

Abstract
Objective
SSc is a rheumatic autoimmune disease affecting roughly 20 000 people worldwide and characterized by excessive collagen accumulation in the skin and internal organs. Despite the high morbidity and mortality associated with SSc, there are no approved disease-modifying agents. Our objective in this study was to explore transcriptomic and model-based drug discovery approaches for SSc.
Methods
In this study, we explored the molecular basis for SSc pathogenesis in a well-studied mouse model of scleroderma. We profiled the skin and lung transcriptomes of mice at multiple timepoints, analysing the differential gene expression that underscores the development and resolution of bleomycin-induced fibrosis.
Results
We observed shared expression signatures of upregulation and downregulation in fibrotic skin and lung tissue, and observed significant upregulation of key pro-fibrotic genes including GDF15, Saa3, Cxcl10, Spp1 and Timp1. To identify changes in gene expression in responses to anti-fibrotic therapy, we assessed the effect of TGF-β pathway inhibition via oral ALK5 (TGF-β receptor I) inhibitor SB525334 and observed a time-lagged response in the lung relative to skin. We also implemented a machine learning algorithm that showed promise at predicting lung function using transcriptome data from both skin and lung biopsies.
Conclusion
This study provides the most comprehensive look at the gene expression dynamics of an animal model of SSc to date, provides a rich dataset for future comparative fibrotic disease research, and helps refine our understanding of pathways at work during SSc pathogenesis and intervention.
Keywords: systemic sclerosis, scleroderma, RNA-seq, fibrosis, bleomycin, ALK5 inhibitor
Rheumatology key messages.
Subcutaneous bleomycin injections induced coordinated pro-fibrotic gene expression changes in skin and lung tissue.
Transcriptomic response to ALK5 inhibition was robust but time-lagged in lung relative to skin.
Transcriptomic profiling from the skin and lung accurately predicted markers of lung function.
Introduction
SSc (the systemic form of scleroderma) is a chronic progressive disease characterized by three main features: vascular injury, immunological abnormalities, and fibrosis of the skin and various internal organs, including the lung. While skin fibrosis is the hallmark feature of SSc, scleroderma interstitial lung fibrosis is responsible for much of the morbidity and mortality associated with this disease [1]. As of 2018, an estimated 19 390 people were living with SSc with significant predicted growth in new cases through 2038 [2]. SSc patients with significant internal organ involvement have a 10-year survival rate of only 38% [3]. The mechanism underlying the development of fibrosis remains unclear, and current therapeutic options are limited and provide only a modest benefit to patients [4, 5].
A well-characterized mouse model of scleroderma involves daily subcutaneous injections of the antitumor antibiotic bleomycin (BLM), which leads to localized dermal fibrosis as well as pulmonary fibrosis [6]. In the present study, we utilized this model, which mimics several key features of human SSc, to examine the pathological mechanisms underlying the development and resolution of fibrosis in SSc. It is well established that the TGF-β signalling pathway is required for bleomycin-induced fibrosis in this model and that genetic or pharmacological inhibition of this pathway causes resolution of dermal and pulmonary fibrosis [7]. Thus, we also used the ALK5 (TGF-β receptor I) inhibitor SB525334 to investigate genome-wide changes that occur during TGF-β inhibitor-mediated resolution.
We characterized the development of dermal and pulmonary fibrosis in the bleomycin-induced mouse model of scleroderma. We compared skin and lung signatures, noting significant overlap between the time-course trajectories of increased and decreased expression, as well as produced a proof-of-concept model for predicting lung function outcomes from lung and skin gene expression data, and report the expression changes in well-known collagen formation and degradation genes in response to the ALK5 inhibitor. Taken together, the data generated in this study and our results provide a trove of resources from which the scientific community can build to better understand the development, progression and treatment of scleroderma.
Results
Experimental design and phenotypic profiling of a bleomycin-induced mouse model of scleroderma
To induce skin and lung fibrosis, female C57BL/6NTac mice were subjected to daily subcutaneous injections of either bleomycin or PBS five times per week for two weeks. Mice were sacrificed on days 7, 14, 21, 28 or 42 following the first bleomycin injections (Fig. 1). For the mice that were sacrificed on days 21 or 28 (Groups 5–10), oral dosing of the ALK5 inhibitor SB525334 was initiated one day after the last bleomycin injection. BID dosing in these groups continued through the day prior to sacrifice. To assess the extent of fibrosis, hydroxyproline analysis was conducted on skin and lung tissues from animals sacrificed at all timepoints. Additionally, flexiVent lung function analysis was performed on mice that were sacrificed on day 21, 28 or 42 (see Methods for details).
Fig. 1.

Overview of study design and animal groups
Bleomycin was dosed at 10 mg/kg/day, and SB525334 was dosed at 30 mg/kg. BID: twice a day; BLM: bleomycin; PO: oral administration; SC: subcutaneous
Bleomycin induced an increase in collagen deposition in the skin and lungs, indicated by increased hydroxyproline content in both tissues (Fig. 2A and B). Skin fibrosis was most prominent at days 14 and 21 and showed evidence of resolution at day 42 (Fig. 2A). Treatment with the ALK5 inhibitor caused a decrease in skin fibrosis at day 21. Lung fibrosis was evident at day 21 and peaked at day 28 (Fig. 2B). Treatment with the ALK5 inhibitor caused a decrease in lung fibrosis at days 21 and 28. Results from flexiVent analysis indicate that lung function was significantly impaired at days 21 and 28 (Fig. 2C). Treatment with the ALK5 inhibitor promoted a modest improvement in lung function at both time points. Representative histopathology images at each stage of the experiment for lung and skin are available in Supplementary Fig. S1, available at Rheumatology online.
Fig. 2.

Effects of bleomycin treatment and ALK5 inhibition on markers of collagen formation and lung function
(A) Hydroxyproline content of skin tissue. (B) Hydroxyproline content of lung tissue (ml: middle lobe; il: inferior lobe; pcl: postcaval lobe). (C) Lung function as assessed by flexiVent analysis. (BLM: bleomycin; ALK5i: the ALK5 inhibitor SB525334). Error bars represent SEM. Statistical significance was assessed using one-way ANOVA with Bonferroni’s correction.
RNA was isolated from skin and lung tissue harvested from mice that were sacrificed at all timepoints, and bulk RNA-seq was conducted on all samples. The resulting gene expression libraries allowed us to investigate transcriptional changes underlying the development and regression of bleomycin-induced fibrosis, as well as the effects of TGF-β pathway inhibition on both tissues. Dimensionality reduction via t-SNE (t-distributed stochastic engineering). [8] revealed stronger separation of gene expression profiles by tissue type than by timepoint, bleomycin/PBS treatment or ALK5 intervention (Supplementary Fig. S2A, available at Rheumatology online). Principal components analysis of individual tissues showed substantial variation by treatment and tissue in the first two principal components for both tissues (Supplementary Fig. S2B, available at Rheumatology online).
Coordinated skin and lung gene expression changes in response to bleomycin injury
We first sought to understand the transcriptional changes underlying bleomycin-induced fibrosis. For each of the five timepoints (days 7, 14, 21, 28 and 42), we compared PBS- and bleomycin- treated mice, and summarized the number of differentially expressed genes identified by limma [9] for each tissue in Fig. 3A. The number of differentially expressed genes increased from day 7 to a maximum at day 14, just after cessation of the bleomycin treatment, and we observed substantial reduction from that maximum by day 42. This pattern of differential gene expression gave us confidence that the observed changes were driven primarily by the bleomycin treatment. For each of these differentially expressed gene sets, we used gene-set enrichment analysis (GSEA) to compute enrichment of pathways in MSigDB including Kyoto Encylopedia of Genes and Genomes (KEGG), Biocarta and Reactome (Supplementary Figs S3 and S4, available at Rheumatology online). We observed significant upregulation of cell cycle, KEGG disease and biosynthesis pathways through early timepoints, giving way to upregulated extracellular matrix formation and degradation pathways in both tissues at days 28 and 42.
Fig. 3.

Integrative differential gene expression analysis between bleomycin-treated mice and control across tissues
(A) Number of differentially expressed genes in lung and skin between bleomycin (BLM) and PBS-treated mice. (B) Cluster-matched patterns of z scores normalized differential expression in skin (top) and lung (bottom). (C) UpSet plot showing the number of genes in each cluster (left) and magnitude of overlaps between clusters (top). (D) Volcano plots of differential expression in skin and lung, coloured by FDR-corrected significance in skin and an absolute log-fold change >=3, with shared genes labelled on the lung volcano plot.
A key objective of our study was to assess the degree and temporal trajectory of change in the transcriptional profiles in skin vs lung. To do this, we performed unsupervised clustering on the differentially expressed genes and explored whether gene expression changes were occurring in the same direction over time for both skin and lung. The clusters that resulted are shown in Fig. 3B. In skin, these clusters roughly represented four unique trajectories, which we have named Skin1–Skin4: persistent upregulation in response to bleomycin, followed by downregulation post-bleo (Skin1); increasing downregulation in response to bleomycin, followed by upregulation post-bleo (Skin2); persistent downregulation in response to bleomycin, followed by upregulation post-bleo (Skin3); and increasing upregulation in response to bleomycin, followed by downregulation post-bleo (Skin4).
Lung clusters were produced and named arbitrarily, and then re-ordered in Fig. 3B according to their closest matching Skin cluster as identified in the UpSet plot in Fig. 3C. The largest overlap occurred between Skin1 and Lung3, meaning that most genes with a persistent upregulation in response to bleomycin followed by down-regulation post-bleo shared that pattern in both lung and skin. Lung cluster trajectories matched skin cluster trajectories in all four cases, indicating that similar sets of genes are being activated or repressed in response to bleomycin in both tissues. A full list of cluster membership is available in Supplementary Table S1 (available at Rheumatology online), and cluster probability distributions are shown in Supplementary Fig. S5B (available at Rheumatology online).
To get a more detailed view of which genes showed the largest coordinated bleomycin-induced changes in both skin and lung, we generated volcano plots of differential expression for skin and lung at day 14, when bleo-induced changes reached an apex in both tissues (Fig. 3D). We observed 261 and 59 genes that displayed differential expression between bleomycin and PBS in skin and lung, respectively (coloured blue, absolute log fold change >3 and False Discovery Rate (FDR)-corrected P-value <0.1; Supplementary Table S2, available at Rheumatology online). Exploration of overlap yielded eight genes differentially expressed in both tissues (labelled on both volcano plots; Timp1 log fold change 2.913 in skin). This provided us with a short list of large, coordinated gene expression changes across both tissues. Notably, many of these genes have been previously shown to be increased in SSc patients (see Discussion for a complete list). Taken together, these results point to shared pathophysiology between the bleomycin-induced mouse model of scleroderma and human SSc.
Time-lagged differential gene expression of ALK5 inhibitor driven intervention
Next, we sought to understand the effect of the ALK5 inhibitor SB525334 on resolution of bleomycin-induced fibrosis by exploring differential expression between SB525334-treated and vehicle-treated mice in both lung and skin at days 21 and 28. Fig. 4A shows that at day 21, there are substantial expression differences in the SB525334-treated mice in skin, but no subsequent changes in lung. By day 28, a comparable number of differentially expressed genes are observable in both tissues, suggesting that systemic effects of oral SB525334 may not be temporally consistent across tissues.
Fig. 4.

Differential gene expression and pathway enrichment in response to ALK5i treatment
(A) Number of differentially expressed genes in lung and skin between ALK5 inhibitor treated and PBS treated mice. (B) UpSet plot showing the overlap of differentially expressed gene sets in the four time/tissue pairs for ALK5 inhibitor vs PBS treated mice. Volcano plots showing shared differentially expressed genes between treated and untreated in skin and lung. (C) Pathway enrichment UpDown plots showing the fraction and number of genes in each pathway upregulated/downregulated in the SB525334 arm compared with vehicle on day 28 for skin and lung (D).
Of the genes showing differential expression in mice treated with SB525334, 1055 were differential at both timepoints in the skin, and 750 were shared between skin and lung on day 28 (Fig. 4B). Analysis of the top differentially expressed genes at day 28 between SB525334 and vehicle treated samples revealed downregulation of several keratin associated genes in skin and one, Krt4, in lung (Fig. 4C). Several collagen genes including Col1a1, Col1a2, Col3a1, Col5a1, Col5a2 and Col12a1 also displayed time-lagged differential expression between bleomycin- and PBS-treated mice, and reductions in response to SB525334 (Supplementary Fig. S6, available at Rheumatology online). GSEA pathway analysis (see Methods) for genes significantly differentially expressed in skin and lung at day 28 revealed downregulation of the Naba collagen pathway in SB525334-treated skin but not lung, suggesting a potential for continued lag in response (Fig. 4D). One gene, Bgn, codes for biglycan and was significantly downregulated in the SB525334 group in all four tissues/timepoints pairs. Biglycan has been previously associated with type I and type II collagens [10], and its synthesis is stimulated by ALK5 [11].
Predicting lung function with lung and skin transcriptional profiles
Of the measurements available for mouse lung function, inspiratory capacity (IC) is considered to be the most analogous to forced vital capacity (FVC). In general, we observed that IC was higher for PBS-treated mice than for bleomycin-treated mice, indicating that bleomycin treatment caused impairment of lung function.
Trained on the lung and skin transcriptional data, we constructed models of IC (see Methods) in order to determine whether lung or skin biopsies could function as surrogate biomarkers of lung function. Due to limited availability of validation data sets, we used repeated cross-validated performance to assess the models.
We observed that ℳung and ℳskin models were equally informative for predicting IC [med(R2lung) ≈ 0.8 and median (R2skin) ≈ 0.8; Fig. 5]. While our sample numbers were limited, these results are encouraging and suggest that RNA-seq from lung or skin could serve as a biomarker of lung function.
Fig. 5.
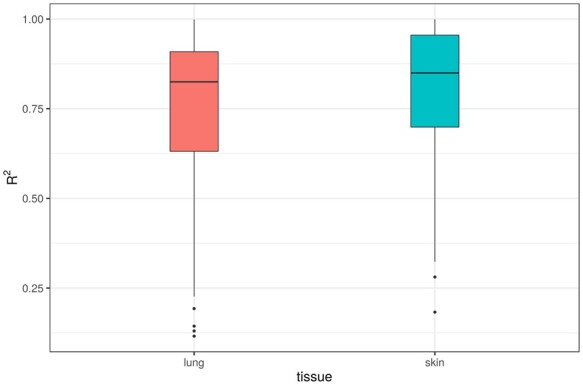
Boxplots of R2 predictive power for elastic net models predicting inspiratory capacity from expression data
Discussion
The subcutaneous bleomycin mouse model is widely used to support efforts to develop novel therapies for scleroderma, including in vivo efficacy studies and biomarker identification and validation. The transcriptomics data presented here represents a novel resource that can be utilized to support ongoing and future studies using this model. The novel design of the present study allows for the comparison of gene expression changes in both skin and lung in response to bleomycin. Our results demonstrate a coordinated response to bleomycin across tissue types, highlighted by the trajectory clusters in Fig. 3B. These findings reflect one of the hallmark features seen in SSc patients: the relatively concurrent development of fibrosis across multiple organs [12]. Thus, our datasets represent a novel resource that can be used to further investigate the systemic development and progression of tissue fibrosis. Further, these data represent the possibility that genetic signatures from peripherally accessible skin biopsies could reflect and/or predict the disease status of the lung. This may represent a significant enhancement over current lung evaluations, which are limited to function imaging-based morphology, due to the risk associated with taking lung tissue samples or lavage.
Additionally, as highlighted in Fig. 3D, a number of genes that exhibited differential expression in both skin and lung in response to bleo have been shown to be increased in SSc patients:
GDF15—GDF15 (growth differentiation factor 15) is a distant member of the TGF-β family. GDF15 is increased in the serum of SSc patients and is correlated with disease severity and extent of organ involvement, particularly with lung fibrosis [13, 14].
CXCL10—CXCL10 (also known as IP-10) is a Th1 chemokine that is induced by IFN-γ [15]. CXCL10 is increased in the skin and serum of SSc patients, and increased serum levels of CXCL10 are associated with disease severity and increased internal organ involvement [16–18].
MMP10—MMP10 (also known as stromelysin 2) is a matrix metalloproteinase that plays a critical role in extracellular matrix (ECM) degradation and remodeling during wound healing and vascular remodeling [19]. MMP10 is increased in the serum and pulmonary arteries of SSc patients with pulmonary hypertension [20].
TIMP-1—TIMP-1 (tissue inhibitor of matrix metalloproteinase-1) levels are increased in the serum and lesional skin of SSc patients [21–23]. Interestingly, TIMP-1 is one of three soluble proteins comprising the ELF (enhanced liver fibrosis) test. The ELF test was recently validated as an SSc biomarker that correlates with both skin and lung involvement [24].
Saa3—Saa3 is a member of the serum amyloid A (SAA) family, which consist of early-phase proteins known to play a key role in inflammation [25]. Serum SAA levels are increased in SSc patients and correlated with the extent of pulmonary involvement [25, 26].
Retnla—Retnla (resistin-like alpha, also known as RELM-α or FIZZ1) has been identified as an M2 macrophage marker in mice [27]. The closest human homologue, RETLN-β, is increased in the lungs of SSc patients with pulmonary hypertension [28].
SPP1—SPP1 (secreted phosphoprotein 1, also known as osteopontin) is a matricellular protein that exhibits proinflammatory and profibrotic properties [29]. Osteopontin is increased in both the serum and lesional skin of SSc patients [29, 30].
These findings support the utility of this model to help identify novel targets and/or biomarkers for SSc. Our data showing that Alk5 inhibition exhibits a time-lagged response in lung vs skin suggests that in vivo compound efficacy studies may need to be designed to allow for analysis of different tissues at multiple timepoints to thoroughly assess systemic treatment effects.
Our preliminary analyses using novel modeling techniques to predict lung function based on gene expression have laid the groundwork for future translational studies. Our findings suggest that there are similarities in gene expression patterns in skin and lung that can be taken advantage of to allow for an increased ability to monitor the fibrotic state of the lungs of SSc patients by assessing gene expression changes in skin biopsies. Analysis of gene expression patterns in the skin may potentially offer insight into which patients will develop SSc-ILD, an aspect of the disease that accounts for a considerable proportion of its morbidity and mortality. The ability to better identify these at-risk patients would allow for earlier treatment and improved patient outcomes.
Methods
Study design
Twelve-week-old female C57BL/6NTac mice (Taconic) were given daily subcutaneous injections of either bleomycin (Hospira; 10 mg/kg per day) or PBS (vehicle control) in a shaved interscapular region. Injections were done using a 27-gauge needle, five times per week for two consecutive weeks (Days 0–4 and 7–11, inclusive). Mice were re-shaved as necessary, and circles were re-drawn every 1–2 days throughout the study [31, 32].
Beginning on day 12, the ALK5 inhibitor SB525334 (30mpk) or vehicle was delivered (PO, BID dosing). Note that Groups 1–2 and Groups 3–4 were sacrificed on day 7 and day 14, respectively, and thus did not receive oral dosing. Groups 5–7 were sacrificed on day 21. The last oral doses were administered on day 27. Groups 8–10 were sacrificed on day 28. Groups 11–12 (which specifically serve to assess resolution in this model, and do not examine the effect of SB525334) were sacrificed on day 42.
Supplementary Fig. S7A (available at Rheumatology online) provides additional information on groups (12 groups, n = 8–10 per group at onset of the study; a total of seven animals died during the study).
Lung function analysis
The mechanical properties of the mouse lungs were determined using the flexiVent apparatus (Scireq, Montreal, QC, Canada). In brief, mice were anesthetized by intraperitoneal administration of Ketamine (91 mg/kg; VedCo) and Xylazine (9.1 mg/kg; Akorn Animal Health), a tracheotomy was performed, and an 18-gauge cannula was inserted into a slit in the trachea and connected to the flexiVent computer-controlled rodent ventilator. After an initial period of ventilation, measurement of lung mechanical properties was initiated by a computer-generated program to measure inspiratory capacity, compliance, lung resistance, tissue elastance and tissue damping. These measurements were repeated three times for each animal.
Analysis of hydroxyproline content
Hydroxyproline analysis was conducted on half of an 8 mm skin biopsy, or on the middle, inferior and post-caval lung lobes. The QuickZyme Total Collagen Assay kit (QuickZyme Biosciences, The Netherlands) was used according to the manufacturer’s instructions. For skin, hydroxyproline levels were normalized to tissue weights to account for potential variability in tissue procurement.
RNA-seq library preparation and pre-processing
Small pieces of tissue (∼5mg) were added to a 2 ml tube with one 5 mm diameter stainless steel bead (Cat # 69989, Qiagen, Valencia, CA, USA) and 1.2 ml Trizol® (Life Technologies, Cat # 15596018, Grand Island, NY, USA). Tissue was homogenized by TissueLyzerII (Qiagen, Valencia, CA, USA) (lung for 1 min, skin for 3 min). 500 μl chloroform was then added to the homogenate. After vortexing for 30 s, samples were centrifuged at 14 000 rpm for 15 min at 4°C. The supernatant layer was collected into a deep 96-well plate, mixed with 50 μl 70% ethanol. The mixtures were transferred to the RNeasy 96 well plate, (RNeasy® 96 Kit, Cat # 74881, Qiagen, Valencia, CA, USA). RNA was isolated by following the manufacturer’s protocol. RNA was eluted in 90 μl H2O then treated with DNaseI by adding 10 μl DNaseI 10X buffer +2 μl DNaseI (ThermoFisher, Cat# AM2222) for 15 min at room temperature. Treated RNA was cleaned up by using RNeasy® 96 Kit following the manufacturer’s protocol. The quality and quantity of the isolated total RNA were evaluated with a NanoDrop 8000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). RNA was then normalized to 50 ng/ul, randomized with DOE, prepared using the Illumina TruSeq Total RNA-Gold kit and sequenced with Illumina HiSeq sequencing.
Raw sequence files were processed by the bcl2fastq software in BaseSpace to generate FASTQ files for each sample. Sequence reads in the FASTQ files were then aligned to the Mouse.B38 genome build using Omicsoft ArrayStudio with the OSA alignment algorithm [33]. Ensembl gene models (version R86) were used by Omicsoft’s reimplementation of the RSEM algorithm to generate gene-level counts for each sample [34, 35]. The resulting gene by sample count matrix was further processed in R using voomWithQualityWeights together with a limma linear modeling workflow to assess differential gene expression [36–38]. Quality control statistics including mapping rate, mitochondrial DNA rate and more are available in Supplementary Table S3, available at Rheumatology online.
RNA quality as measured through RIN score is available in Supplementary Fig. S7B (available at Rheumatology online). Multiple plates were used for RNA-seq library preparation: we used ComBat [39] to correct for this confounding variable and demonstrate the reduction of variance explained by plate pre- and post-run using Variance Partition Analysis (Supplementary Fig. S7C and D, available at Rheumatology online) [40].
Differential gene expression analysis
Differential expression analysis was performed independently for lung and skin samples using limma [9] with plate and RIN score included as covariates. Genes were identified as differentially expressed if their FDR-corrected P-value was <0.1; P-value distributions are available in Supplementary Fig. S5A and C (available at Rheumatology online) for all contrasts studied. All differential expression performed for every tissue and contrast, with gene names, log fold changes and adjusted P-values is summarized in Supplementary Table S4 (available at Rheumatology online).
Time-course clusters were identified using TCseq [41] and UpSet plots were generated using UpSetR [42].
We used the Molecular Signatures Database (MSigDB) [43] and computed Gene Set Enrichment Analysis as described previously [44]. We used the fast implementation fgsea [45].
The GSEA enrichment score (ES) represents the degree to which a gene set is overrepresented at the top or bottom of a ranked list of genes. A positive ES indicates gene set enrichment at the top of the ranked list, and a negative ES indicates gene set enrichment at the bottom of the ranked list. The ES is a function of gene set size, and, therefore, ESs cannot be directly compared across gene sets. Cross-gene set comparisons are facilitated by the normalized enrichment score (NES).
Model construction and optimization
Inspiratory Capacity (IC) was modelled as a continuous response by fitting a linear regression model to the RNA-Seq expression data, computed with an elastic net regularization path. We modelled lung and skin tissues separately yielding two models of IC, ℳung and ℳskin. Linear regression can be unreliable when p > n (relatively few samples with many transcript observations). By linearly combining both l1 and l2 penalties of the lasso and ridge regression methods, respectively, elastic net regularization improves model performance [46–48].
Elastic net training requires the selection of both a lasso and ridge mixing parameter, α, and a penalty strength parameter, λ. To identify the optimal combination with the highest performance, we conducted 10-fold balanced cross-validation for each (α, λ) pair in a grid search on each training set. We chose α =0.95 based on the suggestion in the glmnet documentation to set α =1 −ε for some small ε >0 [46]. The rationale is to improve numerical stability and reduce the degeneracies caused by high correlations between covariates.
We performed 100 repeats of 10-fold cross-validation in caret to select the λ that yielded the highest performing final model (with the lowest mean-squared error) [49].
Supplementary Material
Acknowledgements
The authors would like to thank L. Burns for in vivo support, Yan Zhang for isolating RNA from skin and lung tissue, Manling Ma-Edmonds for conducting the RNA-seq library prep and running the RNA-seq analysis, and anonymous reviewers for their helpful feedback.
This study was conducted with the approval of the Bristol Myers Squibb Internal Animal Care and Use Committee. All protocols and procedures were reviewed and approved prior to the start of the animal experiments.
Funding: All work described in this manuscript was funded by Bristol Myers Squibb.
Disclosure statement: B.E.D., R.A., L.R.-B., J.R.T. and A.V.A. were employees of Bristol Myers Squibb when the work was done and may hold stock in the company.
Data availability statement
All RNA-seq data analysed in this manuscript was deposited in GEO under accession number GSE132869. Code associated with this analysis can be found at www.github.com/bdecato/SSc-Transcriptome-Manuscript-Code.
Supplementary data
Supplementary data are available at Rheumatology online.
Contributor Information
Benjamin E Decato, Research & Early Development, Bristol-Myers Squibb Company, Lawrenceville, NJ, USA.
Ron Ammar, Research & Early Development, Bristol-Myers Squibb Company, Lawrenceville, NJ, USA.
Lauren Reinke-Breen, Research & Early Development, Bristol-Myers Squibb Company, Lawrenceville, NJ, USA.
John R Thompson, Research & Early Development, Bristol-Myers Squibb Company, Lawrenceville, NJ, USA.
Anthony V Azzara, Research & Early Development, Bristol-Myers Squibb Company, Lawrenceville, NJ, USA.
References
- 1. Simeon C, Armadans L, Fonollosa V et al. Mortality and prognostic factors in Spanish patients with systemic sclerosis. Rheumatology 2003;42:71–5. [DOI] [PubMed] [Google Scholar]
- 2. Royle JG, Lanyon PC, Grainge MJ, Abhishek A, Pearce FA. The incidence, prevalence, and survival of systemic sclerosis in the UK Clinical Practice Research Datalink. Clin Rheumatol 2018;37:2103–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Korman BD, Criswell LA. Recent advances in the genetics of systemic sclerosis: toward biological and clinical significance. Curr Rheumatol Rep 2015;17:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Distler JH, Feghali‐Bostwick C, Soare A et al. Frontiers of antifibrotic therapy in systemic sclerosis. Arthritis Rheumatol 2017;69:257–67. [DOI] [PubMed] [Google Scholar]
- 5. Volkmann ER, Varga J. Emerging targets of disease-modifying therapy for systemic sclerosis. Nat Rev Rheumatol 2019;15:208–24. [DOI] [PubMed] [Google Scholar]
- 6. Błyszczuk P, Kozlova A, Guo Z, Kania G, Distler O. Experimental mouse model of bleomycin‐induced skin fibrosis. Curr Protoc Immunol 2019;126:e88. [DOI] [PubMed] [Google Scholar]
- 7. Meng X-M, Nikolic-Paterson DJ, Lan HY. TGF-β: the master regulator of fibrosis. Nat Rev Nephrol 2016;12:325–38. [DOI] [PubMed] [Google Scholar]
- 8. Van der Maaten L, Hinton G. Visualizing data using t-SNE. J Mach Learn Res 2008;9:2579–605. [Google Scholar]
- 9. Ritchie ME, Phipson B, Wu D et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schönherr E, Witsch-Prehm P, Harrach B et al. Interaction of biglycan with type I collagen. J Biol Chem 1995;270:2776–83. [DOI] [PubMed] [Google Scholar]
- 11. Burch ML, Yang SN, Ballinger ML et al. TGF-β stimulates biglycan synthesis via p38 and ERK phosphorylation of the linker region of Smad2. Cell Mol Life Sci 2010;67:2077–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Asano Y, Varga J. Rationally-based therapeutic disease modification in systemic sclerosis: novel strategies. Semin Cell Dev Biol 2020;101:146–60. [DOI] [PubMed] [Google Scholar]
- 13. Yanaba K, Asano Y, Tada Y et al. Clinical significance of serum growth differentiation factor-15 levels in systemic sclerosis: association with disease severity. Mod Rheumatol 2012;22:668–75. [DOI] [PubMed] [Google Scholar]
- 14. Lambrecht S, Smith V, De Wilde K et al. Growth differentiation factor 15, a marker of lung involvement in systemic sclerosis, is involved in fibrosis development but is not indispensable for fibrosis development. Arthritis Rheumatol 2014;66:418–27. [DOI] [PubMed] [Google Scholar]
- 15. Crescioli C, Corinaldesi C, Riccieri V et al. Association of circulating CXCL10 and CXCL11 with systemic sclerosis. Ann Rheum Dis 2018;77:1845–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Antonelli A, Ferri C, Fallahi P et al. CXCL10 (α) and CCL2 (β) chemokines in systemic sclerosis—a longitudinal study. Rheumatology 2008;47:45–9. [DOI] [PubMed] [Google Scholar]
- 17. Rabquer BJ, Tsou P-S, Hou Y et al. Dysregulated expression of MIG/CXCL9, IP-10/CXCL10 and CXCL16 and their receptors in systemic sclerosis. Arthritis Res Ther 2011;13:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu X, Mayes MD, Tan FK et al. Correlation of interferon‐inducible chemokine plasma levels with disease severity in systemic sclerosis. Arthritis Rheum 2013;65:226–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sokai A, Handa T, Tanizawa K et al. Matrix metalloproteinase-10: a novel biomarker for idiopathic pulmonary fibrosis. Respir Res 2015;16:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Avouac J, Guignabert C, Hoffmann‐Vold AM et al. Role of stromelysin 2 (matrix metalloproteinase 10) as a novel mediator of vascular remodeling underlying pulmonary hypertension associated with systemic sclerosis. Arthritis Rheumatol 2017;69:2209–21. [DOI] [PubMed] [Google Scholar]
- 21. Young-Min SA, Beeton C, Laughton R et al. Serum TIMP-1, TIMP-2, and MMP-1 in patients with systemic sclerosis, primary Raynaud's phenomenon, and in normal controls. Ann Rheum Dis 2001;60:846–51. [PMC free article] [PubMed] [Google Scholar]
- 22. Meng C, Chen X, Li J, Wu Y, Liu H. Expression of MMP-9 and TIMP-1 in lesions of systemic sclerosis and its implications. J Huazhong Univ Sci Technolog Med Sci 2008;28:480–2. [DOI] [PubMed] [Google Scholar]
- 23. Kuźnik-Trocha K, Winsz-Szczotka K, Komosińska-Vassev K et al. Plasma glycosaminoglycan profiles in systemic sclerosis: associations with MMP-3, MMP-10, TIMP-1, TIMP-2, and TGF-beta. BioMed Res Int 2020;2020:6416514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abignano G, Blagojevic J, Bissell L-A et al. European multicentre study validates enhanced liver fibrosis test as biomarker of fibrosis in systemic sclerosis. Rheumatology 2019;58:254–9. [DOI] [PubMed] [Google Scholar]
- 25. Lakota K, Carns M, Podlusky S et al. Serum amyloid A is a marker for pulmonary involvement in systemic sclerosis. PLoS One 2015;10:e0110820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lis-Święty A, Widuchowska M, Brzezińska-Wcisło L, Kucharz E. High acute phase protein levels correlate with pulmonary and skin involvement in patients with diffuse systemic sclerosis. J Int Med Res 2018;46:1634–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Manetti M. Deciphering the alternatively activated (M2) phenotype of macrophages in scleroderma. Exp Dermatol 2015;24:576–8. [DOI] [PubMed] [Google Scholar]
- 28. Angelini DJ, Su Q, Yamaji-Kegan K et al. Resistin-like molecule-β in scleroderma-associated pulmonary hypertension. Am J Respir Cell Mol Biol 2009;41:553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu M, Schneider DJ, Mayes MD et al. Osteopontin in systemic sclerosis and its role in dermal fibrosis. J Invest Dermatol 2012;132:1605–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Corallo C, Volpi N, Franci D et al. Is osteopontin involved in cutaneous fibroblast activation? Its hypothetical role in scleroderma pathogenesis. Int J Immunopathol Pharmacol 2014;27:97–102. [DOI] [PubMed] [Google Scholar]
- 31. Bhattacharyya S, Tamaki Z, Wang W et al. FibronectinEDA promotes chronic cutaneous fibrosis through Toll-like receptor signaling. Sci Transl Med 2014;6:232ra50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang J, Beyer C, Palumbo-Zerr K et al. Nintedanib inhibits fibroblast activation and ameliorates fibrosis in preclinical models of systemic sclerosis. Ann Rheum Dis 2016;75:883–90. [DOI] [PubMed] [Google Scholar]
- 33. Hu J, Ge H, Newman M, Liu K. OSA: a fast and accurate alignment tool for RNA-Seq. Bioinformatics 2012;28:1933–4. [DOI] [PubMed] [Google Scholar]
- 34. Li J, Hu J, Newman M, Liu K, Ge H. RNA-seq analysis pipeline based on Oshell environment. IEEE/ACM Trans Comput Biol Bioinform 2014;11:973–8. [DOI] [PubMed] [Google Scholar]
- 35. Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 2010;26:493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Law CW, Chen Y, Shi W, Smyth GK. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014;15:R29–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu R, Holik AZ, Su S et al. Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic Acids Res 2015;43:e97-e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Law CW, Alhamdoosh M, Su S et al. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR . F1000Res 2016;5:1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012;28:882–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hoffman GE, Schadt EE. variancePartition: interpreting drivers of variation in complex gene expression studies. BMC Bioinformatics 2016;17:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wu MG. TCseq: Time course sequencing data analysis. R package version 1.14.0 , 2020. https://rdrr.io/bioc/TCseq.
- 42. Conway JR, Lex A, Gehlenborg N. UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 2017;33:2938–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liberzon A, Birger C, Thorvaldsdóttir H et al. The molecular signatures database hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Subramanian A, Tamayo P, Mootha VK et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sergushichev AA. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. BioRxiv 2016;060012. Preprint; not yet published [Google Scholar]
- 46. Zou H, Hastie T. Regularization and variable selection via the elastic net. J R Stat Soci B 2005;67:301–20. [Google Scholar]
- 47. Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- 48. James G, Witten D, Hastie T, Tibshirani R. An introduction to statistical learning. Vol. 112. New York: Springer, 2013: 18. [Google Scholar]
- 49. Kuhn M. Building predictive models in R using the caret package. J Stat Softw 2008;28:1–26.27774042 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All RNA-seq data analysed in this manuscript was deposited in GEO under accession number GSE132869. Code associated with this analysis can be found at www.github.com/bdecato/SSc-Transcriptome-Manuscript-Code.