

Abstract
The conformational landscape of a protein is constantly expanded by genetic variations that have a minimal impact on the function(s) while causing subtle effects on protein structure. The wider the conformational space sampled by these variants, the higher the probabilities to adapt to changes in environmental conditions. However, the probability that a single mutation may result in a pathogenic phenotype also increases. Here we present a paradigmatic example of how protein evolution balances structural stability and dynamics to maximize protein adaptability and preserve protein fitness. We took advantage of known genetic variations of human alanine:glyoxylate aminotransferase (AGT1), which is present as a common major allelic form (AGT‐Ma) and a minor polymorphic form (AGT‐Mi) expressed in 20% of Caucasian population. By integrating crystallographic studies and molecular dynamics simulations, we show that AGT‐Ma is endowed with structurally unstable (frustrated) regions, which become disordered in AGT‐Mi. An in‐depth biochemical characterization of variants from an anticonsensus library, encompassing the frustrated regions, correlates this plasticity to a fitness window defined by AGT‐Ma and AGT‐Mi. Finally, co‐immunoprecipitation analysis suggests that structural frustration in AGT1 could favor additional functions related to protein–protein interactions. These results expand our understanding of protein structural evolution by establishing that naturally occurring genetic variations tip the balance between stability and frustration to maximize the ensemble of conformations falling within a well‐defined fitness window, thus expanding the adaptability potential of the protein.
Keywords: alanine:glyoxylate aminotransferases, conformational plasticity, protein evolution, protein fitness, structural dynamics
1. INTRODUCTION
Globular proteins are endowed with variable degrees of structural plasticity. Indeed, the native state is only marginally stable and dynamically samples within a wide range of conformers. 1 An extreme consequence of conformational sampling is seen in protein regions that are structurally frustrated and thus explore an excess of flexibility at the edge of disorder (fuzziness). 2 The structural dynamics of globular proteins represents a yin/yang for protein fitness, namely the ensemble of properties contributing to the function(s) of a protein, including stability, activity, and interactions. On the one hand, protein plasticity can be beneficial by promoting the adaptability to environmental changes, expanding the landscape of protein activities, and/or mediating the interaction with multiple binding partners. 3 , 4 , 5 At the same time, increasing frustration and disorder can negatively affect protein stability, which in turn can decrease overall protein fitness in a specific intracellular environment. 6 However, the fundamental biophysical mechanisms underlying globular protein surface plasticity and the evolutionary balance of the dynamics of frustrated regions to maintain intracellular fitness within a defined window are elusive.
The analysis of genetic variations, like polymorphic changes, which are highly abundant in human genome and mainly involve surface residues, can give some clues on the factors that influence protein plasticity. 7 , 8 However, the structural effects of genetic variations are often subtle and difficult to identify. They can contribute to protein adaptability, by widening the protein conformational sampling. 9 Nevertheless, they can also influence the emergence of complex diseases as well as represent modifier factors in the phenotypic expression of rare mutations. In this regard, multiple allelic forms of a protein represent a trade‐off between an advantageous adaptation, when an unusual condition becomes common, and the generation of traits that increase disease susceptibility. 6 , 10 Is it possible to unravel the structural basis of the trade‐off defining the protein fitness window?
Liver alanine:glyoxylate aminotransferase 1 (AGT1) is a dimeric pyridoxal‐5' phosphate (PLP)‐dependent enzyme that transaminates l‐alanine and glyoxylate to pyruvate and glycine, respectively, a reaction crucial for glyoxylate detoxification. 10 AGT1 is peroxisomal in humans, but it localizes to mitochondria in other vertebrates. 9 The targeting evolution has probably occurred as a molecular adaptation to diet‐specific subcellular localization of glyoxylate precursors, with an herbivorous diet favoring peroxisomes and a carnivorous diet favoring mitochondria. 9 , 11 , 12 Numerous naturally occurring variants of AGT1 are known, some of which are associated with the life‐threatening condition named Primary Hyperoxaluria Type I (PH1), characterized by the progressive accumulation of calcium oxalate stones first in the urinary tract, and then at systemic level. 13 , 14 Besides pathogenic forms, it is of note that 20% of the Caucasian population express a polymorphic form of AGT1 called minor allele (AGT‐Mi), which differs from the more common major allele (AGT‐Ma) for the P11L and I340M substitutions. 15 AGT‐Mi is not pathogenic per se, and it is not considered a risk factor for idiopathic kidney stone disease. 16 However, some missense mutations in AGT1 are only pathogenic if inherited on the background of the minor allele polymorphism, a form of genetic epistasis. 17 , 18 As compared to AGT‐Ma, AGT‐Mi displays a slightly reduced catalytic efficiency and thermodynamic stability, as well as increased tendency to aggregation and intracellular degradation. 19 , 20 , 21 Indeed, AGT‐Ma and AGT‐Mi have been proposed as the higher and the lower limits, respectively, for AGT1 thermodynamic and kinetic stability. This view would place AGT‐Mi at the edge of misfolding, so that any further destabilizing mutation leads to a decrease in protein fitness resulting in a pathological phenotype. 20 , 22 , 23 Thus, AGT1 represents an ideal model to understand how nature explores structural dynamics to shape the fitness window in a specific cellular environment.
In this work, we have performed a systematic study showing that AGT1 retains a certain degree of conformational plasticity whose limit is represented by AGT‐Mi. Structural analyses of AGT‐Mi allow to unravel that the plasticity of AGT1 is constituted by three frustrated regions whose dynamics is the result of an enhanced conformational flexibility of the N‐terminal arm that propagates through all the structure. A mainly “anticonsensus” library of AGT‐Ma in these three regions reveals the biochemical and biophysical aspects related to the transition from the fitness of AGT‐Ma to the lowest limit of fitness represented by AGT‐Mi. Finally, studies in eukaryotic cells demonstrate that an altered conformational plasticity affects intracellular stability and interaction with binding partners, suggesting that polymorphic genetic variations can also shape the cellular interactome.
2. RESULTS
2.1. The crystal structure of AGT‐Mi unveils three unexpected frustrated regions and allows to decipher AGT1 hidden plasticity
While several crystal structures of AGT‐Ma were solved, 24 , 25 , 26 , 27 , 28 , 29 , 30 no structural information on AGT‐Mi was available. Therefore, we first solved the structure of AGT‐Mi by x‐ray crystallography at 2.2 Å resolution (Figure 1, Table S1). The I340M mutation was clearly observed as a sharp positive electron density peak corresponding to the sulfur atom position (Figure S1a). On the other hand, no electron density was observed for the N‐terminal residues (residues 1–10: MASHKLLVTP; Figure S1b), indicating that the P11L mutation results in a loss of a defined structure in this region. In the AGT1 dimer, the N‐terminal arm is involved in an important interaction between the monomers, and mutations altering its conformation are known to affect protein thermodynamic and kinetic stability. 22 , 31 , 32 Thus, our data provide a definitive proof for many biochemical and in silico studies performed in the last decade, which have suggested that the lower stability of AGT‐Mi was linked to a disordered N‐terminus. 19 , 32 , 33 , 34 , 35 Surprisingly, we found the presence of another disordered region in AGT‐Mi: an undefined electron density was observed for residues 97–102 (GDSFLV), 121–146 (ARVHPMTKDGGHYTLQEVEEGLAQH), and 170–177 (GELCHRYK), which were not included in the final model (Figure S1c,d). In AGT‐Ma, these regions form two α‐helices and two β‐strands at the periphery of the large domain (the structural unit binding PLP; Figure 1a), which tend to display a higher mobility in all the structures solved to date, with temperature factors (B‐factors) that appear systematically higher than the mean values of each structure (Figure 1b,c). By using multiple in silico disorder prediction softwares, we did not identify the three regions as disordered, although they appear as higher peaks of disorder score compared to the rest of the structure, excluding the N‐terminus (Figure S2). Notably, no significant differences in disorder between AGT‐Mi and AGT‐Ma were observed (Figure S2). These results suggest that AGT‐Ma itself is endowed with an unexpected structural dynamics, which becomes extreme and detectable in AGT‐Mi. Thus, AGT1 samples different conformations whose relative populations are driven by the presence of the two polymorphic changes typical of the minor allele. Ultimately, these mutations induce an increased disorder not only at the N‐terminus, but also in other unrelated regions of AGT1 appearing as structurally frustrated.
FIGURE 1.
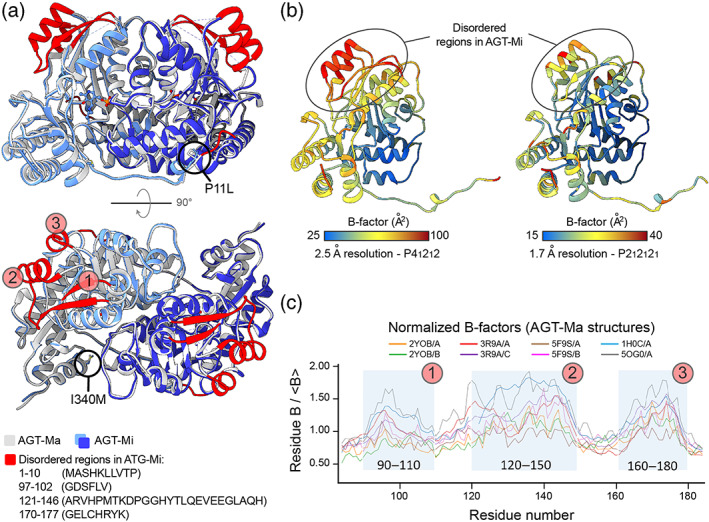
Structure of AGT‐Mi. (a) Cartoon representation of the structure of AGT‐Mi superimposed on that of AGT‐Ma (PDB: 5F9S). The two subunits of AGT‐Mi are shown in dark and light blue while for AGT‐Ma are shown in white and dark gray. The regions that were not included in the model of AGT‐Mi, due to disorder, are colored in red in the structure of AGT‐Ma. Except for the N‐terminus, the other disordered regions are numbered from 1 to 3 and correspond to residue stretches 97–102, 121–146, and 170–177, respectively. (b) Two AGT‐Ma structures solved at different resolution and in different space groups are colored according to temperature factor (B‐factors) values (PDBs: 5OG0 and 5F9S on the left and right side, respectively). Both structures show high B‐factors in the regions that are disordered in AGT‐Mi. (c) Analysis of normalized B‐factors over the range of all available AGT‐Ma structures in different space groups. High B‐factors values correspond to the disordered regions (1, 2, and 3) in AGT‐Mi. These regions are not involved in crystal contacts; therefore, the observed disorder is not a consequence of crystal packing. The B‐factor of the structure of the sigle mutant I340M (PDB: 2YOB) 32 is the lowest, suggesting a destabilizing effect of the P11L mutation balanced by a stabilizing effect of the I340M mutation
2.2. Molecular dynamics allow to study the role of frustrated regions in AGT1
Next, we investigated the AGT1 conformational sampling by molecular dynamics (MD) simulations. We aimed at detecting any difference between the conformers sampled by AGT‐Ma and AGT‐Mi. To be unbiased, we used as a model the stable AGT‐Ma conformation (PDB: 5F9S) as a starting point for the single‐ and double‐mutant models. The single‐mutant models were obtained by individually mutating P11L or I340M, while the double‐mutant model was obtained by mutating both positions together, to achieve the AGT‐Mi model (more details in the Methods). After an equilibration step, the models were run for 200 ns in five repeats (Figure 2a).
FIGURE 2.

Molecular dynamics simulations. (a) Plot of the root mean square fluctuation (RMSF) as a function of residue number: AGT‐Ma (purple) and AGT‐Mi (green). The difference ΔRMSF (RMSF AGT‐Mi – RMSF AGT‐Ma) is mapped on the structure of AGT‐Ma as thickness of the tube‐like representation (obtained by Chimera). 36 The RMSF values used were obtained as the average of five trajectories. The regions of interest are marked with the same colors in the structure and in the plot. The increased mobility of the N‐terminus (in pink) and especially of the residues 48–64 (in magenta) of AGT‐Mi may cause a domino effect that is propagated through the C‐terminus (in yellow) and three structural regions (indicated with a, b, and c) that are directly interacting with PLP and with the disordered regions 1, 2, and 3 (in red). Details on the MD method are reported in Table S2. (b) Free energy landscape as a function of the Φ and Ψ dihedral angles of the backbone of the high B‐factor region 121–133. This region forms a β‐strand that contacts the flexible C‐terminus. (c) AGT‐Ma conformational frustration plot. Green and red colors indicate couples of minimal and maximal frustrated residues, respectively. The configurational frustration index evaluates how the energetic contribution to protein stability of the interaction between residues i and j compares to different possible local environments. The interaction is considered frustrated when other conformations with higher stabilization energy are possible. 37 Areas of high frustration are localized at the interface of the highly fluctuating regions, connecting the N‐terminus to the disordered regions in AGT‐Mi (blue arrow). The same coloring of panel A is used to highlight the different portions of AGT1 in the structure and in the correlation plot. (d) Free Energy Surface (FES) of the five combined simulations for AGT‐Ma and AGT‐Mi. The first 5 ns of each simulation were removed from the computations to remove any bias due to the equilibration of the system. The FES was generated in a similar fashion as in Strodel et al. 38 and using RMSD and gyration radius as the two coordinate systems. On the right, a representative structure of AGT‐Mi in the highlighted region of panel B (with high RMSD and high gyration radius) is shown in pink, superposed with the structure of AGT‐Ma (5F9S) that is depicted with the same coloring pattern of panel A. The loss of secondary structure and displacement (blue arrows) is evident in parts of the disordered regions and in the C‐terminal domain. The displacement also affects the cofactor position (inset)
The simulations confirmed that the stretches of residues identified as disordered in AGT‐Mi by crystallographic studies also fluctuate in AGT‐Ma (Figure 2a), as suggested by B‐factor analysis (Figure 1b,c), indicating that these regions are prone to disorder in AGT‐Ma too. However, in AGT‐Mi, the N‐terminus (residues 6–18) and the helix 48–64 quickly sample a range of alternative conformations due to an increase of the fluctuations in these regions that are not observed in the AGT‐Ma simulations (Figure 2a). Notably, this helix is in close contact with both the very dynamic C‐terminal domain and several regions involved in PLP binding (Figure 2a). From the simulation results, it appears that the multiple conformations adopted by AGT‐Mi result from backbone dynamics initiated by an increased fluctuation of the helix 48–64 and the N‐terminus, which propagates through the C‐terminus and the active site to the frustrated regions (Figure S3a–e). Indeed, comparing the simulations of AGT‐Mi and AGT‐Ma, we observed differences in the distribution of the dihedral angles of the backbone in the second frustrated region (residues 121–131). This region constitutes the β‐strand in contact with the highly flexible C‐terminus (Figure 2b) and the third frustrated region (residues 170–177; Figure S3e). However, the same analysis on the other frustrated regions does not discriminate between AGT‐Mi and AGT‐Ma (Figure S3e). Analysis of protein local frustration nicely correlates with molecular dynamics data. The high conformational frustration cluster in AGT‐Ma pairs with the maximal fluctuation regions and ultimately connect the N‐ and C‐terminus dynamics (Figure 2c, Movie S1). Interestingly, Free Energy Surface (FES) analysis confirmed that AGT‐Mi uniquely explores a set of conformations at high RMSD and gyration radius where the C‐terminal helices and parts of the frustrated regions are notably displaced (Figure 2d). This apparently small detachment of two helices may indeed weaken the hydrophobic interactions that stabilize the structured state leading to a disordered conformation. Furthermore, MD simulations of the single‐mutant P11L showed enhanced fluctuations of the N‐terminal tail (Figure S3b), and a higher occupancy of the conformations at high radius of gyration (Figure S3c). This result highlights the structural determinants of the stabilizing effect of the I340M mutation that we observed in the MD simulation of this single mutant and also in previous studies. 39 In fact, Ile340 is placed at the interface between helix 48–64 and the C‐terminus, and its substitution with a longer hydrophobic side chain partially reduces the dynamics of these regions (Figure S3d).
To test the role of the order/disorder transition in a cellular environment, we rationally introduced four mutations—L101A, H146A, L172A, and C173A—that should enhance the plasticity of frustrated regions by reducing the number of favorable interactions between the side chains. As shown in Figure S4, substitutions in the first and third region (residues 97–102 and 170–177, respectively) do not significantly change AGT1‐specific activity and protein levels, while the H146A mutation of the second region leads to a significant reduction of soluble protein and active enzyme levels, possibly related to the increased backbone flexibility of adjacent regions (121–131) suggested by the MD analysis.
Overall, by combining structural, MD, and cellular data, we highlighted the presence of a structural hotspot that is able to sample multiple conformations for AGT1 in the native state. These results place AGT‐Ma and AGT‐Mi at the boundaries of an unexpectedly wide conformational landscape that is dominated by the structural dynamics of three frustrated regions, with the major allele sampling, on average, ordered conformations, and the minor allele living on the edge of disorder.
2.3. A library of the frustrated regions of AGT1 hints to the advantage of structural plasticity
To better define the role of the three flexible regions (residues 97–102; 121–146; 170–177) on the conformational sampling of AGT1, we generated a library of AGT‐Ma variants in these stretches. The design of the library was based on two main principles.
First, we identified all the positions where the residue conservation is below 80% among the alignment (Table S3). Then we identified “anticonsensus” mutations, namely those causing the change of a residue with another one present in nature in the same position but showing a lower abundance (Figure 3a). This choice was dictated by the need to mimic a subtle destabilization, while avoiding big conformational changes and keeping intact the AGT1 overall structure. Positions 127 and 177 are the only ones in which the human sequence does not show a consensus residue. Therefore, we decided to mutate them to the most represented residue in other species, with the aim of exploring the possibility of achieving a more stable and active structure at the expenses of the above detected structural plasticity.
FIGURE 3.
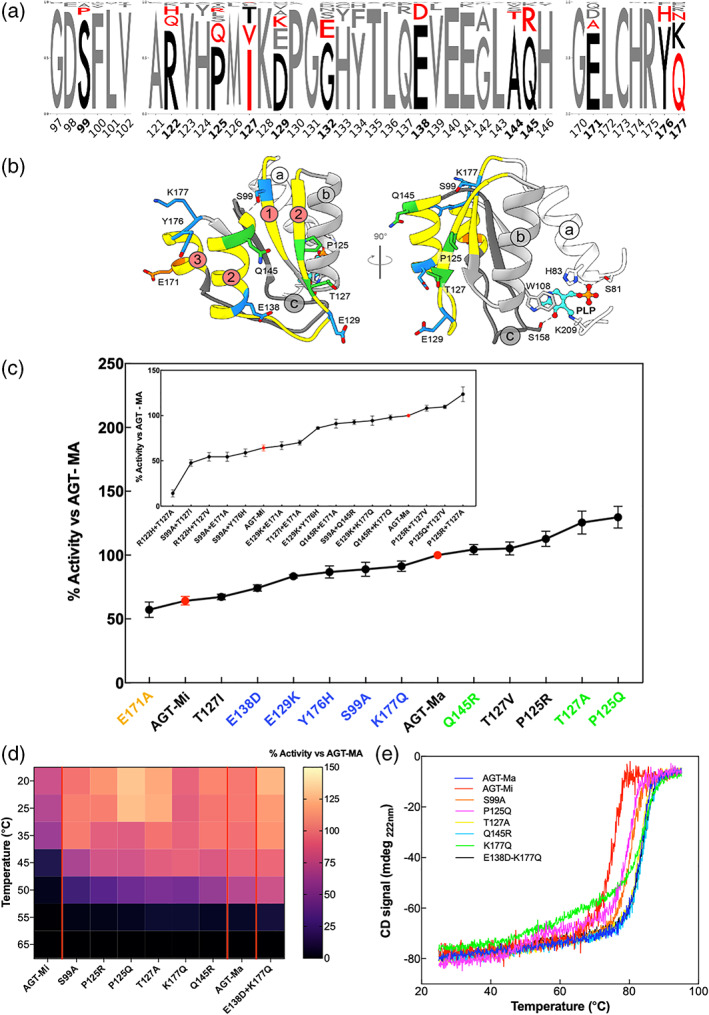
Library construction and biochemical and biophysical characterization of the variants. (a) Consensus logo of the three frustrated regions under study. The sequences (110–130) from the non‐redundant database were collected by blast search and aligned using MUSCLE algorithm on Jalview. 40 The logo was generated using Logo maker (https://logomaker.readthedocs.io/en/latest/). In bold black numbers, the positions that were mutated. In black, the amino acids of the human AGT1 sequence, while in red, the amino acids considered for the library. (b) The mutated residues are plotted over the AGT‐Ma structures in different colors. In orange those with activity below AGT‐Mi, in blue those with activity between AGT‐Mi and AGT‐Ma, and in green those above AGT‐Ma. (c) Residual activity of the indicated single variants as compared to AGT‐Ma. Inset panel C) Residual activity of the indicated double variants as compared to AGT‐Ma. Experiments have been performed in duplicate, and for each single experiments, three technical triplicates have been used. Data are represented as mean values ± SD. (d) Heat map of residual activity of selected mutants obtained after incubating the lysate at different temperatures for 10 min. The color code is the same of panel c. (e) Thermal stability curves of the selected variants represented in panel d. Color code is the same of panel c. Experiments have been performed in duplicate, and for each single experiment, three technical replicates have been used
Second, we used a semi‐rational approach, that is, a strategy based on the random combination of rationally chosen mutations (Section 4). As a result, we obtained a small library (48 variants) bearing single, double, and triple mutations.
Interestingly, although the library was focused on a region distant from the catalytic site (>12 Å, Figure 3b), its importance for the overall AGT1 stability allowed the reconstruction of the putative functional intermediates within and beyond the fitness window defined by AGT‐Ma and AGT‐Mi (Figures 3c and S5). Then, 21% of the mutants showed specific activity below AGT‐Mi, 48% between AGT‐Mi and AGT‐Ma, and 31% above AGT‐Ma. The wide range of activity sampled by this library demonstrates the importance of these three regions on the overall structure and gives further information on the effect of the frustration on AGT1.
To explain the fitness outcomes of the mutations introduced on the surface of the flexible regions, we chose two variants with specific activity between AGT‐Mi and AGT‐Ma (S99A, K177Q) and four with specific activity above that of AGT‐Ma (P125Q, T127A, Q145R, E138A/K177Q) for further characterization in the purified form. Upon confirming by CD studies that none of the introduced substitutions caused gross structural changes in AGT1 (Figure S6a), we determined both the melting temperature (Figure 3d) and the kinetic parameters for the transamination reaction, to get insights on possible alterations of global stability and intrinsic catalytic activity, respectively. The functional characterization of the purified variants revealed no significant changes in the kinetic parameters as reported in Table 1 and Figure S7. In fact, frustrated regions are distant from the catalytic site (>12 Å), and the mutations do not directly influence the microenvironment of the cofactor, as shown by the visible bands of the CD spectra of the corresponding variants (Figure S6b). More interestingly, none of the mutations interferes with the turnover number of the enzyme, thus indicating that the observed changes in specific activity in the bacterial lysate (Figure 3c) can be mainly ascribed to subtle structural alterations that affect protein levels, possibly implying that dynamic surfaces represent critical regions for the overall AGT1 fitness in a cellular environment.
TABLE 1.
Kinetic parameters of AGT‐Ma, AGT‐Mi, and the variants chosen from the AGT library
| Enzyme | Substrate | Cosubstrate | k cat (s−1) | K M (mM) | k cat/K M (s−1 mM−1) |
| AGT‐Ma a | l‐alanine | Glyoxylate | 45 ± 2 | 31 ± 4 | 1.4 ± 0.2 |
| Glyoxylate | l‐alanine | 45 ± 3 | 0.23 ± 0.05 | 196 ± 44 | |
| AGT‐Mi b | l‐alanine | Glyoxylate | 33 ± 5 | 28 ± 2 | 1.2 ± 0.2 |
| Glyoxylate | l‐alanine | 37 ± 1 | 0.22 ± 0.01 | 168 ± 8 | |
| S99A | l‐alanine | Glyoxylate | 36 ± 2 | 29 ± 4 | 1.2 ± 0.18 |
| Glyoxylate | l‐alanine | 34 ± 3 | 0.31 ± 0.05 | 109 ± 20 | |
| P125Q | l‐alanine | Glyoxylate | 43 ± 5 | 55 ± 8 | 0.78 ± 0.14 |
| Glyoxylate | l‐alanine | 35 ± 2 | 0.22 ± 0.03 | 159 ± 22 | |
| T127A | l‐alanine | Glyoxylate | 42 ± 3 | 33 ± 4 | 1.27 ± 0.17 |
| Glyoxylate | l‐alanine | 34 ± 4 | 0.16 ± 0.03 | 212 ± 44 | |
| Q145R | l‐alanine | Glyoxylate | 47 ± 4 | 41 ± 5 | 1.15 ± 0.17 |
| Glyoxylate | l‐alanine | 38 ± 4 | 0.21 ± 0.04 | 181 ± 32 | |
| K177Q | l‐alanine | Glyoxylate | 37 ± 3 | 35 ± 5 | 1.05 ± 0.17 |
| Glyoxylate | l‐alanine | 36 ± 4 | 0.27 ± 0.06 | 133 ± 33 | |
| E138D/K177Q | l‐alanine | Glyoxylate | 44 ± 4 | 53 ± 7 | 0.83 ± 0.13 |
| Glyoxylate | l‐alanine | 39 ± 3 | 0.27 ± 0.04 | 145 ± 24 |
It is known that both AGT‐Ma and AGT‐Mi display a cooperative thermal unfolding process, with AGT‐Mi showing lower melting temperature as compared to AGT‐Ma. 19 , 32 The analysis of the melting curves of the single variants confirmed how AGT1 is exploring a wide window of stability. 19 , 32 The stability of AGT‐Ma was not affected or slightly improved by the T127A, Q145R, and K177Q mutations, but it was reduced by the S99A and P125Q substitutions to values between those of the two polymorphic forms. Notably, the denaturation curves of the P125Q and K177Q variants show the loss of cooperativity on the unfolding of the two domains of AGT1, in line with MD results indicating that frustrated regions on the large domain influence the conformation of the small C‐terminal domain where Ile340 is located. 28 The latter effect on the single K177Q variant is abolished in the E138D/K177Q variant, in agreement with the improved fitness of the double variant upon Escherichia coli expression (Figure 3c). Overall, all the mutants included in the library are functionally active, meaning that mutations of the frustrated regions do not have substantial negative effects on the AGT1 structure. The finding that all the detected changes in stability and activity are subtle suggests that naturally occurring mutations can be tolerated while maintaining the protein within a defined fitness window.
2.4. Expression of library variants in a cellular system highlights their role for AGT1 fitness and interactions
To investigate the effects of the library changes in a mammalian cellular environment, selected variants with single or double mutations in the frustrated regions were expressed in Human Embryonic Kidney 293 (HEK293) cells and analyzed for specific activity and protein levels (Figure 4).
FIGURE 4.
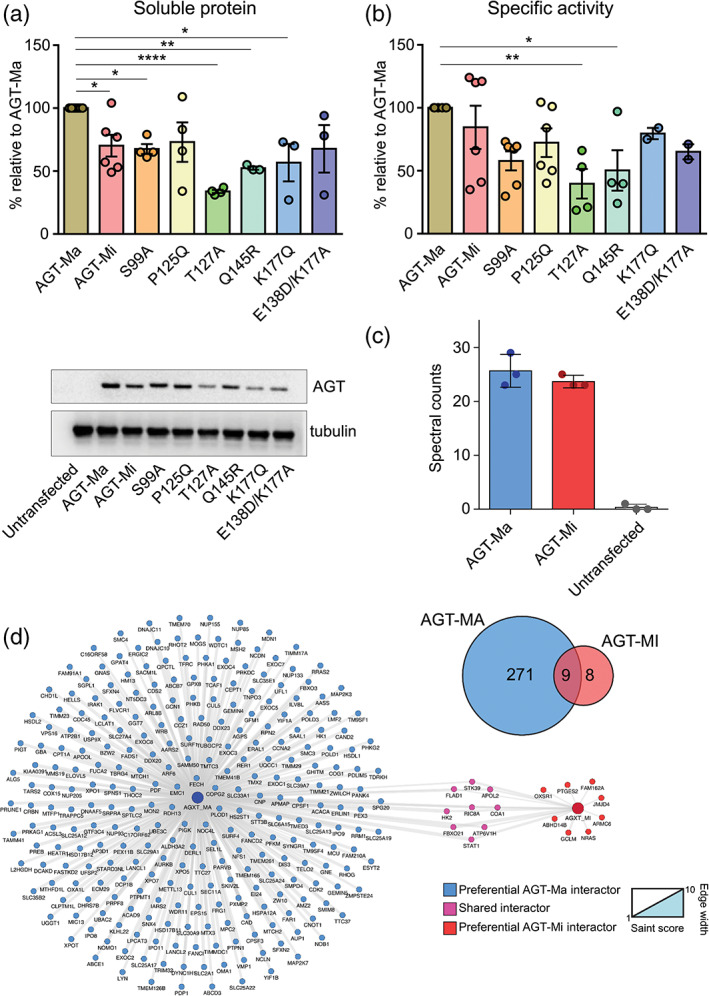
Expression, specific activity, and interactors of AGT‐Ma, AGT‐Mi, and selected variants in human cells. (a) and (b) HEK293 cells expressing the indicated species were harvested and lysed. In panel a, soluble protein levels were quantified by western‐blot. Then, 10 μg of soluble lysate of each sample was subjected to SDS‐PAGE, immunoblotted with anti‐AGT from rabbit (1:10,000), and detected by chemiluminescence. The histogram is representative of immunoblot band volume (as mean ± SEM of at least three independent experiments). In panel b, the specific activity for the transamination reaction was measured by incubating 90 μg of soluble lysate with 0.5 M l‐alanine, 10 mM glyoxylate, and 200 μM PLP at 25°C in 100 mM KP, pH 7.4. c,d) Interactome of AGT‐Ma and AGT‐Mi. Panel c shows that AGT1 protein levels were enriched in AGT‐Ma and AGT‐Mi transfected samples after immunoprecipitation using a pan anti‐AGT1 antibody. Control samples (Ctrl) showed undetectable levels of AGT; Panel d shows Venn diagram of significant interactions compared to controls between AGT‐Ma and AGT‐Mi and the interaction network of AGT‐Ma and AGT‐Mi isoforms. Nodes are representative of proteins described with gene name, edges' width is proportional to SAINT score versus control samples
As expected, cells expressing AGT‐Mi displayed a slightly lower specific activity and protein levels as compared to AGT‐Ma. The S99A mutation in region 1 reduced AGT1 specific activity and protein levels below AGT‐Ma. These data are in line with those obtained upon bacterial expression, where the S99A variant showed intermediate properties between AGT‐Ma and AGT‐Mi when present alone, but all double and triple variants containing the S99A mutation displayed a fitness lower than AGT‐Mi. On the other hand, differences between the bacterial and mammalian environment were observed on variants belonging to the second region. P125Q, T127A, and Q145R variants showed specific activity values higher than AGT‐Ma upon expression in E. coli. However, in analogy with the data obtained upon mutation of His146 (Figure S4), the presence of these mutations strongly impaired the specific activity and relative expression of AGT1 in mammalian cells. Indeed, the P125Q variant showed a behavior comparable to AGT‐Mi, while lower activity and expression levels were observed in the presence of T127A and Q145R mutations. Since Pro125, Thr127, and Gln145 do not affect the kinetic properties of AGT1 or its intrinsic thermodynamic stability, these results indicate that the observed changes are mediated by the mammalian cellular environment. It can be suggested that the conformational change caused by the mutation of Thr127 and Gln145 could favor the interaction with components of the quality control network typical of eukaryotic cells. 41 Finally, mutations in the third region are expected to slightly affect protein fitness with L172A and C173A displaying a specific activity similar, if not higher, compared to AGT‐Ma (Figure S4). However, the K177Q and E138D/K177Q variants showed specific activity and protein levels lower than AGT‐Ma and similar to those of AGT‐Mi, in line with the observed changes of melting temperature caused by the Lys177 mutation.
Overall, these data allow to conclude that introducing mutations in residues belonging to frustrated regions does indeed affect intracellular protein fitness, thus suggesting a potential role of these regions in mediating processes occurring in a cellular environment. In this regard, it is known that frustrated surface regions can create hubs for the interaction network 42 , 43 and that polymorphic and pathogenic variations involving residues located on structurally exposed regions often cause a rewiring of a protein interactome. 44 , 45
Thus, we analyzed the interactome of the two AGT1 isoforms by co‐immunoprecipitation coupled with quantitative mass spectrometry (IP‐MS). Selected clones of HEK293 cells expressing AGT‐Ma or AGT‐Mi did not show any gross changes in morphology or viability as compared to control cells. Moreover, they displayed a similar number of identified proteins compared to controls not expressing AGT (~3,000, data not shown). Label‐free quantification of AGT1 showed a significant enrichment of both AGT‐Ma and AGT‐Mi isoforms compared to control samples in which AGT1 was basically not detectable (Figure 4c). SAINT analysis of the interactions of AGT‐Ma and AGT‐Mi indicated a marked difference in the preferential interactome of the two forms (Table S6). According to our filtering criteria versus control IPs, AGT‐Ma showed 281 significant interactions while AGT‐Mi only 17 (Figure 4d). Overlap analysis of the interactions between the two forms showed a minimal overlap with nine proteins interacting with both AGT‐Ma and AGT‐Mi. A network and pathway analysis of the interactors which are lost by AGT‐Mi and are preferential interactors of AGT‐Ma (Figure S11) provided evidences of enrichment in proteins linked to both mitochondria and peroxisome organization, a finding possibly linked to the dual localization of AGT1 in vertebrates. 12 It is also interesting that AGT‐Mi shows a loss of interactions with some key peroxisomal biogenesis factors (PEX3, PEX11B, and PXMP2) which are retained by AGT‐Ma, in line with the reported tendency of 5% of AGT‐Mi to be imported to mitochondria. 18
3. DISCUSSION
The main finding of this work is that AGT1 is endowed with a high conformational plasticity (fuzziness), due to the presence of structurally frustrated regions. This leads to two main questions: how AGT1 structural frustration has been shaped during evolution? Which are the implications of AGT1 dynamics on the fitness and function at cellular level?
The presence of frustrated regions that can render an enzyme less tolerant to mutations seems counterintuitive, but AGT1 can be considered a paradigmatic example of how a certain level of frustration is retained to possibly mediate processes occurring within the cellular environment. In the subtle trade‐off between a stable rigid structure with high catalytic activity, and a flexible one, endowed with a lower catalytic activity but capable of additional functions, nature finds a compromise by keeping the degree of plasticity within a defined fitness window (Figure 5).
FIGURE 5.
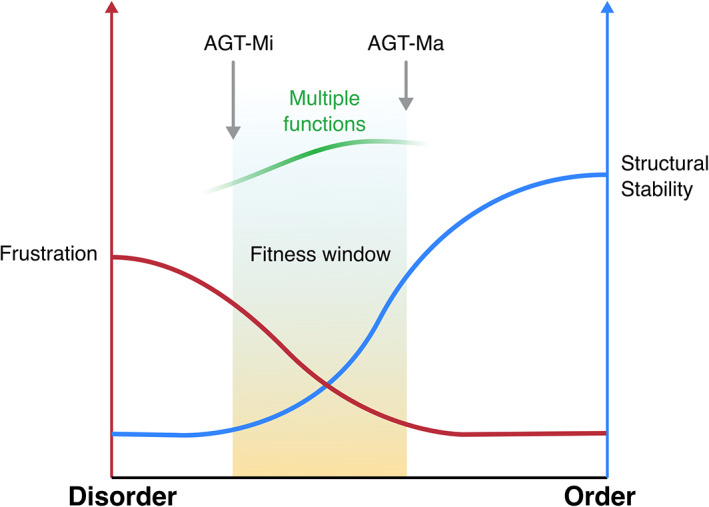
Scheme representing the fitness window of AGT1 as resulting from the balance between structural stability (blue curve) and frustration (red curve)
We immediately recognized the AGT1 polymorphism as a powerful model system to explore how point mutations may affect protein fitness. AGT‐Ma and AGT‐Mi structurally differ for the degree of flexibility of three peripheral regions and represent the limits of the fitness window of AGT1. 20 We explored these limits by constructing a small‐scale “anticonsensus” library and found that not only it sampled the entire fitness range from AGT‐Mi to AGT‐Ma, but also identified variants with higher activity and/or stability than AGT‐Ma. Although it is not possible to rationalize the effect of each mutation, we noticed, as expected, that mutating less frequent residues to the most frequent ones in other species gives rise to advantageous effects, while mutating the most conserved residues to the less conserved ones seems to be generally disadvantageous. Since we were able to sample many AGT1 variants with a stability and activity comparable or even better than AGT‐Ma with just a small library, it is likely that the conformational plasticity of AGT‐Ma due to the presence of frustrated regions may have been conserved by evolution to keep the balance between the advantages of a certain degree of dynamics, and the need to maintain an acceptable level of structural stability. Recent studies indicate that proteins showing flexible regions play a major role during the first stages of speciation due to their tolerability to mutations. 46 Therefore, the evolutionary advantage of a protein endowed with structural dynamics goes probably beyond the catalytic efficiency to encompass the stability/function/interactions in a cellular environment.
In AGT1, the frustrated regions play a crucial role for intracellular stability, with AGT‐Ma maximizing both the interactions and the activity and AGT‐Mi being at the edge of misfolding, thus drastically reducing the possibility to establish stable interactions. Following this interpretation, genetic variations that force the protein outside this fitness window toward an excess of disorder or a too stable structure are not tolerated. This is in line with the finding that additional mutations on the background of the minor allele that further reduce AGT1 fitness lead to pathological phenotypes. 13 , 17 , 20 Our data on the behavior of selected single variants of the library expressed in HEK293 cells agree with this hypothesis, by showing that mutations on the frustrated regions that make AGT1 more stable and active when expressed in bacteria (i.e., T127A and Q145R) can reduce the intracellular stability in human cells in terms of both protein levels and specific activity. At the same time, mutations that shift the equilibrium of the native conformers toward a disordered state may induce aggregation or degradation upon sensing by the quality control systems. 41 Indeed, the majority of protein stretches that confer protein instability, called degrons, are located in disordered surface regions that facilitate local unfolding and binding to the proteasome. 47 Degrons—namely portions of protein that regulate degradation rate—affect protein half‐life and can represent hubs for protein–protein interactions, so that their alterations also lead to interactome rewiring. 47 , 48 This observation is in line with our proteomic data showing that AGT‐Ma is able to establish protein–protein interactions, whose number is remarkably reduced in AGT‐Mi, the form mainly shifted toward disorder. Notably, a common binding partner between the two forms is FBXO21, a E3 ubiquitin ligase, 49 thus further reinforcing the view that frustrated regions in AGT1 play a crucial role in regulating protein turnover.
Besides uncovering an unexpectedly wide structural landscape of AGT1 in the native state, our data combined with previous evidences also definitely place AGT‐Mi as the lower limit of fitness. Interestingly, when looking at the rate of the evolution of the AGT1 structure, positions 11 and 340 are characterized by low evolutionary rate (Figure S8). While Pro11 is well conserved as expected, Ile340 is not conserved, and its substitutions to Met represents a consensus change (Figures S9 and S10). 27 , 39 On this basis, the P11L change is supposed to be highly destabilizing and its effects should be compensated by the I340M. 18 Indeed, under conditions mimicking a physiological environment in terms of pH, ionic strength, and temperature, AGT‐Mi in the native form is more prone to aggregation mediated by electrostatic forces. 39 Moreover, the two polymorphic mutations P11L and I340M typical of the minor allele oppositely affect the propensity of native AGT1 to aggregation and degradation. Our data confirm and structurally explain these observations as a consequence of the increased surface frustration in AGT‐Mi, which is promoted by the P11L mutation at the N‐terminus, and attenuated by the I340M mutation, therefore giving a detailed picture of how the two changes compromise to keep the protein in a strict fitness window. Moreover, the data allow to re‐interpret the effects of two PH1‐associated mutations in residues belonging to region 2 (D129H and E141D) that were predicted to cause the AGT1 deficit by changing the electrostatic potential surface. 39 It is likely that these mutations modify the degree of disorder of region 2 with consequent negative impact on the intracellular stability of the protein.
The latter observations call for a third and last question: why is the minor allele polymorphism retained during evolution?
Although the detailed analysis of the interactors at functional level goes beyond the scope of this work, we observed that AGT1 seems to retain the possibility to interact with partners involved in both peroxisomal organization and mitochondrial protein import and/or assembly (Table S6, Figure S11). It cannot be excluded that this property represents a vestigial of the mitochondrial localization of AGT1 that has been lost as a consequence of an adaptive shift in AGT1 targeting, under the evolutive pressure of a diet change from mainly carnivorous to omnivorous and herbivorous. 12 This concept has been confirmed in the last years by evolutionary studies performed in birds and bats. 9 , 11 Notably, the P11L polymorphism reduces AGT1 half‐life and remodels regions possibly involved in the interaction with binding partners, but also generates a weak N‐terminal mitochondrial targeting sequence that redirects 5% of the protein to mitochondria, where the metabolism of hydroxyproline takes place. 18 It can be speculated that this feature might be conserved in Caucasian population, whose diet includes more meat than African and Asian populations, as an advantage which may in part compensate for the reduced activity and interactions. This view opens new avenues for studies aimed at better understanding glyoxylate/oxalate metabolism in humans and its relation with environmental conditions.
Overall, our work has broader implications into the view of protein folding landscapes as flat‐bottom funnels, where the native state is not unique but fluctuates among different conformers. We showed that the conformational landscape explored by a protein can be greatly influenced by naturally occurring mutations, which can influence the fitness by determining the population of different conformers, a process linked to the necessity of balancing catalysis with networking. Following this view, the fate of a protein would also depend on the equilibrium between “functional” or “potentially noxious” native conformers, which in turn might be determined by mutations involving surface residues, whose importance has been underestimated in classical biochemical and protein folding studies. In this regard, a growing number of reports underline that natural polymorphisms can be key contributors to human diseases, 50 , 51 and that single amino acid changes common in human population often affect binding interfaces with other proteins or nucleic acids. 52
The AGT1 case can thus be viewed as an example of evolutionary “risk‐management”: a certain degree of plasticity, although it may lead to misfunction, is retained and “controlled” in order to achieve additional functions (multitasking).
4. MATERIALS AND METHODS
4.1. Reagents
Pyridoxal phosphate (PLP), l‐alanine, sodium glyoxylate, rabbit muscle l‐lactic dehydrogenase (LDH), β‐Nicotinamide adenine dinucleotide reduced form (NADH), and isopropyl‐β‐d‐thiogalactoside (IPTG) were purchased from Sigma‐Aldrich. Protease Inhibitor Cocktails organic solvents were purchased from Nacalai tesque. All other chemicals, unless otherwise noted, were from Sigma‐Aldrich, Nacalai tesque, or Wako. The anti‐AGT1 antibody from rabbit was provided by Prof. Chris Danpure (University College London).
4.2. Crystallography of AGT‐Mi and structure determination
Single crystals of AGT‐Mi were obtained by vapor diffusion methods (sitting drop) mixing 0.4 μl of a 37 μM protein solution (buffer: 0.1 M potassium phosphate pH = 7.4) with 0.4 μl of the A9 solution of the Morpheus screen (Molecular Dimension), containing: 30 mM MgCl2, 30 mM CaCl2, 50 mM sodium 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonate (HEPES) + 50 mM 3‐(N‐morpholino) propane sulfonic acid (MOPS) at a final pH 7.5, 40% v/v Ethylene glycol; 20% w/v PEG 8 K. Yellow diamond‐shaped crystals of about 100–120 μm grew in 1 week. Crystals were flash frozen without prior addition of cryoprotectant. A complete X‐ray diffraction dataset was collected at the Elettra synchrotron in Trieste (IT) at XRD1 beamline. The data were processed with XDS 53 and scaled with Aimless 54 , 56 , 79 initially in P41212 space group to a final resolution of 2.0 Å. However, the observed intensities were very different from the theorical values suggesting the presence of twinning. Indeed, although a very clear molecular replacement solution was found in P41212—using the AGT‐Ma structure as search model (PDB: 5F9S6) in MOLREP 57 —and the electron density maps were very clear, the model refined to poor R free and geometry values with respect to the resolution of the data. Therefore, different space groups were evaluated and finally the data were reindexed in P41 with merohedral twin operator h, −k, −l and an estimated twin fraction of 0.44 (Phenix.xtriage 58 ) that converged to 0.49 during twin‐refinement in Refmac5 or Phenix.refine. 58 , 59 Given the almost perfect twin, model building was carried out in Coot 60 using electron density maps calculated ignoring twinning, since twin‐refinement may introduce a strong model‐bias, and with an extensive and careful use of omit‐maps. Twin‐refinement was used only in the final refinement cycle. Data collection and refinement statistics are summarized in Table S1, and the quality of the electron density maps is illustrated in Figure S1. Coordinates and structure factors have been deposited in the protein data bank with PDB code: 7NS7. B‐factor analysis of AGT‐Ma solved structures was performed in Phenix GUI using structure comparison tool 58 Figures were rendered using Chimera. 36
4.3. Molecular dynamics of AGT‐Mi and AGT‐Ma
The dimeric structure of AGT‐Ma (PDB ID 5F9S) was used as starting point of MD simulations, and as template for the generation of the mutant structures. The monomeric structure of the mutants was obtained through the Swiss‐model web server, 61 realigned to the reference structure, and manually added of the PLP atoms. PDB file manipulation was performed using pdb‐tools. 62 By following the procedure of Gupta et al., a hybrid residue named “LYP” was created by combining the PLP and the Lysine 209 atoms in the PDB file and manually parametrized in the forcefield using the same values reported in the paper. 63 All simulations were performed using GROMACS version 2020.3 (www.gromacs.org) 64 and CHARMM36‐mar2019 forcefield. 65 The model was solvated in a dodecahedral box using 3 points charge water model (tip3p). 66 After charge, temperature, and pressure equilibration, each simulation run 200 ns in 2 fs steps (four repeats). A modified Berendsen thermostat was used to maintain the temperature at 310 K, 67 and a Parrinello–Rahman barostat was used to maintain a pressure of 1 bar. 68 A Particle Mesh Ewald (PME) was used for long‐range electrostatic interactions. 69 More details about the system setup and simulation parameters are provided as Supporting Information.
4.4. Site‐directed mutagenesis
Mutants were constructed as described in the Q5 Site‐Directed Mutagenesis manual (New England Biolabs). The primers list is reported in Table S4.
4.5. Expression and purification of AGT1 and variants
E. coli BL21(DE3) cells were transformed with the constructs encoding AGT‐Ma, AGT‐Mi, and the selected variants of the library. Expression and purification of the proteins were performed as reported. 10
4.6. Frustration analysis
Analysis of protein frustration was performed by uploading the AGT‐Ma structure (PDB: 5F9S) 26 to the Frustratometer server (http://frustratometer.qb.fcen.uba.ar/). 37 First a list of interacting residues are computed (Qi,j) and the native energy of the structure is calculated according to the AMW energy function. 70 Frustration between a given couple of amino acids (i,j) is then calculated by perturbating the protein sequence and calculating the total energy change. The contact is then considered minimally frustrated, neutral, or highly frustrated according to how favorable the interaction is compared to all possible interaction. Conformational frustration was visualized using VMD molecular graphics software. 71
4.7. Kinetics assays
To determine the kinetic parameters for the transamination reaction, the purified proteins (0.1–0.2 μM) were incubated in presence of 100 mM PLP in KP 0.1 M pH 7.4 at 25°C at increasing alanine concentrations (15–500 mM) or glyoxylate concentrations (0.07–5 mM) and a fixed glyoxylate (10 mM) or alanine (500 mM) concentration. The reaction was quenched by adding TCA 10% (v/v), and pyruvate production was measured using a spectrophotometric assay coupled with LDH following the NADH signal at 340 nm. 10 To determine the activity in crude bacterial cell lysates, a Tecan Spark Microplate Reader (Thermo Fischer Scientific) was used and the cells were grown and lysed as described below under the section “Library Screening.”
To measure the enzymatic activity in HEK293 cellular lysates, 100 μg of lysate were incubated with 0.5 M l‐alanine and 10 mM glyoxylate at 25°C for 20–60 min in 100 mM KP buffer, pH 7.4 in presence of 100 μM PLP. The reactions were stopped by adding TCA 10% (v/v) and pyruvate production was measured using a spectrophotometric assay coupled with lactate dehydrogenase. 72
4.8. Circular dichroism spectroscopy
Circular dichroism (CD) spectra were obtained using a Jasco J‐820 spectropolarimeter with a thermostatically controlled compartment at 25°C. In the near‐UV and visible spectra, protein concentration was 10 μM in a cuvette with a path length of 1 cm. Far‐UV measurements were taken at a protein concentration of 0.1 mg/ml using a cuvette with a path length of 0.1 cm.The thermal unfolding of AGT‐Ma, AGT‐Mi, and variants was monitored by following the loss of CD signal at 222 nm at a protein concentration of 5–8 μM with temperature increasing of 0.5°C/min from 25 to 90°C. The experiments were performed in KP 0.1 M pH 7.4.
4.9. Cloning and library construction
The AGT1 sequence (human) (uniprot: P21549, SPYA_HUMAN) was used for conducting NCBI BLAST searches across the non‐redundant database. Database search conditions were filtered for search results with query coverage >80%, and sequence identity between 70 and 90% were considered for the next step. The collected sequences were then subjected to multiple sequence alignment using MUltiple Sequence Comparison by Log‐Expectation (MUSCLE). The library was prepared using a modified Combinatorial Assembly PCR protocol. The three different regions of interest (97–102, 121–146, and 170–177) were mutated using single‐stranded mutagenic‐degenerated primers (Table S5) that anneal each other at their edges. The PCR fragments of the three regions of interest were cloned in the pTrcHis2A plasmid by Megawhop cloning 73 and mixed between them to obtain random combination of double and triple mutants. The library was then transformed in BL21 (DE3) E. coli strain, and more than 2000 clones were screened.
4.10. Library screening
Library has been cloned into pTrcHis‐2A, transformed into E. coli BL21 (DE3), and plated on ampicillin LB‐agar plates. Colonies were picked into deep 96‐well plates and grown overnight at 30°C in 500 μl LB per well supplemented with 50 μg/ml ampicillin. Wells containing fresh LB/ampicillin (2 ml) were inoculated with 100 μl of this preculture, and protein expression was induced adding 0.1 mM IPTG at 25°C for 16–18 hr. Cells were pelleted by centrifugation and the supernatant removed. Cell lysis was performed by freezing for 60 min at −80°C and resuspending in 200 μl of sodium phosphate buffer pH 7.4 and supplemented with 100 μg/ml lysozyme, proteinase inhibitor cocktail (Nacalai tesque), and 25 U/ml benzonase (Merck). The mixture has been incubated for 30 min at room temperature. Cell debris was pelleted by centrifugation, and 20–100 μl of clarified lysate were removed and tested for AGT1 activity. The activity was tested in presence of 10 μM PLP and saturating concentration of substrate and co‐substrate (10 mM glyoxylate and 500 mM l‐alanine) in 200 μl of KP 0.1 M pH 7.4. Some variants were picked and regrown in triplicate, and the rates re‐measured and the average values determined. Plasmid DNA was isolated, and variants were sequenced. The plasmids were retransformed, and the resulting chosen mutants were used to inoculate new cultures for protein purification.
4.11. Cell culture, transfection, and lysis
Human Embryonic Kidney cells (HEK293 cells) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with fetal bovine serum (FBS) (10%, v/v), penicillin (100 units/ml), and streptomycin (100 μg/ml) at 37°C in a 5% CO2 humidified environment. To test the effects of different AGT1 single aminoacid substitutions on the expression level and enzymatic activity of AGT, cells were seeded in Petri dishes at the density of 2.1 × 104/cm2 in DMEM. After 24 hr of subculture, cells were transfected with different AGT1 constructs using Lipofectamine LTX and PLUS Reagent (Invitrogen) according to the manufacturer's instructions. After 24 h, cells were harvested and used for western blotting analyses and enzymatic activity assays. For Co‐IP experiments, stable clones of HEK293 cells expressing AGT‐Ma or AGT‐Mi were generated by selecting transfected cells with 1 mg/ml G418 in the culture medium for 3 weeks.
For enzymatic activity assays and western‐blot analyses, cell pellets were lysed by freeze/thawing (5 cycles) in PBS (Phosphate Buffer Saline), pH 7.2 supplemented with 100 μM PLP and protease inhibitor cocktail (CompleteMini, Roche). The whole cell extract was separated by centrifugation (12,900×g, 10 min, 4°C) to obtain the soluble fraction. Protein concentration in the soluble fraction was measured using the Bradford protein assay.
4.12. Western blot and co‐immunoprecipitation
Ten micrograms of soluble cell lysate were loaded on 10% SDS‐PAGE, transferred on a nitrocellulose membrane and immunoblotted with anti‐AGT antibody (1:10,000) in 2.5% milk w/v in TTBS (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.1% Tween 20) overnight at 4°C. After three washes in TBST, the membrane was incubated with peroxidase‐conjugated antirabbit immunoglobulin G (IgG) (1:10,000) in 5% milk in TTBS for 1 hr at room temperature. Immunocomplexes were visualized by an enhanced chemiluminescence kit (ECL, Pierce Biotechnology, Rockford, IL). An antibody against human tubulin was used as internal loading control.
Co‐immunoprecipitation was performed using the Pierce™ Co‐Immunoprecipitation Kit according to manufacturer's instructions (Thermo‐Scientific). Briefly, cell pellets were lysed in the IP Lyses/Wash Buffer included in the kit, supplemented with protease inhibitor cocktail (CompleteMini, Roche) and phosphatase inhibitors (Sigma‐Aldrich). The whole cell extract was separated by centrifugation (12,900 × g, 10 min, 4°C) to obtain the soluble fraction. Two milligrams of soluble cell lysate from untransfected (negative control) or transfected cells (AGT‐Ma and AGT‐Mi) were incubated with 10 μg of polyclonal anti‐AGT primary antibody, immobilized on column according to the manufacturer's instructions, overnight at 4°C. The samples were eluted from the column using SDS buffer (2% SDS, 100 mM Tris–HCl pH 7.2, 10 mM DTT). After immunoprecipitation, the protein eluates were precipitated overnight with ice‐cold acetone (1:4 v/v) and subjected to sample preparation for proteomics according to previously published procedures. 74
4.13. Mass spectrometry and data analysis
Peptides derived from tryptic digestion (~1 μg) were separated on a reverse phase Pico Frit column (75 μm ID, 8 μm tip, 250 mm bed packed with Reprosil‐PUR C18‐AQ, 1.9 μm particle size, 120 Å pore size, New Objective, Inc., Woburn, MA, cat. PF7508‐250H363), using an EASY‐nLC™ 1,200 System (Thermo Fisher Scientific, Waltham, MA). Total run time for each sample was 120 min, peptides were separated using a 100 min gradient (4–40% acetonitrile + 0.1% formic acid at 300 nl/min). Eluting peptides were measured on‐line with a Q Exactive HF benchtop mass spectrometer (Thermo Fisher Scientific) operating in data‐dependent acquisition mode (Top20). Peptides were ionized at a potential of +2 kV, intact peptide ions were detected at a resolution of 120,000 (at m/z 200), and fragment ions at a resolution of 15,000 (at m/z 200); AGC Target settings for MS were 3 × 106 charges and for MS/MS 1 × 105 charges. Peptides were selected for Higher‐energy C‐trap dissociation (HCD) fragmentation with a quadrupole isolation window of 1.4 Th, peptides were fragmented at a normalized collision energy of 30. The intensity threshold was set at 2 × 104 and Dynamic exclusion at 30 s. Raw files from MS analysis were processed using the MaxQuant software version 1.6.10.43 22 (Martinsried, Germany). 17 The spectra were searched against a Uniprot human database (release 2020_2, 20,375 sequences, including isoforms). Precursor and fragment mass tolerance were set to 7 and 20 ppm., respectively, while the minimum length of the peptide was set to 7. False discovery rate (FDR) was set to 1% both at the peptide and protein level. Enzyme for in silico digestion were set to trypsin and lysine C, and up to two missed cleavages were allowed. Cysteine carbamidomethylation (Cys, +57.021464 Da) was set as a fixed modification, whereas N‐acetylation of proteins (N‐terminal, +42.010565 Da) and oxidized methionine (Met, +15.994915 Da) were included as variable modifications.
Proteins identified and quantified via LC–MS/MS were analyzed using different bioinformatics tools to distinguish interactors from false positives and obtain a list of candidates constituting the core local network of AGT proteoforms. Label‐free quantification of proteins were performed using spectral counts, while the Significance Analysis of INTeractome (SAINT) approach was used for scoring the significance of the interactions. 75 , 76 , 77 This method estimates the distribution of protein abundances under true and false interaction hypotheses and computes the probability of true interaction, taking into account the information from negative controls included in the experimental setup. Our negative controls were represented by wild‐type HEK293 cells not expressing AGT (Ctrl, n = 3) and HEK293 expressing the AGT‐Ma or AGT‐Mi isoforms subjected to the co‐IP without the AGT antibody. Scored interaction by the SAINT algorithm were filtered using a fold change versus controls >2 and a SAINT score (SP score) >0.7.
Network analysis and visualization was performed using the Cytoscape software as previously reported. 78 The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE 79 partner repository with the dataset identifier PXD025661.
4.14. Phylogenetic analysis
The aligned sequences of 254 annotated vertebrate AGT1 was obtained from the Ensembl database. 7 The best‐fit evolutionary model (JTT + 6R) 8 , 80 was chosen using ModelFinder. 81 The phylogenetic tree and the rates of evolution were calculated using IQTREE software. 82 The Coelacanth sequence ENSLACT00000026005 was used as outgroup for the tree generation.
CONFLICT OF INTEREST
The authors have declared no conflicts of interest for this article.
AUTHOR CONTRIBUTIONS
Mirco Dindo: Conceptualization (equal); data curation (equal); formal analysis (equal); investigation (equal); methodology (equal); validation (equal); writing – original draft (equal); writing – review and editing (equal). Stefano Pascarelli: Data curation (equal); formal analysis (equal); investigation (equal); writing – review and editing (equal). Davide Chiasserini: Data curation (equal); formal analysis (equal); methodology (equal); writing – review and editing (equal). Silvia Grottelli: Data curation (equal); formal analysis (equal); methodology (equal); writing – review and editing (equal). Claudio Costantini: Data curation (equal); formal analysis (equal); methodology (equal); writing – review and editing (equal). Gen‐Ichiro Uechi: Data curation (equal); formal analysis (equal); methodology (equal).
Supporting information
Appendix S1: Supporting Information
ACKNOWLEDGMENTS
The authors would like to thank the Scientific Computing and Data Analysis section of Research Support Division at OIST for the support provided for the molecular dynamics simulations. The authors would also like to thank Stefano Gianni and Benjamin Clifton for critical reading and feedback on the manuscript. Open Access Funding provided by Universita degli Studi di Perugia within the CRUI‐CARE Agreement.
Dindo M, Pascarelli S, Chiasserini D, Grottelli S, Costantini C, Uechi G‐I, et al. Structural dynamics shape the fitness window of alanine:glyoxylate aminotransferase. Protein Science. 2022;31(5):e4303. 10.1002/pro.4303
Review Editor: Nir Ben‐Tal
Giorgio Giardina, Paola Laurino, and Barbara Cellini contributed equally to this study.
Funding Information Sapienza University of Rome no. RP11916B407928AA to Giorgio Giardina; Oxalosis and Hyperoxaluria Foundation to Barbara Cellini; Financial support by the Okinawa Institute of Science and Technology to Paola Laurino is gratefully acknowledged; Mirco Dindo thanks Japan Society for the Promotion of Science (JSPS). Fellowship number: P19764.
Contributor Information
Giorgio Giardina, Email: giorgio.giardina@uniroma1.it.
Paola Laurino, Email: paola.laurino@oist.jp.
Barbara Cellini, Email: barbara.cellini@unipg.it.
REFERENCES
- 1. Chong SH, Ham S. Folding free energy landscape of ordered and intrinsically disordered proteins. Sci Rep. 2019;9:14927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gianni S, Freiberger MI, Jemth P, Ferreiro DU, Wolynes PG, Fuxreiter M. Fuzziness and frustration in the energy landscape of protein folding, function, and assembly. Acc Chem Res. 2021;54:1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Babu MM. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem Soc Trans. 2016;44:1185–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu Z, Huang Y. Advantages of proteins being disordered. Protein Sci. 2014;23:539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Uversky VN. Intrinsically disordered proteins and their “mysterious” (meta)physics. Front Phys. 2019;7:10. [Google Scholar]
- 6. van der Lee R, Lang B, Kruse K, et al. Intrinsically disordered segments affect protein half‐life in the cell and during evolution. Cell Rep. 2014;8:1832–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zerbino DR, Achuthan P, Akanni W, et al. Ensembl 2018. Nucleic Acids Res. 2018;46:D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci. 1992;8:275–282. [DOI] [PubMed] [Google Scholar]
- 9. Liu Y, Xu H, Yuan X, Rossiter SJ, Zhang S. Multiple adaptive losses of alanine‐glyoxylate aminotransferase mitochondrial targeting in fruit‐eating bats. Mol Biol Evol. 2012;29:1507–1511. [DOI] [PubMed] [Google Scholar]
- 10. Cellini B, Bertoldi M, Montioli R, Paiardini A, Borri Voltattorni C. Human wild‐type alanine:Glyoxylate aminotransferase and its naturally occurring G82E variant: functional properties and physiological implications. Biochem J. 2007;408:39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang BJ, Xia JM, Wang Q, Yu JL, Song Z, Zhao H. Diet and adaptive evolution of alanine‐glyoxylate aminotransferase mitochondrial targeting in birds. Mol Biol Evol. 2020;37:786–798. [DOI] [PubMed] [Google Scholar]
- 12. Birdsey GM, Lewin J, Cunningham AA, Bruford MW, Danpure CJ. Differential enzyme targeting as an evolutionary adaptation to herbivory in carnivora. Mol Biol Evol. 2004;21:632–646. [DOI] [PubMed] [Google Scholar]
- 13. Oppici E, Montioli R, Cellini B. Liver peroxisomal alanine:glyoxylate aminotransferase and the effects of mutations associated with primary Hyperoxaluria type I: an overview. Biochim Biophys Acta. 2015;1854:1212–1219. [DOI] [PubMed] [Google Scholar]
- 14. Lorenzo V, Torres A, Salido E. Primary hyperoxaluria. Nefrologia. 2014;34:398–412. [DOI] [PubMed] [Google Scholar]
- 15. Purdue PE, Takada Y, Danpure CJ. Identification of mutations associated with peroxisome‐to‐mitochondrion mistargeting of alanine/glyoxylate aminotransferase in primary hyperoxaluria type 1. J Cell Biol. 1990;111:2341–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Theka T, Rodgers AL, Webber D, O'Ryan C. Variability in kidney stone incidence between black and white south Africans: AGT Pro11Leu polymorphism is not a factor. J Endourol. 2014;28:577–581. [DOI] [PubMed] [Google Scholar]
- 17. Lumb MJ, Danpure CJ. Functional synergism between the most common polymorphism in human alanine:glyoxylate aminotransferase and four of the most common disease‐causing mutations. J Biol Chem. 2000;275:36415–36422. [DOI] [PubMed] [Google Scholar]
- 18. Danpure CJ. Primary hyperoxaluria type 1: AGT mistargeting highlights the fundamental differences between the peroxisomal and mitochondrial protein import pathways. Biochim Biophys Acta. 2006;1763:1776–1784. [DOI] [PubMed] [Google Scholar]
- 19. Cellini B, Lorenzetto A, Montioli R, Oppici E, Voltattorni CB. Human liver peroxisomal alanine:Glyoxylate aminotransferase: Different stability under chemical stress of the major allele, the minor allele, and its pathogenic G170R variant. Biochimie. 2010;92:1801–1811. [DOI] [PubMed] [Google Scholar]
- 20. Mesa‐Torres N, Salido E, Pey AL. The lower limits for protein stability and foldability in primary hyperoxaluria type I. Biochim Biophys Acta. 2014;1844:2355–2365. [DOI] [PubMed] [Google Scholar]
- 21. Pey AL, Albert A, Salido E. Protein homeostasis defects of alanine‐glyoxylate aminotransferase: New therapeutic strategies in primary hyperoxaluria type I. BioMed Res Intl. 2013;2013:687658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cellini B, Montioli R, Voltattorni CB. Human liver peroxisomal alanine:Glyoxylate aminotransferase: Characterization of the two allelic forms and their pathogenic variants. Biochim Biophys Acta. 2011;1814:1577–1584. [DOI] [PubMed] [Google Scholar]
- 23. Oppici E, Dindo M, Conter C, Borri Voltattorni C, Cellini B. Folding defects leading to primary hyperoxaluria. Handb Exp Pharmacol. 2018;245:313–343. [DOI] [PubMed] [Google Scholar]
- 24. Dindo M, Grottelli S, Annunziato G, et al. Cycloserine enantiomers are reversible inhibitors of human alanine:glyoxylate aminotransferase: Implications for primary Hyperoxaluria type 1. Biochem J. 2019;476:3751–3768. [DOI] [PubMed] [Google Scholar]
- 25. Fodor K, Wolf J, Erdmann R, Schliebs W, Wilmanns M. Molecular requirements for peroxisomal targeting of alanine‐glyoxylate aminotransferase as an essential determinant in primary hyperoxaluria type 1. PLoS Biol. 2012;10:e1001309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Giardina G, Paiardini A, Montioli R, Cellini B, Voltattorni CB, Cutruzzola F. Radiation damage at the active site of human alanine:glyoxylate aminotransferase reveals that the cofactor position is finely tuned during catalysis. Sci Rep. 2017;7:11704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mesa‐Torres N, Yunta C, Fabelo‐Rosa I, et al. The consensus‐based approach for gene/enzyme replacement therapies and crystallization strategies: the case of human alanine‐glyoxylate aminotransferase. Biochem J. 2014;462:453–463. [DOI] [PubMed] [Google Scholar]
- 28. Oppici E, Fodor K, Paiardini A, et al. Crystal structure of the S187F variant of human liver alanine:glyoxylate [corrected] aminotransferase associated with primary hyperoxaluria type I and its functional implications. Proteins. 2013;81:1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang X, Roe SM, Hou Y, et al. Crystal structure of alanine:glyoxylate aminotransferase and the relationship between genotype and enzymatic phenotype in primary hyperoxaluria type 1. J Mol Biol. 2003;331:643–652. [DOI] [PubMed] [Google Scholar]
- 30. Djordjevic S, Zhang X, Bartlam M, Ye S, Rao Z, Danpure CJ. Structural implications of a G170R mutation of alanine:glyoxylate aminotransferase that is associated with peroxisome‐to‐mitochondrion mistargeting. Acta Cryst F. 2010;66:233–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hopper ED, Pittman AM, Fitzgerald MC, Tucker CL. In vivo and in vitro examination of stability of primary hyperoxaluria‐associated human alanine:glyoxylate aminotransferase. J Biol Chem. 2008;283:30493–30502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mesa‐Torres N, Fabelo‐Rosa I, Riverol D, et al. The role of protein denaturation energetics and molecular chaperones in the aggregation and mistargeting of mutants causing primary hyperoxaluria type I. PLoS one. 2013;8:e71963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Montioli R, Fargue S, Lewin J, et al. The N‐terminal extension is essential for the formation of the active dimeric structure of liver peroxisomal alanine:glyoxylate aminotransferase. Intl J Biochem Cell Biol. 2012;44:536–546. [DOI] [PubMed] [Google Scholar]
- 34. Dindo M, Mandrile G, Conter C, et al. The ILE56 mutation on different genetic backgrounds of alanine:Glyoxylate aminotransferase: clinical features and biochemical characterization. Mol Genet Metab. 2020;131:171–180. [DOI] [PubMed] [Google Scholar]
- 35. Cellini B, Montioli R, Paiardini A, et al. Molecular defects of the glycine 41 variants of alanine glyoxylate aminotransferase associated with primary hyperoxaluria type I. Proc Natl Acad Sci USA. 2010;107:2896–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Goddard TD, Huang CC, Meng EC, et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Parra RG, Schafer NP, Radusky LG, et al. Protein frustratometer 2: a tool to localize energetic frustration in protein molecules, now with electrostatics. Nucleic Acids Res. 2016;44:W356–W360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Strodel B, Whittleston CS, Wales DJ. Thermodynamics and kinetics of aggregation for the GNNQQNY peptide. J Am Chem Soc. 2007;129:16005–16014. [DOI] [PubMed] [Google Scholar]
- 39. Dindo M, Conter C, Cellini B. Electrostatic interactions drive native‐like aggregation of human alanine:Glyoxylate aminostransferase. FEBS J. 2017;284:3739–3764. [DOI] [PubMed] [Google Scholar]
- 40. Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fernandez‐Higuero JA, Betancor‐Fernandez I, Mesa‐Torres N, Muga A, Salido E, Pey AL. Structural and functional insights on the roles of molecular chaperones in the mistargeting and aggregation phenotypes associated with primary hyperoxaluria type I. Adv Protein Chem Struct Biol. 2019;114:119–152. [DOI] [PubMed] [Google Scholar]
- 42. Uversky VN. Functional roles of transiently and intrinsically disordered regions within proteins. FEBS J. 2015;282:1182–1189. [DOI] [PubMed] [Google Scholar]
- 43. Freiberger MI, Wolynes PG, Ferreiro DU, Fuxreiter M. Frustration in fuzzy protein complexes leads to interaction versatility. J Phys Chem B. 2021;125:2513–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sahni N, Yi S, Taipale M, et al. Widespread macromolecular interaction perturbations in human genetic disorders. Cell. 2015;161:647–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wong ETC, So V, Guron M, et al. Protein‐protein interactions mediated by intrinsically disordered protein regions are enriched in missense mutations. Biomolecules. 2020;10:1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Forcelloni S, Giansanti A. Mutations in disordered proteins as early indicators of nucleic acid changes triggering speciation. Sci Rep. 2020;10:4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guharoy M, Bhowmick P, Sallam M, Tompa P. Tripartite degrons confer diversity and specificity on regulated protein degradation in the ubiquitin‐proteasome system. Nat Commun. 2016;7:10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Niemeyer M, Moreno Castillo E, Ihling CH, et al. Flexibility of intrinsically disordered degrons in AUX/IAA proteins reinforces auxin co‐receptor assemblies. Nat Commun. 2020;11:2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang C, Li X, Adelmant G, et al. Peptidic degron in EID1 is recognized by an SCF E3 ligase complex containing the orphan F‐box protein FBXO21. Proc Natl Acad Sci USA. 2015;112:15372–15377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schuch JB, Paixao‐Cortes VR, Friedrich DC, Tovo‐Rodrigues L. The contribution of protein intrinsic disorder to understand the role of genetic variants uncovered by autism spectrum disorders exome studies. Am J Med Genet B Neuropsychiatr Genet. 2016;171B:479–491. [DOI] [PubMed] [Google Scholar]
- 51. Vacic V, Iakoucheva LM. Disease mutations in disordered regions—Exception to the rule? Mol Biosyst. 2012;8:27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Qiu J, Nechaev D, Rost B. Protein‐protein and protein‐nucleic acid binding residues important for common and rare sequence variants in human. BMC Bioinformatics. 2020;21:452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. W Kabsch. XDS. Acta Crystallographica Section D, 2010;D66, 125–132. http://scripts.iucr.org/cgi-bin/paper?s0907444909047337. 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Philip R, Evans , & Garib N, Murshudov . How good are my data and what is the resolution?. Acta crystallographica. Section D, Biological crystallography, 2013;69(Pt 7), 1204–1214. http://scripts.iucr.org/cgi-bin/paper?ba5190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. CCP4 Collaborative Computational Project. The CCP4 suite: programs for protein crystallography.. Acta Crystallogr D Biol Crystallogr, 1994;50(Pt 5), 760–763. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15299374 [DOI] [PubMed] [Google Scholar]
- 56. Evans PR, & Murshudov GN. How good are my data and what is the resolution?. Acta crystallographica. Section D, Biological crystallography, 2013;69(Pt 7), 1204–12014. http://scripts.iucr.org/cgi-bin/paper?ba5190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vagin A, & Teplyakov A. Molecular replacement with MOLREP.. Acta Crystallogr D Biol Crystallogr, 2010;66(Pt 1), 22–25. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20057045. 10.1107/S0907444909042589 [DOI] [PubMed] [Google Scholar]
- 58. Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd, JJ , Hung Li‐Wei, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, & Zwart PH. PHENIX: a comprehensive Python‐based system for macromolecular structure solution.. Acta crystallographica. Section D, Biological crystallography, 2010;66(Pt 2), 213–221. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2815670&tool=pmcentrez&rendertype=abstract. 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner, RA , Nicholls RA, Winn MD, Long F, & Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures.. Acta crystallographica. Section D, Biological crystallography, 2011;67(Pt 4), 355–367. http://scripts.iucr.org/cgi-bin/paper?ba5152. 10.1107/S0907444911001314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Emsley P, & Cowtan K. Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr, 2004;60(Pt 12), 2126–2132. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15572765. 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 61. Waterhouse A, Bertoni M, Bienert S, et al. SWISS‐MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rodrigues J, Teixeira JMC, Trellet M, Bonvin A. Pdb‐tools: a swiss army knife for molecular structures. F1000Res. 2018;7:1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gupta S, Kelow S, Wang L, Andrake MD, Dunbrack RL Jr, Kruger WD. Mouse modeling and structural analysis of the p.G307S mutation in human cystathionine beta‐synthase (CBS) reveal effects on CBS activity but not stability. J Biol Chem. 2018;293:13921–13931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Abraham MJ, Murtola T, Schulz R, et al. GROMACS: high performance molecular simulations through multi‐level parallelism from laptops to supercomputers. SoftwareX. 2015;1‐2:19–25. [Google Scholar]
- 65. Brooks BR, Brooks CL 3rd, Mackerell AD Jr, et al. CHARMM: The biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Izadi S, Anandakrishnan R, Onufriev AV. Building water models: A different approach. J Phys Chem Lett. 2014;5:3863–3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126:014101. [DOI] [PubMed] [Google Scholar]
- 68. Parrinello M, Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J Appl Phys. 1981;52:7182–7190. [Google Scholar]
- 69. Darden T, York D, Pedersen L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 70. Papoian GA, Ulander J, Eastwood MP, Luthey‐Schulten Z, Wolynes PG. Water in protein structure prediction. Proc Natl Acad Sci USA. 2004;101:3352–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14(33–38):27–38. [DOI] [PubMed] [Google Scholar]
- 72. Oppici E, Roncador A, Montioli R, Bianconi S, Cellini B. Gly161 mutations associated with primary Hyperoxaluria type I induce the cytosolic aggregation and the intracellular degradation of the apo‐form of alanine:Glyoxylate aminotransferase. Biochim Biophys Acta. 2013;1832:2277–2288. [DOI] [PubMed] [Google Scholar]
- 73. Miyazaki K. MEGAWHOP cloning: a method of creating random mutagenesis libraries via megaprimer PCR of whole plasmids. Methods Enzymol. 2011;498:399–406. [DOI] [PubMed] [Google Scholar]
- 74. Macchioni L, Chiasserini D, Mezzasoma L, et al. Crosstalk between long‐term sublethal oxidative stress and detrimental inflammation as potential drivers for age‐related retinal degeneration. Antioxidants (Basel). 2020;10:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Choi H, Larsen B, Lin ZY, et al. SAINT: probabilistic scoring of affinity purification‐mass spectrometry data. Nat Methods. 2011;8:70–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Choi H, Glatter T, Gstaiger M, Nesvizhskii AI. SAINT‐MS1: protein‐protein interaction scoring using label‐free intensity data in affinity purification‐mass spectrometry experiments. J Proteome Res. 2011;11:2619–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Guard SE, Ebmeier CC, Old WM. Label‐free immunoprecipitation mass spectrometry workflow for large‐scale nuclear interactome profiling. J Vis Exp. 2019. 10.3791/60432. [DOI] [PubMed] [Google Scholar]
- 78. Chiasserini D, van Weering JR, Piersma SR, et al. Proteomic analysis of cerebrospinal fluid extracellular vesicles: a comprehensive dataset. J Proteomics. 2014;106:191–204. [DOI] [PubMed] [Google Scholar]
- 79. Perez‐Riverol Y, Csordas A, Bai J, et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Soubrier J, Steel M, Lee MS, et al. The influence of rate heterogeneity among sites on the time dependence of molecular rates. Mol Biol Evol. 2012;29:3345–3358. [DOI] [PubMed] [Google Scholar]
- 81. Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ‐TREE: A fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Mol Biol Evol. 2015;32:268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Cellini B, Montioli R, Paiardini A, Lorenzetto A, Voltattorni CB. Molecular insight into the synergism between the minor allele of human liver peroxisomal alanine:glyoxylate aminotransferase and the F152I mutation. J Biol Chem. 2009;284:8349–8358. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information