

Abstract
Background:
Patients with Parkinson’s disease (PD) show motor symptoms as well as various non-motor symptoms. Postmortem studies of PD have suggested that initial α-Synuclein pathology develops independently in the olfactory bulb and lower brainstem, spreading from there stereotypically. However, it remains unclear how these two pathological pathways contribute to the clinicopathological progression of PD.
Objective:
The objective of this study was to examine the clinicopathological contribution of α-Synuclein spread from the olfactory bulb.
Methods:
We conducted pathological and behavioral analyses of human α-Synuclein bacterial artificial chromosome transgenic mice injected with α-Synuclein preformed fibrils into the bilateral olfactory bulb up to 10 months postinjection.
Results:
α-Synuclein preformed fibril injections induced more widespread α-Synuclein pathology in the transgenic mice than that in wild-type mice. Severe α-Synuclein pathology in the transgenic mice injected with α-Synuclein preformed fibrils was initially observed along the olfactory tract and later in the brain regions which are included in the limbic system and have connections with it. The α-Synuclein pathology was accompanied by regional atrophy, neuron loss, reactive astrogliosis, and microglial activation, which were remarkable in the hippocampus. Behavioral analyses revealed hyposmia, followed by anxiety-like behavior and memory impairment, but not motor dysfunction, depression-like behavior, or circadian rhythm disturbance.
Conclusion:
Our data suggest that α-Synuclein spread from the olfactory bulb mainly affects the olfactory tract and limbic system as well as its related regions, leading to the development of hyposmia, anxiety, and memory loss in PD.
Keywords: Parkinson’s disease, α-Synuclein, Propagation, Olfactory bulb, Behavioral abnormalities
Introduction
Pathological features of Parkinson’s disease (PD) include dopaminergic neuron loss in the substantia nigra pars compacta (SNpc) and intraneuronal α-Syn inclusions, called Lewy bodies (LBs) and Lewy neurites.1 Clinically, dopamine depletion in the nigrostriatal system causes motor symptoms, which can be partially relieved by dopamine replacement therapy. However, patients with PD also show non-motor symptoms such as hyposmia, anxiety, cognitive dysfunction, depression, apathy, rapid eye movement sleep behavior disorder (RBD), urinary dysfunction, constipation, and orthostatic hypotension.1 They often require clinical management, and some of them have been reported to be prodromal symptoms of PD.
In accordance with a variety of symptoms in PD, Lewy pathology is not limited to dopaminergic neurons in the SNpc. Braak et al. systematically analyzed the pathology in subjects with incidental LB pathology and PD, suggesting that Lewy pathology develops in the olfactory bulb (OB) and the dorsal motor nucleus of the vagus nerve (dmX) in the earliest stage and then spreads from there in a stereotypical manner.2 Considering that Lewy pathology starts in the extranigral regions and spreads to a wide range of brain regions,2 the diverse distribution of Lewy pathology may underlie the development of various non-motor symptoms of PD.3 However, it remains unclear how Lewy pathology spreads from the two independent initial regions, the OB and dmX, and how each pathological pathway contributes to the development of clinical symptoms of PD.
Accumulating evidence suggests that templated fibrillization and interneuronal dissemination of α-Syn aggregates underlie the spread of α-Syn pathology in brains, supporting Braak’s hypothesis.4–6 Taking advantage of injection methodology which can trigger pathology in certain brain regions, we and others previously analyzed wild-type (WT) mice injected with α-Syn PFFs into the OB.7–9 In these studies, unilateral OB injections of α-Syn PFFs induced dense α-Syn pathology mainly along the olfactory tract at early time points. Although α-Syn pathology further spread in the brains up to 18 months postinjection, only mild pathology appeared in most of the affected brain regions at later time points.8, 9 Moreover, these mice showed olfactory deficits, but not other behavioral abnormalities up to 12 months postinjection.7 Another group analyzed WT mice injected with α-Syn PFFs into the OB/anterior olfactory nucleus (AON) complex.10, 11 These mice exhibited α-Syn pathology in the limbic structures at 3 months postinjection,10 but it remained centered there up to 10.5 months postinjection.11 These previous data suggest that even long incubation of WT mice after the unilateral OB injections of α-Syn PFFs does not fully recapitulate pathological progression or neurological deficits which may be developed over the years in patients with PD.
We recently generated a novel PD mouse model, i.e. the bacterial artificial chromosome (BAC) transgenic (Tg) mice harboring human α-Syn gene (SNCA) with the A53T mutation and risk SNPs.12 These heterozygous A53T BAC-SNCA Tg mice express ~1.5-fold more α-Syn in the brains compared with WT mice, with the expression pattern resembling that of endogenous mouse α-Syn. In this regard, they have the genetic feature of SNCA duplication. They exhibited RBD-like behavior and hyposmia, both of which are known to be prodromal symptoms of PD. Pathologically, they showed slight dopaminergic neuron loss and proteinase K-resistant α-Syn pathology in a wide range of regions, but not LB-like inclusions.
In the present study, to accelerate pathological progression, we employed the bilateral OB injections of mouse α-Syn PFFs on A53T BAC-SNCA Tg mice. We conducted comprehensive pathological and behavioral analyses of these mice to examine the clinicopathological contribution of α-Syn spread from the OB.
Methods
Animals and ethics statement
Heterozygous A53T BAC-SNCA Tg and WT mice with C57BL/6J background at 2–3 months of age were used in this study.12 All experimental procedures followed national guidelines. The Animal Research Committee of Kyoto University granted ethical approval and permission.
Sequential extraction and western blot analysis
Sequential extraction and western blot analysis were performed as previously described with minor modifications.12 Details are provided in the Supplementary Methods.
Preparation of recombinant α-Syn monomers and preformed fibrils
Mouse α-Syn PFFs were generated as described previously.13 Briefly, mouse α-Syn was expressed in Escherichia coli BL21 (DE3) (BioDynamics Laboratory) and was purified by boiling and ion exchange using Q sepharose fast flow (GE Healthcare). After dialyzed against dialysis buffer (150 mM KCl, 50 mM Tris-HCl, pH 7.5), α-Syn solution (7 mg/ml) was agitated at 37°C at 1,000 rpm for 10 d. After ultracentrifugation at 186,000 g at 20°C for 20 min, the pellets of α-Syn PFFs were resuspended in phosphate-buffered saline (PBS) (5 μg/μl) and then sonicated with a Bioruptor ultrasonic wave disruption system for 2 min before injections. The characterization of α-Syn PFFs before and after sonication was described previously.13, 14
Stereotaxic surgery
Mice anesthetized with Avertin received stereotaxic injections of 0.5 μl of α-Syn PFFs in PBS (5 μg/μl) or PBS into the bilateral OB (coordinates: −1.0 mm relative to the inferior cerebral vein; 0.9 mm from the midline; 1.5 mm beneath the scull surface) using a 33 gauge Neuros syringe (Hamilton).
Histologic and immunohistochemical analysis
Histologic and immunohistochemical analyses were performed as described previously.13 Details are provided in the Supplementary Methods. Primary antibodies used in this study are listed in Table S1. For assessment of α-Syn pathology throughout the brains, every 10th paraffin section was stained with a phosphorylated α-Syn (pSyn) antibody (EP1536Y). Semiquantitative analyses were performed on eight coronal sections (4.28, 2.46, 1.34, 0.26, −0.34, −1.58, −3.16, and −5.40 mm relative to bregma), and color coded onto heat maps. Phosphorylated α-Syn–positive pathology was scored as mild to very severe pathology based on the following criteria: mild, neuritic pathology or 0–1 LB-like inclusion; moderate, 2–3 LB-like inclusions; severe, 4–8 LB-like inclusions; very severe, 9 or more LB-like inclusions seen with a 40× objective lens. Scores were determined by the observations of at least 2 mice for each time point (n = 3, 4, and 2 for 2, 6, and 10 months postinjection, respectively). Quantification of total hippocampal and Nissl-positive areas in the dorsal and ventral hippocampus (−1.94, and −3.28 mm relative to bregma, respectively), thickness of the piriform cortex (−1.94 mm relative to bregma), pSyn-positive, glial fibrillary acidic protein (GFAP)-positive, and ionized calcium-binding adaptor protein-1 (Iba-1)–positive areas was performed with the ImageJ. The number of Nissl-positive neurons with visible nuclei in the dorsal hippocampus was manually counted for sampled neuronal density. The number of choline acetyltransferase (ChAT)-positive cells with visible nuclei in the nucleus basalis magnocellularis (NBM) was manually counted in every 10th section. Sections were examined with a BX43 microscope (Olympus) and an FV-1000 confocal laser scanning microscope (Olympus).
Behavioral analysis
Behavioral analyses were conducted using male mice. For the comparisons among WT mice injected with PBS (WT-PBS mice) and A53T BAC-SNCA Tg mice injected with PBS (Tg-PBS mice) or α-Syn PFFs (Tg-PFF mice), a batch composed of 13, 13, and 14 mice, respectively, was subjected to the smell test at 2 months postinjection and to the other behavioral tests from 7 to 10 months postinjection. One Tg-PFF mouse died at 9 months postinjection. An additional batch composed of 6 mice for each group was subjected to the olfactory preference test using peanut butter (Fig. 4B). Before every test, the mice were habituated to the experimental environment for more than 30 min. The details of each behavioral test are provided in the Supplementary Methods.
Statistical analysis
Statistical calculations were performed with GraphPad Prism Software, Version 7.04. An F test, a Brown–Forsythe test, and a Bartlett’s test were performed to evaluate the differences in variances. Differences with p values of less than 0.05 were considered significant.
Results
Characterization of α-Syn PFF–induced α-Syn pathology in A53T BAC-SNCA Tg mice
To characterize α-Syn PFF–induced α-Syn pathology in A53T BAC-SNCA Tg mice, we injected the Tg mice as well as WT mice with α-Syn PFFs or PBS into the bilateral OB and analyzed them at 2 months postinjection. Consistent with our previous study,9 the WT mice injected with α-Syn PFFs (WT-PFF mice) exhibited pSyn pathology almost restricted to the olfactory tract and the ventral Cornu Ammonis 1 (vCA1), which directly project to the OB (Fig. 1A).15 Given that the α-Syn pathology primarily spreads retrogradely after injections of α-Syn PFFs,16 these affected regions can be considered the first-order regions of α-Syn spread from the injection site. Because A53T BAC-SNCA Tg mice express higher amount of pSyn as well as α-Syn than WT mice,12 pSyn immunohistochemistry revealed stronger pSyn immunoreactivity in the Tg-PBS mice than that in the WT-PBS mice. However, α-Syn PFF–induced α-Syn pathology was clearly distinguished based on its even stronger pSyn staining intensity and morphology. The Tg-PFF mice exhibited α-Syn pathology in the brain regions beyond the olfactory tract and the vCA1, such as the ventral and dorsal dentate gyrus (vDG and dDG, respectively), suggesting α-Syn pathology spread beyond the first-order regions. In terms of severity, the Tg-PFF mice showed comparable or more severe α-Syn pathology in the affected brain regions than the WT-PFF mice except for the AON. The EP1536Y-positive pSyn pathology in the Tg-PFF mice was also detected by other pSyn antibodies and by antibodies to misfolded α-Syn (Fig. 1B). The pSyn-positive pathology in the Tg-PFF mice was also positive for p62, ubiquitin, and thioflavin S (Fig. 1C). We further characterized α-Syn aggregation in the Tg-PFF mice by western blot analyses (Fig. 1D). In Triton-soluble fractions, there were no significant differences in total α-Syn, human α-Syn, or pSyn expression between the WT-PBS and WT-PFF mice and between the Tg-PBS and Tg-PFF mice. In SDS-soluble fractions, high molecular weight bands of total α-Syn were observed only in the WT-PFF and Tg-PFF mice. The WT-PFF mice showed a significant increase in total α-Syn and pSyn expression compared with the WT-PBS mice. Likewise, the Tg-PFF mice showed significant increase in pSyn expression and trend towards increase in total and human α-Syn expression compared with the Tg-PBS mice.
Fig. 1. Characterization of α-Syn pathology induced by α-Syn PFF injections in A53T BAC-SNCA Tg mice.
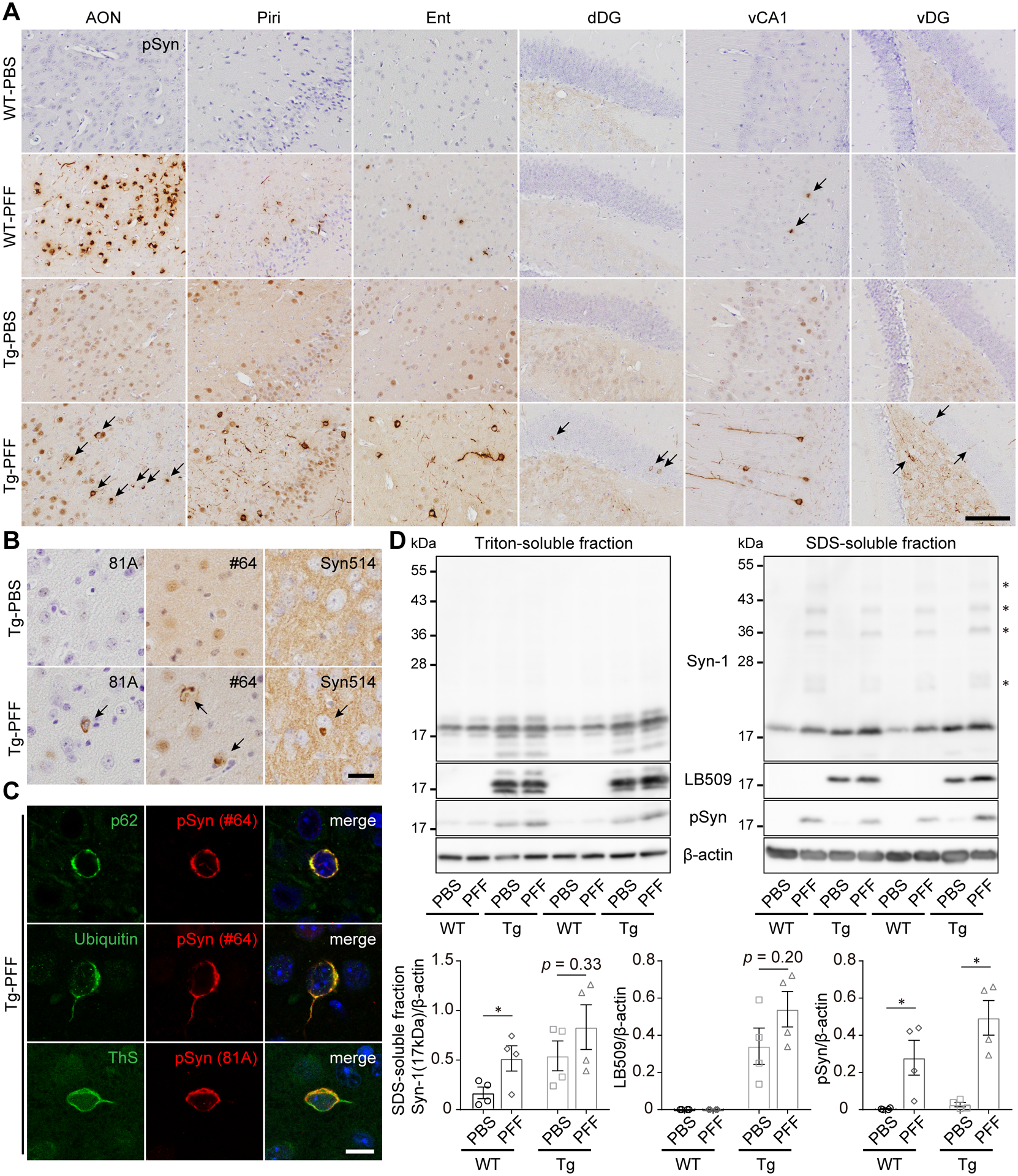
(A–D) The WT-PBS, WT-PFF, Tg-PBS, and Tg-PFF mice were analyzed at 2 months postinjection (MPI). (A) Phosphorylated α-Syn immunohistochemistry (EP1536Y) in the AON, piriform cortex (Piri), entorhinal cortex (Ent), vCA1, vDG, and dDG. Scale bar 100 μm. (B) Immunohistochemistry assessing pSyn (81A and #64) and misfolded α-Syn (Syn514) in the AON shows pSyn-positive and misfolded α-Syn–positive inclusions (arrows) in the Tg-PFF mice. Scale bar 20 μm. (C) Confocal microscope images in the AON of the Tg-PFF mice. The order from top to bottom: p62 (green) and pSyn (#64, red), ubiquitin (green) and pSyn (#64, red), thioflavin S (ThS, green) and pSyn (81A, red). DAPI (blue) in the merged images. Scale bar 10 μm. (D) Western blot analysis of sequentially extracted ventral half of the cortices. Upper panels: Representative immunoblots of Triton-soluble and SDS-soluble fractions with Syn-1 (human + mouse α-Syn), LB509 (human α-Syn), pSyn, and β-actin antibodies. Asterisks indicate high molecular weight bands. Numbers represent molecular weight markers of migrated protein standards (kDa). Lower panels: quantification of Syn-1 (17kDa), LB509, pSyn-positive bands normalized by β-actin in SDS-soluble fractions. For Syn-1 (17kDa) and LB509, a two-tailed unpaired Student’s t-test was performed; *p < 0.05. For pSyn, a Mann-Whitney test was performed; *p < 0.05. WT-PBS (n = 4), WT-PFF (n = 4), Tg-PBS (n = 4) and Tg-PFF (n = 4). Data are the mean ± SEM.
Widespread, severe α-Syn pathology in A53T BAC-SNCA Tg mice injected with α-Syn PFFs
We conducted pathological and behavioral analyses of the Tg-PFF mice together with the WT-PBS and Tg-PBS mice as controls up to 10 months postinjection (Fig. 2A). At 2 months after injections of α-Syn PFFs, moderate to severe α-Syn pathology was observed not only along the olfactory tract but also in the BLA, vCA1, vDG, and dDG, and mild pathology in several brain regions, such as the orbital cortex, cingulate cortex, insular cortex, septal nucleus, nucleus of accumbens (NAcc), diagonal band, preoptic area, ventral pallidum, hypothalamus, and mammillary nucleus (Fig. 2B,C). These brain regions are included in the limbic system or have direct connections with it.17–19 At 6 months after injections of α-Syn PFFs, very severe pathology was observed in the dDG, vDG, and entorhinal cortex. Severe pathology was seen in the AON, NAcc, and vCA1. Mild to moderate pathology appeared in the paraventricular thalamic nucleus (PVA), caudate putamen, and globus pallidus. At 10 months after injections of α-Syn PFFs, very severe pathology in the NAcc and severe pathology in the bed nucleus of stria terminalis (BNST), PVA, dorsal and ventral hippocampus, central amygdala (CeA), and entorhinal cortex were observed. Mild pathology appeared in the SNpc and the pontine tegmentum area, including the locus coeruleus (LC). α-Syn pathology was not identified in the dmX at any time points. Because the association of the ChAT-positive neuron loss in the nucleus basalis of Meynert with cognitive dysfunction has been reported in PD,20 we examined the pathological changes in the NBM of the Tg-PFF mice, showing rare α-Syn pathology without decrease in the number of ChAT-positive neurons (Fig. S1).
Fig. 2. Spatiotemporal distribution of α-Syn pathology in A53T BAC-SNCA Tg mice injected with α-Syn PFFs.
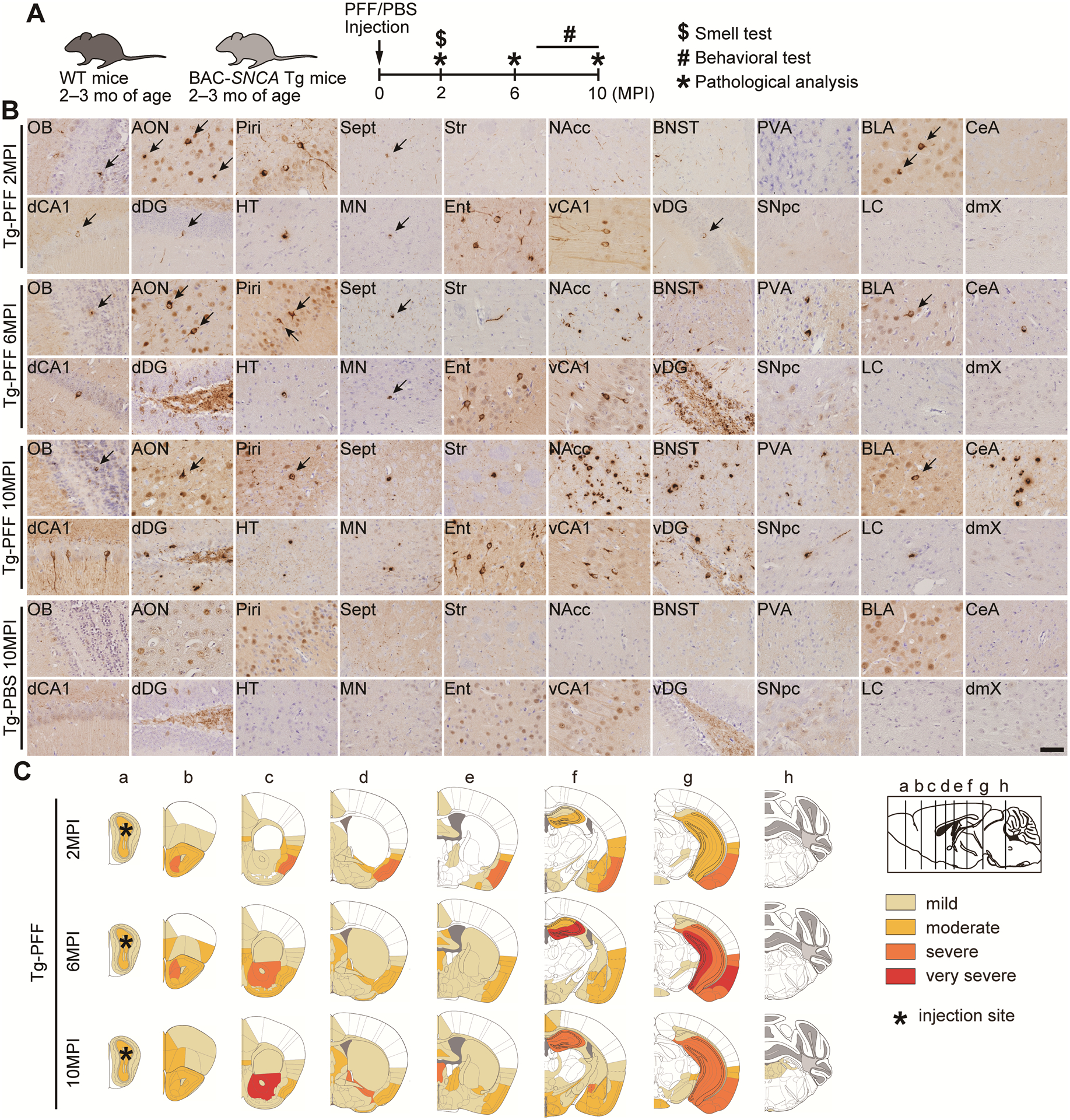
(A) Schematic representation of experimental design. (B) Phosphorylated α-Syn immunohistochemistry (EP1536Y) in the indicated brain regions of the Tg-PFF mice at 2, 6, and 10MPI and the Tg-PBS mice at 10MPI. Arrows indicate pSyn-positive inclusions. Sept, septal nucleus; Str, Striatum; dCA1, dorsal CA1; HT, hypothalamus; MN, mammillary nucleus. Scale bar 50 μm. (C) Heat map colors represent the extent of α-Syn pathology (light yellow, mild pathology; yellow, moderate pathology; orange, severe pathology, red; very severe pathology).
α-Syn pathology is accompanied by atrophy, neuron loss, and gliosis in A53T BAC-SNCA Tg mice injected with α-Syn PFFs
Because we found considerable amount of α-Syn pathology in the hippocampus of the Tg-PFF mice, we focused on this region to examine temporal relationships among α-Syn pathology, atrophy, neuron loss, and gliosis. First, we examined the atrophy and neuron loss by Nissl staining. The total area of ventral and dorsal hippocampus was significantly decreased at 6 and 10 months postinjection, respectively (Fig. 3A,B,S2A,B). Neuron loss was evaluated by the Nissl-positive area of each hippocampal region. Significant neuron loss was seen in the dCA2/3, dDG, and vCA1 at 10 months postinjection and in the vDG at 6 and 10 months postinjection (Fig. 3A,C–E,S2A,C–E). High magnification images and Nissl-positive neuronal density in each subregion of the dorsal hippocampus are provided in Fig. S3.
Fig. 3. Hippocampal atrophy, neuron loss, and gliosis in A53T BAC-SNCA Tg mice injected with α-Syn PFFs.
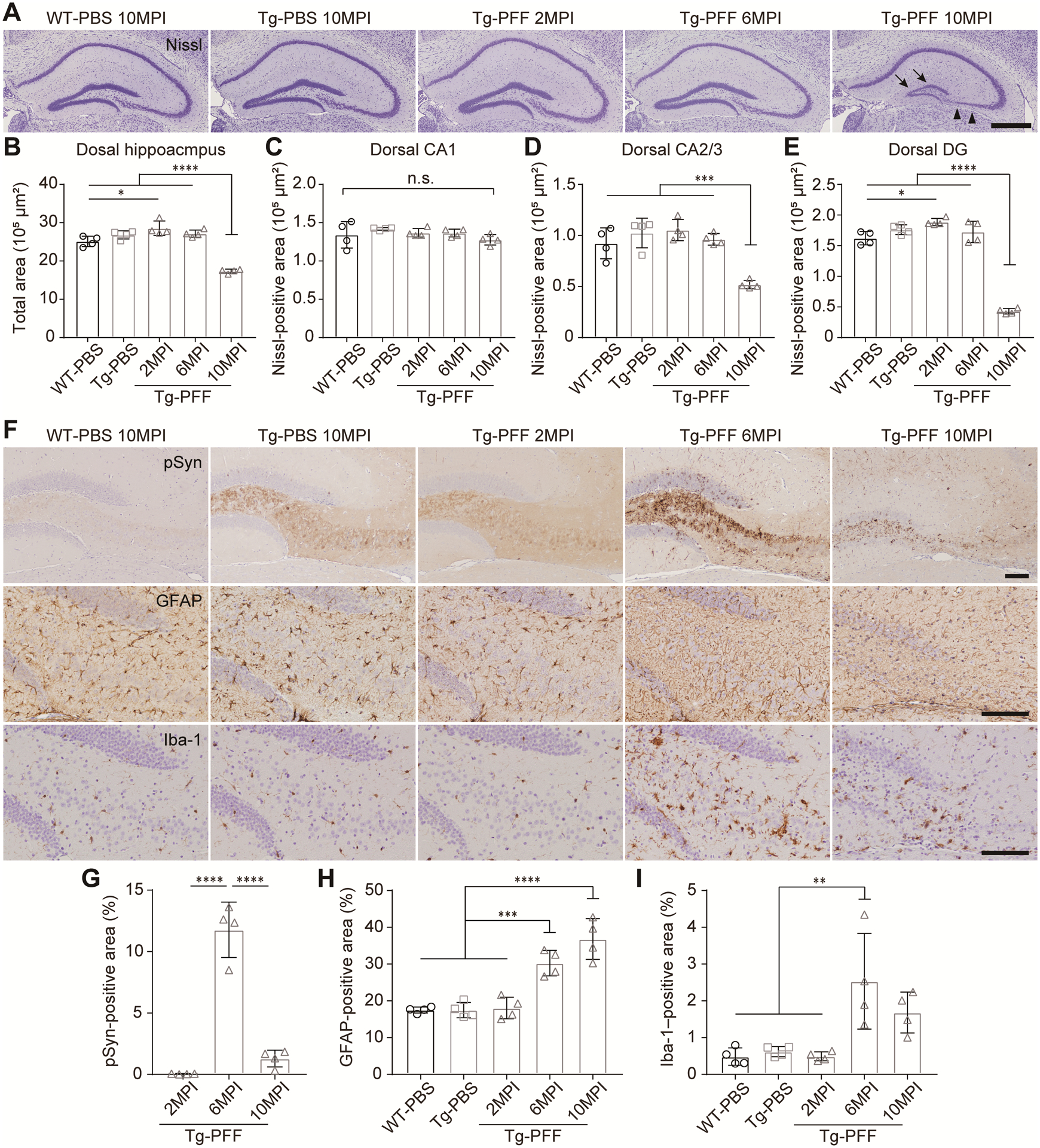
(A) Nissl staining in the dorsal hippocampus. Arrowheads and arrows indicate neuron loss in the CA3 and DG, respectively. Scale bar 500 μm. (B) Total area of the dorsal hippocampus. (C–E) Nissl-positive area in the CA1, CA2/3, and DG of the dorsal hippocampus. (F) Phosphorylated α-Syn (EP1536Y), GFAP, and Iba-1 immunohistochemistry in the region across CA2/3, hilus and DG of the dorsal hippocampus. Scale bar 100 μm. (G) Percentage of pSyn-positive area occupied in this region. One-way ANOVA with Tukey’s post hoc test was performed; ****p < 0.0001. (H) Percentage of GFAP-positive area occupied in this region. One-way ANOVA with Tukey’s post hoc test was performed; ***p < 0.001, ****p < 0.0001. (I) Percentage of Iba-1–positive area occupied in this region. One-way ANOVA with Tukey’s post hoc test was performed; **p < 0.01. WT-PBS (n = 4), Tg-PBS (n = 4) and Tg-PFF (n = 4). Data are the mean ± SEM.
Next, we evaluated α-Syn pathology, astrogliosis, and microglial activation in the dorsal hippocampus of the Tg-PFF mice (Fig. 3F–I). At 2 months postinjection, little pSyn-positive pathology without astrogliosis or microglial activation was observed. At 6 months postinjection, pSyn-positive pathology became abundant, accompanied by reactive astrogliosis and microglial activation. At 10 months postinjection, pSyn-positive pathology became less abundant, but reactive astrogliosis and microglial activation still remained.
We also examined the pathological changes in the piriform cortex (Fig. S4). In accordance with the appearance of severe α-Syn pathology, reactive astrogliosis and microglial activation were observed in the Tg-PFF mice at 2 months postinjection. Reduced thickness with decrease in severity of α-Syn pathology was observed at 10 months postinjection.
Taken together, reactive astrogliosis and microglial activation accompanied the appearance of severe α-Syn pathology, preceding significant neuron loss. These findings suggest that α-Syn aggregation as well as neuron loss induced the glial responses. The decrease in pSyn-positive area in parallel with severe neuron loss at 10 months postinjection probably reflects death of α-Syn inclusion–bearing neurons.
Hyposmia, anxiety-like behavior, and memory impairment in A53T BAC-SNCA Tg mice injected with α-Syn PFFs
We conducted a comprehensive battery of behavioral tests to examine behavioral abnormalities related to PD in the Tg-PFF mice. We performed the olfactory preference test at 2 months postinjection. The WT-PBS and Tg-PBS mice sniffed female mouse urine and peanut butter longer than water, while the Tg-PFF mice did not, suggesting that the Tg-PFF mice could not distinguish the odorants from water (Fig. 4A,B). The differences in sniffing time between the odorants and water indicated worse olfactory function of the Tg-PFF mice than that of the WT-PBS and Tg-PBS mice.
Fig. 4. Hyposmia and anxiety-like behavior in A53T BAC-SNCA Tg mice injected with α-Syn PFFs.

(A,B) Olfactory preference test conducted at 2MPI. (A) Sniffing time on water vs. female urine. WT-PBS (n = 13), Tg-PBS (n = 13), and Tg-PFF (n = 14). Left panel: a two-tailed paired test was performed for each group; *p < 0.05, **p < 0.01, n.s., not significant. Right panel: time difference between water and female urine. One-way ANOVA with Tukey’s post hoc test was performed; *p < 0.05. (B) Sniffing time on water vs. peanut butter. WT-PBS (n = 19), Tg-PBS (n = 19), and Tg-PFF (n = 20). Left panel: a two-tailed paired test was performed for each group; *p < 0.05, n.s., not significant. Right panel: time difference between water and peanut butter. One-way ANOVA with Tukey’s post hoc test was performed; *p < 0.05. (C–E) Light/dark transition test conducted at 7MPI. (C) Time spent in dark and light boxes, respectively. One-way ANOVA was performed within each box; n.s., not significant. (D) Latency to first entry into the light box. A Kruskal-Wallis test with Dunn’s post hoc test was performed; *p < 0.05. (E) Number of transitions between light and dark boxes. One-way ANOVA with Tukey’s post hoc test was performed; *p < 0.05. (F,G) Open field test conducted at 7MPI. (F) Left panel: moving distance plotted every 5 min. Two-way repeated measures (RM) ANOVA was performed; effect of group, F2,37 = 3.24, p = 0.050. One-way ANOVA with Tukey’s post hoc test was performed within each time frame; $ $p < 0.01, WT-PBS vs. Tg-PFF: ##p < 0.01, Tg-PBS vs. Tg-PFF. Middle and right panels: moving distance during the first 10 min and 60 min, respectively. One-way ANOVA with Tukey’s post hoc test was performed; **p < 0.01, n.s., not significant. (G) Center time during the first 10 min. A Kruskal-Wallis test with Dunn’s post hoc test was performed; *p < 0.05. WT-PBS (n = 13), Tg-PBS (n = 13), and Tg-PFF (n = 14). Data are the mean ± SEM.
We conducted other behavioral tests to examine general health, muscle strength, motor function, anxiety-like behavior, depression-like behavior, memory impairment, and circadian rhythm of the Tg-PFF mice from 7 to 10 months postinjection. There were no differences in body weight, rectal temperature, or muscular strength among the groups (Fig. S5). In the light/dark transition test, time spent in either the dark or light box was not significantly different among the groups (Fig. 4C). However, latency to first entry into the light box was significantly longer in the Tg-PFF mice than that in the WT-PBS mice, and number of transitions between the boxes was significantly decreased in the Tg-PFF mice compared with that in the WT-PBS mice (Fig. 4D,E). In the open field test, distance traveled was significantly decreased during first 10 min in the Tg-PFF mice, while that was not different among the groups during 60 min (Fig. 4F). In addition, time spent in the center area was significantly decreased during first 10 min in the Tg-PFF mice, as compared with the WT-PBS mice (Fig. 4G). The light/dark transition test is based on the natural aversion of mice to bright areas and on their spontaneous exploratory behavior in novel environments, while the open field test is based on active exploration of mice in a novel environment and inherent aversion to open spaces.21, 22 Therefore, these results indicate normal locomotion with increased anxiety related to bright areas, new environments, and open spaces in the Tg-PFF mice.
To evaluate memory impairment, we conducted the Y-maze test, Barnes maze test, and fear conditioning test. In the Y-maze test, which assesses short-term memory, spontaneous alteration behavior was not significantly different among the groups (Fig. S6). The Barnes maze test assesses spatial learning and long-term memory (Fig. 5A). During the acquisition trials, the Tg-PFF mice showed trend towards increased latency to reach the target, suggesting possible impairment of spatial learning (Fig. 5B). In the probe test, the Tg-PFF mice spent significantly shorter time in the target zone than the WT-PBS mice and less frequently entered the target zone than the WT-PBS and Tg-PBS mice, suggesting impaired long-term memory of the Tg-PFF mice (Fig. 5C,D). The fear conditioning test assesses fear memory. In the conditioning phase, mice received conditioned stimulus (CS)-unconditioned stimulus (US) parings in a chamber (Fig. 5E). The contextual memory retention test was conducted in the same chamber without the CS, showing no difference in freezing time among the groups (Fig. 5F). However, in the auditory-cued fear retention test conducted in a different chamber with the CS, the WT-PBS and Tg-PBS mice showed significantly increased freezing time, whereas the Tg-PFF mice did not (Fig. 5G). The differences in freezing time between pre-CS and CS was significantly shorter in the Tg-PFF mice than that in the Tg-PBS mice. These results suggest impairment of cued fear memory of the Tg-PFF mice.
Fig. 5. Memory impairment in A53T BAC-SNCA Tg mice injected with α-Syn PFFs.
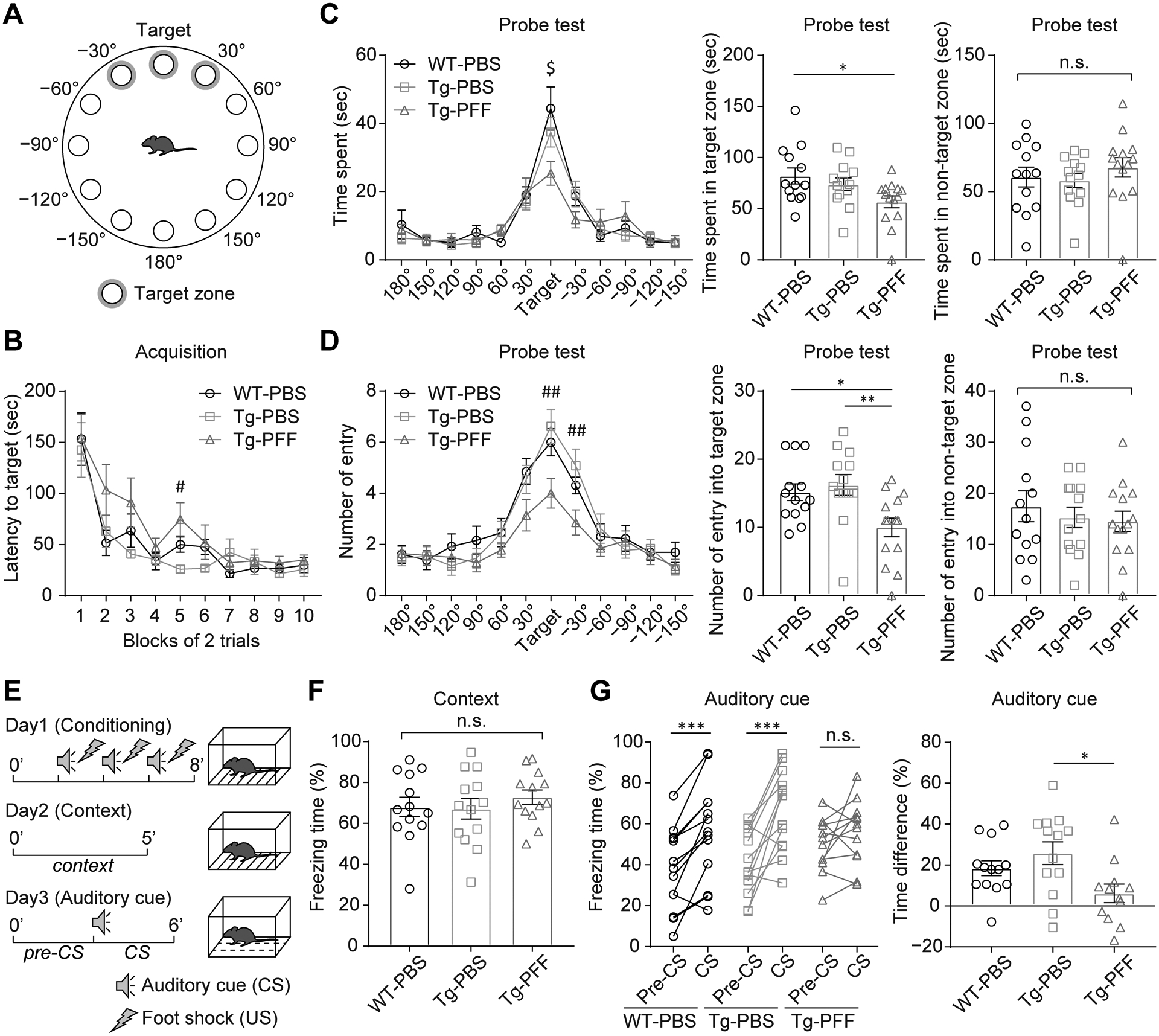
(A–D) Barnes maze test conducted at 8MPI. WT-PBS (n = 13), Tg-PBS (n = 13), and Tg-PFF (n = 14). (A) Schematic showing locations of holes with a shelter under the target hole. Target zone was defined as the vicinities of target, 30°, and −30° holes. (B) Acquisition trials. Latency to enter the shelter at the target hole. Two-way RM ANOVA was performed; effect of group, F2,37 = 2.16, p = 0.13. A Kruskal-Wallis test with Dunn’s post hoc test was performed within each block; #p < 0.05, Tg-PBS vs. Tg-PFF. (C,D) Probe test. (C) Left panel: time spent in the vicinity of each hole. One-way ANOVA with Tukey’s multiple-comparisons test was performed within each hole; $p < 0.05, WT-PBS vs. Tg-PFF. Middle and right panels: time spent in the target and non-target zones, respectively. One-way ANOVA with Tukey’s multiple-comparisons test was performed; *p < 0.05, not significant. (D) Left panel: number of entry in the vicinity of each hole. One-way ANOVA with Tukey’s multiple-comparisons test was performed within each hole; ##p < 0.01, Tg-PBS vs. Tg-PFF. Middle and right panels: number of entry in the target and non-target zones, respectively. One-way ANOVA with Tukey’s multiple-comparisons test was performed; *p < 0.05, **p < 0.01, n.s., not significant. (E–G) Fear conditioning test conducted at 9MPI. WT-PBS (n = 13), Tg-PBS (n = 13), Tg-PFF (n = 13) (E) Schematic representation showing the procedures of fear conditioning test. (F) Contextual fear retention test. One-way ANOVA was performed; n.s., not significant. (G) Auditory-cued fear retention test. Left panel: a two-tailed paired test was performed; ***p < 0.001, n.s., not significant. Right panel: time difference between pre-CS and CS. One-way ANOVA with Tukey’s post hoc test was performed; *p < 0.05. Data are the mean ± SEM.
We also assessed motor function, depression-like behavior, and circadian rhythm, but we did not note any significant abnormalities in the Tg-PFF mice (Fig. S7–9).
Discussion
In the present study, we found that α-Syn PFF injections induced more widespread α-Syn pathology in A53T BAC-SNCA Tg mice than that in WT mice. In addition, compared with the WT mice injected with α-Syn PFFs into the unilateral OB which we previously reported,9 the Tg-PFF mice showed more severe pathology in almost all affected regions throughout the time course. Severe α-Syn pathology in the Tg-PFF mice was initially seen along the olfactory tract and later in the brain regions which are included in the limbic system and have connections with it. The severe α-Syn pathology was accompanied by regional atrophy, neuron loss, reactive astrogliosis, and microglial activation. Behaviorally, the Tg-PFF mice showed hyposmia, followed by anxiety-like behavior and memory impairment. Meanwhile, the Tg-PFF mice did not show motor dysfunction, depression-like behavior, or circadian rhythm abnormality during the time course. These suggest that α-Syn spread from the OB contributes less to the development of those symptoms than to the development of hyposmia, anxiety, and memory impairment in this model. These pathological and behavioral features of the Tg-PFF mice are clearly different from the previous descriptions of WT mice injected with α-Syn PFFs into the unilateral or bilateral OB only showing neuron loss in the AON and olfactory dysfunction.7–9, 23 The previous study analyzing WT mice injected with α-Syn PFFs into the OB/AON complex reported sex differences.11 Male PFF-injected mice developed more severe α-Syn pathology and neuron loss in the olfactory and limbic structures than female ones.11 Male PFF-injected mice exhibited olfactory deficits, while female ones showed motor deficits.11 However, the bilateral OB α-Syn PFF injection model showed a contrasting result; female mice exhibited olfactory deficits, while male mice did not.23 Since we only used male mice for behavioral analyses in the present study, sex differences should be considered in future studies.
Hyposmia has been well described as a prodromal symptom or a risk factor of developing PD.24–26 Given that Lewy pathology is often observed in the OB and primary olfactory cortex in subjects with incidental LB pathology, it is conceivable that those lesions are responsible for hyposmia as a prodromal symptom of PD.2, 27, 28 Indeed, previous studies have suggested the association of incidental LB pathology with hyposmia.29, 30 In support of this notion, the Tg-PFF mice showed hyposmia at 2 months postinjection, when severe α-Syn pathology was confined within the olfactory tract. Moreover, in this sense, this model mimics cases of incidental LB pathology in the olfactory tract and allow us to track the spread of α-Syn pathology from there and development of other behavioral abnormalities.
There are several cross-sectional studies showing the association of hyposmia and other non-motor symptoms, including apathy, anxiety, depression, cognitive dysfunction, psychosis, RBD and autonomic symptoms in patients with PD.31–36 However, it cannot be determined from these studies whether hyposmia serves as a predictor of developing these symptoms. In other words, the causality between hyposmia and these non-motor symptoms remains unknown. Among these non-motor symptoms, longitudinal studies have revealed the hyposmia as a risk of developing dementia in PD.37, 38 For the motor symptoms, there are only cross-sectional studies showing the association of hyposmia with their severity.32, 39 In the present study, our comprehensive behavioral analyses revealed the causal relationship of hyposmia with anxiety and memory impairment, but not with motor dysfunction, depression, or circadian rhythm disturbance in the Tg-PFF mice. Further longitudinal studies are required to confirm how hyposmia works as a predictor of development or progression of other symptoms in patients with PD.
Since Lewy pathology is seen in various brain regions in subjects with PD, it is difficult to disentangle pathological basis of specific symptoms in PD. Animal models like the Tg-PFF mice could provide some insights into the affected brain regions responsible for each symptom. Previous positron emission tomography and single-photon emission computed tomography studies have demonstrated that severity of anxiety in PD patients are inversely correlated with availability of dopamine or noradrenaline transporter in the ventral striatum, caudate, LC, thalamus, amygdala, and anterior putamen.40, 41 Despite these previous studies, little evidence exists to explain the pathological or biochemical basis for the expression of anxiety in PD patients. In the present study, the Tg-PFF mice showed severe pathology in the ventral hippocampus, CeA, and BNST up to 10 months postinjection, which are involved in the known neural circuit for anxiety.42 Patients with PD often develop dementia years after the onset of motor dysfunction, diagnosed as PD with dementia (PDD). Postmortem studies have demonstrated that the severity of Lewy pathology in the cortical areas and hippocampus correlates with cognitive decline in PD.20, 43, 44 The Tg-PFF mice exhibited worse spatial memory than the WT-PBS and Tg-PBS mice in the Barnes maze test. These performances are largely dependent on the integrity of the hippocampus,45 consistent with severe pathological changes in the hippocampus of the Tg-PFF mice. Meanwhile, a neural circuit including the thalamus and amygdala is mainly involved in cued fear learning.42, 46 Among them, the CeA was severely affected in the Tg-PFF mice at 10 months postinjection and could be one of the causes of the impairment of cued fear memory. In addition, among severely affected brain regions up to 10 months postinjection, the PVA and BNST, which interact with the CeA, might also affect cued fear memory of the Tg-PFF mice.47, 48
The Tg-PFF injected mice showed very severe α-Syn pathology in the NAcc at 10 months postinjection. Although we did not conduct a behavioral analysis assessing apathy-like behavior, this pathological change points to the possibility of developing this behavioral abnormality in this model. Indeed, a previous study has reported the association of apathy in PD patients with atrophy of the NAcc.49
This study was conceived from the Braak’s hypothesis about the pathological progression of PD. However, the behavioral abnormalities seen in the Tg-PFF mice resembled the clinical symptoms of dementia with Lewy bodies (DLB).50 Given that Lewy pathology is often seen in the OB and primary olfactory cortex in subjects with incidental LB pathology,2, 27 Lewy pathology spreading from these brain regions may be associated with the development of DLB. However, the pathological progression of DLB remains largely unknown, and frequent concomitant Alzheimer’s disease pathology should be also taken into account for the pathogenesis of DLB.51 Further studies are required to reveal the pathological mechanisms of DLB.
One limitation of the Tg-PFF mice as a PD mouse model is that some of their pathological features are uncommon to sporadic PD. For instance, severe α-Syn pathology accompanied by neuron loss was seen throughout the hippocampus of Tg-PFF mice, while Lewy pathology without neuron loss is seen in the CA2/3 regions in most subjects with PDD.20, 43, 52 Given the genetic features of the A53T BAC-SNCA Tg mice, the Tg-PFF mice may recapitulate the PD subjects with the A53T mutation and/or multiplication of SNCA, in both of which severe neuron loss was documented in the hippocampus.52–55 Defective neurogenesis as well as neurotoxicity of α-Syn aggregation may contribute to the remarkable neuron loss seen in the hippocampus of the Tg-PFF mice.56
In conclusion, our data indicate that α-Syn spread from the OB preferentially affects specific brain regions and neurological functions. Considering Braak’s hypothesis, α-Syn spread from the OB and from the lower brainstem may have distinct clinicopathological roles which define the overall features of PD. The mouse model we created in this study provide a new platform for the better understanding of clinicopathological progression of PD and could contribute to development of new biomarkers and therapies.
Supplementary Material
Fig. S1 No ChAT-positive neuron loss in the NBM of A53T BAC-SNCA Tg mice injected with α-Syn PFFs. (A) ChAT immunohistochemistry in the NBM at 10MPI. Scale bar 200 μm. (B) Number of ChAT-positive cells in the NBM. One-way ANOVA was performed; n.s., not significant. WT-PBS (n = 4), Tg-PBS (n = 4), and Tg-PFF (n = 4). Data are the mean ± SEM.
Fig. S2 Hippocampal atrophy and neuron loss in A53T BAC-SNCA Tg mice injected with α-Syn PFFs. (A) Nissl staining in the ventral hippocampus. Arrowheads and arrows indicate neuron loss in the CA1 and DG, respectively. Scale bar 500 μm. (B) Total area of the ventral hippocampus. (C–E) Nissl-positive area in the CA1, CA2/3, and DG of the ventral hippocampus. One-way ANOVA with Tukey’s post hoc test was performed; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n.s., not significant. WT-PBS (n = 4), Tg-PBS (n = 4) and Tg-PFF (n = 4). Data are the mean ± SEM.
Fig. S3 Nissl-positive neuronal density in the dorsal hippocampus. (A) Nissl staining of the dCA1 and dCA3 pyramidal cells and the dDG granular cells. Scale bar 20 μm. (B–D) Nissl-positive neuronal density in the dCA1, dCA3, and dDG, respectively. One-way ANOVA with Tukey’s post hoc test was performed; ****p < 0.0001, n.s., not significant. WT-PBS (n = 4), Tg-PBS (n = 4) and Tg-PFF (n = 4). Data are the mean ± SEM.
Fig. S4 Atrophy and gliosis in the piriform cortex of A53T BAC-SNCA Tg mice injected with α-Syn PFFs. (A) Nissl staining and GFAP and Iba-1 immunohistochemistry in the piriform cortex. Arrowheads indicate the borders of the piriform cortex. Scale bar 100 μm. (B) Thickness of the piriform cortex. One-way ANOVA with Tukey’s post hoc test was performed; ****p < 0.0001. (C) Percentage of GFAP-positive area. One-way ANOVA with Tukey’s post hoc test was performed; *p < 0.05, **p < 0.01. (D) Percentage of Iba-1–positive area. One-way ANOVA with Tukey’s post hoc test was performed; **p < 0.01, ***p < 0.001. WT-PBS (n = 4), Tg-PBS (n = 4) and Tg-PFF (n = 4). Data are the mean ± SEM.
Fig. S5 General health and muscle strength of A53T BAC-SNCA Tg mice injected with α-Syn PFFs. All analyses were performed at 7MPI. (A) Body weight. (B) Rectal temperature. (C) Wire hang test. (D) Grip strength test. One-way ANOVA was performed; n.s., not significant. WT-PBS (n = 13), Tg-PBS (n = 13), and Tg-PFF (n = 14). Data are the mean ± SEM.
Fig. S6 Y maze test on A53T BAC-SNCA Tg mice injected with α-Syn PFFs. Y maze test conducted at 7MPI. (A) Total distance traveled. (B) Number of arm entries. (C) Number of alternations. (D) Percentage of alternations. One-way ANOVA was performed; n.s., not significant. WT-PBS (n = 13), Tg-PBS (n = 13), and Tg-PFF (n = 14). Data are the mean ± SEM.
Fig. S7 No significant motor impairment in A53T BAC-SNCA Tg mice injected with α-Syn PFFs. Rotarod test conducted at 7MPI. Two-way RM ANOVA was performed; effect of group, F2,37 = 0.79, p = 0.46. One-way ANOVA revealed no significant difference within each trial. WT-PBS (n = 13), Tg-PBS (n = 13), and Tg-PFF (n = 14). Data are the mean ± SEM.
Fig. S8 No significant depression-like behavior in A53T BAC-SNCA Tg mice injected with α-Syn PFFs. (A) Porsolt forced swim test conducted at 7MPI. WT-PBS (n = 13), Tg-PBS (n = 13), and Tg-PFF (n = 14). (B) Tail suspension test conducted at 9MPI. WT-PBS (n = 13), Tg-PBS (n = 13), and Tg-PFF (n = 13). One-way ANOVA was performed; n.s., not significant. Data are the mean ± SEM.
Fig. S9 No significant circadian rhythm abnormality in A53T BAC-SNCA Tg mice injected with α-Syn PFFs. Home cage activity test conducted at 10MPI. Left panel: Moving distance per minute is plotted every hour. Two-way RM ANOVA was performed; effect of group, F2,24 = 4.276, p = 0.026. A Kruskal-Wallis test with Dunn’s post hoc test was performed within each time frame; %p < 0.05, WT-PBS vs. Tg-PBS: $p < 0.05, $ $p < 0.01, WT-PBS vs. Tg-PFF. Middle and right panels: Moving distance per minute during dark and light phase, respectively. A Kruskal-Wallis test was performed; n.s., not significant. WT-PBS (n = 13), Tg-PBS (n = 13), and Tg-PFF (n = 13). Data are the mean ± SEM.
Acknowledgements
We thank Ms. Rie Hikawa for the technical assistance.
Funding sources:
Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS) from Ministry of Education, Culture, Sports Science, MEXT, and the Japan Agency for Medical Research and Development, AMED under the Grant Numbers JP18dm0207020 and JP20dm0207070 (RT), JSPS KAKENHI Grant Numbers JP17H05698 (RT), JP17K16119 (NU), the Takeda Science Foundation (NU), the Kanae Foundation for the Promotion of Medical Science (NU), JSPS Overseas Research Fellowships 201860169 (NU) and 201870008 (MTU), and 1U19AG062418-01A1 (JQT).
Financial Disclosures of all authors (for the preceding 12 months)
N.U. and M.T.U. received Japan Society for the Promotion of Science (JSPS) Overseas Research Fellowships. J.U. has no financial disclosure. T.Y., M.I., and H.Y. received grants from JSPS. M.A. received grants from JSPS and Japan Agency for Medical Research and Development (AMED). J.Q.T. received grants from NIH; revenue from the sale of Avid to Eli Lily as co-inventor on imaging related patents submitted by the University of Pennsylvania; and may accrue revenue in the future on patents submitted by the University of Pennsylvania wherein he is co-inventor. R.T. received consultancies from KAN Research Institute, Inc., Ono Pharmaceutical Co., Ltd., Chugai Pharmaceutical Co., Ltd.; grants/research support from Sumitomo Dainippon Pharma Co., Ltd., Takeda Pharmaceutical Co., Ltd., Eisai Co., Ltd., Kyowa Kirin Co., Ltd., Sanofi K.K., Otsuka Pharmaceutical Co., Ltd., and Nippon Boehringer Ingelheim Co., Ltd.; grants from AMED, JSPS, and Ministry of Education Culture, Sports, Science and Technology Japan; and honoraria from Sumitomo Dainippon Pharma Co., Ltd., Takeda Pharmaceutical Co., Ltd., Novartis Pharma K.K., Kyowa Kirin Co., Ltd., Eisai Co., Ltd., Otsuka Pharmaceutical Co., Ltd., Ono Pharmaceutical Co., Ltd., AbbVie Inc, and Alexion Pharmaceuticals, Inc.
Footnotes
Financial disclosure/Conflict of interest: Nothing to report.
References
- 1.Kalia LV, Lang AE. Parkinson’s disease. The Lancet 2015;386(9996):896–912. [DOI] [PubMed] [Google Scholar]
- 2.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiology of aging 2003;24(2):197–211. [DOI] [PubMed] [Google Scholar]
- 3.Adler CH, Beach TG. Neuropathological basis of nonmotor manifestations of Parkinson’s disease. Mov Disord 2016;31(8):1114–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luk KC, Kehm V, Carroll J, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012;338(6109):949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masuda-Suzukake M, Nonaka T, Hosokawa M, et al. Prion-like spreading of pathological alpha-synuclein in brain. Brain 2013;136(Pt 4):1128–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uemura N, Uemura MT, Luk KC, Lee VM, Trojanowski JQ. Cell-to-Cell Transmission of Tau and α-Synuclein. Trends in molecular medicine 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rey NL, Steiner JA, Maroof N, et al. Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. The Journal of experimental medicine 2016;213(9):1759–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rey NL, George S, Steiner JA, et al. Spread of aggregates after olfactory bulb injection of alpha-synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta neuropathologica 2018;135(1):65–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uemura N, Uemura MT, Lo A, et al. Slow Progressive Accumulation of Oligodendroglial Alpha-Synuclein (alpha-Syn) Pathology in Synthetic alpha-Syn Fibril-Induced Mouse Models of Synucleinopathy. Journal of neuropathology and experimental neurology 2019;78(10):877–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mason DM, Nouraei N, Pant DB, et al. Transmission of alpha-synucleinopathy from olfactory structures deep into the temporal lobe. Molecular neurodegeneration 2016;11(1):49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mason DM, Wang Y, Bhatia TN, et al. The center of olfactory bulb-seeded α-synucleinopathy is the limbic system and the ensuing pathology is higher in male than in female mice. Brain pathology (Zurich, Switzerland) 2019;29(6):741–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taguchi T, Ikuno M, Hondo M, et al. α-Synuclein BAC transgenic mice exhibit RBD-like behaviour and hyposmia: a prodromal Parkinson’s disease model. Brain 2020;143(1):249–265. [DOI] [PubMed] [Google Scholar]
- 13.Uemura N, Yagi H, Uemura MT, Hatanaka Y, Yamakado H, Takahashi R. Inoculation of alpha-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Molecular neurodegeneration 2018;13(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uemura N, Yagi H, Uemura MT, Yamakado H, Takahashi R. Limited spread of pathology within the brainstem of alpha-synuclein BAC transgenic mice inoculated with preformed fibrils into the gastrointestinal tract. Neuroscience letters 2020;716:134651. [DOI] [PubMed] [Google Scholar]
- 15.Padmanabhan K, Osakada F, Tarabrina A, et al. Centrifugal Inputs to the Main Olfactory Bulb Revealed Through Whole Brain Circuit-Mapping. Frontiers in neuroanatomy 2018;12:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henderson MX, Cornblath EJ, Darwich A, et al. Spread of alpha-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nature neuroscience 2019;22(8):1248–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Catani M, Dell’acqua F, Thiebaut de Schotten M. A revised limbic system model for memory, emotion and behaviour. Neuroscience and biobehavioral reviews 2013;37(8):1724–1737. [DOI] [PubMed] [Google Scholar]
- 18.Smith KS, Tindell AJ, Aldridge JW, Berridge KC. Ventral pallidum roles in reward and motivation. Behavioural brain research 2009;196(2):155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roland JJ, Stewart AL, Janke KL, et al. Medial septum-diagonal band of Broca (MSDB) GABAergic regulation of hippocampal acetylcholine efflux is dependent on cognitive demands. J Neurosci 2014;34(2):506–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hall H, Reyes S, Landeck N, et al. Hippocampal Lewy pathology and cholinergic dysfunction are associated with dementia in Parkinson’s disease. Brain 2014;137(Pt 9):2493–2508. [DOI] [PubMed] [Google Scholar]
- 21.Takao K, Miyakawa T. Light/dark transition test for mice. Journal of visualized experiments : JoVE 2006(1):104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shoji H, Takao K, Hattori S, Miyakawa T. Age-related changes in behavior in C57BL/6J mice from young adulthood to middle age. Molecular brain 2016;9:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson ME, Bergkvist L, Mercado G, et al. Deficits in olfactory sensitivity in a mouse model of Parkinson’s disease revealed by plethysmography of odor-evoked sniffing. Scientific reports 2020;10(1):9242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doty RL, Deems DA, Stellar S. Olfactory dysfunction in parkinsonism: a general deficit unrelated to neurologic signs, disease stage, or disease duration. Neurology 1988;38(8):1237–1244. [DOI] [PubMed] [Google Scholar]
- 25.Ponsen MM, Stoffers D, Booij J, van Eck-Smit BL, Wolters E, Berendse HW. Idiopathic hyposmia as a preclinical sign of Parkinson’s disease. Ann Neurol 2004;56(2):173–181. [DOI] [PubMed] [Google Scholar]
- 26.Berg D, Postuma RB, Adler CH, et al. MDS research criteria for prodromal Parkinson’s disease. Mov Disord 2015;30(12):1600–1611. [DOI] [PubMed] [Google Scholar]
- 27.Beach TG, Adler CH, Lue L, et al. Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta neuropathologica 2009;117(6):613–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sengoku R, Saito Y, Ikemura M, et al. Incidence and extent of Lewy body-related alpha-synucleinopathy in aging human olfactory bulb. Journal of neuropathology and experimental neurology 2008;67(11):1072–1083. [DOI] [PubMed] [Google Scholar]
- 29.Ross GW, Abbott RD, Petrovitch H, et al. Association of olfactory dysfunction with incidental Lewy bodies. Mov Disord 2006;21(12):2062–2067. [DOI] [PubMed] [Google Scholar]
- 30.Adler CH, Connor DJ, Hentz JG, et al. Incidental Lewy body disease: clinical comparison to a control cohort. Mov Disord 2010;25(5):642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fullard ME, Tran B, Xie SX, et al. Olfactory impairment predicts cognitive decline in early Parkinson’s disease. Parkinsonism & related disorders 2016;25:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berendse HW, Roos DS, Raijmakers P, Doty RL. Motor and non-motor correlates of olfactory dysfunction in Parkinson’s disease. Journal of the neurological sciences 2011;310(1–2):21–24. [DOI] [PubMed] [Google Scholar]
- 33.Damholdt MF, Borghammer P, Larsen L, Ostergaard K. Odor identification deficits identify Parkinson’s disease patients with poor cognitive performance. Mov Disord 2011;26(11):2045–2050. [DOI] [PubMed] [Google Scholar]
- 34.Morley JF, Weintraub D, Mamikonyan E, Moberg PJ, Siderowf AD, Duda JE. Olfactory dysfunction is associated with neuropsychiatric manifestations in Parkinson’s disease. Mov Disord 2011;26(11):2051–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen W, Kang WY, Chen S, et al. Hyposmia correlates with SNCA variant and non-motor symptoms in Chinese patients with Parkinson’s disease. Parkinsonism & related disorders 2015;21(6):610–614. [DOI] [PubMed] [Google Scholar]
- 36.Hong JY, Sunwoo MK, Ham JH, Lee JJ, Lee PH, Sohn YH. Apathy and olfactory dysfunction in early Parkinson’s disease. Journal of movement disorders 2015;8(1):21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baba T, Kikuchi A, Hirayama K, et al. Severe olfactory dysfunction is a prodromal symptom of dementia associated with Parkinson’s disease: a 3 year longitudinal study. Brain 2012;135(Pt 1):161–169. [DOI] [PubMed] [Google Scholar]
- 38.Domellöf ME, Lundin KF, Edström M, Forsgren L. Olfactory dysfunction and dementia in newly diagnosed patients with Parkinson’s disease. Parkinsonism & related disorders 2017;38:41–47. [DOI] [PubMed] [Google Scholar]
- 39.Tissingh G, Berendse HW, Bergmans P, et al. Loss of olfaction in de novo and treated Parkinson’s disease: possible implications for early diagnosis. Mov Disord 2001;16(1):41–46. [DOI] [PubMed] [Google Scholar]
- 40.Remy P, Doder M, Lees A, Turjanski N, Brooks D. Depression in Parkinson’s disease: loss of dopamine and noradrenaline innervation in the limbic system. Brain 2005;128(Pt 6):1314–1322. [DOI] [PubMed] [Google Scholar]
- 41.Weintraub D, Newberg AB, Cary MS, et al. Striatal dopamine transporter imaging correlates with anxiety and depression symptoms in Parkinson’s disease. Journal of nuclear medicine : official publication, Society of Nuclear Medicine 2005;46(2):227–232. [PubMed] [Google Scholar]
- 42.Tovote P, Fadok JP, Lüthi A. Neuronal circuits for fear and anxiety. Nat Rev Neurosci 2015;16(6):317–331. [DOI] [PubMed] [Google Scholar]
- 43.Irwin DJ, White MT, Toledo JB, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol 2012;72(4):587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Churchyard A, Lees AJ. The relationship between dementia and direct involvement of the hippocampus and amygdala in Parkinson’s disease. Neurology 1997;49(6):1570–1576. [DOI] [PubMed] [Google Scholar]
- 45.Kennard JA, Woodruff-Pak DS. Age sensitivity of behavioral tests and brain substrates of normal aging in mice. Frontiers in aging neuroscience 2011;3:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luchkina NV, Bolshakov VY. Mechanisms of fear learning and extinction: synaptic plasticity-fear memory connection. Psychopharmacology 2019;236(1):163–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Penzo MA, Robert V, Tucciarone J, et al. The paraventricular thalamus controls a central amygdala fear circuit. Nature 2015;519(7544):455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gungor NZ, Paré D. Functional Heterogeneity in the Bed Nucleus of the Stria Terminalis. J Neurosci 2016;36(31):8038–8049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carriere N, Besson P, Dujardin K, et al. Apathy in Parkinson’s disease is associated with nucleus accumbens atrophy: a magnetic resonance imaging shape analysis. Mov Disord 2014;29(7):897–903. [DOI] [PubMed] [Google Scholar]
- 50.Donaghy PC, McKeith IG. The clinical characteristics of dementia with Lewy bodies and a consideration of prodromal diagnosis. Alzheimer’s research & therapy 2014;6(4):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Irwin DJ, Hurtig HI. The Contribution of Tau, Amyloid-Beta and Alpha-Synuclein Pathology to Dementia in Lewy Body Disorders. Journal of Alzheimer’s disease & Parkinsonism 2018;8(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Farrer M, Kachergus J, Forno L, et al. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol 2004;55(2):174–179. [DOI] [PubMed] [Google Scholar]
- 53.Muenter MD, Forno LS, Hornykiewicz O, et al. Hereditary form of parkinsonism--dementia. Ann Neurol 1998;43(6):768–781. [DOI] [PubMed] [Google Scholar]
- 54.Ikeuchi T, Kakita A, Shiga A, et al. Patients homozygous and heterozygous for SNCA duplication in a family with parkinsonism and dementia. Archives of neurology 2008;65(4):514–519. [DOI] [PubMed] [Google Scholar]
- 55.Spira PJ, Sharpe DM, Halliday G, Cavanagh J, Nicholson GA. Clinical and pathological features of a Parkinsonian syndrome in a family with an Ala53Thr alpha-synuclein mutation. Ann Neurol 2001;49(3):313–319. [PubMed] [Google Scholar]
- 56.Winner B, Winkler J. Adult neurogenesis in neurodegenerative diseases. Cold Spring Harbor perspectives in biology 2015;7(4):a021287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 No ChAT-positive neuron loss in the NBM of A53T BAC-SNCA Tg mice injected with α-Syn PFFs. (A) ChAT immunohistochemistry in the NBM at 10MPI. Scale bar 200 μm. (B) Number of ChAT-positive cells in the NBM. One-way ANOVA was performed; n.s., not significant. WT-PBS (n = 4), Tg-PBS (n = 4), and Tg-PFF (n = 4). Data are the mean ± SEM.
Fig. S2 Hippocampal atrophy and neuron loss in A53T BAC-SNCA Tg mice injected with α-Syn PFFs. (A) Nissl staining in the ventral hippocampus. Arrowheads and arrows indicate neuron loss in the CA1 and DG, respectively. Scale bar 500 μm. (B) Total area of the ventral hippocampus. (C–E) Nissl-positive area in the CA1, CA2/3, and DG of the ventral hippocampus. One-way ANOVA with Tukey’s post hoc test was performed; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n.s., not significant. WT-PBS (n = 4), Tg-PBS (n = 4) and Tg-PFF (n = 4). Data are the mean ± SEM.
Fig. S3 Nissl-positive neuronal density in the dorsal hippocampus. (A) Nissl staining of the dCA1 and dCA3 pyramidal cells and the dDG granular cells. Scale bar 20 μm. (B–D) Nissl-positive neuronal density in the dCA1, dCA3, and dDG, respectively. One-way ANOVA with Tukey’s post hoc test was performed; ****p < 0.0001, n.s., not significant. WT-PBS (n = 4), Tg-PBS (n = 4) and Tg-PFF (n = 4). Data are the mean ± SEM.
Fig. S4 Atrophy and gliosis in the piriform cortex of A53T BAC-SNCA Tg mice injected with α-Syn PFFs. (A) Nissl staining and GFAP and Iba-1 immunohistochemistry in the piriform cortex. Arrowheads indicate the borders of the piriform cortex. Scale bar 100 μm. (B) Thickness of the piriform cortex. One-way ANOVA with Tukey’s post hoc test was performed; ****p < 0.0001. (C) Percentage of GFAP-positive area. One-way ANOVA with Tukey’s post hoc test was performed; *p < 0.05, **p < 0.01. (D) Percentage of Iba-1–positive area. One-way ANOVA with Tukey’s post hoc test was performed; **p < 0.01, ***p < 0.001. WT-PBS (n = 4), Tg-PBS (n = 4) and Tg-PFF (n = 4). Data are the mean ± SEM.
Fig. S5 General health and muscle strength of A53T BAC-SNCA Tg mice injected with α-Syn PFFs. All analyses were performed at 7MPI. (A) Body weight. (B) Rectal temperature. (C) Wire hang test. (D) Grip strength test. One-way ANOVA was performed; n.s., not significant. WT-PBS (n = 13), Tg-PBS (n = 13), and Tg-PFF (n = 14). Data are the mean ± SEM.
Fig. S6 Y maze test on A53T BAC-SNCA Tg mice injected with α-Syn PFFs. Y maze test conducted at 7MPI. (A) Total distance traveled. (B) Number of arm entries. (C) Number of alternations. (D) Percentage of alternations. One-way ANOVA was performed; n.s., not significant. WT-PBS (n = 13), Tg-PBS (n = 13), and Tg-PFF (n = 14). Data are the mean ± SEM.
Fig. S7 No significant motor impairment in A53T BAC-SNCA Tg mice injected with α-Syn PFFs. Rotarod test conducted at 7MPI. Two-way RM ANOVA was performed; effect of group, F2,37 = 0.79, p = 0.46. One-way ANOVA revealed no significant difference within each trial. WT-PBS (n = 13), Tg-PBS (n = 13), and Tg-PFF (n = 14). Data are the mean ± SEM.
Fig. S8 No significant depression-like behavior in A53T BAC-SNCA Tg mice injected with α-Syn PFFs. (A) Porsolt forced swim test conducted at 7MPI. WT-PBS (n = 13), Tg-PBS (n = 13), and Tg-PFF (n = 14). (B) Tail suspension test conducted at 9MPI. WT-PBS (n = 13), Tg-PBS (n = 13), and Tg-PFF (n = 13). One-way ANOVA was performed; n.s., not significant. Data are the mean ± SEM.
Fig. S9 No significant circadian rhythm abnormality in A53T BAC-SNCA Tg mice injected with α-Syn PFFs. Home cage activity test conducted at 10MPI. Left panel: Moving distance per minute is plotted every hour. Two-way RM ANOVA was performed; effect of group, F2,24 = 4.276, p = 0.026. A Kruskal-Wallis test with Dunn’s post hoc test was performed within each time frame; %p < 0.05, WT-PBS vs. Tg-PBS: $p < 0.05, $ $p < 0.01, WT-PBS vs. Tg-PFF. Middle and right panels: Moving distance per minute during dark and light phase, respectively. A Kruskal-Wallis test was performed; n.s., not significant. WT-PBS (n = 13), Tg-PBS (n = 13), and Tg-PFF (n = 13). Data are the mean ± SEM.