

Abstract
Hosts rely on the innate immune system to clear pathogens in response to infection. Pathogen-associated molecular patterns (PAMPs) bind to innate immune receptors and engage activation of downstream signaling to initiate a host immune response to fight infection. A key component of this innate response is programmed cell death (PCD). Recent work has highlighted significant crosstalk and functional redundancy between cell death pathways, leading to the discovery of PANoptosis, an inflammatory PCD pathway dependent on PANoptosomes, which are innate immune danger sensing complexes that activate inflammatory cell death and contain caspase(s) with or without inflammasome components and RHIM-containing proteins. While PANoptosis has been characterized in response to a growing number of pathogens, inflammatory diseases and cancer, its role and the functional consequences of PANoptotic component modulation during NLRC4 activation by Pseudomonas aeruginosa infection remain unknown. Here, we show that P. aeruginosa can induce PANoptosis in mouse bone marrow-derived macrophages (BMDMs). Only the combined deletion of caspase-1, -11, -8, and RIPK3 protected mouse BMDMs from cell death. Moreover, we showed that PANoptotic components act in a compensatory manner; in the absence of NAIP5 and NLRC4 during P. aeruginosa challenge, alternative cell death molecules such as RIPK1 and MLKL were activated, while activation of caspase-1, -3, -7, and -8 were reduced in mouse BMDMs. Together, these data highlight the extensive crosstalk between cell death signaling molecules and showcase the plasticity of the system.
Keywords: NAIP5, NLRC4, inflammasome, MLKL, PANoptosis, PANoptosome, Pseudomonas, caspase-1, caspase-8, caspase-3, caspase-7, gasdermin D, RIPK1, RIPK3
INTRODUCTION
Innate immunity plays a crucial role in detecting and eliminating pathogens from an infected host. Programmed cell death (PCD) is a vital innate immune mechanism utilized by a host. Multiple PCD pathways, such as pyroptosis, apoptosis, and necroptosis, have been described with clear links to innate immunity. More recently, evidence of extensive crosstalk between cell death signaling molecules has been found, which has led to the conceptualization of PANoptosis. PANoptosis is defined as an innate immune inflammatory PCD pathway dependent on PANoptosomes, which are caspase(s)-containing complexes with or without inflammasome components and RHIM-containing proteins. The plasticity in this PCD pathway interconnects pyroptosis, apoptosis and necroptosis, but cannot be accounted for by individually (1–19).
Several bacterial, viral, and fungal pathogens induce PANoptosis (7, 12, 14–18, 20). Salmonella enterica serovar Typhimurium, a Gram-negative bacterium, is one such pathogen that has been shown to trigger PANoptosis in macrophages (14, 20). Though S. Typhimurium robustly activates NLR family CARD domain-containing protein 4 (NLRC4), which complexes with NLR family apoptosis inhibitory proteins (NAIPs) to activate the inflammasome and pyroptosis (21, 22), deletion of pyroptotic molecules, as in Casp1/11−/−, or Gsdmd−/− cells, only partially protects from death (14, 20). Cell death is only rescued when PANoptosis is inhibited via combined deletions of caspases-1/11/8 with Ripk3 (14, 20).
Like Salmonella, Pseudomonas aeruginosa, an opportunistic Gram-negative bacteria which causes severe infections in humans (23), can be detected in the cytosol by NLRC4 and activate pyroptosis (22). Upon the Gram-negative bacteria infection, host NAIP5 interacts with flagellin and triggers a NAIP5-NLRC4 interaction which activates caspase-1 (24, 25). Activated caspase-1 cleaves gasdermin D (GSDMD) and pro-inflammatory cytokines, IL-1β and IL-18, which can then be released via activated GSDMD pores (26–30). The interplay between pyroptosis and apoptosis has been studied in several NLRC4 engaging Gram-negative bacteria (20, 22, 31); however, the role of necroptosis as well as PANoptosis have not been well studied in this infection model.
Understanding PCD pathways and crosstalk between them helps identify new drug targets and strategies to circumvent host pathogenesis upon infection. Each bacterial, viral, and fungal trigger can induce a unique immune response; therefore, a comprehensive characterization of cell death, in response to a variety of immune triggers, is warranted. In this study, we show that in primary macrophages, P. aeruginosa infection can induce inflammatory cell death with key features of PANoptosis and that cell death was rescued in PANoptosis-deficient bone marrow-derived macrophages (BMDMs). In addition, we demonstrate that BMDMs deficient in inflammasome activation—NAIP5-, or NLRC4-deficient cells—show enhanced activation of phosphorylated MLKL and RIPK1. This, along with experimental evidence showing that caspase-1 deficiency does not fully rescue BMDMs from cell death, indicates that P. aeruginosa-induced cell death pathway involves key components of pyroptosis, apoptosis, and necroptosis and that MLKL-mediated cell death may be acting as a compensatory mechanism in the absence of NLRC4.
Materials and methods
Mice
C57BL/6J [wild-type (WT)], Naip5−/− (32), Nlrc4−/− (33), Nlrc4−/−Nlrp3−/− (34), and Casp1/11−/−Casp8–/–Ripk3–/– (19) mice have been described previously. All mice were bred and maintained in a specific pathogen-free facility at the Animal Resources Center at St. Jude Children’s Research Hospital and were backcrossed to the C57BL/6 background (J substrain) for at least 10 generations. Age- and sex-matched male and female 6- to 12-week-old mice were used. Animal studies were conducted under protocols approved by the St. Jude Children’s Research Hospital committee on the Use and Care of Animals.
Cell culture
Primary mouse bone marrow-derived macrophages (BMDMs) were generated from the bone marrow of wild-type and indicated mutant mice. Cells were grown for 5–6 days in IMDM (Thermo Fisher Scientific, 12440053) supplemented with 1% non-essential amino acids (Thermo Fisher Scientific, 11140-050), 10% FBS (Biowest, S1620), 30% L929 conditioned media, and 1% penicillin and streptomycin (Thermo Fisher Scientific, 15070-063). BMDMs were then seeded into antibiotic-free media at a concentration of 1 × 106 cells into 12-well plates and incubated overnight.
Bacterial infection
BMDMs were infected with PAO1 strain of P. aeruginosa at an MOI of 1. After 2 h of infection, 50 μg/ml gentamicin (Thermo Fisher Scientific, 15750-060) was added to kill extracellular bacteria, and BMDMs were then incubated for 12 h.
Real-time imagine for cell death
The kinetics of cell death were determined using the IncuCyte SX5 (Essen BioScience) live-cell automated system. BMDMs (5 × 105 cells/well) were seeded in 24-well tissue culture plates. Cells were infected with P. aeruginosa at an MOI of 1 and stained with propidium iodide (PI; Life Technologies, P3566) following the manufacturer’s protocol. The plate was scanned, and fluorescent and phase-contrast images (4 image fields/well) were acquired in real-time every 1 h from 0 to 12 h post-infection. PI-positive dead cells are marked with a red mask for visualization. The image analysis, masking, and quantification of dead cells were performed using the software package supplied with the IncuCyte imager.
Immunoblot analysis
Cell lysates and culture supernatants were combined in caspase lysis buffer (containing 1× protease inhibitors, 1× phosphatase inhibitors, 10% NP-40, and 25 mM DTT) and 4× sample loading buffer (containing SDS and 2-mercaptoethanol) for immunoblot analysis of caspases. For immunoblot analysis of signaling components, supernatants were removed, and cells were washed once with DPBS, followed by lysis in RIPA buffer and sample loading buffer. Proteins were separated by electrophoresis through 8–12% polyacrylamide gels. Following electrophoretic transfer of proteins onto PVDF membranes (Millipore, IPVH00010), nonspecific binding was blocked by incubation with 5% skim milk, then membranes were incubated with primary antibodies against: caspase-8 (AdipoGen, AG-20T-0138-C100, 1:1000), cleaved caspase-8 (CST, #8592, 1:1000), caspase-7 (CST, #9492, 1:1000), cleaved caspase-7 (CST, #9491, 1:1000), caspase-3 (CST, #9662, 1:1000), cleaved caspase-3 (CST, #9661, 1:1000), caspase-1 (AdipoGen, AG-20B-0042, 1:1000), caspase-11 (Novus Biologicals, NB120-10454), GAPDH (CST, #5174, 1:1000), pMLKL (CST, #37333, 1:1000), tMLKL (Abgent, AP14272b, 1:1000), pRIPK1 (CST, #65746, 1:1000), tRIPK1 (CST, #3493, 1:1000), GSDMD (Abcam, ab209845, 1:1000), and β-actin horseradish peroxidase (HRP) (sc-47778, 1:5000). Membranes were then washed and incubated with the appropriate HRP–conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, anti-rabbit [111-035-047; 1:5000] and anti-mouse [315-035-047; 1:5000]). Proteins were visualized using Immobilon Forte Western HRP Substrate (Millipore, WBLUF0500).
Cytokine analysis
TNF cytokine was measured from BMDM culture supernatant according to the manufacturer’s instructions (Invitrogen, BMS607-3).
Lactate dehydrogenase (LDH) assay for cell death
Levels of lactate dehydrogenase released by cells were determined in the supernatant using the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega, G1780) according to the manufacturer’s instructions.
Quantification and Statistical Analysis
GraphPad Prism v8.0 software was used for data analysis. Data are shown as mean ± SEM. Statistical significance was determined by two-way ANOVA. P < 0.05 was considered statistically significant.
RESULTS
Cell death induced by P. aeruginosa infection is abrogated in PANoptosis-deficient BMDMs
To characterize P. aeruginosa-induced cell death, we first examined the activation of cell death signaling molecules in infected wild-type (WT) BMDMs. We observed robust cell death in these cells (Fig. 1A–B), along with activation of caspase-1, GSDMD, caspase-8, caspase-3, and caspase-7 (Fig. 1C). Additionally, phosphorylated MLKL, phosphorylated RIPK1, and an increase in total RIPK1 cleavage were observed in response to infection (Fig. 1D). Under these conditions, these data show that P. aeruginosa activates a combination of molecules indicative of PANoptosis. The activation of a molecular signature consistent with PANoptosis led us to hypothesize that inhibiting PANoptosis, by deleting key components of pyroptosis, apoptosis, and necroptosis, would provide complete protection against cell death. When PANoptosis-deficient BMDMs (Casp1/11–/–Ripk3–/–Casp8–/–; referred to as QKO) were challenged with P. aeruginosa infection, QKO BMDMs were protected from cell death (Fig. 2A–C) and showed impaired activation of caspase-1, GSDMD, caspase-8, and caspase-3 (Fig. 2D). Activation of phosphorylated MLKL were also impaired in QKO BMDMs in response to infection (Fig. 2E). This suggests that PANoptosis is involved in mediating the cell death in response to P. aeruginosa infection.
Figure 1. P. aeruginosa induces PANoptosis in primary macrophages.
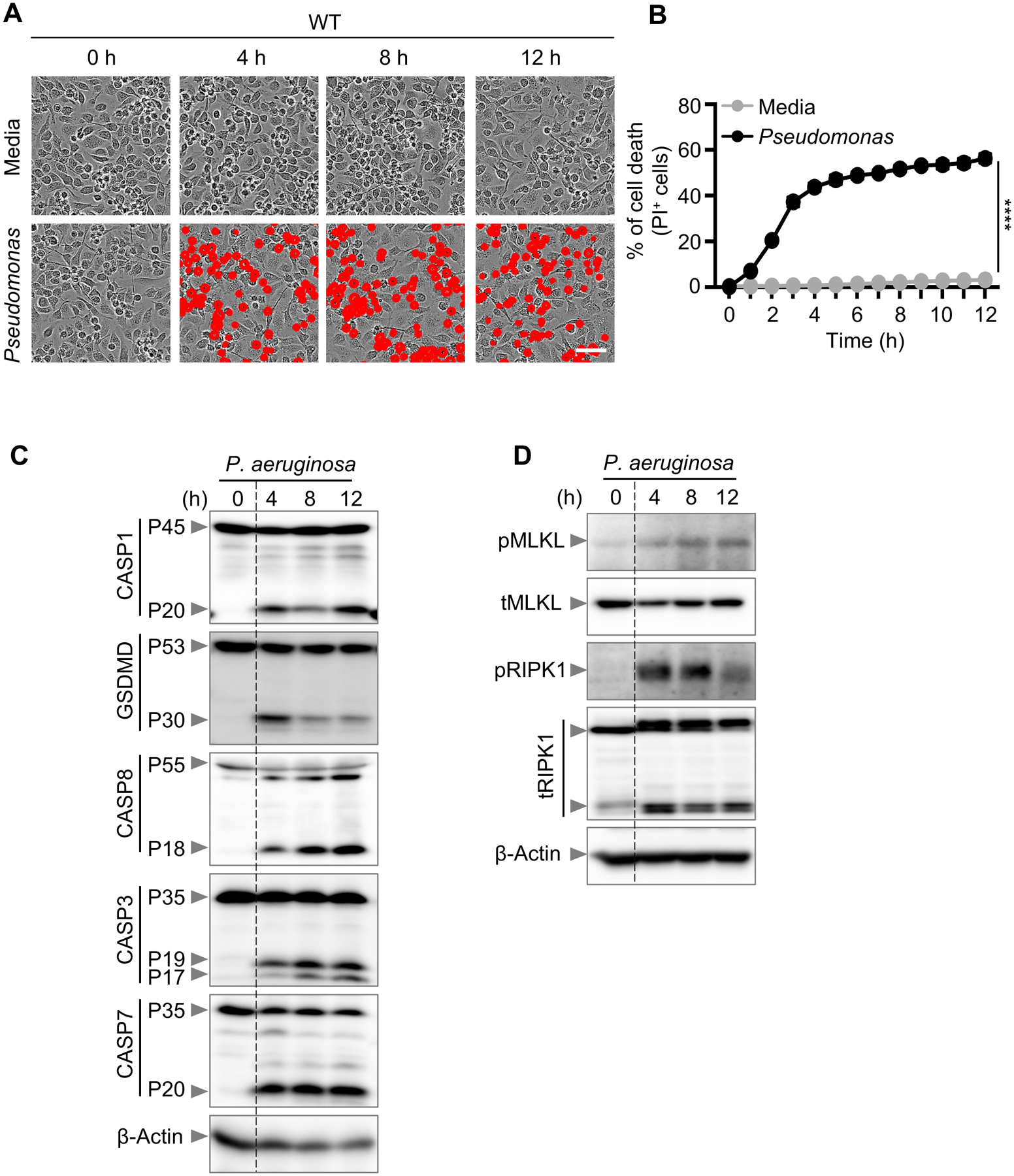
(A) Representative images of cell death in wild type (WT) bone marrow-derived macrophages (BMDMs) in a media control and infected with P. aeruginosa (MOI, 1) for the indicated time; (B) Quantification of cell death from (A); (C-D) Immunoblot analysis of (C) pro- (P45) and activated (P20) caspase-1 (CASP1), pro- (P53) and activated (P30) gasdermin D (GSDMD), pro- (P55) and cleaved CASP8 (P18), pro- (P35) and cleaved CASP3 (P19/17), and pro- (P35) and cleaved CASP7 (P20); (D) phosphorylated MLKL (pMLKL) and total MLKL (tMLKL), and phosphorylated RIPK1 (pRIPK1) and total RIPK1 (tRIPK1) in WT BMDMs infected with P. aeruginosa (MOI, 1) for the indicated time. β-actin was used as the loading control. Data are representative of at least four independent experiments. Scale bar, 50 μm. ****P < 0.0001. Analysis was performed using the two-way ANOVA. Data are shown as mean ± SEM (B).
Figure 2. Cell death induced by P. aeruginosa infection is abrogated in PANoptosis-deficient BMDMs.
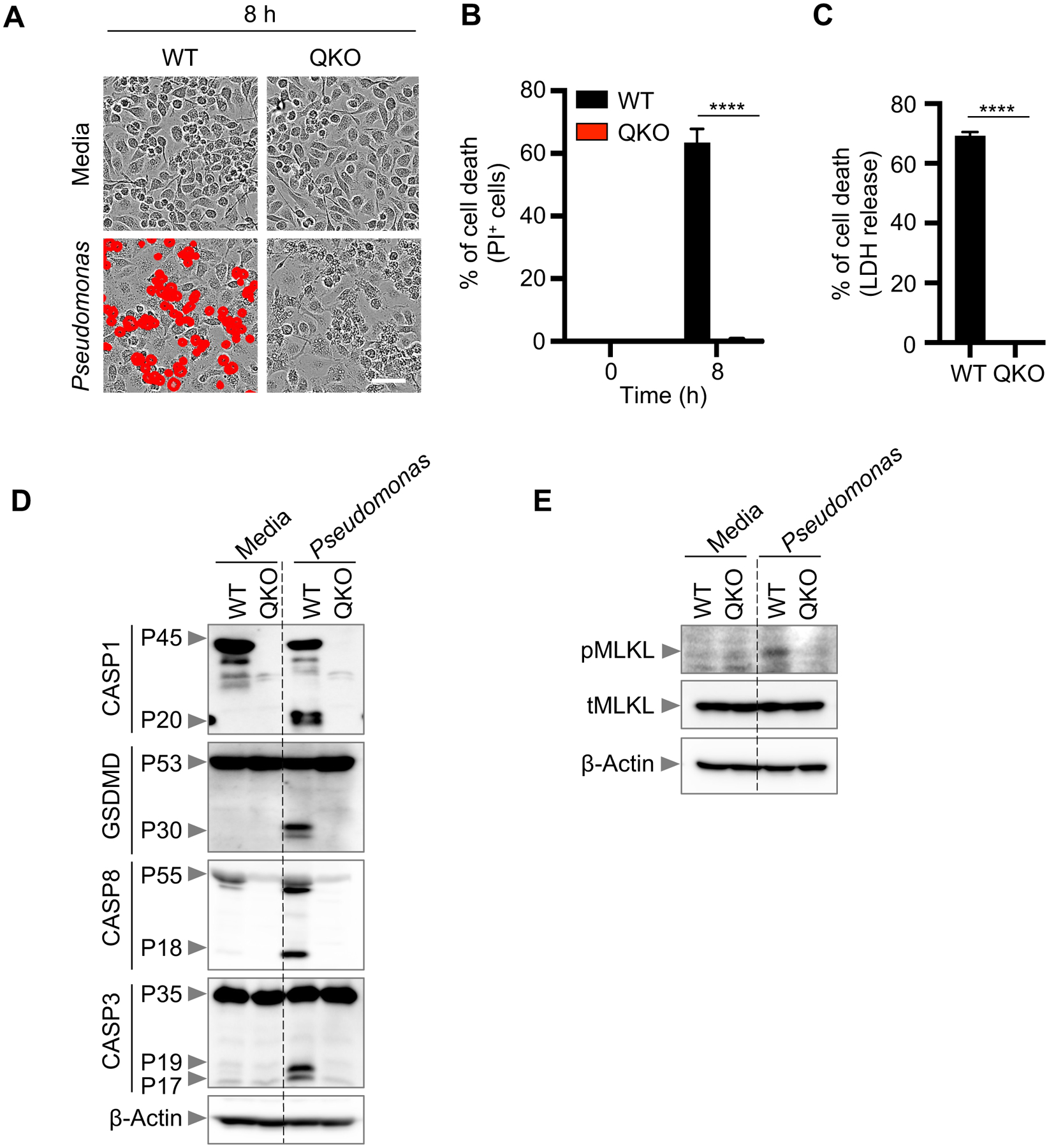
(A) Representative images of cell death in wild type (WT) and Casp1/11−/−Ripk3−/−Casp8−/− (QKO) bone marrow-derived macrophages (BMDMs) infected with P. aeruginosa (MOI, 1) for 8 h; (B) Quantification of cell death from (A); (C) Quantification of cell death by LDH release in WT and QKO BMDMs infected with P. aeruginosa (MOI, 1) for 8 h; (D-E) Immunoblot analysis of (D) pro- (P45) and activated (P20) caspase-1 (CASP1) and pro- (P53) and activated (P30) gasdermin D (GSDMD), pro- (P55) and cleaved CASP8 (P18) and pro- (P35) and cleaved CASP3 (P19/17); (E) phosphorylated MLKL (pMLKL) and total MLKL (tMLKL) in WT and QKO BMDMs infected with P. aeruginosa (MOI, 1) for 8 h. β-actin was used as the loading control. Data are representative of at least four independent experiments. Scale bar, 50 μm. ****P < 0.0001. Analysis was performed using the two-way ANOVA. Data are shown as mean ± SEM (B, C).
BMDMs deficient in NAIP5 or NLRC4 show compensatory activation of RIPK1 and MLKL
Because of the flexibility built into the PANoptotic cell death pathway, with multiple cell death effectors and executioners being activated, then modifying it, through a combination of gene deletions, may result in a compensatory response of other key cell death signaling components. This phenomenon has previously been shown in Salmonella- and Bacillus anthracis lethal toxin-treated cells void of pyroptosis (Casp1−/−, Gsdmd−/−, or Casp1/11−/− cells) where, instead, caspase-8–mediated apoptosis executes a cell death response (20, 31, 35). However, the contribution of necroptosis, in the absence of pyroptosis, is less characterized in infections. To gain insight into the interconnectivity of these key cell death pathways in response to P. aeruginosa infection, we examined necroptotic markers in infected BMDMs deficient in the upstream pyroptotic molecules, NAIP5 and NLRC4. We observed reduced cell death in Naip5–/– and Nlrc4–/– BMDMs compared to WT BMDMs after P. aeruginosa infection (Fig. 3A–B). As infection progressed, the percentage of cells dying increased in Naip5–/– and Nlrc4–/– BMDMs, suggesting that the loss of pyroptotic sensors cannot completely protect against cell death (Fig. 3B). We also monitored the cleavage of caspase-1 and GSDMD and, consistent with the observed partial protection from cell death, Naip5–/– and Nlrc4–/– BMDMs showed impaired activation of these molecules (Fig. 3C). We also observed decreased activation of caspase-8, caspase-3, and caspase-7 in Naip5–/– and Nlrc4–/– BMDMs (Fig. 3C). However, the loss of Naip5–/– and Nlrc4–/– resulted in an increase in the activation of phosphorylated MLKL (Fig. 3D). Consistent with MLKL activation, there was increased phosphorylated RIPK1 and total RIPK1 (Fig. 3D). Components of necroptosis can be activated by tumor necrosis factor-alpha (TNF-α) (36). To determine whether NAIP5 or NLRC4 deficiency triggered enhanced production of TNF-α upon Pseudomonas infection, we measured TNF-α release in the supernatant of WT, Naip5–/–, and Nlrc4–/ BMDMs. All released similar amounts of TNF-α upon Pseudomonas infection (Fig. 3E). Altogether, these data suggest that in response to P. aeruginosa infection, cells deficient in inflammasome sensors modulate their overall cell death response and enhance activation of some PANoptotic effectors when others are impaired.
Figure 3. Pyroptosis deficiency induces enhanced necroptosis.
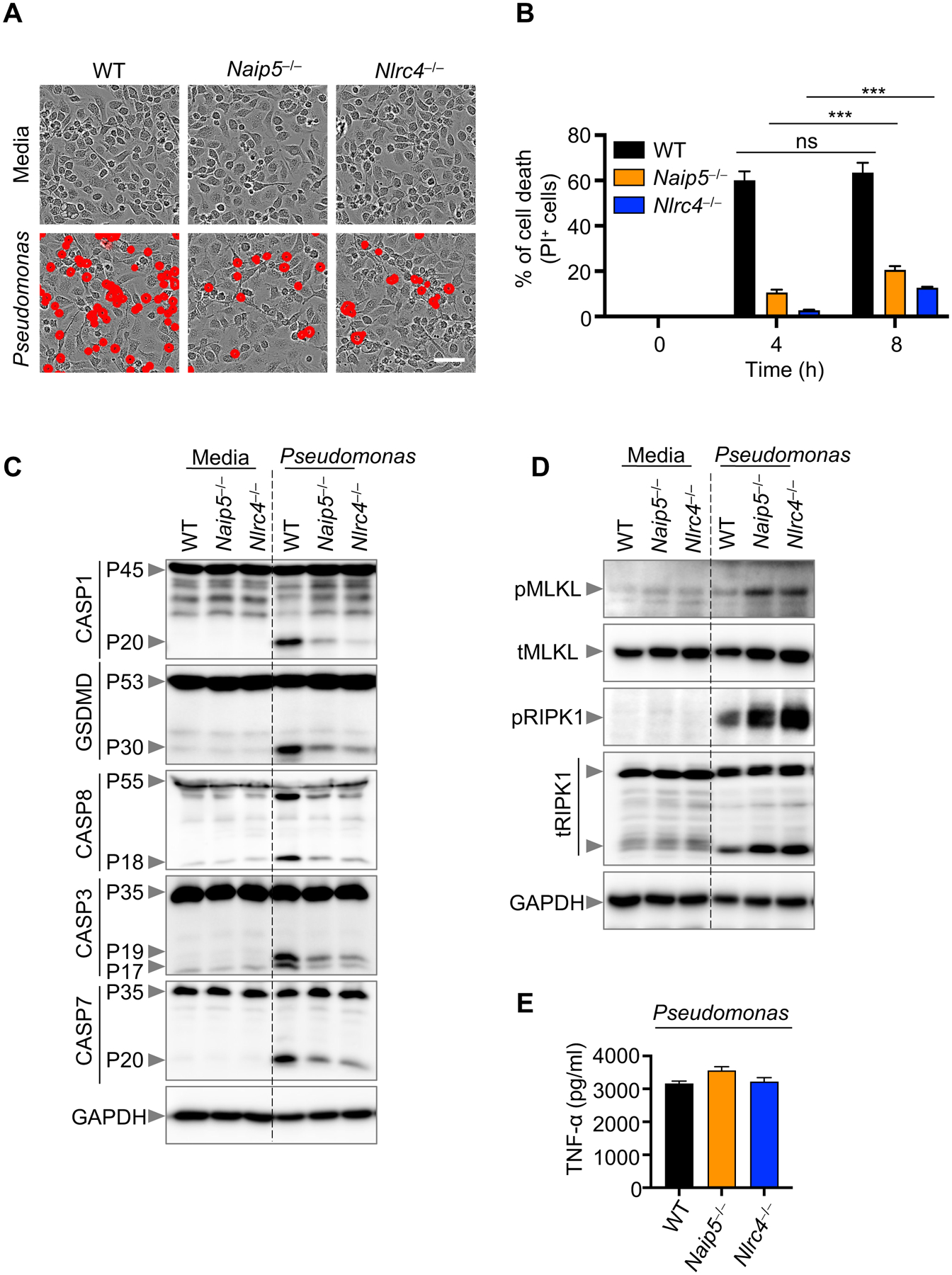
(A) Representative images of cell death in wild type (WT), Naip5−/−, and Nlrc4−/− bone marrow-derived macrophages (BMDMs) infected with P. aeruginosa (MOI, 1) for 8 h; (B) Quantification of cell death from (A) at the indicated times; (C-D) Immunoblot analysis of (C) pro- (P45) and activated (P20) caspase-1 (CASP1), pro- (P53) and activated (P30) gasdermin D (GSDMD), pro- (P55) and cleaved CASP8 (P18), pro- (P35) and cleaved CASP3 (P19/17), and pro- (P35) and cleaved CASP7 (P20); (D) phosphorylated MLKL (pMLKL) and total MLKL (tMLKL), and phosphorylated RIPK1 (pRIPK1) and total RIPK1 (tRIPK1) in WT, Naip5−/−, and Nlrc4−/− BMDMs infected with P. aeruginosa (MOI, 1) for 8 h. GAPDH was used as the loading control. (E) Measurement of TNF-α release from the supernatant of WT, Naip5–/–, and Nlrc4–/– BMDMs upon Pseudomonas infection. Data are representative of at least four independent experiments. Scale bar, 50 μm. ***P < 0.001. Analysis was performed using the two-way ANOVA. Data are shown as mean ± SEM (B).
Blocking pyroptosis does not rescue cell death
Caspase-1 and GSDMD activation were not completely abolished in Naip5−/− and Nlrc4−/− BMDMs (Fig. 3C–D) most likely due to the fact that P. aeruginosa has been shown to activate the NLRP3 inflammasome sensor to induce pyroptosis in response to a bacterial infection (37–41). To determine the contribution of NLRP3 in response to infection, we used Nlrc4–/–Nlrp3–/– BMDMs. Although Nlrc4–/–Nlrp3–/– and Nlrc4–/– BMDMs both had less cell death than WT BMDMs, the loss of both inflammasome sensors resulted in significantly less cell death than the single Nlrc4–/– knockout (Fig. 4A–B). However, Nlrc4–/–Nlrp3–/– BMDMs were still not fully protected from cell death, indicating that other signaling molecules may be responsible for cell death in the absence of canonical P. aeruginosa-induced inflammasome sensors. Notably, activation of molecules involved in necroptosis was markedly enhanced in pyroptosis-deficient BMDMs compared to WT BMDMs (Fig. 4D). There was a complete absence of active caspase-1 in Nlrc4–/–Nlrp3–/– BMDMs, though GSDMD was cleaved (Fig. 4C). This could possibly be due to caspase-11-mediated GSDMD activation (29) and, indeed, we observed activation of caspase-11 in Nlrc4–/–Nlrp3–/– BMDMs (Fig. 4C) upon P. aeruginosa infection. Overall, these results suggest that upon infection, the activation of MLKL-dependent cell death is enhanced in the absence of key P. aeruginosa inflammasome sensors. P. aeruginosa likely induces a PANoptosis response which activates MLKL in a compensatory manner when cells are unable to activate inflammasomes.
Figure 4. Blocking pyroptosis does not fully rescue cell death.
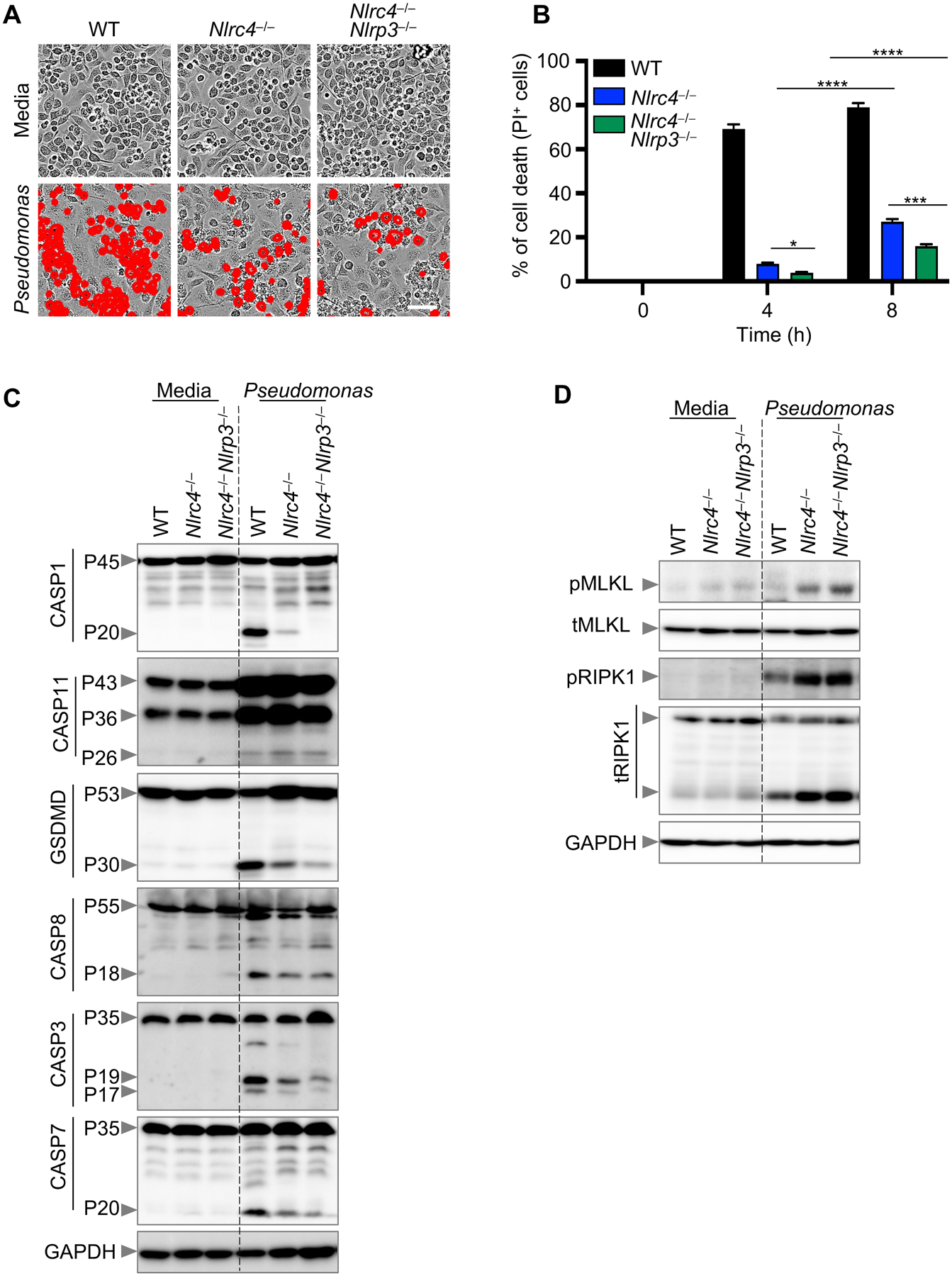
(A) Representative images of cell death in wild type (WT), Nlrc4−/−, and Nlrc4−/−Nlrp3−/− bone marrow-derived macrophages (BMDMs) infected with P. aeruginosa (MOI, 1) for 8 h; (B) Quantification of cell death from (A) at the indicated times; (C-D) Immunoblot analysis of (C) pro- (P45) and activated (P20) caspase-1 (CASP1), pro- (P43, P36) and activated (P26) caspase-11 (CASP11), pro- (P53) and activated (P30) gasdermin D (GSDMD), pro- (P55) and cleaved CASP8 (P18), pro- (P35) and cleaved CASP3 (P19/17), and pro- (P35) and cleaved CASP7 (P20); (D) phosphorylated MLKL (pMLKL) and total MLKL (tMLKL), and phosphorylated RIPK1 (pRIPK1) and total RIPK1 (tRIPK1) in WT, Nlrc4−/−, and Nlrc4−/−Nlrp3−/− BMDMs infected with P. aeruginosa (MOI, 1) for 8 h. GAPDH was used as the loading control. Data are representative of at least four independent experiments. Scale bar, 50 μm. *P < 0.05, ***P < 0.001, ****P < 0.0001. Analysis was performed using the two-way ANOVA. Data are shown as mean ± SEM (B).
DISCUSSION
The critical role of crosstalk between various cell death signaling molecules in the host innate immune response has been increasingly recognized, particularly crosstalk resulting in functional redundancy between pyroptotic and apoptotic molecules. Previous studies have shown an increase in caspase-8–mediated cell death in response to caspase-1, caspase-1/11, or Gsdmd deletions (20, 31, 35). Crosstalk is further highlighted by the pyroptotic caspase-1, which can cleave caspase-7 (10), and inflammasome activation, which can result in the cleavage of the apoptotic substrate PARP (6). In the context of osteomyelitis, the NLRP3 inflammasome or caspase-1 can play a redundant role with caspase-8 in promoting IL-1β-mediated disease (5, 19). Additionally, in GSDMD-deficient macrophages, caspase-3 and caspase-7 are activated (42), showcasing the interplay between pyroptosis and apoptosis to achieve cell death. Enabling apoptosis in lieu of pyroptosis may prevent the host from initiating a hyper-inflammatory response to pathogens.
Crosstalk between apoptosis and necroptosis is also well-understood, as necroptosis functions as the backup cell death pathway when the apoptotic caspase-8 is inhibited (36). Though suspected, few studies have aimed at discovering crosstalk between pyroptosis and molecules associated with necroptosis. In this study, we provide evidence of crosstalk between components involved in pyroptosis and necroptosis in response to P. aeruginosa infection. The NAIP5-NLRC4 cytosolic bacterial sensor complex modulated the biochemical features of cell death induced by P. aeruginosa; its absence activated RIPK1 and phosphorylated MLKL, suggesting that RIPK1 and MLKL activation can serve as a backup for not only the loss of caspase-8 activation but also caspase-1 activation. Non-cleavable caspase-8 (Casp8DA/DA) can facilitate cleavage of RIPK1 in response to TNF and cycloheximide treatment (43). Similarly, we also observed reduced cleavage of caspase-8 and increased cleavage of total RIPK1 in Naip5–/– or Nlrc4–/– BMDMs, suggesting that non-cleaved caspase-8 might still regulate RIPK1 cleavage in Naip5–/– or Nlrc4–/– BMDMs upon Pseudomonas infection. While we observed no difference in the amounts of TNF released between WT and NAIP5- or NLRC4-deficient cells, future studies should be aimed at determining the activation of components of necroptosis in Nlrc4–/–Tnf–/– macrophages to mechanistically define the contribution of TNF in Nlrc4–/– BMDMs upon Pseudomonas infection.
Overall, our results highlight that P. aeruginosa-induced cell death is characterized by a compensatory activation of MLKL in response to a deficiency in inflammasome sensing. Not only do our data lead to a more comprehensive understanding of PCD responses, but they provide more evidence of a unified PCD response, PANoptosis, and showcase the ability of P. aeruginosa to induce a finely tuned cell death response that specifically modulates PANoptosis executioners in a way that dampens components of apoptosis and activates components of necroptosis in BMDMs deficient in inflammasome sensors. In the context of infection, it would benefit the host to respond to pathogens by utilizing a myriad of cell death molecules; pathogens carrying specific inhibitors would be less successful at colonization due to the ability of the host immune system to reroute PCD in real time. PANoptosis is a strategy built on flexible redundancy, allowing for the use of multiple paths to achieve cell death. The connection between components of pyroptosis and necroptosis presented here expands our knowledge of the intricacies of a Gram-negative bacteria induced PANoptosis cell death response.
Acknowledgement
We thank all the members of the Kanneganti laboratory for their comments and suggestions during the development of this manuscript. We also thank J. Gullett, PhD, and R. Tweedell, PhD, for scientific editing and writing support.
Funding
Work from our laboratory is supported by the US National Institutes of Health (AI101935, AI124346, AI160179, AR056296, and CA253095 to T.-D.K.) and the American Lebanese Syrian Associated Charities (to T.-D.K.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
REFERENCES
- 1.Kuriakose T, Man SM, Malireddi RK, Karki R, Kesavardhana S, Place DE, Neale G, Vogel P, and Kanneganti TD. 2016. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malireddi RKS, Karki R, Sundaram B, Kancharana B, Lee S, Samir P, and Kanneganti TD. 2021. Inflammatory Cell Death, PANoptosis, Mediated by Cytokines in Diverse Cancer Lineages Inhibits Tumor Growth. Immunohorizons 5: 568–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kesavardhana S, Malireddi RKS, Burton AR, Porter SN, Vogel P, Pruett-Miller SM, and Kanneganti T-D. 2020. The Zα2 domain of ZBP1 is a molecular switch regulating influenza-induced PANoptosis and perinatal lethality during development. Journal of Biological Chemistry 295: 8325–8330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karki R, Sharma BR, Lee E, Banoth B, Malireddi RKS, Samir P, Tuladhar S, Mummareddy H, Burton AR, Vogel P, and Kanneganti T-D. 2020. Interferon regulatory factor 1 regulates PANoptosis to prevent colorectal cancer. JCI insight 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lukens JR, Gurung P, Vogel P, Johnson GR, Carter RA, McGoldrick DJ, Bandi SR, Calabrese CR, Vande Walle L, Lamkanfi M, and Kanneganti TD. 2014. Dietary modulation of the microbiome affects autoinflammatory disease. Nature 516: 246–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malireddi RK, Ippagunta S, Lamkanfi M, and Kanneganti TD. 2010. Cutting edge: proteolytic inactivation of poly(ADP-ribose) polymerase 1 by the Nlrp3 and Nlrc4 inflammasomes. J Immunol 185: 3127–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malireddi RKS, Kesavardhana S, Karki R, Kancharana B, Burton AR, and Kanneganti TD. 2020. RIPK1 Distinctly Regulates Yersinia-Induced Inflammatory Cell Death, PANoptosis. Immunohorizons 4: 789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, Zheng M, Sundaram B, Banoth B, Malireddi RKS, Schreiner P, Neale G, Vogel P, Webby R, Jonsson CB, and Kanneganti TD. 2021. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 184: 149–168.e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malireddi RKS, Gurung P, Mavuluri J, Dasari TK, Klco JM, Chi H, and Kanneganti TD. 2018. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J Exp Med 215: 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamkanfi M, Kanneganti TD, Van Damme P, Vanden Berghe T, Vanoverberghe I, Vandekerckhove J, Vandenabeele P, Gevaert K, and Nunez G. 2008. Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol Cell Proteomics 7: 2350–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gurung P, Anand PK, Malireddi RK, Vande Walle L, Van Opdenbosch N, Dillon CP, Weinlich R, Green DR, Lamkanfi M, and Kanneganti TD. 2014. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol 192: 1835–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee S, Karki R, Wang Y, Nguyen LN, Kalathur RC, and Kanneganti TD. 2021. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karki R, Sundaram B, Sharma BR, Lee S, Malireddi RKS, Nguyen LN, Christgen S, Zheng M, Wang Y, Samir P, Neale G, Vogel P, and Kanneganti TD. 2021. ADAR1 restricts ZBP1-mediated immune response and PANoptosis to promote tumorigenesis. Cell Rep 37: 109858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christgen S, Zheng M, Kesavardhana S, Karki R, Malireddi RKS, Banoth B, Place DE, Briard B, Sharma BR, Tuladhar S, Samir P, Burton A, and Kanneganti TD. 2020. Identification of the PANoptosome: A Molecular Platform Triggering Pyroptosis, Apoptosis, and Necroptosis (PANoptosis). Front Cell Infect Microbiol 10: 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banoth B, Tuladhar S, Karki R, Sharma BR, Briard B, Kesavardhana S, Burton A, and Kanneganti TD. 2020. ZBP1 promotes fungi-induced inflammasome activation and pyroptosis, apoptosis, and necroptosis (PANoptosis). J Biol Chem 295: 18276–18283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng M, Karki R, Vogel P, and Kanneganti TD. 2020. Caspase-6 Is a Key Regulator of Innate Immunity, Inflammasome Activation, and Host Defense. Cell 181: 674–687 e613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malireddi RKS, Gurung P, Kesavardhana S, Samir P, Burton A, Mummareddy H, Vogel P, Pelletier S, Burgula S, and Kanneganti TD. 2020. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J Exp Med 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng M, Williams EP, Malireddi RKS, Karki R, Banoth B, Burton A, Webby R, Channappanavar R, Jonsson CB, and Kanneganti TD. 2020. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J Biol Chem 295: 14040–14052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gurung P, Burton A, and Kanneganti TD. 2016. NLRP3 inflammasome plays a redundant role with caspase 8 to promote IL-1beta-mediated osteomyelitis. Proc Natl Acad Sci U S A 113: 4452–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doerflinger M, Deng Y, Whitney P, Salvamoser R, Engel S, Kueh AJ, Tai L, Bachem A, Gressier E, Geoghegan ND, Wilcox S, Rogers KL, Garnham AL, Dengler MA, Bader SM, Ebert G, Pearson JS, De Nardo D, Wang N, Yang C, Pereira M, Bryant CE, Strugnell RA, Vince JE, Pellegrini M, Strasser A, Bedoui S, and Herold MJ. 2020. Flexible Usage and Interconnectivity of Diverse Cell Death Pathways Protect against Intracellular Infection. Immunity 53: 533–547 e537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Man SM, and Kanneganti TD. 2015. Regulation of inflammasome activation. Immunol Rev 265: 6–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, and Flavell RA. 2007. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med 204: 3235–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diggle SP, and Whiteley M. 2020. Microbe Profile: Pseudomonas aeruginosa: opportunistic pathogen and lab rat. Microbiology (Reading) 166: 30–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kofoed EM, and Vance RE. 2011. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477: 592–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, and Shao F. 2011. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477: 596–600. [DOI] [PubMed] [Google Scholar]
- 26.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, and Shao F. 2016. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535: 111–116. [DOI] [PubMed] [Google Scholar]
- 27.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, and Lieberman J. 2016. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535: 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Place DE, and Kanneganti TD. 2019. Cell death-mediated cytokine release and its therapeutic implications. J Exp Med 216: 1474–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, and Dixit VM. 2015. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526: 666–671. [DOI] [PubMed] [Google Scholar]
- 30.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F. 2015. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: 660–665. [DOI] [PubMed] [Google Scholar]
- 31.de Vasconcelos NM, Van Opdenbosch N, Van Gorp H, Martin-Perez R, Zecchin A, Vandenabeele P, and Lamkanfi M. 2020. An Apoptotic Caspase Network Safeguards Cell Death Induction in Pyroptotic Macrophages. Cell Rep 32: 107959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lightfield KL, Persson J, Brubaker SW, Witte CE, von Moltke J, Dunipace EA, Henry T, Sun YH, Cado D, Dietrich WF, Monack DM, Tsolis RM, and Vance RE. 2008. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat Immunol 9: 1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, and Dixit VM. 2004. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430: 213–218. [DOI] [PubMed] [Google Scholar]
- 34.Karki R, Lee E, Place D, Samir P, Mavuluri J, Sharma BR, Balakrishnan A, Malireddi RKS, Geiger R, Zhu Q, Neale G, and Kanneganti TD. 2018. IRF8 Regulates Transcription of Naips for NLRC4 Inflammasome Activation. Cell 173: 920–933 e913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Opdenbosch N, Van Gorp H, Verdonckt M, Saavedra PHV, de Vasconcelos NM, Goncalves A, Vande Walle L, Demon D, Matusiak M, Van Hauwermeiren F, D’Hont J, Hochepied T, Krautwald S, Kanneganti TD, and Lamkanfi M. 2017. Caspase-1 Engagement and TLR-Induced c-FLIP Expression Suppress ASC/Caspase-8-Dependent Apoptosis by Inflammasome Sensors NLRP1b and NLRC4. Cell Rep 21: 3427–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, and Tschopp J. 2000. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1: 489–495. [DOI] [PubMed] [Google Scholar]
- 37.Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, and Monack DM. 2010. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med 207: 1745–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gram AM, Wright JA, Pickering RJ, Lam NL, Booty LM, Webster SJ, and Bryant CE. 2021. Salmonella Flagellin Activates NAIP/NLRC4 and Canonical NLRP3 Inflammasomes in Human Macrophages. J Immunol 206: 631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Man SM, Hopkins LJ, Nugent E, Cox S, Gluck IM, Tourlomousis P, Wright JA, Cicuta P, Monie TP, and Bryant CE. 2014. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci U S A 111: 7403–7408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qu Y, Misaghi S, Newton K, Maltzman A, Izrael-Tomasevic A, Arnott D, and Dixit VM. 2016. NLRP3 recruitment by NLRC4 during Salmonella infection. J Exp Med 213: 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Balakrishnan A, Karki R, Berwin B, Yamamoto M, and Kanneganti TD. 2018. Guanylate binding proteins facilitate caspase-11-dependent pyroptosis in response to type 3 secretion system-negative Pseudomonas aeruginosa. Cell Death Discov 4: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taabazuing CY, Okondo MC, and Bachovchin DA. 2017. Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem Biol 24: 507–514 e504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Newton K, Wickliffe KE, Dugger DL, Maltzman A, Roose-Girma M, Dohse M, Komuves L, Webster JD, and Dixit VM. 2019. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 574: 428–431. [DOI] [PubMed] [Google Scholar]