

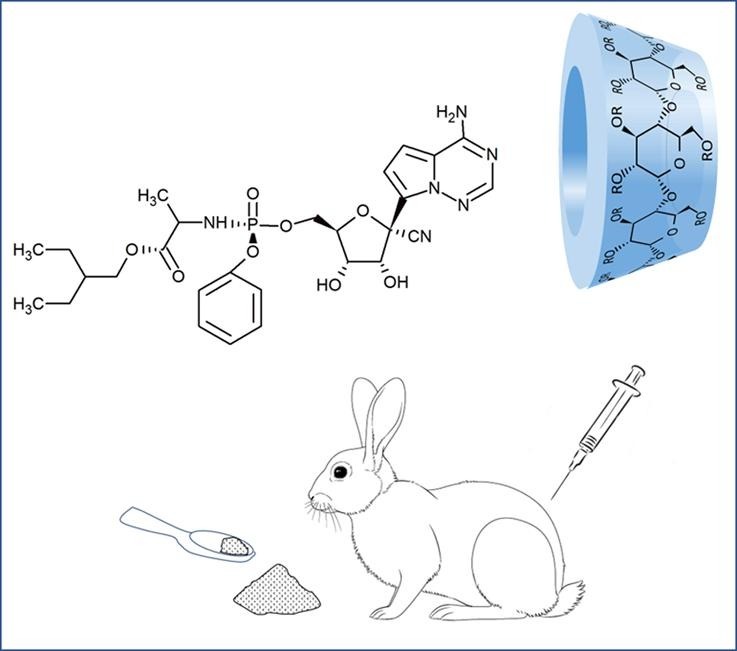
Graphical abstract
Keywords: Buccal administration, Remdesivir, Pharmacokinetic study, Rabbits
Abstract
As remdesivir, the first FDA-approved drug for SARS-CoV-2 infection, can be used only for hospitalized patients due to intravenous administration, there is an urgent need of effective oral antiviral formulations to be used at early stage of infection in an outpatient setting. The present paper reports on the comparative pharmacokinetics of the electrospun nanofiber remdesivir/sulfobutyl ether beta-cyclodextrin formulation after intravenous and buccal administration. It was postulated that oral transmucosal administration avoids remdesivir from metabolic transformation and intact remdesivir can be detected in plasma, but only the active metabolite GS-441524 could be experimentally detected at a significantly lower plasma level, than that provided by the intravenous route. In buccally treated animals, the metabolite GS-441524 appeared only at 1 h after treatment, while in intravenously treated animals, GS-441524 was possible to quantify even at the first time-point of blood collection. Further optimization of formulation is required to improve pharmacokinetics of remdesivir-sulfobutyl ether beta-cyclodextrin formulation upon buccal administration.
1. Introduction
The buccal surface of oral cavity has been reputed to offer a convenient route of drug administration aiming at efficient systemic delivery via circumventing gastro-intestinal tract and liver related first pass drug metabolism. The mucus layer of oral cavity has a rich blood supply and is relatively permeable. Buccal drug delivery is known as one of the most patient-friendly way of drug administration (Dodla and Velmurugan, 2013, Lakshmi et al., 2021). By oral transmucosal drug absorption we can bypass the gastro-intestinal tract and more importantly, avoid the hepatic first-pass metabolism, so that sensitive drug actives show enhanced oral bioavailability. This is one of the main reasons for our studies. We applied this buccal route of administration for remdesivir known to have significant first pass metabolism in liver and consequently not applicable orally, only intravenously.
Buccal penetration can be improved by using permeation enhancing agents for example surfactants, fatty acids, bile salts, chelators, co-solvents and cyclodextrins (CDs) (Guo and Cooklock, 1995). These compounds enhance cell membrane fluidity, extract intercellular lipids, interact with epithelial proteins, and alter mucus layer physics (Sayani and Chien, 1996). Among the currently used buccal permeation enhancers, CDs have been found potent delivery agents, they are multifunctional penetration enhancers: besides providing molecularly dispersed state and improved solubility of drug actives, they temporarily alter the barrier function of absorptive mucosae, by extracting/mobilizing membrane lipids in a reversible manner (Palem et al., 2012).
CDs are cyclic oligosaccharides prepared from starch amylose by enzymatic reaction. The main representatives, α-, β- and γ-CDs consisting of 6, 7 or 8 glucopyranose units, respectively, possess hydrophilic outer surface and less hydrophilic cavities suitable to include hydrophobic molecules via molecular encapsulation (Szejtli, 2004). The cyclodextrin-assisted buccal drug delivery was first reported by J. Pitha who studied buccal absorption of sex hormones such as estradiol, progesterone, and testosterone administered as an inclusion compound with hydroxypropyl- β -cyclodextrin (HPBCD) (Pitha et al., 1986). The sublingual or buccal route was found to result in the effective transfer of the steroids into the systemic circulation followed by only gradual elimination. These drugs are active only by this route and not from the gastrointestinal tract, due to fast metabolism of hormones by liver. Thus, a testosterone compound (hormone, 10 mg) with HPBCD tablet administered sublingually to a Caucasian male with a hypopituitary condition showed a hormone level of 1020 ng/100 mL serum at 2 h after administration compared to 480 ng from a traditional gelatin capsule (Pitha et al., 1986). Similar results were published by Loftsson et al. on the sublingual delivery of 17beta-estradiol in HPBCD-complexed form (Loftsson et al., 2003).
Remdesivir (GS-5734, (2S)-2-{(2R,3S,4R,5R)-[5-(4-Aminopyrrolo[2,1-f] [1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxy-tetrahydro-furan-2-ylmethoxy]phenoxy-(S)-phosphorylamino}propionic acid 2-ethyl-butyl ester), a viral RNAse inhibitor was discovered and developed by Gilead Sciences, Inc. It has recently been introduced as a promising new antiviral agent for the therapy against SARS-CoV-2 in the form of a marketed product, named Veklury™, which is a sulfobutyl ether β -cyclodextrin (SBECD)-enabled remdesivir injectable formulation. SBECD, produced by reacting β -cyclodextrin with butane sultone under alkaline conditions, is an FDA-approved pharmaceutical excipient with average degree of substitution (DS) of 6.2-9 (Puskás et al., 2015; Stella and Rajewski, 2020). The role of SBECD is to improve both the solubility and stability of remdesivir via inclusion complex formation (Pipkin et al., 2020). Molecular dynamic study proved the interaction between remdesivir and SBECD of various DS (Garrido et al., 2020; Piñeiro et al., 2021, Piñeiro et al., 2022). Recent NMR investigations suggested that the protonation state of the aminopyrrolo-triazine moiety of remdesivir can play a key role in the cyclodextrin-remdesivir interaction (Várnai et al., 2022). The resulting electrostatic interaction between the positive charge of remdesivir and negative charge of SBECD induces eightfold enhancement in complex association constant compared to underivatized β -CD.
The formulation contains 3.2% remdesivir and the rest is SBECD. This large excess of SBECD is necessary for solubilizing the drug: 20 w/v% SBECD can dissolve 6.7 mg/mL remdesivir and by lyophilizing this solution a powder with the composition of 3.23% remdesivir and 96.77% SBECD is obtained according to the patent of Gilead Sciences, Inc. (Larsen, 2019). This powder is reconstituted in water before injection.
Remdesivir exhibits potent in vitro activity against SARS-CoV-2 with a half-maximal effective concentration of 0.77 μM (Cao et al., 2020). In the clinical trials, although no benefit in mortality, but higher rate of improvement, faster recovery and significant decrease in the risk of serious adverse events were observed (Jorgensen et al., 2020, Garibaldi et al., 2021, Singh et al., 2021). It received US Food and Drug Administration (FDA) approval for the treatment of CoViD-19 patients (Rubin et al., 2020).
Due to the high first pass liver metabolism of phosphoramidates and low oral bioavailability, remdesivir cannot be administered orally (Cho et al., 2012). After intravenous administration it is rapidly metabolized to GS-441524, its parent nucleoside with high antiviral activity (Xie and Wang, 2021) (Fig. 1 ). Remdesivir is a diastereomer monophosphophoramidate prodrug of the adenine nucleoside analogue GS-441524. The latter is metabolically converted in cells and tissues into the pharmacologically active triphosphate which inhibits viral RNA polymerases, but does not affect host RNA or DNA polymerases (EMEA, 2020). Recent studies have focused on development of some other prodrugs of GS-441524, e.g., isobutyryl esters which resist to first -pass metabolism and demonstrate improved oral bioavailability (Cox et al., 2021, Schäfer et al., 2021, Wei et al., 2021).
Fig. 1.

Structural formulas of Remdesivir (A), GS441524 (B) and GS-441524-triphosphate (C).
Another option to avoid first pass metabolism is the buccal administration which needs orally disintegrating, fast-dissolving formulation, such as the one obtained by electrospinning of SBECD/remdesivir complex (Szente et al., 2021). Although the pioneering patent application related to remdesivir (Clarke et al. ,2017) outlines the possible buccal administration of the compound, the originator has not provided any experimental data to illustrate that utility. A follow-up patent application of Jubilant Generics Limited discloses a sublingual or buccal tablet composition of remdesivir using native β -cyclodextrin amongst various excipients, but the corresponding pharmacokinetic data are not provided by the applicant (Nandi et al., 2021). The presence of SBECD in the formulation has the advantage of improved solubility and reduced aggregation of remdesivir (Piñeiro et al., 2021). A recent paper by Szente et al. (2021) gave a detailed comparison of the physico-chemical characteristics and dissolution behavior of freeze-dried and electrospun nanofiber forms of SBECD-enabled remdesivir formulation. As a follow up of this pharmaceutical technology study, authors hereby report on the comparative in vivo pharmacokinetics of SBECD/remdesivir complex as fast-dissolving electrospun nanofiber in rabbits, following buccal and intravenous administration. To the best of our knowledge, this is the first report on remdesivir absorption and pharmacokinetics from a cyclodextrin-enabled formulation upon buccal administration.
2. Experimental
2.1. Materials
Remdesivir with a chemical purity of 99.2% was purchased from Echemi Pharma, Qingdao, China. The active metabolite (GS-441524) was received from Selleckchem, Houston, TX, USA. Stable isotope labeled remdesivir was purchased from Alsachim (Illkirch-Graffenstaden, France). Remdesivir was stored at room temperature, while GS-441254 and the internal standard (stable isotope labeled remdesivir) were stored in a freezer (−15 °C to −30 °C) protected from light and humidity.
SBECD/remdesivir fast-dissolving formulation was prepared as described earlier (Szente et al., 2021). Briefly, an aqueous solution of SBECD was prepared and the pH of the solution was set to 1.9 by hydrochloric acid solution. Then remdesivir was dissolved under intense agitation and finally the pH was set to approximately 3.5 by adding NaOH solution. The solution was processed by electrospinning to get nanofibers. The product is a white to off-white solid powder, freely soluble in water and PBS solutions. The anhydrous sample contains 3% remdesivir and 97% SBECD by weight.
Acetonitrile and methanol of HPLC Super gradient grade, formic acid for LC-MS and dimethyl sulfoxide (DMSO) of analytical reagent grade were used in the analysis. Ultrapure water was obtained from water purification system (ELGA).
2.2. Methods
2.2.1. Administration of remdesivir formulations
New Zealand white rabbits (6–6 males in the two treatments) with body weights at arrival of 2.5 to 3.0 kg were used after acclimatization for 7–8 days. The weight variation did not exceed ±20% of the mean weight.
The dose of SBECD/remdesivir (95 mg/kg body weight corresponding to 2.9 mg/kg bw remdesivir) was calculated on the basis of human treatment, 200 mg remdesivir/patient (about 200 mg/70 kg) and 30 mg/g content in the formulation. The calculated dose was given in the content of active ingredient. In both intravenous and buccal treated groups, the dose volume was based on the most recent body weight measurement.
The electrospun nanofiber sample was dissolved in sterile distilled water for the intravenous application of 3 animals. For the buccal treatments, SBECD/remdesivir was formulated into a thick paste-like texture with water. 100 mg SBECD/remdesivir (corresponding to 2.9 mg/kg bw remdesivir) was moistened with 0.04 mL distilled water. This paste-like preparation was applied to the sublingual mucosa of the rabbit (3 animals) anesthetized with release injection (with active ingredient: Pentobarbital sodium), with a metal spatula. The test samples were prepared on day of treatment, and administered to the animals within 1 h. Anesthetizing the animals during buccal administration was important in order to place the paste containing the drug formulation precisely on the oral mucosa and avoid the swallowing of the formulation. Absorption through buccal mucosa was ensured this way.
2.2.2. Blood sampling
According to the time schedule of first treatment period, blood sample of approximately 300 µL was obtained from the marginal vein of ear at 0 (pre-value), 1, 2, 4, 6, 9, 12 and 24 h after the treatment. In the second treatment period, blood sample of approximately 300 µL was obtained from the marginal vein of ear at 0 (pre-value), 10, 20, 30, 40, 50 min, 1, 1.5, 2, 2.5, 3, 3.5 and 4 h after the treatment. After the last blood sampling, the animals were anesthetized with Release injection (Pentobarbital sodium), and sacrificed without necropsy.
Due to the large number of blood sampling, clinical observation was not planned.
2.3. Bioanalysis
2.3.1. Calibration, quality control (QC) solutions and samples
Two independently prepared remdesivir and GS-441254 stock solutions of 100 µg/mL for the calibration and QC solutions and the internal standard (IS) stock solution of 100 µg/mL were prepared in DMSO and the solutions were stored in the freezer (−15 °C – −30 °C). Calibration working solutions were prepared in DMSO by serial dilution from the corresponding stock solution to achieve 5 (only for remdesivir), 10, 25, 50, 100, 250, 500, 1000, 2500 and 5000 ng/mL for the analytes, QC working solutions were similarly prepared to achieve 5 (only for remdesivir), 15, 150 and 4000 ng/mL for remdesivir and 25, 150 and 4000 ng/mL for GS-441254, and a working solution of 50 ng/mL was prepared in acetonitrile for the internal standard. Calibration samples for both analytes were prepared at the following nominal concentration levels: 1 (only for remdesivir), 2, 5, 10, 20, 50, 100, 200, 500 and 1000 ng/mL and quality control samples at three concentration levels: 3, 300 and 800 ng/mL for remdesivir and 5, 300 and 800 ng/mL for GS-441254. Calibration and QC samples were prepared by spiking 50 µL of blank rabbit plasma with 10 µL of the corresponding working solution and subsequently subjected to the sample preparation protocol.
2.3.2. Sample preparation
The plasma samples were thawed at room temperature. An aliquot of 50 μL of rabbit plasma sample was transferred into a microcentrifuge tube then 10 µL of DMSO and 150 µL of internal standard working solution were added. Afterwards, the samples were vortexed thoroughly for protein precipitation. Then the samples were centrifuged (Heraeus Biofuge Stratos, Osterode, Germany) at 10,000 rpm at 4 °C for 5 min. Aliquot (100 µL) of the supernatant was collected into autosampler vials.
2.3.3. LC-MS/MS conditions
A Shimadzu UFLC system (Kyoto, Japan) containing two LC-20AD XR pumps, a DGU-20A3R degassing unit, a CTO-20AC column oven, a SIL-20AXR autosampler with a CBM-20A communication bus module coupled to an AB SCIEX 5500 QTrap triple quadrupole tandem mass spectrometer (Vaughan, Canada) was used for the bioanalytical measurements.
Reverse phase HPLC separation on an Inertsil ODS-4 (3.0x150 mm, 3 μm) (GL Sciences, Japan) column with gradient elution was performed at room temperature by injecting 1 μL of sample with a flow rate of 0.4 mL/min. The eluent A was ultrapure water containing 0.1 % V/V formic acid, the eluent B was acetonitrile containing 0.1 % V/V formic acid. The initial eluent composition was 5 % B, then a linear gradient was applied to 100% B until 4.0 min after the injection. This composition was kept for 1.0 min and then the column was re-equilibrated to the initial condition until the stop time of 8.0 min.
Remdesivir, GS-441254 and IS were quantified in MRM (multiple reaction monitoring) mode with electrospray ionization (ESI) in the positive mode at the following transitions (m/z): 603.1 → 402.1, 292.1 → 163.1 and 609.1 → 229.1 for remdesivir, GS-441254 and SIL-remdesivir (internal standard), respectively. The retention time was 4.25 min and 5.45 min for GS-441254 and remdesivir, respectively.
For quantification the response ratio of the analytes to the internal standard versus the concentration of analyte was used with 1/x2 weighting. The concentration range for rabbit plasma was 1 ng/mL − 1000 ng/mL and 2 ng/mL − 1000 ng/mL for remdesivir and GS-441254, respectively. The limit of quantification was determined: 1 ng/mL and 2 ng/mL for remdesivir and GS-441254, respectively.
The chromatograms were analyzed by the Analyst 1.6.2 software.
3. Results
Buccal administration of a fast-dissolving SBECD/remdesivir formulation was evaluated compared to intravenous administration using rabbits. The time schedule of blood sampling selecting 1 h after administration as the first sampling point was not acceptable for the toxicokinetic measurement in the first treatment period. Remdesivir could not be measured in the blood plasma even at the first hour, so the experiment was repeated (second treatment) with a modified time schedule with several sampling points within the first hour. It was also concluded from the failure of the first experiment that it is not enough to measure intact remdesivir in the plasma, so in the second treatment period the analytical measurements were extended to its active metabolite, GS-441254, too. The blood concentrations of both remdesivir and its metabolite for intravenous and buccal treatment groups are shown in Fig. 2, Fig. 3 .
Fig. 2.

Concentration of Remdesivir and GS-441524 (Group 1: intravenous treatment) (The error bars represent SD, n = 3).
Fig. 3.

Concentration of remdesivir and GS-441524 (Group 2: buccal treatment) (The error bars represent SD, n = 3).
In the second treatment period, in the group 1 (intravenously treated group) the highest plasma concentration of remdesivir (383 ng/mL) was measured at 0.17 h, while that of GS-441524 (145 ± 27 ng/mL) was found at 0.5 h after the treatment. The half-life times were 0.85 and 1.45 h, respectively.
In animals of group 2 (treated buccally) remdesivir was not detected and the GS-441524 concentration appeared only at 1, 1.5 and 2.5 h. The highest plasma concentration of GS-441524 (4–5 ng/mL) was found between the 2 and 3 h after the treatment. Approximately 4 h half-life time was calculated.
In the buccally treated group, the plasma concentrations of GS-441524 were significantly lower compared to the intravenously treated group. AUC0-∞ was 263 ± 72 and 26.4 ± 2.3 ng*h/mL for the intravenous and buccal administration, respectively, resulting in 10% bioavailability of this metabolite given buccally compared to intravenous application.
There was no mortality either in the intravenous or buccal treated groups.
4. Discussion
This study aimed at evaluating the feasibility for buccal administration of remdesivir, a potent antiviral drug against SARS-CoV-2 infections. Due to the first pass metabolism in the liver this drug cannot be applied orally, only intravenously for hospitalized patients. But there is a high, still un-met need for a patient-friendly formulation which can be used in prevention and in the treatment of patients with less severe disease not requiring hospital care. Buccal administration is a potential route of administration avoiding first pass metabolism in the liver. Compared to oral, the buccal delivery offers to avoid the first pass metabolism of a drug sensitive to the liver enzymes. At present there is no approved oral remdesivir formulation due to this sensitivity.
The remdesivir formulation approved by Food and Drug Agency of the US (FDA) contains SBECD as excipient. SBECD is an anionic cyclodextrin derivative used to enhance the solubility of poorly soluble compounds (Puskás et al., 2015, Stella and Rajewski, 2020). There are several drugs marketed in SBECD-solubilized form, such as voriconazole, maropitant, aripiprazole, amiodarone, melphalan, etc. (Stella and Rajewski, 2020). As earlier we have developed a fast-dissolving formulation of remdesivir and SBECD of identical composition as the intravenous injection (Szente et al., 2021), we studied the single-dose buccal administration of this formulation in animal experiments and compared the bioavailability with intravenous administration.
While no studies on buccal remdesivir formulations, only a few papers can be found in the scientific literature on buccal administration of SBECD-formulated drugs. SBECD/danazol complex was perorally administered to dogs and compared to buccal administration of this complex formulated into buccal tablets by using mucoadhesive polymers to obtain 64% and 25% absolute bioavailabilities, respectively (Jain et al., 2002). When triclosan complexed with SBECD or HPBCD (2-hydroxypropyl-beta-cyclodextrin, a neutral CD derivative) no difference was found in adhesion to porcine buccal adhesion (Sigurdsson et al., 2002). In a similar study, the authors found only a slightly higher transport of SBECD/roprinolone compared to HPBCD/roprinolone across porcine buccal epithelium: 28 and 24% improvement compared to buffer only (Kontogiannidou et al., 2017).
In our study, rabbit was selected as animal model for the in vivo pharmacokinetic experiments. The oral mucosa of dog, pig and rabbit show the highest similarity in structure and composition to the human buccal mucosa (Harris and Robinson, 1992, Dali et al., 2006). The thickness of the oral epithelium of rabbits is similar to humans (about 600 μm). The extent of keratinization is only a bit higher which is an important factor in mucosal drug delivery as keratinized oral epithelium has low permeability.
The metabolism of remdesivir in the blood is fast in mice resulting in extremely low half-life time (a few minutes) due to enzymes, while the blood level of GS-441524 metabolite remained high even after 24 h (Hu et al., 2021). In humans the median terminal half-lives of remdesivir and GS-441524 were approximately 1 and 27 h, respectively (Health, 2021).
Although we have not found any data on the pharmacokinetics of remdesivir in rabbits, our results are consistent with the human data with the half-life of remdesivir of 0.85 h after intravenous application of SBECD/remdesivir fast-dissolving electrospun formulation. The half-life for GS-441254 metabolite was somewhat longer than that of remdesivir, as expected, but only 1.45 h.
Buccal administration resulted in highly reduced bioavailability of remdesivir and its metabolite. It is a challenge in buccal drug delivery to keep the drug concentration in the oral mucosa for drugs with short half-lives such as remdesivir due to salivary flow, mastication and ingestion of food and beverage.
Remdesivir is a substrate for esterases in plasma and tissue, for drug metabolizing enzymes in liver, such as CYP2C8, CYP2D6, and CYP3A4, and is also a substrate for transporters Organic Anion Transporting Polypeptides 1B1 (OATP1B1) and P-glycoprotein (P-gp (Health, 2021). The complexation of remdesivir with SBECD aims at solubilization but gives hardly any protection to the drug against these enzymes and transporters. The esterases of oral mucosa might hydrolyze the buccally administered formulation, too.
Further studies are needed to understand the delay in the appearance of GS-441254 in the blood plasma after buccal administration. Other studies with propranolol, verapamil and captopril buccally administered to rabbits showed no such delay (Dali et al., 2006).
The formulation should be further developed to improve the pharmacokinetic properties. In addition to solubilization by SBECD, other permeability enhancers and the use of enzyme inhibitors to reduce the enzymatic degradation within the mucosa could help to get a formulation with higher bioavailability. Encapsulation of remdesivir in a polymer matrix, which was found to be useful for intravenous injections (Chakraborty et al., 2021), could be applied for formulations in buccal administration, too.
5. Conclusion
The remdesivir formulation of Gilead Sciences, Inc. marketed as Veklury™ contains SBECD to address the solubility and stability of the drug approved for the treatment of SARS-CoV-2 infected patients. It can be used intravenously only because of its fast decomposition by the liver enzymes if taken orally. Our working hypothesis was to avoid first pass metabolism via buccal administration. The composition of the formulation was identical to the marketed formulation Veklury™, but was prepared by electrospinning instead of lyophilization to obtain a fast-dissolving powder. Rabbits were treated with this formulation in two groups: intravenously and buccally. Rabbit is typically used as buccal drug delivery model due to similarity of its mucosa to human.
Under the conditions of the present study, no intact remdesivir could be detected only the GS-441524 metabolite after buccal administration and in much smaller concentration than after intravenous injection. The 10% bioavailability compared to intravenous administration suggests that the complexation with SBECD cannot protect remdesivir from the effect of esterases of the oral mucosa. The results of this preliminary study indicate that development of a buccal formulation of remdesivir is not an easy task, needs further development, optimization and characterization studies. It seems to be necessary to further develop the formulation enabling fast absorption through the mucosal layer and shielding the drug from the enzymes of the oral cavity.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Cao Y.-C., Deng Q.-X., Dai S.-X. Remdesivir for severe acute respiratory syndrome coronavirus 2 causing COVID-19: An evaluation of the evidence. Travel. Med. Infect. Dis. 2020;35:101647. doi: 10.1016/j.tmaid.2020.101647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty, A., Diwan, A., Arora, V., Thakur, Y., Chiniga, V., Tatake, J., Holkar, P., Holkar, N., Pond, B., 2021. Nanoviricide’s platform technology based NV-CoV-2 polymer increases the half-life of Remdesivir in vivo. bioRxiv 468980. doi: 10.1101/2021.11.17.468980.
- Cho A., Saunders O.L., Butler T., Zhang L., Xu J., Vela J.E., Feng J.Y., Ray A.S., Kim C.U. Synthesis and antiviral activity of a series of 1′-substituted 4-aza-7,9-dideazaadenosine C-nucleosides. Bioorg. Med. Chem. Lett. 2012;22(8):2705–2707. doi: 10.1016/j.bmcl.2012.02.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, M.O.H., Feng, J.Y., Jordan, R., Mackman, R.L., RAY, A.S., Siegel, D., 2017. Methods for treating arenaviridae and coronaviridae virus infections. PCT Pat Appl. WO/2017/049060, 23.03.2017.
- Cox R.M., Wolf J.D., Lieber C.M., et al. Oral prodrug of remdesivir parent GS-441524 is efficacious against SARS-CoV-2 in ferrets. Nat. Commun. 2021;12:6415. doi: 10.1038/s41467-021-26760-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dali M.M., Moench P.A., Mathias N.R., Stetsko P.I., Heran C.L., Smith R.L. A rabbit model for sublingual drug delivery: comparison with human pharmacokinetic studies of propranolol, verapamil and captopril. J. Pharm. Sci. 2006;95(1):37–44. doi: 10.1002/jps.20312. [DOI] [PubMed] [Google Scholar]
- Dodla S., Velmurugan S. Buccal penetration enhancers-an overview. Asian J. Pharm. Clin. Res. 2013;6:39–47. [Google Scholar]
- Jain A.C., Aungst B.J., Adeyeye M.C. Development and in vivo evaluation of buccal tablets prepared using danazol-sulfobutylether 7 β-cyclodextrin (SBE 7) complexes. J. Pharm. Sci. 2002;91(7):1659–1668. doi: 10.1002/jps.10163. [DOI] [PubMed] [Google Scholar]
- EMEA, 2020. Summary on compassionate use of Remdesivir Gilead (EMEA/H/K/005622/CU) https://www.ema.europa.eu/en/documents/other/summary-compassionate-use-remdesivir-gilead_en.pdf. 1 (accessed 12 January 2022).
- Health, 2021. ANNEX I Summary of product characteristics. https://ec.europa.eu/health/documents/community-register/2021/20210624152163/anx_152163_en.pdf (accessed 12 January 2022).
- Garibaldi, B.T., Wang, K., Robinson, M.L., Zeger, S.L., Bandeen-Roche, K., Wang, M.C., Alexander, G.C., Gupta, A., Bollinger, R., Xu, Y., 2021. Comparison of time to clinical improvement with vs without remdesivir treatment in hospitalized patients with COVID-19. JAMA Netw. Open. 4(3):e213071. https://doi.porg/10.1001/jamanetworkopen.2021.3071. [DOI] [PMC free article] [PubMed]
- Garrido P., Calvelo M., Garcia-Fandiño R., Piñeiro Á. Rings, Hexagons, Petals, and Dipolar Moment Sink-Sources: The Fanciful Behavior of Water around Cyclodextrin Complexes. Biomolecules. 2020;10:431. doi: 10.3390/biom10030431. https://www.mdpi.com/2218-273X/10/3/431, March 10, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J.-H., Cooklock K.M. Bioadhesive polymer buccal patches for buprenorphine controlled delivery: solubility consideration. Drug Dev. Ind. Pharm. 1995;21(17):2013–2019. doi: 10.3109/03639049509065885. [DOI] [Google Scholar]
- Harris D., Robinson J.R. Drug delivery via the mucous membranes of the oral cavity. J. Pharm. Sci. 1992;81(1):1–10. doi: 10.1002/jps.2600810102. [DOI] [PubMed] [Google Scholar]
- Hu W.-J., Chang L.u., Yang Y., Wang X., Xie Y.-C., Shen J.-S., Tan B.o., Liu J. Pharmacokinetics and tissue distribution of remdesivir and its metabolites nucleotide monophosphate, nucleotide triphosphate, and nucleoside in mice. Acta Pharmacol. Sin. 2021;42(7):1195–1200. doi: 10.1038/s41401-020-00537-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen S.C.J., Kebriaei R., Dresser L.D. Remdesivir: Review of pharmacology, pre-clinical data, and emerging clinical experience for COVID-19. Pharmacotherapy. 2020;40(7):659–671. doi: 10.1002/phar.2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontogiannidou E., Andreadis D.A., Zografos A.L., Nasar H., Klepetsanois P., van der Merwe S.M., van der Merwe S.M., Fatouros D.G. Ex vivo buccal drug delivery of ropinirole hydrochloride in the presence of permeation enhancers: the effect of charge. Pharm. Dev. Technol. 2017;22:1017–1021. doi: 10.3109/10837450.2015.1135343. [DOI] [PubMed] [Google Scholar]
- Lakshmi V.L., Umashankar M.S., Alagusundaram M. An assessment on buccal mucoadhesive drug delivery system. Int. J. Appl. Pharm. 2021;13:66–74. https://doi.o4g/10.22159/ijap.2021v13i6.42760 [Google Scholar]
- Larsen, N. 2019. Compositions comprising an RNA polymerase inhibitor and cyclodextrin for treating viral infections. US patent US10675296, 21 March 2019.
- Loftsson T., Gudmundsson J.A., Arnadottir R.O., Fridriksdottir H. Sublingual delivery of 17beta-estradiol from cyclodextrin containing tablets. Pharmazie. 2003;58:358–359. [PubMed] [Google Scholar]
- Nandi I., Jain A., Gat G.V. Transmucosal dosage forms of remdesivir. PCT Int. Appl. 2021;WO2021240531:02.12.2021. [Google Scholar]
- Palem C.R., Kumar Battu S., Gannu R., Yamsani V.V., Repka M.A., Yamsani M.R. Role of cyclodextrin complexation in felodipine-sustained release matrix tablets intended for oral transmucosal delivery: In vitro and ex vivo characterization. Pharm. Dev. Technol. 2012;17(3):321–332. doi: 10.3109/10837450.2010.535829. [DOI] [PubMed] [Google Scholar]
- Piñeiro Á., Pipkin J., Antle V., Garcia-Fandino R. Aggregation versus inclusion complexes to solubilize drugs with Cyclodextrins. A case study using sulphobutylether-β-cyclodextrins and remdesivir. J. Mol. Liq. 2021;343:117588. doi: 10.1016/j.molliq.2021.117588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piñeiro Á., Pipkin J., Antle V., Garcia-Fandino R. Remdesivir interactions with sulphobutylether-β-cyclodextrins: A case study using selected substitution patterns. J. Mol. Liq. 2022;346:117157. doi: 10.1016/j.molliq.2021.117157. [DOI] [Google Scholar]
- Pipkin, J., Antle, V., Garcia-Fandiño, R. 2020. “FORMULATION FORUM – Application of Captisol Technology to Enable the Formulation of Remdesivir in Treating COVID-19”. Drug Development & Delivery. June 2020. https://drug-dev.com/formulation-forum-application-of-captisol-technology-to-enable-the-formulation-of-remdesivir-in-treating-covid-19/ (accessed 28 February 2022).
- Pitha J., Harman S.M., Michel M.E. Hydrophilic cyclodextrin derivatives enable effective oral administration of steroidal hormones. J. Pharm. Sci. 1986;75(2):165–167. doi: 10.1002/jps.2600750213. [DOI] [PubMed] [Google Scholar]
- Puskás, I., Varga, E., Tuza, K., Szemán, J., Fenyvesi, É., Sohajda, T., Szente, L., 2015. Sulfobutylether-cyclodextrins: Structure, degree of substitution and functional performance. In: F.G. Ramirez (Ed.) Cyclodextrins. Synthesis, Chemical Applications and Role in Drug Delivery, Nova Science Publishers, New York.
- Rubin D., Chan-Tack K., Farley J., Sherwat A. FDA approval of remdesivir — a step in the right direction. N. Engl. J. Med. 2020;383(27):2598–2600. doi: 10.1056/NEJMp2032369. [DOI] [PubMed] [Google Scholar]
- Sayani A.P., Chien Y.W. Systemic delivery of peptides and proteins across absorptive mucosae. Crit. Rev. Ther. Drug Carrier Syst. 1996;13:85–184. [PubMed] [Google Scholar]
- Schäfer A., Martinez D.R., Won J.J., Moreira F.R., Brown A.J., Gully K.L., et al. Therapeutic efficacy of an oral nucleoside analog of remdesivir against SARS-CoV-2 pathogenesis in mice. BioRxiv. 2021;460111 doi: 10.1101/2021.09.13.460111. [DOI] [Google Scholar]
- Sigurdsson H.H., Knudsen E., Loftsson T., Leeves N., Sigurjonsdottir J.F., Másson M. Mucoadhesive sustained drug delivery system based on cationic polymer and anionic cyclodextrin/triclosan complex. J Incl Phenom. 2002;44:169–172. doi: 10.1023/A:1023098730627. [DOI] [Google Scholar]
- Singh S., Khera D., Chugh A., Khera P.S., Chugh V.K. Efficacy and safety of remdesivir in COVID-19 caused by SARS-CoV-2: a systematic review and meta-analysis. BMJ Open. 2021;11(6):e048416. doi: 10.1136/bmjopen-2020-048416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella V.J., Rajewski R.A. Sulfobutylether-β-cyclodextrin. Int. J. Pharm. 2020;583:119396. doi: 10.1016/j.ijpharm.2020.119396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szejtli, J., 2004. Cyclodextrins and molecular encapsulation. In: Nalwa, H.S. (Ed.), Encyclopedia of Nanoscience and Nanotechnology, vol. 2, American Scientific Publishers, pp. 283–304.
- Szente L., Puskás I., Sohajda T., Varga E., Vass P., Nagy Z.K., Farkas A., Várnai B., Béni S., Hazai E. Sulfobutylether-beta-cyclodextrin-enabled antiviral remdesivir: characterization of electrospun- and lyophilized formulations. Carb. Pol. 2021;264:118011. doi: 10.1016/j.carbpol.2021.118011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Várnai B., Malanga M., Sohajda T., Béni S. Molecular interactions in remdesivir-cyclodextrin systems. J. Pharm. Biomed. Anal. 2022;209:114482. doi: 10.1016/j.jpba.2021.114482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei D., Hu T., Zhang Y., Zheng W., Xue H., Shen J., Xie Y., Aisa H.A. Potency and pharmacokinetics of GS-441524 derivatives against SARS-CoV-2. Bioorg. Med. Chem. 2021;46:116364. doi: 10.1016/j.bmc.2021.116364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J., Wang Z. Can remdesivir and its parent nucleoside GS-441524 be potential oral drugs? An in vitro and in vivo DMPK assessment. Acta Pharm. Sinica B. 2021;11:1607–1616. doi: 10.1016/j.apsb.2021.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]