

Abstract
BACKGROUND:
The integrated stress response (ISR) is an evolutionarily conserved intracellular signaling network that helps the cell, tissue, and organism to adapt to a variable environment and maintain health. In response to different environmental and pathological conditions, including protein homeostasis (proteostasis) defects, nutrient deprivation, viral infection, and oxidative stress, the ISR restores balance by reprogramming gene expression. The various stresses are sensed by four specialized kinases (PERK, GCN2, PKR and HRI) that converge on phosphorylation of a single serine on the eukaryotic translation initiation factor eIF2. eIF2 phosphorylation blocks the action of eIF2’s guanine nucleotide exchange factor termed eIF2B, resulting in a general reduction in protein synthesis. Paradoxically, phosphorylation of eIF2 also triggers the translation of specific mRNAs, including key transcription factors, such as ATF4. These mRNAs contain short inhibitory upstream open reading frames in their 5′-untranslated regions that prevent translation initiation at their canonical AUGs. By tuning down general mRNA translation and up-regulating the synthesis of a few proteins that drive a new transcriptional program, the ISR aims to maintain or reestablish physiological homeostasis. However, if the stress cannot be mitigated, the ISR triggers apoptosis to eliminate the damaged cell.
ADVANCES:
Our understanding of the central mechanisms that govern the ISR has advanced vastly. The ISR’s central regulatory hub lies in the eIF2-eIF2B complex, which controls the formation of the eIF2•GTP•methionyl-intiator tRNA ternary complex (TC), a prerequisite for initiating new protein synthesis. Assembly of functional TC is inhibited by eIF2-P, which blocks eIF2B noncompetitively. In mammalian cells, the phosphorylation of eIF2 is a tightly regulated process. In addition to the four specialized eIF2 kinases that phosphorylate eIF2, two dedicated phosphatases antagonize this reaction. Both phosphatases contain a common catalytic core subunit, the protein phosphatase 1 (PP1), and a regulatory subunit (GADD34 or CReP), which render the phosphatase specific to eIF2. Structural and biophysical approaches have elucidated the mechanism of action of eIF2B and its modulation by ISR inhibitors and activators. Gene expression analyses have revealed complex ISR-driven reprogramming. Although it has been long recognized that, in the brain, long-term memory formation requires new protein synthesis, recent causal and convergent evidence across different species and model systems has shown that the ISR serves as a universal regulator of this process. Briefly, inhibition of the ISR enhances long-term memory formation, whereas activation of the ISR prevents it. Consistent with this notion, unbiased genome-wide association studies have identified mutations in key components of the ISR in humans with intellectual disability. Furthermore, age-related cognitive disorders are commonly associated with the activation of the ISR. Most notably, oxidative stress, misfolded proteins, and other stressors induce the ISR in several neurodegenerative disorders, including Alzheimer’s disease. Recent genetic and pharmacological evidence suggest that tuning the ISR reverses cognitive dysfunction as well as neurodegeneration in a wide range of memory disorders that result from protein homeostasis defects. Thus, long-term memory deficits may primarily results as a consequence of ISR activation rather than from the particular proteostasis defects that lead to its induction. Finally, the ISR is also implicated in the pathogenesis of a plethora of other complex diseases, including cancer, diabetes, and metabolic disorders.
OUTLOOK:
The ISR is emerging as a central regulator of protein homeostasis at both the cellular and organismal level. Mechanistically, much remains to be understood regarding additional inputs into the eIF2B-eIF2 regulatory hub controlling TC concentration, as well as the ISR’s connectivity to other intracellular signaling networks. As yet, little is known about the role of the specific proteins whose synthesis is altered during acute and persistent ISR activation and how these effectors collaborate to compute the life or death decisions cells make upon ISR activation. ISR gene expression signatures and functional consequences will need to be mapped across different tissues, cell types, and developmental stages. In addition, it will be invaluable to generate additional genetic and molecular tools that permit the direct temporal and spatial manipulation of ISR pathway in specific cells and circuits to determine their function. From a medical perspective, the ISR is implicated in the etiology of several disorders, and manipulation of the ISR is emerging as a promising therapeutic avenue for the treatment of a variety of diseases. The use of innovative mouse models, patient-derived induced pluripotent stem cells, and human organoids will greatly enhance our ability to explore the ISR’s clinical relevance further and help define therapeutic windows in which ISR modulation may prove beneficial. Identifying additional specific small-molecule inhibitors and activators of the ISR will offer valuable opportunities to dissect the role of the ISR pharmacologically in health and disease. Finally, discovery and mechanistic understanding of additional ISR modulators will increase the repertoire of therapeutic targets and may further enable clinical development in a wide range of age-related human diseases.
Graphical Abstract
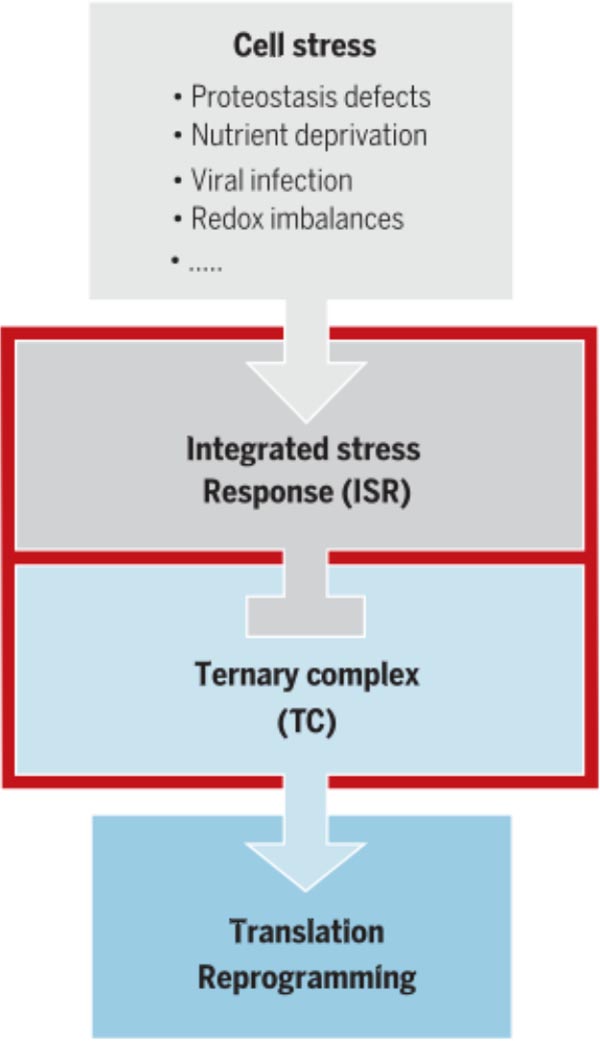
The regulatory network of the ISR. Diverse deviations from homeostasis activate the ISR. The resulting dysregulation of translation contributes to numerous diseases.
Protein quality control is essential for the proper function of cells and the organisms that they make up. The resulting loss of proteostasis, the processes by which the health of the cell’s proteins is monitored and maintained at homeostasis, is associated with a wide range of age-related human diseases. Here, we highlight how the integrated stress response (ISR), a central signaling network that responds to proteostasis defects by tuning protein synthesis rates, impedes the formation of long-term memory. In addition, we address how dysregulated ISR signaling contributes to the pathogenesis of complex diseases, including cognitive disorders, neurodegeneration, cancer, diabetes, and metabolic disorders. The development of tools through which the ISR can be modulated promises to uncover new avenues to diminish pathologies resulting from it for clinical benefit.
To maintain cell health, proteins must be synthesized in proper amounts; folded with high fidelity; and assembled, appropriately localized, and degraded. Specialized mechanisms respond to malfunction in these essential processes to maintain or reestablish protein homeostasis (proteostasis), when intracellular signaling networks are triggered by a variety of stress sensor molecules. The unfolded protein response (UPR) senses misfolded protein accumulation in the endoplasmic reticulum (ER), whereas the heat shock response (HSR) operates from the cytosol, enhancing the cell’s protein folding and degradation capacity (1–3). By contrast, the integrated stress response (ISR), a central and evolutionarily conserved signaling network, responds to stress conditions from both the lumen of the ER and the cytosol (4). Indeed, ISR induction can be coupled to UPR and HSR activation (5, 6). The ISR operates by reprogramming translation, the last step of gene expression. Its name derived from the realization that ISR signaling nucleates diverse stress inputs—including proteostasis defects, nutrient deprivation, viral infection, and redox imbalance—and leads to a central output: the reduction in general translation initiation rates and the increase in translation of specific messenger RNAs (mRNAs). These two processes reprogram gene expression to maintain cellular equilibrium, sustain protein-folding capacity, support differentiation, and respond to injury. When the stress cannot be mitigated, the ISR triggers apoptosis to eliminate the damaged cell. We provide an overview of our current understanding of the ISR pathway and its role in cell and organismal physiology. In addition, we highlight how abnormal ISR signaling contributes to the pathogenesis of complex diseases, including cognitive dysfunction, neurodegeneration, diabetes mellitus, myclination, and metabolic disorders. Finally, we review the new pharmacological tools through which the ISR can be modulated and discuss the potential therapeutic opportunity that targeting the ISR offers.
Mechanism of the integrated stress response
The ISR’s central regulatory switch lies in eIF2 ternary complex formation
The salient feature of ISR signaling lies in the modulation of the cellular concentration of the ternary complex (TC). The TC is composed of the heterotrimeric eukaryotic translation initiation factor eIF2 (consisting of an α, β, and γ subunit), guanosine 5′-triphosphate (GTP), and charged methionyl-initiator tRNA (Met-tRNAi) (Fig. 1). The TC is instrumental in initiating translation of AUG-initiated open reading frames (ORFs) in the cell’s transcriptome (7). AUG codon recognition triggers GTP hydrolysis in the TC, releasing Met-tRNAi to the ribosomal P site. eIF2-GDP (guanosine 5′-diphosphate) then dissociates, which allows the assembly of the ribosome that commences the elongation phase of protein synthesis. Exchange of the eIF2-bound GDP for GTP recycles eIF2 to its active state. This nucleotide exchange reaction is rate limiting for TC formation and hence for AUG-initiated mRNA translation.
Fig. 1. The molecular wiring of ISR control.
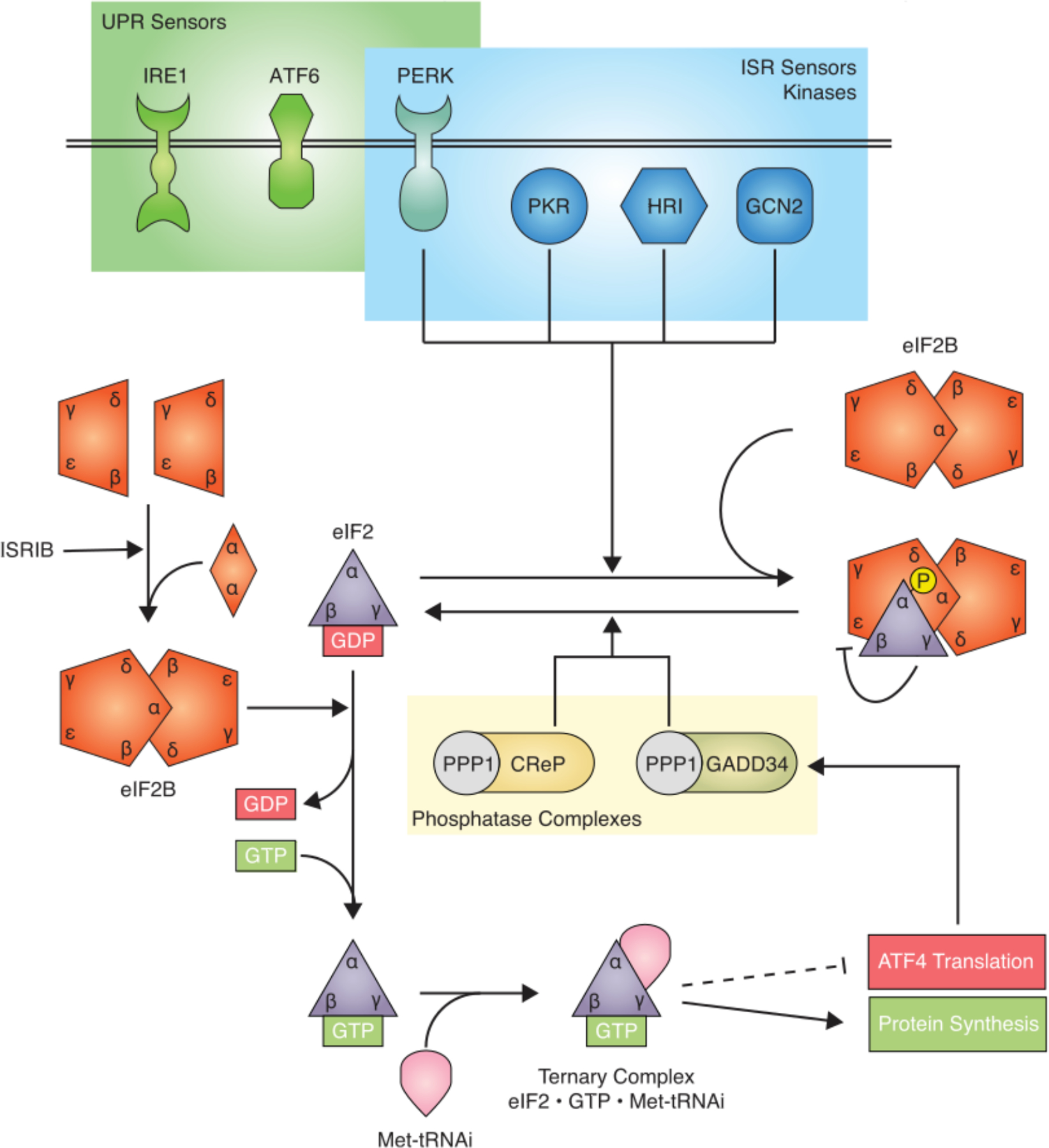
Stress sensing: ISR sensor kinases phosphorylate eIF2 in response to a diverse spectrum of cellular stresses. One of these kinases, PERK, overlaps with the UPR. eIF2B sequestration: Phosphorylation of eIF2 leads to sequestration of inactive eIF2B•eIF2-P, limiting available eIF2B activity in the cell, thus reducing TC concentration. TC control: TC concentration is controlled by GDP/GTP exchange on eIF2, which is catalyzed by eIF2B. The active decameric eIF2B complex is assembled from subcomplexes, a reaction that is facilitated by ISRIB. Translational control: The concentration of TC determines the translational status of general protein synthesis (green) and translation of specific mRNAs, such as ATF4 (red). Feedback regulation: Two phosphatase complexes antagonize the ISR. PP1•CReP in a constitutive regime and PP1•GADD34 in a feedback regime in response to ISR activation.
eIF2 phosphorylation regulates TC formation
For the TC to form, eIF2 must first be charged with GTP (Fig. 1) (8). Because the intrinsic rate of GDP release from eIF2 is low, the exchange reaction is catalyzed by eIF2’s dedicated guanine nucleotide exchange factor (GEF) eIF2B. eIF2B is composed of two copies of five different subunits (α, β, δ, γ, and ε) that assemble into a large twofold symmetric heterodecamer (9, 10) (Fig. 2A). The building blocks of this complex are stable subcomplexes: two heterotetramers (eIF2Bβγδε) and a homodimer (eIF2Bα2) (9, 11) (Fig. 2B). The structure of the eIF2B is highly conserved from yeast to mammals, reflecting its ancient evolutionary origin in eukaryotic cells (10–12).
Fig. 2. The structure and assembly of eIF2B.
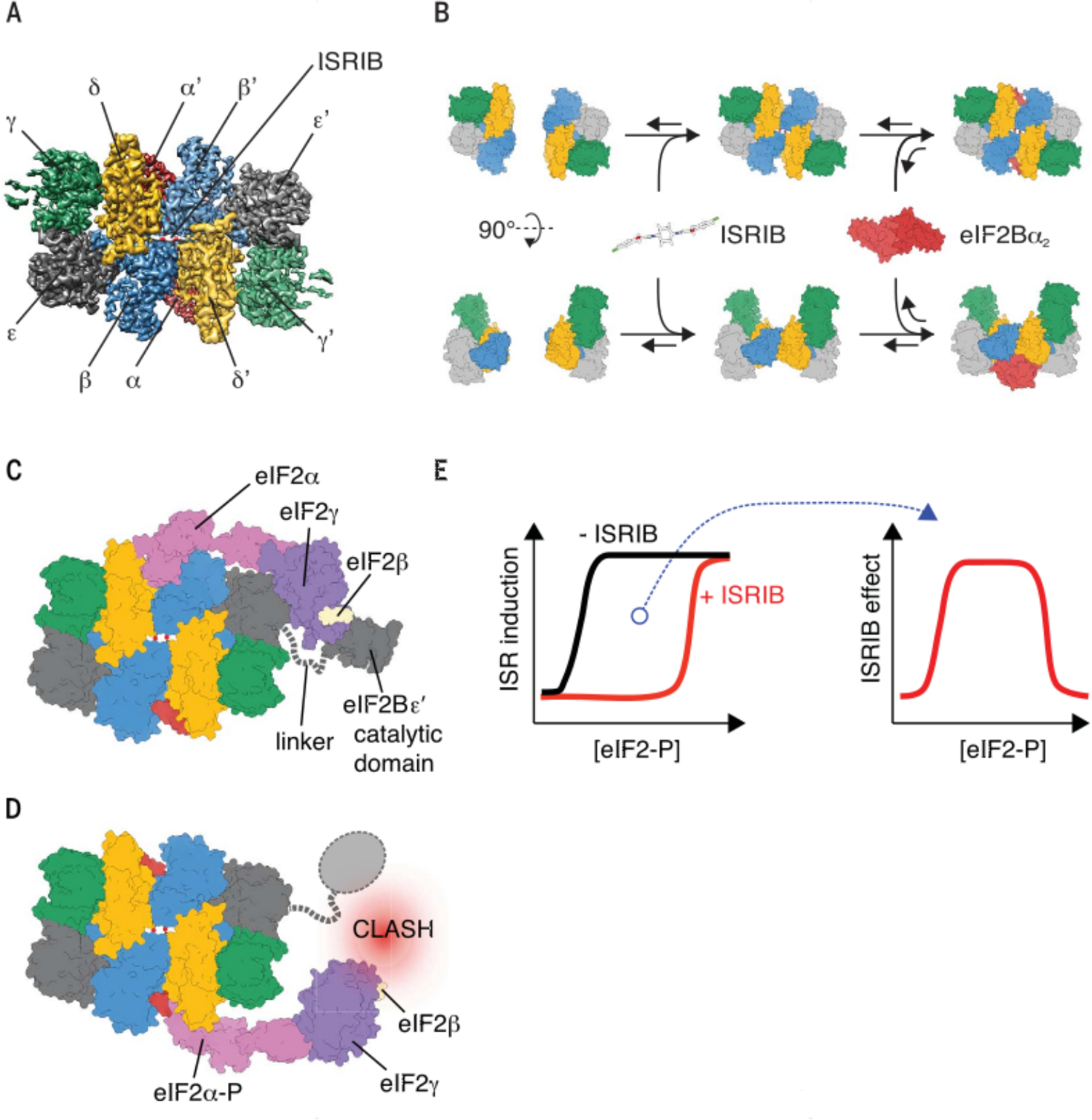
(A) Top view of the human eIF2B decamer bound to ISRIB. The cryo-EM density at a resolution of 2.7 Å shows ISRIB bound in a deep central binding pocket. (B) Model of eIF2B decamer assembly mediated by ISRIB. Top and side views of the same assembly path are shown (11). (C) Model of the eIF2B•eIF2αβγ complex. The interaction stabilizes the open conformation of eIF2γ’s GTP binding site, thus catalyzing GTP exchange. (D) Model of eIF2B bound to its inhibitor eIF2-P. eIF2-P binds to a different site on eIF2B, noncompetitively blocking eIF2B’s GEF activity. (E) Bell-shaped response of ISR inhibition by ISRIB. ISRIB blocks the ISR only at intermediate activation levels, as shown in (22).
eIF2B’s catalytic nucleotide exchange activity resides in eIF2Bε (13), suggesting that the remainder of the complex plays a regulatory role(s). The eIF2ε subunits are positioned at opposing ends of the decamer. Structures of human eIF2B complexed with its substrate, eIF2, illuminate the mechanism of guanine nucleotide exchange and explain why the eIF2B decamer is more active than its incompletely assembled building blocks (14, 15) (Fig. 2C). eIF2B-bound eIF2 adopts an extended conformation. The guanine nucleotide binding site in eIF2γ is sandwiched between two domains of eIF2Bε. This bipartite interaction of eIF2γ with eIF2Bε stabilizes the nucleotide binding pocket in an open state, thus releasing GDP and readying eIF2 for loading with a fresh GTP present in the cytosol in large excess over GDP.
Whereas the eIF2Bε-eIF2γ interaction occurs at either end of the eIF2B decamer, eIF2α interacts at a composite binding site bridging eIF2B’s central symmetry interface. Therefore, the eIF2 binding sites on eIF2B are only complete in the assembled enzyme.
eIF2 phosphorylation transforms eIF2 from an eIF2B substrate into an eIF2B inhibitor
Upon stress, the α subunit of eIF2 is phosphorylated at Ser51, yielding eIF2-P and ISR activation. This modification leads to a pro-found structural rearrangement in eIF2α, forming a hydrophobic surface patch on eIF2-P that displays strong affinity for a different binding site on eIF2B (Fig 2D) (14–18). Binding of eIF2-P to eIF2B sterically interferes with proper positioning of eIF2 to the catalytic domain of eIF2Bε (Fig. 2D). In this way, eIF2-P acts as a potent noncompetitive inhibitor of eIF2B.
The pharmacologic ISR inhibitor ISRIB targets eIF2B
ISRIB is a recently developed drug-like small-molecule ISR inhibitor that is bioavailable in vivo, blood-brain barrier penetrant, and highly potent. ISRIB binds to eIF2B and enhances its activity by promoting its assembly (11, 12, 19–21).
When active eIF2B decamers become depleted during the ISR because of their sequestration into inactive eIF2B•eIF2-P complexes, ISRIB replenishes the stock by assembling new eIF2B decamers from its subcomplexes. As such, ISRIB functions as an eIF2B activator. It accomplishes this task akin to a molecular staple by binding to eIF2B in a deep binding pocket that bridges across the eIF2B tetramer-tetramer symmetry interface (Fig. 2A). This mode of action has intriguing mechanistic consequences because ISRIB can only act as an eIF2B activator if there is a pool of unassembled eIF2B tetramers available. As the concentration of eIF2 phosphorylation increases and more eIF2B becomes trapped in inhibited, stable eIF2B•eIF2-P complexes, the assembly equilibrium of eIF2B shifts, leading to a depletion of eIF2B tetramers. Therefore, as the eIF2-P concentration exceeds a certain threshold—that is, when the ISR is strongly activated—ISRIB can no longer replenish the depleted pool of eIF2B decamers. Under these conditions, therefore, ISRIB does not inhibit the ISR (Fig. 2E) (22, 23). The resulting bell-shaped response to increasing stress explains why ISRIB displays no overt toxicity when administered in vivo—even at saturating concentrations. Thus, ISRIB does not abolish the ISR’s cytoprotective effects in cells in which the ISR is strongly activated.
Four kinases converge on eIF2 to activate the ISR
The ISR responds to a variety of different stress conditions that lead to alterations in cellular homeostasis. In metazoans, these stresses are sensed by at least four different kinases that each phosphorylate Ser51 in eIF2α to activate the ISR (24) (Fig. 1). In the yeast Saccharomyces cerevisiae and mammalian cells, the ancestral kinase Gcn2 (general amino acid control nonderepressible 2) responds to nutrient deprivation (25), whereas in metazoans the repertoire of sensor kinases has expanded to include HRI (heme-regulated inhibitor, gene name EIF2AK1), PKR (double-stranded RNA-dependent protein kinase, gene name EIF2AK2), and PERK (PKR-like ER kinase, gene name EIF2AK3) in addition to GCN2 (gene name EIF2AK4). All four kinases contain both conserved kinase domains and divergent regulatory domains that enable them to respond to different stimuli. Stress signals detected by the regulatory domains trigger kinase activation by dimerization and transautophosphorylation (26).
GCN2 contains a regulatory domain homologous with histidyl-tRNA synthetase and when amino acids are scarce, binding of deacylated His-tRNA triggers its activation (25, 27). GCN2 can also be activated by other stresses, including ultraviolet light, viral infection, serum starvation, and oxidative stress. Recent work shows that GCN2 strongly activates by binding to ribosomal protein uL10, a component of the P1/P2 stalk of the large ribosomal subunit, suggesting that GCN2 actively monitors mRNA translation and not just aminoacyl-tRNA availability (28–30). However, the precise mechanism(s) underlying these vastly different activation modalities remain unknown.
PERK, a transmembrane kinase (31), mediates the translation arm of the UPR (6). Its N-terminal domain is located in the ER lumen and associates with the Hsp70 family chaperone BiP (binding immunoglobulin protein) (32, 33). After BiP release, mis- or unfolded ER proteins can directly activate PERK by binding its ER lumenal domain (34). PERK can also be activated by changes in lipid bilayer fluidity, bypassing the actuator function of its lumenal domain (35).
PKR contains a double-stranded RNA (dsRNA) binding domain. dsRNA of viral origin and secondary structures resembling dsRNA on mRNAs trigger PKR activation (36, 37). In the brain, PKR is activated in a variety of neurological disorders (38, 39); however, the underlying molecular mechanism of this activation remains poorly understood.
HRI contains an N-terminal domain that binds heme. When heme concentrations are low, HRI is activated. Although HRI was long thought to have a specialized role in erythroid cells dedicated to hemoglobin synthesis (40), it is now recognized that HRI is widely expressed in several cell types and organs (41) and responds to multiple other forms of cellular cell stress, such as oxidative and mitochondrial stress, heat shock, and cytosolic protein aggregation (42).
ISR kinases are sentinels for a broad and partially overlapping spectrum of stress conditions, and much remains to be learned about their relative importance in different tissues and cell types. The development of small-molecule inhibitors for all of the eIF2 kinases (23, 43–50) has provided invaluable tools to better understand their cellular function. Unfortunately, their therapeutic promise remains hampered by their relative lack of specificity (51) and substantial toxicitics, such as pancreatic toxicity associated with the use of the PERK inhibitor (52).
The ISR reprograms gene expression at both translational and transcriptional levels
Diminishing TC availability as a consequence of eIF2 phosphorylation leads to reprogramming of protein synthesis. As expected, mRNA translation rates are reduced globally as TCs become rate-limiting for translation initiation (Fig. 1). Paradoxically, however, lowered TC availability also increases the translation of a subset of mRNAs. The mechanism that allows preferential translation upon ISR activation was first elucidated over 25 years ago for the yeast mRNA encoding the stress response transcription factor Gcn4 (25). In metazoans, the ortholog of Gcn4 is ATF4, the best-characterized effector of the ISR (27). GCN4 and ATF4 mRNAs contain short inhibitory upstream open reading frames (uORFs) in their 5′-untranslated regions (5′UTRs) that prevent initiation at their canonical AUGs (25, 53, 54). ISR-induced reduction in TC enables a portion of scanning ribosomes to initiate at the canonical AUG of the GCN4/ATF4 ORFs instead. Other uORF-containing mRNAs are also translated in response to ISR activation, including those encoding ATF5 (55); CHOP (C/EBP-homologous protein) (56); GADD34 (57); and in neurons, OPHN1 (58). However, the precise mechanism by which these mRNAs are translationally controlled remains unclear. Although it is assumed that they reinitiate at downstream codons, nascent peptides that emerge upon translation of uORFs could cause ribosome stalling (59). In the case of mRNAs encoding the transcription regulators ATF4, ATF5, and CHOP, their translational derepression leads to reprogramming of the cell’s transcriptional activities. Such regulation is instrumental in responding to extrinsically induced stresses or developmental cues.
Two phosphatase complexes reset eIF2-P to counteract ISR activation
The dephosphorylation of eIF2-P is a tightly regulated process carried out in mammalian cells by two phosphatase complexes. Both phosphatase complexes contain a common catalytic core, the protein phosphatase 1 (PP1, gene name PPP1CA) and a regulatory subunit, GADD34 (gene name PPP1R15A) or CReP (gene name PPP1R15B), which render the phosphatase specific to eIF2 (Fig. 1).
CReP is constitutively expressed, leading to a slow but steady rate of eIF2-P dephosphorylation (60). By contrast, GADD34 expression is induced as a consequence of increased eIF2-P abundance, thus acting in a feedback loop that antagonizes the relative strength of ISR activation (61). Two synergistic mechanisms increase GADD34 expression as the ISR progresses: (i) The mRNA encoding GADD34 bears uORFs in its 5′-UTR and is translationally controlled in a manner similar to that of CHOP mRNA (62); and (ii) GADD34 mRNA is transcriptionally up-regulated by ATF4, which further increases its cellular abundance and GADD34 protein concentrations (61). Both phosphatase complexes, GADD34•PP1 and CReP•PP1, can be activated by association with G-actin, suggesting that the ISR feedback loop may be tied to cell processes connected to actin polymerization (63, 64).
Small-molecule inhibitors of the phosphatase complexes offer the opportunity to dissect pharmacologically the physiological role of the ISR feedback loops in healthy cells and disease models. Salubrinal (65) and its more potent and soluble derivative Sal003 (66) inhibit both phosphatase complexes (GADD34•PP1 and CReP•PP1), but their precise mechanism of action remains unclear. Inhibitors of the individual phosphatase complexes have also been recently reported: Guanabenz and sephin1 inhibit GADD34•PP1 (67, 68), whereas raphin1 selectively inhibits CReP•PP1 (69). Although these findings are currently under active discussion (70–73), identification of putative other targets for these inhibitors and structural information of inhibitor-bound enzymes might help to resolve this puzzle.
ISR activation leads to complex, cell-wide changes
On the surface, the molecular wiring of the ISR appears seductively simple: A few kinases sense different stresses and converge on phosphorylation of a single serine on eIF2; phosphorylation causes a drop in the cell’s TC concentration, which reprograms translation. However, the resulting consequences are amazingly complex and remain poorly understood. The transcription factors (e.g., ATF4, ATF5, CHOP) that are made in response to ISR activation would be useless, for example, if the mRNAs of their transcriptional targets could not be translated. Therefore, the TC concentration must not fall to zero, and the ISR must act as a rheostat rather than an on-off switch controlling TC abundance.
More recently, genome-wide studies identified 5′ uORFs in ~50% of all mRNAs (74, 75). How only a subset of genes containing 5′ uORFs are translationally controlled by the ISR remains unknown. Similarly, it remains to be examined whether the same set of these mRNAs is affected when the different ISR sensor kinases are activated, and, if not, how this specialization of the ISR output might be orchestrated molecularly. Other signaling networks can also impinge on eIF2B activity—for example, by posttranslationally modifying eIF2B subunits (76) and thus tune TC availability and mRNA translation independent of or together with eIF2 phosphorylation (77).
It likewise remains unknown how mRNA structural features and cis-regulatory elements adjacent to AUG codons affect translation under conditions of limiting TC availability. Additional complexities arise from cell-type differences and the complex kinetic controls imposed by both constitutive slow deactivation operating through CReP•PP1 and the ISR-dependent negative feedback loop operating through GADD34•PP1. In yeast, amino acid deprivation-induced eIF2 phosphorylation leads to increased ribosome footprints at non-AUG sites (74), further adding to the long list of regulatory complexities that should be investigated in both yeast and mammalian cells, when the ISR is activated.
Physiology and pathology of the integrated stress response
Homozygous disruption of eIF2 phosphorylation or ablation of the eIF2 kinase PERK leads to postnatal lethality in mice (78, 79), underscoring the essential role of the ISR in normal physiology and mammalian development. Similarly, persistent activation of the ISR in mice lacking the two eIF2 phosphatase complexes is incompatible with life, likely due to shutdown of protein synthesis during embryogenesis (80). Milder forms of ISR dysregulation often result in disease.
Mutations in ISR components in human disease
The organs that are most affected by alterations in the ISR are the brain and pancreas. Homozygous loss-of-function mutations in the gene encoding CReP are associated with intellectual disability, short stature, and diabetes (81, 82). These rare mutations map to the PP1 binding site of CReP and destabilize the CReP•PP1 phosphatase complex, thereby increasing eIF2-P. Other mutations that reduce TC formation—and consequently induce the ISR’s translational program—similarly cause cognitive dysfunction. Patients with MEHMO (mental deficiency, epilepsy, hypogenitalism, microcephaly, and obesity) syndrome, an X-linked intellectual disability syndrome, carry mutations in the gene encoding eIF2γ (83–86). Although different mutations affect eIF2 function in a mechanistically different manner (some impair the binding of eIF2γ to eIF2β, whereas others impair the binding of Met-tRNAiMet to eIF2), all of them reduced TC formation (87).
Mutations in each of the five subunits of eIF2B occur in a rare autosomal recessive leukoencephalopathy with vanishing white matter (VWM) (88, 89). Studies in yeast models of VWM indicate that eIF2B mutations cause disease by reducing eIF2B activity and, consequently, TC concentrations. VWM is characterized by myelin loss and progressive neurological symptoms, such as ataxia, spasticity, cognitive deterioration and, ultimately, death (90). Notably, the pathologies associated with cells and mice bearing human hypomorphic VWM mutations can be rescued by ISRIB and ISRIB-like molecules (2BAct), which enhance the cellular pool of available TC and restore translation rates (91, 92).
It is not clear why different mutations that reduce TC availability cause diverse pathologies and selectively affect specific tissues and cell types. Mutations in eIF2B selectively lead to myelin loss resulting from ISR activation in oligodendrocytes and astrocytes (91), whereas mutations in eIF2γ or CReP affect a broader spectrum of tissues. Mutations in eIF2 not only decrease TC concentration and consequently general translation, but also reduce the stringency of AUG start codon selection (93), hence leading to translation initiation at noncanonical codons engendering the production of different protein isoforms, of completely different proteins. Whether all mutations that reduce TC complex also alter start codon selection and whether some mRNAs are differentially sensitive to the effect remains unknown. That said, in the context of nucleotide repeat expansions associated with neurodegenerative disorders, activation of the ISR promotes non-AUG (RAN) translation (94, 95). It will be of interest to test whether compounds that inhibit the ISR, such as ISRIB, prevent neurodegeneration in these disorders. In addition, it will be important to examine the expression levels and posttranslational modification status of ISR components in different tissues and cell types in development and adulthood.
Unlike mutations that activate the ISR, loss-of-function mutations in the genes encoding PERK and GCN2 impair its induction. Mutations in PERK are associated with a rare autosomal recessive disease, Wolcott-Rallison syndrome (WRS), characterized by early-onset diabetes and multiple epiphyseal dysplasia (96). PERK knockout mice recapitulate the human phenotype, showing growth retardation, neonatal diabetes, and skeletal malformation (97, 98). Both individuals with WRS and mice lacking PERK are thought to be unable to cope with the stress caused by mis-folded proteins or protein overload in the ER of secretory cells. This may explain why tissues that support high protein secretion loads and that express more PERK are most susceptible to its absence.
Mutations in the gene encoding GCN2 have been identified in a rare form of pulmonary arterial hypertension (99). This pathology arises from disruption of GCN2’s role in promoting angiogenesis in response amino acid deprivation (100). However, the precise mechanism by which these mutations cause disease remains unknown.
The ISR is a molecular rheostat during long-term memory formation
In the brain, the formation of long-term memory occurs in two temporally distinct steps: First, short-term memories lasting minutes to hours are formed; next, these can be consolidated into long-term memories, lasting days, weeks, and even a lifetime (101, 102). De novo protein synthesis is required for forming long-lasting memories in all species (ranging from invertebrates to mammals) (102–106). In response to activity, neurons rapidly regulate local protein synthesis at dendrites without altering mRNA synthesis and/or transport (107). The synthesized proteins in turn modulate the strength of synaptic connections during memory consolidation. Although other translation control mechanisms also modulate long-term memory formation (108, 109), translational reprogramming by the ISR emerges as a primary way to regulate mnemonic processes.
This conclusion rests on numerous, convergent discoveries in several animal models and across different levels of analysis.
For example, genetic inhibition of the ISR in mice lacking GCN2 or PKR, or eIF2 heterozygous knock-in mice, in which in one allele of eIF2(α) Ser51 is replaced by alanine, facilitate long-term memory formation (110–113).
Furthermore, memory enhancement upon genetic ISR inhibition parallels the pharmacological inhibition of different ISR components; PKR and PERK inhibitors, and ISRIB, enhance long-term memory in rodents (19, 112–114) and birds (115).
By contrast, ISR activation by manipulations that promote eIF2-P abundance impairs long-term memory formation: hippocampal or basolateral amygdala injection of Sal003 (65, 66), or pharmacogenetic activation of PKR in the hippocampus, impairs long-term memory in both rodents and birds (111, 115–118).
Accordingly, mutations in key components of the ISR in humans that activate the ISR, such as in the genes encoding eIF2γ and CReP, are linked to intellectual disability (81–86).
Moreover, activity-dependent dependent modulation of synaptic function induced by long-term potentiation (or L-LTP) (110, 119, 120), behavioral training (111, 121–123), or different drugs of abuse (124–127) negatively regulates the ISR, as determined by reduced eIF2-P concentrations in the hippocampus and ventral tegmental area (VTA), the brain regions implicated in memory and addiction, respectively. Accordingly, in the hippocampus, behavioral training induces a protein synthesis signature that mimics that of animals in which the ISR is reduced, as determined by ribosome profiling (121).
Finally, the ISR also modulates the two major forms of synaptic plasticity in the mammalian brain—protein synthesis-dependent LTP and long-term depression (LTD) (58, 111, 117, 120, 124, 128)—that are crucial for long-term memory formation (129). Two mechanisms by which the ISR controls activity-dependent changes in synaptic function in principal neurons are through regulation of translation of (i) OPHN1, a rho-GAP implicated in AMPA receptor down regulation (58, 124,130) and (ii) ATF4, a repressor cAMP response element binding protein (CREB)-mediated gene expression, which is required for L-LTP (102, 131).
Thus, causal and convergent evidence across different species and model systems supports the notion that the ISR serves as a universal protein synthesis regulator of long-term memory formation.
The ISR in cognitive and neurodegenerative disorders
Protein misfolding and aggregation, mitochondrial dysfunction, and oxidative stress are all common features of age-associated protein-opathies. As a consequence, protein synthesis, folding, and degradation are altered in these disorders. Because it acts as a central regulator of protein homeostasis, the ISR is activated in a wide range of disorders of the brain. This activation is evidenced by detection of eIF2-P and phosphorylation of PKR, PERK and GCN2 in post-mortem brains from individuals and animal models of cognitive and neurodegenerative disorders, including Alzheimer’s disease, Parkinson’s disease, Huntington disease, amyotrophic lateral sclerosis (ALS), traumatic brain injury, Down syndrome, and Charcot-Marie-Tooth disease (30, 109, 132–136). Notably, ISR activation causes cognitive defects in mouse models of traumatic brain injury (137, 138), aging (114), and Alzheimer’s disease (139–143), although negative results have also been reported (144, 145).
Genetic and pharmacological inhibition of the upstream ISR-sensor kinase PKR or activation of eIF2B with ISRIB reverses the aberrant translational program and deficits in synaptic plasticity and long-term memory in a mouse model of Down syndrome, the most common genetic cause of intellectual disability in humans (146). Down syndrome is characterized by a high incidence of Alzheimer’s disease, with most Down syndrome individuals developing Alzheimer’s disease-type dementia at age 45 (147). Thus, tuning of the ISR emerges as a promising avenue to reverse the cognitive dysfunction in a wide range of memory disorders that result from disruption in protein homeostasis.
Reversal of ISR-mediated translation reprogramming also results in neuroprotection. In a mouse model of prion disease, genetic and pharmacological suppression of the ISR by either overexpressing GADD34 in the brain, or treatment with ISRIB, PERK inhibitor, or trazodone, an antidepressant claimed to increase TC complex formation by an unknown mechanism, rescued neuronal loss and improved neuronal survival (23, 148–150). Consistent with these data, pharmacological inhibition of PERK was neuroprotective in two different Drosophila models of early-onset Parkinson disease (151), as well as in a mouse model of frontotemporal dementia (152). Moreover, genetic inhibition of PERK prevented neurodegeneration in a mouse model of Alzheimer’s disease (153). Pharmacological and genetic suppression of PKR rendered neurons more resistant to excitotoxicity (154) and β-amyloid accumulation (155). A potential mechanism by which inhibition of the ISR leads to neuroprotection is through inhibition of hypoxia-induced cell death, which has been linked with the pathogenesis of several neurodegenerative disorders (156) and can be prevented by ISRIB in a human cellular three-dimensional (3D) model of hypoxic brain injury (157). Other conditions, including hearing loss and neurodegeneration following NGF withdrawal, also activate the ISR, and its pharmacological inhibition with ISRIB improves the pathology (158, 159). Thus, inhibition of ISR-mediated translational reprogramming emerges as a promising therapeutic avenue for the treatment of cognitive disorders and neurodegenerative disorders (Fig. 3).
Fig. 3. The ISR in brain disorders.
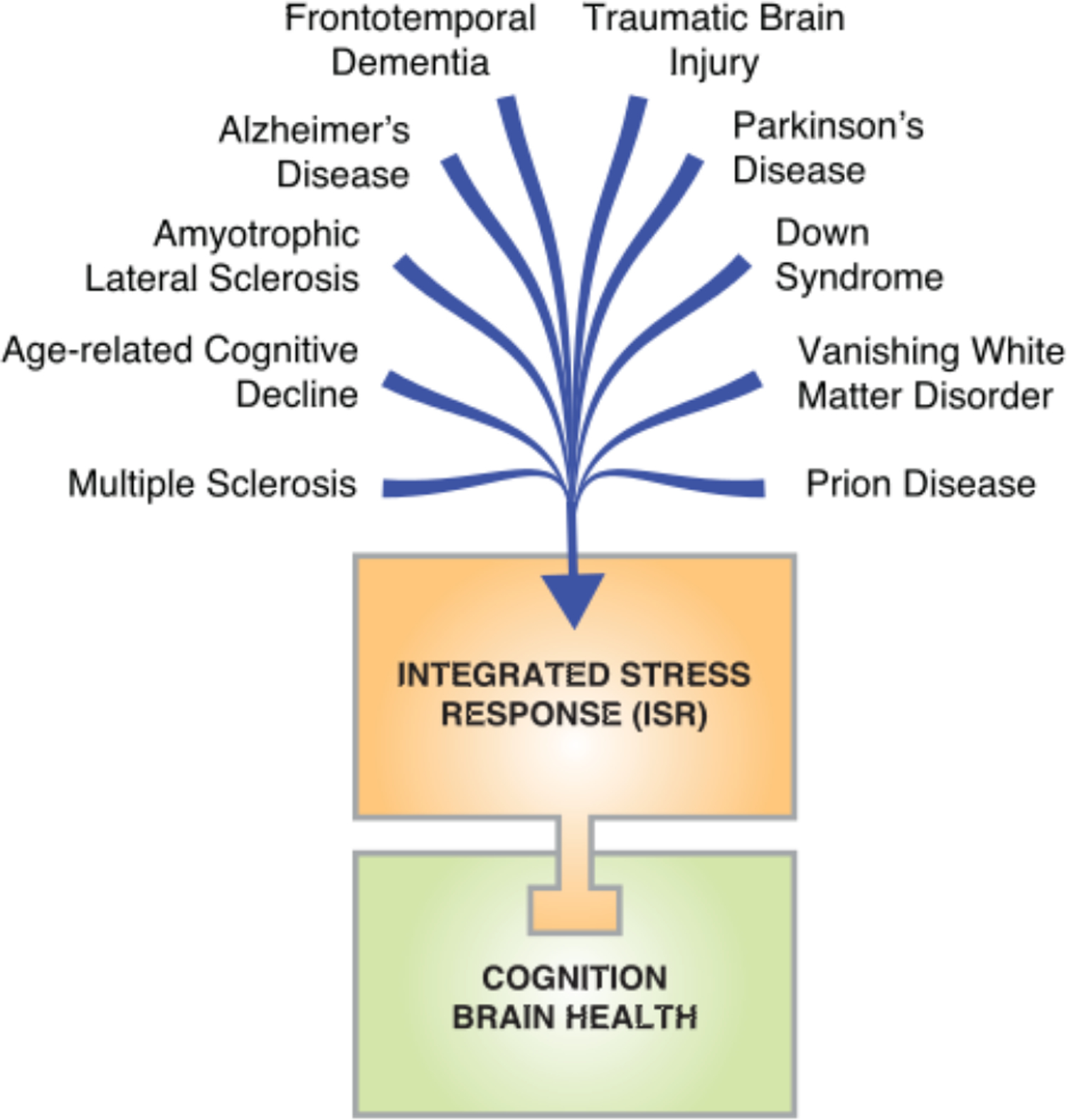
The ISR is a causative mechanism underlying the cognitive deficits and neurodegeneration in a wide broad range of brain disorders.
Given that myelinating cells from either the central or the peripheral nervous system synthesize a large amount of myelin proteins and lipids, they typically accumulate mis-folded or unfolded proteins and activate both the UPR and the ISR. In myelination disorders, the role of the ISR in pathology is complex. In some conditions, activation of the ISR is protective: Whereas GCN2 protects oligodendrocytes and white matter during branch amino acid deficiency (160), PERK protects from demyelination and axonal degeneration in a mouse model of experimental autoimmune encephalomyelitis (161). Correspondingly, in mouse models of Charcot-Marie-Tooth disease that exhibit increased eIF2-P concentrations, germline ablation (162, 163) or pharmacological inhibition of GADD34 with sephin1 improved motor function and reduced demyelination (164). Counterintuitively, in the same model, removing one copy of PERK in all or just in Schwann cells partially reversed the pathology (165, 166). Thus, inhibition of the ISR could have disease-protective or disease-causing effects. However, the molecular mechanisms underlying these two opposing outcomes remain unclear.
Similarly, in a mouse model of ALS, activation and inhibition of specific branches of the ISR by genetic deletion of ATF4, GADD34, CHOP, or PERK, or treatment with salubrinal, guanabenz, or sephin1, led to conflicting results regarding disease onset and survival (167–171). The disparate results obtained by different groups may result from use of different mouse models and transgenic lines, differences in bioavailability and potential off-target effects of the pharmacological compounds, and differential effects of blocking activation of the ISR during development or adulthood. In the future, it will be important to discern how ISR activation or inhibition in different cell types (astrocytes, myelinating cells, and neurons) affects ALS pathology.
The ISR in metabolic disorders
The ISR connects with glucose homeostasis and diabetes development. Recessive loss-of-function mutations in PERK in humans lead to WRS, which is characterized by early-onset diabetes and growth retardation (96). The human phenotype associated with β-cell dysfunction and hyperglycemia is recapitulated in mice in which the ISR has been genetically inhibited (eg., in PERK-deficient mice, or in mice in which eIF2 cannot be phosphorylated) (78, 97, 172, 173). As with mutations in the insulin gene, it is thought that β-cells with a constitutively inhibited ISR cannot properly adjust to an overload of misfolded proteins, and the stress resulting from it compromises the ability of islets to maintain glycemic control.
Permanent activation of the ISR as determined by increased eIF2-P, is also not tolerated by pancreatic β-cells. Patients carrying a specific mutation in eIF2(γ), which results in a decrease in TC and translation fidelity, exhibit hyperglycemia and diabetes (86, 174). Accordingly, reduced TC owing to a loss-of-function mutation in CReP has been associated with diabetes (81). CReP-deficient β-cells are more susceptible to apoptosis: It seems that persistent activation of the ISR and ATF4 translation induces the transcription of the proapoptotic transcription factor CHOP, which promotes apoptosis (175).
Thus, in β-cells, precise ISR-mediated translation regulation is important for adequate ER stress management. The observation that both enhancement and inhibition of the ISR lead to pathology indicates that normal β-cell function requires eIF2-P abundance to be maintained in a narrow range and/or that the ISR needs to switch on and off dynamically. Compound-based strategies that compensate for deviating eIF2-P abundance by adjusting the extent of ISR activation or inhibition will be required to improve pathology associated with these disorders. Thus, depending on the disease, either ISR inhibition or ISR activation may be beneficial therapeutically (Fig. 4).
Fig. 4. Model for proteostasis control by the ISR.
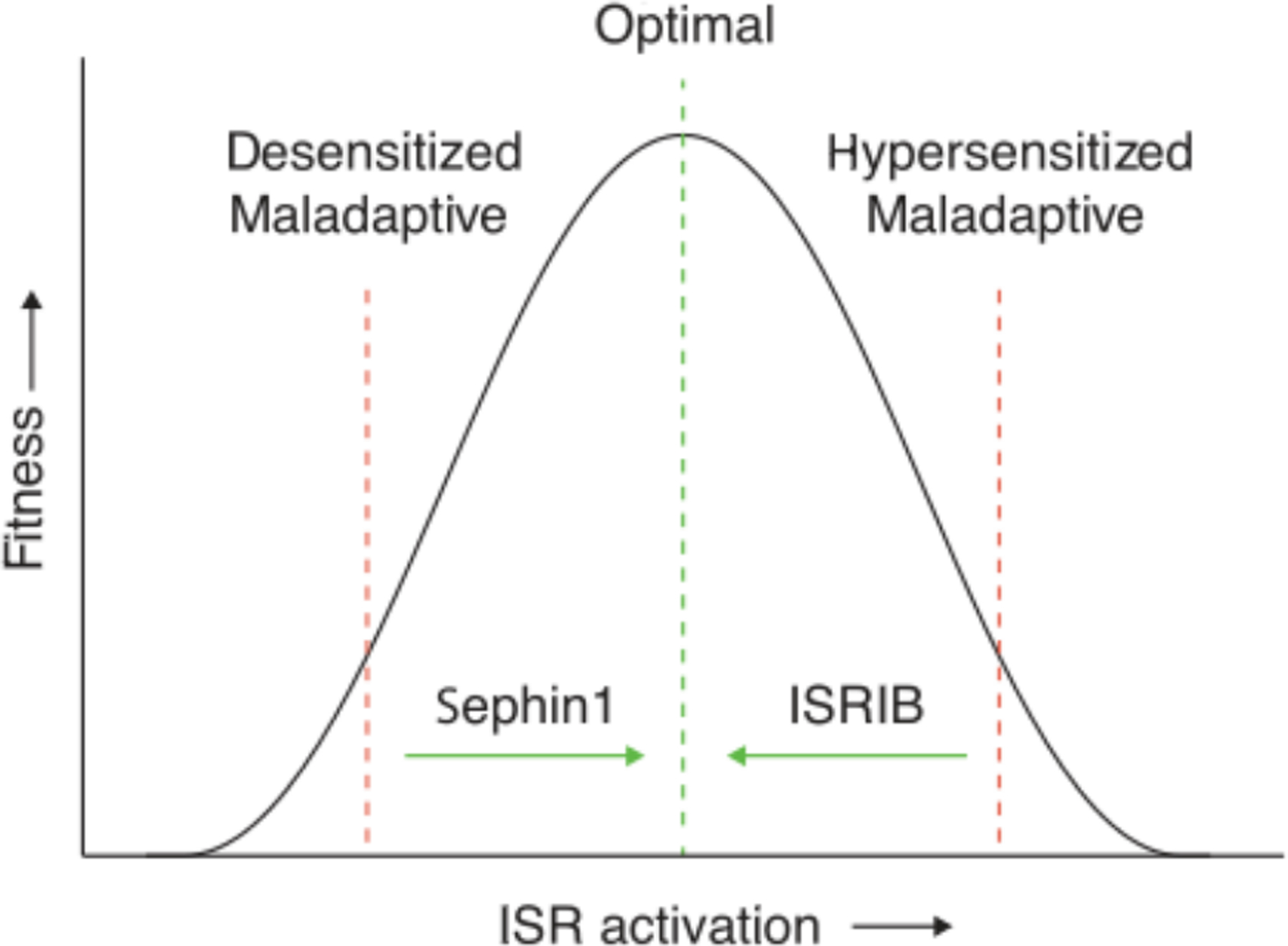
Different pathologies may have distinct homeostatic set points that relate to phenotypic fitness, such as cognition. As considered here, either reduced or increased ISR activation can be maladaptive. Therefore, depending on the disease or pathology and the optimal homeostatic set point for a particular phenotype, activation of the ISR (e.g., with sephin1) or inhibition of the ISR (e.g., with ISRIB) would restore homeostasis to optimal cell fitness.
An intriguing mechanism by which the ISR may control cellular metabolism is through the regulation of mitochondrial function. Mitochondrial stress associated with the loss of the AAA+ mitochondrial protease LONP1, which is essential for mitochondria proteostasis, activates the ISR (176). In addition, mitochondrial dysfunction triggers the induction of mitochondrial chaperones (defined as mitochondrial UPR), a process that is mediated by the transcription factors ATFS-1 and ATF5 in Caenorhabditis elegans and mammals, respectively (177, 178). Of interest, ATF5 induction during mitochondrial stress is required to maintain mitochondrial activity and homeostasis. Future experiments should focus on elucidating how mechanistically the different ISR kinases and effectors signal to the mitochondria and vice versa.
The ISR in cancer
Aberrant cell survival and reduced cell death are hallmarks of cell transformation and cancer progression, and genetic alteration in several translation regulatory proteins has been associated with cancer (179, 180). In addition, most oncogenic pathways found in human cancers lead to various forms of protein synthesis dysregulation. Given that (i) the ISR controls protein synthesis and protcostasis, (ii) over-expression of Met-tRNAi promotes proliferation in human epithelial cells (181), and (iii) the ISR target genes CHOP and ATF4 are crucial regulators of the balance between survival and cell death, it comes as no surprise that the ISR is exploited by cancer cells. Indeed, three ISR kinases (PKR, PERK, and GCN2) have been implicated in cancer. Early studies showed that inhibition of either the PKR branch of the ISR or phosphorylation of eIF2 led to transformation of mouse fibroblasts and increased tumor formation in immune-deficient mice (182–185). Paradoxically, deletion of PKR or overall reduction of eIF2-P in mice was not tumorigenic in vivo (78, 186, 187). The resolution to the paradox may lie in the complexity of ISR regulation, which controls both prosurvival and pro-death mechanisms, including the activation of nuclear factor κB (NF-κB), phosphatidylinositol 3-kinase (PI3K), and c-Jun N-terminal kinase (JNK) pathways (188), which may be differentially important in cancers with diverse origins and/or may present differently in cell culture and animal models.
In many cancers, mutations in tumor suppressor genes, such as PTEN (phosphatase and tensin homolog), or the activation of oncogenes, such as MYC, results in increased protein synthesis, which can saturate the cells’ proteostasis machinery and activate the ISR. As the capacity for protein folding becomes limiting, translation is reduced to rebalance proteostasis. In this way, the ISR acts as a governor keeping protein synthesis in check. If this control fails to cope with the protein-folding problem, the cell enters apoptosis. In the context of a cancer cell and its microenvironment, activation of the ISR by the protein misfolding kinase PERK promotes tumor initiation and progression (189–191). Accordingly, in mouse and human models of aggressive metastatic prostate cancer, in which the tumor suppressor PTEN is ablated and the oncogene MYC is overexpressed, the PERK branch of the ISR is activated and limits protein synthesis rates. Removing this regulation with ISRIB selectively killed patient-derived metastatic prostate cancer cells in xenograft mouse models (192). Similarly, acute deletion of the ISR effector ATF4 substantially delays MYC-driven tumor progression and increases survival in mice (193).
Finally, GCN2, a crucial regulator of amino acid metabolism (194, 195), is necessary for metabolic homeostasis of tumor cells. Tumors lacking GCN2 or ATF4 grow more slowly (196, 197). Thus, in cancers where amino acids are scarce, targeting the GCN2 branch of the ISR may be beneficial. Indeed, combination treatment with l-asparinase and GCN2 inhibitors causes apoptosis in several cancer cell types (43).
In conclusion, depending on the gene mutation and cellular context, targeting the ISR can selectively tilt the balance of cancer cells toward apoptosis, rendering the ISR a promising chemotherapy target for many different types of cancers.
The ISR in immunity
Several lines of evidence indicate that the ISR is intimately embedded in the cell’s innate immune response (198, 199). All four ISR kinases play parts in immunity and inflammation. ISR activation leads to secretion of inflammatory cytokines, such as interleukin 1β (IL-1β) and IL6. In this way, ISR signaling does not remain cell autonomous and promotes communication between neighboring cells in a state of local inflammation. Pharmacologic ISR inhibition may therefore have broadly beneficial anti-inflammatory consequences.
Activation of the proinflammatory transcription factor NF-κB that drives transcription of a large set of proinflammatory genes crucially depends on the activation of the ISR. In its latent state, NF-κB is anchored in the cytoplasm by its inhibitor IκB. Because IκB is a short-lived protein, it needs to be constantly replenished by new synthesis. The translation inhibition exerted by the ISR activates NF-κB activation by lowering IκB’s steady-state concentration. Notably, this control does not require the ATF4 axis of the ISR (200, 201).
In addition, NF-κB activation in response to intracellular pathogens is crucially dependent on the ISR kinase HRI (202). Both liberation of the chaperone HSPB8 from autophosphorylated HRI and the additional transcriptional up-regulation of HSPB8 mRNA downstream of ATF4 lead to HSPB8 binding to the pattern recognition receptors NOD1/2, which in turn assemble into large inflammasome complexes. The inflammasome serves as a scaffold that assembles the molecular machinery, including ubiquitylation enzymes, which accelerate IκB degradation, thus activating NF-κB. Pro-caspase 1 also binds to inflammasome platforms where it activates by autoproteolysis and then cleaves cytoplasmic pools of pro-IL-1β, producing mature IL-1β, which in turn is secreted from the cell by a signal sequence-independent nonconventional mechanism. Another inflammatory cytokine, IL-6, is transcriptionally up-regulated by ATF4 and hence is encoded by a direct ISR target gene (203).
Saturated fatty acids, abundant in Western diets, promote cardiovascular disease, including atherosclerosis. They induce the ISR by causing ER stress that is sensed by the UPR. In bone-derived macrophages, saturated fatty acid treatment induces die UPR and ISR kinase PERK (204), which leads to caspase 1 activation and IL-1β secretion. Notably, inhibition of the ISR with ISRIB blocked lipid-induced inflammasome activation, inflammation, and atherosclerotic progression in mouse models.
Activated PKR also links to inflammasome activation through direct a coassembly with NOD-like receptors (205). This activation mode occurs upstream of PKR’s role in eIF2 phosphorylation. ATF4 however, which is produced downstream of eIF2 phosphorylation, drives transcription of at least one NOD-like receptor (NLRP1) (206). Thus, even in this scenario, bona fide ISR induction is an important facet in regulating the response. In addition, PKR responds to nutrients and ER stress to regulate insulin and metabolism through the proinflammatory kinase JNK (207).
Future perspectives
Research in the past two decades has provided great progress in the identification of key components of the ISR and resulted in a detailed understanding of the mechanism by which the ISR regulates cell physiology and function. It is now clear that the ISR serves as a molecular control center for long-term memory formation. In addition, state-of-the-art pharmacological and genetic manipulations have begun to elucidate how activation of the ISR contributes to different diseases.
Although all four eIF2 kinases converge on eIF2, it is estimated that each kinase in the human kinome phosphorylates on average more than 10 substrates (208). Thus, the paucity of other substrates for the eIF2 kinases is surprising, and it will be important to rule in or out the possibility that other substrates functionally modulate the ISR, such Nrf2, which is phosphorylated by PERK (209). In this light, it will be important to assess whether activation of the different kinases leads to the same translational reprogramming or whether bifurcation of the signal at the level of the ISR kinases modulates the translational outcome in different cell types and pathological contexts. Recent advances in functional genomic tools, including CRISPRi- and CRISPRa-based approaches that allow for silencing or activating any gene in vivo (210), provide opportunities to identify new components and regulatory complexities of the ISR and to test for their effects in an unbiased manner.
In the brain, little is known about the nature of the specific proteins whose synthesis is controlled by the ISR during long-term memory formation. It would be interesting to examine whether the ISR selectively regulates translation locally at dendrites, where most of the mRNAs arc engaged with single ribosomes (monosomes), vis-à-vis the cell body, where mRNAs are bound to multiple ribosomes (211).
Finally, a major focus of ISR research will be to develop new and more specific molecules that tune the activity of the ISR up or down and to address diseases for which such drug-like molecules may open therapeutic windows for ISR manipulation in the clinic. The molecular identification of drug targets, mechanism of action, and structural basis of their activity will crucially enable preclinical development. In addition, the use of convergent models, from mouse models to human derived induced pluripotent stem cells to organoids, will further accelerate the process of linking basic mechanistic discoveries to tangible clinical applications.
ACKNOWLEDGMENTS
We apologize to the authors of the numerous outstanding papers that contributed to the field and could not be cited here owing to space limitations. We thank A. Bertolotti, E. Erbay, S. Matus. D. Ron, S. Rosi, L. Reichardt, D. Ruggero, and members of the Costa Mattioli and Walter labs for their thoughtful comments on the manuscript. We thank A. Anand for his invaluable help in preparing the figures.
Funding: M.C. -M is supported by funding from the NIH and the generous support from Sammons Enterprise. P.W. is supported by funding from the NIH, the Howard Hughes Medical Institute, and Calico.
Footnotes
Competing interests: The authors declare no competing interest. P.W. is an inventor on U.S. Patent 9708247 held by the Regents of the University of California describing ISRIB and its analogs. Rights to the invention have been licensed by Calico.
REFERENCES AND NOTES
- 1.Hipp MS, Kasturi P, Hartl FU, The proteostasis network and its dedine in ageing. Nat. Rev. Mol. Cell Biol 20, 421–435 (2019). doi: 10.1038/s41580-019-0101-y; [DOI] [PubMed] [Google Scholar]
- 2.Labbadia J, Morimoto RI, The biology of proteostasis in aging and disease. Annu. Rev. Biochem 84, 435–464 (2015). doi: 10.1146/annurev-biochem-060614-033955; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walter P, Ron D, The unfolded protein response: From stress pathway to homeostatic relation. Science 334, 1081–1086 (2011). doi: 10.1126/Science.l209038; [DOI] [PubMed] [Google Scholar]
- 4.Harding HP et al. , An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633 (2003). doi: 10.1016/S1097-2765(03)00105-9; [DOI] [PubMed] [Google Scholar]
- 5.Scheper GC et al. , Inadivation of eIF2B and phosphorylation of PHASI in heat shocked rat hepatoma cells. J. Biol. Chem 272, 26850–26856 (1997). doi: 10.1074/jbe.272.43.26850; [DOI] [PubMed] [Google Scholar]
- 6.Harding HP, Zhang Y, Ron D, Protein translation and folding are coupled by an endoplasmic reticulum resident kinase. Nature 397, 271–274 (1999). doi: 10.1038/16729; [DOI] [PubMed] [Google Scholar]
- 7.Hinnebusch AG, Ivanov IP, Sonenberg N, Translational control by 5′ untranslated regions of eukaryotic mRNAs. Science 352, 1413–1416 (2016). doi: 10.1126/science.aad9868; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Algire MA, Maag D, Lorsch JR, Pi release from eIF2. not GTP hydrolysis, is the step controlled by start site selection during eukaryotic translation initiation. Mol. Cell 20, 251–262 (2005). doi: 10.1016/j.molcel2005.09.008; [DOI] [PubMed] [Google Scholar]
- 9.Wortham NC, Martinez M, Gordiyenko Y, Robinson CV, Proud CG, Analysis of the subunit organization of the eIF2B complex reveals new insights into its structure and regulation. FASEB J 28, 2225–2237 (2014). doi: 10.1096/fj.13-243329; [DOI] [PubMed] [Google Scholar]
- 10.Kashiwagi K et al. , Crystal structure of eukaryotic translation initiation factor 2B. Nature 531, 122–125 (2016). doi: 10.1038/nature16991; [DOI] [PubMed] [Google Scholar]
- 11.Tsai JC et al. , Structure of the nucleotide exchange factor eIF2B reveals mechanism of memory enhancing molecule. Science 359, eaaq0939 (2018). doi: 10.1126/science.aaq0939; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zyryanova AF et al. , Binding of ISRIB reveals a regulatory site in the nucleotide exchange factor eIF2B. Science 359, 1533–1536 (2018). doi: 10.1126/science.aar5129; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boesen T, Mohammad SS, Pavitt GD, Andersen GR, Structure of the catalytic fragment of translation initiation factor 2B and identification of a critically important catalytic residue. J. Biol. Chem 279, 10584–10592 (2004). doi: 10.1074/jbc.M311055200; [DOI] [PubMed] [Google Scholar]
- 14.Kenner LR et al. , eIF2B catalyzed nucleotide exchange and phosphoregulation by the integrated stress response. Science 364, 491–495 (2019). doi: 10.1126/science.aaw2922; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kashiwagi K et al. , Structural basis for eIF2B inhibition in integrated stress response. Science 364. 495–499 (2019). doi: 10.1126/science.aaw4104; [DOI] [PubMed] [Google Scholar]
- 16.Bogorad AM, Lin KY, Marintchev A, Novel mechanisms of eIF2B action and relation by eIF2α phosphorylation. Nucleic Acids Res 45, 11962–11979 (2017). doi: 10.1093/nar/gkx845; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adomavicius T et al. , The structural basis of translational control by eIF2 phosphorylation. Nat. Commun 10, 2136 (2019). doi: 10.1038/s41467-01910167-3; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gordiyenko Y, Llácer JL, Ramakrishnan V, Structural basis for the inhibition of translation through eIF2α phosphorylation. Nat. Commun 10, 2640 (2019). doi: 10.1038/s41467-019-10606-1; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sidrauski C et al. , Pharmacological brake release of mRNA translation enhances cognitive memory. eLife 2, e00498 (2013). doi: 10.7554/eLife.00498; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sidrauski C et al. , Pharmacological dimerization and activation of the exchange factor eIF2B antagonizes the integrated stress response. eLife 4, e07314 (2015). doi: 10.7554/eLife.07314; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sekine Y et al. , Stress responses. Mutations in a translation initiation factor identify the target of a memory-enhancing compound. Science 348, 1027–1030 (2015). doi: 10.1126/science.aaa6986; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rabouw HH et al. , Small molecule ISRIB suppresses the integrated stress response within a defined window of activation. Proc. Natl. Acad. Sci. USA 116, 2097–2102 (2019). doi: 10.1073/pnas.l815767116; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Halliday M et al. , Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis 6, e1672 (2015). doi: 10.1038/cddis2015.49; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wek RC Role of eIF2α Kinases in Translational Control and Adaptation to Cellular Stress. Cold Spring Harb. Perspect. Biol 10, a032870 (2018). doi: 10.1101/cshperspect.032870; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hinnebusch AG Translational regulation of GCN4 and the general amino acid control of yeast. Annu. Rev. Microbiol 59, 407–450 (2005). doi: 10.1146/annurev.micro.59.031805.133833; [DOI] [PubMed] [Google Scholar]
- 26.Lavoie H, Li JJ, Thevakumaran N, Therrien M, Sicheri F, Dimerization-induced allostery in protein kinase regulation. Trends Biochem. Sci 39, 475–486 (2014). doi: 10.1016/j.tibs2014.08.004; [DOI] [PubMed] [Google Scholar]
- 27.Harding HP et al. , Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108 (2000). doi: 10.1016/S1097-2765(00)00108-8; [DOI] [PubMed] [Google Scholar]
- 28.Inglis AJ et al. , Activation of GCN2 by the ribosomal P-stalk. Proc. Natl. Acad. Sci. U.S.A 116, 4946–4954 (2019). doi: 10.1073/pnas.1813352116; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harding HP et al. , The ribosomal P-stalk couples amino acid starvation to GCN2 activation in mammalian cells. eLife 8, e50149 (2019). doi: 10.7554/eLife.50149; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishimura R, Nagy G, Dotu I, Chuang JH, Ackerman SL, Activation of GCN2 kinase by ribosome stalling links translation elongation with translation initiation. eLife 5, e14295 (2016). doi: 10.7554/eLifel4295; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi Y et al. , Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase. PEK. involved in translational control. Mol. Cell. Biol 18, 7499–7509 (1998). doi: 10.1128/MCB.18.12.7499; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kopp MC, Larburu N, Durairaj V, Adams CJ, Ali MMU, UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat. Struct. Mol. Biol 26, 1053–1062 (2019). doi: 10.1038/s41594-019-0324-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D, Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol 2, 326–332 (2000). doi: 10.1038/35014014: [DOI] [PubMed] [Google Scholar]
- 34.Wang P, Li J, Tao J, Sha B, The luminal domain of the ER stress sensor protein PERK binds misfolded proteins and thereby triggers PERK oligomerization. J. Biol. Chem 293, 4110–4121 (2018). doi: 10.1074/jbc.RA117.001294; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Volmer R, van der Ploeg K, Ron D, Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. U.S.A 110, 4628–4633 (2013). doi: 10.1073/pnas.1217611110; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dey M et al. , Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell 122, 901–913 (2005). doi: 10.1016/j.cell2005.06.041; [DOI] [PubMed] [Google Scholar]
- 37.García MA, Meurs EF, Esteban M, The dsRNA protein kinase PKR: Virus and cell control. Biochimie 89, 799–811 (2007). doi: 10.1016/j.biochi.2007.03.001; [DOI] [PubMed] [Google Scholar]
- 38.Hugon J, Mouton-Liger F, Dumurgier J, Paquet C, PKR involvement in Alzheimer’s disease. Alzheimers Res. Ther 9, 83 (2017). doi: 10.1186/s13195-017-0308-0; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peel AL, PKR activation in neurodegenerative disease. J. Neuropathoi. Exp. Neurol 63, 97–105 (2004). doi: 10.1093/jnen/63.2.97; [DOI] [PubMed] [Google Scholar]
- 40.Chen JJ, Translational control by heme-regulated eIF2α kinase during erythropoiesis. Curr. Opin. Hematol 21, 172–178 (2014). doi: 10.1097/MOH.0000000000000030: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tabula Muris Consortium et al. , Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372 (2018). doi: 10.1038/s4158601805904: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo X et al. , Mitochondrial dysfunction is signaled to the integrated stress response by OMAL DELEI and HRI. bioRtiv (2019). doi: 10.1101/715896 [DOI] [Google Scholar]
- 43.Nakamura A et al. , Inhibition of GCN2 sensitizes ASNS-low cancer cells to asparaginase by disrupting the amino acid response. Proc. Natl. Acad. Sci. U.S.A 115, E7776–E7785 (2018). doi: 10.1073/pnas.l805523115; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Axten JM et al. , Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}−2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414). a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem 55, 7193–7207 (2012). doi: 10.1021/jm300713s; [DOI] [PubMed] [Google Scholar]
- 45.Rosen MD et al. , Discovery of the first known small-molecule inhibitors of heme-regulated eukaryotic initiation factor 2alpha (HRI) kinase. Bioorg Med. Chem. Lett 19, 6548–6551 (2009). doi: 10.1016/j.bmcl.2009.10.033: [DOI] [PubMed] [Google Scholar]
- 46.Huang JT, Schneida RJ, Adenovirus inhibition of cellular protein synthesis is prevented by the drug 2-aminopurine. Proc. Natl. Acad. Sci. U.S.A 87, 7115–7119 (1990). doi: 10.1073/pnas.87.18.7115; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bryk R et al. , Identification of new inhibitors of protein kinase R guided by statistical modeling. Bioorg. Med. Chem. Lett 21, 4108–4114 (2011). doi: 10.1016/j.bmcl2011.04.149; [DOI] [PubMed] [Google Scholar]
- 48.Robert F et al. , Blocking UV-induced eIF2alpha phosphorylation with small molecule inhibitors of GCN2. Chem. Biol. Drug Des 74, 57–67 (2009). doi: 10.1111/j.1747-0285.2009.00827.x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yefidoff-Freedman R et al. , Development of 1-((1,4-trans)-4-Aryloxycyclohexyl)-3-arylurea Activators of Heme-Regulated Inhibitor as Selective Activators of the Eukaryotic Initiation Factor 2 Alpha (eIF2α) Phosphorylation Arm of the Integrated Endoplasmic Reticulum Stress Response. J. Med. Chem 60, 5392–5406 (2017). doi: 10.1021/acs.jmedchem.7b00059; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hong MN, Nam KY, Kim KK, Kim SY, Kim I, The small molecule ‘1-(4-biphenylylcarbonyl)-4-(5-bromo-2-methoxybenzyl) piperazine oxalate’ and its derivatives regulate global protein synthesis by inactivating eukaryotic translation initiation factor 2 alpha. Cell Stress Chaperones 21, 485–497 (2016). doi: 10.1007/sl219201606775: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rojas-Rivera D. et al. , When PERK inhibitors turn out to be new potent RIPK1 inhibitors: Critical issues on the specificity and use of GSK2606414 and GSK2656157. Cell Death Differ 24, 1100–1110 (2017). doi: 10.1038/cdd2017.58; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu Q et al. , Type I interferons mediate pancreatic toxicities of PERK inhibition. Proc. Natl. Acad. Sci. U.S.A 112, 15420–15425 (2015). doi: 10.1073/pnas.1516362112; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu PD, Harding HP, Ron D, Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol 167, 27–33 (2004). doi: 10.1083/jcb200408003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vattem KM, Wek RC, Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. U.S.A 101, 11269–11274 (2004). doi: 10.1073/pnas.0400541101; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou D et al. , Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. J. Biol. Chem 283, 7064–7073 (2008). doi: 10.1074/jbc.M708530200; [DOI] [PubMed] [Google Scholar]
- 56.Palam LR, Baird TD, Wek RC, Phosphorylation of eIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J. Biol. Chem 286, 10939–10949 (2011). doi: 10.1074/jbc.M110216093; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee YY, Cevallos RC, Jan E, An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2alpha phosphorylation. J. Biol. Chem 284, 6661–6673 (2009). doi: 10.J074/jbc.M806735200; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Di Prisco GV et al. , Translational control of mGluR-dependent long term depression and object-place learning by eIF2α. Nat. Neurosci 17, 1073–1082 (2014). doi: 10.1038/nn.3754; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morris DR Geballe AP Upstream open reading frames as regulators of mRNA translation. Mol. Cell. Biol 20, 8635–8642 (2000). doi: 10.1128/MCB.20.23.8635-8642.2000; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jousse C et al. , Inhibition of a constitutive translation initiation factor 2alpha phosphatase. CReP. promotes survival of stressed cells. J. Cell Biol 163, 767–775 (2003). doi: 10.1083/jcb200308075; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Novoa I, Zeng H, Harding HP, Ron D, Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol 153, 1011–1022 (2001). doi: 10.1083/jcb.1535.1011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Young SK, Wily JA, Wu C, Sachs MS, Wek RC, Ribosome Reinitiation Directs Gene-specific Translation and Regulates the Integrated Stress Response. J. Biol. Chem 290, 28257–28271 (2015). doi: 10.1074/jbc.M115.693184; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen R et al. , G-actin provides substrate-specificity to eukaryotic initiation factor 2α holophosphatases. eLife 4, e04871 (2015). doi: 10.7554/eLife.04871; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chambers JE et al. , Actin dynamics tune the integrated stress response by regulating eukaryotic initiation factor 2α dephosphorylation. eLife 4, e04872 (2015). doi: 10.7554/eLife.04872; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Boyce M et al. , A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307, 935–939 (2005). doi: 10.1126/science.1101902; [DOI] [PubMed] [Google Scholar]
- 66.Robert F et al. , Initiation of protein synthesis by hepatitis C virus is refractory to reduced eIF2 · GTP · Met-tRNAiMet ternary complex availability. Mol. Biol. Cell 17, 4632–4644 (2006). doi: 10.1091/mbc.e06-06-0478; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tsaytler P, Harding HP, Ron D, Bertolotti A, Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 332, 91–94 (2011). doi: 10.1126/science.1201396; [DOI] [PubMed] [Google Scholar]
- 68.Dash PK et al. , Inhibition of Eukaryotic Initiation Factor 2 Alpha Phosphatase Reduces Tissue Damage and Improves Learning and Memory after Experimental Traumatic Brain Injury. J. Neurotrauma 32, 1608–1620 (2015). doi: 10.1089/neu2014.3772; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Krzyzosiak A et al. , Target-Based Discovery of an Inhibitor of the Regulatory Phosphatase PPP1R15B. Cell 174, 1216–1228. el9 (2018). doi: 10.1016/j.cell.2018.06.030; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Crespillo-Casado A et al. , A Sephin1-insensitive tripartite holophosphatase dephosphorylates translation initiation factor 2α. J. Biol. Chem 293, 7766–7776 (2018). doi: 10.1074/jbc.RA118.002325: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Crespillo-Casado A, Chambers JE, Fischer PM, Marciniak SJ, Ron D, PPP1R15A-mediated dephosphorylation of eIF2α is unaffected by Sephin1 or Guanabenz. eLife 6, e26109 (2017). doi: 10.7554/eLife.26109; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dedigama-Arachchige PM, Acharige NPN, Pflum MKH, Identification of PP1-Gadd34 substrates involved in the unfolded protein response using K-BIPS, a method for phosphatase substrate identification. Mol Omics 14, 121–133 (2018). doi: 10.1039/C7M000064B; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen Y et al. , Sephin1, which prolongs the integrated stress response, is a promising therapeutic for multiple sclerosis. Brain 142, 344–361 (2019). doi: 10.1093/brain/awy322; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS, Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223 (2009). doi: 10.1126/science.1168978; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee S et al. , Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. U.S.A 109, E2424–E2432 (2012). doi: 10.1073/pnas.1207846109; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Welsh GI, Proud CG, Glycogen synthase kinase-3 is rapidly inactivated in response to insulin and phosphorylates eukaryotic initiation factor eIF-2B. Biochem. J 294, 625–629 (1993). doi: 10.1042/bj2940625; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Welsh GI, Miyamoto S, Price NT, Safer B, Proud CG, T-cell activation leads to rapid stimulation of translation initiation factor eIF2B and inactivation of glycogen synthase kinase-3. J. Biol. Chem 271, 11410–11413 (1996). doi: 10.1074/jbc.271.19.11410; [DOI] [PubMed] [Google Scholar]
- 78.Scheuner D et al. , Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 7, 1165–1176 (2001). doi: 10.1016/S1097-2765(01)00265-9; [DOI] [PubMed] [Google Scholar]
- 79.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D, Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 5, 897–904 (2000). doi: 10.1016/S1097-2765(00)80330-5; [DOI] [PubMed] [Google Scholar]
- 80.Harding HP et al. , Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2alpha) dephosphorylation in mammalian development. Proc. Natl. Acad. Sci. U.S.A 106, 1832–1837 (2009). doi: 10.1073/pnas.0809632106; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Abdulkarim B et al. , A Missense Mutation in PPP1R15B Causes a Syndrome Including Diabetes, Short Stature, and Microcephaly. Diabetes 64, 3951–3962 (2015). doi: 10.2337/dbl5-0477; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kernohan KD et al. , Homozygous mutation in the eukaryotic translation initiation factor 2alpha phosphatase gene, PPP1R15B. is associated with severe microcephaly, short stature and intellectual disability. Hum. Mol. Genet 24, 6293–6300 (2015). doi: 10.1093/hmg/ddv337; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Borck G et al. , eIF2γ mutation that disrupts eIF2 complex integrity links intellectual disability to impaired translation initiation. Mol. Cell 48, 641–646 (2012). doi: 10.1016/j.molcel.2012.09.005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Skopkova M et al. , EIF2S3 Mutations Associated with Severe X-Linked Intellectual Disability Syndrome MEHMO. Hum. Mutat 38, 409–425 (2017). doi: 10.1002/humu.23170; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gregory LC et al. , Impaired EIF2S3 function associated with a novel phenotype of X-linked hypopituitarism with glucose dysregulation. EBioMedicine 42, 470–480 (2019). doi: 10.1016/j.ebiom.2019.03.013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moortgat S et al. , Two novel EIF2S3 mutations associated with syndromic intellectual disability with severe microcephaly, growth retardation, and epilepsy. Am. J. Med. Genet. A 170, 2927–2933 (2016). doi: 10.1002/ajmg.a.37792; [DOI] [PubMed] [Google Scholar]
- 87.Young-Baird SK, Shin BS, Dever TE, MEHMO syndrome mutation EIF2S3-I259M impairs initiator Met-tRNAiMet binding to eukaryotic translation initiation factor eIF2. Nucleic Acids Res 47, 855–867 (2019). doi: 10.1093/nar/gky1213; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.A Leegwater P et al. , Subunits of the translation initiation factor eIF2B are mutant in leukoencephalopathy with vanishing white matter. Nat. Genet 29, 383–388 (2001). doi: 10.1038/ng764; [DOI] [PubMed] [Google Scholar]
- 89.van der Knaap MS et al. , Mutations in each of the five subunits of translation initiation factor eIF2B can cause leukoencephalopathy with vanishing white matter. Ann. Neurol 51, 264–270 (2002). doi: 10.1002/ana.10112; [DOI] [PubMed] [Google Scholar]
- 90.van der Knaap MS et al. , A new leukoencephalopathy with vanishing white matter. Neurology 48, 845–854 (1997). doi: 10.1212/WNL.48.4.845; [DOI] [PubMed] [Google Scholar]
- 91.Wong YL et al. , eIF2B activator prevents neurological defects caused by a chronic integrated stress response. eLife 8, e42940 (2019). doi: 10.7554/eLife.42940; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wong YL et al. , The small molecule ISRIB rescues the stability and activity of Vanishing White Matter Disease eIF2B mutant complexes. eLife 7, e32733 (2018). doi: 10.7554/eLife.32733; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Williams NP, Hinnebusch AG, Donahue TF, Mutations in the structural genes for eukaryotic initiation factors 2 alpha and 2 beta of Saccharomyces cerevisiae disrupt translational control of GCN4 mRNA. Proc. Natl. Acad. Sci. U.S.A 86, 7515–7519 (1989). doi: 10.1073/pnas.86.197515; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Green KM et al. , RAN translation at C9orf72 associated repeat expansions is selectively enhanced by the integrated stress response. Nat. Commun 8, 2005 (2017). doi: 10.1038/S4M67-017-02200-0; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cheng W et al. , C90RF72 GGGGCC repeat-associated non-AUG translation is upregulated by stress through eIF2α phosphorylation. Nat. Commun 9. 51 (2018). doi: 10.1038/S41467-017-02495-z; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Delépine M et al. , EIF2AK3. encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet 25, 406–409 (2000). doi: 10.1038/78085; [DOI] [PubMed] [Google Scholar]
- 97.Harding HP et al. , Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Mol. Cell 7, 1153–1163 (2001). doi: 10.1016/S1097-2765(01)00264-7; [DOI] [PubMed] [Google Scholar]
- 98.Zhang P et al. , The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol. Cell. Biol 22, 3864–3874 (2002). doi: 10.1128/MCB22.113864-38742002; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Eyries M et al. , EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat Genet 46, 65–69 (2014). doi: 10.1038/ng2844; [DOI] [PubMed] [Google Scholar]
- 100.Longchamp A et al. , Amino Add Restriction Triggers Angiogenesis via GCN2/ATF4 Regulation of VEGF and H2S Production. Cell 173, 117–129 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McGaugh JL, Memory—A century of consolidation. Science 287, 248–251 (2000). doi: 10.1126/science.287.5451248; [DOI] [PubMed] [Google Scholar]
- 102.Kandel ER, The molecular biology of memory storage: A dialogue between genes and synapses. Science 294, 1030–1038 (2001). doi: 10.1126/science.1067020; [DOI] [PubMed] [Google Scholar]
- 103.Agranoff BW, Davis RE, Brink JJ, Memory fixation in the goldfish. Proc. Natl. Acad. Sci. U.S.A 54, 788–793 (1965). doi: 10.1073/pnas.54.3.788; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Barondes SH, Cohen HD, Puromycin effect on successive phases of memory storage. Science 151, 594–595 (1966). doi: 10.1126/science.151.3710.594; [DOI] [PubMed] [Google Scholar]
- 105.Davis HP, Squire LR, Protein synthesis and memory: A review. Psychol. Bull 96, 518–559 (1984). doi: 10.1037/00332909.96.3.518; [DOI] [PubMed] [Google Scholar]
- 106.Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N, Translational control of long-lasting synaptic plasticity and memory. Neuron 61, 10–26 (2009). doi: 10.1016/j.neuron.2008.10.055; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sutton MA, Schuman EM, Dendritic protein synthesis, synaptic plasticity, and memory. Cell 127, 49–58 (2006). doi: 10.1016/j.cell.2006.09.014; [DOI] [PubMed] [Google Scholar]
- 108.Richter JD, Klann E, Making synaptic plasticity and memory last: Mechanisms of translational relation. Genes Dev 23, 1–11 (2009). doi: 10.1101/gad.1735809; [DOI] [PubMed] [Google Scholar]
- 109.Buffington SA, Huang W, Costa-Mattioli M Translational control in synaptic plasticity and cognitive dysfunction. Annu. Rev. Neurosci 37, 17–38 (2014). doi: 10.1146/annurev-neuro-071013-014100; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Costa-Mattioli M et al. , Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature 436, 1166–1170 (2005). doi: 10.1038/nature03897; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Costa Mattioli M et al. , eIF2alpha phosphorylation bidirectionally regulates the switch from short to long term synaptic plasticity and memory. Cell 129, 195–206 (2007). doi: 10.1016/j.cell.2007.01050; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhu PJ et al. , Suppression of PKR promotes network excitability and enhanced cognition by interferon-γ-mediated disinhibition. Cell 147, 1384–1396 (2011). doi: 10.1016/j.cell.2011.11.029; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stem E, Chinnakkaruppan A, David O, Sonenberg N, Rosenblum K, Blocking the eIF2α kinase (PKR) enhances positive and negative forms of cortex-dependent taste memory. J. Neurosci 33, 2517–2525 (2013). doi: 10.1523/JNEUROSCI.2322-12-2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sharma V et al. , Local Inhibition of PERK Enhances Memory and Reverses Age-Related Deterioration of Cognitive and Neuronal Properties. J. Neurosci 38, 648–658 (2018). doi: 10.1523/JNEUROSCI.0628-17.2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Batista G, Johnson JL, Dominguez E, Costa-Mattioli M, Pena JL, Translational control of auditory imprinting and structural plasticity by eIF2α. eLife 5, e17197 (2016). doi: 10.7554/eLife.17197; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jian M et al. , eIF2α dephosphorylation in basolateral amygdala mediates reconsolidation of drug memory. J. Neurosci 34, 10010–10021 (2014). doi: 10.1523/JNEUROSCI.0934-14.2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jiang Z et al. , eIF2alpha Phosphorylation-dependent translation in CA1 pyramidal cells impairs hippocampal memory consolidation without affecting general translation. J. Neurosci 30, 2582–2594 (2010). doi: 10.1523/JNEUROSCI.3971-09.2010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shrestha P, Ayata P, Herrero-Vidal P, Longo F, Gastone A, Ledoux JE, Heintz N, Klann E, Chemogenetic evidence that rapid neuronal de novo protein synthesis is required for consolidation of long-term memory. bioRxiv 704965 (2019). 10.1101/704965. [DOI] [Google Scholar]
- 119.Panja D et al. , Novel translational control in Arc dependent long term potentiation consolidation in vivo. J. Biol. Chem 284, 31498–31511 (2009). doi: 10.1074/jbc.M109056077; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Trinh MA et al. , The eIF2α kinase PERK limits the expression of hippocampal metabotropic glutamate receptor-dependent long-term depression. Learn. Mem 21, 298–304 (2014). doi: 10.1101/lm032219.113; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Eacker SM et al. , Experience-dependent translational state defined by cell type-specific ribosome profiling. bioRxiv 169425 (2017). 10.1101/169425. [DOI] [Google Scholar]
- 122.Jian M et al. , eIF2α dephosphorylation in basolateral amygdala mediates reconsolidation of drug memory. J. Neurosci 34, 10010–10021 (2014). doi: 10.1523/JNEUROSCI.0934-14.2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Werner CT, Stefanik MT, Milovanovic M, Caccamise A, Wolf ME, Protein Translation in the Nucleus Accumbens Is Dysregulated during Cocaine Withdrawal and Required for Expression of Incubation of Cocaine Craving. J. Neurosci 38, 2683–2697 (2018). doi: 10.1523/JNEUROSCI.2412-172018: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Huang W et al. , Translational control by eIF2α phosphorylation regulates vulnerability to the synaptic and behavioral effects of cocaine. eLife 5, e12052 (2016). doi: 10.7554/eLite.12052; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Placzek AN et al. , Translational control of nicotine-evoked synaptic potentiation in mice and neuronal responses in human smokers by eIF2α. eLife 5, e12056 (2016). doi: 10.7554/eLife.12056; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Placzek AN et al. , eIF2α-mediated translational control regulates the persistence of cocaine-induced LTP in midbrain dopamine neurons. eLife 5. e17517 (2016). doi: 10.7554/eLife.17517; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Melas PA et al. , Cannabinoid Modulation of Eukaryotic Initiation Factors (eIF2α and eIF2B1) and Behavioral Cross-Sensitization to Cocaine in Adolescent Rats. Cell Rep 22, 2909–2923 (2018). doi: 10.1016/j.celrep2018.02.065; [DOI] [PubMed] [Google Scholar]
- 128.Rittiner JE et al. , Functional Genomic Analyses of Mendelian and Sporadic Disease Identify Impaired eIF2α Signaling as a Generalizable Mechanism for Dystonia. Neuron 92, 1238–1251(2016). doi: 10.1016/j.neuron.2016.11.012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nabavi S et al. , Engineering a memory with LTD and LTP. Nature 511, 348–352 (2014). doi: 10.1038/naturel3294; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nadif Kasri N, Nakano-Kobayashi A, Van Aelst L, Rapid synthesis of the X-linked mental retardation protein OPHN1 mediates mGluR-dependent LTD through interaction with the endocytic machinery. Neuron 72, 300–315 (2011). doi: 10.1016/j.neuron.2011.09.001; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Silva AJ, Kogan JH, Frankland PW, Kida S, CREB and memory. Annu. Rev. Neurosci 21, 127–148 (1998). doi: 10.1146/annurev.neuro.21.1.127; [DOI] [PubMed] [Google Scholar]
- 132.Chang RC, Wong AK, Ng HK, Hugon J. Phosphorylation of eukaryotic initiation factor-2α (eIF2α) is associated with neuronal degeneration in Alzheimer’s disease. Neuroreport 13, 2429–2432 (2002). doi: 10.1097/00001756-200212200-00011; [DOI] [PubMed] [Google Scholar]
- 133.Hoozemans JJ et al. , Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun 354, 707–711 (2007). doi: 10.1016/j.bbrc.2007.01043: [DOI] [PubMed] [Google Scholar]
- 134.Atkin JD et al. , Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis 30, 400–407 (2008). doi: 10.1016/j.nbd.200802.009; [DOI] [PubMed] [Google Scholar]
- 135.Smith HL, Mallucci GR The unfolded protein response: Mechanisms and therapy of neurodegeneration. Brain 139, 2113–2121 (2016). doi: 10.1093/brain/aww101; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Moon SL, Sonenberg N, Parker R, Neuronal Regulation of eIF2α Function in Health and Neurological Disorders. Trends Mol. Med 24, 575–589 (2018). doi: 10.1016/j.molmed.2018.04.001; [DOI] [PubMed] [Google Scholar]
- 137.Chou A et al. , Inhibition of the integrated stress response reverses cognitive deficits after traumatic brain injury. Proc. Natl. Acad. Sci. U.S.A 114, E6420–E6426 (2017). doi: 10.1073/pnas.1707661114; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sen T, Gupta R, Kaiser H, Sen N, Activation of PERK Elicits Memory Impairment through Inactivation of CREB and Downregulation of PSD95 After Traumatic Brain Injury. J. Neurosci 37, 5900–5911 (2017). doi: 10.1523/JNEUROSCI.2343-16.2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ma T et al. , Suppression of eIF2α kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci 16, 1299–1305 (2013). doi: 10.1038/nn.3486; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tibie M et al. , PKR knockout in the 5xFAD model of Alzheimer’s disease reveals beneficial effects on spatial memory and brain lesions. Aging Cell 18, e12887 (2019). doi: 10.1111/acel.12887; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hwang KD, Bak MS, Kim SJ, Rhee S, Lee YS, Restoring synaptic plasticity and memory in mouse models of Alzheimer’s disease by PKR inhibition. Mol. Brain 10, 57 (2017). doi: 10.1186/s13041-017-0338-3; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Segev Y et al. , PKR Inhibition Rescues Memory Deficit and ATF4 Overexpression in ApoE ε4 Human Replacement Mice. J. Neurosci 35. 12986–12993 (2015). doi: 10.1523/JNEUROSCI.5241-14.2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lourenco MV et al. , TNF-α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s β amyloid oligomers in mice and monkeys. Cell Metab 18, 831–843 (2013). doi: 10.1016/j.cmet.2013.11.002; [DOI] [PubMed] [Google Scholar]
- 144.Paesla K et al. , Limited effects of an eIF2αS51A allele on neurological impairments in the 5xFAD mouse model of Alzheimer’s disease. Neural Plast 2015, 825157 (2015). doi: 10.1155/2015/825157; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Devi L, Ohno M, Deletion of the eIF2α Kinase GCN2 fails to rescue the memory decline associated with Alzheimer’s disease. PLOS CNE 8, e77335 (2013). doi: 10.1371/journal.pone.0077335; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhu PJ et al. , Activation of the ISR mediates the behavioral and neurophysiological abnormalities in Down syndrome. Science 366,843–849 (2019). doi: 10.1126/science.aaw5185; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Dierssen M, Down syndrome: The brain in trisomic mode. Nat. Rev. Neurosci 13, 844–858 (2012). doi: 10.1038/nm3314; [DOI] [PubMed] [Google Scholar]
- 148.Moreno JA et al. , Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 485, 507–511 (2012). doi: 10.1038/nature11058; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Moreno JA et al. , Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med 5, 206ra138 (2013). doi: 10.1126/scitranslmed.3006767; [DOI] [PubMed] [Google Scholar]
- 150.Halliday M et al. , Repurposed drugs targeting eIF2&alpha:-P-mediated translational repression prevent neurodegeneration in mice. Brain 140, 1768–1783 (2017). doi: 10.1093/brain/awx074; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Celardo I et al. , Mitofusin mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis 7. e2271 (2016). doi: 10.1038/cddis.2016.173; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Radford H, Moreno JA, Verity N, Halliday M, Mallucci GR, PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol 130, 633–642 (2015). doi: 10.1007/s00401-015-1487-z; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Devi L, Ohno M, PERK mediates eIF2α phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiol. Aging 35, 2272–2281 (2014). doi: 10.1016/J.neurobiolaging.2014.04.031; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tronel C, Page G, Bodard S, Chalon S, Antier D, The specific PKR inhibitor C16 prevents apoptosis and IL-1β production in an acute excitotoxic rat model with a neuroinflammatory component. Neurochem. Int 64, 73–83 (2014). doi: 10.1016/j.neuint.2013.10.012; [DOI] [PubMed] [Google Scholar]
- 155.Carret-Rebillat AS et al. , Neuroinflammation and Aβ accumulation linked to systemic inflammation are decreased by genetic PKR down-regulation. Sci. Rep 5, 8489 (2015). doi: 10.1038/srep08489; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nalivaeva NN, Rybnikova EA, Editorial: Bran Hypoxia and Ischemia: New Insights Into Neurodegeneration and Neuroprotection. Front. Neurosci 13, 770 (2019). doi: 10.3389/fnins.2019.00770; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Paşca AM et al. , Human 3D cellular model of hypoxic brain injury of prematurity. Nat. Med 25, 784–791 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Li J et al. , Deletion of Tmtc4 activates the unfolded protein response and causes postnatal hearing loss. J. Clin. Invest 128, 5150–5162 (2018). doi: 10.1172/JCI97498; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lahammar M et al. , Dual leucine zipper kinase dependent PERK activation contributes to neuronal degeneration following insult. eLife 6, e20725 (2017). doi: 10.7554/eLife.20725; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.She P et al. , General control nonderepressible 2 (GCN2) kinase protects oligodendrocytes and white matter during branched-chain amino add deficiency in mice. J. Biol. Chem 288, 31250–31260 (2013). doi: 10.1074/jbc.M113.498469; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lin W et al. , The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J. Clin. Invest 117, 448–456 (2007). doi: 10.1172/JC129571; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.D’Antonio M et al. , Resetting translational homeostasis restores myelination in Charcot-Marie-Tooth disease type 1B mica. J. Exp. Med 210, 821–838 (2013). doi: 10.1084/jem.20122005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Pennuto M et al. , Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie- Tooth 1B mice. Neuron 57, 393–405 (2008). doi: 10.1016/j.neuron.2007.12.021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Das I et al. , Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 348, 239–242 (2015). doi: 10.1126/sdence.aaa4484; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Musner N et al. , Perk Ablation Ameliorates Myelination in S63del-Charcot-Marie-Tooth 1B Neuropathy. ASN Neuro 8. 1759091416642351 (2016). doi: 10.1177/1759091416642351; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Sidoli M et al. , Ablation of Perk in Schwann Cells Improves Myelination in the S63del Charcot-Marie-Tooth 1B Mouse. J. Neurosci 36, 11350–11361 (2016). doi: 10.1523/JNEUROSCI.1637-16.2016: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Dzhashiashvili Y, Monckton CP, Shah HS, Kunjamma RB, Popko B, The UPR-PERK pathway is not a promising therapeutic target for mutant SOD1-induced ALS. Neurobiol. Dis 127, 527–544 (2019). doi: 10.1016/j.nbd.2019.03.024; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Matus S, Lopez E, Valenzuela V, Nassif M, Hetz C, Functional contribution of the transcription factor ATF4 to the pathogenesis of amyotrophic lateral sclerosis. PLOS ONE 8, e66672 (2013). doi: 10.1371/journal.pone.0066672; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wang L, Popko B, Roos RP, An enhanced integrated stress response ameliorates mutant SOD1-induced ALS. Hum. Mol. Genet 23, 2629–2638 (2014). doi: 10.1093/hmg/ddt658: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wang L, Popko B, Tixier E, Roos RP, Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1 induced amyotrophic lateral sclerosis. Neurobiol. Dis 71, 317–324 (2014). doi: 10.1016/j.nbd.2014.08.010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Vieira FG et al. , Guanabenz Treatment Accelerates Disease in a Mutant SOD1 Mouse Model of ALS. PLOS ONE 10, e0135570 (2015). doi: 10.1371/journal.pone.0135570; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gao Y et al. , PERK is required in the adult pancreas and is essential for maintenance of glucose homeostasis. Mol. Cell. Biol 32, 5129–5139 (2012). doi: 10.1128/MCB.01009-12; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zhang W et al. , PERK EIF2AK3 control of pancreatic beta cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab 4, 491–497 (2006). doi: 10.1016/j.cmet.2006.11.002; [DOI] [PubMed] [Google Scholar]
- 174.Stanik J et al. , Neonatal hypoglycemia early-onset diabetes and hypopituitarism due to the mutation in EIF2S3 gene causing MEHMO syndrome. Physiol. Res 67, 331–337 (2018). doi: 10.33549/physiolres.933689; [DOI] [PubMed] [Google Scholar]
- 175.Zinszner H et al. , CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 12, 982–995 (1998). doi: 10.1101/gad.12.7.982; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zurita Rendón O, Shoubridge EA, LONP1 Is Required for Maturation of a Subset of Mitochondrial Proteins, and Its Loss Elicits an Integrated Stress Response. Mol. Cell. Biol 38, e00412–17 (2018). doi: 10.1128/MCB.00412-17; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Fiorese CJ et al. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol 26, 2037–2043 (2016). doi: 10.1016/j.cub.2016.06.002; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM, Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337, 587–590 (2012). doi: 10.1126/science.1223560; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Robichaud N, Sonenberg N, Ruggero D, Schneider RJ, Translational Control in Cancer. Cold Spring Harb. Perspect. Biol 11. a032896 (2019). doi: 10.1101/cshperspect.a032896; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Chu J, Cargnello M, Topisirovic I Pelletier J, Translation Initiation Factors: Reprogramming Protein Synthesis in Cancer. Trends Cell Biol 26, 918–933 (2016). doi: 10.1016/j.tcb.2016.06.005; [DOI] [PubMed] [Google Scholar]
- 181.Pavon-Eternod M, Gomes S, Rosner MR, Pan T, Overexpression of initiator methionine tRNA leads to global reprogramming of tRNA expression and increased proliferation in human epithelial cells. RNA 19, 461–466 (2013). doi: 10.1261/rna.037507.112; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Koromilas AE, Roy S, Barber GN, Katze MG, Sonenberg N, Malignant transformation by a mutant of the IFN inducible dsRNA dependent protein kinase. Science 257, 1685–1689 (1992). doi: 10.1126/science.1382315; [DOI] [PubMed] [Google Scholar]
- 183.Donzé O, Jagus R, Koromilas AE, Hershey JW, Sonenberg N, Abrogation of translation initiation factor eIF-2 phosphorylation causes malignant transformation of NIH 3T3 cells. EMBO J 14, 3828–3834 (1995). doi: 10.1002/j.l460-2075.1995.tb00052.x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Barber GN Wambach M, Thompson S, Jagus R, Katze MG, Mutants of the RNA-dependent protein kinase (PKR) lacking double-stranded RNA binding domain I can act as transdominant inhibitors and induce malignant transformation. Mol. Cell. Biol, 15, 3138–3146 (1995). doi: 10.1128/MCB.15.6.3138; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Meurs EF, Galabru J, Barber GN, Katze MG, Hovanessian AG, Tumor suppressor function of the interferon-induced double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. U.S.A 90, 232–236 (1993). doi: 10.1073/pnas.90.1.232; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Yang YL et al. , Deficient signaling in mice devoid of double stranded RNA-dependent protein kinase. EMBO J 14, 6095–6106 (1995). doi: 10.1002/j.1460-2075.1995.tb00300.x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Abraham N et al. , Characterization of transgenic mice with targeted disruption of the catalytic domain of the double-stranded RNA-dependent protein kinase. PKR. J. Biol. Chem 274, 5953–5962 (1999). doi: 10.1074/jbc.274.9.5953; [DOI] [PubMed] [Google Scholar]
- 188.Koromilas AE, M(en)TORship lessons on life and death by the integrated stress response. Biochim. Biophys. Acta Gen. Subj 1863, 644–649 (2019). doi: 10.1016/j.bbagen.2018.12.009; [DOI] [PubMed] [Google Scholar]
- 189.Bobrovnikova-Marjon E et al. , PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 29, 3881–3895 (2010). doi: 10.1038/onc.2010.153; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hart LS et al. , ER stress mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Invest 122, 4621–4634 (2012). doi: 10.1172/JCI62973; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Nagy P, Varga A, Pircs K, Hegedüs K, Juhász G, Myc-driven overgrowth requires unfolded protein response-mediated induction of autophagy and antioxidant responses in Drosophila melanogaster. PLOS Genet 9, e1003664 (2013). doi: 10.1371/journal.pgen.1003664; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Nguyen HG et al. , Development of a stress response therapy targeting aggressive prostate cancer. Sci. Transl. Med 10, eaar2036 (2018). doi: 10.1126/scitranslmed.aar2036; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Tameire F et al. , ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat. Cell Biol 21 889–899 (2019). doi: 10.1038/s41556-019-0347-9; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Maurin AC et al. , The GCN2 kinase biases feeding behavior to maintain amino acid homeostasis in omnivores. Cell Metab 1, 273–277 (2005). doi: 10.1016/j.cmet.2005.03.004; [DOI] [PubMed] [Google Scholar]
- 195.Hao S et al. , Uncharged tRNA and sensing of amino acid deficiency in mammalian piriform cortex. Science 307, 1776–1778 (2005). doi: 10.1126/science.1104882; [DOI] [PubMed] [Google Scholar]
- 196.Ye J et al. , The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J 29, 2082–2096 (2010). doi: 10.1038/emboj.2010.81; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Wang Y et al. , Amino acid deprivation promotes tumor angiogenesis through the GCN2/ATF4 pathway. Neoplasia 15, 989–997 (2013). doi: 10.1593/neo.13262; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Cláudio N Dalet A Gatti E Pierre P Mapping the crossroads of immune activation and cellular stress response pathways. EMBO J 32, 1214–1224 (2013). doi: 10.1038/emboj.2013.80; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Pulendran B The varieties of immunological experience: Of pathogens, stress, and dendritic cells. Annu. Rev. Immunol 33, 563–606 (2015). doi: 10.1146/annurev-immunol-020711-075049; [DOI] [PubMed] [Google Scholar]
- 200.Deng J et al. , Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell. Biol 24, 10161–10168 (2004). doi: 10.1128/MCB.24.23.10161-10168.2004; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Jiang HY Wek RC, Phosphorylation of the alpha-subunit of the eukaryotic initiation factor-2 (eIF2alpha) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J. Biol. Chem 280, 14189–14202 (2005). doi: 10.1074/jbc.M413660200; [DOI] [PubMed] [Google Scholar]
- 202.Abdel-Nour M et al. , The heme-regulated inhibitor is a cytosolic sensor of protein misfolding that controls innate immune signaling. Science 365, eaaw4144 (2019). doi: 10.1126/science.aaw4144; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Iwasaki Y et al. , Activating transcription factor 4 links metabolic stress to interleukin-6 expression in macrophages, Diabetes 63, 152–161 (2014). doi: 10.2337/db13-0757; [DOI] [PubMed] [Google Scholar]
- 204.Onat UI et al. , Intercepting the Lipid-Induced Integrated Stress Response Reduces Atherosclerosis. J. Am. Coll. Cardiol 73, 1149–1169 (2019). doi: 10.1016/j.jacc.2018.12.055; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lu B et al. , Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488, 670–674 (2012). doi: 10.1038/nature11290; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.D’Osualdo A et al. , Transcription Factor ATF4 Induces NLRP1 Inflammasome Expression during Endoplasmic Reticulum Stress. PLOS ONE 10, e0130635 (2015). doi: 10.1371/journal.pone.0130635; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Nakamura T et al. , Double stranded RNA dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell 140, 338–348 (2010). doi: 10.1016/j.cell.2010.01.001; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hu J et al., Global analysis of phosphorylation networks in humans. Biochim. Biophys. Acta 1844 (1 Pt B), 224–231 (2014). doi: 10.1016/j.bbapap.2013.03.009; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Cullinan SB et al. , Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol 23, 7198–7209 (2003). doi: 10.1128/MCB.23.20.7198-7209.2003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Gilbert LA et al. , Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661 (2014). doi: 10.1016/j.cell.2014.09.029; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Biever A et al. , Monosomes actively translate synaptic mRNAs in neuronal processes. Science 367, eaay4991 (2020). doi: 10.1126/science.aay4991; [DOI] [PubMed] [Google Scholar]