

Abstract
Mutations in Cu/Zn–superoxide dismutase 1 (SOD1) are linked to amyotrophic lateral sclerosis (ALS). Using a line of ALS-related mutant human SOD1 (hSOD1) transgenic Caenorhabditis elegans, we determined the effects of metformin on the progression of ALS-like pathological abnormalities. We found that metformin significantly extended the lifespan, improved motor performance, and enhanced antioxidant activity of mutant worms. We further showed that metformin enhanced expression of lgg-1, daf-16, skn-1 and other genes known to regulate autophagy, longevity and oxidative stress in hSOD1 transgenic worms. Accordingly, overexpression of lgg-1 or daf-16 attenuated the aging and pathological abnormalities of mutant human SOD1 worms, while genetic deletion of lgg-1 or daf-16 abolished the beneficial effects of metformin. Collectively, we demonstrate that metformin protects against mutant SOD1-induced cytotoxicity in part through enhancement of autophagy and extends lifespan through daf-16 pathway. Our findings suggest that metformin could be further explored as a potential therapeutic agent in treating ALS.
Keywords: Metformin, Lifespan, Neuroprotection, ALS, Autophagy
1. Introduction
Amyotrophic lateral sclerosis (ALS) is the most common degenerative motor neuron disease [1], Most ALS cases occur sporadically, while approximately 10% are familial [2]. One of the most prevalent familial forms of the disease involves mutations of Cu/Zn–superoxide dismutase 1 (SOD1) [3]. Since the discovery of SOD1 G93A missense mutation in 1993 [4], more than 185 different ALS-related SOD1 mutations have been identified [3], The disease causal effect of SOD1 mutations is attributed to the degeneration of motor neurons [5], Despite decades of research, the pathogenesis of ALS and the mechanism by which SOD1 mediates its toxic effects in motor neurons are not fully understood. Studies on neuronal dysfunction have primarily relied on models overexpressing human SOD1 protein and have supported a role for SOD1 dysfunction in ALS pathogenesis [6], However, these findings have limited translational value in terms of extending the lifespan of ALS patients and the development of effective therapeutics. Although the degeneration of motor neurons in ALS is mainly attributable to the missense mutation in SOD1 and aggregation of SOD1 protein, the specific molecular events and pathways involved have yet to be fully elucidated. As such, there is a need for a more suitable model to investigate the etiopathogenesis of ALS and evaluate the efficacy of potential drugs for its treatment. Although the clinical course is variable, ALS patients generally have short lifespan. The molecular basis for short lifespan and whether it is mediated by pathways known to regulate longevity are open questions. It was reported that a prolonged lifespan depends on the FOXO transcription factor DAF-16 [7] and may be related to autophagy and various aspects of cellular and organismal aging. Furthermore, autophagy has been directly linked to neurodegeneration and ageing by an extensive body of research [8], However, our understanding of the mechanistic link between autophagy and lifespan is limited, particularly with regard to neurodegenerative diseases.
Metformin (metformin hydrochloride) is the most widely prescribed biguanide drug worldwide for the treatment of type II diabetes and metabolic syndromes [9]. Previous research has shown that metformin is associated with a reduced risk of cancer and cardiovascular disease, raising the possibility that it could have beneficial effects in other age-related diseases [10]. Metformin is thought to act by activating the 5′ adenosine monophosphate-activated protein kinase (AMPK) pathway or by inhibiting the mammalian target of rapamycin (mTOR) pathway [11], while some studies have indicated that it also activates the lysosomal degradation pathway [12]. Metformin was reported to slow aging in C. elegans by altering microbial folate and methionine metabolism [13] and prolong lifespan through mitohormesis by modulating peroxiredoxin 2 (PRDX-2) [9]. In addition, it was reported that metformin-bacterial interactions engaged lipid metabolism in C. elegans to extend lifespan [14], Moreover, a recent study has shown that metformin could inhibit RAN translation through PKR pathway [15]. However, whether and how metformin exerts these effects in the context of neurodegenerative diseases and the associated mechanisms are not known.
Transgenic Caenorhabditis elegans (C. elegans) models have been established for a number of neurodegenerative diseases, such as Alzheimer’s disease (AD) [16], Parkinson’s disease (PD) [17], and ALS [18]. It is also a useful tool for evaluating the toxicity of mutant SOD1 in motor neurons and for exploring the mechanisms by which SOD1 mutation affects longevity and other physiologic functions in ALS. SOD1 overexpression models support a role for SOD1-mediated toxicity in the pathogenesis of ALS [19]. In our previous work, we found that hSOD1 aggregates and forms inclusions in the motor neurons of C. elegans [20]. The hSOD1-induced death of motor neurons is independent of the Cell death protein 4 (CED-4)/CED-3 apoptosis pathway [20]. Recent studies in ALS models have demonstrated that mutant SOD1 not only induces toxicity in motor neurons but also reduces longevity, possibly due to mutant SOD1 aggregation [21,22]. Verapamil was shown to inhibit the aggregation of mutant SOD1, promote motor neuron survival, and extend lifespan in SOD1G93A mice by inducing the activation of autophagy [23]; meanwhile, shatavarin IV prevented reactive oxygen species-induced oxidative damage in C. elegans and prolonged lifespan by downregulating the expression of the Leucine-rich repeats, Ras-like domain, kinase 1 (lrk-1) gene [24], These findings provide evidences that mutant SOD1 causes motor neuron damage and short lifespan through complex mechanisms. However, the specific mechanisms underlying these effects remain unknown.
In addition, the insulin-IGF1 signaling pathway is known to be involved in the determination of lifespan. The daf-16 signaling pathway, a homologous to the insulin/IGF-1 signaling network in humans, is critical in endocrine processes that regulate longevity, metabolism, stress resistance, and development [7]. In this study, we generated and characterized a strain of transgenic C. elegans that specifically overexpresses ALS-related SOD1 G93A mutation in motor neurons to systematically investigate the molecular pathogenesis of ALS and evaluate the therapeutic potential of metformin for ALS treatment. We found that metformin had beneficial effects on the lifespan, motor behavior, and oxidative stress response of mutant worms by stimulating autophagy and daf-16 pathways.
2. Materials and methods
2.1. Worm strains and culture conditions
Worms were grown on nematode growth medium (NGM) plates seeded with E. coli strain OP50. The N2 (Bristol) strain was used as nontransgenic control worms. Daf-16 (m26) and DA2123(adIs2122, lgg-1p::GFP::lgg-1+rol-6) strains were provided by the Caenorhabditis Genetics Center (CGC), which is funded by the National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440). The unc-25 promoter was amplified using KOD-Plus Neo DNA polymerase from N2 worm genomic DNA. The unc-25 promoter was inserted into the Hind III and Bam HI sites of pPD95.77 to generate pPD95.77-unc-25::GFP. The plasmids of pPD95.77-unc-25::hSOD1G93A::GFP were made using the same method. The following crossing strains were used in this study: hSOD1G93A::lgg-1::GFP; hSOD1G93A::daf-16::GFP; and lgg-1::daf-16::GFP. All experiments were performed at 20°C according to standard C. elegans culture techniques [25].
2.2. Determination of optimal metformin concentration
Metformin (Solarbio, Beijing, China) was dissolved in sterile water to prepare a 1 M stock solution, which was added to OP50 culture solution (OD 0.3–0.5) to prepare working solutions at concentrations of 25, 50, and 100 mM. OP50 without metformin was used as a control. The working solutions were coated onto the surface of NGM plates. Synchronized L1 larval stage worms were transferred to fresh NGM plates with or without the drug and grown at 20°C for 3 days until they reached the adult stage. The worms were washed and transferred to fresh regular NGM plates and grown at 20°C.
2.3. RNA interference
RNAi experiments were performed as previously described [26]. Briefly, freshly streaked single colonies of HT115 bacteria containing either the empty L4440 vector or hSOD1 RNAi plasmid were grown overnight at 37°C in Luria-Bertani medium supplemented with tetracycline (50 μg/ml). The next day, freshly grown bacteria were seeded onto NGM plates supplemented with tetracycline (50 μg/ml) and isopropyl-β-d-thiogalactopyranoside (IPTG, 0.1 mM). Synchronized L1 larval stage worms were washed from regular NGM plates, transferred to RNAi plates, and grown at 20°C for 3 days until the adult stage. The worms were then washed and transferred to fresh regular NGM plates and grown at 20°C.
2.4. Lifespan assay
Lifespan experiments were performed as previously described [20], with minor modifications. Briefly, synchronized L4 stage worms (n ≈ 100) were placed on NGM plates, and the following day was defined as day 1. The viability of each worm was scored every day thereafter; those that did not respond to external mechanical stimuli or swallow were judged to be dead. Events in which worms crawled off the plate were not recorded. All worms were transferred to new NGM plates every other day until death. The survival curves were analyzed by GraphPad Prism software (GraphPad, La Jolla, CA, USA).
2.5. Behavioral assay
To evaluate locomotion of worms, we monitored their crawling behavior using previously described methods [27]. Worms from different experimental conditions were individually carried out immediately after each worm had been transferred to a fresh NGM plate, and their crawling tracks were monitored for 30 s. A video was collected, and the ratio of the movement distance to the body length was measured by Image J software. A total of 30 worms per experimental group were monitored.
2.6. Stress response assay
To assess resistance to oxidative stress, we transferred adult worms with or without drug treatment or RNAi to fresh NGM plates containing paraquat (3 mM, Solarbio, Beijing, China) at 20°C. The number of dead worms was recorded every hour. Worms without treatment served as the control. The survival curves were analyzed by GraphPad Prism software.
2.7. Quantitative real-time polymerase chain reaction (RT-PCR)
Gene expression levels in worms were quantified by RT-PCR [28]. Total mRNA was extracted from each worm groups using TRIzol reagent (Takara Biomedical Technology, Beijing, China) and reverse-transcribed using the TransScript cDNA Synthesis SuperMix kit (TransGen Biotech Co, Beijing, China). RT-PCR was performed on a 7500 Dx PCR system (Life Technologies, Carlsbad, CA, USA) with a mixture of TransStart Top Green qPCR SuperMix kit (TRAN), 10 ng cDNA, and 0.4 μM each primer in a final volume of 20 μl. The relative expression level of target genes was normalized to that of actin (act-1) and quantified using Applied Biosystems 7500 software v2.0.5 (Life Technologies). Three repeats were performed per condition. The primers used for RT-PCR are shown in Table 1.
Table 1.
Primer sequences of RT-PCR.
| Primers | Forward (5’-3’) | Reverse (5’-3’) |
|---|---|---|
|
| ||
| act-1 | GCTGGACGTGATCTTACTGATTACC | GTAGCAGAGGTTGTCGTTGATGTC |
| hsod-1 | GGTGGGCCAAAGGATGAAGAG | GGACAAGCCAAACGACTTGC |
| lgg-1 | CCGCAGAAAGTACCCAGACC | CTGGACGAAGTTGGATGCGT |
| bec-1 | CCGATGAGGAGAAGGAGC | AACCCAAATGTGGAAGCA |
| daf-16 | GCGAATCGGTTCCAGCAATTCCAA | ATGCACGGACAGTGTTCAAGTGGT |
| daf-2 | GGCCGATGGACGTTATTTTG | TTCCACAGTGAAGAAGCCTGG |
| age-1 | CGGAAAGAGCAAACTTGGGATC | GGTAGGCTTCGACGCATAAGG |
| skn-1 | AGTGTCGGCGTTCCAGATTTC | GTCGACGAATCTTGCGAATCA |
2.8. Fluorescence microscopy
Worms were washed off NGM plates and immobilized in 5 mM sodium azide (Santa Cruz Biotechnology, Santa Cruz, CA, USA) in M9 buffer on glass slides. Images were collected using an A1R multiphoton confocal microscope (Nikon Instruments, Shanghai, China). At least 20 worms per strain were examined.
2.9. Western blotting
To analyze protein expression, adult worms were washed 4 times with M9 buffer and packed worms were lysed by sonication on ice in radioimmunoprecipitation assay buffer (Beyotime, Shanghai, China) containing a 1% protease inhibitor cocktail. The lysates were centrifuged at 13,000×g for 15 min at 4°C using a Sorvall Legend Micro 17R centrifuge (Thermo Fisher Scientific, Waltham, MA, USA). The supernatant was collected and protein concentration was measured with a bicinchoninic acid (BCA) protein assay kit (Beyotime). The remaining supernatant was combined with 5 × sodium dodecyl sulfate (SDS; Beyotime) sample buffer and boiled for 5 min at 95°C. Equal amounts of total protein (40–60 μg) were separated by 10% SDS-polyacrylamide gel electrophoresis and transferred to a 0.45-μm pore size polyvinylidene difluoride membrane (Millipore, Kankakee, MA, USA). After blocking with 5% skimmed milk for 1 h at room temperature, the membrane was incubated overnight (16–18 h) at 4°C with primary antibody, then washed with Tris-buffered saline containing Tween-20 and incubated with secondary antibody. Protein bands were visualized using an enhanced chemiluminescence detection kit (Advansta, San Jose, CA, USA) and signal intensity was normalized to that of actin (clone C4; Millipore, Darmstadt, Germany) using the FluorChem Q system (Protein Simple, San Jose, CA, USA). The experiment was repeated 3 times. The following antibodies were used: anti-SOD1 (Abeam, Cambridge, MA, USA), anti-actin (Millipore, Temecula, CA, USA), goat anti-mouse IgG H&L (Proteintech, Rosemont, II, USA), and goat anti-rabbit IgG H&L (Proteintech).
2.10. Statistical analysis
Data were analyzed using Prism software. Differences between groups were evaluated with the Student’s t-test (2-tailed; 2-sample equal variance): ****P < 0.001; ***P < 0.005; **P < 0.001; *P < 0.01. Lifespan was compared between groups by Kaplan-Meier survival analysis and with the log-rank test. Other data were evaluated by one-way analysis of variance followed by the Tukey post hoc test. P<0.05 was considered statistically significant.
3. Results and discussion
3.1. Overexpression of mutant hSOD1 induces motor neuron degeneration and shortens the lifespan of transgenic worms
To examine the effect of ALS-associated human mutant SOD1 in motor neurons, we generated transgenic C. elegans overexpressing ALS-linked G93A mutant hSOD1 (hSOD1G93A) in motor neurons. In these worms, hSOD1 protein was tagged with green fluorescent protein (GFP). We examined the expression of hSOD1 mRNA and protein in transgenic worms and found expression of hSOD1G93A mRNA and almost 8-fold increase of protein levels against native worm SOD1 protein, respectively (Fig. S1A–B). The hSOD1G93A mutant worms displayed uncoordinated movements starting at age of 3 days and showed markedly reduced locomotor activity at age of 7 days (Fig. S1C). In correlation with the motor deficits, we observed substantial loss of motor neuron axons in hSOD1G93A transgenic worms at age of 7 days (Fig. S1D). We then examined the morphology of remaining motor neurons and found motor neuron in the hSOD1G93A transgenic worms were completely broken down (Fig. S1E). Therefore, we generated a line of motor neuron-specific hSOD1G93A transgenic worms that recapitulate ALS-like motor neurodegeneration.
We next assessed the lifespan of hSOD1G93A transgenic C. elegans by recording the survival rate every day from L4 stage until death. The survival analysis showed that hSOD1G93A transgenic worms had a significantly shorter lifespan than controls, with median survival reduced from 17 to 10 days (Fig. S1F). Thus, in addition to inducing motor neuron degeneration, the hSOD1G93A mutation decreases the longevity of worms.
As C. elegans are well suited to the study of stress response [29], we also investigated the effects of hSOD1G93A mutation on the oxidative stress responses in transgenic worms. As expected, hSOD1G93A worms exposed to paraquat had a shortened lifespan compared to their control counterparts under the same stress (Fig. S1G). Together, these findings indicate that hSOD1G93A mutation decreases longevity and enhances the stress response.
3.2. Metformin extends lifespan and alleviates motor dysfunction in hSOD1 mutant worms
To assess the therapeutic potential of metformin for the treatment of ALS, we mixed metformin with OP50 and fed the mixture to hSOD1G93A transgenic worms. The optimal concentration of metformin was determined to be 50 mM (Fig. S2). The hSOD1 RNA interference (RNAi) served as a positive control. Compared to the hSOD1G93A group, hSOD1 mRNA and protein levels were significantly reduced in worms fed metformin (Fig. 1A–B). Metformin also attenuated the hSOD1G93A-induced degeneration of motor neuron axons, indicating that it has a neuroprotective effect (Fig. 1C). Moreover, compared to untreated hSOD1G93A worms, those fed metformin showed partly restored motor neuron morphology (Fig. 1D). Similarly, hSOD1 knockout by RNAi partly rescued the morphologic defects in motor neurons caused by hSOD1G93A mutation. We also compared lifespan in different groups to determine whether metformin has beneficial effects on the longevity of ALS model worms. Metformin treatment significant prolonged the lifespan of hSOD1G93A worms, with median survival increased from 10 to 13 days (Fig. 1E). Metformin also improved the locomotion of hSOD1G93A worms (Fig. 1F), and extended the lifespan of hSOD1G93A worms under the oxidative stress (Fig. 1G). These results demonstrate that metformin can improve the lifespan, motor activity, motor neuron survival, and stress response of hSOD1G93A worms.
Fig. 1.
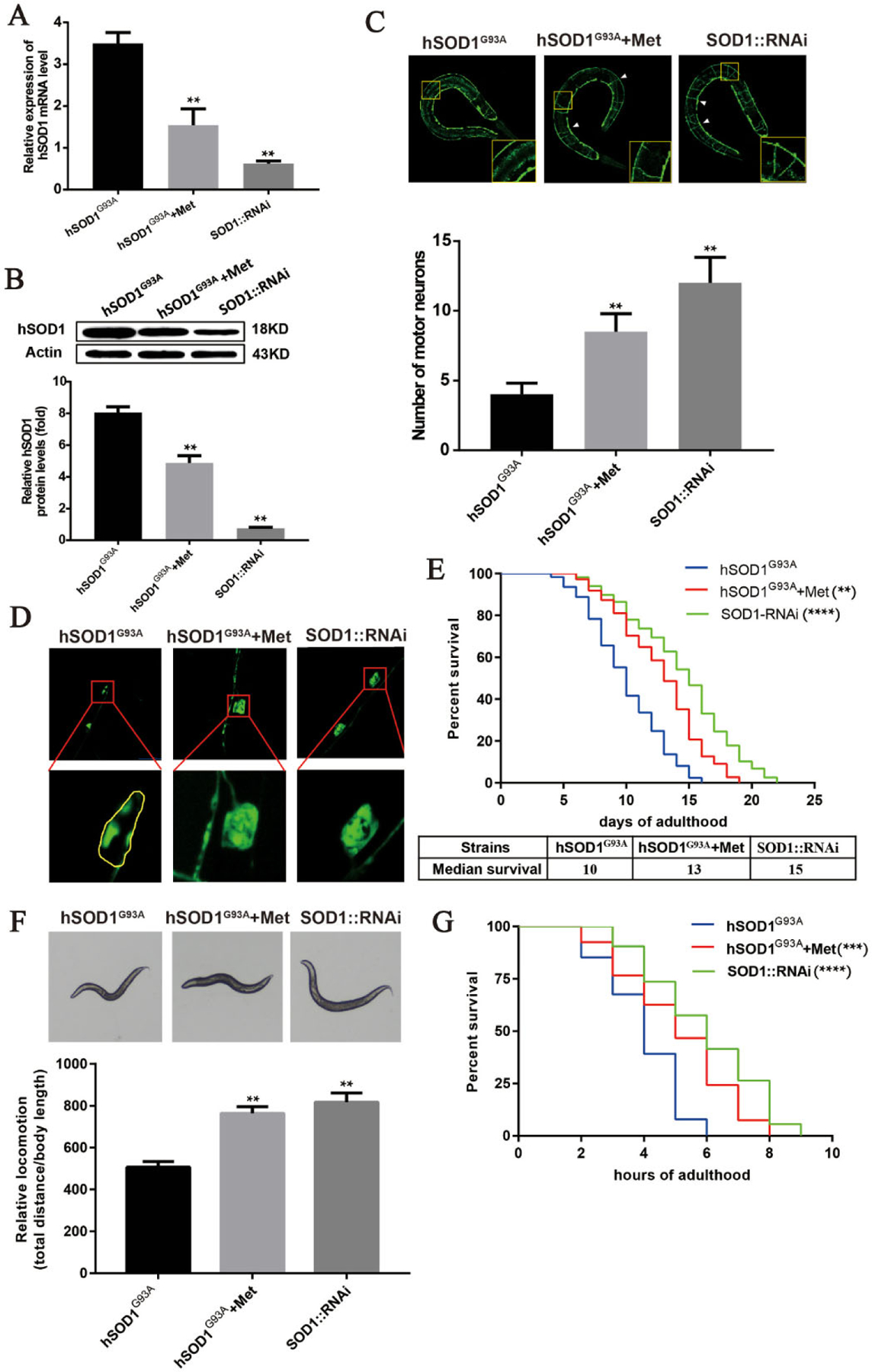
Metformin extends lifespan and alleviates locomotor deficits and motor neuron degeneration in hSOD1G93A mutant C. elegans. (A) Relative expression of hSOD1 mRNA in hSOD1G93A transgenic worms treated with metformin or hSOD1 RNAi. (B) Western blot analysis of hSOD1 protein level in extracts from transgenic strains. hSOD1 expression was lower in hSOD1G93A worms fed metformin than in untreated mutant worms. (C, D) Motor neuron survival and morphology in transgenic worms. Metformin prevented motor neuron degeneration (C) and restored neuronal morphology (D) in hSOD1G93A worms. (E) Suivival analysis. Median suivival of hSOD1G93A worms was increased by metformin treatment (13 days vs 10 days without treatment). (F) Locomotion was partly restored in hSOD1G93A worms treated with metformin. (G) Oxidative stress response in transgenic worms treated with metformin. Metformin prolonged the lifespan of hSOD1G93A transgenic worms exposed to paraquat. **P<0.001, ***P<0.0005, ****p<0.0001 (N ~ 100 worms per condition from 3 independent experiments).
3.3. Metformin increases the expression of daf-16 and lgg-1 genes in hSOD1 mutant worms
It is known that daf-2 modulates the aging process via insulin/insulin-like growth factor (IGF)-1 signaling [7]. Daf-16 and age-1 are downstream proteins of daf-2 involved in regulating the lifespan of C. elegans [30]. Lgg-1 and beclin-1 homolog (.bec-1) genes play critical roles in autophagy that regulates the lifespan of C. elegans [31,32], Furthermore, skn-1 regulates lifespan in C. elegans by conferring protection against stress [33], In the present work, we investigated the effects of hSOD1G93A mutation and metformin on the expression of genes associated with autophagy, aging, and the stress response, with hSOD1 RNAi serving as a positive control. Compared to the control worms, the relative expressions of lgg-1, bec-1, daf-16 and skn-1 were slightly decreased while those of daf-2 and age-1 were significantly increased in hSOD1G93A worms, indicating that autophagy and oxidative stress genes were partly decreased and longevity genes were significantly reduced (Fig. 2A). In hSOD1G93A worms fed metformin, the levels of lgg-1, bec-1 and skn-1 were significantly increased, indicating that autophagy was activated and antioxidative response was intensified. We also tested the relative mRNA expressions of skn-1 and bec-1 in DA2123 worms, and found that lgg-1 increased the mRNA expression of skn-1 and bec-1 in worms, verifying the conclusion that activation of autophagy can improve the antioxidant capacity by increasing skn-1 in worms (Fig. 2B). In addition, the level of daf-16 was notably increased while those of daf-2 and age-1 showed a marked decrease, indicating that daf-16 pathway was activated and longevity genes were significantly increased. Thus, metformin can enhance the expression of genes associated with longevity and the stress response, possibly through activation of genes in the autophagy and daf-16 pathway such as lgg-1 and daf-16.
Fig. 2.
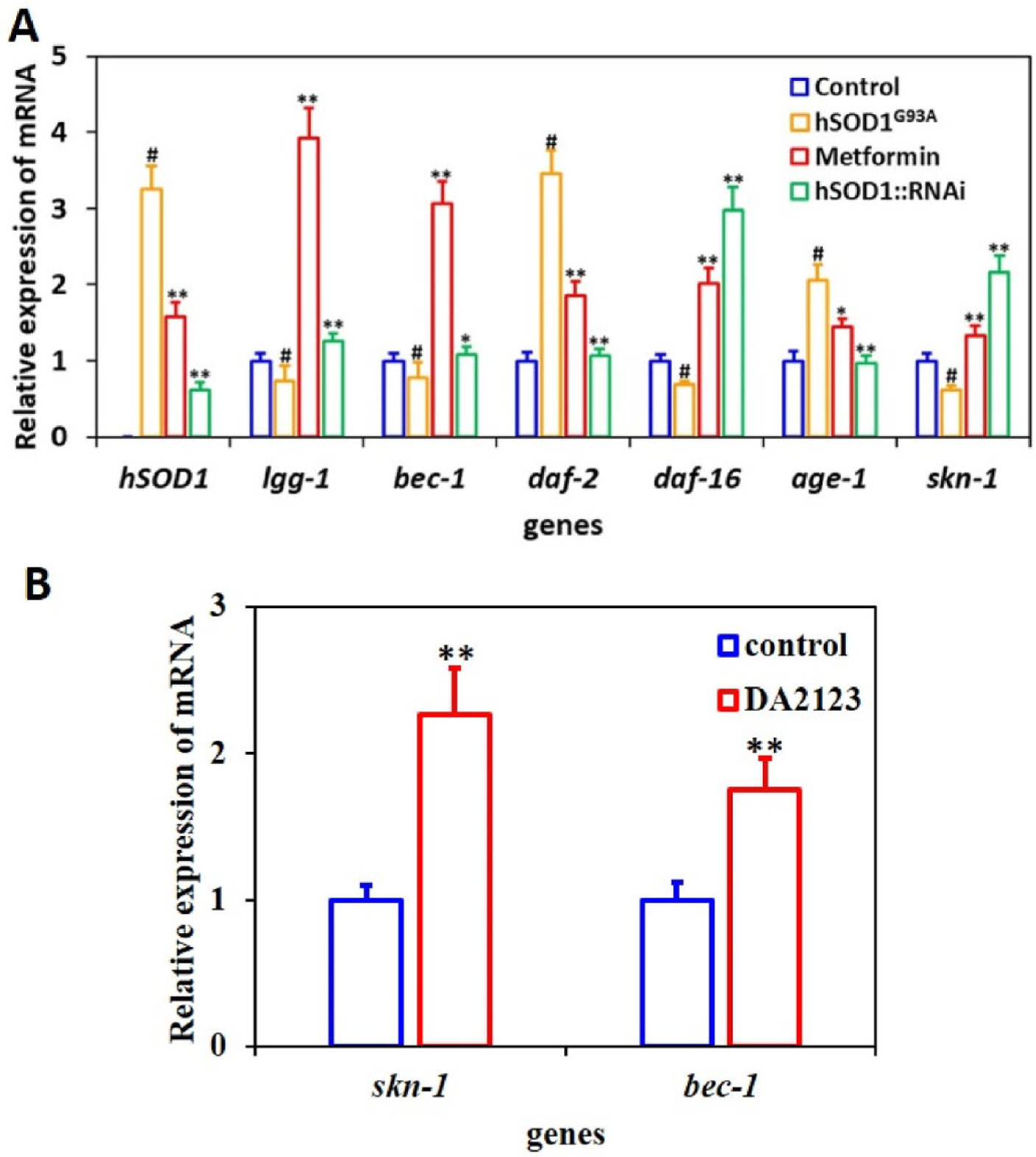
Metformin alters the expression of genes related to autophagy, aging, and stress response in a C. elegans model of ALS. (A) Quantification of gene expression in hS0DlG93A transgenic worms fed metformin or treated with RNAi, hSOD1G93A worms without treatment served as the control. Metformin reduced hSOD1 expression in hSOD1G93A worms and increased Lisal of genes related to autophagy, lifespan, and stress response. (B) Quantification of gene expression in DA2123 worms. #P<0.01 (vs. control), **P<0.001,*P<0.01 (vs. hSOD1G93A). N ~ 1000 worms per condition front 3 independent experiments.
3.4. Overexpression of daf-16 and lgg-1 extends lifespan and oxidative stress in hSOD1G93A worms
To investigate the roles of lgg-1 and daf-16 in ALS, we used hSOD1G93A::daf-16 and hSOD1 G93A::lgg-1 worms, and also generated the lgg-1::daf-16 double transgenic strain to examine the interaction between the two genes. The survival curves showed that the lifespan of hSOD1G93A::daf-16 worms was prolonged compared to hSOD1G93A worms, with an increase in median survival from 10 to 13 days (Fig. 3A). This suggests that activation of the daf-16 signaling pathway is involved in the extension of lifespan in ALS. We next examined whether changes in autophagy contributed to the reduction in longevity caused by hSOD1 mutation. The lifespan of hSOD1G93A::lgg-1 worms was extended relative to that of hSOD1G93A worms, with median survival increased from 10 to 13 days (Fig. 3B), implying that activation of lgg-1 promotes longevity in hSOD1 mutant worms by inducing the degradation of hSOD1 and thereby alleviating hSOD1 aggregation-induced neurotoxicity. In addition, while lgg-1 and daf-16 both prolonged lifespan, there was no additive effect when both genes were simultaneously overexpressed (Fig. 3C). We speculate that the two genes are involved in distinct signaling pathways and thus interact separately with hSOD1 to increase longevity.
Fig. 3.
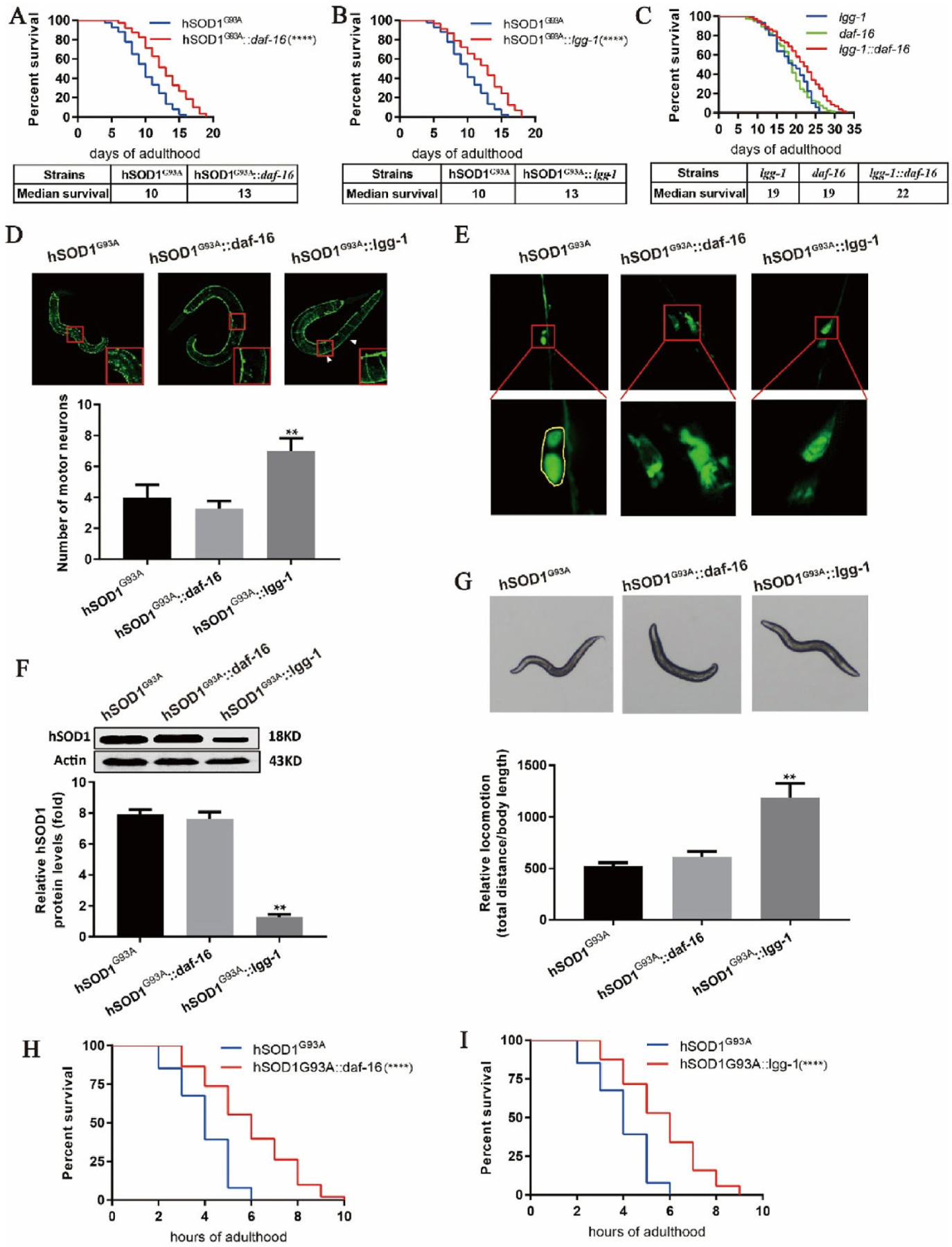
Overexpression of daf-16 and lgg-1 extends lifespan, alleviates locomotor deficits, and improves stress response in hSOD1G93A worms. (A-C) Survival curves for hSOD1G93A::daf-16, hSOD1G93A::lgg-1, and lgg-1::daf-16 worms. (D, E) Motor neuron survival and morphology in transgenic worms. Overexpression of lgg-1 but not daf-16 protected against hSOD1G93A-induced neurotoxicity (D) and restored motor neuron morphology (E) in hSOD1G93A worms. (F) Western blot analysis of protein extracts from hSOD1G93A transgenic strains. Lgg-1 overexpression reduced the level of hSOD1 protein in hSOD1G93A worms, whereas daf-16 overexpression had no effect. (G) Locomotor behavior in hSOD1 transgenic worms overexpressing daf-16 or lgg-1. Locomotion was restored in hSOD1G93A::lgg-1 worms, and to a lesser extent in hSOD1G93A::daf-16 worms. (H, I) Oxidative stress response in hSOD1G93A transgenic worms, hSOD1G93A worms over expressing lgg-1 and exposed to paraquat showed increased longevity compared to hSOD1G93A worms. Overexpression of daf-16 had no obvious effect on hSOD1 protein degradation, but partly restored the oxidative stress response that was diminished by hSOD1G93A mutation. ****P<0.0001, **P <0.001 (N ~ 100 worms per condition from 3 independent experiments).
We also examined the number and morphology of motor neurons, the hSOD1 protein level, and locomotor behavior and stress response in hSOD1G93A::daf-16 and hSOD1G93A::lgg-l worms. Overexpression of daf-16 in hSOD1G93A worms did not abrogate the toxic effects of hSOD1 mutation on motor neurons, while the number of neurons was slightly increased in hSOD1G93A::lgg-1 worms (Fig. 3D). Furthermore, compared to hSOD1G93A worms, motor neurons in hSOD1G93A::lgg-1 worms were morphologically normal (Fig. 3E). The results of the western blot analysis indicated that overexpression of daf-16 in hSOD1 mutants did not induce the degradation of hSOD1 protein, whereas lgg-1 overexpression decreased hSOD1 protein level (Fig. 3F). Moreover, hSOD1G93A::lgg-1 but not hSOD1G93A::daf-16 worms showed clear improvements in locomotion (Fig. 3G). Overexpression of both daf-16 and lgg-1 significantly enhanced the oxidative stress response in ALS model worms (Fig. 3H and I). Thus, lgg-1 overexpression induces the degeneration of hSOD1 protein and alleviates the toxic effects of hSOD1 mutation; however, daf-16 overexpression did not reduce the level of hSOD1 protein, implying that it prolongs lifespan in ALS through a different mechanism.
3.5. Genetic knockout of daf-16 and lgg-1 abrogates the beneficial effects of metformin
To investigate whether the beneficial effect of metformin on the longevity of ALS model worms involves daf-16 and lgg-1, we generated the genetic knockout lines hSOD1G93A::daf-16(RNAi) and hSOD1G93A::lgg-1(RNAi). We first examined the survival curves of worms treated with metformin and found that lifespan was not prolonged (Fig. 4A–B), suggesting that the effect of metformin on the lifespan of ALS model worms is mediated by daf-16 and lgg-1. We also examined the number and morphology of motor neurons in the two strains after treatment with metformin. The number of motor neurons in hSOD1G93A::lgg-1(RNAi) worms fed metformin was slightly increased but neuronal morphology remained abnormal (Fig. 4C–D). In lgg-1-deficient worms, neither the number nor the morphology of motor neurons was restored by metformin. Moreover, metformin did not improve stress tolerance in hSOD1G93A::daf-16(RNAi) and hSOD1G93A::lgg-16(RNAi) worms (Fig. 4E–F) and had no effect on their locomotion (Fig. 4G). Finally, we examined the expression of hSOD1 protein in the two strains by treatment with metformin and found that compared to hSOD1G93A worms, the decrease in hSOD1 protein levels observed in the absence of daf-16 and lgg-1 genes was abrogated (Fig. 4H). Taken together, the above results demonstrate that metformin extends lifespan and alleviates motor deficits in a C. elegans model of ALS by activating autophagy and the daf-16 pathway.
Fig. 4.
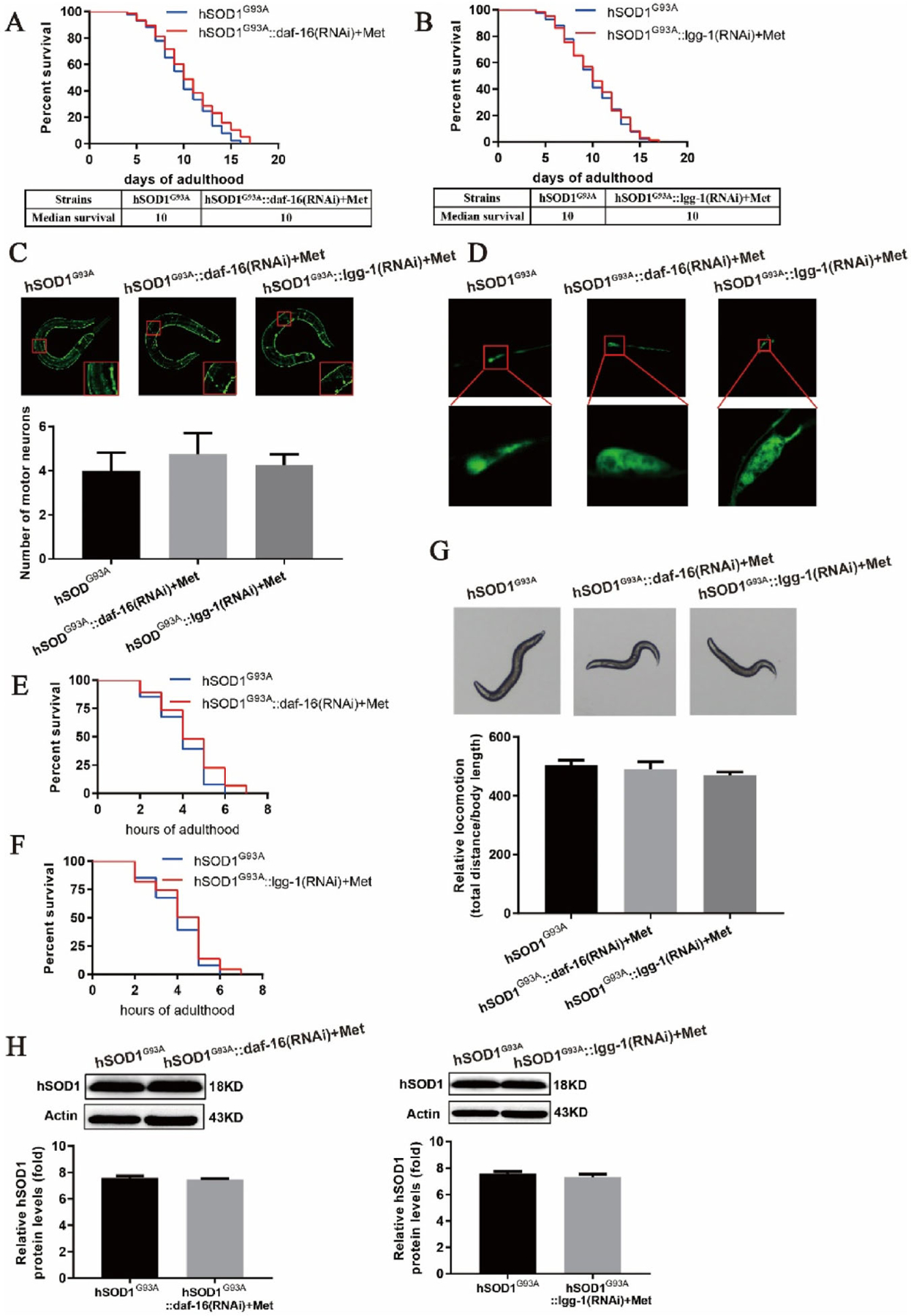
Metformin has no significant effect in daf-16- and lgg-1-deficient hSOD1 mutant C. elegcuis. (A, B) Survival curves for hSOD1G93A worms lacking daf-16 or lgg-1. Metformin did not prolong the lifespan of hSOD1G93A worms lacking the daf-16 or lgg-1 gene. (C, D) Motor neuron survival and morphology in daf-16 and lgg-1 knockout worms treated with metformin. Compared to hSOD1G93A worms, metformin did not alleviate the neurotoxicity (C) or defects in neuronal morphology (D) caused by hSOD1 mutation in hSOD1G93A worms lacking daf-16 and lgg-1. (E, F) Oxidative stress response in hSOD1G93A mutant daf-16 and lgg-1 knockout worms treated with metformin. Metformin did not prolong the lifespan of hSOD1G93A worms lacking the daf-16 or lgg-1 gene and exposed to paraquat. (G) Locomotor behavior in hSOD1G93A worms lacking daf-16 or lgg-1. Metformin had no significant effect on the locomotion of hSOD1G93A mutant daf-16 or lgg-1 knockout worms. (H) Western blot analysis of protein extracts from transgenic strains. hSOD1G93A protein level in hSOD1 mutant worms was not reduced by metformin treatment in the absence of the daf-16 or lgg-1 gene. N ~ 100 worms per condition from 3 independent experiments.
4. Conclusions
In summary, we developed a ALS model by generating a transgenic C. elegans strain that specifically overexpress human SOD1 G93A mutation in motor neurons to study the molecular pathogenesis and evaluate the therapeutic effects of metformin on ALS. hSOD1 mutant worms had a shorter lifespan, and displayed locomotor defects and paralysis in the adult stage, which was accompanied by a reduction in antioxidant activity. We found that treatment with metformin restored lifespan and locomotion and improved the stress response capacity of hSOD1G93A worms, providing evidence for the therapeutic potential of metformin in the treatment of ALS. We also found that treatment with metformin induced the upregulation of genes associated with autophagy. This suggests that metformin can activate the autophagy pathway by which the levels of hSOD1 mRNA is decreased, and promote the degradation of hSOD1 aggregates in motor neurons, thus abrogate their toxicity in ALS. We demonstrated that metformin or hSOD1 RNAi can upregulate daf-16 and downregulate daf-2 in hSOD1G93A worms; metformin can also reverse the negative effect of hSOD1 mutation on lifespan and protect against oxidative stress. These findings imply that the ability of metformin to reverse the decline in lifespan caused by hSOD1 mutation in the C. elegans ALS model involves the activation of the daf-16 pathway. Collectively, our data provide strong evidence for the regulatory effects of autophagy and the daf-16 signaling pathway in ALS (Fig. 5) and suggest that metformin may be an effective drug for delaying age-related ALS.
Fig. 5.
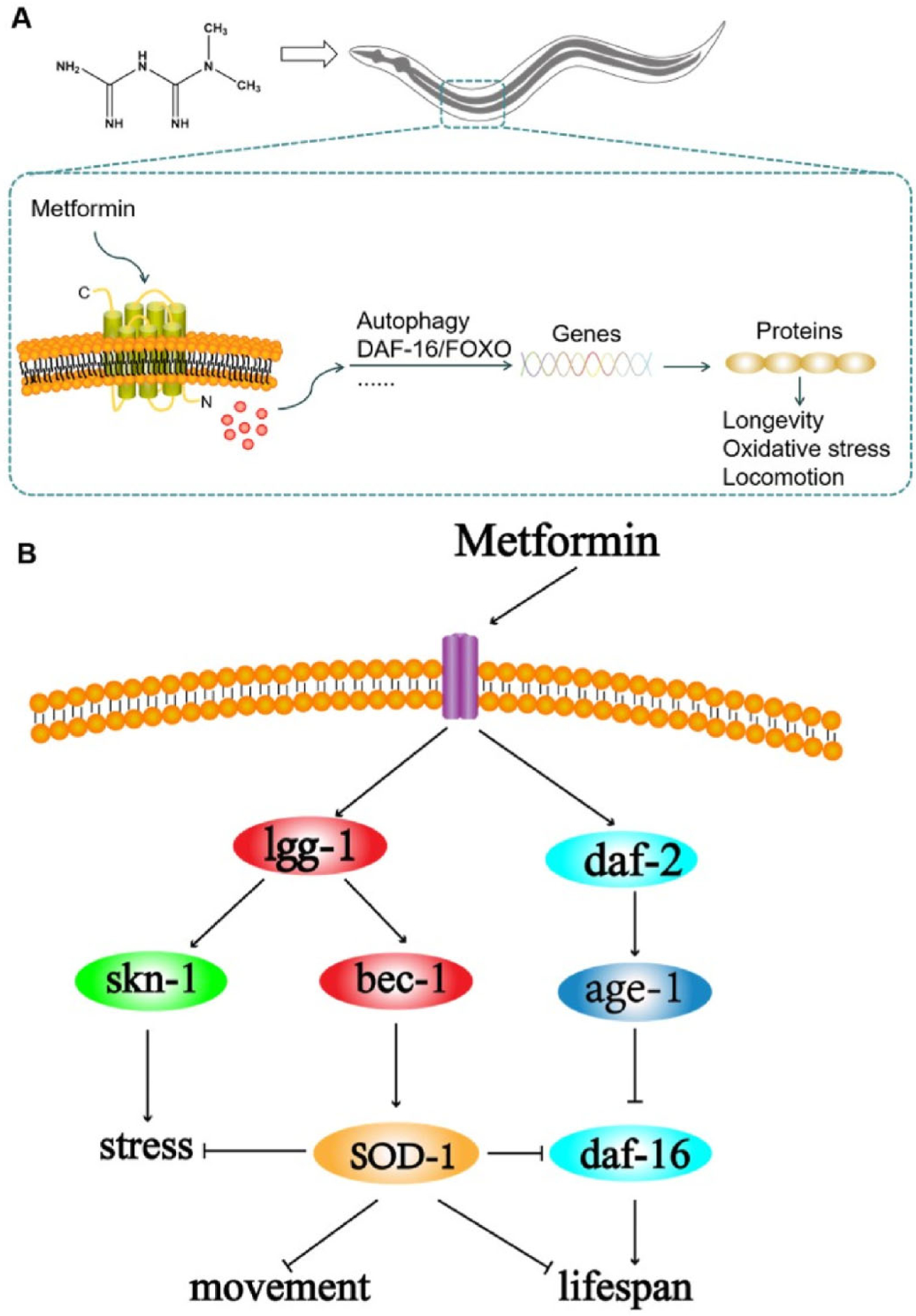
Schematic for the metformin extends lifespan and ameliorates motor deficits in an ALS C. elegans model. (A) The addition of metformin activates the autophagy and daf-16 pathways in C. elegans and upregulates autophagy and longevity genes in vivo, such as lgg-1 and daf-16. Increased autophagic activity promotes the degradation of toxic substances, enhances stress response and promotes longevity. (B) Schematic of interactions between the sod-1, lgg-1 and daf-16 genes in ALS C. elegans.
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (NSFC 81771521 and 31700853), the Guangdong Provincial Key R & D Program (2018B030337001), and in part by the Intramural Research Programs of National Institute on Aging, NIH (HC, ZIA AG000944, AG000928). Strains used in this work were provided by the Caenorhabditis Genetics Center (CGC), which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Abbreviations:
- SOD1
superoxide dismutase 1
- C. elegans
Caenorhabditis elegans
- ALS
amyotrophic lateral sclerosis
- AMPK
adenosine monophosphate-activated protein kinase
- mTOR
mammalian target of rapamycin
- PRDX-2
peroxiredoxin 2
- AD
Alzheimer’s disease
- PD
Parkinson’s disease
- lrk-1
Leucine-rich repeats, Ras-like domain, kinase 1
- NGM
nematode growth medium
- IPTG
isopropyl-β-D-thiogalactopyranoside
- IGF
insulin/insulin-like growth factor
Footnotes
Declaration of competing interest
The authors declare no conflicts of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.freeradbiomed.2022.01.030.
References
- [1].Patten SA, Parker JA, Wen XY, Drapeau P, Simple animal models for amyotrophic lateral sclerosis drug discovery, Expet Opin. Drug Discov. 11 (8) (2016) 797–804. [DOI] [PubMed] [Google Scholar]
- [2].Cleveland DW, Rothstein JD, From charcot to lou gehrig: deciphering selective motor neuron death in als, Nat. Rev. Neurosci. 2 (2001) 806–819. [DOI] [PubMed] [Google Scholar]
- [3].Chen S, Sayana P, Zhang X, Le W, Genetics of amyotrophic lateral sclerosis: an update, Mol. Neurodegener. 8 (2013) 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng H-X, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Bergh R.V.d, Hung W-Y, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, R.H.B. Jr, Mutations in Gu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis, Nature 362 (1993) 59–62. [DOI] [PubMed] [Google Scholar]
- [5].Gill C, Phelan JP, Hatzipetros T, Kidd JD, Tassinari VR, Levine B, Wang MZ, Moreno A, Thompson K, Maier M, Grimm J, Gill A, Vieira FG, SOD1-positive aggregate accumulation in the CNS predicts slower disease progression and increased longevity in a mutant SOD1 mouse model of ALS, Sci. Rep. 9 (1) (2019) 6724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hayashi Y, Homma K, Ichijo H, SOD1 in neurotoxicity and its controversial roles in SOD1 mutation-negative ALS, Adv. Biol. Regul. 60 (2016) 95–104. [DOI] [PubMed] [Google Scholar]
- [7].Senchuk MM, Dues DJ, Schaar CE, Johnson BK, Madaj ZB, Bowman MJ, Winn ME, Van Raamsdonk JM, Activation of DAF-16/FOXO by reactive oxygen species contributes to longevity in long-lived mitochondrial mutants in Caenorhabditis elegans, PLoS Genet. 14 (3) (2018), e1007268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Karabiyik C, Frake RA, Park SJ, Pavel M, Rubinsztein DC, Autophagy in ageing and ageing-related neurodegenerative diseases, Ageing Neurodegener. Dis. 1 (2021), 2. [Google Scholar]
- [9].De Haes W, Frooninckx L, Van Assche R, Smolders A, Depuydt G, Billen J, Braeckman BP, Schoofs L, Temmerman L, Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2, Proc. Natl. Acad. Sci. Unit. States Am. 111 (24) (2014) E2501–E2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, Gomes AP, Ward TM, Minor RK, Blouin MJ, Schwab M, Pollak M, Zhang Y, Yu Y, Becker KG, Bohr VA, Ingram DK, Sinclair DA, Wolf NS, Spindler SR, Bernier M, de Cabo R, Metformin improves healthspan and lifespan in mice, Nat. Commun. 4 (2013) 2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wu L, Zhou B, Oshiro-Rapley N, Li M, Paulo JA, Webster CM, Mou F, Kacergis MC, Talkowski ME, Carr CE, Gygi SP, Zheng B, Soukas AA, An ancient, unified mechanism for metformin growth inhibition in C. elegans and cancer, Cell 167 (7) (2016) 1705–1718, e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chen J, Ou Y, Li Y, Hu S, Shao L-W, Liu1 Y, Metformin extends C. elegans lifespan through lysosomal pathway, Elife 6 (2017) 1–17, e31268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cabreiro F, Au C, Leung KY, Vergara-Irigaray N, Cocheme HM, Noori T, Weinkove D, Schuster E, Greene ND, Gems D, Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism, Cell 153 (1) (2013) 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pryor R, Norvaisas P, Marinos G, Best L, Thingholm LB, Quintaneiro LM, De Haes W, Esser D, Waschina S, Lujan C, Smith RL, Scott TA, Martinez-Martinez D, Woodward O, Bryson K, Laudes M, Lieb W, Houtkooper RH, Franke A, Temmerman L, Bjedov I, Cocheme HM, Kaleta C, Cabreiro F, Host-microbe-drug-nutrient screen identifies bacterial effectors of metformin therapy, Cell 178 (6) (2019) 1299–1312 e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zu T, Guo S, Bardhi O, Ryskamp DA, Li J, Khoramian Tusi S, Engelbrecht A, Klippel K, Chakrabarty P, Nguyen L, Golde TE, Sonenberg N, Ranum LPW, Metformin inhibits RAN translation through PKR pathway and mitigates disease inC9orf72ALS/FTD mice, Proc. Natl. Acad. Sci. Unit. States Am 117 (31) (2020) 18591–18599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].McColl G, Roberts BR, Gunn AP, Perez KA, Tew DJ, Masters CL, Barnham KJ, Cherny RA, Bush AI, The Caenorhabditis elegans A beta 1–42 model of Alzheimer disease predominantly expresses A beta 3–42, J. Biol. Chem. 284 (34) (2009) 22697–22702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cooper JF, Van Raamsdonk JM, Modeling Parkinson’s disease in C. elegans, J. Parkinsons Dis 8 (1) (2018) 17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Aaron C, Beaudry G, Parker JA, Therrien M, Maple syrup decreases TDP-43 proteotoxicity in a Caenorhabditis elegans model of amyotrophic lateral sclerosis (ALS), J. Agric. Food Chem. 64 (17) (2016) 3338–3344. [DOI] [PubMed] [Google Scholar]
- [19].Baskoylu SN, Yersak J, O’Hern P, Grosser S, Simon J, Kim S, Schuch K, Dimitriadi M, Yanagi KS, Lins J, Hart AC, Single copy/knock-in models of ALS SOD1 in C. elegans suggest loss and gain of function have different contributions to cholinergic and glutamatergic neurodegeneration, PLoS Genet. 14 (10) (2018), e1007682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li J, Li T, Zhang X, Tang Y, Yang J, Le W, Human superoxide dismutase 1 overexpression in motor neurons of Caenorhabditis elegans causes axon guidance defect and neurodegeneration, Neurobiol. Aging 35 (4) (2014) 837–846. [DOI] [PubMed] [Google Scholar]
- [21].Yang YQ, Zheng YH, Zhang CT, Liang WW, Wang SY, Wang XD, Wang Y, Wang TH, Jiang HQ, Feng HL, Wild-type p53-induced phosphatase 1 down-regulation promotes apoptosis by activating the DNA damage-response pathway in amyotrophic lateral sclerosis, Neurobiol. Dis. 134 (2020) 104648. [DOI] [PubMed] [Google Scholar]
- [22].Zhang J, Liu Y, Liu X, Li S, Cheng C, Chen S, Le W, Dynamic changes of CX3CL1/CX3CR1 axis during microglial activation and motor neuron loss in the spinal cord of ALS mouse model, Transl. Neurodegener. 7 (2018) 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang X, Chen S, Lu K, Wang F, Deng J, Xu Z, Wang X, Zhou Q, Le W, Zhao Y, Verapamil ameliorates motor neuron degeneration and improves lifespan in the SOD1G93A mouse model of ALS by enhancing autophagic flux, Aging Dis 10 (6) (2019) 1159–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Smita SS, Raj Sammi S, Laxman TS, Bhatta RS, Pandey R, Shatavarin IV elicits lifespan extension and alleviates Parkinsonism in Caenorhabditis elegans, Free Radic. Res. 51 (11–12) (2017) 954–969. [DOI] [PubMed] [Google Scholar]
- [25].Brenner S, The genetics of Caenorhabditis elegans, Genetics 77 (1973) 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Xiao R, Chun L, Ronan EA, Friedman DI, Liu J, Xu XZ, RNAi interrogation of dietary modulation of development, metabolism, behavior, and aging in C. elegans, Cell Rep. 11 (7) (2015) 1123–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang J, Farr GW, Hall DH, Li F, Furtak K, Dreier L, Horwich AL, An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans, PLoS Genet. 5 (1) (2009), e1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kumsta C, Chang JT, Schmalz J, Hansen M, Hormetic heat stress and HSF-1 induce autophagy to improve survival and proteostasis in C. elegans, Nat. Commun. 8 (2017) 14337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gusarov I, Pani B, Gautier L, Smolentseva O, Eremina S, Shamovsky I, Katkova-Zhukotskaya O, Mironov A, Nudler E, Glycogen controls Caenorhabditis elegans lifespan and resistance to oxidative stress, Nat Commun. 8 (2017) 15868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Uno M, Nishida E, Lifespan-regulating genes in C. elegans, NPJ Aging Mech. Dis. 2 (2016) 16010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wu F, Watanabe Y, Guo XY, Qi X, Wang P, Zhao HY, Wang Z, Fujioka Y, Zhang H, Ren JQ, Fang TC, Shen YX, Feng W, Hu JJ, Noda NN, Zhang H, Structural basis of the differential function of the two C. elegans Atg8 homologs, LGG-1 and LGG-2, in autophagy, Mol. Cell 60 (6) (2015) 914–929. [DOI] [PubMed] [Google Scholar]
- [32].Ames K, Da Cunha DS, Gonzalez B, Konta M, Lin F, Shechter G, Starikov L, Wong S, Bulow HE, Melendez A, A non-cell-autonomous role of BEC-1/BECN1/beclin1 in coordinating cell-cycle progression and stem Cell proliferation during germline development, Curr. Biol. 27 (6) (2017) 905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Blackwell TK, Steinbaugh MJ, Hourihan JM, Ewald CY, Isik M, SKN-1/Nrf, stress responses, and aging in Caenorhabditis elegans, Free Radic. Biol. Med. 88 (Pt B) (2015) 290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.