

Abstract
The use of quality control samples in metabolomics ensures data quality, reproducibility, and comparability between studies, analytical platforms, and laboratories. Long-term, stable, and sustainable reference materials (RMs) are a critical component of the quality assurance/quality control (QA/QC) system; however, the limited selection of currently available matrix-matched RMs reduces their applicability for widespread use. To produce an RM in any context, for any matrix that is robust to changes over the course of time, we developed iterative batch averaging method (IBAT). To illustrate this method, we generated 11 independently grown Escherichia coli batches and made an RM over the course of 10 IBAT iterations. We measured the variance of these materials by nuclear magnetic resonance (NMR) and showed that IBAT produces a stable and sustainable RM over time. This E. coli RM was then used as a food source to produce a Caenorhabditis elegans RM for a metabolomics experiment. The metabolite extraction of this material, alongside 41 independently grown individual C. elegans samples of the same genotype, allowed us to estimate the proportion of sample variation in preanalytical steps. From the NMR data, we found that 40% of the metabolite variance is due to the metabolite extraction process and analysis and 60% is due to sample-to-sample variance. The availability of RMs in untargeted metabolomics is one of the predominant needs of the metabolomics community that reach beyond quality control practices. IBAT addresses this need by facilitating the production of biologically relevant RMs and increasing their widespread use.
Graphical Abstract
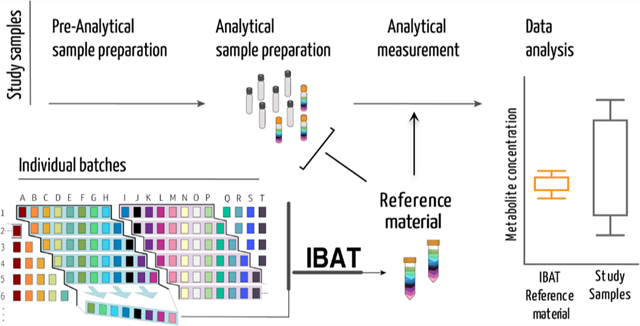
Biological reference materials are needed to compare metabolomics data across multiple instruments, studies, and batches. Whenever there are more samples collected than can be processed in a single “run”, there is an added unwanted variation that if captured can be modeled and removed, leading to more powerful tests.1 Readily available long-term biologically relevant reference materials (RMs) represent a critical component to achieve reproducibility.2,3 Commercially available RMs and standard reference materials (SRMs) address some of these needs but can be expensive to purchase, offer limited quantities and matrix diversity, and have an expiration date.3 The National Institute of Standards and Technology (NIST) has a long history of producing biofluid-based materials, both certified and noncertified, to facilitate standardization and to improve comparability and reproducibility of analytical measurements. The NIST SRMs are trademarked as certified reference materials (CRMs) and are specifically designed to provide quantified metabolite levels to serve strict objectives (i.e., calibration, method validation, measurement accuracy).4−6 Pooled quality control (QC) samples produced from experimental samples are valuable as they capture instrument variation within the experiment but have limited value in comparing across experiments or in synthesizing results from large studies.7,8 The individual variation intrinsic in subjecting biological material to extraction and quantification is not captured by pooled samples or by chemical standards made after extraction. There is a recognized need for matrix-specific stable RMs that can be used to compare data across long-term studies with multiple batches or across different laboratories and instrumention.9
Homogeneous and stable materials that are fit for purpose are reference materials (as per the International Vocabulary of Metrology (VIM)).10 RM does not require a metrologically valid metabolite quantification (certification) and should be straightforward to produce and maintain. For untargeted metabolomics, additional criteria for an RM are important. Namely, it should (i) be made from the same biological matrix as the experimental samples, (ii) have a profile that is as complex as the experimental samples, (iii) be sustainably produced over time, and (iv) facilitate the annotation of known and unknown compounds.
The proteomics community devoted substantial effort to the development and application of RMs, which greatly improved standardization and reproducibility in the field.4,9 The metabolomics community has highlighted the need for RMs as part of the development of resources and practices to measure, detect, and prevent unwanted preanalytical and instrumental variation.2,3,5,8,11
Here, we introduce iterative batch averaging method (IBAT) that can be used to create a stable RM produced over time in any context. The concept is straightforward: multiple small batches of starting material are produced and aliquoted and then pooled to generate the RM. A stable and long-lasting RM can be generated by repeating the process over time, as illustrated in Figure 1.
Figure 1.

Iterative batch average method (IBAT). Batches of material are represented by columns (same-colored squares and letters). Rows represent homogeneous aliquots from each batch. Examples of sequential batch combinations are rows shaded from blue to purple. The right panel illustrates the IBAT-generated pools from individual batches. IBAT is only limited by the number of individual batches produced and can be adjusted to the number of aliquots required and to any material.
IBAT results in an RM that (i) is robust to changes over time, (ii) minimizes variance between batches of RM, (iii) can be used over the course of large-scale experiments, (iv) can be made with a small amount of constant effort and smaller storage space, (v) can be applied to any organism or biological matrix of interest, and (vi) can be used for evaluation of multiple sources of variation at multiple points in a metabolomics experiment. To illustrate IBAT, we made and characterized a Caenorhabditis elegans reference material. C. elegans eats bacteria, which is also subject to variation over time, so to make a stable C. elegans RM, we first needed to make an Escherichia coli RM that can be fed to C. elegans. This two-step IBAT shows the flexibility of the approach, and in the Discussion and Conclusions section, we outline strategies to apply IBAT to create other RMs of interest to metabolomics researchers.
RESULTS
Production and Analysis of an IBAT E. coli as a Food Source for C. elegans.
For this RM, we used a bioreactor to generate large quantities of bacteria in each batch, but the principle holds on a smaller scale with flasks and a shaker/incubator. We grew 11 different 2 L bioreactor batches (columns in Figure 1) that each produced an average of 84 g of bacterial paste. Each batch was continuously mixed to maintain homogeneity and aliquoted into 60−90 tubes (rows in Figure 1) containing 1 g each.
To evaluate the E. coli IBAT process, we selected the first three aliquots (as replicates) from all 11 batches and split each aliquot into two parts. One of the parts from each aliquot was used to make the IBAT samples, according to Table 1.
Table 1.
List of Individual Batches Pooled Together for Each Stable Food Source Iterationa 1
| IBAT iterations | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| combined individual batches | A−E | B−F | C−G | D−H | E−I | F−J | G−K | H−A | I−B | J−C |
This process follows the same methodology described in Figure 1
We compared the 10 different E. coli IBAT samples (Table 1) with the second split part from each of the 11 batches. All samples contained the same volume of bacteria. A total of 33 samples from 11 individual batches of E. coli and 30 IBAT samples were analyzed by nuclear magnetic resonance (NMR) spectroscopy. The IBAT method reduces variance between different tubes of RMs. The NMR spectra for these samples are nearly identical, with a very low variance (Figure 2a). The variance here is due to extraction and quantification. In contrast, the variance between the 33 individual spectra is much larger, reflecting a combination of biological variance and technical variance. To quantify variance, we selected 19 metabolites that we could identify were present in all of the samples and were consistent between replicate measurements. The difference between the observed values among the replicates was small according to the Bland−Altman analysis in Kirpich et al.12 Furthermore, we chose nonoverlapped peaks for accurate quantification. The coefficient of variation (CV − standard deviation/mean) was calculated separately for each metabolite within each group (Figure 2b and Supporting Table 1). Similar to the overlaid NMR spectra (Figure 2a), the CV was lower for IBAT-generated samples (between 0.19 and 0.91) than for individual samples (0.36−1.26). Using the Fligner−Killeen13 test for homogeneity of variances for each of the selected metabolites showed significantly different variances between IBAT-produced samples and individual batch samples (p value < 0.05) except for betaine (p value =0.21).
Figure 2.

(A) Untargeted full-resolution 1H NMR profile of E. coli and spectral expansion between 6.8 and 7.2 ppm. NMR spectra in gray or orange correspond to IBAT or individual batches, respectively. (B) Radial plot representing the coefficient of variation (CV) for annotated metabolites using the same colors. The length of spokes corresponds to the CV of each metabolite. (C) Each data point represents the mean-centered peak height in each sample. Experimental IBAT samples are depicted in orange, and individual batches are depicted in gray. Cyan data points represent the simulated metabolite peak heights per number of averaged batches. Light gray-shaded areas represent ±1 standard deviation from the mean. Iva, isovalerate; Leu, leucine; Val, valine; Ile, isoleucine; 3 Hba, 3-hydroxybutyrate; Lac, lactate; Cad, cadaverine; AcOH, acetate; Glu, glutamate; Met, methionine; Asp, aspartate; Bet, betaine; Rib, ribose; Ura, uracil; Fum, fumarate; Tyr, tyrosine; Phe, phenylalanine; niacin, nicotinic acid; and Form, formate.
The IBAT process depends on pooling batches. We used the individual batch data to simulate the IBAT process. We generated 10 iterations for combining 2−11 individual batches to generate an IBAT compliant RM. We used the individual data to estimate the mean-centered peak heights and respective standard deviations for our 19 metabolites. The variance (10 iterations) decreases as the number of batches used increases (Figures 2c and 1 and Table 1). This is consistent with the predictions of Spearman−Brown.14,15
Production and Application of a C. elegans PD1074 Reference Material.
To create an IBAT C. elegans RM, we used a 2 L bioreactor and fed the worms with the IBAT E. coli RM. Each batch of the bioreactor produced between 40 and 60 million mixed-stage worms. These were harvested and aliquoted with constant mixing into 20−30 tubes so that every tube contained approximately two million worms (by diluted aliquot counts). These were then frozen at −80 °C. After three bioreactor batches, we combined one aliquot from each batch for a total of six million batch-averaged worms. This was divided into 30 aliquots of C. elegans RM with 200 000 worms each and refrozen until use (Figure 3).
Figure 3.

Schematic overview of the C. elegans reference material production. The reference strain PD1074 nematodes were seeded from cryopreserved stocks and fed an E. coli RM (Supporting Methods). The harvested material from each bioreactor was washed, aliquoted, and stored. Aliquots from each reactor iteration were combined to produce a stable C. elegans reference material. This material can be divided into different sized aliquots according to the downstream application needs.
In a metabolomics experiment, there are three main sources of variation: the sample material itself, the extraction, and data acquisition (Supporting Figure 1). An experimental sample will encompass all three of those sources. The IBAT RM reduces the sample material variation, and pooled QC samples average over both the sample variance and the extraction variation. We compared the C. elegans RM to 41 independent samples of the same strain (PD1074).16 These individual samples were prepared in three sets of two extraction blocks. For each set, an equimolar pool was formed from all individual samples for a total of three QC pools. One C. elegans RM aliquot was included in each extraction block. For NMR data collection, one block was analyzed per run. We selected 26 annotated features that were common to all samples and computed pairwise standardized Euclidean distances for each sample (Figure 4). The distances between samples in the IBAT material reflect instrument variability (QC pools) and extraction variability. The distances between individual sample data include extraction and instrument variability but also sample variability. The mean and median distances, minimum and maximum values, and sample distribution for each of these groups allow us to estimate the variability from these different sources of variation. The individual PD1074 samples, which include all three sources of variation, have the largest variability with mean values from 25.5 to 54.1 and the min/max of 8.19 and 66 (blocks 1 through 6 in Figure 4). The IBAT samples, representing the extraction and technical variance, have a smaller range of mean distances (28.2−38.7) and min/max values of 21.3−44.8. As expected, the pooled individual PD1074 samples representing differences in the manual preparation and instrumentation between sets have the smallest range with the respective boxplot bounds between 31.4 and 33.
Figure 4.

Boxplots of pairwise standardized Euclidean distances. Each boxplot represents the distribution of distances from one sample to all of the other samples of the same group. Mean and median distances for each sample are indicated by markers. Blue-colored boxplots represent PD1074 samples that were processed in each block. The three pooled PD1074 samples were created from the samples in blocks 1 + 2, 3 + 4, and 5 + 6, respectively. C. elegans RM samples were generated using IBAT and processed alongside the PD1074 samples, one per block.
DISCUSSION AND CONCLUSIONS
The IBAT process reduces the growth and sampling contributions to variance by creating a common source of material from which homogeneous aliquots are produced. The advantage here is that instead of producing a single large batch, which will have its own challenges in achieving homogeneity, the material is continuously generated over time, with each iteration using only small amounts of new materials, thus capturing small changes over time while having minimal variance between experiments. This minimal variance can be theoretically predicted as a function of the number of distinct batches combined and the variance between the continuously produced material or estimated from empirical data (Figure 2c) to take into account the overlap between iterations. The IBAT process is flexible and can be adjusted to production throughput, the type of material, the quantities produced, the degree of variance reduction, and the metabolomics technology. We demonstrated this concept for two different types of matrices, E. coli and C. elegans. However, the method is general and can be applied to any biological matrix. In nonmodel system studies, it is common to use human plasma or urine or commercially available materials that are aliquoted from a single large batch and frozen. However, when a batch runs out, shifting to a new external standard will often not be comparable to the prior standard. IBAT can be used by making pools from different batches of materials, as illustrated in Figure 1. New batches can be incorporated over time, and this will minimize the change in the RM over time. Similar strategies can be used with diverse applications such as plants or cultured mammalian cells for biotherapeutics. In these scenarios, the main issue is minimizing the freeze−thaw cycles and so the size of the initial aliquots for future blending must be planned.
An RM of the same biological matrix as the study samples together with a carefully planned experimental design can be used to determine the magnitude and variance in the extraction, a major source of variation in metabolomics experiments.3,17 It can also facilitate comparison among separate experiments. IBAT can then be used to separate the extraction variance from the sample-to-sample variance in the individually grown and processed samples, as demonstrated here. The individual C. elegans samples are genetically identical to the RM. Variance in metabolite intensities was larger as a result of sample variation during growth, handling, storage, and sampling, added to the technical variation in extraction and data acquisition. The pooled individual C. elegans PD1074 samples minimize the sample and extraction variance by averaging over both samples and extractions and reflect only the variation in the analytical measurement (which is low for NMR) and the pooling strategy. By processing the experimental replicates and RM aliquots, one can independently estimate the contribution of the metabolite extraction step to individual metabolite variation. The IBAT C. elegans RM samples can be used to estimate variance due to extraction. We find that 40% of the total variance as estimated by the variation between individually grown, extracted, and quantified samples is due to extraction variance and analysis and of that variance approximately 15% is due to technical variation.
IBAT increases the efficacy of quality assurance (QA)/QC and is expected to improve the performance of biological reference materials by allowing estimation of process-derived variance including facilitating studies across multiple labs. Finally, the cost of using an IBAT process should be lower than acquiring a single large batch of reference material, thus enabling labs to amortize the process over time while maintaining the stability of the material and facilitating comparison of experiments conducted months or years apart.
METHODS
E. coli Individual Batch Production and Storage.
To produce a stable and consistent C. elegans food source, batches of E. coli HT115 were grown in bioreactors (Biostat, Sartorius) using standardized protocols (Supporting Methods). A total of 11 batches were produced, and each batch was divided into approximately 60−90 aliquots, flash-frozen, and stored at −80 °C. Each aliquot comprised 2 mL of bacterial suspension (1 g of wet bacterial paste and OD600 ranging from 17.5 to 24).
NMR Sample Preparation of E. coli IBAT and Individual Batches.
All 33 individual batch samples and 30 IBAT-generated samples were prepared for NMR analysis. Approximately 200 μL of 0.7 mm silica beads (BioSpec products) was added to each of the 63 samples. These were homogenized at 1800 rpm for 300 s (FastPrep 96, MPBIO) and centrifuged at 20 000g for 15 min. From each sample, 450 μL of supernatant was transferred to a new tube and 150 μL of deuterated water was added (D2O, D, 99.9%, Cambridge Isotope Laboratories). Each sample was vortex-mixed for 1 min before transferring into 5 mm SampleJet NMR tubes. Details of NMR acquisition and spectra processing can be found in the Supporting Methods.
NMR Sample Preparation of C. elegans Samples.
For the NMR analysis, six IBAT RM aliquots were prepared alongside 41 individual samples of the C. elegans strain PD1074 that were grown according to our previously published method.16 Each of these samples contained approximately 200 000 nematodes. All samples were previously flash-frozen and then lyophilized until dry. Approximately 200 μL of 1 mm zirconia beads (BioSpec products) was added to each dried sample and homogenized at 1800 rpm for a total of 270 s (FastPrep 96, MPBIO). The samples were then delipidated by adding 1 mL of cold (−20 °C) isopropanol (Optima, LC/MS grade, Fisher Scientific) and left overnight (12 h) at −20 °C after a 20 min resting period at room temperature. The supernatant was removed after being centrifuged for 30 min at 20 000g, and 1 mL of cold (4 °C) 80:20 methanol/water (Optima, LC/MS grade, Fisher Scientific) was added to the remaining contents. The tubes were shaken for 30 min at 4 °C and centrifuged at 20 000g for 30 min. The methanol/water supernatant was transferred to new tubes, and these were vacuum-dried using a CentriVac benchtop vacuum concentrator (Labconco). The extracts were reconstituted in 45 μL of deuterated (D2O, D, 99.9%, Cambridge Isotope Laboratories) 100 mM sodium phosphate buffer (mono- and dibasic; Fisher BioReagents) containing 0.11 mM of the internal standard (sodium 2,2-dimethyl-2-silapentane-5-sulfonate (DSS), D6, 98%; Cambridge Isotope Laboratories) at pH 7.0 and vortex-mixed for <1 min prior to transfer into 1.7 mm SampleJet NMR tubes. The three pooled PD1074 samples were created by adding together 6 μL from the samples in each NMR run (12, 14, and 15 samples, respectively), after having been reconstituted in the internal standard containing NMR solvent. Details of NMR acquisition and spectra processing can be found in the Supporting Methods.
DATA ANALYSIS
Following acquisition and processing, spectra were imported into Matlab programming software (MATLAB, MathWorks, R2019a). Using a toolbox developed in-house and available at https://github.com/artedison/Edison_Lab_Shared_Metabolomics_UGA, the following was carried out: plotting, referencing, baseline correction, alignment (CCOW18), and solvent peak removal. Feature detection (peak picking) was automated using a combination of an in-house peak picking function and binning algorithm19 to extract peak heights. Data were exported for the Bland−Altman analysis12 to select features that were consistent between replicates (cutoffs used: a sample flag of 0.2, a feature flag of 0.05, and a residual of 3). We also computed pairwise standardized Euclidean distances using the Southeast Center for Integrated Metabolomics Tools (SECIMTools).12 Coefficient of variation (CV) calculations, variance, %variance, and Fligner−Killeen test were carried out in Matlab.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Institutes of Health under Award Number 1U2CES030167-01. The authors would like to thank Dr. David Blum and Dr. Ron Garrison from the Bio-expression and Fermentation Facility at the University of Georgia for training and advice using the Bioreactors and Pamela Kirby at the Edison Lab for assistance with material handling and storage logistics.
The authors declare no competing financial interest.
All raw and processed data, along with detailed experimental, NMR acquisition, and data analysis methods, are available under project identifier PR001106 at the Metabolomics Workbench (www.metabolomicsworkbench.org). The data can be accessed directly via its project doi: 10.21228/M8R395. Metabolomics Workbench is supported by NIH grant U2C-DK119886.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.1c01294.
Additional methods detailing the bioreactor production of both C. elegans and E. coli, making an E. coli reference material, NMR acquisition and processing, database matching procedures, metabolite summary table, and a figure detailing sources of variance (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.1c01294
Contributor Information
Goncalo J. Gouveia, Department of Biochemistry & Molecular Biology, University of Georgia, Athens, Georgia 30602, United States; Complex Carbohydrate Research Center, University of Georgia, Athens, Georgia 30602, United States.
Amanda O. Shaver, Department of Genetics, University of Georgia, Athens, Georgia 30602, United States; Complex Carbohydrate Research Center, University of Georgia, Athens, Georgia 30602, United States
Brianna M. Garcia, Department of Chemistry, University of Georgia, Athens, Georgia 30602, United States; Complex Carbohydrate Research Center, University of Georgia, Athens, Georgia 30602, United States
Alison M. Morse, Department of Molecular Genetics and Microbiology, University of Florida Genetics Institute, University of Florida, Gainesville, Florida 32610, United States
Erik C. Andersen, Department of Molecular Biosciences, Northwestern University, Evanston, Illinois 60208, United States
Arthur S. Edison, Department of Biochemistry & Molecular Biology, University of Georgia, Athens, Georgia 30602, United States; Department of Genetics, University of Georgia, Athens, Georgia 30602, United States; Complex Carbohydrate Research Center, University of Georgia, Athens, Georgia 30602, United States.
Lauren M. McIntyre, Department of Molecular Genetics and Microbiology, University of Florida Genetics Institute, University of Florida, Gainesville, Florida 32610, United States
REFERENCES
- (1).Cochran WG; Cox GM Experimental Designs, 2nd ed., 1957.
- (2).Dunn WB; Broadhurst DI; Edison A; Guillou C; Viant MR; Bearden DW; Beger RD Metabolomics 2017, 13, No. 50. [Google Scholar]
- (3).Broadhurst D; Goodacre R; Reinke SN; Kuligowski J; Wilson ID; Lewis MR; Dunn WB Metabolomics 2018, 14, No. 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Paulovich AG; Billheimer D; Ham AJ; Vega-Montoto L; Rudnick PA; Tabb DL; Wang P; Blackman RK; Bunk DM; Cardasis HL; Clauser KR; Kinsinger CR; Schilling B; Tegeler TJ; Variyath AM; Wang M; Whiteaker JR; Zimmerman LJ; Fenyo D; Carr SA; Fisher SJ; Gibson BW; Mesri M; Neubert TA; Regnier FE; Rodriguez H; Spiegelman C; Stein SE; Tempst P; Liebler DC Mol. Cell. Proteomics 2010, 9, 242–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Phinney KW; Ballihaut G; Bedner M; Benford BS; Camara JE; Christopher SJ; Davis WC; Dodder NG; Eppe G; Lang BE; Long SE; Lowenthal MS; McGaw EA; Murphy KE; Nelson BC; Prendergast JL; Reiner JL; Rimmer CA; Sander LC; Schantz MM; Sharpless KE; Sniegoski LT; Tai SS; Thomas JB; Vetter TW; Welch MJ; Wise SA; Wood LJ; Guthrie WF; Hagwood CR; Leigh SD; Yen JH; Zhang NF; Chaudhary-Webb M; Chen H; Fazili Z; LaVoie DJ; McCoy LF; Momin SS; Paladugula N; Pendergrast EC; Pfeiffer CM; Powers CD; Rabinowitz D; Rybak ME; Schleicher RL; Toombs BM; Xu M; Zhang M; Castle AL Anal. Chem 2013, 85, 11732–11738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Simón-Manso Y; Lowenthal MS; Kilpatrick LE; Sampson ML; Telu KH; Rudnick PA; Mallard WG; Bearden DW; Schock TB; Tchekhovskoi DV; Blonder N; Yan X; Liang Y; Zheng Y; Wallace WE; Neta P; Phinney KW; Remaley AT; Stein SE Anal. Chem 2013, 85, 11725–11731. [DOI] [PubMed] [Google Scholar]
- (7).Han W; Li L. Mass Spectrom. Rev 2020, 1–22, DOI: 10.1002/mas.21672. [DOI] [PubMed]
- (8).Peng J; Chen Y-T; Chen C-L; Li L. Anal. Chem 2014, 86, 6540–6547. [DOI] [PubMed] [Google Scholar]
- (9).Bunk DM Proteomics 2010, 10, 4220–4225. [DOI] [PubMed] [Google Scholar]
- (10).Köhler R. International Vocabulary of Metrology - Basic and General Concepts and Associated Terms. In Transverse Disciplines in Metrology, 3rd ed., 2012; Vol 1, pp 233–238. [Google Scholar]
- (11).Beger RD; Dunn WB; Bandukwala A; Bethan B;Broadhurst D; Clish CB; Dasari S; Derr L; Evans A; Fischer S; Flynn T; Hartung T; Herrington D; Higashi R; Hsu PC; Jones C; Kachman M; Karuso H; Kruppa G; Lippa K; Maruvada P; Mosley J; Ntai I; O’Donovan C; Playdon M; Raftery D; Shaughnessy D; Souza A; Spaeder T; Spalholz B; Tayyari F; Ubhi B; Verma M; Walk T; Wilson I; Witkin K; Bearden DW; Zanetti KA Metabolomics 2019, 15, No. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kirpich AS; Ibarra M; Moskalenko O; Fear JM; Gerken J; Mi X; Ashrafi A; Morse AM; McIntyre LM BMC Bioinf. 2018, 19, No. 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Conover WJ; Johnson ME; Johnson MM Technometrics 1981, 23, 351–361. [Google Scholar]
- (14).Brown W. Br. J. Med. Psychol 1910, 3, 296–322. [Google Scholar]
- (15).Spearman C. Br. J. Med. Psychol 1910, 3, 271–295. [Google Scholar]
- (16).Shaver AO; Gouveia GJ; Kirby PS; Andersen EC; Edison ASJ Visualized Exp. 2020, No. e61453. [DOI] [PMC free article] [PubMed]
- (17).Liu Q; Walker D; Uppal K; Liu Z; Ma C; Tran V; Li S; Jones DP; Yu T. Sci. Rep 2020, 10, No. 13856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Tomasi G; van den Berg F; Andersson CJ Chemom. 2004, 18, 231–241. [Google Scholar]
- (19).Sousa SAA; Magalhaes A; Ferreira MMC~ Chemom. Intell. Lab. Syst 2013, 122, 93–102. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.