

Abstract
Objectives
Advances in immunotherapy by blocking TNF have remarkably improved treatment outcomes for Rheumatoid arthritis (RA) patients. Although treatment specifically targets TNF, the downstream mechanisms of immune suppression are not completely understood. The aim of this study was to detect biomarkers and expression signatures of treatment response to TNF inhibition.
Methods
Peripheral blood mononuclear cells (PBMCs) from 39 female patients were collected before anti-TNF treatment initiation (day 0) and after 3 months. The study cohort included patients previously treated with MTX who failed to respond adequately. Response to treatment was defined based on the EULAR criteria and classified 23 patients as responders and 16 as non-responders. We investigated differences in gene expression in PBMCs, the proportion of cell types and cell phenotypes in peripheral blood using flow cytometry and the level of proteins in plasma. Finally, we used machine learning models to predict non-response to anti-TNF treatment.
Results
The gene expression analysis in baseline samples revealed notably higher expression of the gene EPPK1 in future responders. We detected the suppression of genes and proteins following treatment, including suppressed expression of the T cell inhibitor gene CHI3L1 and its protein YKL-40. The gene expression results were replicated in an independent cohort. Finally, machine learning models mainly based on transcriptomic data showed high predictive utility in classifying non-response to anti-TNF treatment in RA.
Conclusions
Our integrative multi-omics analyses identified new biomarkers for the prediction of response, found pathways influenced by treatment and suggested new predictive models of anti-TNF treatment in RA patients.
Keywords: rheumatoid arthritis, anti-TNF, methotrexate, treatment response, inflammation, peripheral blood mononuclear cells
Rheumatology key messages.
Expression of the CHI3L1 gene and its protein YKL-40, which regulates T cell activation, was suppressed in rheumatoid arthritis responders following anti-TNF treatment.
Machine learning models using transcriptomic data at baseline showed high predictive utility in classifying rheumatoid arthritis response to future anti-TNF treatment.
Introduction
Rheumatoid arthritis is a systemic autoimmune disease characterized by chronic inflammation in symmetric joints that leads to pain and eventually bone destruction. RA is one of the most common autoimmune diseases, affecting ∼0.5–1% of the world’s population [1]. Currently there is no cure for RA, but several DMARDs are used to treat patients with the disease. During a successful treatment course, inflammation in the joints decreases, resulting in disease remission or low disease activity [2, 3]. Methotrexate (MTX) is recommended as the first-line treatment of early RA. However, at least 30% of patients do not respond to the treatment and significant disease activity remains [4, 5]. The patients who do not respond to first-line treatment are recommended for additional treatment, in most cases with TNF inhibitors. TNF is a pro-inflammatory cytokine secreted mainly by monocytes and macrophages, but also by other immune and non-immune cells, including fibroblasts and endothelial cells, involved in systemic inflammation.
Anti-TNF therapy has been used to treat RA for 2 decades, but one-third of treated patients do not respond or have a poor response [6]. Hence prediction of treatment efficacy before initiating treatment would help patients to start effective treatment and decrease the number of adverse effects.
In the present study, we selected a well-characterized cohort that allowed us to investigate gene expression differences in peripheral blood mononuclear cells (PBMCs), the proportion of different cell types and cell phenotypes in peripheral blood measured using flow cytometry and the level of several proteins in serum. In addition, we developed models using machine learning techniques to predict non-response for the anti-TNF treatment.
Materials and methods
Patient cohort
In this study, patient samples were obtained from the COMBINE cohort, which includes individuals treated with MTX (89 patients), anti-TNF drugs (59 patients) or a second biologic agent (31 patients) and healthy controls (60 individuals) [7]. Healthy controls were recruited from the Swedish Blood Centre in Uppsala and were closely matched for age and sex with the patient groups. The patients who did not respond adequately to MTX treatment (based on the judgment of a local rheumatologist) were prescribed anti-TNF treatment in combination with MTX. Patients included in this study donated blood at the clinic before starting anti-TNF therapy and at the follow-up visit after approximately 3 months. Clinical assessments and routine blood sampling were performed at both visits and were used to calculate the 28-joint DAS with CRP (DAS28-CRP) [8]. Of the 59 patients who started anti-TNF therapy, 3 dropped out before the scheduled 3 month visit and therefore lacked clinical assessment. For RNA-seq analysis, we did not include patient samples with low-quality or low-quantity RNA samples. Due to the small number of male non-responders in our study cohort, we included only female patients (n = 39) in our study (Fig. 1A). The clinical and demographic variables at baseline are shown in Table 1.
Fig. 1.

(A) The design of the current study showing the number of patient samples used for the cross-sectional and longitudinal analysis of RNA-Seq, flow cytometry and protein data analysis. (B) Volcano plot representation of differentially expressed genes in PBMCs between future responders and non-responders before anti-TNF treatment. The top regulated genes are marked in blue (upregulated genes) and red (downregulated genes). The vertical lines correspond to a log2 fold change of 1 (genes are represented in black) and the horizontal line represents a P-value of 0.001. (C) The box plot shows normalized log2 expression values for the differentially expressed genes EPPK1, BCL6-AS1 and CDC20 in PBMCs before treatment
Table 1.
Baseline demographic characteristics of female RA patients treated with anti-TNF
| Characteristics | Values |
|---|---|
| Age, years, median (range) | 57 (19–76) |
| Swedish, n (%) | 34 (82.9) |
| Current smoker, n (%) | 11 (28.2) |
| HLA-DR shared epitope positive, n (%) | 26 (66.6) |
| ACPA positive, n (%) | 29 (74.3) |
| Bone erosions, n (%) | 18 (46.1) |
| DAS28, median (range) | 4.79 (2.49–7.48) |
| 28-joint swollen joint count, median (range) | 6 (1–25) |
| 28-joint tender joint count, median (range) | 8 (1–28) |
| Prednisolone treatment, n (%) | 23 (58.9) |
| Anti-TNF drugs, n | |
| Infliximab | 16 |
| Etanercept | 8 |
| Adalimumab | 11 |
| Golimumab | 2 |
| Certolizumab | 2 |
| CRP, mg/L, median (range) | 2 (0.5–59) |
| Patient global health assessment, median (range) | 50 (5–100) |
| HAQ physical function, median (range) | 0.75 (0–2.6) |
| Health professional global health assessment, median (range) | 45 (11–82) |
Response measures
We used the EULAR response criteria to classify patient response to treatment [9, 10]. In our analysis we considered good and moderate EULAR responders as ‘responders’ and compared these with the EULAR ‘non-responders’.
RNA sequencing
RNA was extracted from PBMCs, freshly isolated using CPT tubes (BD Biosciences, Franklin Lakes, NJ, USA) and sequenced as previously described [7]. Of the 39 female RA patients, we obtained high-quality RNA-seq data from 28 patients at baseline (responders, 10; moderate responders, 9; non-responders, 9) and 32 patients after treatment (responders, 11; moderate responders, 9; non-responders, 12), giving a total of 25 patients with paired RNA-seq samples (both anti-TNF naïve and treated) (Fig. 1A). The sequencing reads were trimmed using Trim Galore (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) and then mapped to the GRCh38 human reference genome; subsequent gene counts were generated using STAR version 2.5.3a [11].
The detailed method for flow cytometry, protein data, machine learning and statistical models used for the analyses are provided in the Supplementary Data S1, available at Rheumatology online.
Results
Gene expression signatures of response to anti-TNF treatment at baseline
The gene expression analysis identified 59 differentially expressed genes between future responders and non-responders, but most of these genes achieved significance [false discovery rate (FDR) ≤0.1] due to a single outlier sample showing a different gene expression profile (Supplementary Fig. S1A and B, available at Rheumatology online). Therefore we decided to exclude this patient sample (both anti-TNF naïve and treated) and a subsequent analysis (without the outlier sample) yielded 192 differentially expressed genes (FDR ≤0.1), including 103 genes with higher expression and 89 genes with lower expression, in future responders. The top differentially expressed genes are presented in Fig. 1B (also see Table 2 and Supplementary Table S1A, available at Rheumatology online).
Table 2.
Differentially expressed genes in future responders and non-responders before anti-TNF treatment
| Genes | Description | Log2 fold change | P-value | Iteration count |
|---|---|---|---|---|
| FOSB | FosB proto-oncogene, AP-1 transcription factor subunit | 3.88 | 6.25E-09 | 28 |
| EPPK1 | Epiplakin 1 | 1.89 | 6.06E-07 | 28 |
| EGR2 | Early growth response 2 | 3.98 | 1.63E-07 | 27 |
| BCL6-AS1 | BCL6 antisense 1 | 2.14 | 2.95E-07 | 27 |
| EGR1 | Early growth response 1 | 3.68 | 5.91E-06 | 27 |
| IGLV10-54 | Immunoglobulin lambda variable 10-54 | −2.73 | 5.46E-06 | 27 |
| IGKV1D-39 | Immunoglobulin kappa variable 1D-39 | −2.68 | 2.13E-05 | 27 |
| PDIA4 | Protein disulfide isomerase family A member 4 | −0.45 | 5.59E-06 | 26 |
| HSP90B1 | Heat shock protein 90 kDa beta member 1 | −0.60 | 1.17E-05 | 26 |
| FAM46C | Family with sequence similarity 46, member C | −1.03 | 1.42E-05 | 26 |
| KDM6B | Lysine demethylase 6B | 0.61 | 1.92E-05 | 26 |
| FBXO7 | F-box protein 7 | −0.37 | 2.57E-05 | 26 |
| PSAT1 | Phosphoserine aminotransferase 1 | −0.78 | 2.49E-05 | 26 |
| CDC20 | Cell division cycle 20 | −1.95 | 8.70E-06 | 25 |
| NDC80 | NDC80 kinetochore complex component | −0.80 | 1.46E-05 | 25 |
| CHEK1 | Checkpoint kinase 1 | −0.80 | 1.91E-05 | 25 |
| ITM2C | Integral membrane protein 2C | −0.89 | 4.86E-05 | 25 |
| SOGA1 | Suppressor of glucose, autophagy associated 1 | 0.66 | 5.61E-05 | 25 |
| TXNDC15 | Thioredoxin domain containing 15 | −0.42 | 6.42E-05 | 25 |
| IGLV3-1 | Immunoglobulin lambda variable 3-1 | −1.80 | 8.19E-05 | 25 |
| MTCO2P12 | MT-CO2 pseudogene 12 | 2.76 | 7.17E-04 | 25 |
The iteration count is the number of LOO iterations where the gene remained significant.
To assess possible heterogeneity and detect consistent differential gene expression, we employed a leave-one-out (LOO) approach, where one sample is removed in each iteration and the association analysis is repeated. The genes were considered if they meet statistical significance (FDR <0.1) in each iteration. The LOO approach yielded two genes, EPPK1 and FOSB, with higher expression in responders in all 28 iterations (Table 2). An additional five genes, EGR1, EGR2, BCL6-AS1, IGLV10-54 and IGKV1D-39, showed significance (FDR <0.1) in 27 LOO iterations. The genes EGR1, EGR2 and BCL6-AS1 had higher expression in future responders, whereas immunoglobulin light chain genes IGLV10-54 and IKV1D-39 had lower expression in future responders. The genes EPPK1, BCL6-AS1 and CDC20 showed a clear expression difference between responders and non-responders (Fig. 1C). We also noticed that six immunoglobulin light chain genes (IGLV10-54, IGKV1D-39, IGKV3-20, IGLV3-1, IGKV1-17 and IGKV2-24) and one heavy chain gene (IGHV5-10-1) had lower expression in responders compared with non-responders; however, these genes did not pass the LOO analysis criteria (Fig. 1B).
The gene set enrichment analysis based on ranking of differentially expressed genes (log2 fold change) identified a total of 127 regulated pathways (Supplementary Table S1B, available at Rheumatology online) between response groups. The pathway analyses reveal that responders were preferentially characterized by higher expression of genes involved in graft-versus-host disease, antigen processing and presentation and the AP-1 transcription factor network, whereas non-responders were characterized by higher expression of genes involved in cell cycle pathways—mainly cell cycle mitotic activity and cell cycle checkpoints. The top 15 most upregulated and downregulated pathways are presented in Supplementary Fig. S2A and Supplementary Table S1B, available at Rheumatology online.
Effect on gene expression in PBMCs during anti-TNF treatment
Since biological and technical confounders between individuals may significantly affect downstream analyses, using paired samples before and after treatment is the preferable approach to address changes related to the treatment. We analysed treatment effects on gene expression using 25 paired RA patients without considering the response. The analysis identified 25 differentially expressed genes, of which 14 genes were suppressed and 11 showed a slight increase in expression (Supplementary Table S1C, available at Rheumatology online). The genes BHLHE40, which controls cytokine production by T cells, and CHI3L1, chitinase-3-like protein, were suppressed during treatment, whereas the B cell novel protein 1 (alias FAM129C) and TTC21A were induced by treatment (Fig. 2A). The pathway analysis of differentially expressed genes did not show any enrichment of gene sets, but when using a less conservative threshold of FDR <0.1, we detected 114 gene sets regulated during the treatment (Supplementary Table S1D, available at Rheumatology online). Induced genes are significantly enriched for genes involved in RNA and protein metabolism and interferon signalling, whereas suppressed genes are predominantly enriched for genes involved in haemostasis and signalling by G protein-coupled receptor (GPCR; Fig. 2B).
Fig. 2.

(A) Box plots showing the expression levels of differentially expressed genes (all RA patients) in PBMCs for baseline vs treated patient samples. The expression levels of responders for selected genes are plotted. (B) The enrichment plot from gene set enrichment analysis represents functional gene sets enriched between baseline and treated RA patients. BHLHE40: basic helix-loop-helix family member E40; CHI3L1: chitinase-3 like-protein-1; FAM129C: family with sequence similarity 129 member C; TTC21A: tetratricopeptide repeat domain 21A
Distinct gene expression signatures for response during anti-TNF treatment
The trajectory of gene expression changes may correlate with measured clinical outcomes. Therefore we separately investigated the transcriptional changes in paired samples of responders (n = 17) and non-responders (n = 8) to anti-TNF treatment. Our analysis identified five genes regulated in responders, whereas no significant regulation was identified in non-responders. Of the five regulated genes in responders, CXCR2, MPO, MYADM and TNFAIP6 were suppressed by treatment, whereas gene FCGR2B was induced during anti-TNF treatment. The gene expression plots for all five genes using normalized counts show a clear difference in responders before and after anti-TNF treatment (Fig. 3A). Interestingly, for these genes we observed a similar trend of regulation in non-responders (same directionality), but it did not reach statistical significance (Supplementary Fig. S3, available at Rheumatology online). The gene set enrichment analysis of differentially expressed genes, ranked based on the fold change, identified 78 regulated pathways in responders (Supplementary Table S1E, available at Rheumatology online). We noticed induction of pathways involved in the regulation of cell cycle mitotic and protein metabolism, whereas genes involved in the extracellular matrix organization, signalling by GPCR and Toll-like receptor cascades were downregulated in responders (Fig. 3B; Supplementary Table S1E, available at Rheumatology online).
Fig. 3.

(A) Box plots showing the expression levels of genes in responders at baseline vs treated patient samples. (B) The enrichment plot from gene set enrichment analysis representing functional gene sets enriched in PBMCs for baseline vs treated RA patients in responders. (C) Bar plot showing the percentage of granulocytes, B cells, T cells, NK cells and monocytes of peripheral blood leucocytes before and after anti-TNF treatment in responders. (D) Box plots showing the YKL-40 protein expression levels in responders before and after anti-TNF treatment. CXCR2: C-X-C motif chemokine receptor 2; MYADM: myeloid associated differentiation marker; TNFAIP6: TNF alpha–induced protein 6; FCGR2B: Fc fragment of IgG receptor IIb
Replication of gene expression associations at baseline
To validate our study, we sought to replicate our findings in an independent cohort (BiOCURA) of gene expression on PBMCs with 80 RA patients before they began adalimumab or etanercept anti-TNF treatment [12]. From the replication cohort we selected gene expression data from only female patients for the analysis. At baseline, we noticed the gene CDC20 showed lower expression in responders compared with non-responders in both the study cohort and the replication cohort (P < 0.087). None of the remaining 20 genes (Table 2) achieved statistical significance (P < 0.1). In the study cohort we reported suppression of the gene CHI3L1 upon anti-TNF treatment in responders; we observed a similar regulation in the replication cohort, with lower expression in responders compared with non-responders (P < 0.096) at baseline.
Changes in cell phenotypes and protein blood plasma during anti-TNF treatment
We studied the changes in 422 immune phenotypes measured by flow cytometry during anti-TNF treatment using paired samples. Analysis of the effect of treatment in responders and non-responders revealed differences in seven cell phenotypes in responders and no significant differences in non-responders (Supplementary Table S1F, available at Rheumatology online). With treatment, responders showed strong suppression of the proportion of granulocytes among leucocytes (defined as CD45+ cells) and of the overall concentration of neutrophils in whole blood. The proportion of T cells (defined as CD3+ cells) and B cells (CD3–CD19+) among leucocytes and the proportion of NKG2A+NKp44+ NK cells out of all NKp44+ NK cells were, in contrast, upregulated (Fig. 3C).
We studied the effect of anti-TNF treatment in 73 proteins (Supplementary Table S1G, available at Rheumatology online) measured from serum (analytes by Myriad (Salt Lake City, Utah) and Crescendo (San Francisco, CA, USA)) and plasma (analytes by ELISA). The longitudinal analysis using paired samples identified regulation of 12 proteins in serum in responders and 1 protein in non-responders. Most of the proteins (CRP, IL-6, MMP-1, MMP-3, SAA, TNF-RI, VEGF, YKL-40, MIG and MIP-1β) were downregulated by anti-TNF treatment, whereas TNFR2 was upregulated (Fig. 3D). Moreover, we identified the downregulation of protein matrix metalloproteinase-3 (MMP-3) measured using two different methods (Supplementary Data S1, available at Rheumatology online), whereas protein adiponectin is induced during anti-TNF treatment in non-responders, while showing a trend for induction in responders (FDR = 0.11). The list of proteins that were regulated during anti-TNF treatment is shown in Supplementary Table S1H, available at Rheumatology online.
Further, our association analysis of immune phenotypes and plasma protein levels to clinically defined response to anti-TNF treatment did not show any significant association between responders and non-responders before treatment.
Classifier performance for anti-TNF response data
We investigated the utility of machine learning methods to predict anti-TNF response using clinical data, flow cytometry measurements, protein measurements and transcriptomic data. At baseline, the linear model predicted response with high accuracy for transcriptomic data [receiver operating characteristics (ROC) area under the curve (AUC) with the standard errors of the mean 0.81 ± 0.17] (Fig. 4), whereas the kernel-based method predicted response with the highest accuracy for clinical data [ROC AUC 0.73 ± 0.17], for flow cytometry [ROC AUC 0.72 ± 0.18] and for protein data [ROC AUC 0.72 ± 0.15] (Fig. 4). We further studied the classifier performance for the anti-TNF-treated data and observed limited classifier utility for models based on flow cytometry, protein and transcriptomic data. In contrast, as expected, the linear model based on clinical data showed high classifier performance with an ROC AUC of 0.85 ± 0.15. For the FACS data, the maximum classifier utility of an ROC AUC of 0.68 ± 0.17 was shown when using the linear model, whereas the non-linear models based on protein and transcriptomic data showed maximum classifier utility with an ROC AUC of 0.73 ± 0.15 and 0.72 ± 0.18, respectively.
Fig. 4.
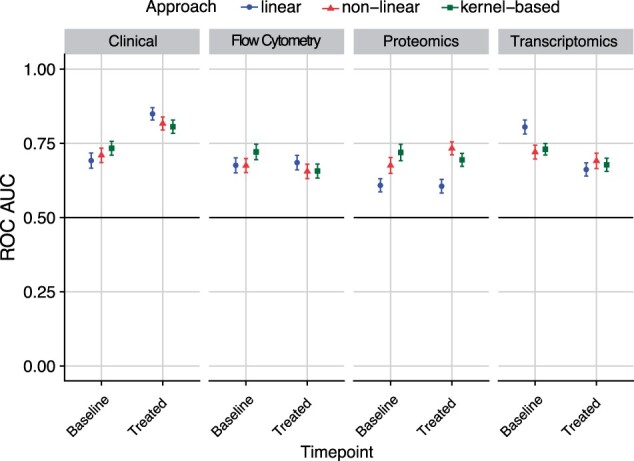
Statistical machine learning models to predict response (evaluated after 3 months) at baseline and anti-TNF-treated using clinical variables, flow cytometry measurements, protein measurements and gene expression data. The y-axis represents the ROC AUCs calculated for estimating the predicted performance
Discussion
This study represents a comprehensive analysis of transcriptomics, proteomics and flow cytometry data analysis of female RA patients treated with anti-TNF. Our analysis was directed towards finding transcriptional and translational regulations during anti-TNF treatment and predicting response using biological measurements collected at two time points. We identified genes that are differentially regulated between responders and non-responders at baseline and reported changes induced by treatment in gene expression, protein measurements and cell phenotypes. Finally, using biological measurements, we employed machine learning models to predict non-response to anti-TNF treatment. Our integration of multi-omics data revealed that the expression of CHI3L1 and its subsequent protein product YKL-40 were suppressed upon anti-TNF treatment. We also observed an increase in the proportion of B cells, T cells and NK cells in responders during treatment, whereas the proportion of granulocytes was strongly suppressed.
The identification of potential biomarkers for RA with prognostic value for response to a given therapy is challenging because RA is a heterogeneous disease by its clinical characteristics and pathological processes. This challenge may also be increased by clinical heterogeneity in RA patient samples and strong confounding (covariates) effects; for example, variation in cell subsets is one of the strong covariates of gene expression [13]. Also, RNA sequencing experiments can often generate outlier read counts in one or several RNA samples and this considerably limits the power of differential testing. Therefore we performed extensive transcriptomic analysis using the LOO approach to find genes with a stable association to response.
The gene expression results at baseline highlighted that cell adhesion gene EPPK1 showed a strong association with response and was expressed more in future responders compared with non-responders. Importantly, the difference we observed in the gene EPPK1 (log2 fold change 1.95, FDR = 5.5E-03) showed similar regulation following the adjustment to CRP measurements in the gene expression model, suggesting that EPPK1 is not a surrogate marker for inflammation in patients. Previous studies have reported the mechanism of infiltration of inflammatory cells into the synovial lining due to deregulation of several adhesion receptors, thus association of response to corresponding genes suggests its role in the pathogenesis of RA and haemophilic arthropathy [14, 15]. When findings from the study cohort were investigated in an independent cohort, we replicated the lower expression of Cell Division Cycle 20 (CDC20) gene in future responders compared with non-responders (P = 0.087). Interestingly, non-responders at baseline showed an increase in cell cycle pathways mainly related to mitotic activity and cell cycle checkpoints. This may partly correspond to the hyperproliferation of cells that leads to the accumulation of pro-inflammatory cytokines in the RA-inflamed joints [16, 17].
Next we extended our transcriptomic analysis using samples from paired patients to study the changes induced by treatment with two different approaches: changes in gene expression patterns in samples from all RA patients not considering response status and treatment effects in responders and non-responders separately to understand differences in gene expression regulation. Our first approach identified 25 genes differentially expressed in RA after treatment, whereas the second approach yielded 5 genes differentially expressed in responders and no significant changes in non-responders. Interestingly, both approaches found four common genes: CXCR2, MPO and MYADM that were downregulated and FCGR2B that was upregulated upon treatment. Also, we observed that TNFAIP6 [also called TNF-stimulated gene 6 (TSG-6)] was suppressed in responders upon treatment. Previous studies have shown that the expression of TSG-6 has a strong correlation with disease severity and is a potential biomarker of inflammation [18–20]. TSG-6 plays a key role in the remodelling of the extracellular matrix, regulation of leucocyte migration and stimulation of cell proliferation during inflammation [21, 22]. These findings correspond well with the pathway analysis that showed downregulation of genes involved in extracellular matrix organization and signalling by interleukins and an upregulation of genes associated with mitotic cell cycle regulation. These results substantially extend our understanding of the transcriptomic profile of responders to anti-TNF treatment.
Our longitudinal studies on cell phenotypes revealed significant changes in seven cell populations in responders, indicating a marked change in the proportions of major cell types during treatment (Fig. 3C). The proportions of B cells and T cells among leucocytes were increased and the proportions of granulocytes were suppressed upon treatment in responders. There was also a decrease in the overall concentration of neutrophils in whole blood in responders as well as in non-responders. The reduction of neutrophils seen by flow cytometry was also associated with a significant reduction in peripheral blood neutrophil count seen by routine blood analysis, leading to 19% of patients becoming neutropenic by clinical judgement. This finding corroborates a recent study that showed patients treated with anti-TNF in combination with MTX had decreased blood neutrophil counts, regardless of their clinical response to therapy [23, 24]. Additionally, our results also corroborate earlier studies showing induction of B cells and NK cells in whole blood following anti-TNF treatment [25].
Similarly, the longitudinal analysis of protein data showed changes in plasma protein levels in responders. The expression of the protein YKL-40 was suppressed upon treatment (β = −0.26) and the gene encoding for YKL-40, CHI3L1, was also down-regulated (Figs 2A and 3D). The model including only responders showed similar suppression of CHI3L1 (log2 fold change = −1.07) with a P-value <1.73E-04 and FDR <0.21. Interestingly, we also observed that the gene CHI3L1 had lower expression in responders compared with non-responders (P < 0.096) at baseline in the replication cohort. The CHI3L1 gene may negatively regulate T cell activation and may also inhibit Th1 differentiation via the IFN-γ–STAT1 pathway [26]. We also showed that treatment with TNF inhibitors leads to a larger proportion of T cells among responders (β = 8.16).
It is tempting to link changes in gene expression patterns between responders and non-responders after anti-TNF treatment to known inflammatory markers or other clinical phenotypes in RA patients. However, our data do not reveal such clear links. Also, when adjusted for the level of CRP, the gene expression analysis did not change the overall results. This suggests that more complex relationships may exist and should be investigated in larger cohorts of RA patients. Nevertheless, these results substantially extend our understanding of the transcriptomic profile of responders to anti-TNF treatment.
Interestingly, two proteins, soluble TNFR2 and adiponectin, showed increased plasma levels in responders and non-responders during treatment. The result of our study replicates previously shown similar induction of adiponectin after treatment with tocilizumab, an anti-IL-6R [27, 28]. Similarly, an increase in TNFR2 signalling leads to the activation and proliferation of Tregs and promotes tissue regeneration in RA patients [29–31].
Our machine learning models showed high predictive utility in classifying non-response prior to anti-TNF treatment. The linear model based on transcriptomic data at baseline found good predictability using the presently applied gene expression classifier. To our surprise, the models with transcriptomic data predict response with higher accuracy compared with models with clinical data. Interestingly, we also found clinical variables add less further utility to the transcriptomic-based predictive models at baseline (data not shown). Previously a similar outcome was observed in a study where models using transcriptomic data alone predicted fibrous cap thickness response to statin treatment with better accuracy than the models with clinical data or transcriptomic plus clinical variables [32]. Our prediction model should be used with caution and these findings need to be validated with an independent study with a larger patient cohort before its potential use in clinical settings.
The major limitation of this study is the small sample size. The current study addressed multi-omics profiling of RA patients treated with anti-TNF with relatively low statistical power and can capture only major effects. Second, the genes that are regulated in responders upon treatment showed similar direction of regulation in non-responders with low statistical significance, and this might be partly explained due to the low number of non-responders included in the current study. Thus, in future studies, it will be especially important to include a higher number of non-responders to treatment. Third, only non-responders to MTX treatment were included in the current study, which is a limitation for understanding the true mechanisms of treatment. Finally, with the goal of decreasing cohort heterogeneity, we studied only female patients, which precludes generalization of results to broader patient populations. Extended future studies with a larger patient cohort are warranted to confirm results from the current investigation.
In summary, this integrative multi-omics study expands our growing knowledge of the biology of anti-TNF treatment in RA patients by identifying a number of changes in gene expression, protein and cell phenotypes during treatment in anti-TNF responders. We report genes that show distinct expression in responders and non-responders before treatment initiation. Our analyses demonstrate that treatment causes a major regulation of cell subsets, which are also mirrored in the protein analysis. Also, our study highlights the machine learning predictive utility of transcriptomic data at baseline in stratifying patients and/or predicted response before anti-TNF treatment initiation. We envision that future machine learning models based on multi-omics data could predict response to anti-TNF treatment and be of great value to rheumatologists making decisions about personalized treatment.
Supplementary Material
Acknowledgements
We wish to thank all the participating patients and healthy controls. We acknowledge support and resources provided by the Swedish National Infrastructure for Computing through Uppsala Multidisciplinary Centre for Advanced Computational Science and National Genomics Infrastructure Sweden. We would like to thank Janet Ahlberg for English-language editing. N.Y., B.B., A.C., L.B., L.P. and L.K. conceived the study. M.M.U., P.S. and N.V. performed wet lab experiments. N.Y., B.B., A.C., L.F. and L.P. contributed to the data preparation. N.Y., B.B. and M.M. performed the data analysis. N.Y., B.B., M.M., D.Z., L.P. and L.K. interpreted the data. N.Y. and M.M. prepared the figures. N.Y. wrote the manuscript with input from B.B., L.P., L.K., S.J. and L.B. All authors read and approved the final version of the manuscript. The COMBINE biobank was generated after written informed consent from all participants had been obtained according to the Declaration of Helsinki and with approval by the Stockholm (2010-351-31-2) and Uppsala (2009-013) Regional Ethics Committees.
Funding: Part of the study received support from Novo Nordisk and Pfizer. L.P. was supported by a grant from the Swedish Council.
Disclosure statement: The authors have declared no conflicts of interest.
Data availability statement
The datasets may contain personal details of the participants and will be available on request.
Supplementary data
Supplementary data are available at Rheumatology online.
Contributor Information
Niyaz Yoosuf, Division of Rheumatology, Department of Medicine Solna, Karolinska Institutet and Karolinska University Hospital; Translational Epidemiology, Institute of Environmental Medicine, Karolinska Institutet, Stockholm, Sweden.
Mateusz Maciejewski, Pfizer, Cambridge, MA, USA.
Daniel Ziemek, Pfizer, Cambridge, MA, USA.
Scott A Jelinsky, Pfizer, Cambridge, MA, USA.
Lasse Folkersen, Danish National Genome Center, Copenhagen, Denmark.
Malin Müller, Division of Rheumatology, Department of Medicine Solna, Karolinska Institutet and Karolinska University Hospital.
Peter Sahlström, Division of Rheumatology, Department of Medicine Solna, Karolinska Institutet and Karolinska University Hospital.
Nancy Vivar, Division of Rheumatology, Department of Medicine Solna, Karolinska Institutet and Karolinska University Hospital.
Anca Catrina, Division of Rheumatology, Department of Medicine Solna, Karolinska Institutet and Karolinska University Hospital.
Louise Berg, Division of Rheumatology, Department of Medicine Solna, Karolinska Institutet and Karolinska University Hospital.
Lars Klareskog, Division of Rheumatology, Department of Medicine Solna, Karolinska Institutet and Karolinska University Hospital.
Leonid Padyukov, Division of Rheumatology, Department of Medicine Solna, Karolinska Institutet and Karolinska University Hospital.
Boel Brynedal, Translational Epidemiology, Institute of Environmental Medicine, Karolinska Institutet, Stockholm, Sweden.
References
- 1. Klareskog L, Catrina AI, Paget S. Rheumatoid arthritis. Lancet 2009;373:659–72. [DOI] [PubMed] [Google Scholar]
- 2. Schett G, Emery P, Tanaka Y et al. Tapering biologic and conventional DMARD therapy in rheumatoid arthritis: current evidence and future directions. Ann Rheum Dis 2016;75:1428–37. [DOI] [PubMed] [Google Scholar]
- 3. Smolen JS, Breedveld FC, Burmester GR et al. Treating rheumatoid arthritis to target: 2014 update of the recommendations of an international task force. Ann Rheum Dis 2016;75:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Smolen JS, Landewé R, Breedveld FC et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Ann Rheum Dis 2014;73:492–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Singh JA, Furst DE, Bharat A, Curtis JR et al. 2012 update of the 2008 American College of Rheumatology recommendations for the use of disease-modifying antirheumatic drugs and biologic agents in the treatment of rheumatoid arthritis. Arthritis Care Res (Hoboken) 2012;64:625–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mewar D, Wilson AG. Treatment of rheumatoid arthritis with tumour necrosis factor inhibitors. Br J Pharmacol 2011;162:785–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Folkersen L, Brynedal B, Diaz-Gallo LM et al. Integration of known DNA, RNA and protein biomarkers provides prediction of anti-TNF response in rheumatoid arthritis: results from the COMBINE study. Mol Med 2016;22:322–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fransen J, van Riel PLCM. The Disease Activity Score and the EULAR response criteria. Rheum Dis Clin North Am 2009;35:745–57. [DOI] [PubMed] [Google Scholar]
- 9. van der Heijde DM, van ’t Hof MA, van Riel PL et al. Judging disease activity in clinical practice in rheumatoid arthritis: first step in the development of a disease activity score. Ann Rheum Dis 1990;49:916–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prevoo ML, van ’t Hof MA, Kuper HH et al. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum 1995;38:44–8. [DOI] [PubMed] [Google Scholar]
- 11. Dobin A, Davis CA, Schlesinger F et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tao W, Concepcion AN, Vianen M et al. Multiomics and machine learning accurately predict clinical response to adalimumab and etanercept therapy in patients with rheumatoid arthritis. Arthritis Rheumatol 2020;73:212–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Whitney AR, Diehn M, Popper SJ et al. Individuality and variation in gene expression patterns in human blood. Proc Natl Acad Sci USA 2003;100:1896–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burkhardt J, Blume M, Petit-Teixeira E et al. Cellular adhesion gene SELP is associated with rheumatoid arthritis and displays differential allelic expression. PLoS One 2014;9:e103872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McMurray RW. Adhesion molecules in autoimmune disease. Semin Arthritis Rheum 1996;25:215–33. [DOI] [PubMed] [Google Scholar]
- 16. Weyand CM, Zeisbrich M, Goronzy JJ. Metabolic signatures of T-cells and macrophages in rheumatoid arthritis. Curr Opin Immunol 2017;46:112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goronzy JJ, Weyand CM. Successful and maladaptive T cell aging. Immunity 2017;46:364–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nagyeri G, Radacs M, Ghassemi-Nejad S et al. TSG-6 protein, a negative regulator of inflammatory arthritis, forms a ternary complex with murine mast cell tryptases and heparin. J Biol Chem 2011;286:23559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Milner CM, Day AJ. TSG-6: a multifunctional protein associated with inflammation. J Cell Sci 2003;116:1863–73. [DOI] [PubMed] [Google Scholar]
- 20. Mahoney DJ, Swales C, Athanasou NA et al. TSG-6 inhibits osteoclast activity via an autocrine mechanism and is functionally synergistic with osteoprotegerin. Arthritis Rheum 2011;63:1034–43. [DOI] [PubMed] [Google Scholar]
- 21. Bayliss MT, Howat SL, Dudhia J, Murphy JM et al. Up-regulation and differential expression of the hyaluronan-binding protein TSG-6 in cartilage and synovium in rheumatoid arthritis and osteoarthritis. Osteoarthritis Cartilage 2001;9:42–8. [DOI] [PubMed] [Google Scholar]
- 22. Wisniewski HG, Maier R, Lotz M et al. TSG-6: a TNF-, IL-1-, and LPS-inducible secreted glycoprotein associated with arthritis. J Immunol 1993;151:6593–601. [PubMed] [Google Scholar]
- 23. Hastings R, Ding T, Butt S et al. Neutropenia in patients receiving anti-tumor necrosis factor therapy. Arthritis Care Res (Hoboken) 2010;62:764–9. [DOI] [PubMed] [Google Scholar]
- 24. Farutin V, Prod’homme T, McConnell K et al. Molecular profiling of rheumatoid arthritis patients reveals an association between innate and adaptive cell populations and response to anti-tumor necrosis factor. Arthritis Res Ther 2019;21:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Conigliaro P, Triggianese P, Perricone C et al. Restoration of peripheral blood natural killer and B cell levels in patients affected by rheumatoid and psoriatic arthritis during etanercept treatment. Clin Exp Immunol 2014;177:234–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim D-H, Park H-J, Lim S et al. Regulation of chitinase-3-like-1 in T cell elicits Th1 and cytotoxic responses to inhibit lung metastasis. Nat Commun 2018;9:503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fioravanti A, Tenti S, Bacarelli MR et al. Tocilizumab modulates serum levels of adiponectin and chemerin in patients with rheumatoid arthritis: potential cardiovascular protective role of IL-6 inhibition. Clin Exp Rheumatol 2019;37:293–300. [PubMed] [Google Scholar]
- 28. Liu D, Luo S, Li Z. Multifaceted roles of adiponectin in rheumatoid arthritis. Int Immunopharmacol 2015;28:1084–90. [DOI] [PubMed] [Google Scholar]
- 29. Yang S, Wang J, Brand DD, Zheng SG. Role of TNF-TNF receptor 2 signal in regulatory T cells and its therapeutic implications. Front Immunol 2018;9:784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ahmad S, Azid NA, Boer JC et al. The key role of TNF-TNFR2 interactions in the modulation of allergic inflammation: a review. Front Immunol 2018;9:2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yang N, Huang J, Frits M et al. Interference of tumor necrosis factor inhibitor treatments on soluble tumor necrosis factor receptor 2 levels in rheumatoid arthritis. Pract Lab Med 2019;16:e00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Johnson KW, Glicksberg BS, Shameer K et al. A transcriptomic model to predict increase in fibrous cap thickness in response to high-dose statin treatment: validation by serial intracoronary OCT imaging. EBioMedicine 2019;44:41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets may contain personal details of the participants and will be available on request.