane Prostatic Diseases and Urologic Cancers Group search strategy.
Sources
Online databases: the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, and EMBASE. The main search terms were: 'cancer, renal cell/' with 'randomized controlled trial(pt)' or 'random(tw)'. We have also used a comprehensive search strategy developed for a kidney cancer policy document and kindly provided by Dr Mike Shelley (NICE 2002*; Thompson Coon 2010a*).
Handsearching of abstracts in the proceedings of the periodic meetings of the American Urologic Association, ECCO ‐ the European Cancer Conference, ESMO ‐ the European Society of Medical Oncology, and ASCO ‐ the American Society of Clinical Oncology (the general meeting held annually, and the genitourinary meeting held annually since 2008).
Handsearching of the bibliography of each primary reference and of recent reviews of renal cell cancer.
Published/unpublished material: all study identification is from published resources. Where studies were identified as meeting abstracts, the presented material was included if publicly accessible on the Internet.
Period of review: January 2000 through June 2010. Language: there was no language restriction for searching. Inclusion of otherwise eligible studies depended on the availability of translation resources for the language(s) concerned. Duplicate search: databases were independently searched by two reviewers; systematic handsearching was conducted by one reviewer (CC).
Data collection and analysis
Inclusion and exclusion of studies
Abstracts of potentially relevant studies identified by the search strategy were surveyed as well as the full text where available electronically. All study reports that met basic eligibility criteria (randomized trial of drug therapy for renal cell carcinoma) were assessed for inclusion or exclusion by two reviewers independently (CC, LL). Studies were evaluated for quality using the CONSORT statement (Moher 2001*). Faulty randomization methodology was not identified. Post‐randomization patient exclusions or inappropriate statistical methods did not necessarily exclude studies if the minimum required data for our analysis was provided, but were considered in the interpretation of results.
Completeness of ascertainment of eligible studies
The clinical trials database clinicaltrials.gov (initiated 2002) was searched for all trials of kidney or renal cancer, then limited to randomized trials that were terminated, or were still active but closed to accrual and candidates for publication. Trials are herein referred to by their eight‐digit NCT number where known. This database lists protocol elements for each trial including the estimated primary completion date, defined as the final data collection date for the primary outcome measure. Also listed is the name of the sponsor (such as the co‐operative research group or sponsoring pharmaceutical company) and the principal contact (principal investigator or sponsoring company). Trials that were terminated or were active and closed for more than one year, and not identified by the search process for published studies, were deemed missing and a potential source of publication bias. This source was also used in preparing the risk of bias tables for each study. Attempted search of additional trials databases was not productive, in some cases because they are access restricted (eg EudraCT.emea.europa.edu).
Data extraction
Data from included studies were independently extracted onto a standardized form by two reviewers (LL, CC), and any discrepancies resolved by consensus. Data extraction fields for each study included: (a) patient eligibility criteria and accrual by arm for age, race, gender, performance status, prior nephrectomy, prior systemic therapy, histologic subtype, and prognostic risk distribution; (b) stratification parameters if any; (c) stated primary and secondary outcome measures; (d) sample‐size calculation; (e) detailed interventions, including criteria for discontinuing therapy and crossover to the investigational arm; (f) treatment delivery evaluation; (g) primary and secondary outcome measures by treatment allocation; (h) adverse events.
Planned comparisons (original protocol)
Individual targeted single agents versus non‐targeted therapies (including classic immunotherapies, hormone therapies, or placebo)
Non‐targeted therapies with or without a targeted agent
Individual targeted agents with or without another agent
Other comparisons
Revised comparisons
Comparisons of different dose and/or schedule of the same agent(s) to clarify the dose‐response‐toxicity relationship for further study
Comparison of targeted agent(s) with a placebo or hormonal control, to identify agents with clinical activity
Comparison of targeted agent(s) with a cytokine control, interferon‐alfa being usual first‐line standard care until approximately 2008
Comparison of targeted agents(s) with a targeted agent control, to identify a superior agent or combination of agents
Since the original version of this review in 2008, targeted agents have become common first‐line therapy for advanced clear‐cell renal cancer, and consequently the appropriate control arm for targeted trials is evolving. In the current analysis, comparisons have been based on the nature of the control arm as in our original protocol (instead of being based on indication as in the 2008 version of this review). Meta‐analyses of groups of agents were only considered if class homogeneity was convincingly demonstrated. Studies that randomized to three or more intervention arms were considered as a set of multiple two‐arm comparisons.
Statistical analysis
We anticipated analysis of four types of outcomes: categorical outcomes, such as tumour remission; single time‐dependant outcomes, such as overall survival; quality‐of‐life surveys; and toxicity tables. Of these, methods for analysis of dichotomous outcomes were fully covered by standard Cochrane Collaboration procedures (Higgins 2005*); there was no need for other measures to be converted into dichotomous outcomes. Multidimensional quality‐of‐life and toxicity outcomes were considered individually. Time‐dependent outcomes were potentially problematic. Where only a single study was available for a comparison, any standard statistical analysis, such as the log‐rank test used by the author, was accepted. For meta‐analysis of multiple studies of the same type, extraction of a dichotomous endpoint such as survival at one year from randomization could be used but was not required.
Methodologic issues arising
Outcome statistics: the author‐quoted outcome statistics were used for the individual studies, except for remissions that were entered as a dichotomous outcome. All authors used the log‐rank test and quoted P values are all two‐sided.
Multi‐arm studies: studies with more than two arms have been approached as comparisons of pairs of trial arms of interest (Atkins 2004; Hudes 2010; Yang 2003). One additional 3‐arm study randomized patients 2:1:1 to an investigational arm and to 2 control arms considered of similar efficacy (Escudier(5) 2010).
Remission rate: major remission by RECIST (Response Evaluation Criteria in Solid Tumors) (Therasse 2000*) or WHO (World Health Orginization) (Miller 1981*) criteria as specified by the study, divided by number of patients evaluable for this endpoint according to investigators (patients with measurable/evaluable disease).
Overall survival as an endpoint: The original intent of this review was to focus on overall survival as definitive endpoint. However, some of the recently available targeted agents described herein have proven or probable survival benefit. Consequently their use in control arm patients within studies (crossover) or before/after study participation may confound potential differences in overall survival. For this reason regulatory bodies may approve marketing of new agents on the basis of improvement in progression‐free survival (PFS). We therefore modified the original protocol to include PFS for standardized data extraction. The interpretation of survival data is discussed in several trials reports and reviews (Escudier(3) 2010; Pal 2010*).
Risk of bias tables were constructed for each study using consistent definitions extended from the general principles described in the Cochrane Handbook (for example, "patients were randomized" was not considered sufficient to qualify as low risk of bias for either the generation sequence or allocation concealment but listed as unclear).
Nomenclature for studies
Study names in this version are based on the name of the principal investigator for each study (usually the first author of related key publications), combined with the year of the most recent related publication. Where the same investigator has led more than one study, the study names include a sequential number e.g. Escudier(1), Escudier(2), etc.
Results
Description of studies
Completeness of Ascertainment
Search of the trials database clinicaltrials.gov identified 19 of the 25 included studies described below. Eight additional studies were identified, five that had been terminated before completion, and three that had been closed for over a year as of the search cutoff date (June 2010). Early termination trials included poor accrual (NCT00110344, NCT00678288), poor tolerance (NCT00491738), a Phase IV trial (NCT00352859), and a placebo‐controlled Phase III trial of bevacizumab after prior cytokine terminated for unknown reasons (NCT00061178). Further efforts failed to identify reports from three completed studies closed for over one year, these being two randomized discontinuation trials of VEGFR‐TKIs (vascular endothelial growth factor receptor‐tyrosine‐kinase inhibitors) cediranib (NCT00423332) and pazopanib (NCT00244764), and a comparison of standard or low dose sorafenib with placebo (NCT00606866).
Survey of Included Studies
Additionally, see 'Table 3' ('Summary of included studies'), and 'Characteristics of included studies'. Note: for consistency across studies, for each comparison 'Arm 1' is the investigational or higher dose arm, 'Arm 2' is the control or lower‐dose arm, and these designations may differ from those used by the study authors. For three‐arm studies, comparisons (A), (B), (C) refers to pairs of arms as defined in the 'Characteristics of included studies'.
1. Summary of Included studies.
| Study Name | Publication Status |
Accrual Started |
Total Patients/ Measured for ORR |
RCC Subtype CC = Clear Cell Component |
Prior Prior Systemic Therapy |
This Review "Arm 1" (investigational) |
This Review "Arm 2" (and "Arm 3" if any) |
| Atkins 2004 | Peer reviewed journal | 2000 | 111 / 111 | any | cytokine | Temsirolimus 250 mg | Temsirolimus 75 mg or Temsirolimus 25 mg (Arm 3) |
| Bhargava 2010 | Abstract+slides | 2007 | 272 / 258 | any, 83% CC | naive 54% | Tivozanib (AV‐951) | Placebo |
| Bracarda 2010 | Abstract+slides | 2006 | 101 / 101 | CC required | naive | Sorafenib + Interferon 3MUx5 | Sorafenib + Interferon 9MUx3 |
| Bukowski 2007 | Peer reviewed journal | 2004 | 104 / 103 | CC required | naive | Bevacizumab + Erlotinib | Bevacizumab |
| Ebbinghaus 2007 | Abstract+slides | 2003 | 103 / 103 | any | naive | ABT‐510 100 mg | ABT‐510 10 mg |
| Escudier(1) 2007 | Abstract; Polish Jnl | 2000 | 305 / 0 | any | cytokine | AE‐941 | Placebo |
| Escudier(2) 2010 | Peer reviewed journal | 2003 | 903 / 903 | CC required | cytokine | Sorafenib | Placebo |
| Escudier(3) 2010 | Peer reviewed journal | 2004 | 649 / 649 | CC required | naive | Bevacizumab + Interferon | Placebo + Interferon |
| Escudier(4) 2009 | Peer reviewed journal | 2005 | 189 / 170 | CC required | naive | Sorafenib | Interferon |
| Escudier(5) 2010 | Abstract+slides | 2008 | 171 / 171 | any except papillary | naive | Bevacizumab + Temsirolimus | Bevacizumab+Interferon or Sunitinib (combined for this analysis) |
| Gordon 2004 | Abstract+slides | 2000 | 342 / 342 | any | naive | Interferon + Thalidomide | Interferon |
| Hudes 2010 | Peer reviewed journal | 2003 | 626 / 626 | any | naive | Temsirolimus | Interferon alone or +Temsirolimus (Arm 3) |
| Jonasch 2010 | Peer reviewed journal | 2005 | 80 / 80 | CC required | naive | Sorafenib + Interferon | Sorafenib |
| Lee 2006 | Peer reviewed journal | 2000 | 60 / 48 | any | 10% naive | Thalidomide | Hormone |
| Madhusudan 2004 | Abstract only | 2002 | 34 / 20 | not stated | naive | Interferon + Thalidomide | Interferon |
| Motzer(1) 2010 | Peer reviewed journal | 2004 | 750 / 750 | CC required | naive | Sunitinib | Interferon |
| Motzer(2) 2010 | Peer reviewed journal | 2006 | 410 / 334 | CC required | sunitinib &/or sorafenib |
Everolimus | Placebo |
| Procopio 2010 | Abstract+slides | 2006 | 128 / 128 | any | naive | Sorafenib + Interleukin‐2 | Sorafenib |
| Ratain 2006 | Peer reviewed journal | 2002 | 65 / 0 | any | naive 16% | Sorafenib | Placebo |
| Ravaud 2008 | Peer reviewed journal | 2002 | 416 / 416 | any, CC 87% | cytokine | Lapatinib | Hormone |
| Rini 2010 | Peer reviewed journal | 2003 | 732 / 639 | CC required | naive | Bevacizumab + Interferon | Interferon |
| Srinivas 2005 | Peer reviewed journal | 1999 | 14 / 14 | any | naive 29% | Thalidomide 800 mg | Thalidomide 200 mg |
| Stadler 2005 | Peer reviewed journal | 2000 | 368 / 0 | any | naive 41% | Carboxyaminoimidazole | Placebo |
| Sternberg 2010 | Peer reviewed journal | 2006 | 435 / 435 | CC required | naive 54% | Pazopanib | Placebo |
| Yang 2003 | Peer reviewed journal | 1998 | 116 / 116 | CC required | IL2 93% | Bevacizumab 10 mg/kg | Bevacizumab 3 mg/kg or Placebo (Arm 3) |
As of the search cutoff date of 30 June 2010, 26 potentially eligible studies were identified and defined as randomized controlled trials, including one or more targeted drugs in patients with advanced renal cell cancer. One study was excluded because there was no reported outcome data by allocation (Escudier(6) 2009); this study of continuous daily sunitinib randomized patients to morning or evening dosing and its omission is not likely to have biased the conclusions of this review. The remaining 25 studies were found to be fully eligible for inclusion (see 'Table 3' for current publication status, year that accrual commenced, total number of patients randomized, histologic subtype, prior systemic therapy, and study treatment summary).
Since the original version of this Review (literature search 2000 to 2007), five new eligible studies were now available for analysis (Bhargava 2010; Escudier(5) 2010; Motzer(2) 2010; Procopio 2010; Rini 2010); also, a previously ineligible study (Hutson 2007*), has been updated and now eligible (Sternberg 2010), and nine of nineteen previously reviewed eligible studies have been updated so that only one study is limited to a meeting abstract with no additional material (Madhusudan 2004).
Study design was standard 2‐arm with 1:1 randomization in the majority. Three studies used a randomized discontinuation design that randomized patients with stable disease after an induction period of 3 to 4 months (Bhargava 2010; Ratain 2006; Stadler 2005). Targeted therapy, in the sense described above, is a relatively new concept. Randomized trials of targeted therapy for advanced renal cancer first commenced accrual in October 1998 (Yang 2003). Since that time, the research effort in this field has accelerated rapidly, as judged by the number of abstracts submitted for presentation at the annual meeting of the American Society of Clinical Oncology (ASCO). In order to give a sense of the chronological development of this rapidly moving field, this report considered studies within each analytic group in the approximate order in which they began accrual.
A survey of reported outcomes is informative ('Table 4'). Major remissions (usually based on the RECIST criterion) were reported in 21 of 22 studies (not relevant in the 3 randomized discontinuation studies) and were listed as the primary or co‐primary endpoint in 5. The hard endpoint of overall survival has so far been reported in 15 of the 25 studies but was a reported primary endpoint of only 7. Progression‐free survival has become the most commonly used endpoint, justified mainly because of the use of crossover and multiple lines of therapy that makes evaluation of overall survival problematic, and because remission has been an unreliable predictor of clinical benefit. Formal quality‐of‐life data have been reported in some studies (Bukowski 2007; Escudier(2) 2010; Escudier(4) 2009; Gordon 2004; Hudes 2010; Motzer(1) 2010; Motzer(2) 2010; Sternberg 2010) and illuminate the clinical relevance of differences in remission rates or freedom from disease progression, most clearly in studies using a placebo control to reduce the risk of patient bias (Escudier(2) 2010). Most studies have reported safety data with details of toxicities encountered in each arm but this material will be selectively reviewed when it bears on study interpretation and application.
2. Outcomes reported by allocation.
| Study Name | Comparison |
Analysis Group |
Pts | ORR | PFS | OS | QOL | Other |
| Atkins 2004 | Temsirolimus (3 doses) | 1 | 111 | yes | yes | yes | no | |
| Bhargava 2010 | Tivozanib vs Placebo (RDT) | 2 | 272 | no | yes | no | no | 12‐week PFS |
| Bracarda 2010 | IFNa+Sorafenib (2 regimens) | 1 | 101 | yes | yes | no | no | |
| Bukowski 2007 | Bevacizumab + Erlotinib | 4 | 104 | yes | yes | yes | yes | |
| Ebbinghaus 2007 | ABT‐510 (2 doses) | 1 | 103 | yes | yes | yes | no | |
| Escudier(1) 2007 | AE‐941 vs Placebo | 2 | 305 | no | no | yes | no | |
| Escudier(2) 2010 | Sorafenib vs Placebo | 2 | 903 | yes | yes | yes | yes | |
| Escudier(3) 2010 | IFNa+Bev vs IFNa+Placebo | 2 | 649 | yes | yes | yes | no | |
| Escudier(4) 2009 | Sorafenib vs IFNa | 3 | 189 | yes | yes | no | yes | |
| Escudier(5) 2010 | Tem+Bev vs IFNa+Bev vs Sunit | 4 | 171 | yes | yes | no | no | 48‐week PFS |
| Gordon 2004 | IFNa + Thalidomide | 3 | 342 | yes | yes | yes | yes | |
| Hudes 2010 | Temsirolimus vs IFNa vs both | 3,4 | 626 | yes | yes | yes | yes | |
| Jonasch 2010 | Sorafenib + IFNa | 4 | 80 | yes | yes | yes | no | biomarker |
| Lee 2006 | Thalidomide vs Hormone | 2 | 60 | yes | yes | yes | no | |
| Madhusudan 2004 | IFNa + Thalidomide | 3 | 34 | yes | no | no | no | |
| Motzer(1) 2010 | Sunitinib vs IFNa | 3 | 750 | yes | yes | yes | yes | |
| Motzer(2) 2010 | Everolimus vs Placebo | 2 | 410 | yes | yes | yes | yes | |
| Procopio 2010 | Sorafenib + IL2 | 4 | 128 | yes | yes | no | no | |
| Ratain 2006 | Sorafenib vs Placebo (RDT) | 2 | 65 | no | yes | no | no | |
| Ravaud 2008 | Lapatinib vs Hormone | 2 | 416 | yes | yes | yes | no | |
| Rini 2010 | IFNa + Bevacizumab | 3 | 732 | yes | yes | yes | no | |
| Srinivas 2005 | Thalidomide (2 doses) | 1 | 14 | yes | no | yes | no | |
| Stadler 2005 | CAI vs Placebo (RDT) | 2 | 368 | no | yes | no | no | |
| Sternberg 2010 | Pazopanib vs Placebo | 2 | 435 | yes | yes | interim | yes | |
| Yang 2003 | Bevacizumab (2 doses) vs Placebo | 1,2 | 116 | yes | yes | yes | no |
Groups 1 to 4 results: see Tables 6‐9; primary outcome(s) are in bold;
RDT = randomized discontinuation trial; pts = total patients randomized; ORR = objective remission rate; PFS = progression‐free survival; OS = overall survival; QOL = formal quality‐of‐life assessment
Patient and disease characteristics
(see table 'Characteristics of included studies')
All studies included adult men and women in approximately the ratio expected for renal cancer (2:1), and age range was broad. Patients with brain metastases were usually excluded. Most studies restricted entry to patients with ambulatory performance status, and other prognostic factors were in approximately the expected distribution, with the exception of one study specifically designed for patients with poor prognosis (Hudes 2010). The majority of patients had undergone prior nephrectomy and some studies required this. With regard to disease profile, histology was restricted to renal cancers with a clear‐cell component in most studies of anti‐angiogenic agents. In the absence of routine gene profiling of primary cancers, histologic subtype represented a practical way of selecting patients with cancers likely to have VHL gene inactivation, resulting in constitutive activity of the hypoxia‐inducible pathway, including angiogenesis promoting factors. Some studies required measurable disease: this may select for a less generalizable patient group but is otherwise unlikely to be an important study distinction because major remissions were generally infrequent and of questionable clinical relevance. Extent of disease, directly determined or as reflected by patient performance status and laboratory correlates, is the main determinant of prognosis and was frequently used for stratification purposes or during analysis. The Memorial Sloan‐Kettering Cancer Center (MSKCC) prognostic risk stratification system (Motzer 1999*; Motzer 2002*; Leibovich 2005*) is most commonly used and was based on patients treated with cytokines. A similar system is now validated in patients treated with targeted agents with the addition of elevated neutrophil or platelet counts as adverse factors (Heng 2009*). The number of involved organ sites may provide additional prognostic power and has been incorporated into systems developed at the Cleveland Clinic in the USA and by the French collaborative Groupe Francais d'Immunotherapie (Mekhail 2005*; Negrier 2005*). These variations require care in the application of study results and in making cross‐study comparisons.
The extent of prior systemic treatment was a key factor in large Phase III studies designed to establish a new standard of care. The three major categories were (a) systemically untreated, (b) second‐line after cytokine therapy, and (c) second‐line after targeted therapy. The approach taken in this review considered the validated patient population (in 'Discussion') should a new intervention be shown superior to control (in 'Results'). In some placebo‐controlled studies of new agents designed to assess activity, mixed populations of untreated and pre‐treated patients have been examined (Sternberg 2010; Bhargava 2010).
Targeted agents tested
A total of 13 targeted agents have been tested in the included studies ('Table 5'). Nine agents were considered to have anti‐angiogenic action, of special interest in renal cell cancer, especially of the clear‐cell histologic subtype. Of these anti‐angiogenic agents, five have known specific targets (bevacizumab binds extracellular VEGF; sorafenib, sunitinib, pazopanib and tivozanib inhibit VEGF cell surface receptor tyrosine kinases with or without additional targets), and four have undefined or putative anti‐angiogenic action (thalidomide, AE‐941, carboxyaminoimidazole CAI, ABT‐510). The VEGFR (vascular endothelial growth factor receptor) tyrosine kinase inhibitors vary somewhat in their spectrum of action against the three VEGF receptor subtypes and against additional targets (Chow 2007*). Two other agents target EGFR (epidermal growth factor receptor inhibitors, lapatinib and erlotinib), and two inhibit mTOR (temsirolimus, everolimus). A recent review of kinase targets in RCC is available (Furge 2010*).
3. Targeted agents in Included studies.
| Generic Name | Code Name | Trade Name | Company | Route | Group | Class | Primary Target | Secondary Target(s) | Studies |
| bevacizumab | Avastin® | Genentech | iv | anti‐angiogenic | humanized monoclonal antibody | VEGF | Bukowski 2007; Escudier(3) 2010; Escudier(5) 2010; Rini 2010; Yang 2003 | ||
| sunitinib | Sutent® | Pfizer | oral | anti‐angiogenic | tyrosine kinase inhibitor | VEGF receptor | PDGFR, c‐kit, flt3 | Escudier(5) 2010; Motzer(1) 2010 | |
| sorafenib | Nexavar® | Bayer | oral | anti‐angiogenic | tyrosine kinase inhibitor | VEGF receptor | PDGFR, Raf kinase | Bracarda 2010;Escudier(2) 2010; Escudier(6) 2009; Jonasch 2010; Procopio 2010; Ratain 2006 | |
| pazopanib | GW786034 | Votrient® | GSK | oral | anti‐angiogenic | tyrosine kinase inhibitor | VEGF receptor | PDGFR, c‐kit | Sternberg 2010 |
| tivozanib | AV‐951 | Aveo | oral | anti‐angiogenic | tyrosine kinase inhibitor | VEGF receptor | Bhargava 2010 | ||
| thalidomide | Thalomid® | Celegene | oral | anti‐angiogenic | small synthetic molecule | not defined | Gordon 2004; Lee 2006; Madhusudan 2004; Srinivas 2005 | ||
| AE‐941 | Neovastat® | Aeterna | oral | anti‐angiogenic | shark cartilage derived | not defined | Escudier(1) 2007 | ||
| ABT‐510 | ‐ | Abbott | sc | anti‐angiogenic | thrombospondin‐like | not defined | Ebbinghaus 2007 | ||
| carboxyaminoimidazole | CAI | ‐ | ‐ | oral | anti‐angiogenic | small synthetic molecule | not defined | Stadler 2005 | |
| lapatinib | GW572016 | Tykerb® | GlaxoSmithKline | oral | anti‐growth factor | tyrosine kinase inhibitor | EGF receptor | Her2 | Ravaud 2008 |
| erlotinib | OSI774 | Tarceva® | Roche | oral | anti‐growth factor | tyrosine kinase inhibitor | EGF receptor | Bukowski 2007 | |
| temsirolimus | CCI‐779 | Torisel® | Wyeth | iv | mTOR inhibitor | macrolide antibiotic | mammalian target of rapamycin | Atkins 2004; Escudier(5) 2010; Hudes 2010 | |
| everolimus | RAD001 | Afinitor® | Novartis | po | mTOR inhibitor | macrolide antibiotic | mammalian target of rapamycin | Motzer(2) 2010 |
Studies listed by target and accrual order. VEGF = vascular endothelial growth factor; EGF = epidermal growth factor
Risk of bias in included studies
In general, the methodologic quality of studies in oncology often cannot be completely assessed by standard CONSORT (Consolidated Standards of Reporting Trials) criteria (Moher 2001*), such as the mechanism by which allocation concealment is achieved, since this level of detail is infrequently provided in published reports. To some extent it must be taken on trust that multicentre trials have unbiased central randomization procedures and data repositories. Close data monitoring is to be expected to ensure that protocol procedures are rigorously followed that will satisfy regulatory authorities. The large Phase III studies providing the most interesting results in the following analysis had independent data and safety monitoring committees and/or submitted their outcome data for blinded review by observers unaware of the treatment allocation of each patient (see tables 'Characteristics of included studies' and 'Table 1'). In addition, eight studies included a placebo component. Since the first edition of this review concerning eligible studies available to the end of 2007, most included studies previously available only from preliminary meeting reports have now been fully published in peer‐reviewed journals with substantially improved level of methodologic detail.
Summary of findings for the main comparison. Phase III studies of Targeted Therapy versus Standard of Care.
| Study Name | Prior Drug Therapy | Comparison | Randomization and Allocation Issues |
PBO Control |
BIA |
Remission Odds Ratio |
Progression Hazard Ratio |
Mortality Hazard Ratio |
QOL |
| Rini 2010 | none | Bevacizumab+IFN vs IFN | None | No | No | 2.28* | 0.71* | 0.86# | nr |
| Escudier(3) 2010 | none | Bevacizumab+IFN vs PBO+IFN | None | Yes | Yes | 3.11 | 0.61 | 0.86# | nr |
| Motzer(1) 2010 | none | Sunitinib vs IFN | None | No | Yes | 6.34 | 0.54 | 0.82 | * |
| Hudes 2010 | none | Temsirolimus vs IFN | None | No | Yes | ns | nr | 0.73 | * |
| Escudier(2) 2010 | cytokine | Sorafenib vs PBO | None | Yes | Yes | ns | 0.44 | ns | +ve |
| Motzer(2) 2010 | anti‐VEGFR | Everolimus vs PBO | None | Yes | Yes | ns | 0.30 | ns | ns |
Only statistically significant results are shown numerically, all favour investigational arms; ns = not statistically significant; IFN = interferon‐alfa; PBO = placebo; nr = not reported; QOL = formal quality‐of‐life evaluation; BIA = blinded imaging assessment (for response and progression). The primary outcome for each study is indicated in bold type.
Hudes 2004 restricted to patients with 3+ adverse factors, all histologies; all other studies restricted to clear‐cell subtype
*= risk of bias because the outcome was observed by investigators or patients in an open‐label study; #= consistent and significant when considered together
Another measure of study quality is study size as surrogate for power to detect differences. The number of patients per arm in ascending order was as follows: 7, 17, 30, 32, 37, 38, 40, 50, 51, 52, 64, 85, 94, 136, 152, 171, 184, 205, 208, 208, 217, 324, 366, 375, 451. Studies at or below the median of 94 patients per arm would have low power, and little can be said of negative small studies.
Risk of bias tables were constructed for each study and tabulated in 'Table 2'. In many fields, the risk of bias was unclear because of failure to provide sufficient detail. Study outcomes were considered at high risk of bias if they were open label and assessed by investigators (remission or progression) and/or by patients (toxicity or formal quality‐of‐life assessment), except that overall survival was considered reliable. A further concern was involvement of an industry sponsor in the randomization process, data management, or publication of reports, risk of bias listed as "unclear" in these circumstances. We did not find the GRADEpro software suitable for this review where the studies were mostly of different comparisons and important outcomes were time‐dependent.
Summary of findings 2. Study quality (from risk of bias tables for individual studies).
|
Allocation Sequence |
Randomization Concealment |
Blinding (objective outcomes) |
Blinding (QOL outcomes) |
Incomplete Data (objective outcomes) |
Incomplete Data (QOL outcomes) |
Selective Reporting |
Other Risks | |
| Atkins 2004 | ? | ? | X | √ | √ | ? | ||
| Bhargava 2010 | ? | ? | √ | ? | ? | ? | ||
| Bracarda 2010 | ? | ? | X | ? | ? | ? | ||
| Bukowski 2007 | ? | √ | √ | ? | √ | ? | √ | ? |
| Ebbinghaus 2007 | ? | ? | X | ? | √ | ? | ||
| Escudier(1) 2007 | ? | ? | √ | √ | √ | ? | ||
| Escudier(2) 2010 | √ | ? | √ | ? | √ | √ | √ | ? |
| Escudier(3) 2010 | √ | √ | √ | √ | √ | ? | ||
| Escudier(4) 2009 | √ | ? | √ | X | ? | ? | √ | ? |
| Escudier(5) 2010 | √ | ? | X | ? | ? | ? | ||
| Gordon 2004 | √ | √ | ? | ? | ? | ? | √ | √ |
| Hudes 2010 | ? | ? | √ | X | √ | √ | √ | ? |
| Jonasch 2010 | √ | √ | √ | √ | √ | √ | ||
| Lee 2006 | ? | ? | X | √ | √ | √ | ||
| Madhusudan 2004 | ? | ? | X | ? | ? | ? | ||
| Motzer(1) 2010 | ? | ? | √ | X | √ | ? | √ | ? |
| Motzer(2) 2010 | ? | ? | √ | √ | √ | ? | √ | ? |
| Procopio 2010 | ? | ? | X | ? | ? | ? | ||
| Ratain 2006 | √ | √ | √ | √ | √ | ? | ||
| Ravaud 2008 | √ | √ | √ | √ | √ | ? | ||
| Rini 2010 | √ | √ | ? | √ | √ | √ | ||
| Srinivas 2005 | ? | ? | ? | √ | ? | ? | ||
| Stadler 2005 | √ | √ | √ | ? | √ | √ | ||
| Sternberg 2010 | √ | √ | √ | √ | √ | √ | √ | ? |
| Yang 2003 | √ | ? | √ | √ | √ | √ |
√ = low risk of bias; ? = unclear risk of bias; X = high risk of bias
Effects of interventions
Approach to detailed comparisons
The overall intent was to be able to divide studies into meaningful groups that would easily incorporate future studies as they are reported. After consideration of alternatives, such as grouping by type of agent or by prior therapy, we have grouped studies based on the type of control used. Detailed outcome data are provided in 'Table 6' (Overall survival), 'Table 7' (Progression‐free survival), and 'Analysis 1.1' (Major objective remission rate). Summarized outcome data are provided for study Groups 1 to 4 in 'Additional tables' 6 to 9, respectively.
4. Overall survival (OS).
| Study Name | "Arm 1" Median OS | "Arm 2" Median OS | Third Arm Median OS |
Author HR (95%CI) |
Author P value | Comment |
| Atkins 2004(A) | 17.5 months | 11.0 months | 13.8 months | 0.66 | ||
| Bhargava 2010 | randomized discontinuation trial | |||||
| Bracarda 2010 | not reported | |||||
| Bukowski 2007 | 20 months | not reached | 0.16 | interim analysis | ||
| Ebbinghaus 2007 | 26.1 months | 27.8 months | 0.59 | |||
| Escudier(1) 2007 | 12.5 months | 12.4 months | 0.51 | |||
| Escudier(2) 2010 | 17.8 months | 15.2 months | 0.88 (.74‐1.04) |
0.15 | see text for secondary analysis | |
| Escudier(3) 2010 | 23.3 months | 21.3 months | 0.86 (.72‐1.04) |
0.13 (stratified) | see also Rini 2010 see text for results for cross over and reduced‐dose patients |
|
| Escudier(4) 2009 | not reported | |||||
| Escudier(5) 2010 | not reported | |||||
| Gordon 2004 | 10.8 months | 13.1 months | 0.88 | |||
| Hudes 2010(A) | 10.9 months | 7.3 months | 0.73 (.58‐.92) |
0.008 | ||
| Jonasch 2010 | 27.04 months | not reached | 1.94 .84‐4.52 2.172 .92‐5.12 |
0.1219 (univariate) 0.0764 (multivariate) |
||
| Lee 2006 | 8.2 months | 4.8 months | 0.88 (.67‐1.94) |
0.62 | ||
| Madhusudan 2004 | not reported | |||||
| Motzer(1) 2010 | 26.4 months | 21.8 months | 0.82 (.67‐1.00) |
0.049 (stratified) |
||
| Motzer(2) 2010 | 14.8 months | 14.4 months | 0.87 (.65‐1.17) |
0.18 | update from Hutson 2009 see text for x‐over analysis |
|
| Procopio 2010 | not reported | |||||
| Ratain 2006 | randomized discontinuation trial | |||||
| Ravaud 2008 | 46.9 weeks | 43.1 weeks | 0.88 (.69‐1.12) |
0.29 | see also Escudier(3) 2010 see text for EGFR 3+ subset |
|
| Rini 2010 | 18.3 months | 17.4 months | 0.86 (.73‐1.01) |
0.069 (stratified) |
||
| Srinivas 2005 | 6 months | 16 months | not available | 0.04 | 14 pts total | |
| Stadler 2005 | randomized discontinuation trial | |||||
| Sternberg 2010 | interim analysis | |||||
| Yang 2003(A) | not reported | not reported | > 0.2 |
All P values are base on the log‐rank test. HR = hazard ratio; CI = confidence interval
5. Progression‐free survival (PFS).
| Study Name | "Arm 1" median PFS | "Arm 2" median PFS | Third Arm median PFS |
Author HR (95%CI) |
Author P value | Comment |
| Atkins 2004 | 5.2 months | 6.7 months | 6.3 months | 0.93 | ||
| Bhargava 2010 | not reached | 2.1 months | 0.006 | randomized discontinuation trial | ||
| Bracarda 2010 | 8.6 months | 7.9 months | 0.049 | |||
| Bukowski 2007 | 9.9 months | 8.5 months | 0.58 | |||
| Ebbinghaus 2007 | 3.3 months | 4.2 months | 0.80 | |||
| Escudier(1) 2007 | not reported | |||||
| Escudier(2) 2010 | 5.5 months | 2.8 months | 0.44 (.35‐.55) |
<0.000001 | ||
| Escudier(3) 2010 | 10.2 months | 5.4 months | 0.61 (0.51‐0.73) |
<0.0001 stratified | ||
| Escudier(4) 2009 | 5.7 months | 5.6 months | 0.88 (.61‐1.27) |
0.50 | independent assessment | |
| Escudier(5) 2010 | 8.2 months | 16.8 months | 8.2 months | arms not compared | ||
| Gordon 2004 | 2.8 months | 2.8 months | 0.33 (stratified) | |||
| Hudes 2010(A) | 3.8 months | 1.9 months | not reported | P<0.05 based on non‐overlapping 95% CIs |
investigator assessed | |
| Jonasch 2010 | 7.56 months | 7.39 months | 0.85 (.51‐1.42) |
0.53 (univariate & multivariate) | ||
| Lee 2006 | 2.4 months | 2.8 months | 0.89 | time to failure | ||
| Madhusudan 2004 | not reported | |||||
| Motzer(1) 2010 | 11 months | 5 months | 0.54 (.45‐.64) |
<0.001 | central review | |
| Motzer(2) 2010 | 4.0 months | 1.9 months | 0.30 (.22‐.40) |
<0.0001 | central review | |
| Procopio 2010 | 8.8 months | 6.9 months | 0.22 | |||
| Ratain 2006 | 5.5 months | 1.4 months | 0.0087 | randomized discontinuation trial | ||
| Ravaud 2008 | 3.6 months | 3.6 months | 0.94 (.75‐1.18) |
0.60 | time to progression | |
| Rini 2010 | 8.5 months | 5.2 months | 0.71 (.61‐.83) |
<0.0001 | investigator assessed | |
| Srinivas 2005 | too small to analyze | |||||
| Stadler 2005 | 2.8 months | 4.2 months | 'no difference' | randomized discontinuation trial | ||
| Sternberg 2010 | 9.2 months | 4.2 months | 0.46 (.34‐.62) |
<0.0001 | ||
| Yang 2003 | 4.8 months | 3.0 months | 2.5 months | 0.39 (95%CI n/a) |
<0.001 | arm 1 vs 3 |
All P values are base on the log‐rank test. HR = hazard ratio; CI = confidence interval
1.1. Analysis.
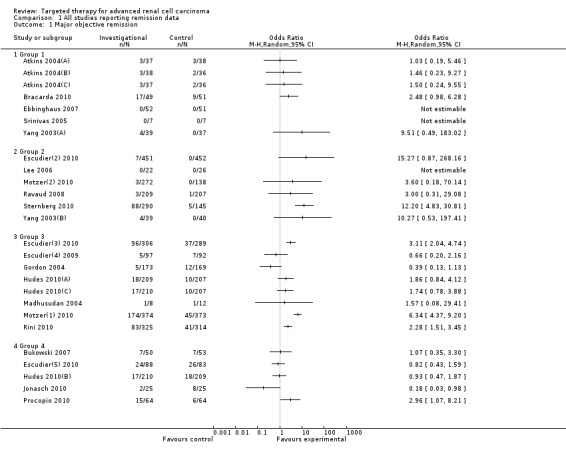
Comparison 1 All studies reporting remission data, Outcome 1 Major objective remission.
Group 1: Same agents in investigational and control arms
Dose‐ and schedule‐finding studies (5 studies, 'Table 6')
Included studies/comparisons: Atkins 2004 (temsirolimus, 3 dose levels); Bracarda 2010 (sorafenib/interferon); Ebbinghaus 2007 (ABT‐510); Yang 2003(A) (bevacizumab); Srinivas 2005 (thalidomide).
This group is presented first as any differences in outcome for the same agent at different dose (or schedule) would influence the interpretation of other studies of that agent. Unlike cytotoxic agents, where the maximum tolerated dose is usually utilized, targeted agents are most rationally used at the lowest dose that reliably blocks the target. In some instances this dose may be determined in vivo as has been done for the mTOR inhibitors temsirolimus (Harding 2003*) and everolimus (Tanaka 2008*). In general an empirical approach to dose optimization is required. Except for bevacizumab dose based on body weight, targeted agents are given initially at fixed dose not adjusted for body size, then adjusted downward for toxicity if necessary. There is emerging evidence for a dose‐response relationship for the VEGFR inhibitors sunitinib (Houk 2010*) and sorafenib (Escudier(4) 2009). Dose titration to tolerance in each patient has been carried out in non‐randomized studies (Amato 2007a*; Rini 2009*) but not in the randomized studies reviewed here.
Bevacizumab is a humanized monoclonal antibody that binds circulating VEGF often found in high levels in patients with clear cell renal cancer. In a three arm placebo‐controlled study, biweekly intravenous bevacizumab was tested at two dose levels: 3 mg/kg (milligrams per kilogram) and 10 mg/kg (Yang 2003). The trial was stopped after accrual of 116 patients when a planned interim analysis showed the higher dose bevacizumab arm to be superior to placebo. Cox modelling gave a time‐to‐progression ratio of 2.55 for placebo versus high dose bevacizumab (P < 0.001) but only 1.26 versus the low‐dose arm (P = 0.053). The median PFS was longer for bevacizumab 10 mg/kg than 3 mg/kg (Yang 2003(A)), at 4.8 versus 3.0 months, respectively, but the authors do not provide statistics for this comparison.
Srinivas 2005 (14 patients total) was considered too small for further consideration. One study compared two schedules of interferon along with standard sorafenib 400 mg twice daily: as such, it was of interest with regard to optimizing subcutaneous interferon dose/schedule and suggested an advantage to treating more frequently despite lower weekly dose (3 MU three times per week compared to the usual 9 MU three times per week, median PFS 8.6 versus 7.9 months, respectively (P = 0.049), and trends to higher objective response rate and reduced incidence of fatigue) (Bracarda 2010). The remaining two studies in Group 1 did not demonstrate significant differences in generally low remission rates for different doses of the same drug, in progression‐free survival, or overall survival (where reported). These studies are small with low power to detect outcome differences.
Conclusions from Group 1 Dose/Schedule Studies: 1. Bevacizumab 10 mg/kg given intravenously on alternate weeks has been validated as the dose and schedule taken forward for additional study. 2. When combined with a targeted agent, interferon may be more effective if given more frequently. 3. The additional agents tested in this group did not demonstrate efficacy differences in the evaluated outcomes (see 'Table 8'); possible reasons for this lack of difference include low study power, lack of efficacy, or lack of a dose‐response relation between the doses tested. ABT‐510 has not been further evaluated. Temsirolimus, despite low remission rates across a 10‐fold dose range, has subsequently shown efficacy at the lowest dose utilized here (Hudes 2010, see 'Group 3' below).
6. Group 1 outcomes summary: dose/schedule studies.
| Agent | Study | ORR | PFS | OS |
| Bevacizumab 10 vs 3 mg/kg | Yang 2003(A) | ns | +ve | ns |
| Thalidomide high vs low dose | Srinivas 2005 | ns | ‐ve* | |
| Temsirolimus (3 dose levels) |
Atkins 2004 | ns | ns | ns |
| ABT‐510 100 vs 10 mg | Ebbinghaus 2007 | ns | ns | ns |
| Sorafenib/IFN (2 schedules) | Bracarda 2010 | ns | +ve |
ns = not statistically different; +ve = statistically favours investigational arm; ‐ve = statistically favours control arm
* very small study suggesting detrimental effect of high dose thalidomide
Group 2: Targeted agent versus inert control
(10 studies, 'Table 9')
7. Group 2 Outcomes Summary: Targeted Agent vs Placebo/Hormone Control.
| Agent | Study | ORR | PFS | OS | QOL |
| Bevacizumab | Yang 2003(B) | ns | +ve | ns | |
| CAI | Stadler 2005 | ns | |||
| AE‐941 | Escudier(1) 2007 | ns | |||
| Thalidomide | Lee 2006 | ns | ns | ns | |
| Lapatinib | Ravaud 2008 | ns | ns | ||
| Sorafenib | Ratain 2006 | +ve | |||
| Sorafenib | Escudier(2) 2010 | ns | +ve | ns | +ve* |
| Everolimus | Motzer(2) 2010 | ns | +ve | ns | ns |
| Pazopanib | Sternberg 2010 | +ve | +ve | ns | |
| Tivozanib | Bhargava 2010 | +ve |
ns = not statistically different; +ve = statistically favours investigational arm; *= placebo‐controlled
(Inert control = placebo or hormone)
Included studies/comparisons: Bhargava 2010 (tivozanib); Escudier(1) 2007 (AE‐941); Escudier(2) 2010 (sorafenib); Lee 2006 (thalidomide); Motzer(2) 2010 (everolimus); Ratain 2006 (sorafenib); Ravaud 2008 (lapatinib); Stadler 2005 (carboxyaminoimidazole); Sternberg 2010 (pazopanib); Yang 2003(B) (bevacizumab).
Nine different targeted single agents have been compared to placebo (8 studies) or hormone control (2 studies). Hormone therapy has no demonstrated objective efficacy and is considered equivalent to placebo. Additional Table 7 lists these studies in approximate accrual order with an efficacy summary. The majority of these studies have been in patients previously treated with a cytokine before this approach was displaced from first‐line by targeted therapy; two recent studies are with new VEGFR inhibitors pazopanib (Sternberg 2010) and tivozanib (Bhargava 2010). Patient selection has increasingly focused on the clear‐cell subtype of renal cancer, especially for treatment with VEGFR inhibitors. Placebo controlled trials are particularly useful for documentation of adverse effects and impact on quality‐of‐life of new agents, but do not permit reliable efficacy comparisons of different agents.
a) Randomized discontinuation trial design (3 studies)
Three studies used a randomized discontinuation trial (RDT) design intended to detect objective stabilization of disease should the agent be cytostatic rather than cytoreductive. After a 12‐to‐16 week run‐in period on the active agent, patients with stable disease were randomly assigned to placebo or to continue the active agent (responding patients continue active therapy). These RDT studies are discussed separately because this strategy is somewhat different than the classic RCT design, and may be attractive for recruitment of untreated patients in future studies since all patients begin with active drug on the open‐label initial phase.
Stadler 2005 (carboxyaminoimidazole, CAI) and Ratain 2006 (sorafenib) from the University of Chicago Medical Center successfully implemented the RDT approach. The majority of patients had received prior systemic therapy with cytokines. The outcome of interest is progression‐free survival. CAI was inactive by this criterion; Bayesian probability of greater disease stability was less than 9% (Stadler 2005). Sorafenib was superior to placebo with a median progression‐free period of 5.5 months on continuing therapy versus 1.4 months for placebo, P = 0.0087; placebo patients went back on sorafenib at progression (Ratain 2006) potentially confounding any further outcome differences. Sorafenib was further evaluated in a large placebo‐controlled study using conventional randomization discussed below (Escudier(2) 2010).
Tivozanib, a new small molecule oral inhibitor of VEGFR, has also been assessed by the RDT technique (Bhargava 2010). Tivozanib inhibits all three VEGFR kinases. In a study of 272 patients, of whom half had no prior systemic therapy, tivozanib was initially given open label for a run‐in period of 16 weeks: 25% of patients demonstrated at least 25% tumour shrinkage by independent review, 8% progressed, and the remainder were stable. One hundred eleven consenting patients with stable disease were then randomized to continue tivozanib 1.5 mg daily or to placebo. Progression‐free survival at a protocol‐specified further 12 weeks was better on continued tivozanib than on placebo (59% versus 38%, P = 0.029). The common toxicities were hypertension (54%, all grades) and dysphonia (21%). Information is not provided as to whether prior systemic therapy influenced the chance of benefit. A Phase III comparison of tivozanib with sorafenib is in progress in the first‐line setting (NCT01030783).
b) Standard randomized controlled trial design (7 studies)
Agents are presented in the approximate order that included studies commenced accrual.
Bevacizumab
In the bevacizumab dose‐finding study mentioned in 'Results' 'Group 1', progression‐free survival was significantly better for bevacizumab 10 mg/kg than for placebo, median PFS 4.8 versus 2.5 months, respectively (hazard ratio (HR) 0.39, P < 0.001) (Yang 2003(B)), but supporting clinical outcome data such as symptom improvement was not provided. The combination of bevacizumab and interferon alfa is discussed under 'Group 3' studies.
Thalidomide
Thalidomide was not beneficial compared to hormone therapy (Lee 2006) and had substantial toxicity.
AE‐941
A large placebo‐controlled study of AE‐941, a derivative of shark cartilage, did not improve the primary outcome of overall survival (Escudier(1) 2007).
Lapatinib
Lapatinib is an oral inhibitor of EGFR (ErbB1) and HER‐2 (ErbB2). Lapatinib 1250 mg daily did not improve progression‐free or overall survival compared to hormone control on an intent‐to‐treat basis (Ravaud 2008). Remissions were rare. However a pre‐planned subset analysis examined outcomes in the 56% of patients with tumours strongly overexpressing the main target for this tumour type ErbB1 (3+ by IHC), a factor that was shown in exploratory analysis to be both prognostic (adverse) and predictive for benefit. Progression‐free survival showed borderline improvement for lapatinib in this subset (HR 0.76, P = 0.06), while median overall survival was increased from 8.7 months for hormone to 10.6 months for lapatinib (HR 0.66, P = 0.012), consistent with improved survival in the ErbB1 3+ setting. The main toxicities of lapatinib were diarrhea and rash (40% and 44% respectively, all grades).
Sorafenib
Sorafenib is an oral small molecule inhibitor of VEGF receptor tyrosine kinase, a key locus in the angiogenesis pathway. A pivotal study of second‐line sorafenib after cytokine failure has been extensively analyzed (see Escudier(2) 2010 references). Sorafenib 400 mg twice daily doubled median progression‐free survival versus placebo (Escudier(2) 2010), 5.5 versus 2.8 months, respectively, as assessed by independent review (HR 0.44, 95% CI 0.35 to 0.55, P < 0.000001). Subset analysis suggested a similar PFS benefit in different subgroups, including patients over 65 years of age without greater apparent toxicity (Eisen 2008*). There may also be a reduced incidence of brain metastases, an uncommon but devastating complication of the disease (3% and 12% for sorafenib and placebo respectively, P < 0.05, Massard 2006*). Most types of severe adverse events grades 3 or 4 were more common on the sorafenib arm than placebo, including cardiac ischemia or hypertension (4% versus < 1%), diarrhea (2% versus 1%), or hand‐foot syndrome (6% versus 0%), though bone pain was more common on placebo. Grade 1 to 2 adverse events were common but their clinical impact is better evaluated by patient self‐reporting: the authors documented symptomatic improvement of sorafenib over placebo on formal quality‐of‐life assessment with validated QOL (quality of life) measures FACT‐G and FKSI (Bukowski 2007*). Skeletal muscle wasting with sorafenib has been reported and sarcopenia may be a feature of multi kinase inhibitors (Antoun 2010*).
The study was closed to accrual following a planned interim analysis because PFS was clearly better for the sorafenib arm. There was an initial trend to improved overall survival at study closure (HR 0.71, P = 0.015) but this failed to reach the prespecified boundary and has disappeared with further follow up (Escudier 2009*). There was no significant difference for the ITT (intention to treat) final overall survival analysis: median OS was 17.8 months for sorafenib versus 15.2 months for placebo (HR 0.88, P = 0.15). At study closure, surviving placebo‐assigned patients were offered crossover to sorafenib, and 48% of all placebo‐assigned patients did so. In an attempt to assess the impact of crossover on overall survival, a pre‐planned secondary survival analysis that censored placebo patients at the time of crossover observed a difference of overall survival (median 17.8 versus 14.3 months, HR 0.78, 95% CI 0.62 to 0.97, P = 0.029, O'Brien‐Fleming boundary P = 0.037). However, placebo‐assigned patients who were censored at crossover to sorafenib were different from those who did not cross over and were not censored, being nearly twice as likely to be ECOG 0 or low MSKCC risk at baseline, and this censoring difference might account for the observed survival difference rather than a salvage effect of crossover. In this regard, the uncensored survival trend of the interim survival analysis is more suggestive of a crossover effect but the hypothesis remains unproven.
Everolimus
Following the publication of evidence for superior outcomes with sunitinib or sorafenib over first‐line interferon alfa (discussed in 'Group 3' below), the need for a new second line of therapy became evident. Everolimus is an oral inhibitor of mTOR (see also temsirolimus below) (Coppin 2010*) and therefore has a different mechanism of action than VEGFR inhibitors with potential for non‐crossresistance. Following encouraging non‐randomized studies in this setting, everolimus was compared to placebo in 410 heavily pretreated ambulatory patients with disease progression on or within 6 months of sunitinib and/or sorafenib (Motzer(2) 2010). The primary endpoint of progression‐free survival was improved from a median 1.9 months for placebo to 4.0 months for everolimus (HR = 0.30, P < 0.0001), with an associated 2‐month delay in decline of performance status and no detriment to overall quality‐of‐life from toxicity. The probability of remaining progression‐free at 10 months on study was 25% on everolimus versus < 2% for placebo. The remission rate was very low. The main concerns with this agent are reversible immunosuppression, non‐infectious pneumonitis, and hyperglycemia. Overall survival was essentially the same in both arms and at this time there is no generally agreed method to correct for the possible impact of crossover to everolimus of most placebo‐assigned patients at disease progression (see references in Motzer(2) 2010 for examples of techniques used to correct overall survival for crossover).
Pazopanib
Pazopanib is an oral antiangiogenesis multi‐kinase inhibitor targeting VEGFR as well as c‐Kit (also known as mast/stem cell growth factor receptor (SCFR) or CD117) and PDGFR. A substantial study randomized 435 patients to pazopanib 800 mg daily or placebo in a 2:1 ratio; 54% had not received prior systemic therapy and the remainder had prior cytokine therapy (Sternberg 2010). The primary outcome of progression‐free survival was median 9.2 months for pazopanib and 4.2 months for placebo (HR = 0.46, P < 0.0001), and was significantly improved for both the cytokine pretreated and naive subpopulations as well as those with favourable or intermediate prognostic scores. In addition, the major objective remission rate was 30% with pazopanib compared to 3% on placebo (P < 0.001). Unfortunately this benefit did not translate into any improvement in formal quality‐of‐life measurement. Interim overall survival did not cross the prespecified difference threshold and final results are awaited, noting that 48% of placebo‐assigned patients crossed over to pazopanib after progression. A Phase III comparison of pazopanib with sunitinib in the first‐line setting is in progress (NCT00720941).
Conclusions from 'Group 2 placebo‐controlled studies': 1. Nonspecific antiangiogenesis agents AE‐941, thalidomide, and carboxyaminoimidazole have no demonstrated benefit over placebo in these studies. 2. All tested VEGF receptor kinase inhibitors have shown some evidence of benefit compared to placebo and have completed or are undergoing comparative evaluation against other agents in the first‐line setting as single agents (except bevacizumab used in combination with interferon alfa). 3. Lapatinib, an EGFR inhibitor, may have activity against tumours that strongly express the primary target but is not reported to be undergoing further evaluation for advanced renal cancer. 4. An oral mTOR inhibitor, everolimus, can delay disease progression compared to placebo following sunitinib and/or sorafenib failure and is a reasonable comparator for additional agents to be tested in this setting.
Group 3: First‐line targeted agent versus cytokine control
(8 comparisons, 'Table 10')
8. Group 3 Outcomes Summary: Targeted Agent vs Cytokine Control.
| Agent | Study | ORR | PFS | OS | QOL |
| Thalidomide+IFN | Gordon 2004 | ns | ns | ns | ‐ve |
| Thalidomide+IFN | Madhusudan 2004 | ns | |||
| Temsirolimus | Hudes 2010(A) | ns | +ve | +ve | +ve |
| Temsirolimus+IFN | Hudes 2010(C) | ns | ns | ns | |
| Bevacizumab+IFN | Rini 2010 | +ve | +ve | ns | |
| Bevacizumab+IFN | Escudier(3) 2010 | +ve | +ve | ns | |
| Sunitinib | Motzer(1) 2010 | +ve | +ve | +ve | +ve |
| Sorafenib | Escudier(4) 2009 | ns | ns | ns |
ns = not statistically different; +ve = statistically favours investigational arm; ‐ve = statistically favours control arm
Included studies/comparisons (all versus interferon‐alfa): Gordon 2004, Madhusudan 2004 (thalidomide + interferon); Hudes 2010(A) (temsirolimus); Hudes 2010(C) (temsirolimus + interferon); Rini 2010, Escudier(3) 2010 (bevacizumab + interferon); Motzer(1) 2010 (sunitinib); Escudier(4) 2009 (sorafenib).
Interferon‐alfa has been shown to modestly improve overall survival (see Coppin 2006*) and became an accepted standard of care suitable as a first‐line trial control arm in both the USA (Motzer 2002*) and Europe (Mickisch 2003*). This somewhat simplified the situation compared to the previous decade when high dose interleukin‐2 was approved in the USA on the basis of occasional durable remissions in the minority of patients fit enough to receive it. High dose IL‐2 has since been shown to have no impact on median overall survival compared to interferon‐alfa (McDermott 2005*). First‐line targeted studies versus interferon‐alfa provided the main focus of interest until recently when targeted agents were shown superior and became the new first‐line standard in North America and Europe. The following studies have all used the standard dose, route, and schedule of interferon‐alfa 9 MU by subcutaneous injection three times per week. In order to provide some chronological perspective, agents will be presented in the approximate order in which accrual commenced in the related randomized studies.
Thalidomide
This oral agent, withdrawn as a sedative following discovery of teratogenicity in the 1960s, has found a new indication as an anti‐cancer agent, initially for multiple myeloma. Thalidomide is a glutamide derivative that has a broad spectrum of cellular action including an anti‐angiogenic effect of interest in RCC. The drug has dose‐limiting neuropathy requiring very careful monitoring. Thalidomide has been added to first‐line interferon in two RCTs (Gordon 2004; Madhusudan 2004). The large ECOG study (Gordon 2004) escalated thalidomide dose to individual MTD (median 400 mg daily, range < 100 to 1000 mg). There was no significant difference in disease‐related endpoints but the detrimental effect of therapy on global quality‐of‐life (FACT‐G assay) was greater for the combined arm than interferon alone. The other small study, reported in abstract only, did not provide additional insight (Madhusudan 2004).
Temsirolimus
Temsirolimus is metabolized to sirolimus, a macrolide antibiotic and immunosuppressive drug also known as rapamycin and derived from a Streptomyces species. Both analogues are inhibitors of an intracellular kinase called mTOR, resulting in disruption of cell cycle progression as well as inhibition of angiogenesis. A large Phase III study has compared temsirolimus with interferon‐alfa in RCC patients with three or more of six adverse prognostic factors for survival: metastasis‐free interval from diagnosis less than one year, impaired performance status (Karnofsky 60 or 70), metastases to multiple organs, elevated LDH (lactate dehydrogenase), low hemoglobin, or high corrected serum calcium (Hudes 2010). It should be noted that this definition of adverse prognosis is slightly different from the MSKCC poor risk group, and the study included 28% patients with intermediate risk as well as patients with poor risk by MSKCC criteria (Dutcher 2007*). All histologic subtypes of renal cell cancer were eligible. The single‐agent temsirolimus arm used a dose of 25 mg IV weekly, which was the lowest dose previously tested in a dose‐finding study (Atkins 2004; see Group 1 above). Temsirolimus has diverse toxicities but at this dose, severe adverse reactions were more common in the interferon arm (78% interferon patients versus 67% temsirolimus patients experienced one or more grade 3/4 adverse events (P = 0.02). The types of adverse events were different, the main toxicity of interferon being asthenia, whereas rash, edema and stomatitis were more common with temsirolimus. Remissions were infrequent and not statistically different, though minor tumour shrinkage was more commonly seen with temsirolimus (Dutcher 2009*). Progression‐free survival, as determined by the investigators and taking into account symptomatic deterioration, was superior for temsirolimus versus interferon (median 3.8 versus 1.9 months, respectively, non‐overlapping 95% confidence intervals). On the average, the additional time progression‐free is spent without symptoms or toxicity: a Q‐TWiST (quality adjusted time without symptoms or toxicity) analysis observed that the median time without grade 3/4 treatment toxicity or recurrence was 6.5 months for temsirolimus versus 4.7 months for interferon (Zbrozek 2010*). Importantly, overall survival was improved for temsirolimus versus interferon, median 10.9 versus 7.3 months respectively, HR 0.73, 95%CI 0.58‐0.92, P = 0.008. A third trial arm randomized patients to receive the combination of interferon‐alfa plus temsirolimus 15 mg/dose; however, outcomes were not superior to interferon alone and there was greater toxicity with the combination.
An exploratory analysis of this study by histologic subtype has been published (Dutcher 2009*). Clear cell cancers constituted 82% of the total, and the overall survival for this subtype showed a trend in favour of temsirolimus over interferon although did not quite reach statistical significance (HR 0.82, P = 0.06, unstratified Cox model). Patients with cancers having non‐clear cell histology, 18% of the total population, had a statistically better survival with temsirolimus than with interferon (HR 0.49, P < 0.05). Similarly, for progression‐free survival, the benefit of temsirolimus over interferon may be greater for non‐clear cell tumours (HR 0.38) than for clear cell tumours (HR 0.76). These observations are of interest because non‐clear cell histologies have been excluded from most studies of angiogenesis inhibitors considered unlikely to be efficacious against RCC histologies that lack the VHL mutation.
Bevacizumab + Interferon
The demonstration of improved progression‐free survival by bevacizumab in cytokine‐pretreated patients was discussed above (Yang 2003(B)). The effect of the addition of bevacizumab to interferon‐alfa has been examined in a placebo‐controlled study of patients with clear cell‐predominant renal cancer and no prior systemic therapy (Escudier(3) 2010). Using blinded independent radiologic assessment, standard interferon‐alfa plus intravenous bevacizumab 10 mg/kg every two weeks gave a major remission rate of 31% compared with 13% with interferon plus placebo, P < 0.0001 (OR (odds ratio) 3.1, 95% CI 2.0 to 4.7). Of time‐dependent outcomes, overall survival was originally selected as the primary endpoint but with the new availability of second‐line angiogenesis inhibitors that might obscure the result, the protocol was modified to permit final analysis of PFS as a secondary endpoint before overall survival was mature. Median progression‐free survival with the addition of bevacizumab to interferon was nearly doubled from 5.4 to 10.2 months (final progression analysis, stratified HR 0.61, 95% CI 0.51 to 0.73, P < 0.0001 log‐rank). As expected, toxicity was greater for patients on the combination arm and 28% of patients had to discontinue a component of combination therapy versus 12% for interferon plus placebo. Bevacizumab‐related toxicities (grade 3/4) were as expected, including proteinuria (5%), hypertension (2%), grade 3+ bleeding (3%), grade 3+ thromboembolism (2%), and GI perforation (1%); death, possibly related to bevacizumab toxicity, was <1%. Interferon‐related toxicities were 10% higher on the combination arm. Recently the final analysis of the primary endpoint of overall survival has been fully published. The stratified HR was 0.86 in favour of the combination but not reaching statistical significance (P = 0.13). The majority of patients received post protocol systemic therapy, and over one third were treated with sunitinib and/or sorafenib that became available during the study, possibly confounding a more substantial survival benefit of the first‐line therapy. Quality‐of‐life evaluation was incorporated into the study and will be of special interest in this placebo‐controlled design, but our literature search has not yet identified a report of QOL findings.
A second mature study of interferon with or without bevacizumab at standard dose and schedule has been completed (Rini 2010). This study was not placebo‐controlled and did not include blinded assessment but otherwise was of very similar design and quality to the European‐based study discussed above. Results are here presented in comparison to Escudier(3) 2010 noting that this is the only regimen in this review tested in more than one large trial. Accrual was similar (Rini: 732 patients accrued from 10/2003 to 7/2005; Escudier: 649 patients accrued from 6/2004 to 10/2005). Objective remissions occurred significantly more often in the combination arm compared to the single agent interferon arm (Rini 25.5% versus 13.1%; Escudier 31% versus 13%). The progression‐free survival was also significantly superior for the combination in both studies (Rini: HR 0.71; Escudier: HR 0.61) and overall survival showed an identical trend (Rini: stratified HR 0.86. Escudier: stratified HR 0.86). A meta‐analysis of these two studies would be feasible: since the P value for overall survival with the Rini study alone is 0.069, the combined data would be expected to show a statistically significant survival benefit for the combination, whether using the published data (Parmar 1998*) or preferably an IPD (individual patient data) analysis, though perhaps such an analysis is unnecessary given the remarkable similarity of the results. In associated publications, the occurrence of hypertension on study correlated with time‐dependent outcomes in the Rini study but not in the Escudier study.
Sunitinib
Like sorafenib, sunitinib is an oral small molecule inhibitor of VEGF receptor tyrosine kinase. They differ in regards to additional sites of action. Because of some unusual toxicities seen in preclinical testing, sunitinib 50 mg was approved for trial in a schedule of 4 weeks of daily therapy followed by a two week break before the next 6‐week cycle. After impressive evidence of action against clear cell RCC in non‐randomized studies, sunitinib was directly tested against interferon‐alfa in a large study of systemically untreated patients (Motzer(1) 2010). Eligibility was restricted to patients with renal cancers with a clear cell component. The investigator‐determined chance of major remission was 46.5% for sunitinib versus 12% for interferon, adjusted on independent blinded review to 38.9% versus 8.4%, respectively (OR 6.34, 95% CI 4.4 to 9.2, P < 0.000001 Pearson Chi2 test). Of severe clinical adverse events, grade 3/4 treatment‐related fatigue was more frequent in patients on interferon (12 versus 7 adverse events), whereas other toxicities were more frequent for sunitinib (diarrhea 5 versus 0 events, vomiting 4 versus 1 event, hypertension 8 versus 1 event, hand‐foot syndrome 5 versus 0 events, all P < 0.05 Fisher's exact test for 715 patients). Formal health‐related quality‐of‐life assessment analysis shows trends or statistically significantly better QOL scores for sunitinib over interferon in multiple dimensions in both the European and the US patient populations (for details, see multiple reports listed in the study references). However, in this unblinded study, the origin of the QOL outcome difference is unclear and could reflect convenience of treatment or other biases. Progression‐free survival, the primary endpoint, was approximately double with sunitinib treatment than with interferon (median 11 versus 5 months respectively by independent central review, HR 0.54, 95% CI 0.44 to 0.66, P < 0.001). Median PFS was superior for patients with either good or intermediate prognostic factors. Improved median PFS could not be separately confirmed for the poor prognostic subset but power was low (only 6% of enrolled patients were poor prognosis by MSKCC criteria). Overall survival showed a strong trend in favour of sunitinib using standard intent‐to‐treat methodology (median 26.4 versus 21.8 months, HR 0.82, P = 0.049 stratified log‐rank). After correction for seven known prognostic factors in a Cox model, the survival benefit was significant (HR 0.76, P < 0.01). An exploratory analysis looked at the possible negating impact of crossover to sunitinib by patients assigned to interferon.
Sorafenib
Following demonstration of activity of sorafenib after prior cytokine therapy (Escudier(2) 2010), a Phase II study directly compared sorafenib with first‐line interferon‐alfa in patients with clear cell renal cancers (Escudier(4) 2009). At the standard dose of sorafenib 400 mg po bid (by mouth twice daily), there was no significant difference in PFS. Hardly any patients had a major remission with this dose of sorafenib, though 68% had minor tumour shrinkage compared with 39% on interferon. Sorafenib was associated with better symptom control and patient satisfaction but, as with sunitinib, this observation is difficult to interpret in an open‐label study with different routes of administration. It is possible that the 400 mg dose is too low as there was further tumour shrinkage after progression on this arm in 42% of patients who were escalated to 600 mg po bid in a second phase of the same study. Indeed, a Phase II study was able to escalate sorafenib dose as high as 1200 to 1600 mg po bid in over 90% of patients and saw an unusually high major response rate of 55% including some complete remission (Amato 2007a*; Amato 2007b*).
Conclusions from Group 3 studies: Compared to interferon, the standard of care for advanced renal cancer in the pre‐targeted therapy era, two targeted options have demonstrated superior remissions and progression‐free survival and probably overall survival. These are oral sunitinib taken daily for four weeks out of six, or bevacizumab given intravenously every two weeks along with subcutaneous interferon thrice weekly. Toxicities and convenience are somewhat different for these two options (see 'Discussion').
Nephrectomy Status
Stratification was considered the most convincing method of pre specification for the purposed of subset analysis. Five studies in Group 3 stratified patients by nephrectomy status, and three of these also provided information on hazard ratios by nephrectomy status for overall survival. For temsirolimus versus interferon (Hudes 2010), HR for death was 0.84 (95% CI 0.79 to 1.12) for nephrectomized patients (n = 278) and 0.61 (95%CI 0.41‐0.91) for patients who had not undergone nephrectomy (n = 138, data extracted from published forest plot). For bevacizumab plus interferon versus interferon monotherapy (Rini 2010), the HR for death was 0.91 (95%CI 0.76‐1.09) after nephrectomy (n = 620) and 0.65 (95% CI 0.43 to 0.98) if no nephrectomy. For sunitinib versus interferon (Motzer(1) 2010), the HR for death was 0.80 (95%CI 0.61‐1.03) after nephrectomy (n = 674) and 0.79 (95% CI 0.47 to 1.33) if no nephrectomy (n = 76). Therefore the lack of nephrectomy was associated with at least as substantial mortality reduction as following nephrectomy and in two studies was statistically significant despite small numbers.
Group 4: Targeted agent(s) versus targeted control
(5 studies, 'Table 11')
9. Group 4 Outcomes Summary: Targeted Agent vs Targeted Control.
| Investigational | Control(s) | Study | ORR | PFS | OS | QOL |
| Temsirolimus+IFN | Temsirolimus | Hudes 2010(B) | ns | ns | ||
| Bevacizumab+Erlotinib | Bevacizumab | Bukowski 2007 | ns | ns | ns | |
| Sorafenib+IFN | Sorafenib | Jonasch 2010 | ns | ns | ns | |
| Sorafenib+IL2 | Sorafenib | Procopio 2010 | ns | ns | ||
| Bevacizumab+Temsirolimus | Bevacizumab+IFN Sunitib |
Escudier(5) 2010 | ns | ns |
ns = not statistically different
Targeted agents have become the developing standard of care for first‐line systemic therapy of advanced clear cell renal cancer, but efficacy remains modest and complete remissions are rare. Consequently efforts to increase efficacy by evaluating combinations of agents have become of increasing interest. The currently included studies fall into two subgroups: a. combinations of targeted agents and cytokines, and b. combinations of multiple targeted agents.
a. Combinations of targeted agents and cytokines
Included studies: Hudes 2010(B) (temsirolimus + interferon); Jonasch 2010 (sorafenib + interferon); Procopio 2010 (sorafenib + IL2).
The previously discussed three‐arm comparison of temsirolimus, interferon‐a, or both together, permits a comparison of the combination with temsirolimus monotherapy (Hudes 2010(B)). However, the combination offered no benefit over single agent therapy; for example the median quality‐adjusted time without toxicity or relapse was 7.0 months for temsirolimus and 6.1 months for temsirolimus plus interferon (Zbrozek 2010*). Sorafenib at standard dose and schedule has been combined with interferon‐alfa using a small frequent dosing schedule that did not improve any outcome and showed a trend to decreased overall survival (Jonasch 2010). The addition of low dose interleukin‐2 to sorafenib was toxic at the original dose and schedule (Procopio 2010), and had to be reduced after 20 patients were enrolled on the combination arm; these early higher dose patients showed a trend to longer freedom from progression but the small numbers preclude definite conclusions. In general, these studies do not encourage further pursuit of this approach. An exception might be to assess the role of interferon in the bevacizumab plus interferon combination by comparing bevacizumab with or without interferon, rather than interferon with or without bevacizumab discussed in Group 3.
b. Combinations of multiple targeted agents
A number of combinations of targeted agents have been tested in nonrandomized Phase I/II trials beyond the scope of this review, and toxicity has been a substantial problem. At the present time, combinations of bevacizumab with agents that have different targets have been found feasible for further testing. Included studies have tested bevacizumab with an EGFR inhibitor (erlotinib) and with an mTOR inhibitor (temsirolimus).
(i) Bevacizumab with or without erlotinib
Bevacizumab plus erlotinib was chosen for study because of potential synergy based on theoretical considerations as well as preclinical and preliminary clinical observations. However the combination did not give better outcomes than with bevacizumab alone.
(ii) Bevacizumab + temsirolimus versus sunitinib versus bevacizumab + interferon
There is potential for synergy by combining an mTOR and a VEGFR inhibitor due to pathway interaction. Bevacizumab and temsirolimus were successfully combined at full standard doses with manageable toxicity in a preliminary Phase I study (Merchan 2007*). In a randomized three‐arm Phase II study, the bevacizumab and temsirolimus combination has been compared with the targeted treatments in most common first‐line use in North America (sunitinib) and in Europe (bevacizumab plus interferon‐alfa) in a 2:1:1 allocation. In a recent preliminary report, the toxicity of the novel combination arm was higher than expected leading to a high drop‐out rate, and no evidence of synergistic or even additive efficacy was observed for the primary endpoint of non‐progression at 48 weeks (Escudier(5) 2010). Exposure to the experimental therapy was limited by adverse events: 41% of patients allocated to bevacizumab plus temsirolimus discontinued one or both agent because of toxicity, compared with 17% for the combined control arms. This study is not designed to compare the two control arms: because of small numbers, differences in outcome are likely due to differences in prognostic factor distribution.
Conclusions from Group 4: Thus far, combinations of targeted agents with either cytokines or with other targeted agents have been disappointing, yielding increased toxicity without evidence of increased efficacy. However this topic remains an active area of research and a number of studies are ongoing and will be incorporated into this review as they are reported.
Discussion
The first preliminary presentations of trials in this review were as recent as 2002 (Atkins 2004; Yang 2003). In only eight years, there has been an explosion of interest in renal cancer, long considered resistant to drug therapy, and for which interferon‐alfa was the one generally available treatment that had only marginal efficacy (Coppin 2006*). This review now surveys the reports of 25 mostly mature randomized controlled trials of targeted agents in advanced renal cancer. Eight additional trials were identified from a search of the publicly accessible database clinicaltrials.gov and none of these were suggestive of publication bias, that is, non‐publication of a contradictory result. Risk of bias has been formally assessed, and no pivotal practice‐changing studies were considered high risk though the involvement of sponsoring pharmaceutical companies in the publication authorship is of some concern.
Non‐specific agents such as thalidomide have been replaced by highly specific targeted agents. Representatives of two classes of agent, targeting VEGFR or mTOR, have now demonstrated superiority in one or more outcomes compared to the appropriate standard comparator. The following discussion mainly considers the clinical application of the positive trial results presented above.
Dose and schedule effects
There is insufficient evidence to make firm statements about optimal dose for most of the agents reviewed in this systematic survey. Bevacizumab substantially improved progression‐free survival over placebo at a dose of 10 mg/kg but not at 3 mg/kg (Yang 2003), thus suggesting 10 mg/kg as the appropriate dose used in subsequent studies (Rini 2010; Bukowski 2007; Escudier(3) 2010). There is some non‐randomized evidence for a dose‐response effect for sorafenib, a drug that has been tested well below its maximum tolerated dose (Escudier(2) 2010; Amato 2007b*). When combined with sorafenib, more frequent dosing of lower dose interferon appears beneficial for the combination (Bracarda 2010), noting however that very low‐dose interferon added to sorafenib was not useful (Jonasch 2010). Particularly lacking are completed controlled trials of dose and schedule effects for sunitinib, the agent in predominant first‐line use in North America, though correlative data is suggestive of a dose‐response relationship (Houk 2010*). Unlike the empiric maximum tolerated dose approach required with chemotherapy agents, at least some targeted agents may be optimized by pharmacodynamic measurements of the activity of the molecular target (Harding 2003*) preferably followed by empirical verification.
Targeted therapy for systemically untreated patients
Four pivotal studies have demonstrated outcomes improved by targeted agents in first‐line systemic treatment compared to the prior standard of thrice‐weekly subcutaneous interferon‐alfa monotherapy (Group 3, 'Table 10'). These studies are approximately contemporaneous with overlapping accrual periods, and are fully published (Escudier(3) 2010; Hudes 2010; Motzer(1) 2010; Rini 2010). With the recent availability of multiple lines of drug therapy, as well as crossover to the investigational arm at disease progression in most studies, the ability to demonstrate improvement in overall survival has become potentially more difficult. Consequently, improvement in progression‐free survival is being increasingly used to demonstrate biologic superiority but raises questions regarding clinical utility unless corroborated with quality‐of‐life benefits in a placebo‐controlled setting.
Compared with interferon, daily oral sunitinib, given for 4 weeks out of 6, improved the chance of major remission four‐fold (46.5% versus 12.1%), and doubled median progression‐free survival from 5 to 11 months (Motzer(1) 2010). Eligibility was confined to patients with excellent performance status (ECOG 0 to 1) and renal cancers having a clear cell component. In this open‐label study, quality‐of‐life was better on oral sunitinib. A benefit to overall survival was statistically significant when stratified for known risk factors (HR 0.82, P = 0.049). Oral sunitinib has subsequently been approved for use in clear cell renal cancers by regulatory authorities in many countries including in North America and in the European Union. The use of continuous daily sunitinib at reduced dose is being explored (Escudier(6) 2009). Sorafenib 400 mg bid, another oral VEGFR inhibitor with activity in cytokine‐treated patients, unfortunately did not improve progression‐free survival over interferon at the dose and schedule used in a randomized Phase II study (Escudier(4) 2009).
In two separate studies of substantial size (Rini 2010; Escudier(3) 2010), the addition of biweekly intravenous bevacizumab to subcutaneous interferon‐alfa was found to provide benefits over interferon monotherapy of similar magnitude to the comparison of sunitinib versus interferon‐alfa. The chance of major remission was improved two to three‐fold, and the time to disease progression nearly doubled. Although the survival trend did not quite reach statistical significance in either study, both observed an identical 14% mortality reduction in the stratified analysis. The treated population was ambulatory and eligibility confined to patients with a clear cell component or clear cell predominant disease. The bevacizumab‐interferon parenteral regimen has not been directly compared with oral sunitinib: both have been included as comparators in a Phase II "non‐comparative" study (Escudier(5) 2010) that might provide some information on relative safety and tolerance if not efficacy. In patients with three or more adverse prognostic factors, the intravenous mTOR inhibitor temsirolimus has demonstrated improved survival over interferon, median survival 10.9 versus 7.3 months respectively, HR 0.73 (Hudes 2010). Remissions were infrequent in a population of patients typically symptomatic from their disease, and improved strategy is needed for this situation.
Second‐line targeted agent after cytokine failure
Sorafenib 400 mg twice daily is the proven choice as 2nd‐line therapy after failure of cytokine therapy for advanced RCC, yielding improved progression‐free survival and quality‐of‐life over placebo (Escudier(2) 2010). Overall survival was not statistically improved on an intent‐to‐treat basis and again is problematic because of crossover from placebo to active treatment after study closure. Bevacizumab 10 mg/kg improves progression‐free survival after interleukin‐2 but the clinical utility of this observation is less clear without reporting clinical outcomes (Yang 2003). In an exploratory analysis of a larger study, lapatinib improved overall survival in patients with renal cancers that strongly over‐expressed ErbB1(EGFR), an interesting observation tempered with concerns that progression‐free survival was only marginally improved and that this was a subset analysis, albeit pre‐planned (Ravaud 2008). Cytokine monotherapy is now infrequently used as first‐line therapy, consequently this situation is becoming less common. The efficacy of sunitinib after cytokine therapy is based on non‐randomized Phase II data not included in this analysis.
Second‐line targeted agent after VEGFR inhibitor failure
An increasingly relevant question is the value of second‐line agents after initial targeted therapy. No identified studies address this question for patients receiving initial bevacizumab plus interferon. One large study enrolled patients progressing on or within six months of the oral VEGFR inhibitors sunitinib or sorafenib, comparing the oral mTOR inhibitor everolimus with placebo, the appropriate comparator in this setting (Motzer(2) 2010). Patients were required to have renal cancers with a clear cell component but could also have received cytokine therapy. Median progression‐free survival was prolonged from 1.9 to 4 months (HR 0.30), accompanied by delayed decline in performance status without adverse effect on quality‐of‐life. The majority of placebo‐assigned patients were switched to everolimus at further disease progression. A survival benefit could not be demonstrated on an intent‐to‐treat basis; a final analysis of survival is awaited.
The Influence of nephrectomy status
Lack of nephrectomy did not preclude risk reduction for death, and was significant with temsirolimus (Hudes 2010), with bevacizumab plus interferon (Rini 2010), and as a trend with sunitinib (Motzer(1) 2010), all versus interferon as control. The exclusion of patients without nephrectomy in some studies no longer appears appropriate.
Extrapolation to broader patient populations
With the exception of temsirolimus (Hudes 2010), first‐line targeted studies resulting in improved outcomes required patients to have good or excellent performance status, and restricted renal cell histology to clear cell containing or clear cell predominant tumours. Following release of sunitinib and sorafenib in North America and Europe in 2006, expanded access studies ensued that had broader eligibility criteria. An international expanded access study of over 4300 patients treated with sunitinib included patients with non‐clear cell histology, poorer performance status (ECOG 2, 3), age over 65, and with brain metastases (Gore 2009*); the safety profile was manageable in all these subgroups, though the major response rate was substantially lower than seen in the trial population. A similar expanded access program for sorafenib has been published (Stadler 2010*).
Temsirolimus has proven efficacy in patients with adverse prognostic factors, including borderline performance status (Hudes 2010). This study accrued patients of all histologic subtypes and 18% were non‐clear cell, mostly papillary. An exploratory analysis suggested that the benefits of temsirolimus might possibly be greater for the non‐clear cell subset (progression‐free HR 0.38) though ideally this observation should be independently verified (Dutcher 2009*).
Comparison with other systematic reviews
Other systematic reviews of this topic were identified by conducting a search of the same databases as in the protocol of this review, and combining renal cell cancer (MeSH (medical subject headings) term) with systematic or meta‐analysis (text words in title or abstract). The systematic reviews were required to have full peer‐reviewed publication, prespecified methods, systematic searching of multiple databases, data extraction by more than one author, quality assessment of identified studies, and standardized analytical methods. Three reviews qualified for comparison (Mills 2009*; Thompson Coon 2009*; Thompson Coon 2010a*). These reviews all had a literature cutoff in 2008 and evaluated the benefit of bevacizumab, sorafenib, sunitinib, or temsirolimus compared to interferon‐alfa or placebo. For direct comparisons, compared to the present review, there were no substantive differences in findings for these agents from studies available in 2008. These systematic reviews also extended their analyses to make indirect efficacy comparisons beyond the scope of this review, using either an adjusted indirect comparison method or a Bayesian approach (see Nuijten 2010* and Thompson Coon 2010b* for informative correspondence regarding these methodologies). The extensive analysis of Thompson Coon and colleagues was commissioned by the UK National Institute of Clinical Effectiveness and addresses cost‐effectiveness and policy issues also beyond the scope of the present work.
Authors' conclusions
Implications for practice.
'Table 1'
A. Choice of therapy sequence for clear cell cancers
Since this review was first published in 2008, targeted therapy has come of age for advanced renal cancer. The demonstration in 2003 of prolongation of progression‐free survival by bevacizumab 10 mg/kg was regarded as proof‐of‐principle but not practice‐changing. In 2006, sunitinib and sorafenib were both released in North America and in Europe for use in patients with advanced renal cell cancer who had failed cytokine therapy. The recent mature reporting of pivotal first‐line studies of sunitinib and of bevacizumab plus interferon have validated these regimens as suitable choices for ambulatory patients with clear cell renal cancers. Local clinical practice guidelines must also consider regulatory status and cost, issues beyond the scope of this review. Of these two choices, oral sunitinib is more convenient than parenteral bevacizumab plus interferon for both patients and facilities. Furthermore, the bevacizumab‐interferon combination includes the toxicities of both drugs whereas sunitinib appears better tolerated than interferon alone at least in most patients with good performance status. In the second‐line setting, everolimus is a validated option after sunitinib but of uncertain efficacy after bevacizumab‐interferon. Asymptomatic patients with slowly growing lung metastases can be considered for initial cytokine‐based therapy (interferon with or without bevacizumab) and then move on to a VEGFR kinase inhibitor followed by everolimus where appropriate, thereby extending options. The overall implication for clinical practice is that patients and oncologists for the first time have choices of active systemic agents that can be tailored to the particular institutional and patient setting.
B. Special situations
In patients with non‐clear cell histology or with poor prognostic factors, temsirolimus is a choice supported by an exploratory analysis of a controlled trial. Remissions are also sometimes seen with other agents including VEGFR kinase inhibitors. In the absence of comparative data, treatment selection remains a matter of availability and opinion‐based choice.
Implications for research.
The first round of Phase III comparative trials are now mature and published, with validated first‐ and second‐line targeted therapy options for patients with advanced clear cell subtype that comprise the majority of renal cancers. The main areas for ongoing research may be summarized as follows.
More effective use of current treatment strategies to minimize adverse effects, and reduce costs to patients and healthcare: these include clarification of the dose intensity‐efficacy relation for different classes of targeted therapy, comparison of continuous versus interrupted cycles of therapy, improved standardized management of individual toxicities, early recognition of non‐responders and their management. Timeframe: current research, e.g., next 1 to 3 years.
Improved patient selection by use of clinical parameters and tumour/serological biomarkers to predict optimized individual patient management: predictors of targeted drug resistance and cross‐resistance for drug selection and sequencing including cytokines and surgical intervention, both for initial decision making and in light of ongoing response or adverse events. Timeframe: medium term, e.g., next 3 to 5 years.
Improved strategies for disadvantaged patient groups: patients with advanced burden of disease, patients with co‐morbidities, patients with non‐clear cell renal cancers. Timeframe: ongoing.
Development of novel strategies with the objective of complete and more durable remissions: these include identifying agents against new targets, combining agents targeting the same or different pathways, and use in earlier stages of disease. These studies should be increasingly based on understanding of the molecular basis of response and resistance (Rini 2010*). Timeframe will be relatively long, perhaps 5 to 10 years.
What's new
| Date | Event | Description |
|---|---|---|
| 1 July 2010 | New search has been performed | Complete update with additional studies, revised analysis, risk of bias assessment, and revised conclusions. In more detail: search updated from end of 2007 to June 2010 with 5 new eligible studies identified; analyses are now based on the nature of the control arm. Targeted agents have now been validated as first and second‐line therapy choices for patients with advanced renal cancers of the clear cell subtype. |
History
Protocol first published: Issue 2, 2006 Review first published: Issue 2, 2008
| Date | Event | Description |
|---|---|---|
| 8 April 2010 | New search has been performed | Converted to new review format. |
| 14 January 2008 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
We thank Dr Michael Shelley for his valuable review of the manuscript, and James Tacklind for collaboration on behalf of the Prostatic Diseases and Urologic Cancers Cochrane Review Group. We also thank Dr Franz Porzsolt for making this a better review.
Data and analyses
Comparison 1. All studies reporting remission data.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Major objective remission | 26 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Group 1 | 7 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Group 2 | 6 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Group 3 | 8 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Group 4 | 5 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [author‐defined order]
Atkins 2004.
| Methods | Multicentre RCT Phase: 2 Accrual period: 2000/04 to 2000/10 Blinding: none Strata: none IMC (independent monitoring committee): data not found Crossover: data not found |
|
| Participants | Histology: RCC NOS
Prior cytokine therapy: required
Measurable disease: On‐study disease progression: required M/F = 77/34 Eligible PS = 0 to 1; actual PS(0) = 69% Age (range) = data not found Prior nephrectomy = 80% Prognostic strata: system, % good/intermediate/poor risk = MSKCC; 8/46 /47 |
|
| Interventions | TEMSIROLIMUS IV weekly: 250 mg; 75 mg; 25 mg; see Atkins 2004(A); Atkins 2004(B); Atkins 2004(C) for paired comparisons from this 3‐arm study | |
| Outcomes | RR: primary endpoint (WHO)
PFS: yes
OS: yes
Toxicity table: yes
QOL: no Other: no Outcome(s) reported by prognostic group: |
|
| Notes | Publication: peer‐reviewed journal Trial name or number: not identified in clinicaltrials.gov/ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned", presumed central randomization |
| Allocation concealment (selection bias) | Unclear risk | not stated, presumed central randomization |
| Blinding (performance bias and detection bias) Objective outcomes | High risk | no blinding of patients, providers, or outcome assessors for primary endpoint |
| Incomplete outcome data (attrition bias) Objective outcomes | Low risk | 9% missing data for primary endpoint, evenly distributed across arms |
| Selective reporting (reporting bias) | Low risk | all protocol endpoints reported |
| Other bias | Unclear risk | completed planned accrual; industry sponsored and co‐authored (Wyeth) |
Atkins 2004(A).
| Methods | see Atkins 2004 | |
| Participants | see Atkins 2004 | |
| Interventions | TEMSIROLIMUS (1) 250 mg/dose; (2) 75 mg/dose; see Atkins 2004 for details | |
| Outcomes | see Atkins 2004 | |
| Notes | Comparison "A" of 3‐arm study | |
Atkins 2004(B).
| Methods | see Atkins 2004 | |
| Participants | see Atkins 2004 | |
| Interventions | TEMSIROLIMUS (1) 75 mg/dose; (2) 25 mg/dose; see Atkins 2004 for details | |
| Outcomes | see Atkins 2004 | |
| Notes | Comparison "B" of 3‐arm study | |
Atkins 2004(C).
| Methods | see Atkins 2004 | |
| Participants | see Atkins 2004 | |
| Interventions | TEMSIROLIMUS (1) 250 mg/dose; (2) 25 mg/dose; see Atkins 2004 for details | |
| Outcomes | see Atkins 2004 | |
| Notes | Comparison "C" of 3‐arm study | |
Bhargava 2010.
| Methods | Multicentre RCT Phase: randomized discontinuation trial Accrual period: 2007/10 to 2008/7 Blinding: double blind, placebo‐controlled Strata: none IMC: independent radiologic assessment Crossover: after progression on placebo |
|
| Participants | Histology: RCC NOS (83% clear cell)
Prior cytokine therapy: permitted (54% naive) Measurable disease: data not found On‐study disease progression: M/F = 191/81 Eligible PS = > 70% KPS; actual PS(0) = 49% Age (range) = 56 (26 to 79) Prior nephrectomy = 72% Prognostic strata: system, % good/intermediate/poor risk = MSKCC, 30/57/8% Stable disease (+ 25%) on study drug during 16 week run‐in period |
|
| Interventions | (1) tivozanib (AV‐951, Aveo Pharmaceuticals) 1.5 mg po daily d1‐21 q28d x 12 weeks (3 weeks out of four, 3 cycles), vs (2) identical oral placebo | |
| Outcomes | RR: not applicable
PFS: yes
OS: no
Toxicity table: yes
QOL: no
Other: 12‐week percent progression‐free (primary endpoint) Outcome(s) reported by prognostic group: |
|
| Notes | Publication: meeting abstract + slides Trial name or number: NCT00502307 (clinicaltrials.gov) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | meeting report; unable to assess |
| Allocation concealment (selection bias) | Unclear risk | meeting report; unable to assess |
| Blinding (performance bias and detection bias) Objective outcomes | Low risk | "double‐blind placebo controlled"; independent radiology assessment |
| Incomplete outcome data (attrition bias) Objective outcomes | Unclear risk | meeting report; unable to assess |
| Selective reporting (reporting bias) | Unclear risk | meeting report, unable to assess |
| Other bias | Unclear risk | unable to assess; industry sponsored and co‐authored (AVEO) |
Bracarda 2010.
| Methods | Multicentre RCT Phase: 2 Accrual period: 2006/01 to 2007/01 Blinding: open‐label Strata: none IMC: Crossover: none |
|
| Participants | Histology: clear cell > 50%
Prior therapy: not permitted Measurable disease: required On‐study disease progression: M/F = 71/29 Eligible PS = 0 to 2; actual PS(0) = 61% in arm (1), 78% in arm (2) Age (range) = 63 (34 to 80) Prior nephrectomy = required Prognostic strata: system, % good/intermediate/poor risk = arm(1) 27/63/10; arm(2) 28/68/6 |
|
| Interventions | SORAFENIB (Bayer) 400 mg BID and either (1) IFN 3 MU SC x5/week, or (2) IFN 9 MU SC x3/week | |
| Outcomes | RR: secondary endpoint (by RECIST)
PFS: primary endpoint
OS: secondary endpoint
Toxicity: co‐primary endpoint in abstract but secondary endpoint in slides
QOL: not assessed
Other: time to progression Outcome(s) reported by prognostic group: no |
|
| Notes | Publication: meeting abstract + slides Trial name or number: RAPSODY; GOIRC 0681 (Italian oncology group for clinical research); not identified in clinicaltrials.gov | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | meeting report; unable to assess |
| Allocation concealment (selection bias) | Unclear risk | meeting report; unable to assess |
| Blinding (performance bias and detection bias) Objective outcomes | High risk | open‐label study; investigator endpoint assessment |
| Incomplete outcome data (attrition bias) Objective outcomes | Unclear risk | meeting report; unable to assess |
| Selective reporting (reporting bias) | Unclear risk | meeting report; unable to assess |
| Other bias | Unclear risk | unable to assess |
Bukowski 2007.
| Methods | Multicentre RCT Phase: 2 Accrual period: 2004/03 to 2004/10 Blinding: yes Strata: ECOG, DFI IMC: data not found Crossover: data not found |
|
| Participants | Histology: clear cell > 50%
Prior therapy: not permitted Measurable disease: required On‐study disease progression: M/F = 73/31 Eligible PS = 0 to 1; actual PS(0) = 62% Age (range) = 64 (35 to 86) Prior nephrectomy = required Other: good or intermediate MSK prognosis Prognostic strata: system, % good/intermediate/poor risk = MSKCC; 34/66/0 |
|
| Interventions | BEVACIZUMAB 10 mg/kg IV q2w (every 2 weeks) plus either (1) ERLOTINIB 150 mg po daily, or (2) placebo | |
| Outcomes | RR: co‐primary endpoint (RECIST)
PFS: co‐primary endpoint
OS: yes
Toxicity table: yes
QOL: no
Other: no Outcome(s) reported by prognostic group: |
|
| Notes | Publication: peer‐reviewed journal Trial name or number: NCT00081614 (clinicaltrials.gov) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | presumed central randomization |
| Allocation concealment (selection bias) | Low risk | used "an interactive voice response service" |
| Blinding (performance bias and detection bias) Objective outcomes | Low risk | "double‐blind placebo controlled" |
| Blinding (performance bias and detection bias) Patient reported outcomes | Unclear risk | success of blinding not reported |
| Incomplete outcome data (attrition bias) Objective outcomes | Low risk | all patients accounted for, only one patient excluded |
| Incomplete outcome data (attrition bias) Patient reported outcomes | Unclear risk | minimally reported, unable to assess |
| Selective reporting (reporting bias) | Low risk | all intended outcomes reported; overall survival, a secondary endpoint, still "immature" |
| Other bias | Unclear risk | completed planned accrual; industry sponsored and co‐authored (Genentech) |
Ebbinghaus 2007.
| Methods | Multicentre RCT Phase: 2 Accrual period: 2003/6 to 2004/7 Blinding: no Strata: none IMC: none Crossover: none |
|
| Participants | Histology: RCC NOS
Prior therapy: not permitted Measurable disease: required On‐study disease progression: yes M/F = 67/36 Eligible PS = 0 to 1; actual PS(0) = 70% Age (range) = 59 (20 to 80) Prior nephrectomy = 92% Prognostic strata: system, % good/intermediate/poor risk = 33/60/7 |
|
| Interventions | ABT‐510 sc bid (subcutaneous twice daily) (1) 100 mg/dose; (2) 10 mg/dose | |
| Outcomes | RR: yes (RECIST)
PFS: primary endpoint
OS: secondary endpoint
Toxicity table: yes
QOL: no
Other: no Outcome(s) reported by prognostic group: overall, not by arm |
|
| Notes | Publication: full publication Trial name or number: NCT00073125 (clinicaltrials.gov) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "patients were randomized"; possible central randomization |
| Allocation concealment (selection bias) | Unclear risk | not stated; possible central randomization |
| Blinding (performance bias and detection bias) Objective outcomes | High risk | no blinding of patients, providers, or outcome assessors for primary endpoint |
| Incomplete outcome data (attrition bias) Objective outcomes | Unclear risk | unable to assess |
| Selective reporting (reporting bias) | Low risk | all planned endpoints reported |
| Other bias | Unclear risk | completed planned accrual; industry sponsored and co‐authored (Abbott) |
Escudier(1) 2007.
| Methods | Multicentre RCT Phase: 3 Accrual period: 2000/05 to 2002/01 Blinding: yes Strata: PS, Met sites IMC: yes Crossover: data not found |
|
| Participants | Histology: RCC
Prior therapy: immunotherapy Measurable disease: required On‐study disease progression: yes M/F = 222/80 (+5 excluded from analysis) Eligible PS = 0 to 1; actual PS(0) = 52% Age (range) = 61 (25 to 81) Prior nephrectomy = 94% Prognostic strata: system, % good/intermediate/poor risk = data not found |
|
| Interventions | (1) AE‐941 120 mL oral bid; (2) placebo | |
| Outcomes | RR: no
PFS: no
OS: primary endpoint
Toxicity table: no
QOL: no
Other: no Outcome(s) reported by prognostic group: |
|
| Notes | Publication: meeting abstracts; full paper in Polish with English abstract Trial name or number: NCT00005995 (clinicaltrials.gov) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "were randomized", possible central randomization |
| Allocation concealment (selection bias) | Unclear risk | not stated, possible central randomization |
| Blinding (performance bias and detection bias) Objective outcomes | Low risk | "double blind placebo controlled" |
| Incomplete outcome data (attrition bias) Objective outcomes | Low risk | all randomized patients included in primary endpoint analysis |
| Selective reporting (reporting bias) | Low risk | overall survival was the only planned and reported endpoint |
| Other bias | Unclear risk | unable to assess (detailed report in Polish); industry sponsored and co‐authored (AEterna) |
Escudier(2) 2010.
| Methods | Multicentre RCT Phase: 3 Accrual period: 2003/11 to 2005/03 Blinding: yes Strata: country, prognostic score IMC: yes Crossover: yes |
|
| Participants | Histology: clear cell
Prior therapy: cytokine required Measurable disease: required On‐study disease progression: yes M/F = 655/248 Eligible PS = 0 to 1; actual PS(0) = 47% Median age = 59 (19 to 86) Prior nephrectomy = 93% Prognostic strata: system, % good/intermediate/poor risk = MSKCC, 51/49/‐ |
|
| Interventions | 1) SORAFENIB 400 mg oral bid; (2) placebo | |
| Outcomes | RR: yes (RECIST)
PFS: yes
OS: primary endpoint
Toxicity table: yes
QOL: yes (FACT‐G; FKSI)
Other: no Outcome(s) reported by prognostic group: no |
|
| Notes | Publication: peer‐reviewed journal+meeting update Trial name or number: TARGET; NCT000073307 (clinicaltrials.gov) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "were randomized"; probable central randomization with stratification |
| Allocation concealment (selection bias) | Unclear risk | not stated; probable central randomization, small block size (four) |
| Blinding (performance bias and detection bias) Objective outcomes | Low risk | "double‐blind placebo controlled" with independent radiologic assessment |
| Blinding (performance bias and detection bias) Patient reported outcomes | Unclear risk | success of blinding not reported |
| Incomplete outcome data (attrition bias) Objective outcomes | Low risk | fully addressed in on‐line supplementary data from Escudier et al (2009) |
| Incomplete outcome data (attrition bias) Patient reported outcomes | Low risk | fully addressed in Bukowski et al (2007) |
| Selective reporting (reporting bias) | Low risk | all planned outcomes reported |
| Other bias | Unclear risk | early patient un blinding and crossover option for placebo‐assigned patients at recommendation of independent monitoring committee based on first preplanned analysis of PFS, a secondary endpoint; investigators remained blinded; industry sponsored and co‐authored (Bayer) |
Escudier(3) 2010.
| Methods | Multicentre RCT Phase: 3 Accrual period: 2004/06 to 2005/10 Blinding: yes Strata: country, risk group IMC: yes Crossover: not planned but at preplanned interim OS analysis, difference in PFS clinically and statistically significant and DSMB recommended that patients in the control group who had not experienced progression should cross over to receive bevacizumab |
|
| Participants | Histology: clear cell predominant
Prior therapy: not permitted Measurable disease: yes by RECIST On‐study disease progression: yes M/F = 457/192 Eligible KPS > 60; actual KPS(100) = 41% Age (range) = 61(18 to 82) Prior nephrectomy = required Prognostic strata: system, % good/intermediate/poor risk = MSKCC, 30/61/9 |
|
| Interventions | Interferon‐a2a 9 MU sc tiw (subcutaneous thrice weekly) plus either (1) BEVACIZUMAB 10 mg/kg IV q2w, or (2) placebo [crossed over at final PFS analysis if not progressed] | |
| Outcomes | RR: yes (RECIST)
PFS: yes [protocol modified to permit final PFS analysis before OS mature]
OS: primary endpoint
Toxicity table: yes
QOL: no
Other: no Outcome(s) reported by prognostic group: yes |
|
| Notes | Publication: peer‐reviewed journal Trial name or number: AVOREN; NCT00738530 (clinicaltrials.gov) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomization was done centrally" |
| Allocation concealment (selection bias) | Low risk | clearly described; interactive voice recognition system |
| Blinding (performance bias and detection bias) Objective outcomes | Low risk | "double blind placebo controlled"; success of blinding not reported (PFS investigator assessed) |
| Incomplete outcome data (attrition bias) Objective outcomes | Low risk | 4% each arm lost or withdrew consent |
| Selective reporting (reporting bias) | Low risk | all protocol endpoints reported |
| Other bias | Unclear risk | early unblinding and add bevacizumab recommendation for unprogressed placebo‐assigned patients by independent monitoring committee based on unplanned final PFS and preplanned interim survival analysis; industry sponsored and co‐authored (Hoffman La Roche) |
Escudier(4) 2009.
| Methods | Multicentre RCT Phase: 2 Accrual period: 2005/07 to 2005/09 Blinding: radiology Strata: MSKCC classification, region IMC: yes Crossover: "period 2" ‐ those who progressed on sorafenib were dose escalated and those who progressed on IFN were switched to sorafenib |
|
| Participants | Histology: clear cell
Prior therapy: not permitted Measurable disease: yes On‐study disease progression: yes M/F = 117/72 Eligible PS = 0 to 1; actual PS(0) = 49% Age (range) = 18 to 80 Prior nephrectomy = 94% Prognostic strata: system, % good/intermediate/poor risk/missing = MSKCC, 52/47/0.5/0.5% |
|
| Interventions | (1) SORAFENIB 400 mg PO BID (dose escalation to 600 mg after progression); (2) Interferon‐alfa 9 MU SC TIW (crossover to SORAFENIB 400 mg after progression) | |
| Outcomes | RR: yes ‐ reported for period 1 & 2
PFS: primary endpoint
OS: no
Toxicity table: yes
QOL: FKSI‐15, FACT‐BRM
Other: no Outcome(s) reported by prognostic group: no |
|
| Notes | Publication: peer‐reviewed journal Trial name or number: NCT00117637 (clinicaltrials.gov) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "were randomized"; presumed central randomization |
| Allocation concealment (selection bias) | Unclear risk | not described; presumed central randomization |
| Blinding (performance bias and detection bias) Objective outcomes | Low risk | independent blinded radiologic assessment of response and PFS |
| Blinding (performance bias and detection bias) Patient reported outcomes | High risk | open‐label study; patients were aware of assignment |
| Incomplete outcome data (attrition bias) Objective outcomes | Unclear risk | unable to assess |
| Incomplete outcome data (attrition bias) Patient reported outcomes | Unclear risk | completeness of QOL data and approach to missing data not described |
| Selective reporting (reporting bias) | Low risk | survival not assessed but not a primary or secondary protocol outcome |
| Other bias | Unclear risk | completed planned accrual; industry sponsored and co‐authored (Bayer) |
Escudier(5) 2010.
| Methods | Multicentre RCT Phase: "non‐comparative" Phase 2 Accrual period: 2008/03 to 2009/05 Blinding: radiology Strata: centre; PS 0 to 1 vs 2 IMC: yes Other: 3‐arm, ratio = 2 (experimental arm): 1:1 Crossover: data not found |
|
| Participants | Histology: all RCC except papillary
Prior therapy: not permitted Measurable disease: required On‐study disease progression: M/F = 119/52 Eligible PS 0 to 2; actual PS(0) = ? (PS0‐1 = 88%) Age (range) = 62 yrs Prior nephrectomy = 85% Prognostic strata: system, % good/intermediate/poor risk = MKSCC, 33/54/13% |
|
| Interventions | BEVACIZUMAB 10 mg/kg iv q2w (1) with TEMSIROLIMUS 25 mg IV q1w, or (2) with Interferon‐alfa 9 MU sc tiw, vs (3) Sunitinib 50 mg po daily 4 wks on, 2 wks off [2 control arms (2) & (3) combined for this analysis] | |
| Outcomes | RR: yes
PFS: yes
OS: planned
Toxicity table: yes
QOL: no
Other: non‐progression at 48 weeks (primary endpoint); biomarkers; contrast‐enhanced ultrasound Outcome(s) reported by prognostic group: |
|
| Notes | Publication: meeting abstract and virtual presentation Trial name or number: TORAVA; NCT00619268 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "were randomized"; presumed central randomization |
| Allocation concealment (selection bias) | Unclear risk | meeting report; unable to assess |
| Blinding (performance bias and detection bias) Objective outcomes | High risk | open label study |
| Incomplete outcome data (attrition bias) Objective outcomes | Unclear risk | meeting report; unable to assess |
| Selective reporting (reporting bias) | Unclear risk | meeting report; unable to assess |
| Other bias | Unclear risk | unexpected toxicity in the investigational arm, 43% stopped treatment without progression; independent sponsor (Centre Leon Berard) |
Gordon 2004.
| Methods | Multicentre RCT Phase: 3 Accrual period: 2000/10 to 2002/12 Blinding: no Strata: nephrectomy, PS, DFI IMC: data not found Crossover: data not found |
|
| Participants | Histology: RCC NOS
Prior therapy: not permitted Measurable disease: required On‐study disease progression: M/F = 228/114 Eligible PS = 0 to 2; actual PS(0) = 42% Age (range) = 59 Prior nephrectomy = 74% Prognostic strata: system, % good/intermediate/poor risk = data not found |
|
| Interventions | Interferon‐alfa2b 1 MU SC BID (1) with, or (2) without, THALIDOMIDE 200 mg (escalating to 1000, median 400 mg) PO daily | |
| Outcomes | RR: yes (criteria not stated)
PFS: co‐primary endpoint
OS: co‐primary endpoint
Toxicity table: yes
QOL: yes (FACT‐G)
Other: no Outcome(s) reported by prognostic group: |
|
| Notes | Publication: meeting abstract+slides Trial name or number: ECOG E2898; NCT00005966 (clinicalgrials.gov) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | central randomization by a major cooperative group (ECOG) |
| Allocation concealment (selection bias) | Low risk | central randomization by a major cooperative group (ECOG) |
| Blinding (performance bias and detection bias) Objective outcomes | Unclear risk | open label study, overall survival primary endpoint |
| Blinding (performance bias and detection bias) Patient reported outcomes | High risk | open label study |
| Incomplete outcome data (attrition bias) Objective outcomes | Unclear risk | meeting report only |
| Incomplete outcome data (attrition bias) Patient reported outcomes | Unclear risk | meeting report only |
| Selective reporting (reporting bias) | Low risk | all protocol endpoints reported in publicly available meeting slides |
| Other bias | Low risk | completed planned accrual; independent sponsor (ECOG) |
Hudes 2010.
| Methods | Multicentre RCT Phase: 3 Accrual period: 2003/07 to 2005/04 Blinding: imaging Strata: subcontinent, nephrectomy IMC: yes Crossover: no |
|
| Participants | Histology: RCC NOS
Prior therapy: not permitted Measurable disease: yes (RECIST) On‐study disease progression: yes M/F = 432/194 Eligible KPS > 50; actual KPS( > 70) = 17% Age (range) = 59 (23 to 86) Prior nephrectomy = 67% Other: > 2 of 6 adverse prognostic factors Brain mets permitted Prognostic strata: system, % good/intermediate/poor risk = MSKCC, ‐/26/74% |
|
| Interventions | TEMSIROLIMUS 25 mg IV weekly, Interferon‐a2a 3‐18 MU sc tiw, or Both; see Hudes 2010(A); Hudes 2010(B); Hudes 2010(C) for paired comparisons from this 3‐arm study | |
| Outcomes | RR: yes (RECIST)
PFS: yes
OS: primary endpoint
Toxicity table: yes
QOL: yes
Other: no Outcome(s) reported by prognostic group: no |
|
| Notes | Publication: peer‐reviewed journal Trial name or number: Global ARCC Trial; NCT00065468 (clinicaltrials.gov) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "were randomly assigned"; possibly by the sponsor |
| Allocation concealment (selection bias) | Unclear risk | "were stratified according to geographic location...with permuted blocks of three" |
| Blinding (performance bias and detection bias) Objective outcomes | Low risk | secondary endpoints by blinded independent radiologic assessment and unblinded clinical investigator assessment; primary endpoint was survival |
| Blinding (performance bias and detection bias) Patient reported outcomes | High risk | open label study |
| Incomplete outcome data (attrition bias) Objective outcomes | Low risk | 3% lost to follow up, slightly more from the control group |
| Incomplete outcome data (attrition bias) Patient reported outcomes | Low risk | addressed in detail in Yang et al (2010) |
| Selective reporting (reporting bias) | Low risk | all protocol outcomes reported |
| Other bias | Unclear risk | completed planned accrual; industry sponsored and co‐authored (Wyeth) |
Hudes 2010(A).
| Methods | see Hudes 2010 | |
| Participants | see Hudes 2010 | |
| Interventions | (1) TEMSIROLIMUS versus (2) Interferon‐a; see Hudes 2010 for details | |
| Outcomes | see Hudes 2010 | |
| Notes | Comparison "A" of 3‐arm study | |
Hudes 2010(B).
| Methods | see Hudes 2010 | |
| Participants | see Hudes 2010 | |
| Interventions | TEMSIROLIMUS (1) with, or (2) without, interferon‐a; see Hudes 2010 for details | |
| Outcomes | see Hudes 2010 | |
| Notes | Comparison "B" of 3‐arm study | |
Hudes 2010(C).
| Methods | see Hudes 2010 | |
| Participants | see Hudes 2010 | |
| Interventions | Interferon‐a (1) with, or (2) without TEMSIROLIMUS; see Hudes 2010 for details | |
| Outcomes | see Hudes 2010 | |
| Notes | ||
Jonasch 2010.
| Methods | Multicentre RCT Phase: 2 Accrual period: 2005/6 to 2007/6 Blinding: independent audit of 20 patients' scans Strata: none IMC: none Crossover: none |
|
| Participants | Histology: clear cell (conventional) RCC
Prior therapy: not permitted Measurable disease: required On‐study disease progression: M/F = 61/19 Eligible PS = 0 to 1; actual PS(0) = 50 Age (range) = 43 to 83 yrs Prior nephrectomy = 98.8% Prognostic strata: system, % good/intermediate/poor risk = retrospective MSKCC, 51/46/3% Results reported by prognostic group ‐ multivariate analysis |
|
| Interventions | SORAFENIB 400 mg oral bid (1) with, or (2) without, Interferon‐alfa 0.5 MU sc bid | |
| Outcomes | RR: co‐primary endpoint (criteria not stated)
PFS: co‐primary endpoint
OS: too early
Toxicity table: yes
QOL: no
Other: biomarker analysis Outcome(s) reported by prognostic group: |
|
| Notes | Publication: peer‐reviewed journal Trial name or number: NCT00126594 (clinicaltrials.gov) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "were randomized", single centre but only a pharmacist had access to the randomization program |
| Allocation concealment (selection bias) | Low risk | "were randomized", only a pharmacist had access to the randomization program |
| Blinding (performance bias and detection bias) Objective outcomes | Low risk | open label study with blinded radiologic assessment of a 25% sample |
| Incomplete outcome data (attrition bias) Objective outcomes | Low risk | patients off study prior to first evaluation were "included in the analysis as treatment failures" |
| Selective reporting (reporting bias) | Low risk | all protocol endpoints reported |
| Other bias | Low risk | completed planned accrual; independent sponsor (NCI‐CTEP) |
Lee 2006.
| Methods | Multicentre RCT Phase: 2 Accrual period: 2000/07 to 2002/12 Blinding: no Strata: data not found IMC: data not found Crossover: data not found |
|
| Participants | Histology: RCC NOS
Prior systemic therapy: 90% Measurable disease: required On‐study disease progression: M/F = 39/21 Eligible PS = 0 to 2; actual PS(0) = 18% Age (range) = 60(24 to 80) Prior nephrectomy = 80% Other: maximum 3 sites of disease Prognostic strata: system, % good/intermediate/poor risk = data not found |
|
| Interventions | (1) THALIDOMIDE (100‐) 400 mg oral daily; (2) medroxyprogesterone acetate 300 mg oral daily | |
| Outcomes | RR: primary endpoint (WHO)
PFS: yes
OS: yes
Toxicity table: yes
QOL: no
Other: no Outcome(s) reported by prognostic group: |
|
| Notes | Publication: peer‐reviewed journal Trial name or number: not identified in clinicaltrials.gov Phase II, did not proceed with Phase III | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "were randomly assigned" |
| Allocation concealment (selection bias) | Unclear risk | not described |
| Blinding (performance bias and detection bias) Objective outcomes | High risk | open label; investigator‐assessed primary outcome |
| Incomplete outcome data (attrition bias) Objective outcomes | Low risk | described in detail and approximately evenly distributed |
| Selective reporting (reporting bias) | Low risk | planned outcomes reported |
| Other bias | Low risk | completed planned accrual; independently conducted |
Madhusudan 2004.
| Methods | Multicentre RCT Phase: 2 Accrual period: not stated Blinding: no Strata: no IMC: data not found Crossover: data not found |
|
| Participants | Histology: data not found
Prior therapy: not permitted Measurable disease: On‐study disease progression: M/F = 22/12 Eligible PS = ; actual PS(0) = Age (range) = 57 (44 to 76) Prior nephrectomy = data not found Prognostic strata: system, % good/intermediate/poor risk = data not found |
|
| Interventions | Interferon‐alfa 9 MU sc tiw, (1) with, or (2) without, THALIDOMIDE (100‐)200 mg oral daily | |
| Outcomes | RR: yes (criteria not stated)
PFS: no
OS: no
Toxicity table: yes
QOL: not reported
Other: no Outcome(s) reported by prognostic group: |
|
| Notes | Publication: meeting abstract of ongoing study Trial name or number: not identified in clinicaltrials.gov | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | meeting report, unable to assess |
| Allocation concealment (selection bias) | Unclear risk | meeting report, unable to assess |
| Blinding (performance bias and detection bias) Objective outcomes | High risk | open label study, investigator assessed primary outcome |
| Incomplete outcome data (attrition bias) Objective outcomes | Unclear risk | meeting report, unable to assess |
| Selective reporting (reporting bias) | Unclear risk | unable to assess |
Motzer(1) 2010.
| Methods | Multicentre RCT Phase: 3 Accrual period: 2004/08 to 2005/10 Blinding: imaging Strata: LDH, PS, nephrectomy IMC: yes Crossover: study amended when sunitinib approved in January 2006 to allow crossover of patients on IFN on documented disease progression as primary endpoint of PFS had been met ‐ agreed with IMC |
|
| Participants | Histology: clear cell component
Prior therapy: not permitted Measurable disease: required On‐study disease progression: yes M/F = 536/214 Eligible PS = 0 to 1; actual PS(0) = 61% Age (range) = 61 (27 to 87) Prior nephrectomy = 90% Prognostic strata: system, % good/intermediate/poor risk = retrospective MSKCC, 37/56/7% |
|
| Interventions | (1) SUNITINIB 50 mg oral daily for four weeks of six week cycle; (2) Interferon‐alfa2a 9 MU sc tiw (with cross‐over to SUNITINIB at disease progression, after second interim analysis) | |
| Outcomes | RR: yes (RECIST)
PFS: primary endpoint
OS: yes
Toxicity table: yes
QOL: yes (FACT‐G, FKSI)
Other: cross‐over post‐study Outcome(s) reported by prognostic group: no |
|
| Notes | Publication: peer‐reviewed journal + meeting update Trial name or number: NCT00083889 (clinicaltrials.gov) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "patients were randomly assigned", presumed central randomization |
| Allocation concealment (selection bias) | Unclear risk | presumed central randomization, permuted blocks of 4 within strata |
| Blinding (performance bias and detection bias) Objective outcomes | Low risk | open label study with independent radiologic evaluation up to interim analysis and crossover |
| Blinding (performance bias and detection bias) Patient reported outcomes | High risk | open label study |
| Incomplete outcome data (attrition bias) Objective outcomes | Low risk | 8% patients withdrew consent on the control arm (vs 1%, P<.001) but primary endpoint was analyzed by allocation |
| Incomplete outcome data (attrition bias) Patient reported outcomes | Unclear risk | completion rates were different on the two arms (investigational 96%, control 88%) |
| Selective reporting (reporting bias) | Low risk | all planned outcomes reported |
| Other bias | Unclear risk | crossover permitted after second interim analysis but planned accrual had been completed; industry sponsored and co‐authored (Pfizer) with "final decisions..content of the manuscript were made by the academic principal investigator in consultation with the other authors" |
Motzer(2) 2010.
| Methods | Multicentre RCT Phase: 3 Accrual period: 2006/9 to 2007/10 Blinding: triple blind ie placebo‐controlled, independent radiologic assessment Strata: prognostic score(3); number prior VEGFR TKIs (1 vs 2) IMC: yes Crossover: yes |
|
| Participants | Histology: RCC with clear cell component
Prior cytokine therapy: permitted Prior TKI (sunitinib/sorafenib/both): required Measurable disease: required (RECIST criteria) On‐study disease progression: required on or within 6 months of TKI therapy M/F = 317/93 Eligible PS = > 70% KPS Actual KPS > 90 = 64% Age (range) = 61 (27 to 85) Prior nephrectomy = 96% Prognostic strata: system, % good/intermediate/poor risk = MSKCC; 26/56/15 |
|
| Interventions | (1) EVEROLIMUS (Afinitor, Novartis) 10 mg PO daily, vs (2) matching PLACEBO (2:1 randomization) | |
| Outcomes | RR: yes
PFS: primary
OS: yes
Toxicity table: yes
QOL: yes
Other: no Outcome(s) reported by prognostic group: yes |
|
| Notes | Publication: peer‐reviewed journal Trial name or number: RECORD‐1; NCT00410124 (clinicaltrials.gov) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "were randomly assigned"; presumed central randomization |
| Allocation concealment (selection bias) | Unclear risk | presumed central randomization; permuted blocks of 6 within strata |
| Blinding (performance bias and detection bias) Objective outcomes | Low risk | double‐blind placebo controlled; independent radiologic evaluation |
| Blinding (performance bias and detection bias) Patient reported outcomes | Low risk | double‐blind placebo controlled using identical tablets, but asymmetric subjective toxicities eg stomatitis (investigational 40%, control 8%) |
| Incomplete outcome data (attrition bias) Objective outcomes | Low risk | small data losses described, intent‐to‐treat analysis |
| Incomplete outcome data (attrition bias) Patient reported outcomes | Unclear risk | unable to assess (no full report of subjective outcomes identified for this study) |
| Selective reporting (reporting bias) | Low risk | all planned outcomes reported in at least preliminary form |
| Other bias | Unclear risk | completed planned accrual but terminated at second interim analysis on recommendation of independent monitoring committee; industry sponsored and co‐authored (Novartis) but corresponding author "had final responsibility for the decision to submit for publication" |
Procopio 2010.
| Methods | Multicentre RCT Phase: 2 Accrual period: 2006/10 to 2008/2 Blinding: none Strata: prognostic score; histology clear vs non‐clear IMC: data not found Crossover: data not found |
|
| Participants | Histology: RCC NOS
Prior cytokine therapy: not permitted Measurable disease: required On‐study disease progression: M/F = 94/34 Eligible PS = 0 to 2 Age (range) = 61 (30 to 80) Prior nephrectomy = 92% Prognostic strata: system, % good/intermediate/poor risk = MSKCC, 49/42/9% |
|
| Interventions | SORAFENIB 400 mg po bid, (1) with versus (2) without INTERLEUKIN‐2 4.5 MIU sc d1‐5, 6 weeks of 8 week cycle (after 40 pts, IL‐2 reduced to 3 MIU sc d1‐5, 2 wks on, 2 wks off) | |
| Outcomes | RR: yes (RECIST)
PFS: primary
OS: no
Toxicity table: yes
QOL: no
Other: no Outcome(s) reported by prognostic group: |
|
| Notes | Publication: abstract + slides Trial name or number: ROSORC; NCT00609401 (clinicaltrials.gov) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | meeting report, unable to assess |
| Allocation concealment (selection bias) | Unclear risk | unable to assess |
| Blinding (performance bias and detection bias) Objective outcomes | High risk | open label study, investigator assessed primary outcome |
| Incomplete outcome data (attrition bias) Objective outcomes | Unclear risk | unable to assess |
| Selective reporting (reporting bias) | Unclear risk | unable to assess |
| Other bias | Unclear risk | unable to assess |
Ratain 2006.
| Methods | Multicentre RCT Phase: 2 Accrual period: 2002/09 to ? Blinding: yes Strata: no IMC: data not found Other: randomized discontinuation trial Crossover: data not found |
|
| Participants | Histology: RCC NOS
Prior therapy: 84% but not required Measurable disease: required On‐study disease progression: stable on initial 12 weeks of sorafenib M/F = 47/18 Eligible PS = 0 to 1; actual PS(0) = 55% Age (range) = 59 (23 to 76) Prior nephrectomy = 89% Prognostic strata: system, % good/intermediate/poor risk = MSKCC; 34/60/3 |
|
| Interventions | (1) continue SORAFENIB 400 mg oral bid, or (2) switch to placebo, after 12‐week initial run‐in | |
| Outcomes | RR: not relevant
PFS: primary endpoint
OS: no
Toxicity table: yes
QOL: no
Other: no Outcome(s) reported by prognostic group: |
|
| Notes | Publication: peer‐reviewed journal Trial name or number: NCT00079612 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | central randomization described |
| Allocation concealment (selection bias) | Low risk | "central allocation via a telephone randomization system" |
| Blinding (performance bias and detection bias) Objective outcomes | Low risk | "assigned to ... or matching placebo in double blind fashion" |
| Incomplete outcome data (attrition bias) Objective outcomes | Low risk | all patients accounted for in both run‐in and randomized study phases |
| Selective reporting (reporting bias) | Low risk | planned single outcome study |
| Other bias | Unclear risk | accrual less than indicated in protocol for reasons not clarified in report; industry sponsored and co‐authored (Bayer) |
Ravaud 2008.
| Methods | Multicentre RCT Phase: 3 Accrual period: 2002/12 to 2005/02 Blinding: no Strata: PS, number of metastatic sites IMC: yes Crossover: no |
|
| Participants | Histology: clear cell 87%, papillary 6%, chromophobe 2%, collecting duct 0.2%, unclassified 4.8%
Prior therapy: cytokine required Patients with tumours showing no EGFR or HER‐2 expression were excluded Measurable disease: data not found On‐study disease progression: yes M/F = 305/111 (+1 excluded from analysis) KPS 90 to 100%: 59%, KPS 70 to 80%: 41% Age (range) = 62(19 to 83) Prior nephrectomy = 94% Proportion (good/intermediate/poor risk) = data not found Prognostic stratification: retrospective by MSKCC criteria Risk level: favourable 39%, intermediate 30%, poor 27% EGFR 3+ by IHC: 58% HER‐2 0 by IHC: 94% |
|
| Interventions | (1) LAPATINIB 1250 mg PO daily; (2) hormone (tamoxifen, megestrol) | |
| Outcomes | RR: not relevant
PFS: primary endpoint
OS: yes
Toxicity table: yes
QOL: no
Other: no Outcome(s) reported by prognostic group: data not found |
|
| Notes | Publication: peer‐reviewed journal Trial name or number: EGF20001; not identified in clinicaltrials.gov | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomization was done centrally" |
| Allocation concealment (selection bias) | Low risk | "via an interactive voice response system" |
| Blinding (performance bias and detection bias) Objective outcomes | Low risk | open label study with independent radiologic blind evaluation |
| Incomplete outcome data (attrition bias) Objective outcomes | Low risk | all patients accounted for with small symmetric losses |
| Selective reporting (reporting bias) | Low risk | all protocol endpoints reported |
| Other bias | Unclear risk | completed 2‐phase accrual; industry sponsored and co‐authored (GlaxoSmithKline) |
Rini 2010.
| Methods | Multicentre RCT Phase: 3 Accrual period: 2003/10 to 2005/7 Blinding: none Strata: nephrectomy status; prognostic risk (3) IMC: yes Crossover: data not found |
|
| Participants | Histology: RCC with clear cell component
Prior cytokine therapy: not permitted Measurable disease: data not found On‐study disease progression: data not found M/F = 508/224 Eligible PS = > 70% KPS Actual PS(0) = 62% Age (range) = 62 Prior nephrectomy = 85% Prognostic strata: system, % good/intermediate/poor risk = MSKCC; 26/64/10 |
|
| Interventions | INTERFERON alfa‐2b (Intron, Schering‐Plough) 9 MU SC TIW (1) with, or (2) without, BEVACIZUMAB 10 mg/kg IV Q2weeks | |
| Outcomes | RR: yes
PFS: yes
OS: primary
Toxicity table: yes
QOL: no
Other: no Outcome(s) reported by prognostic group: |
|
| Notes | Publication: peer‐reviewed journal Trial name: CALGB 90206; NCT00072046 (clinicaltrials.gov) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | central randomization by cooperative groups (CALGB and NCI‐Canada) |
| Allocation concealment (selection bias) | Low risk | central randomization |
| Blinding (performance bias and detection bias) Objective outcomes | Unclear risk | open label study, investigator assessment, overall survival primary outcome assumed reliable |
| Incomplete outcome data (attrition bias) Objective outcomes | Low risk | all patients accounted for with small symmetric losses |
| Selective reporting (reporting bias) | Low risk | all protocol outcomes reported |
| Other bias | Low risk | planned accrual completed; independently sponsored and conducted |
Srinivas 2005.
| Methods | Multicentre RCT Phase: 2 Accrual period: 1999/09 to 2001/11 Blinding: no Strata: no IMC: data not found Crossover: data not found |
|
| Participants | Histology: RCC NOS
Prior therapy: 10 of 14 Measurable disease: data not found On‐study disease progression: data not found M/F = 11/3 Eligible KPS > 60; actual PS(0) = 50% Age (range) = 63 (51 to 80) Prior nephrectomy = 79% Prognostic strata (system; prospective/retrospective/none) = Proportion (good/intermediate/poor risk) = |
|
| Interventions | THALIDOMIDE oral daily (1) 800 to 1200 mg, or (2) 200 mg | |
| Outcomes | RR: yes (WHO)
PFS: no
OS: yes
Toxicity table: yes
QOL: no
Other: no Outcome(s) reported by prognostic group: |
|
| Notes | Publication: peer‐reviewed journal Trial name or number: not identified in clinicaltrials.gov | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | not described, presumed single centre study |
| Allocation concealment (selection bias) | Unclear risk | not described |
| Blinding (performance bias and detection bias) Objective outcomes | Unclear risk | open label study, overall survival presumed reliable |
| Incomplete outcome data (attrition bias) Objective outcomes | Low risk | all 14 patients in survival curves |
| Selective reporting (reporting bias) | Unclear risk | unable to assess |
| Other bias | Unclear risk | unable to assess |
Stadler 2005.
| Methods | Multicentre RCT Phase: randomized discontinuation trial Accrual period: 2000/12 to 2002/07 Blinding: yes Strata: no IMC: data not found Crossover: data not found |
|
| Participants | Histology: RCC NOS
Prior therapy: 59% Measurable disease: required On‐study disease progression: remained stable on initial 12 weeks of therapy M/F = 257/111 Eligible PS = 0 to 2; actual PS(0) = 41% Age (range) = 61 (20 to 87) Prior nephrectomy = 80% Prognostic strata: system, % good/intermediate/poor risk = data not found |
|
| Interventions | (1) continue CARBOXYAMINOIMIDAZOLE 250 mg oral daily, or (2) switch to placebo, after 16‐week initial run‐in | |
| Outcomes | RR: not relevant
PFS: primary endpoint
OS: no
Toxicity table: yes
QOL: no
Other: no Outcome(s) reported by prognostic group: |
|
| Notes | Publication: peer‐reviewed journal Trial name or number: CALGB 69901; NCT00006486 (clinicaltrials.gov) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomly assigned by the CALGB Statistical Center" |
| Allocation concealment (selection bias) | Low risk | central randomization |
| Blinding (performance bias and detection bias) Objective outcomes | Low risk | double blind placebo control |
| Incomplete outcome data (attrition bias) Objective outcomes | Unclear risk | only 17% of run‐in phase patients were randomized, 30% withdrew for reasons other than disease progression or response |
| Selective reporting (reporting bias) | Low risk | planned protocol outcome reported |
| Other bias | Low risk | study closed early at recommendation of monitoring committee following a futility analysis; independently sponsored and conducted |
Sternberg 2010.
| Methods | Multicentre RCT Phase: 3 Accrual period: 2006/4 to 2007/4 Blinding: double‐blind placebo‐control, independent radiologic review Strata: PS 0vs1; Nx status; prior cytokine IMC: yes Crossover: 48% of placebo‐assigned patients |
|
| Participants | Histology: RCC clear cell
Prior cytokine therapy: one permitted 46%, 54% naive Measurable disease: required On‐study disease progression: M/F = 71%M Eligible PS = 0 to 1; actual PS(0) = 41% Age (range) = 59 (25 to 85) Prior nephrectomy = 89% Prognostic strata: system, % good/intermediate/poor risk = 39/54/3 |
|
| Interventions | (1) PAZOPANIB 800 mg PO daily, vs (2) matched PLACEBO (2:1 randomization) Crossover 48% |
|
| Outcomes | RR: yes
PFS: primary
OS: pending
Toxicity table: yes
QOL: yes
Other: no Outcome(s) reported by prognostic group: |
|
| Notes | Publication: peer‐reviewed journal Trial name or number: VEG10192; NCT00334282 (clinicaltrials.gov) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | central randomization |
| Allocation concealment (selection bias) | Low risk | central randomization |
| Blinding (performance bias and detection bias) Objective outcomes | Low risk | double blind placebo controlled, independent radiologic blind assessment |
| Blinding (performance bias and detection bias) Patient reported outcomes | Low risk | double blind with matching placebo |
| Incomplete outcome data (attrition bias) Objective outcomes | Low risk | all patients accounted for, losses 6% (investigational) and 3% (control) unlikely significant |
| Incomplete outcome data (attrition bias) Patient reported outcomes | Low risk | "completion rates > 90% across most of the assessment timepoints" |
| Selective reporting (reporting bias) | Low risk | all protocol endpoints reported (interim overall survival) |
| Other bias | Unclear risk | planned accrual completed; industry sponsored and co‐authored (GlaxoSmithKline) |
Yang 2003.
| Methods | Multicentre RCT Phase: 2 Accrual period: 1998/10 to 2001/09 Blinding: yes Strata: no IMC: data not found Crossover: data not found |
|
| Participants | Histology: clear cell
Prior therapy: IL‐2 (93%) Measurable disease: required On‐study disease progression: yes M/F = 75/41 Eligible PS = 0 to 2; actual PS(0) = 78% Age (range) = 53 ( ‐ ) Prior nephrectomy = 91% Prognostic strata (system; prospective/retrospective/none) = Proportion (good/intermediate/poor risk) = |
|
| Interventions | BEVACIZUMAB 10 mg/kg IV q2w, BEVACIZUMAB 3 mg/kg IV q2w; or placebo; see Yang 2003(A); Yang 2003(B); Yang 2003(C) for paired comparisons from this 3‐arm study | |
| Outcomes | RR: co‐primary endpoint (WHO)
PFS: co‐primary endpoint
OS: yes
Toxicity table: yes
QOL: no
Other: no Outcome(s) reported by prognostic group: data not found |
|
| Notes | Publication: peer‐reviewed journal Trial name: NCT00019539 (clinicaltrials.gov) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | randomly assigned with support of NCI‐US biostatistics section |
| Allocation concealment (selection bias) | Unclear risk | not described |
| Blinding (performance bias and detection bias) Objective outcomes | Low risk | double‐blind placebo controlled 3‐arm study with post‐study independent blind radiologic assessment |
| Incomplete outcome data (attrition bias) Objective outcomes | Low risk | limited information but only one patient lost to follow‐up |
| Selective reporting (reporting bias) | Low risk | all protocol outcomes reported |
| Other bias | Low risk | completed planned accrual; independently sponsored and conducted (NCI‐CTEP) |
Yang 2003(A).
Yang 2003(B).
Yang 2003(C).
| Methods | see Yang 2003 | |
| Participants | see Yang 2003 | |
| Interventions | (1) BEVACIZUMAB 3 mg/kg IV q2w; (2) placebo; see Yang 2003 for details | |
| Outcomes | see Yang 2003 | |
| Notes | Comparison "C" of 3‐arm study | |
All fields: if blank, the data was not identified in the available publication(s). Methods: RCT = randomized controlled trial; IMC = independent monitoring committee; DFI = disease‐free interval. Participants: RCC NOS = renal cell carcinoma not otherwise specified; prior therapy refers to eligibility requirement to have received or not received prior drug therapy, usually immunotherapy; M/F = number of males/number of females; PS = performance status (Eastern Oncology Cooperative Group scale). Interventions: TARGETED AGENT(S) listed in capital (upper case) letters, using the generic name (if any), see also Additional Table 04; for consistency, the experimental arm (1) is listed before a control arm (2) and higher dose (1) before lower dose (2) arms; (1), (2) refers to the arm number used in this review for each paired comparison and may differ from the number assigned by the investigators; comparisons (A), (B), (C) are used to denote paired comparisons in three arm trials; Outcomes: RR = major remission rate; PFS = progression‐free survival; OS = overall survival; Toxicity = tabulation of major (grade 3+) toxicities; QOL = quality‐of‐life instrument; 'yes' = reported by arm; 'no' = not reported by arm. Notes: publication (known status as of search cutoff date in Methods) is the first of the following sequence ‐ peer‐reviewed journal (with or without updates); meeting abstract + presentation material available in public domain; meeting abstract only; other.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Escudier(6) 2009 | No outcome data by randomization arm |
Contributions of authors
Coppin: concept, protocol, literature search, duplicate data extraction, draft text
Le: duplicate literature search, primary data extraction
Porzsolt: critical appraisal of protocol and review
Wilt: Cochrane review group direction, critical appraisal of protocol and review
Kollmannsberger: content expertise, critical appraisal of review
Sources of support
Internal sources
BC Cancer Agency, Canada.
External sources
Prostate Diseases and Urologic Cancers CRG, USA.
Department of Veterans Affairs Health Services Research and Development (HSRD) Office, USA.
Cochrane Urological Cancers Subgroup, Velindre NHS Trust, Cardiff, UK.
Declarations of interest
To be declared by each reviewer.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Atkins 2004 {published data only}
- Atkins MB, Hidalgo M, Stadler W, et al. A randomized double‐blind phase 2 study of intravenous CCI‐779 administered weekly to patients with advanced renal cell carcinoma. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2002; Vol. 20 suppl:abstr 36.
- Atkins MB, Hidalgo M, Stadler WM, et al. Randomized phase II study of multiple dose levels of CCI‐779, a novel mammalian target of rapamycin inhibitor, in patients with advanced refractory renal cell carcinoma. Journal of Clinical Oncology 2004;22:909‐18. [DOI] [PubMed] [Google Scholar]
- Hidalgo M, Atkins MB, Stadler W, et al. A randomized double‐blind phase 2 study of intravenous CCI‐779 administered weekly to patients with advanced renal cell carcinoma: prognostic factor analysis. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2003; Vol. 21 suppl:abstr 804.
Atkins 2004(A) {published data only}
- see Atkins 2004.
Atkins 2004(B) {published data only}
- see Atkins 2004.
Atkins 2004(C) {published data only}
- see Atkins 2004.
Bhargava 2010 {published data only}
- Bhargava P, Esteves B, Al‐Adhami M, et al. Activity of Tivozanib (AV‐951) in patients with renal cell carcinoma: subgroup analysis from a phase II randomized discontinuation trial. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2010; Vol. 28 suppl:abstr 4599.
- Bhargava P, Esteves B, Al‐Adhami M, et al. Effect of hypertension, nephrectomy, and prior treatment on the efficacy of tivozanib (AV‐951) in a phase II discontinuation trial in patients with renal cell carcinoma. Genitourinary Cancers Symposium, American Society of Clinical Oncology. 2010:abstr 342.
- Bhargava P, Esteves B, Lipatov ON, et al. Activity and safety of AV‐951, a potent and selective VEGFR1, 2, and 3 kinase inhibitor, in patients with renal cell carcinoma: interim results of a phase II randomized discontinuation trial. Genitourinary Cancers Symposium, American Society of Clinical Oncology. 2009:abstr 283.
- Bhargava P, Esteves B, Nosov DA, Lipatov ON, et al. Updated activity and safety results of a phase II randomized discontinuation trial of AV‐951, a potent and selective VEGFR1, 2, and 3 kinase inhibitor, in patients with renal cell carcinoma. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2009; Vol. 27 suppl:abstr 5032.
Bracarda 2010 {published data only}
- Bracarda S, Ludovini V, Porta C, et al. Serum thrombospondin, vascular endothelial growth factor, VEGF receptor‐2, and basic‐fibroblast growth factor as predictive factors for sorafenib plus interferon‐alfa‐2a in metastatic renal cell carcinoma: biologic results from the randomized phase II RAPSODY trial. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2010; Vol. 28 suppl:abstr 4628.
- Bracarda S, Porta C, Boni C, et al. Randomized prospective phase II trial of two schedules of sorafenib daily and interferon‐a2a in metastatic renal cell carcinoma (RAPSODY): GOIRC Study 0681. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2007; Vol. 25 suppl:abstr 5100.
- Bracarda S, Porta C, Boni C, et al. Randomized prospective phase II trial of two schedules of sorafenib daily and interferon‐a2a in metastatic renal cell carcinoma (RAPSODY): GOIRC study 0681. Genitourinary Cancers Symposium, American Society of Clinical Oncology. 2008:abstr 357.
Bukowski 2007 {published data only}
- Bukowski RM, Kabbinavar F, Figlin RA, et al. Bevacizumab with or without erlotinib in metastatic renal cell carcinoma. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2006; Vol. 24 suppl:abstr 4523.
- Bukowski RM, Kabbinavar F, Figlin RA, et al. Bevacizumab with or without erlotinib in metastatic renal cell carcinoma. http://www.asco.org/ASCOv2/Meetings/Abstracts?&vmview=abst_detail_view&confID=40&abstractID=33238 [accessed 10 January 2007] 2006.
- Bukowski RM, Kabbinavar FF, Figlin RA, et al. Randomized phase II study of erlotinib combined with bevacizumab compared with bevacizumab alone in metastatic renal cell cancer. Journal of Clinical Oncology 2007;25:4536‐41. [DOI] [PubMed] [Google Scholar]
Ebbinghaus 2007 {published data only}
- Ebbinghaus S, Hussain M, Tannir N, et al. Phase 2 study of ABT‐510 in patients with previously untreated advanced renal cell carcinoma. Clinical Cancer Research 2007;13:6689‐95. [DOI] [PubMed] [Google Scholar]
- Ebbinghaus SW, Hussain M, Tannir NM, et al. A randomized phase II study of the thrombospondin‐mimetic peptide ABT‐510 in patients with previously untreated advanced renal cell carcinoma. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2005; Vol. 23 suppl:abstr 4607.
Escudier(1) 2007 {published data only}
- Escudier B, Choueiri TK, Oudard S, et al. Prognostic factors of metastatic renal cell carcinoma after failure of immunotherapy: new paradigm from a large phase III trial with shark cartilage extract AE 941. Journal of Urology 2007;178:1901‐5. [DOI] [PubMed] [Google Scholar]
- Escudier B, Venner P, Buckowski R, et al. Phase III trial of Neovastat in metastatic renal cell carcinoma patients refractory to immunotherapy. Journal of Oncology, ASCO Annual Meeting Proceedings. 2003; Vol. 22 suppl:abstr 844.
- Escudier B, Venner P, Bukowski R, et al. Phase III trial of Neovastat in metastatic renal cell carcinoma patients refractory to immunotherapy. European Journal of Cancer Supplements, ECCO 12 Meeting Proceedings. 2003; Vol. 1:abstr 1098.
- Nurzynski P, Zolnierek J, Korniluk J, et al. Cartilage in therapy of renal cell cancer [Preparaty chrzastki w terapii raka nerki [Polish]]. Wspolczesna Onkologia 2005;9:136‐9. [Google Scholar]
Escudier(2) 2010 {published data only}
- Antoun S, Birdsell L, Sawyer MB, Venner P, Baracos V. Association of skeletal muscle wasting with treatment with sorafenib in patients with advanced renal cell carcinoma: results from a placebo‐controlled study. Journal of Clinical Oncology 2010;28:1054‐60. [DOI] [PubMed] [Google Scholar]
- Bukowski R, Cella D, Gondek K, Escudier B. Effects of sorafenib on symptoms and quality of life. Results from a large randomized placebo‐controlled study in renal cancer. American Journal of Clinical Oncology 2007;30:220‐7. [DOI] [PubMed] [Google Scholar]
- Bukowski R, Eisen TE, Stadler WS, et al. Efficacy and safety of sorafenib in patients with advanced clear‐cell renal‐cell carcinoma with bone metastases: results from the phase III target study. European Journal of Cancer Supplements, ECCO 15 Meeting Proceedings. 2009; Vol. 7:abstr 7130.
- Bukowski RM, Eisen T, Szczylik C, et al. Final results of the randomized phase III trial of sorafenib in advanced renal cell carcinoma: survival and biomarker analysis. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2007; Vol. 25 suppl:abstr 5023.
- Bukowski RM, Eisen T, Szczylik C, et al. Final results of the randomized phase III trial of sorafenib in advanced renal cell carcinoma: survival and biomarker analysis. www.asco.org/virtual meeting [accessed 10 January 2007] 2007.
- Dhanda R, Gondek K, Song J, et al. A quality of life, symptoms and survival comparison in kidney cancer patients receiving sorafenib versus placebo. Annals of Oncology, ESMO Meeting Proceedings. 2006; Vol. 17 suppl:abstr 447PD.
- Eisen T, Bukowski RM, Staehler M, et al. Randomized phase III trial of sorafenib in advanced renal cell carcinoma: impact of crossover on survival. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2006; Vol. 24 suppl:abstr 4524.
- Eisen T, Oudard S, Szczylik C, et al. Clinical benefit of sorafenib in the elderly with advanced renal cell carcinoma: subgroup analysis of the approaches in renal cancer global evaluation trial (TARGETs). Annals of Oncology, ESMO Meeting Proceedings. 2006; Vol. 17 suppl:abstr 446PD.
- Eisen T, Oudard S, Szczylik C, et al. Sorafenib for older patients with renal cell carcinomas: subset analysis from a randomized trial. Journal of the National Cancer Institute 2008;100:1454‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escudier B, Eisen T, Stadler WM, et al. Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the Phase III Treatment Approaches in Renal cancer Global Evaluation Trial. Journal of Clinical Oncology 2009;27:3312‐8. [DOI] [PubMed] [Google Scholar]
- Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear‐cell renal‐cell carcinoma. New England Journal of Medicine 2007;356:125‐34. [DOI] [PubMed] [Google Scholar]
- Massard C, Zonierek J, Laplanche A, et al. Incidence of brain metastasis in advanced renal cell carcinoma among patients randomized in a phase III trial of sorafenib, an oral multi‐kinase inhibitor. Annals of Oncology, ESMO Meeting Proceedings. 2006; Vol. 17 suppl:abstr 454P.
- Negrier S, Jager E, Port C, et al. Sorafenib in patients with advanced renal cell carcinoma and prior cytokine therapy: subgroup analysis of TARGETs. Annals of Oncology, ESMO Meeting Proceedings. 2006; Vol. 17 suppl:abstr 4390.
Escudier(3) 2010 {published data only}
- Bajetta E, Ravaud A, Bracarda S, et al. Efficacy and safety of first‐line bevacizumab plus interferon‐a2a in patients ≥ 65 years with metastatic renal cell carcinoma. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2007; Vol. 26 suppl:abstr 5095.
- Bellmunt J, Melichar B, Bracarda S, et al. Bevacizumab and interferon therapy does not increase risk of cardiac events in metastatic renal cell carcinoma. European Journal of Cancer Supplements, ECCO 15 Meeting Proceedings. 2009; Vol. 7:abstr 7121.
- Bracarda S, Bellmunt J, Negrier SN, et al. What is the impact of subsequent anti‐neoplastic therapy on overall survival following first‐line bevacizumab/interferon‐alpha2a in metastatic renal cell carcinoma ‐ experience from AVOREN. European Journal of Cancer Supplements, ECCO 15 Meeting Proceedings. 2009; Vol. 7:abstr 7127.
- Bracarda S, Koralewski P, Pluzanska A, et al. Bevacizumab/interferon‐alpha2a provides a progression‐free survival benefit in all prespecified patient subgroups as first‐line treatment of metastatic renal cell carcinoma (AVOREN). European Journal of Cancer Supplements, ECCO 14 Meeting Proceedings. 2007; Vol. 5:abstr 4008.
- Escudier B, Koralewski P, Pluzanska A, et al. A randomized, controlled, double‐blind phase III study (AVOREN) of bevacizumab/interferon‐a2a vs placebo/interferon‐α2a as first‐line therapy in metastatic renal cell carcinoma. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2007; Vol. 25 suppl:abstr 3.
- Escudier B, Koralewski P, Pluzanska A, et al. A randomized, controlled, double‐blind phase III study (AVOREN) of bevacizumab/interferon‐a2a vs placebo/interferon‐α2a as first‐line therapy in metastatic renal cell carcinoma. www.asco.org/virtual meeting [accessed 10 January 2007] 2007.
- Escudier B, Pluzanska A, Koralewski P, et al. Bevacizumab plus interferon alfa‐2a for treatment of metastatic renal cell carcinoma: a randomized double‐blind phase III trial. Lancet 2007;370:2103‐11. [DOI] [PubMed] [Google Scholar]
- Escudier BB, Bellmunt J, Negrier S, et al. Phase III trial of bevacizumab plus interferon alfa‐2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. Journal of Clinical Oncology 2010;28:2144‐50. [DOI] [PubMed] [Google Scholar]
- Escudier BJ, Bellmunt J, Negrier, et al. Final results of the phase III, randomized, double‐blind AVOREN trial of first‐line bevacizumab + interferon‐α2a in metastatic renal cell carcinoma. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2009; Vol. 27 suppl:abstr 5020.
- Escudier BJ, Negrier S, Chevreau C, et al. Analysis of prognostic risk categories in the phase III AVOREN trial of first‐line bevacizumab plus interferon‐α2a in patients with metastatic renal cell carcinoma: French prognostic scoring system. Genitourinary Cancers Symposium, American Society of Clinical Oncology. 2010:abstr 417.
- Escudier BJ, Ravaud A, Negrier S, et al. Update on AVOREN trial in metastatic renal cell carcinoma: efficacy and safety in subgroups of patients and pharmacokinetic analysis. Genitourinary Cancers Symposium, American Society of Clinical Oncology. 2008:abstr 5025.
- Karakiewicz P, Sun M, Sneller V, Escudier B. Use of a nomogram to quantify overall survival (OS) benefit in patients with metastatic renal cell carcinoma receiving bevacizumab (BEV) with interferon (IFN) versus IFN alone. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2010 suppl; Vol. 28:abstr 4592.
- Karakiewicz PI, Sun M, Sneller V, et al. Use of a nomogram to quantify progression‐free survival benefit in metastatic renal cell carcinoma patients receiving bevacizumab plus interferon or interferon alone. Genitourinary Cancers Symposium, American Society of Clinical Oncology. 2010:abstr 392.
- Melichar B, Koralewski P, Pluzanska A, et al. First‐line bevacizumab improves progression‐free survival with lower doses of interferon‐α2a in the treatment of patients with metastatic renal cell carcinoma (AVOREN). European Journal of Cancer Supplements. 2007; Vol. 5:abstr 4518.
- Melichar B, Koralewski P, Ravaud A, et al. First‐line bevacizumab combined with reduced dose interferon‐α2a is active in patients with metastatic renal cell carcinoma. Annals of Oncology 2008;19:1470‐6. [DOI] [PubMed] [Google Scholar]
Escudier(4) 2009 {published data only}
- Escudier B, Szczylik C, Demkow T, et al. Randomized phase II trial of the multi‐kinase inhibitor sorafenib versus interferon in treatment‐naïve patients with metastatic renal cell carcinoma. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2006; Vol. 24 suppl:abstr 4501.
- Escudier B, Szczylik C, Hutson TE, et al. Randomized phase II trial of first‐line treatment with sorafenib versus interferon alfa‐2a in patients with metastatic renal cell carcinoma. Journal of Clinical Oncology 2009;27:1280‐9. [DOI] [PubMed] [Google Scholar]
- Szczylik C, Demkow T, Staehler M, et al. Randomized phase II trial of first‐line treatment with sorafenib versus interferon in patients with advanced renal cell carcinoma: final results. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2007; Vol. 25 suppl:abstr 5025. [DOI] [PubMed]
- Szczylik C, Demkow T, Staehler M, et al. Randomized phase II trial of first‐line treatment with sorafenib versus interferon in patients with advanced renal cell carcinoma: final results. www.asco.org/virtual meeting [accessed 10 January 2007] 2007. [DOI] [PubMed]
Escudier(5) 2010 {published data only}
- Escudier BJ, Negrier S, Gravis G, et al. Can the combination of temsirolimus and bevacizumab improve the treatment of metastatic renal cell carcinoma? Results of the randomized TORAVA phase II trial. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2010; Vol. 28 suppl:abstr 4516.
Gordon 2004 {published data only}
- Gordon MS, Manola J, Fairclough D, et al. Low dose interferon‐α2b + thalidomide in patients with previously untreated renal cell cancer. Improvement in progression‐free survival but not quality of life or overall survival. A phase III study of the Eastern Cooperative Oncology Group (E2898). Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2004; Vol. 22 suppl:abstr 4516.
- Gordon MS, Manola J, Fairclough D, et al. Low dose interferon‐α2b + thalidomide in patients with previously untreated renal cell cancer. Improvement in progression‐free survival but not quality of life or overall survival. A phase III study of the Eastern Cooperative Oncology Group (E2898). www.asco.org/virtual meeting [accessed 10 January 2007] 2004.
Hudes 2010 {published data only}
- Alemao E, Rajagopalan S, Yang S, et al. Quality adjusted survival in randomized, controlled trials: application of inverse probability weighting in renal cell cancer. Genitourinary Cancers Symposium, American Society of Clinical Oncology. 2010; Vol. 28 suppl:abstr 399.
- Armstrong AJ, George DJ, Halabi S. Serum lactate dehydrogenase as a biomarker for survival with mTOR inhibition in patients with metastatic renal cell carcinoma. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2010; Vol. 28 suppl:abstr 4631.
- Dutcher JP, Szczylik C, Tannir N, et al. Correlation of survival with tumor histology, age, and prognostic risk group for previously untreated patients with advanced renal cell carcinoma receiving temsirolimus or interferon‐alpha. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2007; Vol. 25 suppl:abstr 5033.
- Dutcher JP, Szczylik C, Tannir N, et al. Correlation of survival with tumor histology, age, and prognostic risk group for previously untreated patients with advanced renal cell carcinoma receiving temsirolimus or interferon‐alpha. www.asco.org/virtual meeting [accessed 10 January 2007] 2007.
- Dutcher JP, Souza P, Figlin R, et al. Effect of temsirolimus versus interferon‐α on survival of patients with advanced renal cell carcinoma of different histologies. Genitourinary Cancers Symposium, American Society of Clinical Oncology. 2008:abstr 384.
- Dutcher JP, Souza P, McDermott D, et al. Effect of temsirolimus versus interferon‐α on outcome of patients with advanced renal cell carcinoma of different histologies. Medical Oncology 2009;26:202‐9. [DOI] [PubMed] [Google Scholar]
- Hudes G, Carducci M, Tomczak P, et al. A phase 3, randomized, 3‐arm study of temsirolimus or interferon‐alpha or the combination in the treatment of first‐line, poor‐risk patients with advanced renal cell carcinoma. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2006; Vol. 24 suppl:abstr LBA4.
- Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal‐cell carcinoma. New England Journal of Medicine 2007;356:2271‐81. [DOI] [PubMed] [Google Scholar]
- Logan T, McDermott D, Dutcher J, et al. Exploratory analysis of the influence of nephrectomy status on temsirolimus efficacy in patients with advanced renal cell carcinoma and poor‐risk features. Genitourinary Cancers Symposium, American Society of Clinical Oncology. 2009:abstr 281.
- Pablo M, Hudes G, Dutcher J, et al. Radiographic findings of drug‐induced pneumonitis and clinical correlation in patients with advanced renal cell carcinoma treated with temsirolimus. European Journal of Cancer Supplements, ECCO 15 Meeting Proceedings. 2009; Vol. 7:abstr 7113.
- Parasuraman S, Hudes G, Levy D, et al. Comparison of quality‐adjusted survival in patients with advanced renal cell carcinoma receiving first‐line treatment with temsirolimus (TEMSR) or interferon‐α (IFN) or the combination of IFN+TEMSR. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2007; Vol. 25 suppl:abstr 5049.
- Parasuraman S, Hudes G, Levy D, et al. Comparison of quality‐adjusted survival in patients with advanced renal cell carcinoma receiving first‐line treatment with temsirolimus (TEMSR) or interferon‐α (IFN) or the combination of IFN+TEMSR. www.asco.org/virtual meeting [accessed 15 January 2007] 2007.
- Yang S, Hudes G, Souza P, et al. Evaluation of quality of life in patients with advanced renal cell carcinoma treated with temsirolimus vs interferon‐alfa: results from a phase III randomized trial. European Journal of Cancer Supplements, ECCO 15 Meeting Proceedings. 2009; Vol. 7:abstr 7134.
- Zbrozek AS, Hudes G, Levy D, et al. Q‐TWiST analysis of patients receiving temsirolimus or interferon alpha for treatment of advanced renal cell carcinoma. Pharmacoeconomics 2010;28:577‐84. [DOI] [PubMed] [Google Scholar]
Hudes 2010(A) {published data only}
- see Hudes 2010.
Hudes 2010(B) {published data only}
- see Hudes 2010.
Hudes 2010(C) {published data only}
- see Hudes 2010.
Jonasch 2010 {published data only}
- Jonasch E, Corn P, Asche RG, et al. Randomized phase II study of sorafenib with or without low‐dose IFN in patients with metastatic renal cell carcinoma. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2007; Vol. 25 suppl:abstr 5104.
- Jonasch E, Corn P, Pagliaro LC, et al. Upfront, randomized, phase 2 trial of sorafenib versus sorafenib and low‐dose interferon alfa in patients with advanced renal cell carcinoma. Cancer 2010;116:57‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannir NM, Zurita AJ, Heymach JV, et al. A randomized phase II trial of sorafenib versus sorafenib plus low‐dose interferon‐alfa: clinical results and biomarker analysis. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2008; Vol. 26 suppl:abstr 5093.
Lee 2006 {published data only}
- Lee CP, Patel PM, Selby PJ, et al. Randomized phase II study comparing thalidomide with medroxyprogesterone acetate in patients with metastatic renal cell carcinoma. Journal of Clinical Oncology 2006;24:898‐903. [DOI] [PubMed] [Google Scholar]
Madhusudan 2004 {published data only}
- Madhusudan S, Protheroe A, Vasey P, et al. A randomized phase II study of interferon alpha alone or in combination with thalidomide in metastatic renal cancer. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2004; Vol. 22 suppl:abstr 4742.
Motzer(1) 2010 {published data only}
- Castellano D, Garcia del Muro X, Perez‐Gracia JL, et al. Patient‐reported outcomes ina phase III, randomized study of sunitinib versus interferon‐a as first‐line systemic therapy for patients with metastatic renal cell carcinoma in a European population. Annals of Oncology 2009;20:1803‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella D, Cappelleri JC, Bushmakin A, et al. Quality of life predicts progression‐free survival in patients with renal cell carcinoma treated with sunitinib versus interferon alfa. Journal of Oncology Practice 2009;5:66‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella D, Li JZ, Cappelari JC, et al. Quality of life in patients with metastatic renal cell carcinoma treated with sunitinib or interferon alfa: results from a phase III randomized trial. Journal of Clinical Oncology 2008;26:3763‐9. [DOI] [PubMed] [Google Scholar]
- Cella D, Li JZ, Cappelleri JC, et al. Quality of life predicts for progression‐free survival in patients with metastatic renal cell carcinoma treated with sunitinib compared to interferon‐alpha. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2007; Vol. 25 suppl:abstr 6594.
- Cella D, Li JZ, Cappelleri JC, et al. Quality of life predicts for progression‐free survival in patients with metastatic renal cell carcinoma treated with sunitinib compared to interferon‐alpha. www.asco.org/virtual meeting [accessed 10 January 2007] 2007.
- Cella D, Michaelson MD, Bushmakin AG, et al. Health‐related quality of life in patients with metastatic renal cell carcinoma treated with sunitinib vs interferon‐a in a phase III trial: final results and geographical analysis. British Journal of Cancer 2010;102:658‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figin RA, Hutson TE, Tomczak P, et al. Overall survival with sunitinib versus interferon‐alfa as first‐line treatment of metastatic renal cell carcinoma. Journal of Clinical Oncology, American Society of Clinical Oncology Annual Meeting Proceedings. 2008; Vol. 26 suppl:abstr 5024.
- Motzer RJ, Bukowski RM, Figlin RA, et al. Prognostic nomogram for sunitinib in patients with metastatic renal cell carcinoma. Cancer 2008;11:1552‐8. [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Figlin RA, Hutson TE, et al. Sunitinib versus interferon‐alfa as first‐line treatment of metastatic renal cell carcinoma: updated results and analysis of prognostic factors. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2007; Vol. 25 suppl:abstr 5024.
- Motzer RJ, Figlin RA, Hutson TE, et al. Sunitinib versus interferon‐alfa as first‐line treatment of metastatic renal cell carcinoma: updated results and analysis of prognostic factors. www.asco.org/virtual meeting [accessed 10 January 2007] 2007.
- Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. Journal of Clinical Oncology 2009;27:3584‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzer RJ, Hutson TE, Tomczak P, et al. Phase III randomized trial of sunitinib malate (SU11248) versus interferon‐alfa as first‐line systemic therapy for patients with metastatic renal cell carcinoma. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2006; Vol. 24 suppl:abstr LBA3.
- Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal‐cell carcinoma. New England Journal of Medicine 2007;356:115‐24. [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Michaelson MD, Hutson TE, et al. Sunitinib versus interferon‐alfa as first‐line treatment of metastatic renal cell carcinoma: updated efficacy and safety results and further analysis of prognostic factors. European Journal of Cancer Supplements. 2007; Vol. 5:abstr 4509.
- Patil S, Figlin RA, Hutson TE, et al. TWiST analysis to estimate overall benefit for metastatic renal cell carcinoma patients treated in a phase III trial with sunitinib versus interferon‐alfa. Journal of Clinical Oncology, ASCO Annual Meeting proceedings. 2010; Vol. 28 suppl:abstr 4594.
- Wilkerson J, Stein WD, Kim ST, et al. Validation of kinetic analysis of renal cancer regression and growth following treatment with sunitinib and interferon‐alfa: analysis of the pivotal randomized trial. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2010; Vol. 28 suppl:abstr 4597.
Motzer(2) 2010 {published data only}
- Beaumont J, Cella D, Hollaender N, et al. Results from additional analyses of patient reported outcomes in RECORD‐1 ‐ a randomized trial of everolimus with metastatic renal cell carcinoma patients. European Journal of Cancer Supplements, ECCO 15 Meeting Proceedings. 2009; Vol. 7:abstr 7127.
- Beaumont J, Cella D, Hutson T, et al. Patient‐reported outcomes in a randomized trial of everolimus with metastatic renal cell carcinoma patients. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2009; Vol. 27 suppl:abstr e17516.
- Escudier B, Ravaud A, Oudard S, et al. Phase‐3 randomized trial of everolimus (RAD001) vs placebo in metastatic renal cell carcinoma. Annals of Oncology, ESMO Meeting Proceedings. 2008; Vol. 18 suppl:abstr 720.
- Hutson T, Negrier S, Kay A. Randomized placebo‐controlled, phase 3 study of everolimus, a novel therapy for patients with metastatic renal cell carcinoma: subgroup analysis of patients progressing on prior bevacizumab therapy. European Journal of Cancer Supplements, ECCO 15 Meeting Proceedings. 2009; Vol. 7:abstr 7136.
- Hutson TE, Calvo E, Escudier BJ, et al. Everolimus in elderly patients with metastatic renal cell carcinoma: an exploratory analysis of the RECORD‐1 study. Genitourinary Cancers Symposium Proceedings, American Society of Oncology. 2010:abstr 362.
- Kay A, Motzer R, Figlin R, et al. Updated data from a phase III randomized trial of everolimus (RAD001) versus PBO in metastatic renal cell carcinoma. Genitourinary Cancers Symposium Proceedings, American Society of Oncology. 2009:abstr 278.
- Korhonen P, Haas T, Zuber E, et al. Overall survival among metastatic renal cell carcinoma patients corrected for crossover using a rank preserving structural failure time model: analyses from the everolimus phase III trial. European Journal of Cancer Supplements, ECCO 15 Meeting Proceedings. 2009; Vol. 7:abstr 7155.
- Korhonen P, Malangone E, Sherman S, et al. Overall survival of metastatic renal cell carcinoma patients corrected for crossover using inverse probability of censoring wights and rank preserving structural failure time models: two analyses from the RECORD‐1 trial. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2010; Vol. 28 suppl:abstr 4595.
- Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double‐blind, randomized, placebo‐controlled phase III trial. Lancet 2008;372:449‐56. [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Escudier B, Oudard S, et al. RAD001 vs placebo in patients with metastatic renal cell carcinoma after progression on VEGFr‐TKI therapy: results from a randomized, double‐blind, multicenter phase‐III study. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2008; Vol. 26 suppl:abstr LBA5026.
- Osanto S, Hutson TE, Calvo E, et al. Efficacy and safety of everolimus in elderly patients with metastatic renal cell carcinoma. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2010; Vol. 28 suppl:abstr 4608.
- White DA, Camus P, Endo M, et al. Noninfectious pneumonitis after everolimus therapy for advanced renal cell carcinoma. American Journal of Respirology and Critical Care Medicine 2010;182:396‐403. [DOI] [PubMed] [Google Scholar]
- Wiederkehr D, Howe CJ, Casciano R, et al. Overall survival among metastatic renal cell carcinoma patients corrected for crossover using inverse probability of censoring weights: analyses from the everolimus phase III trial. European Journal of Cancer Supplements, ECCO 15 Meeting Proceedings. 2009; Vol. 7:abstr 7131.
- Zuber E, Korhonen P, Branson M, et al. Correcting overall survival effect for the impact of cross‐over via rank preserving structural failure time model: case of mRCC RECORD‐1 trial of everolimus. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2010; Vol. 28 suppl:abstr 4690.
Procopio 2010 {published data only}
- Procopio G, Verzoni E, Bracarda S, et al. A randomized, prospective, phase 2 study, with sorafenib (So) and interleukin‐2 versus So alone as first line treatment in advanced renal cell cancer: ROSORC trial. European Journal of Cancer Supplements, ECCO 15 Meeting Proceedings. 2009; Vol. 7:abstr 7107.
- Procopio G, Verzoni E, Bracarda S, et al. Subgroup analysis and updated results of the randomized study comparing sorafenib (So) plus interleukin‐2 versus So alone as first‐line treatment in metastatic renal cell carcinoma. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2010; Vol. 28 suppl:abstr 4589.
Ratain 2006 {published data only}
- Ratain MJ, Eisen T, Stadler WM, et al. Phase II placebo‐controlled randomized discontinuation trial of sorafenib in patients with metastatic renal cell carcinoma. Journal of Clinical Oncology 2006;24:2505‐12. [DOI] [PubMed] [Google Scholar]
Ravaud 2008 {published data only}
- Hawkins RE, Ravaud A, Masse H, et al. Lapatinib extends survival in patients with high ErbB1 (EGFR) tumour expression: subgroup results of a phase III trial in advanced renal cell carcinoma. Annals of Oncology, ESMO Meeting Proceedings. 2006; Vol. 17 suppl:abstr 440O.
- Ravaud A, Gardner J, Hawkins R, et al. Efficacy of lapatinib in patients with high tumor EGFR expression: results of a phase III trial in advanced renal cell carcinoma. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2006; Vol. 24 suppl:abstr 4502.
- Ravaud A, Gardner J, Hawkins R, et al. Efficacy of lapatinib in patients with high tumor EGFR expression: results of a phase III trial in advanced renal cell carcinoma. www.asco.org/virtual meeting [accessed 10 January 2007] 2006.
- Ravaud A, Hawkins R, Gardner JP, et al. Lapatinib versus hormone therapy in patients with advanced renal cell carcinoma: a randomized phase III clinical trial. Journal of Clinical Oncology 2008;26:2285‐91. [DOI] [PubMed] [Google Scholar]
Rini 2010 {published data only}
- Halabi S, Rini BI, Stadler WM, Small EJ. Use of progression‐free survival to predict overall survival in patients with metastatic renal cell carcinoma. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2010; Vol. 28 suppl:abstr 4525.
- Harzstark AL, Halabi S, Stadler WM, et al. Hypertension is associated with clinical outcome for patients with metastatic renal cell carcinoma treated with interferon and bevacizumab on CALGB 90206. Genitourinary Cancers Symposium, American Society of Clinical Oncology. 2010:abstr 351.
- Rini BI, Halabi S, Rosenberg J, et al. Bevacizumab plus interferon‐alfa versus interferon‐alfa monotherapy in patients with metastatic renal cell carcinoma: results of overall survival for CALGB 90206. Journal of Clinical Oncology, 2009 American Society of Clinical Oncology Annual Meeting Proceedings. 2009; Vol. 27 suppl:abstr LBA5019.
- Rini BI, Halabi S, Rosenberg JE, et al. Bevacizumab plus interferon alfa compared with interferon alfa monotherapy in patients with metastatic renal cell carcinoma: CALGB 90206. Journal of Clinical Oncology 2008;26:5422‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rini BI, Halabi S, Rosenberg JE, et al. CALGB 90206: a phase III trial of bevacizumab plus interferon‐alfa versus interferon‐alfa monotherapy in metastatic renal cell carcinoma. Genitourinary Cancers Symposium, American Society of Clinical Oncology. 2008:abstr 350.
- Rini BI, Halabi S, Taylor J, Small EJ, Schilsky RL. Cancer and Leukemia Group B 90206: a randomized phase III trial of interferon‐a or interferon‐a plus anti‐vascular endothelial growth factor antibody (bevacizumab) in metastatic renal cell carcinoma. Clinical Cancer Research 2004;10:2584‐6. [DOI] [PubMed] [Google Scholar]
Srinivas 2005 {published data only}
- Srinivas S, Guarding AE. A lower dose of thalidomide is better than a high dose in metastatic renal cell carcinoma. BJU International 2005;96:536‐9. [DOI] [PubMed] [Google Scholar]
Stadler 2005 {published data only}
- Stadler WM, Rosner G, Small E, et al. Successful implementation of the randomized discontinuation trial design: an application to the study of the putative antiangiogenic agent carboxyaminoimidazole in renal cell carcinoma ‐CALGB 69901. Journal of Clinical Oncology 2005;23:3726‐32. [DOI] [PubMed] [Google Scholar]
Sternberg 2010 {published data only}
- Hutson TE, Bukowski RM. A phase II study of GW786034 using a randomized discontinuation design in patients with locally recurrent or metastatic clear‐cell renal cell carcinoma. Clinical Genitourinary Cancer 2006;4:296‐8. [DOI] [PubMed] [Google Scholar]
- Hutson TE, Davis ID, Machiels JP, et al. Pazopanib (GW786034) is active in metastatic renal cell carcinoma: interim results of a phase II randomized discontinuation trial. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2007; Vol. 25 suppl:abstr 5031.
- Hutson TE, Davis ID, Machiels JP, et al. Pazopanib (GW786034) is active in metastatic renal cell carcinoma: interim results of a phase II randomized discontinuation trial. www.asco.org/virtual meeting [accessed 10 January 2007] 2007.
- Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. Journal of Clinical Oncology 2010;28:1061‐8. [DOI] [PubMed] [Google Scholar]
- Sternberg CN, Szczylik C, Lee E, et al. A randomized, double‐blind phase III study of pazopanib in treatment‐naive and cytokine‐pretreated patients with advanced renal cell carcinoma. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2009; Vol. 27 suppl:abstr 5021.
Yang 2003 {published data only}
- Elaraj DM, White DE, Steinberg SM, et al. A pilot study of anti‐angiogenic therapy with bevacizumab and thalidomide in patients with metastatic renal cell carcinoma. Journal of Immunotherapy 2004;27:259‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JC, Haworth L, Sherry RM, et al. A randomized trial of bevacizumab, an anti‐vascular endothelial growth factor antibody, for metastatic renal cancer. New England Journal of Medicine 2003;349:427‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JC, Haworth L, Steinberg S, Rosenberg SA, Novotny W. A randomized double‐blind placebo‐controlled trial of bevacizumab (anti‐VEGF antibody) demonstrating a prolongation in time to progression in patients with metastatic renal cancer. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2002; Vol. 20 suppl:abstr 15.
Yang 2003(A) {published data only}
- see Yang 2003.
Yang 2003(B) {published data only}
- see Yang 2003.
Yang 2003(C) {published data only}
- see Yang 2003.
References to studies excluded from this review
Escudier(6) 2009 {published data only}
- Escudier B, Roigas J, Gilleson S, et al. Phase II study of sunitinib administered in a continuous once‐daily dosing regimen in patients with cytokine‐refractory metastatic renal cell carcinoma. Journal of Clinical Oncology 2009;27:4068‐75. [DOI] [PubMed] [Google Scholar]
- Escudier B, Srinivas S, Roigas J, et al. A phase II study of continuous daily administration of sunitinib in patients with cytokine‐refractory metastatic renal cell carcinoma ‐ final results. European Journal of Cancer Supplements, ECCO 14 Meeting Proceedings. 2007; Vol. 5:abstr 4504.
- Srinivas S, Roigas J, Gillessen S, et al. Continuous daily administration of sunitinib in patients with cytokine‐refractory metastatic renal cell carcinoma. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings 2007;25 suppl:abstr 5040. [DOI] [PubMed] [Google Scholar]
Additional references
Adjei 2005*
- Adjei AA, Hidalgo M. Intracellular signal transduction pathway proteins as targets for cancer therapy. Journal of Clinical Oncology 2005;23:5386‐5403. [DOI] [PubMed] [Google Scholar]
Amato 2007a*
- Amato RJ, Harris P, Dalton M, et al. A phase II trial of intra‐patient dose‐escalated sorafenib in patients with metastatic renal cell cancer. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2007; Vol. 25 suppl:abstr 5026.
Amato 2007b*
- Amato RJ, Harris P, Dalton M, et al. A phase II trial of intra‐patient dose‐escalated sorafenib in patients with metastatic renal cell cancer. www.asco.org/virtual meeting [accessed 10 January 2007] 2007.
Antoun 2010*
- Antoun S, Birdsell L, Sawyer MB, et al. Association of skeletal muscle wasting with treatment with sorafenib in patients with advanced renal cell carcinoma: results from a placebo‐controlled study. Journal of Clinical Oncology 2010;28:1054‐60. [DOI] [PubMed] [Google Scholar]
Bergsland 2006*
- Bergsland EK. When does the presence of the target predict response to the targeted agent?. Journal of Clinical Oncology 2006;24:1‐4. [DOI] [PubMed] [Google Scholar]
Bukowski 2007*
- Bukowski R, Cella D, Gondek K, Escudier B. Effects of sorafenib on symptoms and quality of life. Results from a large randomized placebo‐controlled study in renal cancer. American Journal of Clinical Oncology 2007;30:220‐7. [DOI] [PubMed] [Google Scholar]
Chow 2007*
- Chow LQM, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. Journal of Clinical Oncology 2007;25:884‐96. [DOI] [PubMed] [Google Scholar]
Coppin 2006*
- Coppin C, Porzsolt F, Awa A, Kumpf J, Coldman A, Wilt T. Immunotherapy for advanced renal cell cancer. Cochrane Database of Systematic Reviews 2006, Issue 1. [DOI: 10.1002/14651858.CD001425.pub2] [DOI] [PubMed] [Google Scholar]
Coppin 2010*
- Coppin C. Everolimus: the first approved product for patients with advanced renal cell cancer after sunitinib and/or sorafenib. Biologics: Targets and Therapy 2010;4:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Deiniger 2005*
- Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood 2005;105:2640‐53. [DOI] [PubMed] [Google Scholar]
Dutcher 2007*
- Dutcher JP, Szczylik C, Tannir N, et al. Correlation of survival with tumor histology, age, and prognostic risk group for previously untreated patients with advanced renal cell carcinoma receiving temsirolimus or interferon‐alpha. www.asco.org/virtual meeting [accessed 10 January 2007]. 2007:abstract A5033.
Dutcher 2009*
- Dutcher JP, Souza P, McDermott D, et al. Effect of temsirolimus versus interferon‐α on outcome of patients with advanced renal cell carcinoma of different tumor histologies. Medical Oncology 2009;26:202‐9. [DOI] [PubMed] [Google Scholar]
Eisen 2008*
- Eisen T, Oudard S, Szczylik C, et al. Sorafenib for older patients with renal cell carcinoma: subset analysis from a randomized trial. Journal of the National Cancer Institute 2008;100:1454‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Escudier 2009*
- Escudier B, Eisen T, Stadler WM, Szczylik C, et al. Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. Journal of Clinical Oncology 2009;27:3312‐8. [DOI] [PubMed] [Google Scholar]
Furge 2010*
- Furge KA, MacKeigan JP, Teh BT. Kinase targets in renal‐cell carcinomas: reassessing the old and discovering the new. Lancet Oncology 2010;11:571‐8. [DOI] [PubMed] [Google Scholar]
Gleave 1998*
- Gleave ME, Elhilali M, Fradet Y, Davis I, Venner P, Saad F, et al. Interferon gamma‐1b compared with placebo in metastatic renal‐cell carcinoma. New England Journal of Medicine 1998;338:1265‐71. [DOI] [PubMed] [Google Scholar]
Goffin 2005*
- Goffin J, Baral S, Tu D, Nomikos D, Seymour L. Objective responses in patients with malignant melanoma or renal cell cancer in early studies do not predict regulatory approval. Clinical Cancer Research 2005;11:5928‐34. [DOI] [PubMed] [Google Scholar]
Gore 2009*
- Gore ME, Szczylik C, BracardaS, et al. Safety and efficacy of sunitinib for metastatic renal‐cell carcinoma: an expanded‐access trial. Lancet Oncology 2009;10:757‐63. [DOI] [PubMed] [Google Scholar]
GRADEpro [Computer program]
- GRADEpro. Version 3.2 for Windows, Jan Brozek, Andrew Oxman, Holger Schünemann, 2008.
Hacker 2010*
- Hacker KE, Rathmell WK. Emerging molecular classification in renal cell carcinoma: implications for drug development. Targeted Oncology 2010;5:75‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Harding 2003*
- Harding MW. Immunophilins, mTOR, and pharmacodynamic strategies for a targeted cancer therapy. Clinical Cancer Research 2003;9:2882‐6. [PubMed] [Google Scholar]
Heng 2009*
- Heng DYC, Xie W, Regan MM, et al. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor‐targeted agents: results from a large, multicenter study. Journal of Clinical Oncology 2009;27:5794‐9. [DOI] [PubMed] [Google Scholar]
Hicklin 2005*
- Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. Journal of Clinical Oncology 2005;23:1011‐27. [DOI] [PubMed] [Google Scholar]
Higgins 2005*
- Higgins JPT, Green S, (editors). Cochrane Handbook for Systematic Reviews of Interventions 5.0.1 [updated September 2008]. The Cochrane Collaboration, 2008. Available from www.cochrane‐handbook.org [accessed 24 November 2010].
Houk 2010*
- Houk BE, Bello CL, Poland B, Rosen LS, Demetri GD, Motzer RJ. Relationship between exposure to sunitinib and efficacy and tolerability endpoints in patients with cancer: results of a pharmacokinetic/pharmacodynamic meta‐analysis. Cancer Chemotherapy Pharmacology 2010;66:357‐71. [DOI] [PubMed] [Google Scholar]
Hutson 2007*
- Hutson TE, Davis ID, Machiels JP, et al. Pazopanib (GW786034) is active in metastatic renal cell carcinoma: interim results of a phase II randomized discontinuation trial. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2007; Vol. 25 suppl:abstr 5031.
Kaelin 2008*
- Kaelin WG Jr. Kidney cancer: now available in a new flavor. Cancer Cell 2008;14:423‐4. [DOI] [PubMed] [Google Scholar]
Leibovich 2005*
- Leibovich BC, Cheville JC, Lohse CM, et al. A scoring algorithm to predict survival for patients with metastatic clear cell renal cell carcinoma: a stratification tool for prospective clinical trials. Journal of Urology 2005;174:1759‐63. [DOI] [PubMed] [Google Scholar]
Linehan 2005*
- Linehan WM, Grubb RL, Coleman JA, Zbar B, Walther MM. The genetic basis of cancer of the kidney: implications for gene‐specific management. BJU International 2005;95(S2):2‐7. [DOI] [PubMed] [Google Scholar]
Massard 2006*
- Massard C, Zonierek J, Laplanche A, et al. Incidence of brain metastasis in advanced renal cell carcinoma among patients randomized in a phase III trial of sorafenib, an oral multi‐kinase inhibitor. Annals of Oncology, ESMO Meeting Proceedings. 2006; Vol. 17(S9):abstr 454P.
McDermott 2005*
- McDermott DF, Regan MM, Clark JI, Flaherty LE, Weiss GR, Logan TF, et al. Randomized phase III trial of high‐dose interleukin‐2 versus subcutaneous interleukin‐2 and interferon in patients with metastatic renal cell carcinoma. Journal of Clinical Oncology 2005;23:133‐41. [DOI] [PubMed] [Google Scholar]
Mekhail 2005*
- Mekhail TM, Abou‐Jawde RM, BouMerhi G, et al. Validation and extension of the Memorial Sloan‐Kettering prognostic factors model for survival in patients with previously untreated metastatic renal cell carcinoma. Journal of Clinical Oncology 2005;23:832‐41. [DOI] [PubMed] [Google Scholar]
Merchan 2007*
- Merchan JR, G. Liu, T. Fitch, et al. Phase I/II trial of CCI‐779 and bevacizumab in stage IV renal cell carcinoma: phase I safety and activity results. Journal of Clinical Oncology, ASCO Annual Meeting Proceedings. 2007; Vol. 25 suppl:abstr 5034.
Mickisch 2003*
- Mickisch GHJ. Rational selection of a control arm for randomized trials in metastatic renal cell carcinoma. European Urology 2003;43:670‐9. [DOI] [PubMed] [Google Scholar]
Miller 1981*
- Miller AB, Hoogstraten B, Staquet M, Winkler A. Reporting results of cancer treatment. Cancer 1981;47:207‐14. [DOI] [PubMed] [Google Scholar]
Mills 2009*
- Mills EJ, Rachlis B, O'Regan C, Thabane L, Perri D. Metastatic renal cell cancer treatments: an indirect comparison meta‐analysis. BMC Cancer 2009;9:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Moher 2001*
- Moher D, Schulz KF, Altman D. The CONSORT statement: revised recommendations for improving the quality of reports of parallel‐group randomized trials. Journal of the American Medical Association 2001;285:1987‐91. [DOI] [PubMed] [Google Scholar]
Motzer 1999*
- Motzer RJ, Mazumdar M, Bacik J, et al. Survival and prognostic stratification of 670 patients with advanced renal cell carcinoma. Journal of Clinical Oncology 1999;17:2530‐40. [DOI] [PubMed] [Google Scholar]
Motzer 2002*
- Motzer RJ, Bacik J, Murphy BA, Russo P, Mazumdar M. Interferon‐alfa as a comparative treatment for clinical trials of new therapies against advanced renal cell carcinoma. Journal of Clinical Oncology 2002;20:289‐96. [DOI] [PubMed] [Google Scholar]
Motzer 2004*
- Motzer RJ, Bacik J, Schwartz LH, Rueter V, Russo P, Marion S, Mazumdar M. Prognostic factors for survival in previously treated patients with metastatic renal cell carcinoma. Journal of Clinical Oncology 2004;22:454‐63. [DOI] [PubMed] [Google Scholar]
Negrier 2005*
- Negrier S, Gomez F, Douillard J‐Y, et al. Prognostic factors of response or failure of treatment in patients with metastatic renal carcinomas treated by cytokines: a report from the Groupe Francais d'Immunotherapie. World Journal of Urology 2005;23:161‐5. [DOI] [PubMed] [Google Scholar]
NICE 2002*
- Mason M, Shelley M, Court S, Burgon K. Guidance on cancer services: improving outcomes in urological cancers ‐ the research evidence. National Institute for Clinical Excellence, UK. National Institute for Clinical Excellence, 2002:300‐26. [Google Scholar]
Nuijten 2010*
- Nuijten M, Mickisch G. SUN vs BEV + IFN in first‐line mRCC therapy: no evidence for a statistically significant difference in progression‐free survival. British Journal of Cancer 2010;102:232‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Oliver 1989*
- Oliver RTD, Nethersell ABW, Bottomly JM. Unexplained spontaneous regression and alpha‐interferon as treatment for metastatic renal carcinoma. British Journal of Urology 1989;63:128‐31. [DOI] [PubMed] [Google Scholar]
Pal 2010*
- Pal SK, Figlin RA. Targeted therapies for renal cell carcinoma: understanding their impact on survival. Targeted Oncology 2010;5:131‐8. [DOI] [PubMed] [Google Scholar]
Parmar 1998*
- Parmar MKB, Torri V, Stewart L. Extracting summary statistics to perform meta‐analysis of the published literature for survival endpoints. Statistics in Medicine 1998;17:2815‐34. [DOI] [PubMed] [Google Scholar]
Patil 2010*
- Patil S, Ishill N, Deluca J, Motzer RJ. Stage migration and increasing proportion of favorable‐prognosis metastatic renal cell carcinoma patients. Cancer 2010;116:347‐54. [DOI] [PubMed] [Google Scholar]
Rini 2005*
- Rini B, Small EJ. Biology and clinical development of vascular endothelial growth factor‐targeted therapy in renal cell carcinoma. Journal of Clinical Oncology 2005;23:1028‐43. [DOI] [PubMed] [Google Scholar]
Rini 2009*
- Rini BI, Wilding G, Hudes G, et al. Phase II study of axitinib in sorafenib‐refractory metastatic renal cell carcinoma. Journal of Clinical Oncology 2009;27:4462‐8. [DOI] [PubMed] [Google Scholar]
Rini 2010*
- Rini BI. New strategies in kidney cancer: therapeutic advances through understanding the molecular basis of response and resistance. Clinical Cancer Research 2010;16:1348‐54. [DOI] [PubMed] [Google Scholar]
Roberts 2003*
- Roberts TG Jr, Lynch TJ Jr, Chabner BA. The phase III trial in the era of targeted therapy: unravelling the "go or no go" decision. Journal of Clinical Oncology 2003;21:3683‐95. [DOI] [PubMed] [Google Scholar]
Sawyers 2004*
- Sawyers C. Targeted cancer therapy. Nature 2004;432:294‐7. [DOI] [PubMed] [Google Scholar]
Stadler 2006*
- Stadler WM. New targets, therapies, and toxicities: lessons to be learned. Journal of Clinical Oncology 2006;24:1‐2. [DOI] [PubMed] [Google Scholar]
Stadler 2010*
- Stadler WM, Figlin RA, McDermott DF, et al. Safety and efficacy results of the advanced renal cell carcinoma sorafenib expanded access program in North America. Cancer 2010;116:1270‐80. [DOI] [PubMed] [Google Scholar]
Tanaka 2008*
- Tanaka C, O'Reilly T, Kovarik JM, et al. Identifying optimal biologic doses of everolimus (RAD001) in patients with cancer based on the modeling of preclinical and pharmacokinetic and pharmacodynamic data. Journal of Clinical Oncology 2008;26:1596‐1602. [DOI] [PubMed] [Google Scholar]
Therasse 2000*
- Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. Journal of the National Cancer Institute 2000;92:205‐16. [DOI] [PubMed] [Google Scholar]
Thompson Coon 2009*
- Thompson Coon JS, Liu Z, Hoyle M, et al. Sunitinib and bevacizumab for first‐line treatment of metastatic renal cell carcinoma: a systematic review and indirect comparison of clinical effectiveness. British Journal of Cancer 2009;101:238‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Thompson Coon 2010a*
- Thompson Coon J, Hoyle M, Green C, et al. Bevacizumab, sorafenib tosylate, sunitinib and temsirolimus for renal cell carcinoma: a systematic review and economic evaluation. Health Technology Assessment 2010;14(2):1‐224. [DOI] [PubMed] [Google Scholar]
Thompson Coon 2010b*
- Thompson Coon JS, Liu Z, Hoyle M, et al. Reply: SUN vs BEV/IFN in first‐line mRCC therapy. British Journal of Cancer 2010;102:234‐5. [Google Scholar]
Uzzo 2003*
- Uzzo RG, Cairns P, Al‐Saleem T, Hudes G, Haas G, Greenberg RE, Kolenko V. The basic biology and immunobiology of renal cell carcinoma: considerations for the clinician. Urologic Clinics of North America 2003;30:423‐36. [DOI] [PubMed] [Google Scholar]
Young 2009*
- Young AC, Craven RA, Cohen D, et al. Analysis of VHL gene alterations and their relationship to clinical parameters in sporadic conventional renal cell carcinoma. Clinical Cancer Research 2009;15:7582‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zbrozek 2010*
- Zbrozek AS, Hudes G, Levy D, et al. Q‐TWiST analysis of patients receiving temsirolimus or interferon alpha for treatment of advanced renal cell carcinoma. Pharmacoeconomics 2010;28:577‐84. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Coppin 2008
- Coppin C, Le L, Porzsolt F, Wilt T. Targeted therapy for advanced renal cell carcinoma. Cochrane Database of Systematic Reviews 2008, Issue 2. [DOI: 10.1002/14651858.CD006017.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]