

Abstract
HIV‐1 reverse transcriptase (RT) is a heterodimer comprised p66 and p51 subunits (p66/p51). Several single amino acid substitutions in RT, including L289K, decrease p66/p51 dimer affinity, and reduce enzymatic functioning. Here, small‐angle X‐ray scattering (SAXS) with proton paramagnetic relaxation enhancement (PRE), 19F site‐specific NMR, and size exclusion chromatography (SEC) were performed for the p66 monomer with the L289K mutation, p66L289K. NMR and SAXS experiments clearly elucidated that the thumb and RNH domains in the monomer do not rigidly interact with each other but are spatially close to the RNH domain. Based on this structural model of the monomer, p66L289K and p51 were predicted to form a heterodimer while p66 and p51L289K not. We tested this hypothesis by SEC analysis of p66 and p51 containing L289K in different combinations and clearly demonstrated that L289K substitution in the p51 subunit, but not in the p66 subunit, reduces p66/p51 formation. Based on the derived monomer model and the importance of the inter‐subunit RNH‐thumb domain interaction in p66/p51, validated by SEC, the mechanism of p66 homodimer formation was discussed.
Keywords: HIV‐1, NMR, paramagnetic relaxation enhancement, reverse transcriptase, SAXS, structure
Abbreviations
- dMTSL
(1‐acetyl‐2,2,5,5‐tetramethyl‐D3‐pyrroline‐3‐methyl) methane‐thiosulfonate
- MTSL
S‐(1‐oxyl‐2,2,5,5‐tetramethyl‐2,5‐dihydro‐1H‐pyrrol‐3‐yl) methyl methanesulfonothioate
- PRE
paramagnetic relaxation enhancement
- RNH
ribonuclease H
- RT
reverse transcriptase
- SAXS
small‐angle X‐ray scattering
- SEC
size exclusion chromatography
- tfmF
4‐trifluoromethyl phenylalanine
1. INTRODUCTION
HIV‐1 reverse transcriptase (RT) is a heterodimer composed of p66 and p51 subunits (p66/p51). 1 , 2 The p51 subunit is generated upon proteolytic removal of the ribonuclease H (RNH) domain from p66. The structure of p66/p51 RT has been well studied, 3 , 4 primarily as a target for drug development against HIV‐1, 5 , 6 , 7 , 8 showing three major contact areas between the two RT subunits; specifically, dimerization is mediated by interactions between the fingers domain of p51 and the palm of p66, between the connection domain of each subunit, and between the p51 thumb and the RNH domain of p66. 3 , 4 Single amino‐acid mutations in RT, particularly in the inter‐subunit interface, are known to significantly lower the dimerization affinity, mostly by altering the dimer interface or domain orientations. 9 , 10 , 11 , 12 , 13 Attempts to understand the mechanisms of p66/p51 heterodimer formation, which is essential for RT function, have been made by several groups. 12 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 However, our current understanding of the heterodimer formation mechanism is limited due to a lack of RT monomer structures.
Among the single amino acid substitutions in RT that weaken heterodimer formation, L289K is known to significantly impact RT dimerization and viral replication, 9 , 22 and is thus a suitable mutant protein to study the monomer structure. Previously, size‐exclusion chromatography (SEC) studies of p66 and p51, with either or both containing the L289K mutation, concluded that L289K in the p66 subunit abrogates p66/p51 formation, as well as its homodimerization. The study also concluded that L289K present in p51 subunit does not abrogate p66/p51 formation; in this case, it is located in the p66 and p51 dimerization interface of p66/p51. 9 Since L289 in the p66 subunit does not interface with the p51 subunit in p66/p51, this SEC conclusion, if correct, is not simply explained by mutation‐induced dimer dissociation. To understand the p66L289K structure, London's group compared NMR spectra of L289K p66, p66L289K, with that of p51; their data indicate that the p51 domain in p66L289K is similar to that of p51 monomer, that is, in a closed conformation similar to that of the p51 subunit in p66/p51. 23 Based on a small angle X‐ray scattering (SAXS) study of p66L289K, they suggested a model in which the polymerase domain is in a p51‐type conformation and linked to a variably positioned RNH domain. 23 , 24 While these SAXS data indicate that a subset of the individual RNH domain orientations are a better fit than the ensemble of RNH positions, the orientation of the RNH domain relative to the p51 domain was not determined. 23 Taken together, despite these previous efforts, the conformational basis for why L289K dissociates RT dimer, that is, p66/p66 or p66/p51, is still unknown.
Here, we extend our understanding of p66 monomer by applying SAXS combined with proton paramagnetic relaxation enhancement (PRE), site‐specific 19F NMR and SEC to characterize p66L289K. Our PRE data clearly demonstrate that the thumb domain in p66L289K monomer is spatially close to the RNH domain and, when combined with SAXS data, indicate the relative orientation of thumb and RNH domain in p66L289K. The observation was further verified with 19F NMR of RTs. Since residue 289 is located near the RNH domain but is exposed to solution in our model structure, our structural model suggests that L289K affects the inter‐subunit thumb‐RNH interaction during p66 homodimer formation, rather than inducing undesired interactions that prevent dimerization. Indeed, our SEC experiment, with validation by mass spectrometry experiments, clearly indicates that the p51 L289K, which is located at the dimer interface, disrupts p66/p51 formation more significantly than that in the p66 subunit. Based on the monomer structure model and the known p66/p51 structure, we discuss the p66/p66 homodimer formation.
2. RESULTS
2.1. Restricted orientation of the thumb domain in p66L289K
We hypothesized two possible thumb domain orientations in p66, based on the known p66/p51 structure (Figure 1a): the thumb domain is oriented similar to that observed in the p66 subunit of p66/p51 and interacts with the fingers domain (Figure 1b), or the thumb domain is oriented similar to that in the p51 subunit in p66/p51 and interacts with the RNH domain (Figure 1c). To investigate whether p66L289K exhibits one of these hypothesized structures or a distinct relative domain orientation, we performed PRE experiments by introducing a paramagnetic label at residue 24 in the fingers domain (Figure 1b) or residue 435 in the RNH domain (Figure 1c), to detect long‐range distance information to the thumb domain. Specifically, we introduced a nitroxide spin label, S‐(1‐oxyl‐2,2,5,5‐tetramethyl‐2,5‐dihydro‐1H‐pyrrol‐3‐yl) methyl methanesulfonothioate (MTSL), or, as a control, a diamagnetic analogue, (1‐acetyl‐2,2,5,5‐tetramethyl‐D3‐pyrroline‐3‐methyl) methane‐thiosulfonate (dMTSL), by conjugation of the label to Cys‐substituted residues in p66L289K, specifically W24C or V435C.
FIGURE 1.
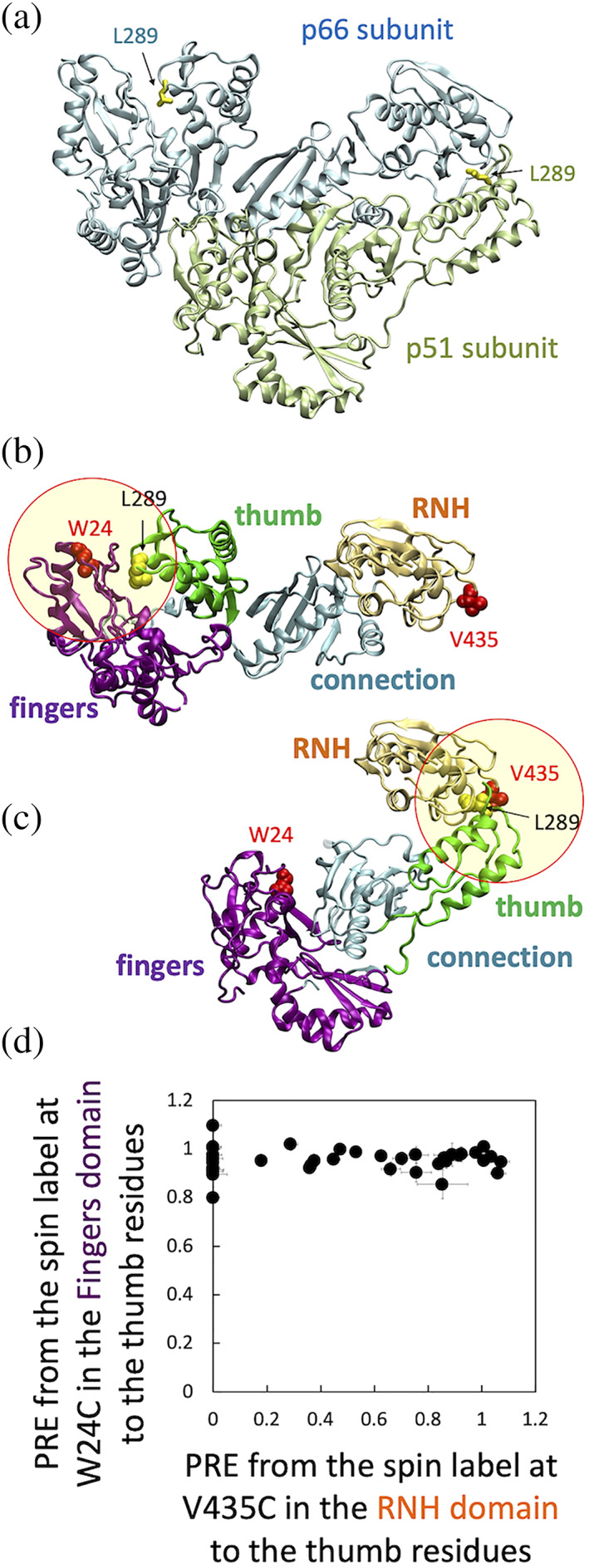
(a) Location of residue L289 in the p66/p51 crystal structure, (b, c) two hypothesized thumb domain orientations based on the p66/p51 crystal structure, (d) PRE, defined as intensity ratio of I para /I dia (Section 5), to thumb residues from the MTSL‐labeled W24C fingers domain relative to those from MTSL‐labeled V435C RNH domain. In (a), p66 subunit (light cyan) and p51 subunit (light green), with L289 side chain in each subunit (yellow sticks), are shown with ribbon presentation using PDB 1DLO. 26 Panels (b) and (c) show RT domains in the same orientation as that of panel (a), in a way to have one p51 and one RNH to generate one p66 monomer. In (b), the p66 subunit in p66/p51 structure is shown with highlight of fingers domain residue 24 (red van der Waal spheres). In (c), the p51 subunit in p66/p51 structure and the RNH domain in p66 are shown with highlight of RNH domain residue 435 (red van der Waal spheres). In (b) and (c), fingers‐palm, thumb, connection, and RNH domains are colored with purple, green, cyan, and light orange, respectively. The circles indicate a ~20 Å radius, at which proton PRE is sensitive (Note, since these circles are drawn in two‐dimensions, the distance is only approximate). In (d), normalized PREs from residue 24 to thumb residues are plotted on the Y‐axis while PREs from residue 435 to the same thumb residues are plotted on the X‐axis
Comparison of 1H‐15N TROSY HSQC spectra of p66L289K/W24C and p66L289K/V435C and those of MTSL‐ and dMTSL‐labeled proteins did not show significant differences, verifying that the labeling does not alter the protein structure (Figure S1A,B). Chemical shifts of the thumb domain region in p66L289K proteins were estimated by superimposing the spectra of the isolated thumb domain protein on to the spectrum of p66L289K, both recorded in the same buffer and the same experimental conditions (Figure S2). 25 The intensity of backbone amides in the thumb domain of p66L289K changed only when the MTSL was introduced to the RNH domain but not the fingers domain (Figure 1d). This clear difference suggests that even though both the thumb and RNH domains are mobile in p66L289K the preferred relative orientation of the two is that shown in Figure 1c, prompting us to further study the p66L289K structure in solution.
2.2. SAXS indicates a monomer size of p66
SAXS curves for p66L289K with a deletion of the C‐terminal four residues and lacking a terminal tag sequence, p66L289Ktr, were recorded at three different concentrations. We used this truncated construct based on a crystal structure of p66/p51 that exhibits coordinates until residue 556 (PDB 1DLO 26 ), with the expectation of a better fit for the RNH domain in the SAXS envelop analysis. No concentration‐dependent anomaly was observed among the three SAXS data sets, indicating that the protein is monomer and monodisperse in solution on the basis of SAXS parameters (Figure S3A), consistent with previous observations. 9 The radius of gyration obtained by Guinier analysis, ~32 Å, and the maximum size of a protein, ~107 Å, are at the lower end of those described previously (Figure S3B, Table 1). 23 The three data sets were extrapolated to infinite dilution for further analysis.
TABLE 1.
SAXS‐derived structural parameters for p66L289Ktr
| Concentration (mg/ml) | Guinier analysis | Distance distribution analysis | Molecular weight a | ||||||
|---|---|---|---|---|---|---|---|---|---|
| I 0 | R g (Å) | I 0 | R g (Å) | D max (Å) | Porod volume b | Volume of correlation b | Size & Shape b | MoW2 c | |
| 1.0 | 0.029 | 31.72 ± 0.63 | 0.029 | 32.07 ± 0.43 | 107 | 67.3 | 62.6 | 79.2 | 71.5 |
| 2.0 | 0.058 | 31.95 ± 0.40 | 0.058 | 32.04 ± 0.31 | 108 | 66.4 | 61.9 | 67.0 | 72.1 |
| 4.3 | 0.13 | 32.26 ± 0.26 | 0.134 | 32.56 ± 0.12 | 107 | 65.6 | 59.1 | 70.1 | 72.8 |
The theoretical molecular mass of L289Ktr monomer is 64.4 kDa.
Determined using primusqt from the ATSAS 2.8 software package.
Determined using SAXS MoW2.
2.3. Relative RNH domain orientation against thumb, elucidated by NMR PRE, and SAXS experiments
To further verify the model in Figure 1c, as suggested by our initial PRE experiments, and to better illuminate the relative orientation between the thumb and RNH domains, additional PRE experiments were performed by introducing the MTSL tag at residue 280 or 291 in the thumb domain, or at residue 435 or 534 in the RNH domain in p66L289K (Figure 2, S1B‐S1D). Reduction of the 1H‐15N TROSY‐HSQC cross‐peaks in the MTSL spectra (red in Figure 2) but not in dMTSL spectra (green in Figure 2) were seen, depending on the MTSL‐labeled sites. Mostly, when the MSTL labels were introduced to S280C or E291C site in the thumb domain, thumb domain signals, such as G262 and L296, exhibited reduced signal intensities. When the spin labels were introduced to V435C or A534C site in the RNH domain, RNH domain signals, such as F440 and I495, exhibited decreased signals intensities (red in Figure 2). However, inter‐domain signal reductions were also observed. The signal intensity of G262 was reduced by MTSL‐labeling at V435C or A534C, and a slight reduction of F440 and I495 intensities by labeling at S280C or E291C site was observed, demonstrating that PRE could be used as domain distance constraints. Overall, over 40 distance constraints were detected in each set of the PRE experiments (Figure S4).
FIGURE 2.
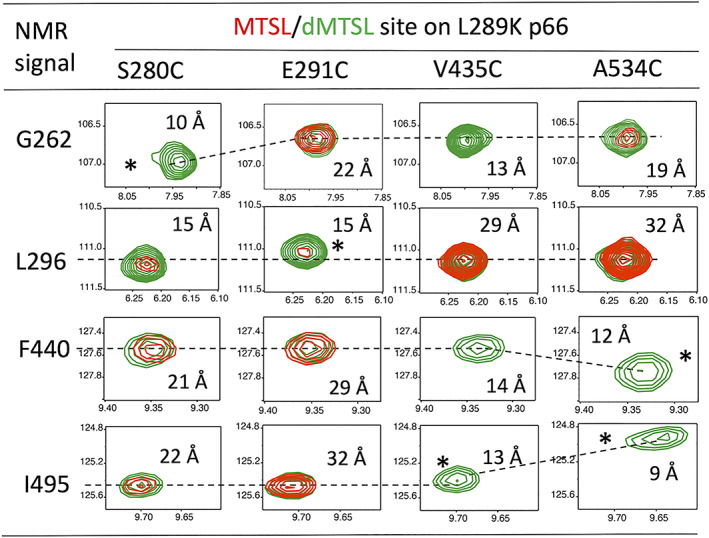
1H‐15N TROSY‐HSQC cross‐peaks of selected residues, G262 and L296 in the thumb domain and F440 and I495 in the RNH domain, in p66L289K monomers that were labeled with the MTSL (red) or dMTSL (green) at S280C, E291C, V435C, and A534C sites. In each panel, the horizontal and vertical scales are 1H (ppm) and 15N (ppm), respectively. Asterisks indicate amides that experience chemical‐shift changes due to close proximity to the spin‐labeled sites. Approximate distances from the amide proton to the Sγ2 position of the labels, obtained in the final model structure in Figure 3, are shown. Positions of the peaks in the entire spectrum are marked in Figure S2
The distance constraints obtained from these PRE data were then combined with the SAXS curve to better determine relative RNH domain orientation against the thumb domain with the fixed p51 subunit conformation. Given the high degree of similarity between the p51 domain signals in p66L289K and those of the p51 monomer, 23 we utilized the p51 subunit structure in p66/p51, that is, the p51 closed conformation, in the same manner as previously published, 23 with the RNH placed in a starting conformation and the relative thumb‐RNH orientation determined based on the obtained constraints. The back‐calculated SAXS intensity, using the best optimized structure, agreed with the experimental data at the normalized χ2, 0.35 (Figure 3b). The observed and calculated PRE values of the lowest energy structure showed reasonable agreement with the intra‐ and inter‐domain Q‐factor at 0.108 and 0.119, respectively (Figure 3c,d). Intra‐domain and inter‐domain distance correlations between the observed PRE‐derived and calculated structures were 0.82 and 0.70, respectively.
FIGURE 3.
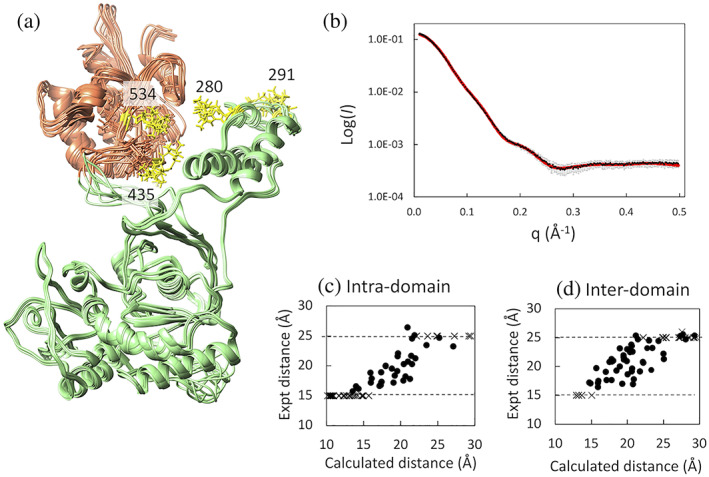
(a) Overview of the 10 energy‐minimized structures of p66L289Ktr, optimized with SAXS and NMR PRE data, (b) plots of experimental (black) and calculated (red) SAXS intensity curves for p66L289Ktr, agreeing with the experimental data at the normalized χ2, 0.35, and comparison of MTSL‐proton distances in the best‐energy minimized structure with those experimentally obtained from PRE, for (c) intra‐domain and (d) inter‐domain residues in p66L289K. In panel (a), the RNH domain is shown by light orange, p51 domain is shown by light green, and MTSL‐tags at residues 280, 291, 435, and 534, with three different orientations for each, are shown by yellow sticks. In panels (c) and (d), distances directly obtained from experimental PREs are shown by black spheres within upper (25 Å) and lower (15 Å) boundaries. Distances above or below the boundaries are shown by cross marks
Given that the structural elements are fixed in the model calculation, the root‐mean‐square‐deviation of the coordinates may not be the best measure of model quality. For this reason, we visually present whether the observed PRE reasonably reflects the structure in each experiment (Figure 4). PRE effects from residue 280, which is located in a short α‐helix of the thumb domain, to the RNH domain (Figure 4a) were more pronounced, compared to those from residue 291, which is located at the tip of the thumb domain (Figure 4b). Similarly, PRE effects from residue 435, which is located at a loop region in the RNH domain, to the thumb domain (Figure 4c) were more pronounced, compared to those from residue 534, which is located at a β‐strand in the RNH domain (Figure 4d). Thus, labeling location is reasonably reflected to the PRE values. Also, consistency of these PRE data confirms the signal assignments that were qualitatively done based on chemical shift similarity between those in isolated domains and in p66L289K.
FIGURE 4.
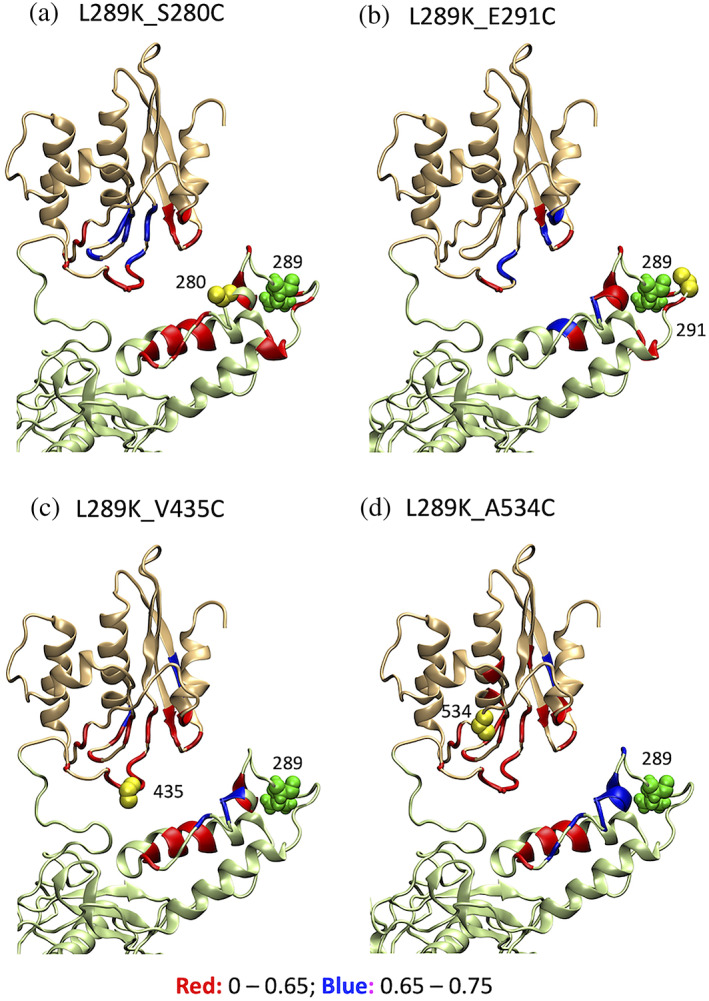
Visual presentation of the PRE effect from MTSL‐tags (yellow spheres) at residues (a) 280, (b) 291, (c) 435, and (d) 534 in p66L289K on the energy‐minimized structure. In each panel, thumb and RNH domains are shown by light green and light orange colors, respectively; residue 289 location is shown by green spheres; I para /I dia less than 0.65, that is, stronger PRE effect, is shown in red ribbon and 0.65–0.75, weaker PRE effect, by blue ribbons
2.4. Characteristics of NMR chemical shifts of 19F‐probes in thumb and RNH domains
The structure calculated using PRE and SAXS indicates that a direct interaction between the thumb domain and RNH domain does not occur, with the closest distance between the two at ~10 Å. The paramagnetic tag is very unlikely to prevent a direct interaction of the two domains because the PRE experiments were performed using four sets of proteins labeled at different sites (Figure 4) and show essentially similar NMR spectra (Figure S1B–D). Still, to verify that a direct interaction does not happen even without the MTSL‐tag, we site‐specifically labeled residue 259 or 532 with a 4‐trifluoromethyl phenylalanine (tfmF) and recorded 19F NMR spectra. In the p66 subunit of p66/p51, residue 259, located in the thumb domain, interfaces with the fingers domain and, in the p51 subunit of p66/p51, it interfaces with the RNH domain (Figure 5a). Residue 532 in the RNH domain interfaces with the thumb domain in the p51 subunit in p66/p51 and is ~3 Å distance from residue 259 (Figure 5a).
FIGURE 5.
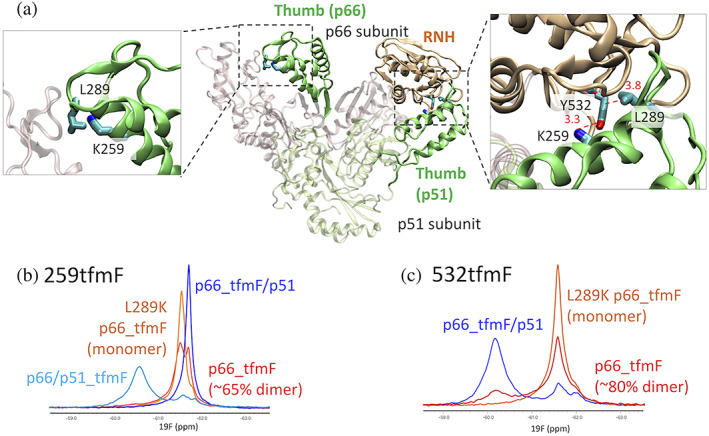
(a) Highlight of the K259 and Y532, sites labeled with tfmF for 19F NMR, on p66/p51 structure and 19F NMR spectra of (b) 259tfmF and (c) 532tfmF, recorded at 298 K. In (b), the spectra are shown for p66/p51259tfmF (light blue), p66259tfmF/L289K (orange), p66259tfmF (red), and p66259tfmF/p51 (blue). In (c), the spectra are shown for p66532tfmF/L289K (orange), p66532tfmF (red), and p66532tfmF/p51 (blue). In these panels, subscript notations were avoided for readability
NMR spectrum of p66/p51 with the tfmF‐labeling at position 259 in the p51 subunit, p66/p51259tfmF, was compared with that obtained when the label was in the p66 subunit, p66259tfmF/p51 (Figure 5b). The 19F resonance in the p66/p51259tfmF spectrum was observed at −60.6 ppm, while that in the p66tfmF/p51 spectrum was −61.7 ppm. NMR spectra of p66 and p66L289K with tfmF‐labeling at position 259, p66259tfmF, and p66259tfmF/L289K, respectively, were both observed at positions similar to the p66259tfmF/p51 spectrum and not at positions similar to the p66/p51259tfmF spectrum (Figure 5b). Based on the homodimer dissociation constant of p66259tfmF [~20 μM (Figure S5)], approximately 65% of the protein is expected to be dimer at the concentration used; thus, the minor peak in the p66259tfmF spectrum (Figure 5b) may represent a different conformation of the homodimer or a second distinct subunit environment. In any case, these data indicate that residue 259 in the thumb domain in p66 or p66L289K is quite different from that in the p51 subunit in p66/p51 and is probably more exposed to solution.
A complementary spectral pattern was observed when the tfmF‐label was at position 532: the resonance of 532 in p66532tfmF/p51 was −60.2 ppm, while in p66532tfmF and p66532tfmF/L289K the 532 resonance was at −61.6 ppm (Figure 5c). Given that the neighboring residue, 532, in the p66 subunit directly interfaces with residue 259 in the p51 subunit at ~3 Å distance in p66/p51, the similarity of 19F chemical shifts of position 259 in p66/p51tfmF259 and position 532 in p66tfmF532/p51 is reasonable, reflecting the same chemical environment. Importantly, the 19F resonances of p66532tfmF and p66532tfmF/L289K did not show a large low‐field shift, indicating a similar 532 environment in these two proteins. No sign of RNH unfolding was observed in p66532tfmF homodimer from these shift comparisons. Thus, thumb domain residue 259 and RNH domain residue 532 do not directly interface in p66L289K, different from the wild‐type heterodimer interaction between the RNH domain in p66 and thumb domain in p51.
2.5. L289K in p51 subunit alters p66/p51 formation
The above observations suggest that L289K weakens either intra‐ or inter‐RNH‐thumb interactions. Indeed, in the p66/p51 structure (Figure 1a), L289 in the p51 subunit interacts with the RNH domain in the p66 subunit, deriving a hypothesis that the mutation weakens the inter‐subunit RNH‐thumb interaction in p66/p51. To test this hypothesis, we prepared p66, p51, p66L289K, and p51L289K and analyzed dimer formation among various combinations of these proteins by SEC. Control proteins without L289K substitution eluted accordingly to their molecular sizes: p66/p51 (molecular weight, 120 kDa) at 13.4 ml, p66 monomer (66 kDa) at 14.3 ml, and p51 (51 kDa) at 15.2 ml, respectively (Figure 6a). The SEC profile of p66 showed an additional small elution peak slightly earlier than 13.4 ml, due to p66 homodimer formation, 130 kDa (Figure 6a). A mixture of equal amounts of p66L289K and p51L289K showed two peaks at 66 and 51 kDa positions, indicating that these proteins do not form a heterodimer or homodimers (Figure 6b, solid line). SEC elution of p66L289K or p51L289K was confirmed by injecting each protein individually (Figure S6A). Consistent SDS gel profiles for each of the three samples were observed (Figure 6c), and eluted species were confirmed by mass spectrometry (Table S1).
FIGURE 6.
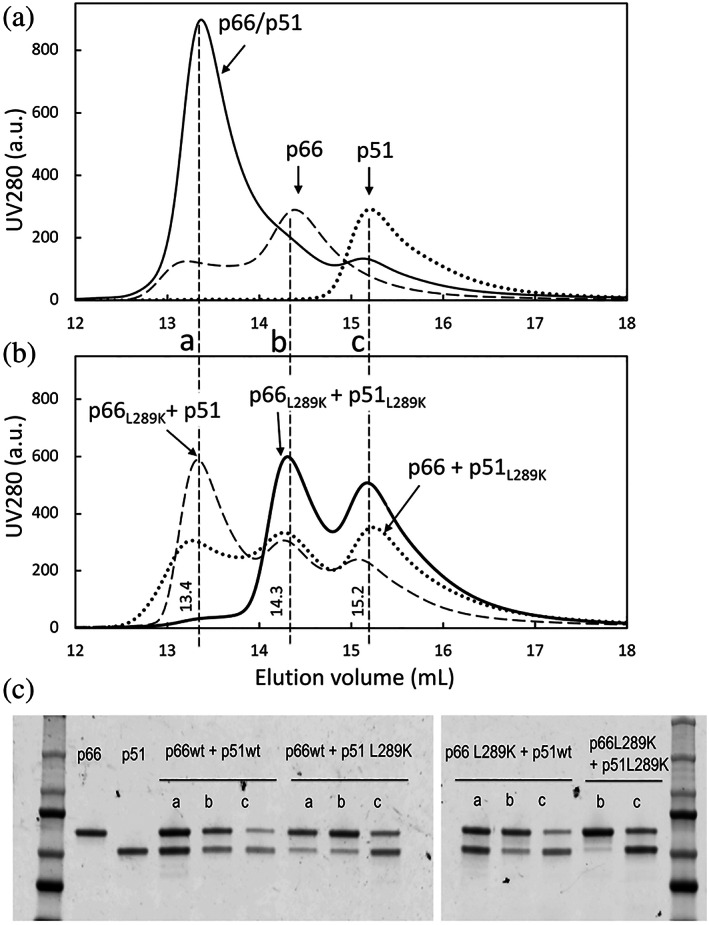
SEC elution profiles of (a) p66/p51 (solid line), p51 (dotted line) and p66 (dashed line), and of (b) p66L289K and p51L289K mixture (solid line), p66 and p51L289K mixture (dotted line), and p66L289K and p51 mixture (dashed line), and (c), SDS‐PAGE of the corresponding peaks. In panel (a) below, the fractions analyzed by SDS‐PAGE are marked with a (at ~13.4 ml), b (at ~14.3 ml), and c (at 15.2 ml). In panel (c), from left to right are: a marker ladder, p66 control, p51 control, and samples from the a, b, or c elution positions, as indicated, for p66/p51, p66 and p51L289K mixture, p66L289K and p51 mixture, and p66L289K and p51L289K mixture, with the ladder for the second PAGE in the last lane
When a mixture with equal amounts of p66L289K and p51 was injected to the SEC, three elution peaks at p66L289K/p51, p66L289K, and p51 were observed (Figure 6b, dashed line). SDS gel clearly indicates almost equal amounts of p66L289K and p51 at 13.2 ml (Figure 6c). Mass spectrometry confirmed that these are p66L289K and p51 and not p66 and p51 (Table S1). When a mixture of equal amounts of p66 and p51L289K was injected to the SEC, three elution peaks were again observed (Figure 6b, dotted line), but the first peak eluted earlier than 13.4 ml, with a lower intensity compared to p66L289K/p51 and in position similar to p66/p66 homodimer. SDS‐PAGE of the elution position showed a stronger band for p66 compared to p51, indicative of p66/p66 in this mixture (Figure 6c). However, SDS‐PAGE also has a faint band corresponding to p51, which might indicate some p66/p51L289K formation (Figure 6c). Experiments were repeated to verify the observations (Figures S6 and S7).
3. DISCUSSION
In this study, we aimed to gain insight into the monomer structure of p66L289K. Our PRE data indicate weak interaction between the RNH and thumb domains in the monomer (Figures 1c and 4). Consistent with this observation, 19F NMR showed that the thumb‐RNH interaction is different in p66L289K compared to p66/p51 in which they directly interact with each other (Figure 5). The SAXS data indicate a monomer molecular size of p66 (Table 1) and, when combined with the PRE data, a possible RNH domain orientation, against the thumb domain, was determined (Figure 3a). Overall, our model structure suggests that the two domains are located relatively close to each other in the monomer (Figure 7a). The data do not have the sensitivity to permit discussion of other minor conformers. Our SAXS data are consistent with those of London's group but with a slightly higher sensitivity. 27 In addition, with the constraints from PRE, we were able to narrow down the conformation.
FIGURE 7.
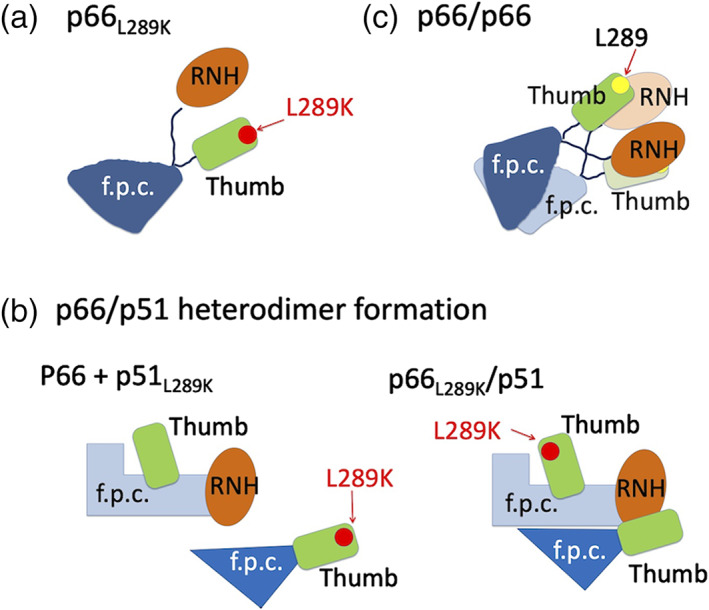
Cartoon indicating (a) our structural model of monomer p66L289K, (b) L298K mutation effect on p66/p51, and (c) a model for p66/p66 homodimer formation as discussed in the text. In all panels, green rounded rectangle and orange ellipsoid indicate thumb and RNH domains, respectively. “f.p.c.” indicates fingers/palm/connection domains. Yellow and red dots indicate L289 and K289 positions, respectively
In our model structure, residue 289 in the monomer is exposed to solution (Figure 4). Importantly, this model is confirmed by our PRE study and the observation that paramagnetic labeling at E291C position, near residue 289, produces the lowest PRE among the four labeling sites (Figure 4). These observations suggest that L289K mutation does not induce an undesired interaction within p66 monomer, but instead disturbs necessary interaction(s) for dimerization. Based on the known inter‐subunit thumb‐RNH interactions in p66/p51 (Figure 1c), one simple hypothesis is that L289K mutation weakens inter‐subunit interaction between the thumb and RNH domains in p66/p51 heterodimer. We tested this hypothesis by mixing L289K and WT proteins and confirmed that p66L289K and p51 form a heterodimer while p66 and p51L289K do so at significantly lesser degree (Figure 6).
Our SEC data are not consistent with the previous SEC experiments by Wilson's group, in which p66L289K and p51 did not form heterodimer while p66 and p51L289K did so. 9 This inconsistency may simply depend on differences in protein concentrations or the presence of minor contaminants in the experiments. In addition, given that changing residues located at or near the dimer interface, for example, W401A and W414L, are known to disrupt dimer formation, 12 , 13 our conclusion that L289K when located at the dimer interface in the p51 subunit (Figure 1c) impacts dimerization more than if it is located in the p66 subunit (Figure 1b) makes sense (Figure 7b). Increases in the p66/p66 homodimer dissociation constant when tfmF‐labeling is introduced at residue 259 or 532, by ~1.2 and ~4.5 times, respectively (Figure S5A–C), also suggest that the RNH and thumb domain interaction, either inter‐subunit or intra‐subunit (discussed below), contributes to homodimerization. These dimer formations are not due to nucleic acid contamination, since comparison of UV 254 and 280 nm gives a ratio less than 0.5 (Figure S5D–F).
With the monomer structural model (Figure 3a) now in hand, combined with the known p66/p51 structure (Figure 1a), we can extrapolate mechanisms of p66/p66 formation. In our structural model of the monomer, the thumb domain residue 289 does not directly face the intra‐subunit RNH domain, but these are spatially close to each other (Figure 4). In contrast, in the p66/p51 structure, L289 in the p51 subunit interacts with the RNH domain in the p66 subunit, resulting in the chemical environments of 259 being similar to that of 532, that is, in the RNH‐thumb locked conformation (Figure 5). In p66/p66 homodimer, the thumb and RNH domain interaction, involving residues 259 and 532, either inter‐subunit or intra‐subunit, contributes to the dimerization (Figure S5). The binding affinity of p66/p66, ~4 μM, is ~10‐fold higher than that of p66/p51. 14 , 25 , 28 , 29 Overall, we postulate that the RNH‐thumb interface orientation, particularly L289‐RNH orientation, in p66/p66 differs from that of p66/p51 and makes the dimerization weaker than that in p66/p51. Our data do not directly indicate whether inter‐subunit or intra‐subunit domain interaction affects the p66/p66 homodimerization. Depending on the protein, intra‐subunit interaction may facilitate dimer formation as much or more than inter‐subunit interactions: for example, in HIV‐1 protease, intra‐subunit interaction between D29 and R87 more significantly contributes dimer formation than inter‐subunit interaction between D29 and R8. 30 However, given that the inter‐subunit interaction is formed between RNH and the thumb domains in p66/p51, the importance of the RNH‐thumb domain interaction for dimerization of p66/p66 is expected to be mediated by an inter‐subunit interaction (Figure 7c). In addition, the interaction must be weak in that it allows the folded RNH and thumb domains to exhibit domain motion. 25
4. CONCLUSIONS
We determined the relative orientation of the RNH and thumb domain within a monomer, based on SAXS with proton PRE, 19F site‐specific NMR and SEC. In the new model, the thumb domain in p66L289K monomer is spatially close to the RNH domain with a limited relative orientation. We also provided SEC data, with validation using mass spectrometry, showing that L289K at the dimer interface affects dimer formation. Based on the monomer structural model, dimer dissociation constants of the tfmF‐labeled p66, and the difference in the thumb–RNH interaction between p66/p66 and p66/p51, we discussed how homodimer of p66 is formed in solution.
5. MATERIALS AND METHODS
5.1. Protein preparation and purification for proton PRE NMR experiment
An expression of RT p66 was performed as described previously, 25 with codon‐optimized C280S/C38V clone and a hexa‐histidine tag at the C‐terminus, purchased at DNA 2.0 (now ATUM, Newark, CA). Dimerization of this p66 was confirmed in a previous study. 25 L289K substitution, as well as additional cysteine‐substitutions, at W24, S280, E291, V435, or A534, for the PRE experiments were introduced by site‐directed mutagenesis (QuikChange II, Agilent, Santa Clara, CA). All DNA primers were custom synthesized at IDT (Integrated DNA Technologies, Coralville, Iowa). The presence of desired substitutions was confirmed by DNA sequencing (Genewiz, South Plainfield, NJ). Protein was expressed in E. coli Rosetta 2(DE3) cells in a minimal medium containing 15NH4Cl as the sole nitrogen source and purified, first using a HisTrap HP column (GE Healthcare, Chicago, IL), followed by a Superdex75 26/60 column (GE Healthcare, Chicago, IL) in 25‐mM sodium phosphate buffer at pH 6.8, 100‐mM NaCl, 0.02% NaN3. For freeze storage, 50% glycerol was added. Isolated thumb and RNH domains, used for qualitative assignments of p66L289K resonances, were prepared as described previously. 25 The final product was verified by mass spectrometry using a Bruker LC‐ESI‐TOF system (Bruker Daltonics Inc., Billerica, MA, Table S2).
For PRE experiments, p66 proteins containing a single Cys were modified with MTSL and the diamagnetic analogue of MTSL, dMTSL (Toronto Research Chemicals), in parallel reactions. The single‐cysteine proteins were purified as described above, but in the presence of 10‐mM DTT to ensure that all the protein was completely reduced. Before the labeling reaction, the protein solution was exchanged with the buffer without DTT. Excess amount of unreacted MTSL or dMTSL was removed by dialysis.
5.2. Protein preparation and purification for proton 19F NMR experiment
The same codon optimized clones that were used for the PRE experiments were used to prepare proteins for 19F NMR experiments. For p51 production, the optimized p51 with a Strep‐tag in the N‐terminus was used. 25 As described previously, competent E. coli BL21 AI cells (Thermo Fisher Scientific, Waltham, MA) were co‐transformed with the vector encoding the constructs, containing TAG at the tfmF labeling position, and the pDule2 RS vector encoding the orthogonal amber tRNA/tRNA synthetase pair, as described previously. 31 , 32 , 33 tfmF‐labeled p66 and p66L289K, either at position 259 or 532, p66259tfmF or p66532tfmF and p66259tfmF/L289K or p66532tfmF/L289K, were purified similarly as described above. To prepare tfmF‐labeled heterodimer (p66tfmF/p51 or p66/p51tfmF), cell pellets for tfmF‐labeled p66 (or p51) were mixed with equivalent amounts of cell pellets containing unlabeled p51 (or p66) before cell lysis, and purified using a HisTrap HP column and a StrepTrap column to extract only the p66 and p51 complex. The effect of the tfmF labeling on p66 dimerization was assessed by running a SEC for p66, p66259tfmF, and p66532tfmF.
5.3. Protein preparation and purification for SAXS experiments
The coding sequence for amino acids 1–556 of p66L289K was cloned into pET‐15b vector, with a hexa‐histidine tag followed by the TEV protease cleavage site at the N‐terminus of the protein. The protein was produced in E. coli Rosetta 2(DE3) cells and purified from the clarified lysate using a HisTrap HP column (GE healthcare), with the loading and elution buffers containing 20‐mM and 500‐mM imidazole, respectively, in 50‐mM sodium phosphate at pH 7.5, 250‐mM NaCl and 0.02% NaN3. The eluted protein was concentrated and then diluted in 25‐mM sodium phosphate buffer at pH 6.8, containing 100‐mM NaCl, 0.02% NaN3 and 2‐mM DTT, to make the final imidazole concentration smaller than 20 mM. The reaction was incubated at 4°C overnight, and the cleaved protein was separated with a second passage over the HisTrap HP column (GE healthcare), from which the flow‐through was collected. The cleaved protein, collected in the flow‐through fraction, was further purified by gel filtration on a Superdex75 26/60 column (GE healthcare) in 25‐mM sodium phosphate buffer at pH 6.8, 100 mM NaCl, 0.02% NaN3.
5.4. Proton PRE NMR experiment
All NMR experiments were performed using ~65‐μM protein in 25‐mM sodium phosphate buffer, containing 100 mM NaCl, 0.02% NaN3, and 5% D2O at pH 6.8 at 295 K. Protein concentration was determined from UV measurement, assuming A280 (g/L) = 2.1 that was calculated using ProtParam tool. 34 Prior to PRE measurements, 2D 1H−15N TROSY‐HSQC data were recorded for five single‐Cys labeled p66L289K proteins to ensure that each substitution did not alter protein folding. Then, for PRE experiments, a set of 2D 1H−15N TROSY‐HSQC spectra were recorded for each protein: (i) with MTSL‐tag, (ii) dMTSL‐tag, and (iii) without any of these tags. All these experiments were performed at 295 K on Bruker 900‐MHz AVANCE spectrometer. Spectra were processed with NMRPipe and analyzed using NMRDraw. 35 Due to low signal‐to‐noise ratios, we used a qualitative estimation of the PRE of p66L289K 36 , 37 : peak heights, as representative of peak intensities, of the assigned cross‐peaks in 1H−15N TROSY‐HSQC spectra of the paramagnetic (I para) and diamagnetic (I dia) states were extracted using NMRDraw. Data were deposited to Mendelay data (https://data.mendeley.com/datasets/7vrvryyghx/2). PRE was calculated as I para/I dia for each residue. 36 , 37 , 38
5.5. 19F NMR experiments
19F NMR resonances of tfmFs at position 259 were recorded for p66259tfmF/L289K, p66259tfmF, p66259tfmF/p51, and p66/p51259tfmF. Similarly, 19F NMR resonances of tfmFs at position 532 were recorded for p66532tfmF/L289K, p66532tfmF, and p66532tfmF/p51. These experiments were performed with ~65‐μM protein in 25‐mM sodium phosphate buffer, containing 100‐mM NaCl, 0.02% NaN3, and 5% D2O at pH 6.8 at 295 K. Spectra were recorded on a 600‐MHz Bruker AVANCE spectrometer, at 19F resonance frequency, 564.65 MHz, equipped with a CP TXO F/C‐H‐D triple‐resonance z‐axis gradient cryoprobe (Bruker Biospin, Billerica, MA), without 1H decoupling and at the total number of scans approximately 4,096. A 1H sodium trimethylsilylpropanesulfonate (DSS) spectrum was recorded each time a set of experiments was performed, and this was used as a reference of 19F resonances. Final spectra were plotted using MestReNova (Escondido, CA).
5.6. SAXS experiments
SAXS samples were prepared in 50‐mM Bis‐Tris buffer, pH 6.8, 100‐mM NaCl, 1% glycerol, 0.02% NaN3 at three different concentrations. All SAXS data were recorded at the 12‐ID‐B beamline of Advanced Photon Source, Argonne National Lab, Argonne, IL, USA, with a photon energy of 13.3 keV. For each measurement, 45 individual exposures of 1 s each were collected, then selected and averaged to yield the final scattering curves. Buffer scattering measurements were performed in an equivalent fashion and subtracted from the protein scattering data. All data were processed and analyzed using tools from the ATSAS software package (version 2.8.4) including PRIMUS, GNOM, ALMERGE, CRYSOL, and BUNCH. Molecular masses, R g values, and P(r) distribution functions were determined with PRIMUS and GNOM. The molecular masses were also determined using SAXS MoW2. The data were deposited to be deposited at SASBDB (accession numbers: SASDNH3, SASDNJ3, SASDNK3, and SASDNL3).
5.7. Determination of relative RNH domain orientation using SAXS and PRE
To investigate whether there is a restriction in the relative RNH domain orientation against the thumb domain, we conducted structural calculations using the SAXS and PRE data. For this analysis, SAXS data were extrapolated to zero concentration and merged with the highest concentration data using ALMERGE. The program BUNCH was used for initial modeling to generate the starting coordinate of p66L289Ktr. For the modeling, the coordinates of the p51 subunit (amino acids 1–417) and isolated RNH domain (amino acids 430–556) in the crystal structure of RT (PDB 1DLO) were used for simulation. The position of p51 subunit was fixed, while the configuration of the RNH domain was searched to fit the SAXS data.
The ratio I para/I dia changes almost linearly within the range of 0.15 < PRE < 0.85. 37 Thus, three types of PRE distance restraints were employed for structure calculation, similar to the previously published protocol. 37 , 38 (i) For amide protons with PRE intensity ratios between 0.15 and 0.85, distance restraints were set with an error margin of ±4 Å. (ii) For amide protons with PRE intensity ratios less than 0.15, distance restraints were set as from 1.8 Å to 15 Å. (iii) For amide protons with PRE intensity more than 0.85, distance restraints were set as from 25 Å to 225 Å, as repulsive distance restraints. For each PRE data set, 43–48 restraints, a total of 186 restraints, were used for the calculation.
Structures of p66L289Ktr were calculated using Xplor‐NIH version 2.52 with the starting coordinate generated using BUNCH. Protons and MTSL tags were then added to the starting model, followed by adding three conformers of each MTSL probe to all the cysteine mutated sites, to include ensemble conformations of the flexible MTSL tag. 39 For structure calculation, the starting structure was energy minimized against the PRE distance restraints and SAXS data in the range q ≤ 0.5 Å−1. In this process, the backbone atomic coordinates of the p51 subunit (residues 1–218, 231–286, and 296–417) were held fixed, while the backbone atomic coordinates of the RNH domain (residues 430–556) were allowed to move freely as a rigid body with the natural p66 amino acids as linkers between them. Energy terms included the NOE potential for PRE distance restraints, the SAXS term, the TorsionDB term for all active torsion angles, and the RepelPot repulsive term for non‐bonded interactions. Additional energy terms, the bond length (BOND), bond angle (ANGL), and improper dihedral (IMPR) potentials, were also employed. Initially, a run of the shorter of 100 ps or 1,000 steps of high temperature dynamics at 3,000 K were performed. The simulated annealing protocol employed initial and final temperatures of 3,000 K and 25 K, respectively, in 12.5 K increments. After simulated annealing, final torsion angle and Cartesian coordinate energy minimizations were performed. All molecular dynamics and minimization steps used the internal variable module (IVM). One hundred structures were calculated, and the 10 lowest energy structures were used for final analysis. For PRE data, the Q factor was calculated according to,
where r exp and r calc are the experimental and calculated PRE distances, respectively.
5.8. SEC experiments of p66 and p51 with/without L289K mutation
The experiments were performed for p66 and p51, either with or without L289K mutations using a 24‐ml analytical Superdex 200 Increase 10/300 GL column (GE Healthcare), equilibrated with a 25‐mM Bis‐tris buffer, pH 7.0, containing 100‐mM NaCl with 0.02% sodium azide, at a flow rate of 0.5 ml/min. In each experiment, an 80‐μl sample, containing 40‐μM each protein, was injected, and protein elution was monitored by UV absorbance at 254 and 280 nm. SDS‐PAGE and mass spectrometry were run for each fraction eluted at 13.4, 14.3, or 15.2 ml. SDS‐PAGE used precast 4–15% Tris‐glycine gels (Bio‐Rad) stained with Bio‐safe Coomassie stain (Bio‐Rad). Mass spectrometry for each fractionated peak was run on a Bruker LC‐ESI‐TOF system (Bruker Daltonics Inc., Billerica, MA).
CONFLICT OF INTEREST
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
Zhaoyong Xi: Conceptualization (equal); investigation (equal); writing – original draft (equal). Tatiana V. Ilina: Conceptualization (equal); investigation (equal); writing – original draft (equal). Michel Guerrero: Formal analysis (equal); investigation (equal); writing – review and editing (equal). Lixin Fan: Investigation (equal); validation (equal). Nicolas Sluis‐Cremer: Conceptualization (equal); funding acquisition (equal); supervision (equal); writing – review and editing (equal). Yun‐Xing Wang: Funding acquisition (equal); investigation (equal); validation (equal); writing – review and editing (equal). Rieko Ishima: Conceptualization (equal); funding acquisition (equal); supervision (equal); writing – original draft (equal).
Supporting information
Data S1: A pdf file that contains the following data is available free of charge
Figure S1. 1H‐15N NMR spectra of p66L289K and the mutants, used for PRE experiments
Figure S2. 1H‐15N NMR spectra of p66L289K overlaid with those of the isolated domains
Figure S3. SAXS Guinier plots and the pair‐distance distribution function for p66L289Ktr
Figure S4. Plots of PRE for residues in the thumb and RNH domains
Figure S5. SEC elution profiles of p66 and the tfmF‐labeled p66
Figure S6. SEC elution profiles of WT and L289K mutant mixtures of p66 and p51
Figure S7. The entire SDS gel shown in Figure 5c and the repeat
Table S1. Molecular masses of the elution peaks in the SEC in Figure 5
Table S2. Selected mass spectrometry results to identify the samples prepared for the study.
ACKNOWLEDGMENTS
We thank Angela Gronenborn and Naima Sharaf for technical support, and Teresa Brosenitsch for critical reading of the manuscript, and acknowledge financial support from National Institutes of Health, P50 AI150481, and University of Pittsburgh. We also acknowledge the use of the SAXS Core facility of Center for Cancer Research (CCR), National Cancer Institute (NCI). The SAXS core has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract 75N91019D00024. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This Research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. This research used 12‐ID‐B beamline of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE‐AC02‐06CH11357.
Xi Z, Ilina TV, Guerrero M, Fan L, Sluis‐Cremer N, Wang Y‐X, et al. Relative domain orientation of the L289K HIV‐1 reverse transcriptase monomer. Protein Science. 2022;31(5):e4307. 10.1002/pro.4307
Zhaoyong Xi and Tatiana V. Ilina contributed equally to this study.
Review Editor: Aitziber Cortajarena
Funding information National Institutes of Health, Grant/Award Numbers: AI150481, 75N91019D00024
REFERENCES
- 1. Katz RA, Skalka AM. The retroviral enzymes. Annu Rev Biochem. 1994;63:133–173. [DOI] [PubMed] [Google Scholar]
- 2. Coffin JM, Hughes SH, Varmus HE. Retroviruses. New York: Cold Spring Harbor Laboratory Press, 1997. [PubMed] [Google Scholar]
- 3. Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal structure at 3.5 A resolution of HIV‐1 reverse transcriptase complexed with an inhibitor. Science. 1992;256:1783–1790. [DOI] [PubMed] [Google Scholar]
- 4. Jacobo‐Molina A, Ding J, Nanni RG, et al. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double‐stranded DNA at 3.0 A resolution shows bent DNA. Proc Natl Acad Sci USA. 1993;90:6320–6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Arnold E, Das K, Ding J, et al. Targeting HIV reverse transcriptase for anti‐AIDS drug design: Structural and biological considerations for chemotherapeutic strategies. Drug Des Discov. 1996;13:29–47. [PubMed] [Google Scholar]
- 6. Erickson JW, Burt SK. Structural mechanisms of HIV drug resistance. Annu Rev Pharmacol Toxicol. 1996;36:545–571. [DOI] [PubMed] [Google Scholar]
- 7. Jochmans D. Novel HIV‐1 reverse transcriptase inhibitors. Virus Res. 2008;134:171–185. [DOI] [PubMed] [Google Scholar]
- 8. Ilina T, Parniak MA. Inhibitors of HIV‐1 reverse transcriptase. Adv Pharmacol. 2008;56:121–167. [DOI] [PubMed] [Google Scholar]
- 9. Goel R, Beard WA, Kumar A, et al. Structure/function studies of HIV‐1(1) reverse transcriptase: Dimerization‐defective mutant L289K. Biochemistry. 1993;32:13012–13018. [DOI] [PubMed] [Google Scholar]
- 10. Ghosh M, Jacques PS, Rodgers DW, Ottman M, Darlix JL, leGrice SFJ. Alterations to the primer grip of p66 HIV‐1 reverse transcriptase and their consequences for template‐primer utilization. Biochemistry. 1996;35:8553–8562. [DOI] [PubMed] [Google Scholar]
- 11. Tachedjian G, Aronson HEG, Goff SP. Analysis of mutations and suppressors affecting interactions between the subunits of the HIV type 1 reverse transcriptase. Proc Natl Acad Sci USA. 2000;97:6334–6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tachedjian G, Aronson HE, de los Santos M, Seehra J, McCoy JM, Goff SP. Role of residues in the tryptophan repeat motif for HIV‐1 reverse transcriptase dimerization. J Mol Biol. 2003;326:381–396. [DOI] [PubMed] [Google Scholar]
- 13. Wapling J, Moore KL, Sonza S, Mak J, Tachedjian G. Mutations that abrogate human immunodeficiency virus type 1 reverse transcriptase dimerization affect maturation of the reverse transcriptase heterodimer. J Virol. 2005;79:10247–10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Restle T, Muller B, Goody RS. Dimerization of human immunodeficiency virus type 1 reverse transcriptase. A target for chemotherapeutic intervention. J Biol Chem. 1990;265:8986–8988. [PubMed] [Google Scholar]
- 15. Divita G, Restle T, Goody RS, Chermann JC, Baillon JG. Inhibition of human‐immunodeficiency‐virus type‐1 reverse‐transcriptase dimerization using synthetic peptides derived from the connection domain. J Biol Chem. 1994;269:13080–13083. [PubMed] [Google Scholar]
- 16. Depollier J, Hourdou ML, Aldrian‐Herrada G, Rothwell P, Restle T, Divita G. Insight into the mechanism of a peptide inhibitor of HIV reverse transcriptase dimerization. Biochemistry. 2005;44:1909–1918. [DOI] [PubMed] [Google Scholar]
- 17. Sluis‐Cremer N, Hamamouch N, San Felix A, Velazquez S, Balzarini J, Camarasa MJ. Structure‐activity relationships of [2′,5′‐bis‐O‐(tert‐butyldimethylsilyl)‐beta‐D‐ribofuranosyl]‐3′‐spiro‐5′‐(4′‐amino‐1′,2′‐oxathiole‐2′,2′‐dioxide)thymine derivatives as inhibitors of HIV‐1 reverse transcriptase dimerization. J Med Chem. 2006;49:4834–4841. [DOI] [PubMed] [Google Scholar]
- 18. Camarasa MJ, Velazquez S, San‐Felix A, Perez‐Perez MJ, Gago F. Dimerization inhibitors of HIV‐1 reverse transcriptase, protease and integrase: A single mode of inhibition for the three HIV enzymes? Antiviral Res. 2006;71:260–267. [DOI] [PubMed] [Google Scholar]
- 19. Grohmann D, Corradi V, Elbasyouny M, et al. Small molecule inhibitors targeting HIV‐1 reverse transcriptase dimerization. ChemBioChem. 2008;9:916–922. [DOI] [PubMed] [Google Scholar]
- 20. Agopian A, Gros E, Aldrian‐Herrada G, Bosquet N, Clayette P, Divita G. A new generation of peptide‐based inhibitors targeting HIV‐1 reverse transcriptase conformational flexibility. J Biol Chem. 2009;284:254–264. [DOI] [PubMed] [Google Scholar]
- 21. Sanchez‐Murcia PA, de Castro S, Garcia‐Aparicio C, et al. Peptides mimicking the beta7/beta8 loop of HIV‐1 reverse transcriptase p51 as “hotspot‐targeted” dimerization inhibitors. ACS Med Chem Lett. 2020;11:811–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sanyal A, Mailliard RB, Rinaldo CR, et al. Novel assay reveals a large, inducible, replication‐competent HIV‐1 reservoir in resting CD4(+) T cells. Nat Med. 2017;23:885–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zheng X, Pedersen LC, Gabel SA, et al. Selective unfolding of one ribonuclease H domain of HIV reverse transcriptase is linked to homodimer formation. Nucleic Acids Res. 2014;42:5361–5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zheng X, Perera L, Mueller GA, DeRose EF, London RE. Asymmetric conformational maturation of HIV‐1 reverse transcriptase. Elife. 2015;4:e06359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sharaf NG, Poliner E, Slack RL, et al. The p66 immature precursor of HIV‐1 reverse transcriptase. Proteins. 2014;82:2343–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hsiou Y, Ding J, Das K, Clark ADJ, Hughes SH, Arnold E. Structure of unliganded HIV‐1 reverse transcriptase at 2.7 A resolution: Implications of conformational changes for polymerization and inhibition mechanisms. Structure. 1996;4:853–860. [DOI] [PubMed] [Google Scholar]
- 27. Zheng X, Mueller GA, Cuneo MJ, Derose EF, London RE. Homodimerization of the p51 subunit of HIV‐1 reverse transcriptase. Biochemistry. 2010;49:2821–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sluis‐Cremer N, Dmitrienko GI, Balzarini J, Camarasa MJ, Parniak MA. Human immunodeficiency virus type 1 reverse transcriptase dimer destabilization by 1‐{spiro[4″‐amino‐2″2″‐dioxo‐1″,2″‐oxathiole‐5″,3′‐[2′,5′‐bis‐O‐(tert‐butyldimethylsilyl)‐beta‐D‐ribofuranosyl]]}‐3‐ethylthymine. Biochemistry. 2000;39:1427–1433. [DOI] [PubMed] [Google Scholar]
- 29. Venezia CF, Howard KJ, Ignatov ME, Holladay LA, Barkley MD. Effects of efavirenz binding on the subunit equilibria of HIV‐1 reverse transcriptase. Biochemistry. 2006;45:2779–2789. [DOI] [PubMed] [Google Scholar]
- 30. Louis JM, Ishima R, Nesheiwat I, et al. Revisiting monomeric HIV‐1 protease. Characterization and redesign for improved properties. J Biol Chem. 2003;278:6085–6092. [DOI] [PubMed] [Google Scholar]
- 31. Sharaf NG, Gronenborn AM. (19)F‐modified proteins and (19)F‐containing ligands as tools in solution NMR studies of protein interactions. Methods Enzymol. 2015;565:67–95. [DOI] [PubMed] [Google Scholar]
- 32. Sharaf NG, Ishima R, Gronenborn AM. Conformational plasticity of the NNRTI‐binding pocket in HIV‐1 reverse transcriptase—A fluorine NMR study. Biochemistry. 2016;55:3864–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sharaf NG, Xi Z, Ishima R, Gronenborn AM. The HIV‐1 p66 homodimeric RT exhibits different conformations in the binding‐competent and ‐incompetent NNRTI site. Proteins. 2017;85:2191–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gasteiger E, Hoogland C, Gattiker A, et al. Protein identification and analysis tools on the ExPASy server. In: Walker JM, editor. The proteomics protocols handbook. Totowa, NJ: Humana Press, 2005; p. 571–607. [Google Scholar]
- 35. Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. [DOI] [PubMed] [Google Scholar]
- 36. Xi Z, Whitley MJ, Gronenborn AM. Human betaB2‐crystallin forms a face‐en‐face dimer in solution: An integrated NMR and SAXS study. Structure. 2017;25:496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Battiste JL, Wagner G. Utilization of site‐directed spin labeling and high‐resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–5365. [DOI] [PubMed] [Google Scholar]
- 38. Tzeng SR, Pai MT, Kalodimos CG. NMR studies of large protein systems. Methods Mol Biol. 2012;831:133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Iwahara J, Schwieters CD, Clore GM. Ensemble approach for NMR structure refinement against (1)H paramagnetic relaxation enhancement data arising from a flexible paramagnetic group attached to a macromolecule. J Am Chem Soc. 2004;126:5879–5896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: A pdf file that contains the following data is available free of charge
Figure S1. 1H‐15N NMR spectra of p66L289K and the mutants, used for PRE experiments
Figure S2. 1H‐15N NMR spectra of p66L289K overlaid with those of the isolated domains
Figure S3. SAXS Guinier plots and the pair‐distance distribution function for p66L289Ktr
Figure S4. Plots of PRE for residues in the thumb and RNH domains
Figure S5. SEC elution profiles of p66 and the tfmF‐labeled p66
Figure S6. SEC elution profiles of WT and L289K mutant mixtures of p66 and p51
Figure S7. The entire SDS gel shown in Figure 5c and the repeat
Table S1. Molecular masses of the elution peaks in the SEC in Figure 5
Table S2. Selected mass spectrometry results to identify the samples prepared for the study.