

Abstract
Studies in recent years have significantly expanded, refined, and redefined the repertoire of transporters and other proteins involved in iron and manganese (Mn) transport and homeostasis. In this review, we discuss highlights of the recent literature on iron and Mn transport, focusing on the roles of membrane transporters and related proteins. Studies are considered from the vantage point of main organs, tissues, and cell types that actively control whole-body iron or Mn homeostasis, with emphasis on studies in which in vivo metal transport was measured directly or implicated by using knockout mouse models. Overviews of whole-body and cellular iron and Mn homeostasis are also provided to give physiological context for key transporters and to highlight how they participate in the uptake, intracellular trafficking, and efflux of each metal. Important similarities and differences in iron and Mn transport are noted, and future research opportunities and challenges are identified.
Keywords: Iron, Manganese, Transport protein, Membrane transport, SLC39, SLC30
1. Introduction
Iron and manganese (Mn), the first and third most abundant transition metal elements in the Earth’s crust, are similar in many respects. As transition elements, they can exist in multiple oxidation states and are redox active, a property that is harnessed and exploited in enzymes such as Mn-dependent superoxide dismutase (MnSOD) and catalase, which utilizes iron (as heme) in its active site. In their reduced state, both metals are divalent (i.e., Fe2+ and Mn2+), water soluble, and are thus available for transport by divalent metal-ion transporters (Table 1). In their oxidized trivalent state (i.e., Fe3+ and Mn3+), they readily precipitate unless bound to ligands such as transferrin, the main iron- and Mn-binding protein in plasma. Given these similarities, it is hardly surprising that DMT1 (divalent metal-ion transporter-1), commonly referred to as an iron transporter because of its essential role in intestinal iron absorption, can also transport Mn2+, as shown when it was discovered in 1997. However, whether DMT1 has an essential role in intestinal Mn absorption—or not—remained in question for nearly 20 years until definitive studies were performed in 2015 [1]. Recent years have witnessed the identification of three essential Mn transporters in humans: SLC30A10, SLC39A8, and SLC39A14 [2]. Two of these (SLC39A14 and SLC39A8) can also transport iron [3,4], with SLC39A14 functioning as an essential iron transporter in vivo in iron overload conditions [5]. Thus, considering that some “iron” transporters and “Mn” transporters may have a dual function, a review of both iron and Mn transport may help to bring into relief similarities, differences, and possible points of convergence in the transport and homeostasis of the two metals. The aim of this review therefore is to summarize and highlight recent advances in mammalian iron and Mn transport, focusing on transport proteins and their roles in tissues and cell types involved in maintaining whole-body homeostasis of each metal. Emphasis is placed on advances made in the past 7 years, but due to the breadth of the literature, the topics and references are not comprehensive, but rather selective and representative of the authors’ point of view. Moreover, although the majority of body iron in mammals is in the form of heme (e.g., in hemoglobin, myoglobin, cytochromes, and heme-containing enzymes), the present review focuses on the transport of iron as non-heme iron, mainly as the hydrated Fe2+ ion. For reviews of heme transport and trafficking, the reader is referred elsewhere [6,7]. Other topics notably omitted from the review include iron and Mn transport across the blood-brain barrier and in the brain, which have been covered recently and comprehensively by others [8–10].
Table 1.
Main proteins involved in iron and/or Mn transport and trafficking.
| Protein name (aliases) | Gene symbol | Subcellular localization | Primary metal substrate | Secondary metal substrate(s) | Function | Phenotypes of KO or mutant mice | Human disease and hallmarks of phenotype |
|---|---|---|---|---|---|---|---|
| Transferrin | TF | – | Fe3+ | Mn3+ | Plasma transport | Severe anemia; iron overload | Atransferrinemia. (OMIM 209300). Severe anemia, iron overload |
| Transferrin receptor 1 | TFRC | Plasma membrane, endosome | – | – | Uptake | Embryonic lethal | – |
| DMT1 (Nramp2) | SLC11A2 | Plasma membrane, endosome, lysosome | Fe2+ | Mn2+ | Uptake | Severe anemia | Anemia, hypochromic microcytic, iron overload (OMIM 206100) |
| Ferroportin | SLC40A1 | Plasma membrane | Fe2+ | Mn2+? | Export | Embryonic lethal | Hemochromatosis, Type 4; tissue iron overload (OMIM 606069) |
| DCYTB | CYBRD1 | Plasma membrane | Fe3+ | Cu2+ | Uptake | None | – |
| Steap3 | STEAP3 | Endosome | Fe3+ | Cu2+ | Uptake | Moderate anemia | Anemia |
| Hephaestin | HEPH | Plasma membrane | Fe2+ | Mn2+ | Export | Microcytic anemia | – |
| Ceruloplasmin | CP | – | Fe2+ | Mn2+ | Export | Mild anemia | Aceruloplasminemia; iron overload (OMIM 604290) |
| PCBP1 PCBP2 | PCBP1 PCBP2 | Cytosolic | Fe2+ | – | Cytosolic transport | Embryonic lethal; anemia | – |
| Mitoferrin 1 Mitoferrin 2 | SLC25A37 SLC25A28 | Inner mitochondrial membrane | Fe2+ | Mn2+ | Uptake | embryonic lethal; Inducible KO causes anemia | – |
| SLC39A14 (ZIP14) | SLC39A14 | Plasma membrane, endosome, lysosome | Mn2+ | Fe2+, Zn2+ | Uptake | Mn overload except for liver; Impaired NTBI uptake | Hypermanganesemia with dystonia-2, HMNDYT2 (OMIM 617013). Brain Mn overload and parkinsonism |
| SLC39A8 (ZIP8) | SLC39A8 | Plasma membrane, endosome, lysosome | Mn2+ | Fe2+, Zn2+ | Uptake | Embryonic lethal; Inducible KO results in Mn deficiency | Congenital disorder of glycosylation, type IIn (OMIM 616721); hypomanganesemia, skeletal abnormalities |
| SLC30A10 (ZnT10) | SLC30A10 | Plasma membrane, Golgi, endosome | Mn2+ | – | Export | Mn overload in all tissues | Hypermanganesemia with dystonia-1, HMNDYT1 (OMIM 613280) |
| ATP13A2 (Park9) | ATP13A2 | Lysosome | Mn2+ | Zn2+ | Export | Increased sensitivity to Mn exposure | Kufor-Rakeb syndrome (OMIM 606693); juvenile- onset atypical Parkinson’s disease |
| TMEM165 | TMEM165 | Golgi | Ca2+ | Mn2+ | Export | Skeletal abnormalities; Impaired Ca, Mn transport into milk | Congenital disorder of glycosylation, type IIk (OMIM 614727); skeletal anomalies |
| SPCA1 | ATP2C1 | Golgi | Ca2+ | Mn2+ | Export | Impaired Ca transport into milk | Hailey-Hailey disease (OMIM 169600); blistering skin |
| LTCC | CACNA1C | Plasma membrane | Ca2+ | Fe2+, Mn2+ | Uptake | Embryonic lethal | Cardiac abnormalities; Timothy syndrome |
2. Overview of iron transport
2.1. Whole-body iron transport, metabolism, and distribution
Dietary non-heme iron in the small intestine is taken up as Fe2+ via DMT1 on the apical membrane of enterocytes (Fig. 1). As a proton-coupled symporter, DMT1 functions optimally in the acidic microclimate at the absorptive surface of the proximal small intestine. Most dietary non-heme iron exists as Fe3+ and therefore must be reduced to Fe2+ prior to uptake via DMT1. Ferrireduction in the intestinal brush border is catalyzed by apical DCYTB (cytochrome B reductase 1) and/or other mechanisms. Iron taken up into the enterocyte moves through the cytosol to the basolateral membrane, where the metal is transported into capillary blood via ferroportin, the sole known iron export protein. Fe2+ transported via ferroportin is oxidized to Fe3+ and becomes bound by transferrin, the primary transporter of iron in the plasma. Ferroxidation is catalyzed by enterocyte hephaestin, a membrane-anchored ferroxidase, and/or ceruloplasmin in the plasma. Transferrin-bound iron in the capillary blood travels to the liver via the portal vein. Under normal circumstances, <5% of transferrin-bound iron in portal blood is taken up by the liver during first-pass extraction. Hepatic uptake of transferrin-bound iron is usually low because hepatocytes and hepatic macrophages (Kupffer cells), which comprise >90% of the cell mass of the liver, are normally iron replete and thus express very little transferrin receptor 1 (TFR1), the primary uptake mechanism for transferrin. Thus, most transferrin-bound iron originating from the gut passes through the liver and enters the systemic circulation, where it delivers iron to cells in proportion to their levels of TFR1. In an adult human, an estimated 80% of total body cellular TFR1 is located in developing erythroid cells in bone marrow. The assimilation of iron from endocytosed transferrin requires DMT1 and the endosomal reductase STEAP3. Each day, erythroid precursor cells take up approximately 25 mg of iron, which is incorporated into protoporphyrin IX to form heme, mostly for hemoglobin. At the end of their ~120-day lifespan, red blood cells are cleared from the circulation by resident macrophages of the liver (Kupffer cells), spleen, and bone marrow—collectively referred to as the “reticuloendothelial system”. Senescent red blood cells are internalized and degraded within the phagolysosome, resulting in the breakdown of hemoglobin and the release of heme, which is transported into the cytosol via HRG1 (see Hamza and Reddy in this volume) and catabolized, releasing iron. The liberated Fe2+ is transported by ferroportin out of the macrophage and into the circulation, where it is oxidized (by ceruloplasmin) and loaded onto transferrin, which returns the metal to the bone marrow for reincorporation into newly synthesized heme, thus efficiently recycling red cell iron. For additional details of iron and heme handling in enterocytes, hepatocytes, erythroid precursor cells, and macrophages, the reader is referred elsewhere [11].
Fig. 1.
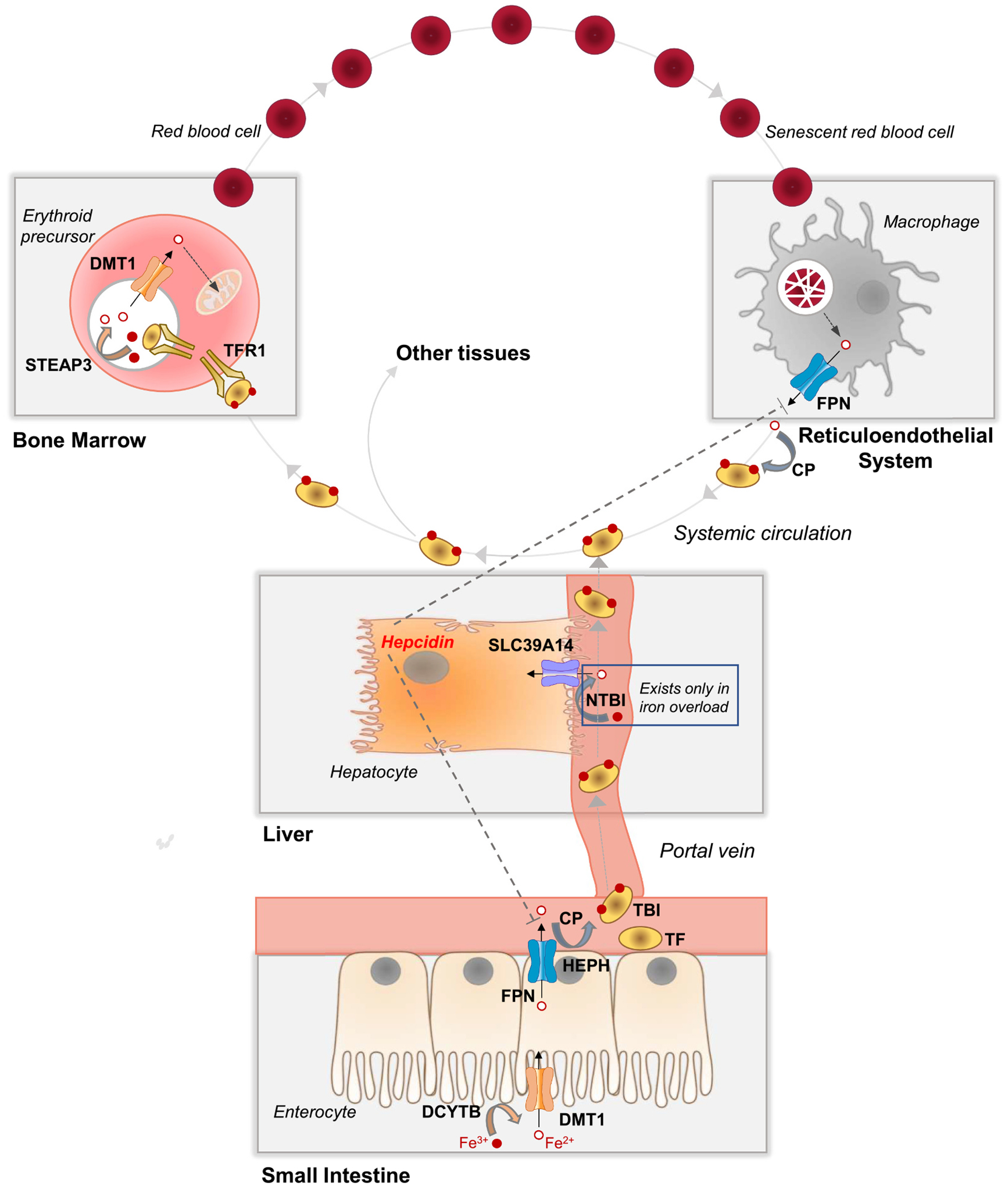
Whole-body iron transport, metabolism, and distribution. In the small intestine, dietary non-heme iron, which exists mainly as Fe3+, is converted to Fe2+ by a reductase (e.g., DCYTB, cytochrome B reductase 1) prior to transport into the enterocyte via DMT1 (divalent metal-ion transporter-1) at the apical membrane. FPN (ferroportin) at the enterocyte basolateral membrane transports Fe2+ into capillary blood, where it is oxidized by a ferroxidase (e.g., HEPH, hephaestin, or CP, ceruloplasmin) to Fe3+, which binds to plasma TF (transferrin) to become TBI (transferrin-bound iron). TBI travels via the portal vein through the liver and into systemic circulation, where it delivers iron to the bone marrow and other tissues. Bone marrow erythroid precursor cells take up TBI via TFR1 (transferrin receptor 1)-mediated endocytosis. Endosomal acidification and reduction by STEAP3 (six-transmembrane epithelial antigen of the prostate 3) releases Fe2+, which is transported out of the endosome via DMT1. Fe2+ is taken up via mitoferrin 1 into the mitochondria, where it is incorporated into protoporphyrin IX to become heme, which is used mainly for hemoglobin synthesis in immature red blood cells. Red blood cells circulate for ~120 days and are then cleared from the circulation via macrophages of the reticuloendothelial system (i.e., macrophages of the liver, spleen, and bone marrow). Senescent red blood cells are taken up by macrophages via phagocytosis. Internalized red blood cells are degraded in the lysosome and heme is degraded to release Fe2+, which is transported via FPN into the plasma, where it binds to TF, thereby recycling red blood cell iron. In iron overload, the iron-carrying capacity of TF become exceeded giving rise to NTBI (non-transferrin-bound iron), a pathological form of iron that is a major contributor to tissue iron loading. Additional details are described in Section 2.1.
A notable aspect of mammalian iron metabolism—unique among nutritional trace elements—is that whole-body iron balance is maintained nearly exclusively by regulating intestinal iron absorption. This is because the body lacks active mechanisms for excreting iron. Iron absorption is regulated chiefly by hepcidin, a peptide hormone produced by hepatocytes. Circulating hepcidin inhibits iron absorption by binding to intestinal ferroportin, causing it to be internalized and degraded, thereby preventing iron from entering portal blood. Hepcidin also binds to ferroportin in macrophages, inhibiting iron release into the plasma. Overproduction of hepcidin causes hypoferremia and the anemia of inflammation/chronic disease. Impaired hepcidin production or function results in increased iron absorption and hyperferremia, leading to the iron overload disorder hereditary hemochromatosis (see Muck-enthaler in this volume).
2.2. Cellular iron transport and trafficking
Most cell types acquire iron primarily from transferrin via receptor-mediated endocytosis (Fig. 2). When diferric transferrin (TF-(Fe3+)2) binds to transferrin receptor 1 (TFR1) at the cell surface, the ligand-receptor complex is internalized into endosomes. Acidification of the endosome causes transferrin to release its Fe3+, which is converted to Fe2+ by an endosomal reductase and transported into the cytosol via DMT1. Iron entering the cytosol binds to PCBP1 and PCBP2, iron chaperones that deliver iron to ferritin (for storage) and to iron-containing enzymes. The mobilization of iron from ferritin is mediated by NCOA4, a selective cargo receptor that binds to ferritin, delivering it to an autophagosome and eventually to the lysosome, where it is degraded. Iron retrieved from degraded ferritin is transported out of the lysosome via DMT1. Iron is taken up into the mitochondria via mitoferrin 1 and mitoferrin 2. Iron export is mediated by ferroportin.
Fig. 2.
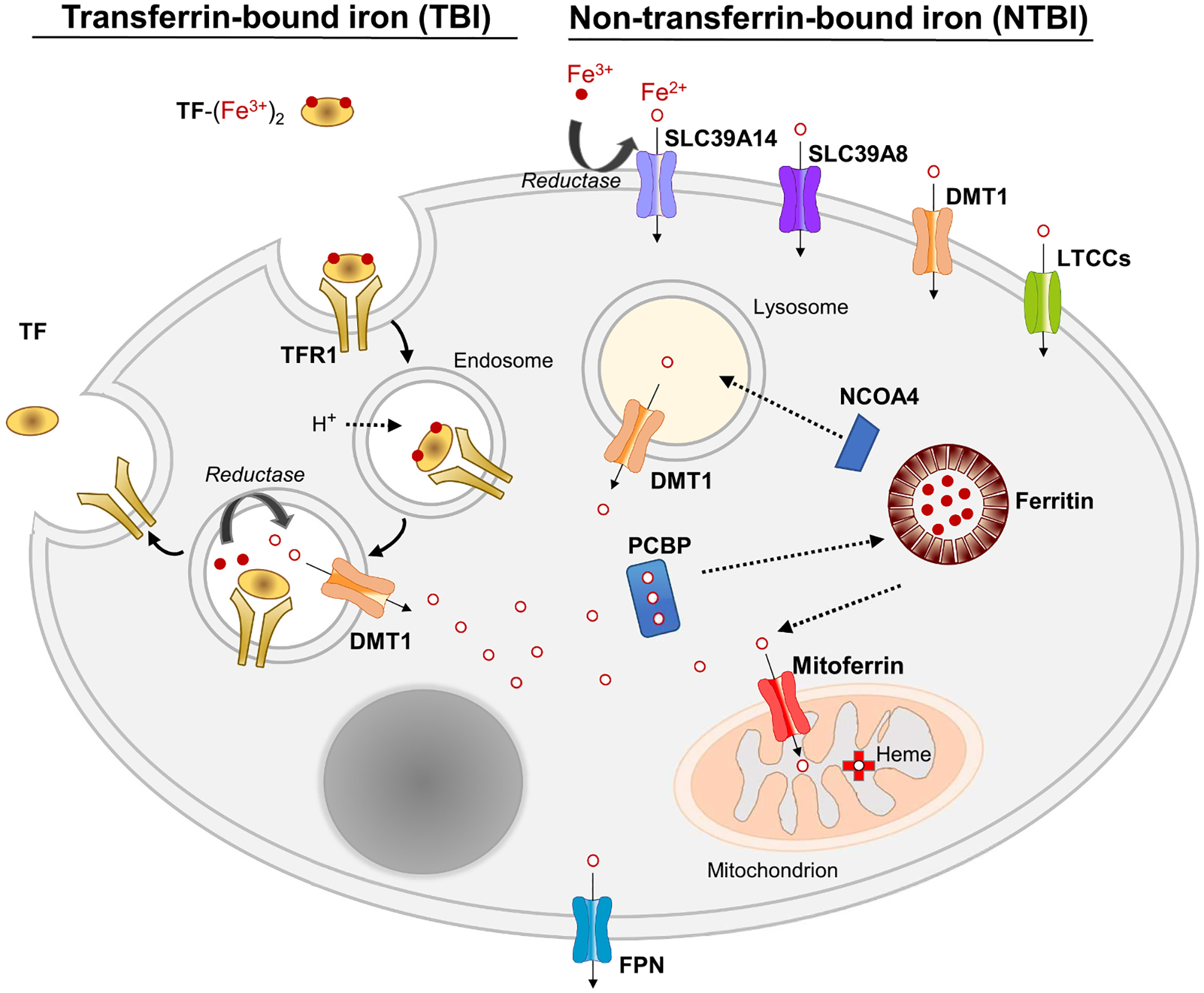
Cellular iron transport and trafficking. A model of a generic cell is depicted. Cells can take up iron from TF (transferrin) as TBI (transferrin-bound iron) or from NTBI (non-transferrin-bound iron). Uptake of TBI is mediated by TFR1 (transferrin receptor 1)-mediated endocytosis as described in Section 2.2. NTBI uptake can be mediated, depending on the cell type, by SLC39A14, SLC39A8, DMT1, or LTCCs (L-type Ca2+ channels). Since most NTBI in plasma is present as Fe3+ species, and NTBI is transported as Fe2+, a reduction step, likely catalyzed by a reductase, precedes transport. Iron trafficking in the cytosol is mediated by PCBP1/2 (Poly (RC) Binding Protein 1/2), which delivers iron to ferritin, the iron storage protein, and several iron-containing enzymes (not shown). Iron is taken up into the mitochondrion via mitoferrin 1/2. Mobilization of iron from ferritin storage is mediated by NCOA4 (Nuclear Receptor Coactivator 4), which directs ferritin to the lysosome for degradation. Cellular iron export is mediated by FPN (ferroportin). Additional details are described in Section 2.2.
When cellular iron concentrations increase, the expression of TFR1 decreases to limit further uptake of iron, preventing it from accumulating to potentially toxic levels. However, some cell types (e.g., hepatocytes, pancreatic acinar cells, cardiomyocytes, and neurons) possess mechanisms to take up non-transferrin-bound iron (NTBI), which appears in the plasma when the iron-carrying capacity of transferrin is exceeded, such as during iron overload. The term “NTBI” refers to a poorly characterized, heterogeneous, and variable pool of labile Fe3+ bound to small molecules (e.g., citrate, acetate, phosphate) or to higher molecular weight proteins such as albumin. Operationally, NTBI has recently been defined as “circulating iron that is not bound to transferrin, ferritin, or heme”. Transport proteins that have been shown to mediate the uptake of NTBI include SLC39A14, SLC39A8, DMT1, and LTCCs (L-type Ca2+ channels) (reviewed in [12]).
3. Recent advances in iron transport
3.1. Intestinal iron absorption
The main proteins that participate in intestinal iron transport were identified by the use of mutant or knockout mouse models between the years 1997 and 2001 during what has been referred to as the “golden age of iron biology” [13]. By 2005, the current model of iron absorption (Fig. 1) was firmly established by the generation of intestine-specific Dmt1 and Fpn knockout (KO) mice, which were found to develop severe iron-deficiency anemia within 2 months of age [14,15]. In recent years, studies have utilized a variety of conditional KO mouse models to better define the enterocyte-specific contributions of proteins to intestinal iron transport and overall iron economy, while other studies have revealed roles for other proteins in the fine tuning of iron absorption. These animal models, as well as other key mouse models discussed in the present review, are summarized in Table 2.
Table 2.
Key recent mouse model studies of iron and/or Mn transport and trafficking.
| Protein | Mouse model(s) | Metal(s) studied | Major finding(s) | Key reference (s) |
|---|---|---|---|---|
| DMT1 | Intestine-specific Dmt1 KO | Fe, Mn | DMT1 is essential for intestinal iron absorption, but is not required for Mn absorption. | [1] |
| DMT1 | Hepatocyte-specific Dmt1 KO | Fe | Hepatocyte DMT1 is dispensable for the uptake of plasma NTBI and iron loading of the liver. | [26] |
| DMT1 | Hepatocyte-specific Dmt1 KO | Fe | Hepatocyte DMT1 is required for iron mobilization from the liver. In this process, DMT1 likely functions in the lysosome, where it transports iron from degraded ferritin into the cytosol. | [29] |
| SLC39A14 | Slc39a14 KO, Hfe;Slc39a14 DKO, Hjv;Slc39a14 DKO | Fe | SLC39A14 is required for the uptake of plasma NTBI and iron loading of the liver (hepatocytes) and pancreas (acinar cells) in mouse models of genetic and dietary iron overload. | [5] |
| SLC39A14 | Slc39a14 KO | Mn | SLC39A14 is required for the uptake of plasma Mn by the liver and pancreas and for gastrointestinal Mn excretion. Mn accumulates in the blood, brain, bone, and kidney. | [71–73] |
| SLC39A14 | Intestine-specific Slc39a14 KO | Mn | Intestinal SLC39A14 localizes to the basolateral membrane of enterocytes, where it takes up Mn from the plasma to promote Mn elimination. | [75,85] |
| SLC39A8 | Slc39a8 KO, Hepatocyte-specific Slc39a8 KO | Mn | SLC39A8 localizes to the hepatocyte apical membrane, where it reclaims Mn from the bile to protect against Mn deficiency. | [68] |
| SLC30A10 | Slc30a10 KO | Mn | SLC30A10 deficiency results in marked increases in Mn levels in liver, blood, and brain, similar to human patients. | [58] |
| SLC30A10 | Slc30a10 KO, Slc30a10-GFP, Hepatocyte-specific Slc30a10 KO, Enterocyte-specific Slc30a10 KO, Hepatocyte- and enterocyte-specific Slc30a10 DKO | Mn | SLC30A10 localizes to the hepatocyte apical membrane, where it exports Mn into bile, and to the duodenal enterocyte apical membrane, where it exports Mn into the lumen to promote Mn elimination. Combined Slc30a10 deficiency in the liver and enterocyte indicate that additional sites of SLC30A10 contribute to Mn homeostasis. | [62] |
| Ferroportin | Erythroblast-specific ferroportin KO | Fe | Erythrocyte ferroportin functions to export iron in mature red blood cells. | [41] |
| Ferroportin | Tmprss6 KO (hepcidin overexpressing), Intestine-specific ferroportin KO | Fe, Mn | Ferroportin plays a major role in iron, but not Mn, transport in vivo. Intestinal ferroportin is not required for Mn absorption. | [84] |
| PCBP1 | Pcbp1 KO | Fe | PCBP1 in erythroid cells delivers iron to ferritin and is required for erythroid iron homeostasis. | [33] |
| Hephaestin, Ceruloplasmin | Heph KO, Cp KO, Heph;Cp DKO, Intestine-specific Heph; Cp DKO | Fe | Hephaestin and ceruloplasmin are not essential for intestinal iron absorption but are required for normal systemic iron distribution. | [21] |
Using intestine-specific Dmt1 KO mice, Shawki et al. [1] determined that loss of intestinal Dmt1 decreases enterocyte uptake and absorption of an intragastric dose of 59Fe by ~90% relative to control mice, thus formally demonstrating that the primary defect in these mice is impaired iron uptake at the enterocyte apical membrane. DMT1 functions as a proton-coupled metal-ion symporter in which the proton motive force is harnessed to drive cytoplasmic import of metal ions [16]. The protons in the acidic microclimate of the brush border are likely provided, at least in part, by Na+/H+ exchanger 3 (NHE3), as Nhe3 KO mice show impairments in the absorption of orally administered 59Fe and depleted iron stores [17]. NHE3 localizes to the apical membrane of surface enterocytes, where it is primarily responsible for intestinal sodium absorption.
3.1.1. Iron absorption during the suckling period
Although the primary mechanisms mediating iron absorption are well understood in adult animals, less has been known about how these mechanisms contribute to iron absorption during the suckling period when the efficiency of absorption is very high. Intestine-specific Fpn KO mice examined at 15 days of age were found to absorb only 10% of an 59Fe test dose compared to 73% in wild-type control animals, indicating that ferroportin plays a major role in intestinal iron absorption during suckling [18]. Intestine-specific inactivation of the basolateral ferroxidase hephaestin (Heph) resulted in ~30% reductions in hemoglobin levels and 59Fe absorption in 21-day-old mice, indicating that intestinal HEPH is required for optimal iron absorption at weaning and during suckling [19]. The observation that intestine-specific Dmt1 KO mice are moderately anemic at weaning (20 days of age) implies that intestinal DMT1 is also operative during suckling, yet measurements of iron absorption were not performed [20].
3.1.2. Ferroxidases are dispensable for iron absorption
In intestine-specific Heph KO mice at 6–10 weeks of age, 59Fe absorption and levels of hemoglobin are normal, indicating that intestinal HEPH is not required for iron absorption in adult mice [19,21]. To test the hypothesis that plasma ceruloplasmin (CP) may compensate for loss of intestinal HEPH, Fuqua et al. [21] crossed intestine-specific Heph KO mice with Cp KO mice to generate double-mutant mice. Interestingly, iron absorption was not impaired in mice lacking CP and intestinal HEPH, indicating that both proteins are dispensable for iron absorption. By extension, it also implies that the iron transport activity of intestinal ferroportin does not require the ferroxidase activity provided by HEPH and CP.
3.1.3. Iron trafficking in the enterocyte and transport to the liver
In the current model of iron absorption, it remains unknown how iron taken up by DMT1 at the enterocyte apical membrane traffics through the cytosol to reach ferroportin at the basolateral side. Cell culture studies raise the possibility that the iron chaperone PCBP2 is involved. Using HEp-2 cells, a human epithelial cell line, Yanatori et al. [22] found that PCBP2 physically interacts with DMT1 and is required for the uptake of 59Fe-NTBI, suggesting that PCBP2 facilitates iron flux through DMT1 by serving as an intracellular iron acceptor. A subsequent study by the same group demonstrated an interaction between PCBP2 and ferroportin and that silencing of PCBP2 decreased ferroportin-dependent iron export [23]. Together, these results suggest that PCBP2 provides an iron transport pipeline through the cytosol, but whether this occurs in enterocytes or other cell types is yet to be shown.
Another important recent advance has been the demonstration that iron entering the blood from the intestine binds to transferrin before reaching the liver via the portal vein. Although it has long been assumed that intestinal iron entering the blood is rapidly oxidized (catalyzed by HEPH, CP, or other ferroxidases) and bound to transferrin for transport to the liver, direct evidence for this has been lacking. Moreover, mathematical models derived from novel mouse pup-swapping experiments using 57Fe and Mossbauer spectroscopy supported the idea that iron was transported from the gut to the liver as NTBI rather than transferrin-bound iron (TBI) [24]. The same group of investigators subsequently tested their hypothesis by using an iron-deficient pig model, in which portal blood was sampled (via a surgically implanted catheter) after the animal was given an intragastric dose of 57Fe [25]. Contrary to expectations, they found that newly absorbed 57Fe was present in portal blood nearly exclusively bound to transferrin and not as NTBI.
3.2. Liver iron transport
The liver plays a central role in iron metabolism by sensing body iron status, synthesizing the iron regulatory hormone hepcidin, and serving as the main site of iron storage. Nonetheless, our understanding of iron transport in this organ has historically lagged behind that of other organs/tissues such as the intestine and bone marrow. Fortunately, this knowledge gap has started to close in the past 7 years, particularly with respect to iron transport in hepatocytes, which make up 70–85% of the liver’s cell mass.
3.2.1. DMT1 is dispensable for hepatic uptake of NTBI and liver iron loading
The role of DMT1 in hepatic uptake of TBI and NTBI, as well as in liver iron loading, was investigated in studies of hepatocyte-specific Dmt1 KO mice [26]. Radiotracer studies using intravenous 59Fe-transferrin demonstrated that hepatocyte-specific Dmt1 KO mice took up 40% less TBI into the liver than did control mice, suggesting that DMT1 participates in hepatic uptake of TBI, though it is not absolutely required [26]. Despite the lower hepatic TBI uptake in hepatocyte-specific Dmt1 KO mice, liver non-heme iron concentrations and other indicators of iron status were normal, implying that hepatocyte DMT1 is not essential for the maintenance of liver iron stores or for systemic iron homeostasis, even during iron deficiency, which was also assessed. Hepatic uptake of intravenous 59Fe-NTBI was found to be normal in hepatocyte-specific Dmt1 KO mice, indicating that hepatocyte DMT1 is dispensable for NTBI clearance by the liver. The role of DMT1 in liver iron loading was assessed by crossing hepatocyte-specific Dmt1 KO mice with Hfe KO mice or hypotransferrinemic (Hpx) mice, models of moderate and severe iron overload, respectively. Loss of hepatocyte DMT1 in these iron overload mouse models had no effect on the amount or distribution of iron in the liver, indicating that DMT1 is not required for hepatic iron loading.
3.2.2. SLC39A14 is required for hepatic uptake of NTBI and liver iron loading
The finding that DMT1 is dispensable for NTBI uptake and iron loading of the liver seems reasonable given that DMT1 levels have been shown to be markedly diminished in iron-loaded liver [27]. By contrast, levels of SLC39A14 (in that same study) were found to be elevated in iron-loaded liver. The iron-related increases in hepatic levels of SLC39A14, together with its ability to mediate the uptake of NTBI in cells [3], strongly implicated SLC39A14 in NTBI uptake in vivo. In subsequent studies using Slc39a14-null mice, Jenkitkasemwong et al. [5] found that SLC39A14 deficiency was associated with ~70% lower uptake of 59Fe-NTBI by the liver and isolated primary hepatocytes, whereas hepatic uptake from 59Fe-transferrin was unaffected. Moreover, Slc39a14-null mice, when fed an iron-loaded diet or crossed with Hfe KO or hemojuvelin (Hjv) KO mouse models of hereditary hemochromatosis, exhibited greatly diminished iron loading of the liver with virtually no iron accumulation in hepatocytes. Collectively, these observations indicate that SLC39A14 is the major mechanism for NTBI uptake by the liver and is essential for hepatocellular iron loading in iron overload. Under normal circumstances, however, only minor perturbations in iron metabolism have been documented in Slc39a14-null mice indicating that SLC39A14 is largely dispensable for normal iron homeostasis.
3.2.3. Role of TFR1 in liver iron transport and homeostasis
The requirement for TFR1 in hepatocellular and systemic iron homeostasis was investigated in studies of hepatocyte-specific TFR1-(Tfrc) KO mice [28]. The mice displayed ~50% lower concentrations of hepatic nonheme iron, 25% lower serum iron levels, and mild microcytosis without anemia. Uptake of 59Fe-transferrin was 40% lower in the liver of hepatocyte-specific Tfrc KO mice (after intraperitoneal injection of 59Fe-transferrin), and 15% lower in primary hepatocytes isolated from these mice. The lower hepatic iron concentrations in hepatocyte-specific Tfrc KO mice were not associated with lower hepcidin levels, indicating dysregulated hepcidin expression. Accordingly, hepatocyte-specific Tfrc KO mice were more susceptible to develop iron deficiency in response to an iron-deficient diet. However, in response to a high-iron diet, the mice accumulated iron normally in the liver, indicating that iron can load in the liver independently of hepatocyte TFR1 (likely via SLC39A14-mediated NTBI uptake as discussed above).
3.2.4. Mobilization of hepatocyte iron stores
When systemic iron demands increase, such as after blood loss or during growth or pregnancy, iron is mobilized from iron stored in ferritin in hepatocytes and macrophages. In studies of the role of DMT1 in ferritin iron mobilization by hepatocytes, La et al. [29] found that treatment of iron-loaded HepG2 hepatoma cells with the iron chelator desferrioxamine resulted in a rapid and 10-fold increase in DMT1 colocalization with the lysosomal marker LAMP2. Using mice, they determined that stimulation of iron mobilization by erythropoietin treatment decreased hepatic ferritin and iron levels in wild-type mice, whereas in hepatocyte-specific Dmt1 KO mice, ferritin levels were decreased (consistent with degradation) yet hepatic iron levels did not decrease (consistent with impaired mobilization). Together, the cell culture and in vivo data are consistent with hepatocyte DMT1 functioning in the lysosome to transport iron from degraded ferritin into the cytosol, thus making it available for export from the cell. The export and mobilization of iron from hepatocytes in response to increased erythropoietic activity requires ferroportin, as demonstrated by studies using hepatocyte-specific Fpn KO mice [30].
3.3. Erythroid iron uptake and trafficking
Developing erythroid cells in the bone marrow, the major consumers of iron in the body, acquire iron nearly exclusively via TFR1-mediated endocytosis of plasma transferrin. Although the principal components of this cellular uptake pathway are well known, recent studies have revealed additional proteins that modulate the assimilation of iron from transferrin. A genome-wide CRISPR screen in K562 cells, a human erythroleukemic cell line, identified CCDC115 to be required for TBI-dependent cell growth [31]. CCDC115 is known to serve as an assembly factor for the vacuolar-type H+-ATPase (VATPase) complex, a proton pump that acidifies endosomes and lysosomes. Uptake assays in CCDC115-null K562 cells confirmed diminished 59Fe uptake from 59Fe-transferrin. As depletion of CCDC115 has been shown to impair acidification of transferrin-containing endosomes [32], it is likely that the decreased acidification prevented the release of iron from transferrin and/or decreased proton-coupled DMT1-mediated transport of Fe2+ into the cytosol, resulting in impaired assimilation of TBI.
3.3.1. Iron trafficking in erythroid cells
Although more than 90% of transferrin iron taken up by developing erythroid cells ends up in the mitochondria where it is inserted into protoporphyrin IX to form heme, precisely how it gets there is not well understood. Recent studies by Ryu et al. [33] provide evidence that the iron chaperone PCBP1 and the ferritin cargo receptor NCOA4 are involved in at least part of the iron trafficking pathway from endosome to mitochondria. Their data, obtained by using G1E-ER4 cells, a model of terminal red blood cell development, support a model in which PCBP1 delivers transferrin-derived iron initially to ferritin for temporary storage. When the stored iron is needed for heme synthesis, ferritin is captured by NCOA4 and directed to the autophagosome and eventually the lysosome where the ferritin is degraded. The lysosomal iron is then transferred to the mitochondria by an undetermined mechanism. They additionally confirmed the involvement of PCBP1 in erythroid iron handling in vivo by demonstrating that Pcbp1 KO mice developed microcytic anemia, and that erythroid precursors isolated from these animals recapitulated the effects seen in G1E-ER4 cells.
Alternate models of intracellular iron trafficking posit that transferrin-derived iron does not enter the cytosol, but is rather transferred via direct interaction between intracellular vesicles and mitochondria. In the “kiss-and-run” model, first proposed in 2005 [34], transient interactions between endosomes and mitochondria allow for direct transfer of iron between these organelles. Additional support for this model was recently provided by Hamdi et al. [35] who used 3-dimensional confocal microscopy to document spatial interactions between transferrin-endosomes and mitochondria in reticulocytes and a novel flow cytometry-based method to quantify these interactions over time. Also consistent with this model is the finding of protein-protein interactions between components of the mitochondrial heme biosynthetic pathway and endosomal proteins TFR1 and STEAP3 in a study characterizing an assembly of proteins associated with heme synthesis in reticulocytes [36]. In another model, TBI is taken up via TFR2, which is also expressed in erythroid progenitors. However, unlike the canonical TFR1-mediated endosomal recycling pathway, the transferrin-TFR2 uptake pathway in erythroid cells traffics to the lysosome, which transfers transferrin-iron directly to the mitochondria via contacts between these two organelles. In this model, iron is transferred out of the lysosome via TRPML1/mucolipin-1, which has been shown to be a lysosomal iron transporter [37].
3.3.2. Iron export from eythroid cells
A surprising finding in the past decade has been the demonstration that erythroid precursor cells, despite having the greatest uptake and need for iron in the body, express abundant amounts of ferroportin and export iron in a hepcidin-dependent manner [38,39]. Such a function may serve to limit erythropoiesis during iron deficiency and allow erythroblasts to release some of their iron to the plasma for distribution to other cells having a more critical need for the metal, such as cardiomyocytes or neurons. Consistent with this proposed role, erythroblasts from erythroblast-specific Fpn KO mice were protected from losing iron and hemoglobin after the mice were switched to an iron-deficient diet [40]. Erythroblast ferroportin may additionally serve a protective role by allowing the cell to divest itself of any excess, potentially toxic redox-active iron, such as iron released from the autooxidation of hemoglobin. Ferroportin is also abundantly expressed in mature red blood cells and is required for iron export, as shown by 55Fe-transport studies using Fpn KO erythrocytes [41]. Considering the number of red blood cells in the circulation, the amount of iron released by these cells likely contributes significantly to plasma iron levels and the overall iron economy.
4. Overview of manganese transport
The year 2012 represents a turning point in our understanding of Mn transport and homeostasis. In February of that year, back-to-back papers published in the American Journal of Human Genetics reported the first hereditary disorder of Mn metabolism characterized by hypermanganesemia, Mn accumulation in the liver and brain, dystonia, polycythemia, and chronic liver disease [42,43]. The syndrome was found to arise from homozygous mutations in the SLC30A10 gene encoding SLC30A10, commonly known at that time as ZnT10, a member of the ZnT (SLC30) family of membrane proteins classified as zinc efflux transporters. Yet, the clinical phenotype of patients with SLC30A10 mutations, together with studies performed in yeast showing that SLC30A10 protected cells from Mn toxicity, pointed to SLC30A10 functioning primarily as a physiological Mn transporter rather than a zinc transporter, a role that was later established. This finding is particularly important in Mn biology because it represents the first identification of a transporter as essential for whole-body Mn homeostasis in mammals. In 2015, mutations in the SLC39A8 gene encoding ZIP8 (SLC39A8), a member of the ZIP (SLC39) family of metal-ion importers, were also shown to majorly disrupt Mn homeostasis, but in this case causing a severe Mn deficiency disorder characterized by abnormal glycosylation patterns, skeletal abnormalities, and global psychomotor disability [44,45]. Less than a year later, SLC39A14 was implicated in Mn homeostasis, as mutations in SLC39A14 were found to cause hypermanganesemia, brain Mn accumulation, dystonia, and juvenile-onset parkinsonism [46]. Thus, in a span of only four short years, three proteins essential for Mn transport were identified, revealing a panoply of new molecular mechanisms governing Mn homeostasis. These discoveries, which have energized, expanded, and accelerated research on Mn transport and metabolism, may very well signal what could be considered a golden age of Mn biology research. Highlights of current knowledge of where and how these and other transporters participate in whole-body Mn homeostasis are indicated in Fig. 3 and discussed in more detail in Section 5.
Fig. 3.
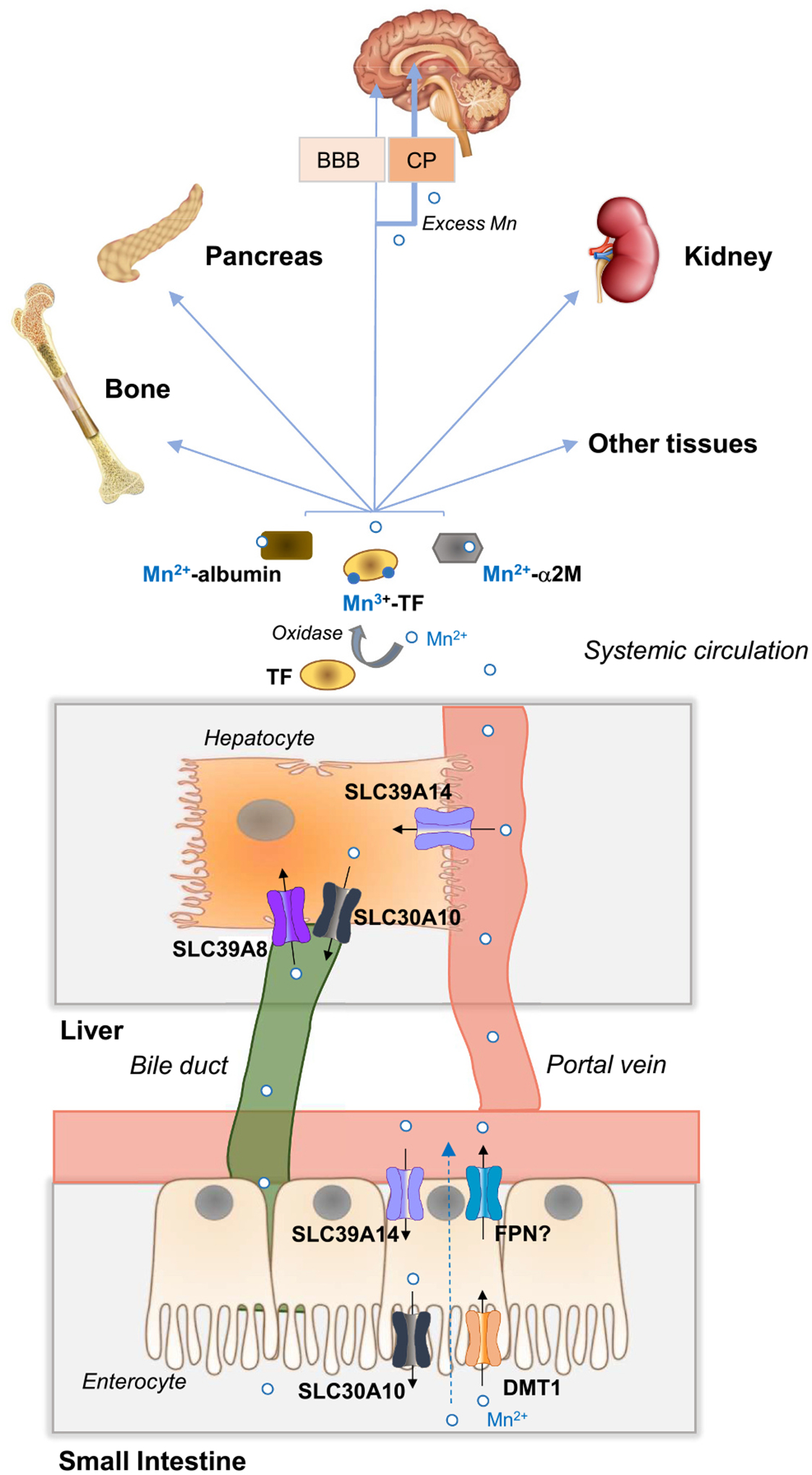
Whole-body Mn transport, metabolism, and distribution. In the small intestine, dietary Mn (Mn2+) is absorbed by incompletely characterized mechanisms that do not require DMT1 or FPN (dashed arrow). DMT1 may contribute significantly to Mn uptake in iron deficiency when DMT1 levels are upregulated. FPN may function in Mn export, but its exact contribution remains to be established. Mn2+ in portal blood is mostly taken up into the liver via SLC39A14 at the basolateral membrane of hepatocytes. Mn in plasma is found as Mn2+ associated with citrate and proteins such as albumin and α2M (α2-macroglobulin) or as Mn3+ bound to transferrin. The oxidation of Mn2+ to Mn3+ does not readily occur and therefore is likely catalyzed by an oxidase. Mn in plasma is taken up by all tissues, but primarily by the liver, pancreas, kidney, brain, and bone. Excess Mn accumulates preferentially in the bone, liver, kidney, pancreas, and brain, with net accumulation reflecting a balance between uptake and efflux of the metal. To enter the brain, Mn must cross the BBB (blood-brain-barrier) or the CP (choroid plexus). When plasma Mn levels are high, excess Mn enters the brain predominantly via the CP. In the liver, Mn that is not utilized by the hepatocyte is excreted into the bile via SLC30A10 located at the apical canalicular membrane. Hepatocyte SLC39A8 at the apical canalicular membrane reclaims Mn from the bile to prevent Mn deficiency. Mn in bile is secreted into the small intestine where it may undergo enterohepatic recirculation or be subsequently eliminated via feces. Mn in blood plasma can also be excreted directly by the intestine via SLC39A14 located on the enterocyte basolateral membrane and SLC30A10 located at the enterocyte apical membrane. Additional details are described in Section 4.1.
4.1. Whole-body Mn transport, metabolism, and distribution
Dietary manganese, which exists mainly as Mn2+, is absorbed in the small intestine by incompletely defined mechanisms. DMT1 at the apical membrane of enterocytes likely participates in intestinal Mn uptake although it is not required (Fig. 3). How Mn traverses the cytosol and is transported across the basolateral membrane is unknown. Once absorbed into capillary blood, Mn2+ may remain free or become bound to α2-macroglobulin or albumin during transport to the liver, where it is extracted from portal blood with high efficiency by SLC39A14 located on the basolateral membrane of hepatocytes. A fraction of absorbed Mn enters the systemic circulation, where it is predominantly associated with carrier proteins such as transferrin, albumin, and α2-macroglobulin [47]. Mn bound to transferrin is in the Mn3+ state indicating that some Mn2+ in the plasma is oxidized, yet where and how this occurs is not known [48]. Mn is rapidly cleared from the circulation primarily by the liver, pancreas, kidney, and intestine. Excess Mn accumulates preferentially in the bone, liver, kidney, pancreas, and brain, with net accumulation reflecting a balance between uptake and efflux of the metal. To enter the brain, Mn must first cross endothelial cells of the blood-brain barrier (BBB) or epithelial cells of the choroid plexus (CP). In Mn excess, non-protein-bound Mn enters mainly via the CP. Although cells can take up transferrin-bound Mn via TFR1-mediated endocytosis, transferrin is not required for the distribution and acquisition of Mn by tissues, indicating that non-transferrin-dependent Mn uptake mechanisms exist (Fig. 4). Surplus Mn in the hepatocyte is excreted into the bile via SLC30A10 localized to the apical (canalicular) membrane. Bile containing Mn is released into the small intestine, where the metal can either be reabsorbed via enterohepatic circulation or subsequently eliminated via feces. Hepatocyte SLC39A8, situated at the apical canalicular membrane, reclaims Mn from the bile to protect against deficiency. Mn excretion also occurs directly via the intestine by enterocyte SLC39A14 at the basolateral membrane (facing the blood) and by SLC30A10 at the enterocyte apical membrane (facing the lumen). In this process, SLC39A14 takes up Mn from the blood into the enterocyte and SLC30A10 exports it into the intestinal lumen for subsequent elimination via feces. Whole-body Mn balance is maintained by regulating gastrointestinal absorption and excretion, although the underlying molecular mechanisms are unclear. Very little Mn is excreted in the urine, even in conditions of Mn excess.
Fig. 4.
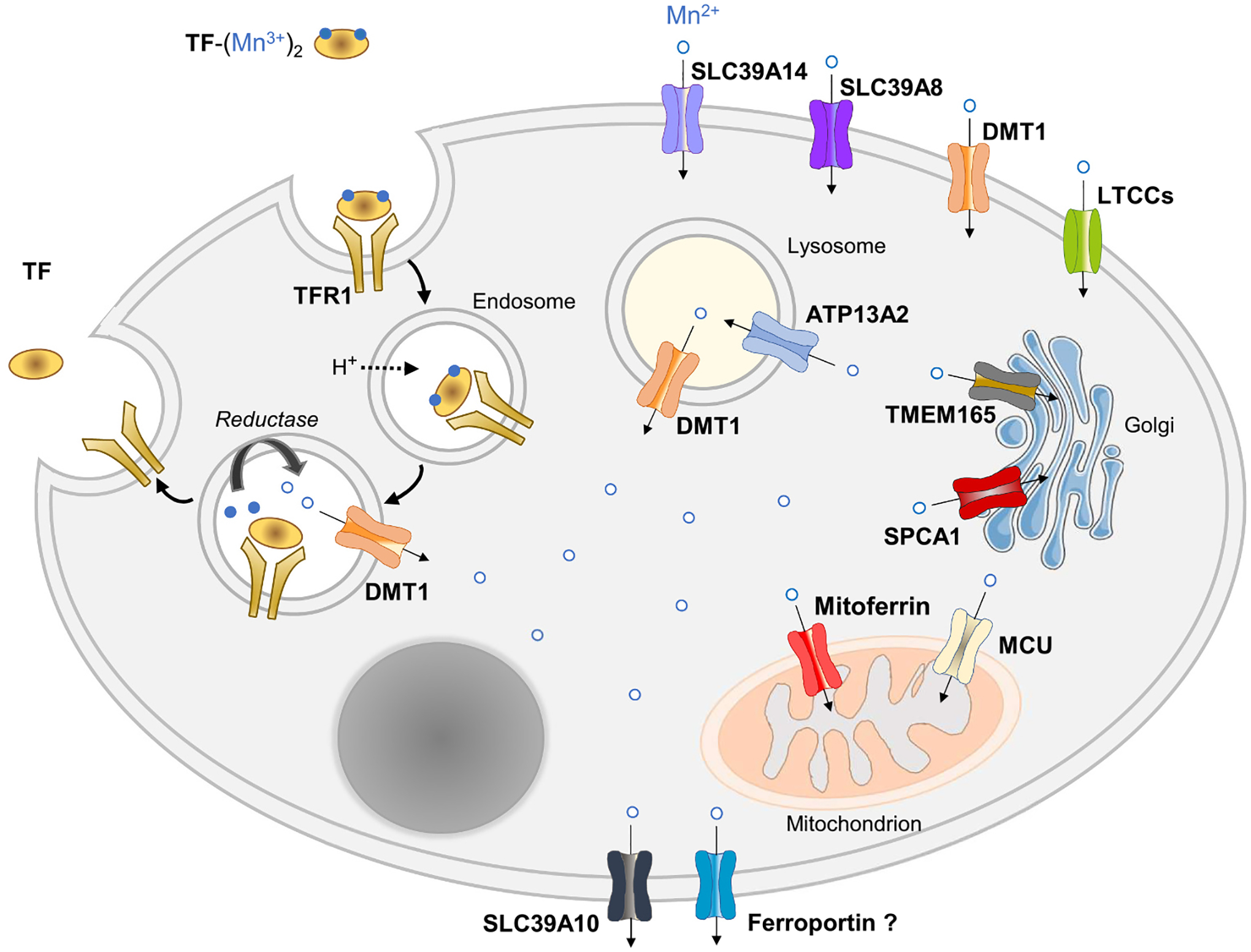
Cellular Mn transport and trafficking. A model of a generic cell is depicted. Cells can take up Mn3+ from TF via TFR1-mediated endocytosis or Mn2+ via SLC39A14, SLC39A8, DMT1, or LTCCs, depending on the cell type. Mn is taken up into the mitochondrion via the MCU (mitochondrial uniporter complex) or possibly by mitoferrin 1/2. ATP13A2 (ATPase Cation Transporting 13A2) transports Mn into the lysosome. Mn is taken up into the Golgi apparatus via TMEM165 (Transmembrane Protein 165) or SPCA1 (secretory pathway Ca2 + -ATPase pump type 1). Cellular Mn export is mediated by SLC30A10 or possibly by FPN. Additional details are described in Section 4.2.
4.2. Cellular Mn transport and trafficking
Transferrin-bound Mn, TF-(Mn3+)2, can be taken up via TFR1-mediated endocytosis (Fig. 4). For endosomal transport via DMT1, internalized Mn3+ would need to be converted to Mn2+ for transport via DMT1, possibly by a STEAP metalloreductase. Conversion to the more stable Mn2+ seems likely given that Mn3+ is a strong oxidizing agent. Mn2+ may be taken up at the cell surface via metal-ion transporters SLC39A14, SCL39A8, DMT1 or by voltage-gated Ca2+ channels. Most cellular Mn is taken up into mitochondria, likely via MCU (mitochondrial uniporter complex) and possibly by mitoferrin 1. Mn uptake into the Golgi is mediated by SPCA1 and TMEM165 (transmembrane protein 165) (see below). Transport of Mn into the lysosome may be mediated by ATP13A2, whereas lysosomal Mn export occurs via DMT1. Cellular efflux of cytosolic Mn is mediated by SLC30A10 and possibly by ferroportin, although Mn transport by ferroportin remains controversial.
4.2.1. TMEM165 and Golgi Mn transport
The Golgi protein TMEM165 merits additional discussion as many studies in the past seven years have been directed at determining its transport function(s) and role in glycosylation. In 2012, deficiencies in TMEM165, a Golgi-localized protein of unknown function, were reported to cause a congenital disorder of glycosylation (CDG) in humans [49]. The protein was first proposed to serve as a calcium (Ca2+) transporter based on its predicted topology and similarity to members of the cation/Ca2+ (CaCA) exchanger superfamily [50]. High luminal Ca2+ concentrations are required in the Golgi and other parts of the secretory pathway for glycosylation, protein synthesis, sorting, and cleavage. Studies of Gdt1p (Gcr1 dependent translation factor 1), the yeast orthologue of TMEM165, demonstrated Ca2+ transport activity and that the protein was required for N-linked and O-linked glycosylation [51]. Interestingly, these studies and others [52] also revealed that glycosylation defects of Gdt1p mutants could be rescued by the addition of high concentrations of Mn. In human cells, depletion of TMEM165 decreased Golgi Mn concentrations and impaired the glycosylation and function of Golgi Mn-dependent enzymes, effects that were reversed with Mn supplementation [53]. Mn transport by both Gdt1p and TMEM165 was confirmed by heterologously expressing these proteins in Lactococcus bacteria and measuring Mn2+-dependent quenching of the fluorescent dye Fura-2/AM [54,55]. Given the Mn transport activity of TMEM165, and the fact that Mn serves as a cofactor for many Golgi glycosyltransferase enzymes, it is not surprising that TMEM165 deficiency results in a congenital disorder of glycosylation [56].
5. Recent advances in Mn transport
5.1. Liver Mn transport
The liver plays a primary role in Mn homeostasis by taking up Mn from portal blood, thereby regulating the amount of Mn that enters the circulation for delivery to other organs. The liver also regulates the transport of Mn into and out of the bile, thus fine-tuning how much metal is excreted via the hepatobiliary route. This regulation occurs in the hepatocyte and is carried out by SLC30A10, SLC39A8, and SLC39A14, which all have essential and non-redundant Mn transport roles that directly affect whole-body Mn balance.
5.1.1. Role of SLC30A10 in hepatocyte Mn export into bile
In the original studies that linked SLC30A10 mutations to hypermanganesemia [42,46], the idea that SLC30A10 functions as a Mn efflux transporter was supported by several observations: patients accumulated Mn in the liver, which is known to secrete (efflux) Mn into bile; SLC30A10 is abundantly expressed in the liver; the protein localizes to hepatocytes and bile duct epithelium; and SLC30A10 was already classified as an efflux transporter—albeit for Zn. Experimental support for SLC30A10 functioning in Mn efflux was provided by expression studies in yeast, which demonstrated that human SLC30A10 protected cells from Mn toxicity [43].
The ability of SLC30A10 to transport Mn in mammalian cells was first investigated using HeLa cells overexpressing SLC30A10 [57]. After exposure to high concentrations of Mn, cells overexpressing SLC30A10 had lower total levels of Mn than did control cells, suggesting that SLC30A10 exports Mn from the cell. Interestingly, SLC30A10 overexpression did not decrease cellular Zn concentrations in cells exposed to high Zn, despite SLC30A10 (aka ZnT10) being included in the SLC30 (ZnT) family of Zn efflux transporters. This study also demonstrated that SLC30A10 localizes to the cell surface and that SLC30A10 mutants that cause disease prevented the protein from reaching the cell surface and rendered cells more susceptible to Mn-induced cell death.
That SLC30A10 functions as a Mn transporter in vivo was confirmed by the generation and characterization of Slc30a10 KO mice [58]. Slc30a10 KO mice appear normal during early development, but fail to gain weight after weaning. At 6 weeks of age, Slc30a10 KO mice have markedly elevated levels (~50–100× normal) of Mn in the blood, liver, and brain, whereas levels of zinc, iron and copper are normal, consistent with Mn being the primary physiological substrate of SLC30A10. Conditional inactivation of Slc30a10 in hepatocytes resulted in only modest elevations in Mn levels in the blood and liver (~2× normal) and brain (~20% higher than normal) [59], indicating that other excretory mechanisms can nearly fully compensate for lack of hepatocyte SLC30A10. The mild phenotype of the hepatocyte-specific Slc30a10 KO mice was initially surprising given the well-known role of the liver in excreting Mn in bile. However, the phenotype was consistent with classic Mn excretion studies conducted in the 1960’s, which demonstrated that bile-duct ligation in rats impairs, but does not prevent Mn excretion via the gastrointestinal tract [60] and that the intestine serves as the auxiliary excretory route when the hepatic route is blocked [61]. Accordingly, endoderm-specific Slc30a10 KO mice, which lack SLC30A10 not only in hepatocytes, but also in all other epithelial cells lining the digestive tract, display much greater Mn accumulation than do hepatocyte-specific Slc30a10 KO mice [59]. Indeed, Mn levels in the liver, blood, and brain in endoderm-specific Slc30a10 KO mice are nearly (i.e., ~80%) as high as those in whole-body Slc30a10 KO mice.
The requirement for hepatocyte SLC30A10 for hepatobiliary Mn excretion in vivo was investigated by Mercadante et al., [62] who assessed biliary metal levels, biliary flow rates, and Mn transport into bile in hepatocyte-specific Slc30a10 KO mice. Bile was collected from anesthetized mice by ligation of the common bile duct and cannulation of the gallbladder. In hepatocyte-specific Slc30a10 KO mice, biliary copper levels were normal, but biliary Mn was virtually undetectable, consistent with a defect in Mn excretion. In control mice, biliary Mn levels were on average twice as high as biliary copper levels. Radiotracer 54Mn injected into the portal vein was measurable in bile of control mice by 5 min post-injection and its levels steadily increased over time; by 60 min, up to 15% of the total injected 54Mn was transferred to bile. By contrast, in hepatocyte-specific Slc30a10 KO mice, no quantifiable 54Mn was detected in bile, even by 60 min after portal vein injection of 54Mn. Likewise, essentially no 54Mn was recovered in bile collected from hepatocyte-specific Slc30a10 KO mice 16 h after 54Mn was administered systemically via retroorbital injection. However, livers from hepatocyte-specific Slc30a10 KO mice did contain significantly increased amounts of 54Mn, likely representing the hepatocyte pool of 54Mn that could not be transferred into bile in these animals. Collectively, these studies unequivocally demonstrate that hepatocyte SLC30A10 is required for hepatobiliary Mn excretion in vivo, thus establishing an essential role for SLC30A10 in Mn transport by the liver.
5.1.2. SLC39A8 and Mn reclamation from the bile
Interestingly, the next protein implicated in Mn homeostasis by human studies, SLC39A8, has a role in the liver that is precisely opposite to that of SLC30A10, although its function was not readily predictable from the phenotype of patients with SLC39A8 mutations. SLC39A8 was first identified in 2002 in a screen of genes induced in monocytes challenged with bacterial cell wall components [63]. Studies of SLC39A8-mediated metal transport documented its ability to promote cellular uptake of zinc [63], manganese [64], and iron [4], thus stimulating speculation over the years on which metal is its main physiologic substrate [64,65]. Slc39a8 hypomorphic mice expressing <10% of normal Slc39a8 levels exhibit severe iron-deficiency anemia in utero and do not survive after birth [66], suggesting that SLC39A8 functions in hematopoiesis or placental iron transport. Transcriptomic and proteomic analyses of differentiating erythroid cells implicated SLC39A8 in mediating increases in intracellular zinc needed for erythroblast survival [67]. Clinical descriptions of the first patients with SLC39A8 mutations failed to provide clarity on the issue as patients were reported to have low circulating levels of Mn or Zn, or in some cases, low levels of both metals [44,45]. Insight into the main physiological substrate of SLC39A8 was provided by Slc39a8 KO mice—more specifically Slc39a8 inducible KO (Slc39a8 iKO) mice because constitutive Slc39a8 KO in mice leads to embryonic lethality. Slc39a8 iKO mice, generated starting at 8 weeks of age and sacrificed 5 weeks later, were found to have lower tissue levels of Mn, but not zinc (or iron) [68]. Compared with control mice, Mn levels in Slc39a8 iKO mice were decreased by 80% in liver and 30–50% in kidney, brain, and heart. The subsequent generation of hepatocyte-specific Slc39a8 KO mice revealed that loss of hepatocyte SLC39A8 recapitulated the Mn deficiency seen in whole-body Slc39a8 KO mice, thus identifying hepatocyte SLC39A8 as an essential regulator of whole-body Mn homeostasis. Immunofluorescence analysis of mouse liver sections localized SLC39A8 to the hepatocyte apical canalicular membrane, suggesting that SLC39A8 functions to take up (reclaim) Mn from the bile. Such a model is supported by the observation that bile Mn levels are elevated (+55%) in hepatocyte-specific Slc39a8 KO mice and by the demonstration that AAV-mediated restoration of hepatic SLC39A8 expression in these mice decreased bile Mn levels by 76%. It should also be noted that both the Slc39a8 iKO mice and the hepatocyte-specific Slc39a8 KO mice displayed defective protein glycosylation, similar to individuals with SLC39A8 mutations [45], thus underscoring the importance of Mn for glycosylation.
5.1.3. Hepatocyte SLC39A14 and Mn uptake by the liver
Like SLC39A8, the main physiological substrate of SLC39A14 was not easily predictable based on cell-based transport studies. SLC39A14 was first shown to function as a Zn2+ (influx) transporter [69], then as a Fe2+/Zn2+ transporter [3], a Cd2+ or Mn2+/bicarbonate symporter [64], and most recently as a Zn2+/Mn2+/Fe2+/Cd2+ transporter [70]. Under physiological conditions, Mn2+ is no doubt an important substrate, as revealed by the hypermanganesemia and tissue Mn accumulation in individuals with SLC39A14 mutations [46] and Slc39a14 KO mice [71–73]. The Mn-loading phenotype in SLC39A14 deficiency is similar to that in SLC30A10 deficiency yet differs in one respect: in SLC30A10 deficiency, the liver accumulates large amounts of Mn, whereas in SLC39A14 deficiency, the liver has less Mn, suggesting impaired Mn uptake by this organ. Consistent with this proposed function, SLC39A14 in the liver is most abundantly expressed in hepatocytes, where it localizes to the basolateral membrane facing nutrient-rich portal blood [27]. Radiotracer studies using intravenously or orally administered 54Mn demonstrate that Mn uptake is markedly impaired in the liver of Slc39a14 KO mice [72]. The disposition of orally administered 54Mn is more physiologically relevant as Mn normally enters the body via the oral route (e.g., from food or water). Two hours after 54Mn was given via orogastric gavage to control mice, ~80% of the total 54Mn that was absorbed/retained by the body was found in the liver, consistent with the well-known highly efficient hepatic first-pass extraction of Mn. By contrast, in Slc39a14 KO mice, less than 10% of the total 54Mn that was absorbed/retained by the body was found in the liver, whereas markedly more was found in extrahepatic tissues such as kidney, brain, and bone. Taken together, these data indicate that the primary function of SLC39A14 in the liver is to take up (extract) Mn2+ from portal blood. In SLC39A14 deficiency, Mn that is absorbed cannot be taken up into the liver (and subsequently excreted in the bile) and therefore enters the circulation where it can build up and deposit in extrahepatic tissues. Mice deficient in both Slc39a14 and Slc30a10 have elevated Mn levels in the blood and brain, but not the liver, strengthening the conclusion that hepatocyte SLC39A14 and SLC30A10 act synergistically to mediate Mn excretion via the liver [74]. However, hepatocyte-specific Slc39a14 KO mice do not accumulate Mn in extrahepatic tissues as do whole-body Slc39a14 KO mice [73,75], indicating that SLC39A14 function is necessary for Mn excretion by other tissues.
5.2. Intestinal Mn absorption and excretion
In contrast to current understanding of the fundamental molecular mechanisms that mediate intestinal iron absorption, comparatively little is known about how Mn is absorbed. This difference is surprising given that iron deficiency is well known to enhance Mn absorption, possibly by sharing common intestinal transport pathways [76].
5.2.1. Role of DMT1 in Mn absorption
It is widely believed that increased Mn absorption in iron deficiency is mediated by DMT1, whose expression increases dramatically in iron deficiency. Although this is very likely the case in iron-deficient conditions, the role of DMT1 in intestinal Mn absorption when iron status is normal has been less clear. This issue was recently addressed in studies that used intestine-specific Dmt1 KO mice to determine which metals rely upon DMT1 for their absorption [1]. Mn absorption in control and intestine-specific Dmt1 KO mice was assessed by measuring the rate of appearance of 54Mn in the blood 15 min to 4 h after mice were fed a dose of 54Mn via orogastric gavage. The amount of 54Mn in enterocytes and liver was also determined (4 h post dose). Prior to the absorption studies, intestine-specific Dmt1 KO mice were made iron replete by iron dextran injections, which normalized their levels of plasma iron and hemoglobin. No differences were observed between intestine-specific Dmt1 KO mice and controls in intestinal 54Mn absorption or levels of 54Mn in enterocytes and liver, indicating that Mn absorption does not rely upon DMT1. Moreover, ICP-MS analysis of metals in various tissues (liver, heart, spleen, muscle, kidney) from intestine-specific Dmt1 KO mice and controls at 4 months of age revealed no differences in tissue Mn content aside from a 30% decrease in the kidney. As a reference, and as expected, tissue iron levels were low in all tissues from mice lacking intestinal DMT1.
5.2.2. Role of ferroportin in Mn absorption
The possibility that ferroportin, which is essential for iron absorption, participates in intestinal Mn absorption was raised by studies of flatiron mice, a model of ferroportin deficiency [77]. Radiotracer studies using intragastrically administered 59Fe or 54Mn in 12-week-old flatiron and control mice revealed that ferroportin deficiency was associated with reduced absorption of not only iron but also Mn (55% and 65% less, respectively). The reduced absorption of Mn in flatiron mice was associated with ~50% lower levels of Mn in the bile, ~10% lower levels of Mn in the blood and liver, and ~ 30% lower MnSOD activity in blood at three different ages. Although the findings from the flatiron mouse are consistent with a role for ferroportin in Mn transport/homeostasis, the ability of ferroportin to transport Mn is controversial. In studies of Mn transport by human ferroportin expressed in Xenopus oocytes, one group reported that ferroportin can function as a Mn exporter [78], whereas another group subsequently concluded that it does not [79]. The latter study estimated the specificity of ferroportin was around three orders of magnitude lower for Mn than for iron, implying that any ferroportin-mediated Mn transport in vivo would likely be trivial. Studies of Mn transport/toxicity and ferroportin overexpression in mammalian cells are also inconsistent [80–83].
A recent study examined the role of ferroportin in Mn homeostasis by using Tmprss6 KO mice, which express high levels of hepcidin and therefore have very low ferroportin levels in their tissues [84]. Levels of Mn and iron were determined in serum, liver, kidney, spleen, heart, and brain from male mice at 2, 4, 8, and 12 weeks of age. Whereas iron levels in serum, liver, heart, and brain were lower in Tmprss6 KO mice than in control mice at all ages but one (2 weeks), corresponding Mn levels in these tissues were not lower, suggesting that ferroportin is not a major regulator of Mn absorption and tissue distribution as it is for iron. Such a conclusion is additionally supported by the levels of Mn and iron in liver from intestine-specific ferroportin KO mice at 8–10 weeks of age. In intestine-specific ferroportin KO mice, liver iron levels were 70% lower than those in control mice, whereas liver Mn levels did not differ between groups. These observations indicate that intestinal ferroportin is required for the body to procure iron but not Mn.
5.2.3. Enterocyte SLC30A10 and SLC39A14 and Mn excretion
While the mechanisms of Mn absorption remain unclear and unresolved, significant progress has been made regarding the mechanisms of Mn excretion via enterocytes. As noted earlier, endoderm-specific Slc30a10 KO mice accumulate more Mn than do hepatocyte-specific Slc30a10 mice cell [59] indicating that SLC30A10 in the gastrointestinal tract participates in Mn excretion, likely in the proximal small intestine, where it is most abundantly expressed [62]. Fluorescence analysis of duodenal sections from Slc30a10-GFP mice revealed that SLC30A10 localizes to the apical membrane of enterocytes [62], where it can mediate the efflux of Mn from the cell [59]. Accordingly, enterocyte-specific Slc30a10 KO mice at 4 months of age were found to accumulate Mn preferentially in the duodenum followed by the liver and bone, thus demonstrating enterocyte SLC30A10 is essential for Mn excretion and whole-body Mn homeostasis. An essential role for enterocyte SLC39A14 in Mn excretion has also been established. SLC39A14 is abundantly expressed in the small intestine [3], where it localizes to the basolateral membrane of enterocytes [75]. Such a function likely occurs in vivo as enterocyte-specific Slc39a14 KO mice at 3 weeks of age display elevated levels of Mn in the liver and brain [75]. A similar phenotype for enterocyte-specific Slc39a14 KO was later reported in an independent study, which additionally demonstrated that uptake/retention of 54Mn in the intestine in enterocyte-specific Slc39a14 KO mice decreased after systemic but not oral 54Mn administration, indicating that enterocyte SLC39A14 enhances intestinal Mn excretion by taking up Mn from the systemic circulation rather than from freshly absorbed Mn [85].
6. Perspectives and future directions
Much progress has been made in the past 7 years in our understanding of iron and Mn transport and homeostasis, allowing for a clearer picture to emerge as to how the transport and handling of these two metals are similar and different. Overall, it seems that whereas iron and Mn may utilize the same transporters under certain circumstances (e.g., DMT1 in iron deficiency), the two metals under normal circumstances appear to be largely handled differently, transported by different transporters, and regulated by independent homeostatic signals. For example, iron absorption requires DMT1 and ferroportin, whereas Mn absorption does not. After absorption into portal blood, iron readily binds to transferrin, whereas Mn does not. The binding of iron to transferrin allows it to pass through the liver (which normally does not need more iron) and enter the systemic circulation to deliver the iron to tissues that need it (e.g., bone marrow). By contrast, Mn not bound to transferrin in portal blood is readily taken up into the liver via SLC39A14, thus enabling control of how much Mn enters the systemic circulation. How much iron enters the systemic circulation is controlled by hepcidin, which prevents intestinal absorption of iron, but not Mn.
Among the transporters that have been proposed to transport both iron and Mn in vivo, the best evidence so far is for SLC39A14. Under normal conditions, SLC39A14 in the liver functions to take up Mn from the plasma, whereas in iron overload, it can also take up NTBI—and in both situations, its role is essential [5,72]. Radiotracer studies show that SLC39A14 in the pancreas functions similarly [5,72,85], yet they further reveal that most other tissues have SLC39A14-independent uptake mechanisms for NTBI and Mn. Therefore, a future challenge will be to identify these mechanisms. High-priority tissues for future research include the brain, kidney, and lung. The brain is the main target organ of Mn toxicity, but the molecular mechanisms mediating brain Mn uptake have not been well characterized. Recent studies using an in vitro model of the blood-brain barrier suggest that both SLC39A8 and SLC39A14 participate [86]. Moreover, how and why Mn in the brain preferentially accumulates in the basal ganglia remains a major unanswered question in the Mn field. As for the kidney, very little iron or Mn is excreted in the urine, even in conditions of excess, indicating that kidney actively re-absorbs these metals after they are filtered by the glomerulus. DMT1, SLC39A8, and SLC39A14 are all expressed in the kidney proximal tubule, [87,88], but their in vivo roles remain to be determined. The lung is important because inhalation of airborne Mn is the most common cause of occupational Mn intoxication. DMT1 appears to be dispensable for pulmonary Mn absorption [89], whereas recent cell culture studies implicate both SLC39A8 and SLC39A14 [90]. The available data suggest functional redundancies among transporters in these tissues and should thus be considered in transport studies using knockout mouse models.
Acknowledgements
This work was supported by National Institutes of Health grant DK080706 to Mitchell D. Knutson.
Footnotes
CRediT authorship contribution statement
Qingli Liu: Writing - original draft, Writing - review & editing. Saiid Barker: Writing - review & editing. Mitchell D. Knutson: Conceptualization, Writing - original draft, Writing - review & editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Cell Biology of Metals III edited by Roland Lill and Mick Petris.
References
- [1].Shawki A, Anthony SR, Nose Y, Engevik MA, Niespodzany EJ, Barrientos T, Ohrvik H, Worrell RT, Thiele DJ, Mackenzie B, Intestinal DMT1 is critical for iron absorption in the mouse but is not required for the absorption of copper or manganese, Am. J. Physiol. Gastrointest. Liver Physiol 309 (2015) G635–G647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Anagianni S, Tuschl K, Genetic disorders of manganese metabolism, Curr. Neurol. Neurosci. Rep 19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Liuzzi JP, Aydemir F, Nam H, Knutson MD, Cousins RJ, Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells, Proc. Natl. Acad. Sci. U.S.A 103 (2006) 13612–13617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wang CY, Jenkitkasemwong S, Duarte S, Sparkman BK, Shawki A, Mackenzie B, Knutson MD, ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular Iron loading, J. Biol. Chem 287 (2012) 34032–34043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jenkitkasemwong S, Wang CY, Coffey R, Zhang W, Chan A, Biel T, Kim JS, Hojyo S, Fukada T, Knutson MD, SLC39A14 is required for the development of hepatocellular iron overload in murine models of hereditary hemochromatosis, Cell Metab 22 (2015) 138–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Reddi AR, Hamza I, Heme mobilization in animals: a metallolipid’s journey, Acc. Chem. Res 49 (2016) 1104–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Donegan RK, Moore CM, Hanna DA, Reddi AR, Handling heme: the mechanisms underlying the movement of heme within and between cells, Free Radic. Biol. Med 133 (2019) 88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Balachandran RC, Mukhopadhyay S, McBride D, Veevers J, Harrison FE, Aschner M, Haynes EN, Bowman AB, Brain manganese and the balance between essential roles and neurotoxicity, J. Biol. Chem 295 (2020) 6312–6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Qian ZM, Ke Y, Brain iron transport, Biol. Rev. Camb. Philos. Soc 94 (2019) 1672–1684. [DOI] [PubMed] [Google Scholar]
- [10].Wade QW, Chiou B, Connor JR, Iron uptake at the blood-brain barrier is influenced by sex and genotype, Adv. Pharmacol 84 (2019) 123–145. [DOI] [PubMed] [Google Scholar]
- [11].Knutson MD, Iron transport proteins: gateways of cellular and systemic iron homeostasis, J. Biol. Chem 292 (2017) 12735–12743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Knutson MD, Non-transferrin-bound iron transporters, Free Radic. Biol. Med 133 (2019) 101–111. [DOI] [PubMed] [Google Scholar]
- [13].Andrews NC, Forging a field: the golden age of iron biology, Blood 112 (2008) 219–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, Andrews NC, The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis, Cell Metab 1 (2005) 191–200. [DOI] [PubMed] [Google Scholar]
- [15].Gunshin H, Fujiwara Y, Custodio AO, Direnzo C, Robine S, Andrews NC, Slc11a2 is required for intestinal iron absorption and erythropoiesis but dispensable in placenta and liver, J. Clin. Invest 115 (2005) 1258–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shawki A, Knight PB, Maliken BD, Niespodzany EJ, Mackenzie B, H(+)-coupled divalent metal-ion transporter-1: functional properties, physiological roles and therapeutics, Curr. Top. Membr 70 (2012) 169–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Shawki A, Engevik MA, Kim RS, Knight PB, Baik RA, Anthony SR, Worrell RT, Shull GE, Mackenzie B, Intestinal brush-border Na+/H+ exchanger-3 drives H+-coupled iron absorption in the mouse, Am. J. Physiol. Gastrointest. Liver Physiol 311 (2016) G423–G430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Frazer DM, Wilkins SJ, Darshan D, Mirciov CSG, Dunn LA, Anderson GJ, Ferroportin is essential for iron absorption during suckling, but is hyporesponsive to the regulatory hormone hepcidin, Cell Mol. Gastroenterol. Hepatol 3 (2016) 410–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Doguer C, Ha JH, Gulec S, Vulpe CD, Anderson GJ, Collins JF, Intestinal hephaestin potentiates iron absorption in weanling, adult, and pregnant mice under physiological conditions, Blood Adv 1 (2017) 1335–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ramakrishnan SK, Anderson ER, Martin A, Centofanti B, Shah YM, Maternal intestinal HIF-2alpha is necessary for sensing iron demands of lactation in mice, Proc. Natl. Acad. Sci. U. S. A 112 (2015) E3738–E3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fuqua BK, Lu Y, Frazer DM, Darshan D, Wilkins SJ, Dunn L, Loguinov AV, Kogan SC, Matak P, Chen H, Dunaief JL, Vulpe CD, Anderson GJ, Severe iron metabolism defects in mice with double knockout of the multicopper ferroxidases hephaestin and ceruloplasmin, Cell Mol. Gastroenterol. Hepatol 6 (2018) 405–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yanatori I, Yasui Y, Tabuchi M, Kishi F, Chaperone protein involved in transmembrane transport of iron, Biochem. J 462 (2014) 25–37. [DOI] [PubMed] [Google Scholar]
- [23].Yanatori I, Richardson DR, Imada K, Kishi F, Iron export through the transporter ferroportin 1 is modulated by the iron chaperone PCBP2, J. Biol. Chem 291 (2016) 17303–17318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chakrabarti M, Barlas MN, McCormick SP, Lindahl LS, Lindahl PA, Kinetics of iron import into developing mouse organs determined by a pup-swapping method, J. Biol. Chem 290 (2015) 520–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dziuba N, Hardy J, Lindahl PA, Low-molecular-mass iron complexes in blood plasma of iron-deficient pigs do not originate directly from nutrient iron, Metallomics 11 (2019) 1900–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang CY, Knutson MD, Hepatocyte divalent metal-ion transporter-1 is dispensable for hepatic iron accumulation and non-transferrin-bound iron uptake in mice, Hepatology 58 (2013) 788–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Nam H, Wang CY, Zhang L, Zhang W, Hojyo S, Fukada T, Knutson MD, ZIP14 and DMT1 in the liver, pancreas, and heart are differentially regulated by iron deficiency and overload: implications for tissue iron uptake in iron-related disorders, Haematologica 98 (2013) 1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Fillebeen C, Charlebois E, Wagner J, Katsarou A, Mui J, Vali H, Garcia-Santos D, Ponka P, Presley J, Pantopoulos K, Transferrin receptor 1 controls systemic iron homeostasis by fine-tuning hepcidin expression to hepatocellular iron load, Blood 133 (2019) 344–355. [DOI] [PubMed] [Google Scholar]
- [29].La A, Nguyen T, Tran K, Sauble E, Tu D, Gonzalez A, Kidane TZ, Soriano C, Morgan J, Doan M, Tran K, Wang CY, Knutson MD, Linder MC, Mobilization of iron from ferritin: new steps and details, Metallomics 10 (2018) 154–168. [DOI] [PubMed] [Google Scholar]
- [30].Zhang Z, Zhang F, Guo X, An P, Tao Y, Wang F, Ferroportin1 in hepatocytes and macrophages is required for the efficient mobilization of body iron stores in mice, Hepatology 56 (2012) 961–971. [DOI] [PubMed] [Google Scholar]
- [31].Sobh A, Loguinov A, Zhou J, Jenkitkasemwong S, Zeidan R, El Ahmadie N, Tagmount A, Knutson M, Fraenkel PG, Vulpe CD, Genetic screens reveal CCDC115 as a modulator of erythroid iron and heme trafficking, Am. J. Hematol (2020), 10.1002/ajh.25899. [DOI] [PubMed] [Google Scholar]
- [32].Miles AL, Burr SP, Grice GL, Nathan JA, The vacuolar-ATPase complex and assembly factors, TMEM199 and CCDC115, control HIF1alpha prolyl hydroxylation by regulating cellular iron levels, Elife 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ryu MS, Zhang D, Protchenko O, Shakoury-Elizeh M, Philpott CC, PCBP1 and NCOA4 regulate erythroid iron storage and heme biosynthesis, J. Clin. Invest 127 (2017) 1786–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sheftel AD, Zhang AS, Brown C, Shirihai OS, Ponka P, Direct interorganellar transfer of iron from endosome to mitochondrion, Blood 110 (2007) 125–132. [DOI] [PubMed] [Google Scholar]
- [35].Hamdi A, Roshan TM, Kahawita TM, Mason AB, Sheftel AD, Ponka P, Erythroid cell mitochondria receive endosomal iron by a “kiss-and-run” mechanism, Biochim. Biophys. Acta 1863 (2016) 2859–2867. [DOI] [PubMed] [Google Scholar]
- [36].Medlock AE, Shiferaw MT, Marcero JR, Vashisht AA, Wohlschlegel JA, Phillips JD, Dailey HA, Identification of the mitochondrial heme metabolism complex, PLoS One 10 (2015), e0135896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Dong XP, Cheng X, Mills E, Delling M, Wang F, Kurz T, Xu H, The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel, Nature 455 (2008) 992–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhang DL, Hughes RM, Ollivierre-Wilson H, Ghosh MC, Rouault TA, A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression, Cell Metab 9 (2009) 461–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhang DL, Senecal T, Ghosh MC, Ollivierre-Wilson H, Tu T, Rouault TA, Hepcidin regulates ferroportin expression and intracellular iron homeostasis of erythroblasts, Blood 118 (2011) 2868–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhang DL, Ghosh MC, Ollivierre H, Li Y, Rouault TA, Ferroportin deficiency in erythroid cells causes serum iron deficiency and promotes hemolysis due to oxidative stress, Blood 132 (2018) 2078–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhang DL, Wu J, Shah BN, Greutelaers KC, Ghosh MC, Ollivierre H, Su XZ, Thuma PE, Bedu-Addo G, Mockenhaupt FP, Gordeuk VR, Rouault TA, Erythrocytic ferroportin reduces intracellular iron accumulation, hemolysis, and malaria risk, Science 359 (2018) 1520–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Quadri M, Federico A, Zhao T, Breedveld GJ, Battisti C, Delnooz C, Severijnen LA, Mammarella LDT, Mignarri A, Monti L, Sanna A, Lu P, Punzo F, Cossu G, Willemsen R, Rasi F, Oostra BA, van de Warrenburg BP, Bonifati V, Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease, Am. J. Hum. Genet 90 (2012) 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Tuschl K, Clayton PT, Gospe SM Jr., Gulab S, Ibrahim S, Singhi P, Aulakh R, Ribeiro RT, Barsottini OG, Zaki MS, Del Rosario ML, Dyack S, Price V, Rideout A, Gordon K, Wevers RA, Chong WK, Mills PB, Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man, Am. J. Hum. Genet 90 (2012) 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Boycott KM, Beaulieu CL, Kernohan KD, Gebril OH, Mhanni A, Chudley AE, Redl D, Qin W, Hampson S, Kury S, Tetreault M, Puffenberger EG, Scott JN, Bezieau S, Reis A, Uebe S, Schumacher J, Hegele RA, McLeod DR, Galvez-Peralta M, Majewski J, Ramaekers VT, C. Care4Rare Canada, Nebert DW, Innes AM, Parboosingh JS, Abou Jamra R, Autosomal-recessive intellectual disability with cerebellar atrophy syndrome caused by mutation of the manganese and zinc transporter gene SLC39A8, Am. J. Hum. Genet 97 (2015) 886–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Park JH, Hogrebe M, Gruneberg M, DuChesne I, von der Heiden AL, Reunert J, Schlingmann KP, Boycott KM, Beaulieu CL, Mhanni AA, Innes AM, Hortnagel K, Biskup S, Gleixner EM, Kurlemann G, Fiedler B, Omran H, Rutsch F, Wada Y, Tsiakas K, Santer R, Nebert DW, Rust S, Marquardt T, SLC39A8 deficiency: a disorder of manganese transport and glycosylation, Am. J. Hum. Genet 97 (2015) 894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tuschl K, Meyer E, Valdivia LE, Zhao N, Dadswell C, Abdul-Sada A, Hung CY, Simpson MA, Chong WK, Jacques TS, Woltjer RL, Eaton S, Gregory A, Sanford L, Kara E, Houlden H, Cuno SM, Prokisch H, Valletta L, Tiranti V, Younis R, Maher ER, Spencer J, Straatman-Iwanowska A, Gissen P, Selim LA, Pintos-Morell G, Coroleu-Lletget W, Mohammad SS, Yoganathan S, Dale RC, Thomas M, Rihel J, Bodamer OA, Enns CA, Hayflick SJ, Clayton PT, Mills PB, Kurian MA, Wilson SW, Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia, Nat. Commun 7 (2016) 11601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Michalke B, Fernsebner K, New insights into manganese toxicity and speciation, J. Trace Elem. Med. Biol 28 (2014) 106–116. [DOI] [PubMed] [Google Scholar]
- [48].Jursa T, Smith DR, Ceruloplasmin alters the tissue disposition and neurotoxicity of manganese, but not its loading onto transferrin, Toxicol. Sci 107 (2009) 182–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Foulquier F, Amyere M, Jaeken J, Zeevaert R, Schollen E, Race V, Bammens R, Morelle W, Rosnoblet C, Legrand D, Demaegd D, Buist N, Cheillan D, Guffon N, Morsomme P, Annaert W, Freeze HH, Van Schaftingen E, Vikkula M, Matthijs G, TMEM165 deficiency causes a congenital disorder of glycosylation, Am. J. Hum. Genet 91 (2012) 15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Demaegd D, Foulquier F, Colinet AS, Gremillon L, Legrand D, Mariot P, Peiter E, Van Schaftingen E, Matthijs G, Morsomme P, Newly characterized Golgi-localized family of proteins is involved in calcium and pH homeostasis in yeast and human cells, Proc. Natl. Acad. Sci. U. S. A 110 (2013) 6859–6864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Colinet AS, Thines L, Deschamps A, Flemal G, Demaegd D, Morsomme P, Acidic and uncharged polar residues in the consensus motifs of the yeast Ca(2+) transporter Gdt1p are required for calcium transport, Cell Microbiol 19 (2017). [DOI] [PubMed] [Google Scholar]
- [52].Potelle S, Morelle W, Dulary E, Duvet S, Vicogne D, Spriet C, Krzewinski-Recchi MA, Morsomme P, Jaeken J, Matthijs G, De Bettignies G, Foulquier F, Glycosylation abnormalities in Gdt1p/TMEM165 deficient cells result from a defect in Golgi manganese homeostasis, Hum. Mol. Genet 25 (2016) 1489–1500. [DOI] [PubMed] [Google Scholar]
- [53].Potelle S, Dulary E, Climer L, Duvet S, Morelle W, Vicogne D, Lebredonchel E, Houdou M, Spriet C, Krzewinski-Recchi MA, Peanne R, Klein A, de Bettignies G, Morsomme P, Matthijs G, Marquardt T, Lupashin V, Foulquier F, Manganese-induced turnover of TMEM165, Biochem. J 474 (2017) 1481–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Stribny J, Thines L, Deschamps A, Goffin P, Morsomme P, The human Golgi protein TMEM165 transports calcium and manganese in yeast and bacterial cells, J. Biol. Chem 295 (2020) 3865–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Thines L, Deschamps A, Sengottaiyan P, Savel O, Stribny J, Morsomme P, The yeast protein Gdt1p transports Mn(2+) ions and thereby regulates manganese homeostasis in the Golgi, J. Biol. Chem 293 (2018) 8048–8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Foulquier F, Legrand D, Biometals and glycosylation in humans: congenital disorders of glycosylation shed lights into the crucial role of Golgi manganese homeostasis, Biochim. Biophys. Acta Gen. Subj 2020 (1864) 129674. [DOI] [PubMed] [Google Scholar]
- [57].Leyva-Illades D, Chen P, Zogzas CE, Hutchens S, Mercado JM, Swaim CD, Morrisett RA, Bowman AB, Aschner M, Mukhopadhyay S, SLC30A10 is a cell surface-localized manganese efflux transporter, and parkinsonism-causing mutations block its intracellular trafficking and efflux activity, J. Neurosci 34 (2014) 14079–14095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hutchens S, Liu C, Jursa T, Shawlot W, Chaffee BK, Yin W, Gore AC, Aschner M, Smith DR, Mukhopadhyay S, Deficiency in the manganese efflux transporter SLC30A10 induces severe hypothyroidism in mice, J. Biol. Chem 292 (2017) 9760–9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Taylor CA, Hutchens S, Liu C, Jursa T, Shawlot W, Aschner M, Smith DR, Mukhopadhyay S, SLC30A10 transporter in the digestive system regulates brain manganese under basal conditions while brain SLC30A10 protects against neurotoxicity, J. Biol. Chem 294 (2019) 1860–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Papavasiliou PS, Miller ST, Cotzias GC, Role of liver in regulating distribution and excretion of manganese, Am. J. Phys 211 (1966) 211–216. [DOI] [PubMed] [Google Scholar]
- [61].Bertinchamps AJ, Miller ST, Cotzias GC, Interdependence of routes excreting manganese, Am. J. Phys 211 (1966) 217–224. [DOI] [PubMed] [Google Scholar]
- [62].Mercadante CJ, Prajapati M, Conboy HL, Dash ME, Herrera C, Pettiglio MA, Cintron-Rivera L, Salesky MA, Rao DB, Bartnikas TB, Manganese transporter Slc30a10 controls physiological manganese excretion and toxicity, J. Clin. Invest 129 (2019) 5442–5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Begum NA, Kobayashi M, Moriwaki Y, Matsumoto M, Toyoshima K, Seya T, Mycobacterium bovis BCG cell wall and lipopolysaccharide induce a novel gene, BIGM103, encoding a 7-TM protein: identification of a new protein family having Zn-transporter and Zn-metalloprotease signatures, Genomics 80 (2002) 630–645. [DOI] [PubMed] [Google Scholar]
- [64].Girijashanker K, He L, Soleimani M, Reed JM, Li H, Liu Z, Wang B, Dalton TP, Nebert DW, Slc39a14 gene encodes ZIP14, a metal/bicarbonate symporter: similarities to the ZIP8 transporter, Mol. Pharmacol 73 (2008) 1413–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Jenkitkasemwong S, Wang CY, Mackenzie B, Knutson MD, Physiologic implications of metal-ion transport by ZIP14 and ZIP8, Biometals 25 (2012) 643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Galvez-Peralta M, He L, Jorge-Nebert LF, Wang B, Miller ML, Eppert BL, Afton S, Nebert DW, ZIP8 zinc transporter: indispensable role for both multiple-organ organogenesis and hematopoiesis in utero, PLoS One 7 (2012), e36055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Tanimura N, Liao R, Wilson GM, Dent MR, Cao M, Burstyn JN, Hematti P, Liu X, Zhang Y, Zheng Y, Keles S, Xu J, Coon JJ, Bresnick EH, GATA/heme multi-omics reveals a trace metal-dependent cellular differentiation mechanism, Dev. Cell 46 (2018) 581–594 ( e584). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Lin W, Vann DR, Doulias PT, Wang T, Landesberg G, Li XL, Ricciotti E, Scalia R, He M, Hand NJ, Rader DJ, Hepatic metal ion transporter ZIP8 regulates manganese homeostasis and manganese-dependent enzyme activity, J. Clin. Invest 127 (2017) 2407–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Taylor KM, Morgan HE, Johnson A, Nicholson RI, Structure-function analysis of a novel member of the LIV-1 subfamily of zinc transporters, ZIP14, FEBS Lett 579 (2005) 427–432. [DOI] [PubMed] [Google Scholar]
- [70].Pinilla-Tenas JJ, Sparkman BK, Shawki A, Illing AC, Mitchell CJ, Zhao N, Liuzzi JP, Cousins RJ, Knutson MD, Mackenzie B, Zip14 is a complex broad-scope metal-ion transporter whose functional properties support roles in the cellular uptake of zinc and nontransferrin-bound iron, Am. J. Physiol. Cell Physiol 301 (2011) C862–C871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Aydemir TB, Kim MH, Kim J, Colon-Perez LM, Banan G, Mareci TH, Febo M, Cousins RJ, Metal transporter Zip14 (Slc39a14) deletion in mice increases manganese deposition and produces neurotoxic signatures and diminished motor activity, J. Neurosci 37 (2017) 5996–6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Jenkitkasemwong S, Akinyode A, Paulus E, Weiskirchen R, Hojyo S, Fukada T, Giraldo G, Schrier J, Garcia A, Janus C, Giasson B, Knutson MD, SLC39A14 deficiency alters manganese homeostasis and excretion resulting in brain manganese accumulation and motor deficits in mice, Proc. Natl. Acad. Sci. U. S. A 115 (2018) E1769–E1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Xin Y, Gao H, Wang J, Qiang Y, Imam MU, Li Y, Wang J, Zhang R, Zhang H, Yu Y, Wang H, Luo H, Shi C, Xu Y, Hojyo S, Fukada T, Min J, Wang F, Manganese transporter Slc39a14 deficiency revealed its key role in maintaining manganese homeostasis in mice, Cell Discov 3 (2017) 17025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Liu C, Hutchens S, Jursa T, Shawlot W, Polishchuk EV, Polishchuk RS, Dray BK, Gore AC, Aschner M, Smith DR, Mukhopadhyay S, Hypothyroidism induced by loss of the manganese efflux transporter SLC30A10 may be explained by reduced thyroxine production, J. Biol. Chem 292 (2017) 16605–16615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Scheiber IF, Wu Y, Morgan SE, Zhao N, The intestinal metal transporter ZIP14 maintains systemic manganese homeostasis, J. Biol. Chem 294 (2019) 9147–9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ye Q, Park JE, Gugnani K, Betharia S, Pino-Figueroa A, Kim J, Influence of iron metabolism on manganese transport and toxicity, Metallomics 9 (2017) 1028–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Seo YA, Wessling-Resnick M, Ferroportin deficiency impairs manganese metabolism in flatiron mice, FASEB J 29 (2015) 2726–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Madejczyk MS, Ballatori N, The iron transporter ferroportin can also function as a manganese exporter, Biochim. Biophys. Acta 1818 (2012) 651–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Mitchell CJ, Shawki A, Ganz T, Nemeth E, Mackenzie B, Functional properties of human ferroportin, a cellular iron exporter reactive also with cobalt and zinc, Am. J. Physiol. Cell Physiol 306 (2014) C450–C459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Choi EK, Nguyen TT, Iwase S, Seo YA, Ferroportin disease mutations influence manganese accumulation and cytotoxicity, FASEB J 33 (2019) 2228–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Nishito Y, Tsuji N, Fujishiro H, Takeda TA, Yamazaki T, Teranishi F, Okazaki F, Matsunaga A, Tuschl K, Rao R, Kono S, Miyajima H, Narita H, Himeno S, Kambe T, Direct comparison of manganese detoxification/efflux proteins and molecular characterization of ZnT10 protein as a manganese transporter, J. Biol. Chem 291 (2016) 14773–14787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Thompson KJ, Hein J, Baez A, Sosa JC, Wessling-Resnick M, Manganese transport and toxicity in polarized WIF-B hepatocytes, Am. J. Physiol. Gastrointest. Liver Physiol 315 (2018) G351–G363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Yin ZB, Jiang HY, Lee ESY, Ni MW, Erikson KM, Milatovic D, Bowman AB, Aschner M, Ferroportin is a manganese-responsive protein that decreases manganese cytotoxicity and accumulation, J. Neurochem 112 (2010) 1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Jin L, Frazer DM, Lu Y, Wilkins SJ, Ayton S, Bush A, Anderson GJ, Mice overexpressing hepcidin suggest ferroportin does not play a major role in Mn homeostasis, Metallomics 11 (2019) 959–967. [DOI] [PubMed] [Google Scholar]
- [85].Aydemir TB, Thorn TL, Ruggiero CH, Pompilus M, Febo M, Cousins RJ, Intestine-specific deletion of metal transporter Zip14 (Slc39a14) causes brain manganese overload and locomotor defects of manganism, Am. J. Physiol. Gastrointest. Liver Physiol 318 (2020) G673–G681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Steimle BL, Smith FM, Kosman DJ, The solute carriers ZIP8 and ZIP14 regulate manganese accumulation in brain microvascular endothelial cells and control brain manganese levels, J. Biol. Chem 294 (2019) 19197–19208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].van Raaij S, van Swelm R, Bouman K, Cliteur M, van den Heuvel MC, Pertijs J, Patel D, Bass P, van Goor H, Unwin R, Srai SK, Swinkels D, Tubular iron deposition and iron handling proteins in human healthy kidney and chronic kidney disease, Sci. Rep 8 (2018) 9353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].van Raaij SEG, Srai SKS, Swinkels DW, van Swelm RPL, Iron uptake by ZIP8 and ZIP14 in human proximal tubular epithelial cells, Biometals 32 (2019) 211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Heilig EA, Thompson KJ, Molina RM, Ivanov AR, Brain JD, Wessling-Resnick M, Manganese and iron transport across pulmonary epithelium, Am. J. Physiol. Lung Cell Mol. Physiol 290 (2006) L1247–L1259. [DOI] [PubMed] [Google Scholar]
- [90].Scheiber IF, Alarcon NO, Zhao N, Manganese Uptake by A549 Cells Is Mediated by both ZIP8 and ZIP14, Nutrients, 11, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]