

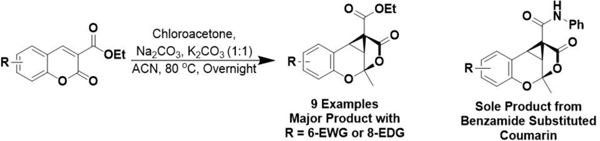
Abstract
Synthesis of highly strained fused substituted dihydrobenzopyran cyclopropyl lactones derived from coumarin carboxylates are reported. Substrate scope tolerates a variety of 6- and 8-substituents on the coumarin ring. Substitution at the 5- or 7-position is resistant to tricyclic lactone formation except with 7-methyl substitution. Benzamide-containing coumarins afford the tricyclic ketal. A plausible mechanism is proposed for the formation of the fused lactone; intramolecular rearrangement of trans cyclopropyl methyl ketones with phenolic acetate via the formation of a hemiacetal.
Graphical Abstract
The search for novel chemical space in pharmaceutical discovery has correlated with increasing three-dimensional character. Thus, sp3 rich scaffolds have garnered increased attention from the medicinal chemistry community, leading to a new predictive metric Fsp3; the fraction of sp3 carbon atoms within a compound.1–3 Of specific interest has been the introduction of multi-cyclic, fused, bridged, or spiro-fused rings.4–6
The cyclopropane ring is a constituent of many compounds with biological activity, including numerous natural products and pharmaceuticals used in the clinical management of cardiovascular, inflammatory diseases, diabetes and cancer.7 Furthermore, coumarin derivatives have been extensively studied due to their wide-ranging biological profiles, including activity as anticancer, anti-inflammatory, antibacterial, antifungal, and anticoagulant agents.8 Despite the potential for increased biological activity, cyclopropanated derivatives of coumarins have not been extensively investigated.
In the course of our continued efforts to identify new aldo-keto reductase 1C3 (AKR1C3) inhibitors,9–14 we desired access to cyclopropyl-fused coumarins. Thus, we investigated the utility of a Michael-initiated ring closure (MIRC) reaction. Prior precedent demonstrates cyclopropanation can be achieved at α,β-unsaturated alkenes, such as those present in the coumarin structure, with activated methylene substrates via the MIRC reaction,15–17 which includes the use of 1,2-dicarboxylate substituted alkenes,18 1,1,2-trisubstituted alkenes,19 and haloketones, among others.20, 21 However, the reaction with highly activated halomethylene ketones has been understudied due to high reactivity and the potential for self-polymerization.22
While this work was in progress, Sun et. al. reported the reaction of a coumarin carboxylate with tert-butyl 2-bromo-acetate using a chiral Lewis base catalyst and Cs2CO3 provided polycyclic ketones in predominantly high yields (Figure 1).23 Guo et. al. reported an enatioselective cyclopropanation reaction employing a phenyliodonium ylide that was tolerative of unsubstituted or 6-methyl substituted coumarins.24 The synthesis of functionalized strained bicylic ketals, where n = 1 or 2 (Figure 1), from bis-homopropargylic diols via use of a AuI or AuIII catalyst was reported by Antoniotti et. al.25
Figure 1.
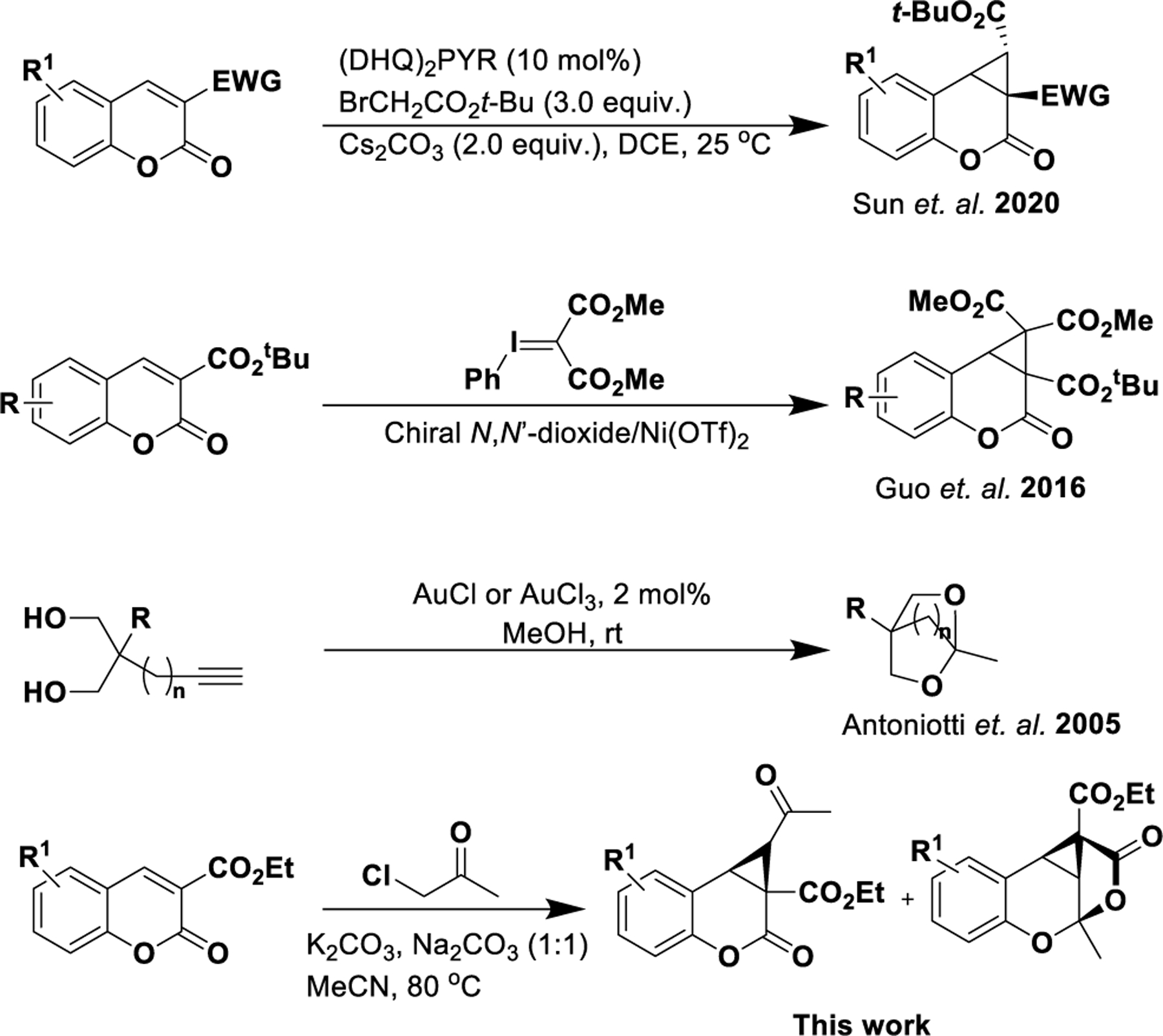
Comparison of the reported work and that of cyclopropanation of coumarins (Sun et al. and Guo et al.) and the formation of strained bicyclic ketals (Antoniotti et al.). Sun and Antoniotti schemes reproduced from references 23 and 25, Copyright 2020 and 2005 respectively, American Chemical Society.
The reaction of coumarin carboxylates with bromoacetophenones leads to the formation of cyclopropanated benzophenone and/or benzofuran derivatives.26, 27 During the course of our synthetic efforts to access cyclopropanated coumarins we intriguingly isolated and characterized a unique tricyclic ketal that, to the best of our knowledge, has not previously been disclosed. An initial version of this work was deposited in ChemRxiv on July 7th 2021.28
Our efforts to access cyclopropanated coumarins began with exposure of 7-hydroxy coumarin ethyl ester (1) to base and stoichiometric amounts of chloroacetone. Total consumption of the starting material was not observed and so addition of excess chloroacetone and K2CO3 was performed. These conditions led to the expected cyclopropanated methyl ketone derivative (1a) upon completion of the reaction between chloroacetone and phenol to afford the respective ether (Scheme 1).
Scheme 1.
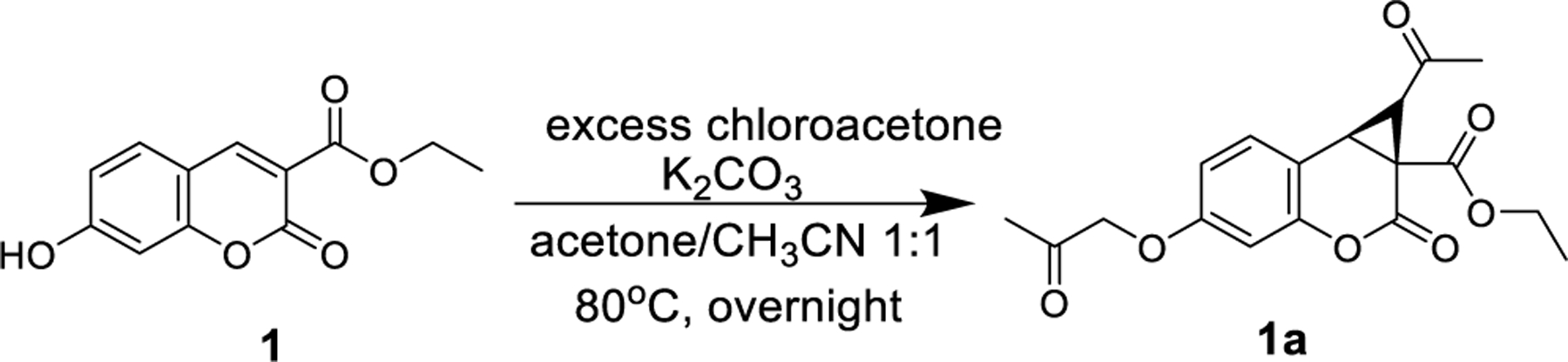
Cyclopropanation of Hydroxy Coumarin Carboxylic Acid Ethyl Ester
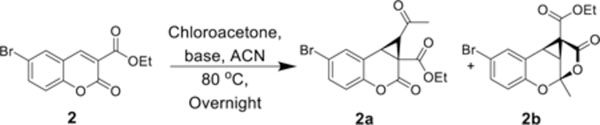
Upon employing 6-bromocoumarin ethyl ester (2) as starting material, a complex mixture of products was generated which included a novel highly strained tricyclic ketal (2b) as the major product alongside fused cyclopropyl coumarin 2a. Intrigued by the novelty of this structure, we investigated conditions that would offer greater yield. Optimization of reaction conditions began with a base scan (Table 1). However, no, to low yields, of product were formed with the use of Na2CO3, CaCO3, NaOMe, NaOEt, KOEt, NaHCO3, or KHCO3. Furthermore, no significant improvement in yield was observed with additives including a 2:1 ratio of Na2CO3+DABCO, Na2CO3+Et3N, or 1:1 ratio of Na2CO3+K2CO3. In all cases, final products were isolated via silica gel column chromatography and recrystallized in 20% EtOAc:Hexane. The structures of 2a and 2b were confirmed by 1H and 13C spectroscopy. Representative examples for the structures of type a and b, and cis and trans isomers of a were confirmed by 2D NMR and X-ray crystallography. We assigned cis and trans according to the orientation of the two hydrogen positions on the cyclopropyl ring.
Table 1.
Reaction Condition Optimization for Cyclopropanation
| ||
|---|---|---|
| Entry | Base | Combined yield |
| 1 | Na2CO3 | no product/trace |
| 2 | CaCO3 | no product/trace |
| 3 | K2CO3 | 40% |
| 4 | Cs2CO3 | 38% |
| 5 | Na2CO3+DABCO (2:1) | 40% |
| 6 | Na2CO3+Et3N (2:1) | 35% (difficulty in isolation) |
| 7 | Na2CO3+K2CO3 (1:1) 3 Hours | Incomplete reaction |
| 8 | Na2CO3+K2CO3 (1:1) 12 Hours | 40% |
| 9 | Na2CO3+K2CO3 (1:1) 24 Hours | 40% |
| 10 | NaHCO3 | no product/trace |
| 11 | KHCO3 | no product/trace |
| 12 | NaOMe | no product/trace |
| 13 | NaOEt | no product/trace |
| 14 | KOEt | no product/trace |
| 15 | KOtBu | 15% (difficulty in isolation) |
| 16 | Et3N | no product/trace |
| 17 | Aqueous K2CO3 | Polymerized products |
To investigate substrate scope, K2CO3 and Na2CO3 (1:1) was employed due to the apparent ease of purification and isolation of products. Synthesis of a library of derivatives from substituted coumarins including unsubstituted (3), electron-donating groups; 7-methyl (4), 8-methoxy (5), 6-methoxy (6), 4-methoxy (16), 8-tert-butyl (7), 7-diethylamine (15) and electron-withdrawing groups; 7-OH (1), 6-fluoro (8), 6-chloro (9), 6-bromo (2), 6,8-dibromo (10), 6-nitro (11) and 7-O-benzyl (12) were utilized. These substrates resulted in the consistent formation of the expected fused cyclopropyl coumarin and the tricyclic ketal in low to moderate, but still synthetically useful, yields. Starting material bearing 6-, 7-, 8- or 6,8-substitution formed the tricyclic product except when lone pair-containing 5- or 7-substitution was present. To further probe the substrate scope, unsubstituted coumarin benzamide (13), 6-methoxy coumarin benzamide (17), 8-tert-butyl coumarin benzamide (18), and quinolin-2(1H)-one (14) were also investigated as starting material (Figure 2).
Figure 2:
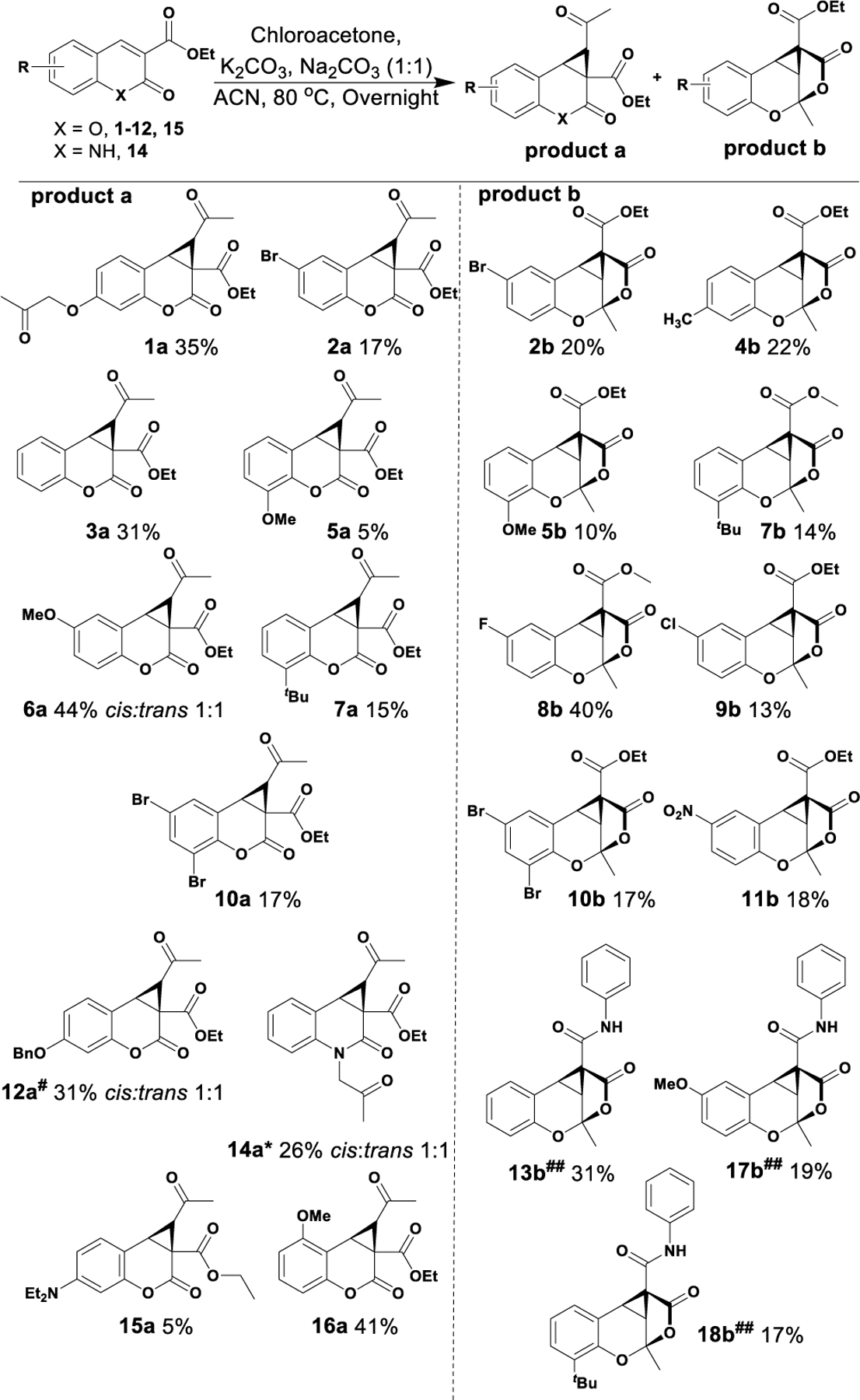
Substrate scope and isolate product yields. *From quinolin-2(1H)-one substrate (14). #See Scheme 2 for conditions. ##From coumarin phenylamide substrates.
7-Hydroxy, saturated, and 7-OBn coumarins (1, 3 and 12 respectively) resulted in the isolation of the cyclopropanated coumarin (overall 31%, 35% and 31% yield) as the major product with only traces of the tricyclic product detected. The 7-methyl (4), 6-fluoro (8), 6-chloro (9) substituted coumarins, and all benzamides (13, 17 and 18) preferentially yielded the tricyclic ketal as the sole isolated product (22%, 40%, 13%, 31%, 19% and 31% yield, respectively). It should be noted that when the 6-fluoro coumarin cyclopropylated product was purified by silica gel chromatography without a methanol wash, decomposition of the product was observed. However, washing the product with methanol followed by silica gel chromatography resulted in trans-esterification of ethyl ester to methyl ester 8b. Similar results were observed with 8-tert butyl isomerized methyl ester 7b.
Interestingly switching coumarin ethyl esters to the respective benzamides (13, 17 and 18) resulted in a switch of major product from the cyclopropylated coumarin to the tricyclic ketal as the sole product (Figure 2), allowing isolation of this novel compound when none was detected with the ethyl ester (compare 3a to 13b, 6a to 17b, and 7a to 18b).
The 8-methoxy (5), 6-bromo (2) and 6,8-dibromo (10) substituted coumarin substrates provided a mixture of fused cyclopropyl coumarin and tricyclic ketal. Lower equivalents of chloroacetone did not lead to the consumption of starting material in the case of 7-hydroxy and 6-nitro coumarins; hence, excess chloroacetone (5 equiv.) was used for these reactions. Difficulty in purification was observed if the starting material was not consumed. Use of excess chloroacetone with 6-nitro coumarin 11 led to the formation of the tricyclic ketal 11b and a ring-opened alkylated diester 11c (supporting information).
Interestingly, 6-methoxy substituted starting material (6) afforded the cyclopropyl coumarin as the major and only product in a 1:1 mixture of cis and trans isomers as confirmed by X-ray crystallography (Supporting Information Figure S1). Substrate scope was further explored with quinolin-2(1H)-one (14), which afforded a mixture of the respective N-alkylated compound and the cyclopropyl coumarin (14a) in equivalent yield but with no tricyclic ketal isolated.
Cyclopropanation of 7-diethylamine coumarin 15 yielded no isolated product under the conditions depicted in Figure 2. However, when the conditions were modified to KOtBu in THF at −78 °C, the cyclopropyl coumarin was isolated in poor yield with no evidence of ketal formation (Scheme 2), suggesting that lone pair-containing substitution at the 7-position is unfavorable for tricyclic ketal formation. The structures of the 6,8-dibromo cyclopropylated coumarin 10a, 6,8-dibromo tricyclic ketal 10b (Figure 3), and cis and trans 6-methoxy cyclopropylated coumarins 6a and 6a’ were confirmed using X-ray crystallography (Supporting Information Figure S1). In most cases, formation of the trans-cyclopropylated product along with isomerized tricyclic ketal derivatives was observed. However, in the case of 6-methoxy, both cis and trans ketones were isolated, leading us to postulate that the trans-ketones undergo intermolecular rearrangement resulting in the formation of the tricyclic ketal derivative by a potential mechanism delineated in Figure 3.
Scheme 2:
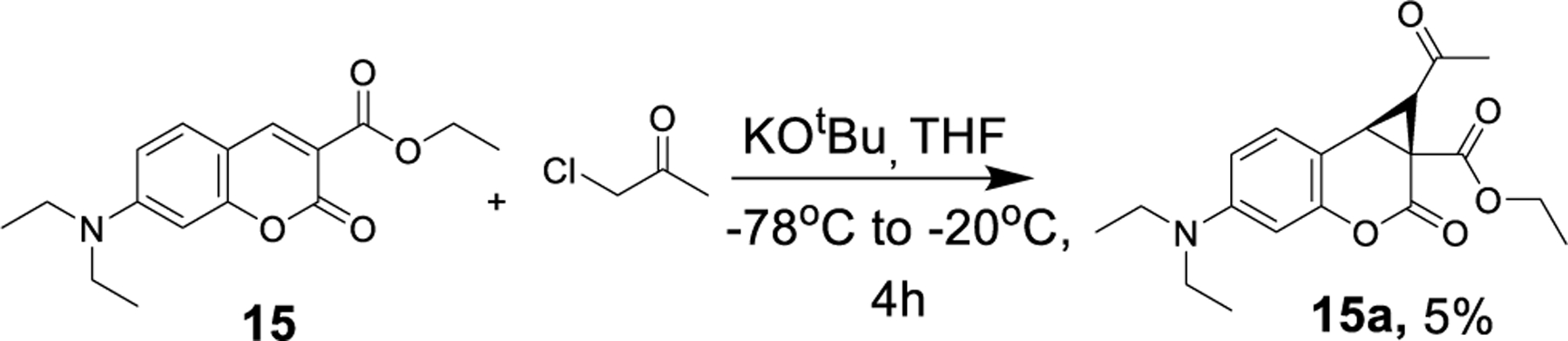
Cyclopropanation of N, N-diethyl coumarin carboxylic acid ethyl ester
Figure 3:
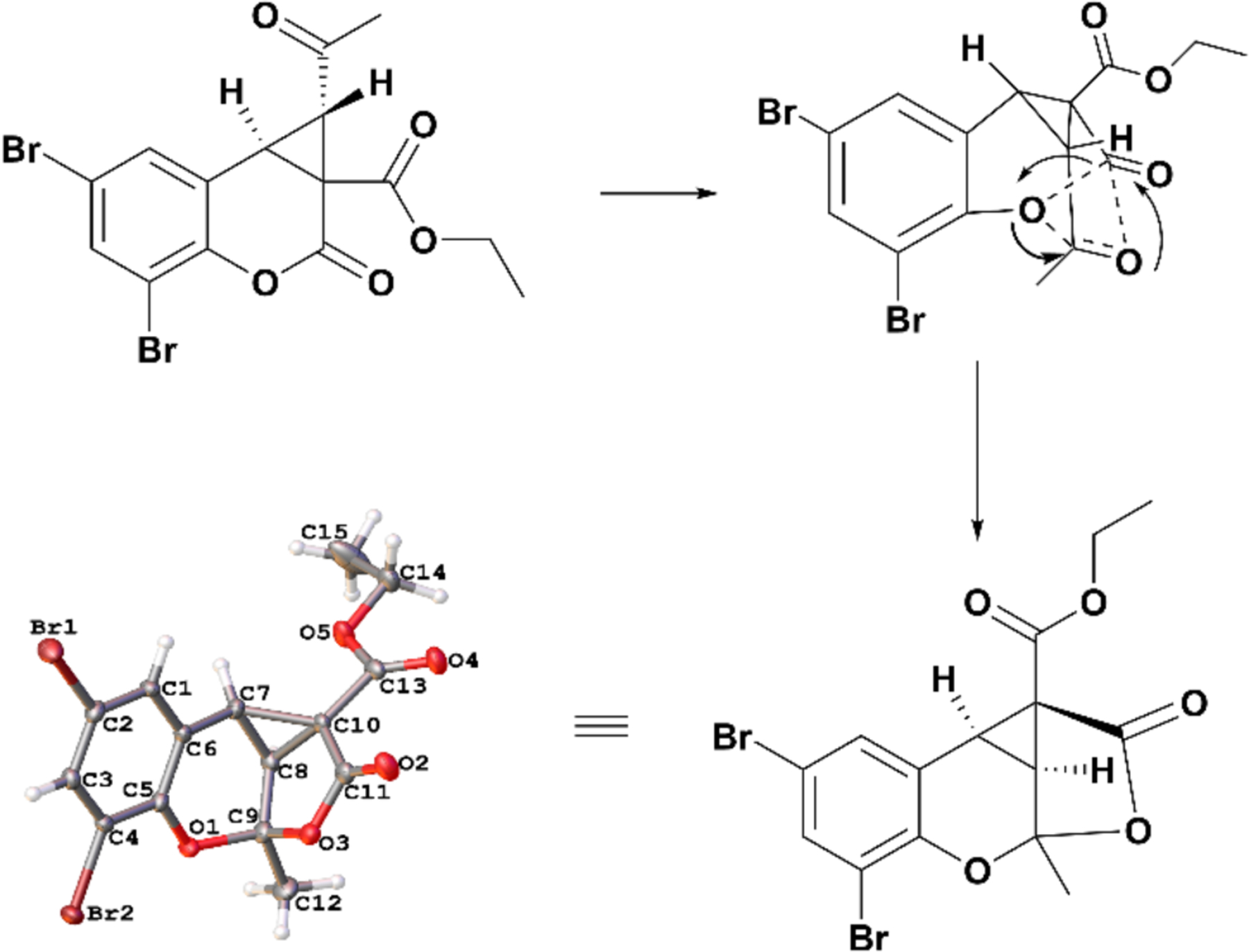
Possible mechanism for the formation of tricyclic ketals from the cyclopropyl carboxylate trans isomer. Thermal ellipsoids were drawn at the 50% probability level in the crystal structure.
To understand the potential antineoplastic effects of these scaffolds, representative compounds were tested for cytotoxicity in the triple negative breast cancer cell line MDA-MB-468 using an MTS assay (Supporting Information Figure S2). A selection of 10 compounds showed dose-dependant reduction of cell proliferation at 100 and 50 µM. 7-Diethylamine 15a showed greatest potency with an IC50 = 21 µM. Tricyclic ketal 11b was submitted to the NCI-60 screen for evaluation of effect at 10 µM in 60 cancer cell lines (Supporting Information Figure S3),29 affording activity in breast and renal cancer cells.
In conclusion, we have identified a synthetically simple, single-step route for the synthesis of previously unreported highly strained tricyclic ketals based around the bioactive coumarin ring structure.28 Substrate scope at the 6-position tolerates a variety of electron-withdrawing groups and alkyl electron-donating groups, that leads to preferential formation of the tricyclic ketal. At the 8-position, electron-donating groups are tolerated but with a mixture of products formed. Saturation, or substitution at the 5- or 7-position fails to afford the tricyclic ketal product expect with 7-methyl substitution. Benzamide-containing coumarins afford the tricyclic ketal in isolation and with increased yield. We propose a plausible mechanism of their formation involving intermolecular rearrangement of the cyclopropyl trans ketone. While the products are formed in low to moderate (10–40%) yield, the single step is synthetically advantageous when compared to the multistep route likely needed to access these types of complex structures.
EXPERIMENTAL SECTION
General experimental methods.
Solvents and reagents of commercial-grade were purchased from Fisher Scientific, VWR, Millipore-Sigma or AK Scientific and were used without additional purification. All reactions were performed in oven-dried flasks under a nitrogen atmosphere. Reaction progress was monitored using thin-layer chromatography (TLC) on Aluminium-backed 20 µm silica plates supplied by Silicycle (TLA-R10011B-323) and visualized by UV (254 nm) or staining agent (ninhydrin solution, phosphomolybdic acid or iodine vapor). Flash column chromatography was performed on silica gel (40 – 63 µm, 60 Å) with the indicated mobile phase. NMR spectrometric analysis were carried out using the indicated solvent on a Bruker Avance III HD spectrometer at 400 or 500 MHz for proton (1H) and 100 or 126 MHz for carbon (13C), respectively. Chemical shifts (δ) are recorded in parts per million (ppm) and reported relative to solvents; coupling constants (J) are reported in hertz (Hz). Splitting of signal peaks are indicated by s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), m (multiplet), and br (broad). High-resolution mass spectrometry (HRMS) was carried out on an Agilent 1200 time-of-flight mass spectrometer equipped with an electrospray ionization source. High-performance liquid chromatography (HPLC) was performed on an Agilent 1220, equipped with a 254 nm UV detector (VWD), employing a Phenomenex C18, Polar-RP column (4µm, 250 × 4.6 mm) or RP column (5µm, 250 × 4.6 mm). Purifications were performed using methanol : water (0.05 % TFA) as mobile phase. The purity of all final compounds was determined as ≥95%, unless otherwise specified. X-ray crystallography data was collected on an Oxford Diffraction Xcalibur, Onyx, Ultra diffractometer.
Representative synthetic procedure.
In a round bottom flask, coumarin carboxylate ethyl ester (2 mmol) was dissolved in acetonitrile (10 mL), followed by the addition of chloroacetone (4 mmol), potassium carbonate (3 mmol) and sodium carbonate (3 mmol) at room temperature while stirring. The reaction mixture in the round bottom flask was refluxed at 80 °C overnight using an oil bath. The contents were filtered and washed with methanol and evaporated, and the products were isolated via silica gel column chromatography using 20–40% EtOAc:Hexanes, and recrystallized in EtOAc:Hexanes. All crystals were grown by dissolving the product in warm EtOAc (20–30%) and adding warm Hexanes followed by sitting at room temperature to initiate crystallization.
Cell lines and culture conditions.
MDA-MB-468 cells were purchased from ATCC and were cultured in Dulbecco’s Modified Eagle Medium (Fisher Scientific, Cat# 50–188-267FP) supplemented with 10% Fetal Bovine Serum (ATCC®, Cat# 30–2020) and 1% penicillin-streptomycin (50 U/mL, 50 µg/mL, Corning™, Cat# MT30001CI). Cells were maintained in a humidified chamber with 5% CO2 atmosphere at 37 °C.
Cytotoxicity using MTS assay.
Briefly, 10,000 cells/well were seeded in 96-well plate and allowed to adhere for 18–24 hours. Test compounds were dissolved in DMSO at 1000x concentration and further diluted in growth medium to final concentrations of 100 and 50 µM and added to the wells. Plates were incubated for 72 hours in the presence of test compounds, at which time MTS was added according to the manufacturer’s protocol. Absorbance was recorded at 570 nm, and % cell survival was calculated using DMSO as reference for 100% cell survival, and a graph was generated using Graphpad prism software.
Product characterization.
Ethyl 1-acetyl-2-oxo-5-(2-oxopropoxy)-1,7b-dihydrocyclopropa[c]chromene-1a(2H)-carboxylate (1a). Note 5 equivalents of chloroacetone were used in the synthetic route. Beige solid (242mg, 35%). 1H NMR (CDCl3, 400 MHz): δ 7.24 (d, J = 8.4 Hz, 1H), 6.70–6.67 (dd, J = 6.60 (d, J = 2.4 Hz, 1H), 4.54 (s, 2H), 4.34–4.23 (m, 2H), 3.50 (d, J = 9.6 Hz, 1H), 3.23 (d, J = 9.6 Hz, 1H), 2.29 (s, 3H), 2.22 (s, 3H), 1.32 (t, J = 7.0 Hz, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 204.3, 199.6, 167.1, 158.6, 152.7, 129.5, 111.5, 106.7, 102.6, 73.0, 63.1, 37.1, 34.1, 32.7, 31.2, 26.6, 14.0; HRMS (ESI) m/z: [M + H]+ Calcd for C18H18O7 347.1125; found 347.1132
Ethyl 1-acetyl-6-bromo-2-oxo-1,7b-dihydrocyclopropa[c]chromene-1a(2H)-carboxylate (2a). Off white solid, clear crystal (120 mg, 17%). 1H NMR (CDCl3, 400 MHz): δ 7.47 (s, 1H), 7.44–7.41 (dd, J = 8.8 Hz, 2.4 Hz, 1H), 7.01 (d, J = 8.8 Hz, 1H), 4.34–4.25 (m, 2H), 3.53 (d, J = 9.6 Hz, 1H), 3.22 (d, J = 9.6 Hz, 1H), 2.27 (s, 3H), 1.33 (t, J = 7.2 Hz, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 199.5, 166.7, 160.0, 150.9, 132.7, 131.2, 118.5, 116.7, 115.67, 63.3, 36.8, 34.1, 32.4, 31.4, 14.0; HRMS (ESI) m/z: [M + H]+ Calcd for C15H13BrO5 353.0020; found 353.0025
Ethyl 6-bromo-2a-methyl-1-oxo-2a1,7b-dihydro-1H-2,3-dioxabenzo[f]cyclopropa[cd] indene-7c(2aH)-carboxylate (2b). White solid (141 mg, 20%). 1H NMR (CDCl3, 400 MHz): δ 7.60 (d, J = 2.4 Hz, 1H), 7.39–7.36 (dd, J = 2.4 Hz, 8.8 Hz, 1H), 6.84 (d, J = 8.8 Hz, 1H), 4.37–4.32 (q, 2H), 3.35 (d, J = 8.0 Hz, 1H), 1.97 (s, 3H), 1.39 (t, J = 7.0 Hz, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 166.3, 165.6, 149.8, 133.4, 133.2, 119.5, 117.8, 115.4, 102.3, 62.8, 38.2, 30.0, 28.6, 25.2, 14.1; HRMS (ESI) m/z: [M + H]+ Calcd for C15H13BrO5 353.0020; found 353.0029
Ethyl 1-acetyl-2-oxo-1,7b-dihydrocyclopropa[c]chromene-1a(2H)-carboxylate (3a). 1H NMR (CDCl3, 400 MHz): Beige solid (169 mg, 31%). δ 7.34–7.31 (m, 2H), 7.15–7.11 (m, 2H), 4.32–4.28 (q, J = 7.2 Hz, 2H), 3.54 (d, J = 9.6 Hz, 1H), 3.26 (d, J = 9.6 Hz, 1H), 2.23 (s, 3H), 1.34 (t, J = 7.0 Hz, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 199.4, 167.1, 160.6, 151.7, 129.7, 128.6, 124.4, 116.8, 113.6, 63.1, 36.9, 34.3, 33.0, 31.2, 30.9, 14.0; HRMS (ESI) m/z: [M + H]+ Calcd for C15H14O5 275.0914; found 275.0958
Ethyl 2a,5-dimethyl-1-oxo-2a1,7b-dihydro-1H-2,3-dioxabenzo[f]cyclopropa[cd]indene-7c(2aH)-carboxylate (4b). Beige solid (126%, 22%). 1H NMR (CDCl3, 400 MHz): δ 7.32 (d, J = 8.0 Hz, 1H), 6.87 (dd, J = 0.8 Hz, 7.6 Hz, 1H), 6.76 (s, 1H), 4.37–4.31 (m, 2H), 3.35 (d, J = 8.0 Hz, 1H), 3.06 (d, J = 8.0 Hz, 1H), 2.32 (s, 3H), 1.96 (s, 3H), 1.38 (t, J = 7.2 Hz, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 166.9, 166.1, 150.5, 140.7, 130.6, 124.2, 118.0, 112.5, 102.3, 62.5, 38.2, 30.4, 29.4, 25.4, 21.4, 14.1; HRMS (ESI) m/z: [M + H]+ Calcd for C16H16O5 289.1071; found 289.1059
Ethyl 1-acetyl-4-methoxy-2-oxo-1,7b-dihydrocyclopropa[c]chromene-1a(2H)-carboxylate (5a). Grey solid (30 mg, 5%). 1H NMR (CDCl3, 400 MHz): δ 7.06 (t, J = 8.0 Hz, 1H), 6.90 (d, J = 8.8 Hz, 2H), 4.31–4.25 (m, 2H), 3.91 (s, 3H), 3.52 (d, J = 10.0 Hz, 1H), 3.24 (d, J = 9.6 Hz, 1H), 2.23 (s, 3H), 1.32 (t, J = 6.8 Hz, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 199.2, 167.2, 160.0, 147.2, 141.3, 124.2, 119.8, 114.4, 112.4, 63.0, 56.2, 36.7, 34.1, 33.1, 31.2, 13.9; HRMS (ESI) m/z: [M + H]+ Calcd for C16H16O6 305.1020; found 305.1010
Ethyl 4-methoxy-2a-methyl-1-oxo-2a1,7b-dihydro-1H-2,3-dioxabenzo[f]cyclopropa[cd]indene-7c (2aH)-carboxylate (5b). Off white solid (60 mg, 10%). 1H NMR (CDCl3, 400 MHz): δ 7.07–6.98 (m, 2H), 6.89–6.87 (m, 1H), 4.37–4.32 (q, J = 7.2 Hz, 2H), 3.87 (s, 3H), 3.39 (d, J = 8.0 Hz, 1H), 3.10 (d, J = 8.0 Hz, 1H), 2.05 (s, 3H), 1.38 (t, J = 7.2 Hz, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 166.7, 166.0, 148.8, 140.1, 122.9, 122.4, 116.6, 112.8, 102.3, 62.6, 56.2, 38.1, 30.3, 29.3, 25.4, 14.1; HRMS (ESI) m/z: [M + H]+ Calcd for C16H16O6 305.1020; found 305.1026
Ethyl 1-acetyl-6-methoxy-2-oxo-1,7b-dihydrocyclopropa[c]chromene-1a(2H)-carboxylate (6a trans). Yellowish solid, clear crystal (134 mg, 22%). 1H NMR (CDCl3, 400 MHz): δ 6.96 (d, J = 8.8 Hz, 1H), 6.90 (d, J = 2.8 Hz, 1H), 6.82–6.79 (dd, J = 9.0 Hz, 3.0 Hz, 1H), 4.27–4.22 (q, J = 7.1 Hz, 2H), 3.80 (s, 3H), 3.50 (d, J = 4.8 Hz, 1H), 2.59 (d, J = 5.2 Hz, 1H), 2.42 (s, 3H), 1.29 (t, J = 7.0 Hz, 1H); 13C{1H} NMR (CDCl3, 100 MHz): δ 199.3, 167.2, 160.8, 156.0, 145.7, 117.6, 116.6, 114.9, 114.2, 113.4, 63.0, 55.7, 36.9, 34.1, 33.2, 31.3, 14.0; HRMS (ESI) m/z: [M + Na]+: Calcd for C16H16O6 327.0839; found 327.0841
Ethyl 1-acetyl-6-methoxy-2-oxo-1,7b-dihydrocyclopropa[c]chromene-1a(2H)-carboxylate, (6a cis). Yellowish solid, pale green crystal (134 mg, 22%). 1H NMR (CDCl3, 400 MHz): δ 7.04 (d, J = 8.8 Hz, 1H), 6.86–6.83 (m, 2H), 4.32–4.24 (m, 2H), 3.78 (s, 3H), 3.52 (d, J = 9.6 Hz, 1H), 3.22 (d, J = 9.6 Hz, 1H), 2.23 (s, 3H), 1.32 (t, J = 7.0 Hz, 1H); 13C{1H} NMR (CDCl3, 100 MHz): δ 199.3, 167.1, 160.8, 156.0, 145.7, 117.6, 114.9, 114.2, 113.4, 63.0, 55.7, 36.9, 34.1, 33.2, 31.3, 14.0; HRMS (ESI) m/z: [M + Na]+ Calcd for C16H16O6 327.0839; found 327.0819
Ethyl 1-acetyl-4-(tert-butyl)-2-oxo-1,7b-dihydrocyclopropa[c]chromene-1a(2H)-carboxylate (7a). viscous gum (100 mg, 15%). 1H NMR (CDCl3, 400 MHz): δ 7.30–7.28 (dd, J = 8.0 Hz, 1.6 Hz, 1H), 7.20–7.17 (dd, J = 7.2 Hz, 1.2 Hz, 1H), 7.03 ((t, J = 7.6 Hz, 1H), 4.31–4.21 (m, 2H), 3.50 (d, J = 9.6 Hz, 1H), 3.29 (d, J = 9.6 Hz, 1H), 2.16 (s, 3H), 1.47 (s, 9H), 1.30 (t, J = 7.0 Hz, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 199.4, 167.0, 160.1, 150.1, 137.5, 127.0, 126.7, 123.8, 113.8, 62.9, 36.7, 34.9, 34.2, 33.5, 31.1, 29.9, 14.00; HRMS (ESI) m/z: [M + H]+ Calcd for C19H22O5 331.1540; found 331.1546
Methyl 4-(tert-butyl)-2a-methyl-1-oxo-2a1,7b-dihydro-1H-2,3-dioxabenzo[f]cyclopropa[cd]indene-7c(2aH)-carboxylate (7b). viscous gum (88 mg, 14%). 1H NMR (CDCl3, 400 MHz): δ7.27 (t, J = 8.4 Hz, 2H), 6.97 (t, J = 7.8 Hz, 1H), 3.86 (s, 3H), 3.39 (d, J = 8.0 Hz, 1H), 3.13 (d, J = 8.0 Hz, 1H), 1.99 (3H), 1.36 (s, 9H); 13C{1H} NMR (CDCl3, 100 MHz): δ 167.0, 166.6, 149.1, 138.8, 128.8, 127.5, 122.7, 115.9, 102.0 53.2, 37.9, 34.7, 30.7, 30.3, 29.7, 25.4; HRMS (ESI) m/z: [M + H]+ Calcd for C18H20O5 317.1384; found 317.1385
Methyl 6-fluoro-2a-methyl-1-oxo-2a1,7b-dihydro-1H-2,3-dioxabenzo[f]cyclopropa[cd] indene-7c(2aH)-carboxylate (8b). Off white solid (222 mg, 40%). 1H NMR (CDCl3, 400 MHz): δ 7.16 (d, J = 2.8 Hz, 1H), 7.01–6.89 (m, 2H), 3.89 (s, 3H), 3.36 (d, J = 8.0 Hz, 1H), 3.09 (d, J = 8.0 Hz, 1H), 1.96 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 166.4, 166.2, 159.3, 156.9, 146.7, 146.6, 119.0, 118.9, 117.4, 117.2, 117.1, 116.9, 102.5, 53.4, 38.2, 30.1, 29.2, 29.2, 25.2; HRMS (ESI) m/z: [M + H]+ Calcd for C14H11FO5 279.0664; found 279.0640
Ethyl 6-chloro-2a-methyl-1-oxo-2a1,7b-dihydro-1H-2,3-dioxabenzo[f]cyclopropa[cd] indene-7c(2aH)-carboxylate (9b). Off white solid (80 mg, 13%). 1H NMR (CDCl3, 400 MHz): δ 7.46 (d, J = 2.4 Hz, 1H), 7.24–7.22 (dd, J = 2.4 Hz, 8.8 Hz, 1H), 6.89 (d, J = 8.8 Hz, 1H), 4.37–4.31 (q, J = 7.2 Hz, 2H), 3.35 (d, J = 8.0 Hz, 1H), 3.09 (d, J = 8.0 Hz, 1H), 1.97 (s, 3H), 1.38 (t, J = 7.0 Hz, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 166.3, 165.6, 149.3, 130.5, 130.3, 128.2, 119.1, 117.3, 102.4, 62.7, 38.2, 30.0, 28.7, 25.2, 14.1; HRMS (ESI) m/z: [M + H]+ Calcd for C15H13ClO5 309.0525; found 309.0520
Ethyl 1-acetyl-4,6-dibromo-2-oxo-1,7b-dihydrocyclopropa[c]chromene-1a(2H)-carboxylate (10a). Pale yellow, clear crystal (147 mg, 17%). 1H NMR (CDCl3, 400 MHz): δ 7.70 (d, J = 3.0 Hz, 1H), 7.41 (d, J = 2.4 Hz, 1H), 4.35–4.25 (m, 2H), 3.54 (d, J = 9.6 Hz, 1H), 3.22 (d, J = 9.6 Hz, 1H), 2.29 (s, 3H), 1.34 (t, J = 7.2 Hz, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 199.4, 166.4, 159.1, 148.3, 135.8, 130.3, 117.0, 116.5, 111.4, 63.4, 36.8, 34.4, 32.6, 31.5, 14.0; HRMS (ESI) m/z: [M + H]+: Calcd for C15H12Br2O5 432.9104; found 432.9080
Ethyl 4,6-dibromo-2a-methyl-1-oxo-2a1,7b-dihydro-1H-2,3-dioxabenzo[f]cyclopropa[cd] indene-7c(2aH)-carboxylate (10b). Pale yellow, clear crystal (147 mg, 17%). 1H NMR (CDCl3, 400 MHz): δ 7.65 (d, J = 2.4 Hz, 1H), 7.56 (d, J = 2.8 Hz, 1H), 4.37–4.32 (q, 2H), 3.37 (d, J = 8.0 Hz, 1H), 3.13 (d, J = 7.6 Hz, 1H), 2.04 (s, 3H), 1.39 (t, J = 7.2 Hz, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 165.9, 165.2, 147.3, 136.2, 132.5, 119.1, 115.4, 112.8, 102.8, 62.9, 38.2, 30.3, 28.6, 25.1, 14.1; HRMS (ESI) m/z: [M + H]+ Calcd for C15H12Br2O5 432.9104; found 432.9114
Ethyl 2a-methyl-6-nitro-1-oxo-2a1,7b-dihydro-1H-2,3-dioxabenzo[f]cyclopropa[cd]indene −7c(2aH)-carboxylate (11b). White solid (115 mg, 18%). Note 5 equivalents of chloroacetone were used in the synthetic route. 1H NMR (CDCl3, 400 MHz): δ 8.41 (d, J = 2.4 Hz, 1H), 8.19–8.16 (dd, J = 2.8 Hz, 9.2 Hz, 1H), 7.08 (d, J = 8.8 Hz, 1H), 4.38–4.33 (q, 2H), 3.47 (d, J = 8.0 Hz, 1H), 3.18 (d, J = 8.0 Hz, 1H), 2.02(s, 3H), 1.38(t, J =7.2 Hz, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 165.9, 165.2, 155.8, 143.2, 126.9, 126.0, 118.6, 116.8, 102.6, 63.0, 37.9, 29.7, 28.2, 25.1, 14.1; HRMS (ESI) m/z: [M + Na]+ Calcd for C15H13NO7 342.0585; found 342.0601
1-ethyl 1-(2-oxopropyl) 2-acetyl-3-(5-nitro-2-(2-oxopropoxy)phenyl)cyclopropane-1,1-dicarboxylate (11c). Off white solid (147 mg, 10%). 1H NMR (CDCl3, 400 MHz): δ 8.19–8.16 (dd, J = 2.8 Hz, 9.2 Hz, 1H), 8.06 (d, J = 2.4 Hz, 1H), 6.82 (d, J = 9.2 Hz, 1H), 4.79–4.46 (m, 4H), 4.28–4.22 (q, J = 7.1 Hz, 2H), 3.77 (d, J = 8.0 Hz, 1H), 3.53 (d, J = 7.6 Hz, 1H), 2.52 (s, 3H), 2.43 (s, 3H), 2.00 (s, 3H), 1.29 (t, J = 7.2 Hz, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 203.9, 201.5, 199.3, 165.1, 165.0, 161.9, 141.4, 125.9, 125.5, 123.1, 111.1, 73.6, 69.2, 62.2, 44.7, 37.8, 32.4, 31.7, 27.0, 25.8, 13.9; HRMS (ESI) m/z: [M + Na]+ Calcd for C21H23NO10 472.1214; found 472.1237
Ethyl 1-acetyl-5-(benzyloxy)-2-oxo-1,7b-dihydrocyclopropa[c]chromene-1a(2H)-carboxylate (12a cis). Off white solid (117 mg, 15%). 1H NMR (CDCl3, 400 MHz): δ 7.42–7.36 (m, 5H), 7.22 (d, J = 8.0 Hz, 1H), 6.77–6.73 (m, 2H), 5.05 (d, J = 3.2 Hz, 2H), 4.28 (t, J = 7.4 Hz, 2H), 3.50 (d, J = 9.2 Hz, 1H), 3.23 (d, J = 9.6 Hz, 1H), 2.23 (s, 3H), 1.33 (t, J = 7.0 Hz, 3H)
Ethyl 1-acetyl-5-(benzyloxy)-2-oxo-1,7b-dihydrocyclopropa[c]chromene-1a(2H)-carboxylate (12a trans). Viscous liquid (117 mg, 15%). 1H NMR (CDCl3, 400 MHz): δ 7.42–7.34 (m, 5H), 7.30 (d, J = 8.4 Hz, 1H), 6.84–6.80 (m, 1H), 6.67 (d, J = 2.4 Hz, 1H), 5.07 (s, 2H), 4.30–4.24 (q, J = 7.2 Hz, 2H), 3.51 (d, J = 5.2 Hz, 1H), 2.57 (d, J = 5.2 Hz, 1H), 2.42 (s, 3H), 1.31 (t, J = 7.2 Hz, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 200.8, 163.6, 161.5, 159.2, 150.7, 136.1, 128.7, 128.6, 128.3, 127.5, 127.4, 112.3, 110.9, 103.6, 62.7, 40.3, 37.9, 31.8, 30.6, 13.9
2a-methyl-1-oxo-N-phenyl-2a1,7b-dihydro-1H-2,3-dioxabenzo[f]cyclopropa[cd]indene-7c(2aH)-carboxamide (13b). Off white solid (200 mg, 31%). 1H NMR (CDCl3, 400 MHz): δ 9.68 (s, 1H), 7.63 (d, J = 8.8 Hz, 2H), 7.48–7.28 (m, 4H), 7.20–6.98 (m, 3H), 3.44 (d, J = 8.0 Hz, 1H), 3.30 (d, J = 8.0 Hz, 1H), 2.02 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 172.1, 162.4, 150.6, 137.2, 130.9, 130.3, 129.1, 124.9, 123.5, 120.0, 117.5, 116.0, 104.0, 39.5, 31.0, 29.2, 25.3; HRMS (ESI) m/z: [M + H]+ Calcd for C19H15NO4 322.1074; found 322.1071
Ethyl 1-acetyl-2-oxo-3-(2-oxopropyl)-1,2,3,7b-tetrahydro-1aH-cyclopropa[c]quinoline-1a-carboxylate (14a trans). White solid (85 mg, 13%). 1H NMR (CDCl3, 400 MHz): δ 7.42 (d, J = 6.8 Hz, 1H), 7.24 (t, J = 7.4 Hz, 1H), 7.10 (t, J = 7.4 Hz, 1H), 6.63 (d, J = 8.4 Hz, 1H), 4.96 (d, J = 18.4 Hz, 1H), 4.65 (d, J = 18.4 Hz, 1H), 4.26–4.21 (m, 2H), 3.61 (d, J = 4.8 Hz, 1H), 4.46 (d, J = 4.8 Hz, 1H), 2.41 (s, 3H), 2.24 (s, 3H), 1.29 (t, J = 7. Hz, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 202.4, 201.9, 164.6, 163.6, 136.0, 129.1, 128.3, 123.7, 120.4, 114.1, 62.2, 51.6, 42.7, 35.2, 32.0, 31.9, 27.2, 13.9; HRMS (ESI) m/z: [M + H]+ Calcd for C18H19NO5 330.1336; found 330.1306
Ethyl 1-acetyl-2-oxo-3-(2-oxopropyl)-1,2,3,7b-tetrahydro-1aH-cyclopropa[c]quinoline-1a-carboxylate (14a cis). White solid (85 mg, 13%). 1H NMR (CDCl3, 400 MHz): δ 7.30 (d, J = 1.2 Hz, 1H), 7.24–7.20 (m, 1H), 7.04–7.00 (m, 1H), 6.66 (d, J = 8.0 Hz, 1H), 5.12 (d, J = 18.0 Hz, 1H), 4.38–4.26 (m, 1H), 4.25–4.17 (m, 2H), 3.39 (d, J = 9.6 Hz, 1H), 3.20 (d, J = 10.0 Hz, 1H), 2.22 (s, 3H), 2.15 (s, 3H), 1.25 (t, J = 7.0 Hz, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 204.8, 200.1, 167.9, 162.3, 138.9, 129.8, 129.0, 123.1, 116.5, 114.5, 62.6, 52.9, 36.9, 33.9, 33.1, 31.8, 27.0, 14.0; HRMS (ESI) m/z: [M + H]+ Calcd for C18H19NO5 330.1336; found 330.1369
Ethyl 1-acetyl-5-(diethylamino)-2-oxo-1,7b-dihydrocyclopropa[c]chromene-1a(2H)-carboxylate (15a). Pale green solid, pale green crystal (34 mg, 5%). 1H NMR (CDCl3, 400 MHz): δ 7.09 (d, J = 8.4 Hz, 1H), 6.40–6.36 (m, 2H), 4.29–4.24 (m, 2H), 3.46 (d, J = 9.2 Hz, 1H), 3.35–3.30 (q, J = 6.9 Hz, 4H), 3.17 (d, J = 9.2 Hz, 1H), 2.21 (s, 3H), 1.31 (t, J = 7.2 Hz, 3H), 1.16 (t, J = 7.0 Hz, 6H); 13C{1H} NMR (CDCl3, 100 MHz): δ 199.7, 167.6, 161.5, 153.0, 148.9, 129.1, 107.8, 99.2, 98.7, 62.8, 44.4, 37.6, 34.2, 33.4, 31.2, 14.0, 12.5; HRMS (ESI) m/z: [M + H]+ Calcd for C19H23NO5 346.1649; found 346.1678
Ethyl 1-acetyl-7-methoxy-2-oxo-1,7b-dihydrocyclopropa[c]chromene-1a(2H)-carboxylate (16a). White solid (250 mg, 41%). 1H NMR (CDCl3, 400 MHz): δ 7.26 (t, J = 8.4 Hz, 1H), 6.75 (d, J = 8.0 Hz, 1H), 6.65 (d, J = 8.0 Hz, 1H), 4.32–4.25 (m, 2H), 3.89 (s, 3H), 3.57 (d, J = 9.6 Hz, 1H), 3.40 (d, J = 9.6 Hz, 1H), 2.23 (s, 3H), 1.33 (t, J = 7.2 Hz, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 199.8, 167.2, 160.9, 157.9, 152.6, 129.8, 109.0, 105.8, 103.0, 62.9, 55.9, 36.4, 33.5, 31.0, 29.6, 14.0; HRMS (ESI) m/z: [M + H]+ Calcd for C16H16O6 305.1020; found 305.1018
Ethyl 1-acetyl-7-methoxy-2-oxo-1,7b-dihydrocyclopropa[c]chromene-1a(2H)-carboxylate (16a cis+trans). White solid (85 mg, 13%). 1H NMR (CDCl3, 400 MHz): δ 7.28–7.21 (m, 2H), 6.74–6.64 (m, 4H), 4.29 (m, 4H), 3.89–3.88 (two s, 6H), 3.82 (d, J = 5.2 Hz, 1H), 3.57 (d, J = 9.6 Hz, 1H), 3.39 (d, J = 10.0 Hz, 1H), 2.54 (d, J = 5.2 Hz, 1H), 2.42 (s, 3H), 2.21 (s, 3H), 1.33–1.29 (m, 6H); 13C{1H} NMR (CDCl3, 100 MHz): δ 200.9, 199.8, 167.2, 163.7, 161.5, 160.9, 157.9, 157.4, 152.6, 150.8, 129.8, 129.1, 109.3, 109.0, 108.0, 106.6, 105.8, 103.0, 62.9, 62.6, 56.0, 39.8, 37.2, 36.4, 33.5, 31.7, 31.0, 29.6, 26.6, 14.0, 13.8; HRMS (ESI) m/z: [M + H]+ Calcd for C16H16O6 305.1020; found 305.1021
6-methoxy-2a-methyl-1-oxo-N-phenyl-2a1,7b-dihydro-1H-2,3-dioxabenzo[f]cyclopropa[cd]indene-7c(2aH)-carboxamide (17b). White solid (133 mg, 19%). 1H NMR (CDCl3, 400 MHz): δ 9.69 (s, 1H), 7.62 (d, J = 8.0 Hz, 2H), 7.392 (t, J = 7.8 Hz, 2H), 7.18 (t, J = 7.4 Hz, 1H), 6.99–6.84 (m, 3H), 3.80 (s, 3H), 3.40 (d, J = 8.0 Hz, 1H), 3.27 (d, J = 8.0 Hz, 1H), 2.01 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 172.1, 162.4, 155.4, 144.3, 137.2, 129.1, 124.9, 119.9, 118.4, 116.6, 116.4, 114.9, 104.2, 55.7, 39.7, 31.3, 29.2, 25.3; HRMS (ESI) m/z: [M + H]+ Calcd for C20H17NO5 352.1180; found 352.1187
4-(tert-butyl)-2a-methyl-1-oxo-N-phenyl-2a1,7b-dihydro-1H-2,3-dioxabenzo[f]cyclopropa[cd]indene-7c(2aH)-carboxamide (18b). White solid (128 mg, 17%). 1H NMR (CDCl3, 400 MHz): δ 9.69 (s, 1H), 7.62 (d, J = 8.0 Hz, 2H), 7.392 (t, J = 7.8 Hz, 2H), 7.18 (t, J = 7.4 Hz, 1H), 6.99–6.84 (m, 3H), 3.80 (s, 3H), 3.40 (d, J = 8.0 Hz, 1H), 3.27 (d, J = 8.0 Hz, 1H), 2.01 (s, 3H); 13C{1H} NMR (CDCl3, 100 MHz): δ 172.1, 162.4, 150.6, 137.2, 130.9, 130.3, 129.1, 124.9, 123.5, 120.0, 117.5, 116.0, 104.0, 39.5, 31.0, 29.2, 25.3; HRMS (ESI) m/z: [M + H]+ Calcd for C19H15NO4 378.1700;, found 378.1698
Supplementary Material
ACKNOWLEDGMENT
This project was supported by the National Cancer Institute of the National Institute of Health under award number R01 CA226436 (PCT). We are grateful to Dr. Pierre LeMagueres (Rigaku) for helpful discussions regarding twin refinement.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/
X-ray crystallography data, NCI-60 screen and NMR spectra (PDF).
Accession Codes
CCDC 2093798, 2093799, 2093800 and 2093801 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/dat_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Center, 12 Union Road, Cambridge, CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no conflicts of interest.
REFERENCES
- 1.Lovering F; Bikker J; Humblet C, Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem 2009, 52 (21), 6752–6756. [DOI] [PubMed] [Google Scholar]
- 2.Wei W; Cherukupalli S; Jing L; Liu X; Zhan P, Fsp(3): A new parameter for drug-likeness. Drug Discov. Today 2020. [DOI] [PubMed]
- 3.Meyers J; Carter M; Mok NY; Brown N, On the origins of three-dimensionality in drug-like molecules. Future Med. Chem 2016, 8 (14), 1753–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou J; Campbell-Conroy EL; Silina A; Uy J; Pierre F; Hurley DJ; Hilgraf N; Frieman BA; DeNinno MP, Synthesis of fused bicyclic piperidines: potential bioactive templates for medicinal chemistry. J. Org. Chem 2015, 80 (1), 70–79. [DOI] [PubMed] [Google Scholar]
- 5.Cox B; Duffy J; Zdorichenko V; Bellanger C; Hurcum J; Laleu B; Booker-Milburn KI; Elliott LD; Robertson-Ralph M; Swain CJ; Bishop SJ; Hallyburton I; Anderson M, Escaping from Flatland: Antimalarial Activity of sp(3)-Rich Bridged Pyrrolidine Derivatives. ACS Med. Chem. Lett 2020, 11 (12), 2497–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng Y; Tice CM; Singh SB, The use of spirocyclic scaffolds in drug discovery. Bioorg. Med. Chem. Lett 2014, 24 (16), 3673–3682. [DOI] [PubMed] [Google Scholar]
- 7.Talele TT, The “Cyclopropyl Fragment” is a Versatile Player that Frequently Appears in Preclinical/Clinical Drug Molecules. J. Med. Chem 2016, 59 (19), 8712–8756. [DOI] [PubMed] [Google Scholar]
- 8.Kupeli Akkol E; Genc Y; Karpuz B; Sobarzo-Sanchez E; Capasso R, Coumarins and Coumarin-Related Compounds in Pharmacotherapy of Cancer. Cancers (Basel) 2020, 12 (7), 1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zang T; Verma K; Chen M; Jin Y; Trippier PC; Penning TM, Screening baccharin analogs as selective inhibitors against type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3). Chem. Biol. Interact 2015, 234, 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verma K; Zang T; Gupta N; Penning TM; Trippier PC, Selective AKR1C3 Inhibitors Potentiate Chemotherapeutic Activity in Multiple Acute Myeloid Leukemia (AML) Cell Lines. ACS Med. Chem. Lett 2016, 7 (8), 774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verma K; Gupta N; Zang T; Wangtrakluldee P; Srivastava SK; Penning TM; Trippier PC, AKR1C3 Inhibitor KV-37 Exhibits Antineoplastic Effects and Potentiates Enzalutamide in Combination Therapy in Prostate Adenocarcinoma Cells. Mol. Cancer Ther 2018, 17 (9), 1833–1845. [DOI] [PubMed] [Google Scholar]
- 12.Verma K; Zang T; Penning TM; Trippier PC, Potent and Highly Selective Aldo-Keto Reductase 1C3 (AKR1C3) Inhibitors Act as Chemotherapeutic Potentiators in Acute Myeloid Leukemia and T-Cell Acute Lymphoblastic Leukemia. J. Med. Chem 2019, 62 (7), 3590–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morsy A; Trippier PC, Reversal of Apalutamide and Darolutamide Aldo-Keto Reductase 1C3-Mediated Resistance by a Small Molecule Inhibitor. ACS Chem. Biol 2020, 15 (3), 646–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Penning TM; Jonnalagadda S; Trippier PC; Rižner TL, Aldo-Keto Reductases and Cancer Drug Resistance. Pharmacol. Rev 2021, 73 (3), 1150–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Little RD; Dawson JR, MIRC (Michael Initiated Ring Closure) Reactions Formation of Three, Five, Six and Seven Membered Rings. Tetrahedron Lett 1980, 21 (27), 2609–2612. [Google Scholar]
- 16.Das I; Pal TK; Pathak T, Diastereoselective michael initiated ring closure on vinyl sulfone-modified carbohydrates: a stereospecific and general route to alpha-substituted cyclopropanes. J. Org. Chem 2007, 72 (24), 9181–9189. [DOI] [PubMed] [Google Scholar]
- 17.Papageorgiou CD; Cubillo de Dios MA; Ley SV; Gaunt MJ, Enantioselective Organocatalytic Cyclopropanation via Ammonium Ylides. Angew. Chem. Int. Ed 2004, 43 (35), 4641–4644. [DOI] [PubMed] [Google Scholar]
- 18.Kozhushkov SI; Leonov A; de Meijere A, Simple Large-Scale Preparation of 1,2,3-Tris-Acceptor Substituted Cyclopropanes. Synthesis 2003, 06, 2003. [Google Scholar]
- 19.Xin X; Zhang Q; Liang Y; Zhang R; Dong D, Tandem halogenation/Michael-initiated ring-closing reaction of alpha,beta-unsaturated nitriles and activated methylene compounds: one-pot diastereoselective synthesis of functionalized cyclopropanes. Org. Biomol. Chem 2014, 12 (15), 2427–2435. [DOI] [PubMed] [Google Scholar]
- 20.Yuan WC; Lei CW; Zhao JQ; Wang ZH; You Y, Organocatalytic Asymmetric Cyclopropanation of 3-Acylcoumarins with 3-Halooxindoles: Access to Spirooxindole-cyclopropa[c]coumarin Compounds. J. Org. Chem 2021, 86 (3), 2534–2544. [DOI] [PubMed] [Google Scholar]
- 21.Shchepin VV; Silaichev PS; Kodess MI, Reaction of zinc enolates prepared from 2,2-dibromoindan-1-one or 2,2-dibromo-1-tetralone and zinc with 2-oxochromen-3-carboxylic acid derivatives. Russ. J. Org. Chem 2007, 43 (10), 1441–1445. [Google Scholar]
- 22.Slates HL; Weber S; Wendler NL, Reactions of chloroacetone in basic media. J. Org. Chem 1969, 34 (2), 457–459. [Google Scholar]
- 23.Sun JC; Wang XH; Ji CB; Peng YY; Zeng XP, Enantioselective Construction of Chiral Cyclopropa[c]coumarins via Lewis Base-Catalyzed Cyclopropanation. J. Org. Chem 2020, 85 (23), 14963–14970. [DOI] [PubMed] [Google Scholar]
- 24.Guo J; Liu Y; Li X; Liu X; Lin L; Feng X, Nickel(ii)-catalyzed enantioselective cyclopropanation of 3-alkenyl-oxindoles with phenyliodonium ylide via free carbene. Chem. Sci 2016, 7 (4), 2717–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antoniotti S; Genin E; Michelet V; Genet JP, Highly efficient access to strained bicyclic ketals via gold-catalyzed cycloisomerization of bis-homopropargylic diols. J. Am. Chem. Soc 2005, 127 (28), 9976–9977. [DOI] [PubMed] [Google Scholar]
- 26.Traven VF; Tolmachev AY; Podhaluzina NY; Kanevskii DS; Solovieva NP, New ways of lactone ring shortening and cylcopropanation in coumarin derivatives. Heterocycl. Commun 1999, 5, 69–76. [Google Scholar]
- 27.A., B.; Trendafilova A; Ivanov C; Rodios NA, Cyclopropanation reaction of 3-Acyl-2H-1-benzopyran-2-ones with phenacylbromide in phase transfer systems. Tetrahedron 1993, 49, 2275–2286. [Google Scholar]
- 28.Jonnalagadda S; Huwaimel B; Jonnalagadda S; Garrison JC; Trippier PC, Expedient Single Step Access to Strained Tricyclic Ketals Published as a preprint on ChemRxiv 2021.
- 29.Shoemaker RH, The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6 (10), 813–823. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.