

Abstract
The TB Portals program is an international collaboration for the collection and dissemination of tuberculosis data from patient cases focused on drug resistance. The central database is a patient-oriented resource containing both patient and pathogen clinical and genomic information. Herein we provide a summary of the pathogen genomic data available through the TB Portals and show one potential application by examining patterns of genomic pairwise distances. Distributions of pairwise distances highlight overall patterns of genome variability within and between Mycobacterium tuberculosis phylogenomic lineages. Closely related isolates (based on whole-genome pairwise distances and time between sample collection dates) from different countries were identified as potential evidence of international transmission of drug-resistant tuberculosis. These high-level views of genomic relatedness provide information that can stimulate hypotheses for further and more detailed research.
Keywords: Mycobacterium tuberculosis, Multi-drug resistant, Extensively-drug resistant
1. Introduction
The TB Portals Program is an international collaboration (currently including 13 countries) whose participants typically are under a heavy burden of drug-resistant tuberculosis (DR-TB) (1). Several different terms are used to describe the extent of drug resistance in tuberculosis isolates. The World Health Organization defines multidrug-resistant TB (MDR-TB) as a positive TB culture demonstrating resistance to the first-line drugs rifampicin and isoniazid. The next defined level of drug resistance is pre-XDR (extensively drug resistant), which is MDR with additional resistance to one of the fluoroquinolone drugs (moxifloxicin or levofloxacin). Extensively drug-resistant TB (XDR-TB) is defined as MDR-TB with additional resistance to at least one fluoroquinolone and at least one of the antibiotics bedaquiline or linezolid(2). Other terms used to describe drug resistant tuberculosis are mono-resistant TB for cultures with demonstrated resistance to only one first-line antibiotic and poly-resistant TB when there is demonstrated resistances to multiple first-line drugs (rifampicin, isoniazid, pyrazinamide, and ethambutol) but not both rifampicin and isoniazid.
While drug-sensitive tuberculosis numbers are diminishing worldwide, and effectiveness of treatment is above 90%, the threat of drug-resistant tuberculosis is growing and the success rate of DR-TB treatment (usually expensive and toxic) is about 60% (2). To this end, one of the permanent goals of TB Portals is to collect, study, and make public a body of information about drug-resistant tuberculosis. A major part of this effort has been the establishment of the TB Portals database, an anonymized patient-centric resource containing multi-domain metadata, including patient images (chest x-rays and CT scans) and pathogen single-nucleotide polymorphisms (SNP) associated with drug resistance (DR), which are collected, curated, and made available to the research community. As part of this international collaboration the TB Portals program has collected and processed genome sequences for more than 2200 Mycobacterium tuberculosis (Mtb) samples.
Compared to drug-sensitive TB, DR-TB usually occurs at a lower frequency, although in some countries secondary TB infections are very likely to be drug-resistant (3). Building the capability to collect and manage a broad variety of patient and pathogen data was considered an important first step for understanding the ecology of DR-TB. Previous analyses of TB Portals data have included country-specific analyses of DR molecular evolution (4) and genomic evaluation of relapse/reinfection status (5), polyclonal infection among lung resection samples (6), clinical metadata correlation with treatment outcomes (7), and prediction of DR status from patient imaging data via machine learning (8).
As a demonstration of the utility of the breadth of genomic data maintained in TB Portals, we analyzed a cohort of all published and publicly available records with genomic data from the TB Portals Data Exploration POrTal (DEPOT) to assess patterns of diversity within and between countries and also phylogenomic lineages. For this cohort we calculated pairwise distances among the samples using SNP panels derived from full genome sequences. The combination of genomic variability with curated patient metadata (especially sample collection dates) allowed us to address one of the persistent problems of healthcare: tracking the sources of drug-resistant TB and identifying local and global pathways of DR-TB transmission.
2. Methods
2.1. Processing of SRA records to generate genomic SNP panels
A snakemake(9) pipeline was created to download genome sequence data files from the NCBI Sequence Read Archive (SRA) database (https://trace.ncbi.nlm.nih.gov/Traces/sra/). These files were then aligned to the standard Mtb H37Rv reference genome (NC_000962.3) using the BWA software (10). The resulting bam files were ordered and indexed with the samtools software (11) and then processed with the pilon software (12) to call variants and generate vcf files. Vcf files were annotated using the SnpEff software (13) and merged into a single vcf file which was then processed to include only variable sites. The multi-sample vcf file was also deposited in the TB Portals Genomic Analysis Portal (GAP) for use with genomic analysis tools. A summary distribution of phylogenomic sublineage frequencies by country was generated using the dplyr data wrangling and ggplot2 data visualization packages in the RStudio statistical analysis software.
2.2. Calculation of pairwise distances
The Plink2 (14) make-king-table function was used to calculate the number of pairwise SNP differences among all samples in our cohort. The KING robust kinship analysis (15) implemented in Plink2 was used to calculate the IBS0 statistic for each pair of samples to further characterize variation among Mtb isolates. Many of these records represent multiple samples from individual patients, including some from the same infection event, which could potentially bias downstream results. To control for this issue, we calculated pairwise distances between samples and only kept the most recent sample for cases where the distance among samples from the same patient was less than 10 SNPs.
The R package ggplot2 was used to generate frequency histograms of the filtered pairwise distance data. For samples with a pairwise distance of 10 SNPs or less the number of expected SNPs between closely related sample pairs was calculated using the lineage-specific substitution rates estimates of Menardo, et al. (16). All of these closely-related samples were from lineages 2 (East Asian) or 4 (Euro-American) and all pairs were from the same sublineage. The specific substitution rates used were the rates estimated by separate BEAST analyses using the 1/x prior on the clock rate on random collections of 300 samples from lineages 2.2.1 (Beijing) and 4 (Euro-American) (16).
3. Results
When this analysis was performed, 2082 genomes (from Azerbaijan, Belarus, Georgia, Kazakhstan, Moldova, and Romania) were available as “published data” for analysis and had genomic data available (presence/absence of DR SNPs) via the TB Portal. After filtering out samples from potentially the same infection event within a patient, 1884 genomes remained in the cohort analyzed for this study.
3.1. Phylogenomic sublineage distributions within countries
The distribution of phylogenomic sublineages (17) within sample cohorts for the countries included in this study are presented in Figure 1. Most countries had the majority of their samples belonging to sublineage 2.2.1. The Moldovan samples had a roughly equal distribution between sublineages 2.2.1 and 4.2.1. A previous analysis of the Moldovan samples (5) found that these sublineage 4.2.1 samples formed a shallow phylogenetic clade which was consistent with local person-to-person transmission of closely related DR isolates. In contrast, compared to samples from other countries, Romanian isolates were mostly represented by sublineages 4.1.2.1 and 4.8 with a reduced frequency of sublineage 2.2.1.
Figure 1.
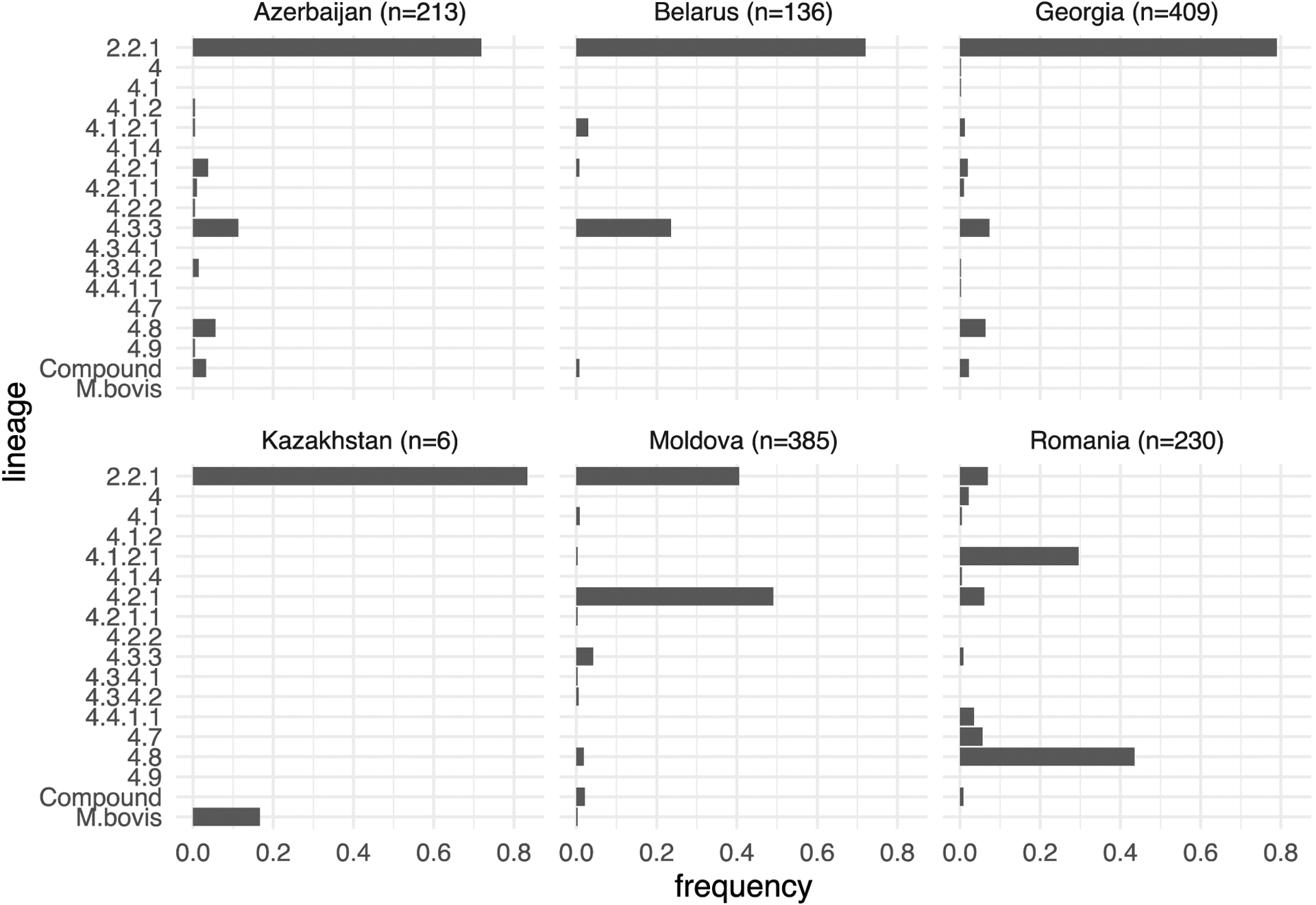
Frequency distributions of phylogenomic sublineages for each country in the data. Total number of samples per country is shown in the header for each individual plot. Samples assigned to multiple sublineages are collected into the category “Compound”.
3.2. Pairwise distance distributions for samples in TB Portals
For the full matrix of pairwise distances we calculated an overall frequency distribution and frequency distributions broken down by lineage and country. The overall distribution of pairwise distances was multimodal, with three distinct peaks at 60, 180, and 240 SNPs, and two large peaks at 1000 (900–1100) and 1500 (1350–1550) SNPs (Figure 2). In the smaller distance peaks (60, 180, and 240 SNPs) all the pairwise distances were between samples from the same lineage (or to a sample with a compound lineage that includes the main lineage). In the two peaks of greater distances (1000 and 1500 SNPs) there were very few pairs from within the same lineage.
Figure 2.
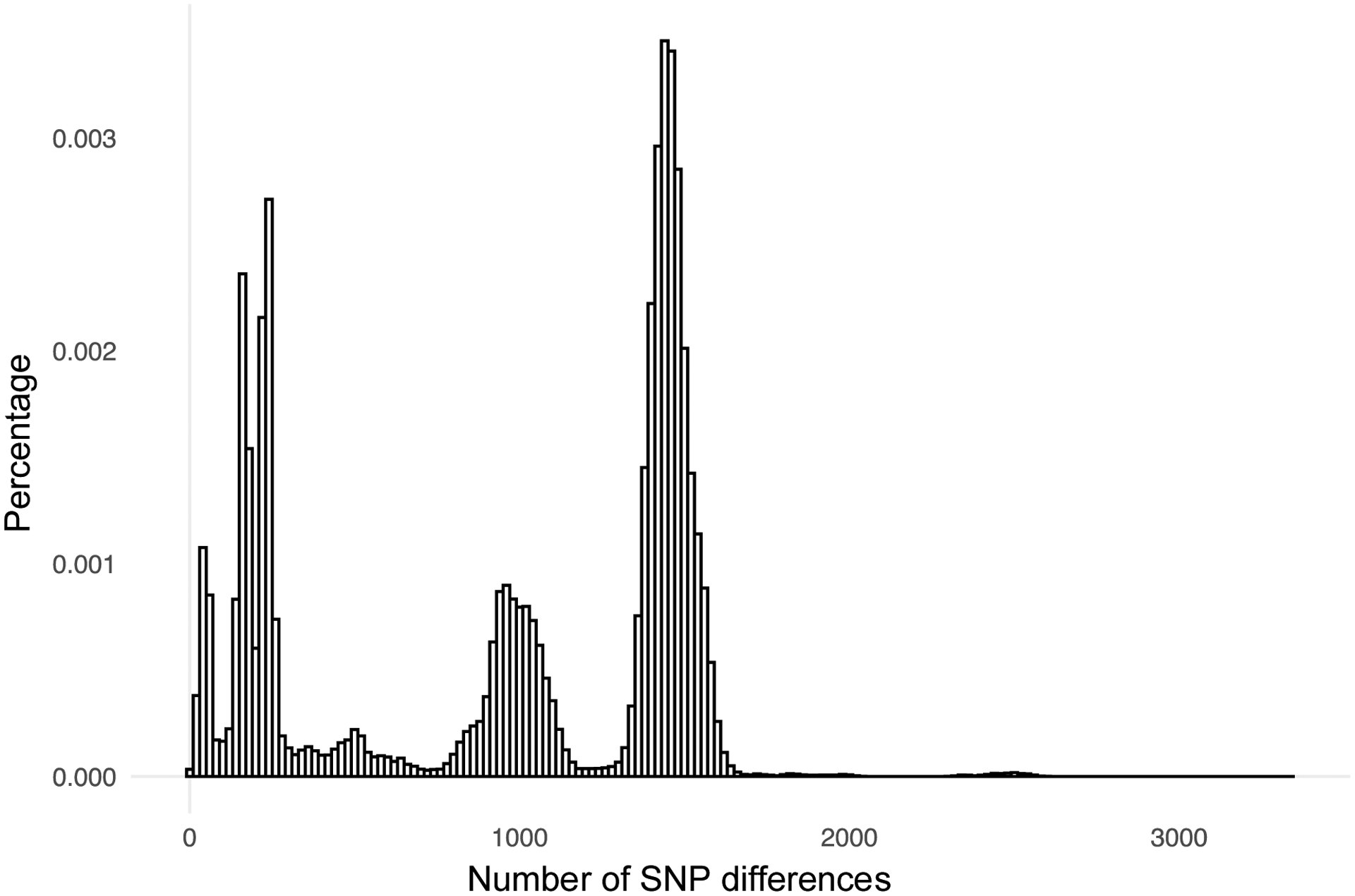
Distribution of pairwise distances for 2225 TB Portals samples. Distances represent number of different genomic SNPs between individual Mtb genomes. PE/PPE loci were not filtered out from the data used for these calculations.
The smaller and broader peak at 1000 SNPs in the overall distribution was primarily composed of paired samples from sublineages 4.1.2.1, 4.2.1, 4.3.3 or 4.8 (87.5%), including many 4.2.1+4.8 pairs. A small number of pairwise distances in this range involved samples from sublineages 4.2.1.1 or 4.8 with samples from sublineage 2.2.1 (9 total pairwise distances). These pairs involved a specific individual sublineage 4.2.1.1 or 4.8 sample with multiple sublineage 2.2.1 samples. In each of the individual country distributions (Supplemental Figure 1) this peak at 1000 SNPs was also present and was the largest peak in the pairwise distance distribution for samples from Romania which reflects the overall predominance of lineage 4 isolates in the Romanian cohort. This peak also contained three instances of pairwise distances between samples from the same lineage 4 sublineages. These associations are described in more detail below.
The large peak at 1500 SNPs was predominantly composed of sublineage 2.2.1 samples paired with samples from lineage 4 sublineages (49.7% of all pairwise distances, with the next highest sublineage at 17.5%). The peak at 1500 SNPs contained two instances of distances between samples from within the same sublineage. These two cases were due to two individual samples, one from sublineage 4.3.3 and the other from sublineage 4.8, paired with multiple samples from the same sublineage. Further investigation of these isolates indicated that these large distances were not due to data quality issues or sample contamination but additional analysis of the cause was beyond the scope of this analysis.
The distributions of pairwise distances within countries (Supplementary Figure 1) followed the overall pattern of pairwise distances. For countries with a substantial number of lineage 2 samples there were multiple peaks at small distances and a large peak centered around a distance of 1500 SNPs. As with the overall distribution, these peaks represented pairwise distances within lineage 2 and between lineage 2 and lineage 4 samples, respectively. The distribution for samples from Kazakhstan was qualitatively different due to the low number of samples from that country. The peak at 1500 SNPs was much smaller for the Romanian isolates due to the overall lower frequency of lineage 2 in the samples from this country.
The distributions of pairwise distances/differences within phylogenomic sublineages were consistently multimodal (Supplemental Figure 2). As most lineage 2 samples were from sublineage 2.2.1 (1006 of 1044 samples) these pairwise distance distributions were nearly identical. They had the same median value (189 SNPs) but the overall lineage 2 distribution had a larger maximum value (889 SNPs versus 728 SNPs for sublineage 2.2.1). Despite the overwhelming number of samples from sublineage 2.2.1 (906, 48.1% of all samples) the maximum pairwise distance was only 889 SNPs, indicating that the lineage 2 isolates are overall less divergent in comparison to the lineage 4 isolates present in our samples.
The lineage 4 pairwise distance distribution had a large peak centered at 1000 SNPs. This peak was predominantly made up of pairwise distances of sublineage 4.2.1 and 4.8 samples with other lineage 4 samples (49% of pairwise distances in this peak). Three lineage 4 samples (one from lineage 4 [no sublineage designation], one from sublineage 4.3.3, and one from sublineage 4.8) had pairwise distances of 900–1100 SNPs with samples of the same sublineage. The sample from sublineage 4.8 was the same sample having within-sublineage distances in the peak at 1500 SNPs. Similar to what was found previously these large within-sublineage distances for these samples do not appear to be due to sequence data quality or contamination and additional analysis of the cause of these differences was beyond the scope of this analysis.
Sublineage 4.2.1 was the least divergent sublineage, with a maximum value of 547 SNPs and a median of 43 SNPs. This low median value is the result of most samples having a pairwise distance of 20–49 SNPs. The majority of samples from this sublineage are from Moldova (214 of 275 samples). These Moldovan sublineage 4.2.1 isolates were part of a very closely related clade of samples that were consistent with high levels of person-to-person transmission of one clonal lineage or a few closely related lineages of DR TB (5). As these samples were selected as part of a study of relapse vs. reinfection in persistent DR TB, there are many paired samples from single patients with recurring DR TB diagnoses in the TB Portals database. Due to the filtering of closely related samples from the same patient, the distribution in Supplementary Figure 2 does not reflect single-patient relapse cases but does include closely related samples from separate patients.
The pairwise distance distributions for sublineages 4.1.2.1, 4.3.3, and 4.8 had much larger maximum (772 SNPs, 1849 SNPs, and 1561 SNPs, respectively) and median (256 SNPs, 168 SNPs, and 412 SNPs, respectively) values, indicating higher levels of among sample diversity in these sublineages. The sublineage 4.8 samples had the most divergent distribution of pairwise differences with a large median (412 SNPs), although the maximum value (1561 SNPs) was just less than the maximum values for sublineage 4.3.3 (1849 SNPs). Based on the median value, sublineage 4.8 has the most divergent isolates among the samples in the TB Portals database.
3.3. Relationships of closely-related samples
We then filtered the complete matrix of distances to only include samples separated by 10 genomic SNPs or less to investigate patterns among closely related samples (Figure 3). Several very closely related and highly internally connected clusters emerged from this analysis. These clusters generally consist of samples from one country, but there were exceptions. There was one sample from Romania that clustered within the main cluster of samples from Moldova. No other Romanian samples had close relationships with samples from other countries in the database. Two clusters of samples from Belarus had close connections to separate samples from Azerbaijan. In the center of Figure 3 there was a loose cluster consisting mostly of samples from Georgia but also including individual samples from Belarus, Azerbaijan, and Moldova. This cluster also had a connection through one sample to a tight cluster of several Georgian samples. For samples from Belarus and Moldova it was shown that these closely related samples were consistent with person-to-person transmission of DR TB (4,5).
Figure 3.
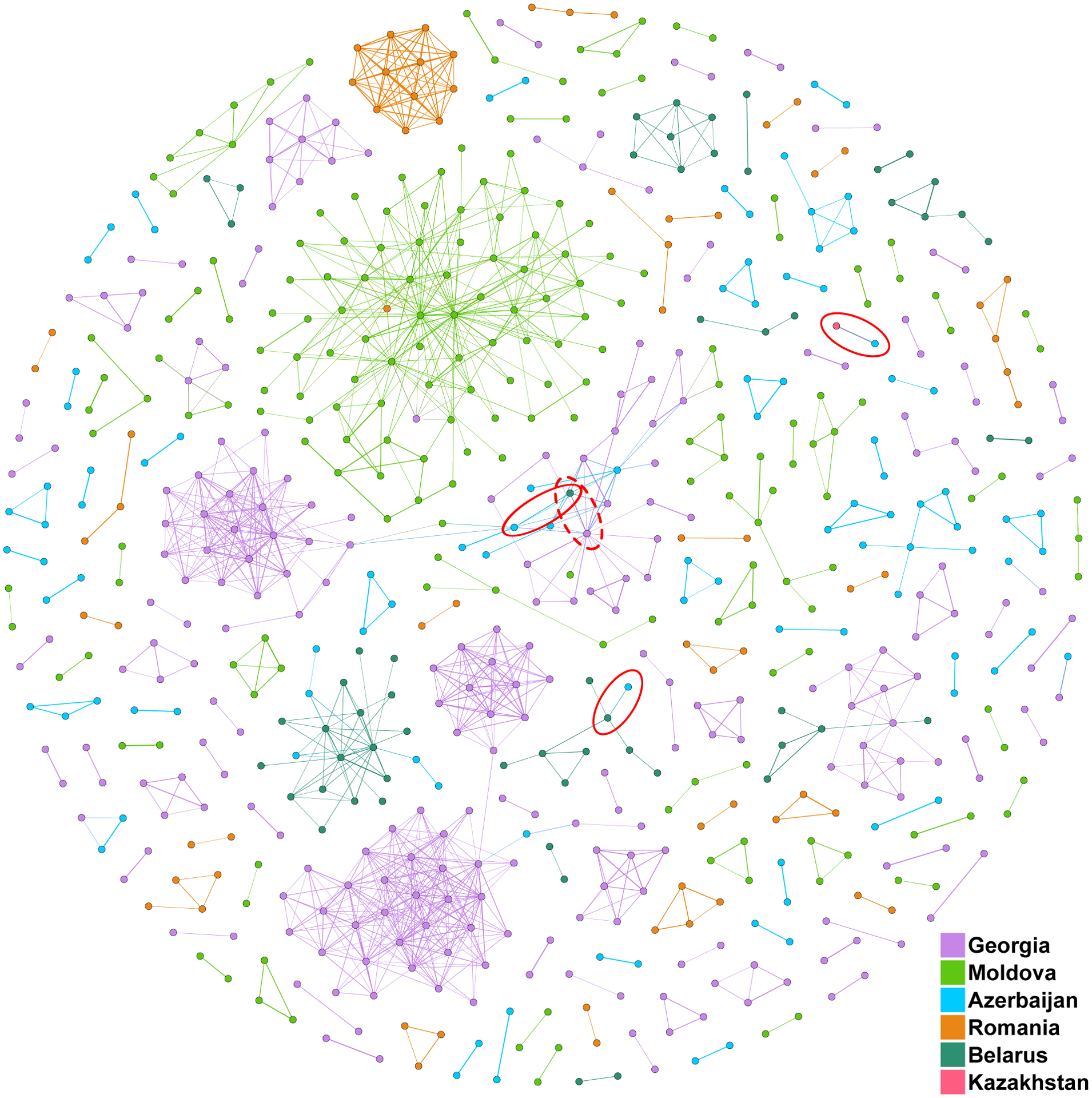
Network diagram of samples separated by 10 genomic SNPs or less. Pairs of samples with number of SNP difference consistent with direct transmission indicated by red ovals. The dotted oval encloses the highly similar pair that includes the sample from Georgia that is potentially a mixed-infection sample.
For this analysis we identified several samples from different countries that were also very closely related (Table 1). To better understand the relationships among these samples, we calculated the lower and upper bounds of expected number of SNPs between samples using the sample collection dates and lineage-specific substitution rates (16). In most cases the number of expected SNPs between closely related pairs was much smaller than the number of SNPs between the two genomes. This pattern is consistent with these samples being related through a common ancestor that is older than the current set of samples. In four cases the actual number of SNPs fell within the range of expected number of SNPs calculated using the 95% highest posterior density of the estimated lineage-specific substitution rates (16). This pattern is consistent with a close genealogical relationship among the samples from these patients. These four pairs of samples are highlighted in Table 1.
Table 1.
Closely related pairs of samples from separate countries. Column “D” is the number of genomic SNPs between this pair of samples. “Lin” indicates phylogenomic lineage for both samples in the pair. “DR Class” indicates drug resistance classification. The substitution rate used for Lineage 2.2.1 samples was 2.0175×10−7 substitutions/site/year ([1.1115×10−7,3.003×10−7] 95% HPD). The substitution rate used for Lineage 4 samples was 5.7694×10−8 substitutions/site/year ([4.0324×10−8,7.3311×10−8] 95% HPD). “Exp SNPs” are the lower and upper bounds of the 95% HPD for expected number of SNPs between pairs of samples. Boldface rows are paired samples where number of genomic SNPs between samples is within the 95% HPD limits of the expected number of SNPs for a direct ancestral relationship.
| SRR ID1 | SRR ID2 | D | Country 1 | Country 2 | Lin | DR Class 1 | DR Class 2 | ΔT - years | Exp SNPs, Lower | Exp SNPs, Upper |
|---|---|---|---|---|---|---|---|---|---|---|
| SRR7655790 | SRR10303352 | 1 | Azerbaijan | Kazakhstan | 2.2.1 | MDR | MDR | 0.79 | 0.77 | 2.09 |
| SRR7654034 | SRR11033773 | 2 | Azerbaijan | Georgia | 4.8 | Sensitive | Sensitive | 1.19 | 0.42 | 0.77 |
| SRR6356966 | SRR10397096 | 3 | Azerbaijan | Georgia | 2.2.1 | MDR | MDR | 0.95 | 0.93 | 2.51 |
| SRR1159053 | SRR10397096 * | 5 | Belarus | Georgia | 2.2.1 | XDR | MDR | 3.30 | 3.23 | 8.73 |
| SRR7516307 | SRR10379886 | 6 | Georgia | Moldova | 2.2.1 | MDR | MDR | 2.15 | 2.11 | 5.69 |
| SRR1159053 | SRR6384965 | 7 | Belarus | Azerbaijan | 2.2.1 | XDR | MDR | 4.50 | 4.41 | 11.90 |
| SRR5153222 | SRR5153718 | 7 | Georgia | Azerbaijan | 2.2.1 | MDR | MDR | 0.09 | 0.09 | 0.23 |
| SRR5153222 | SRR6356966 | 7 | Georgia | Azerbaijan | 2.2.1 | MDR | MDR | 0.19 | 0.19 | 0.51 |
| SRR6356966 | SRR5153279 | 7 | Azerbaijan | Georgia | 2.2.1 | MDR | MDR | 0.04 | 0.04 | 0.10 |
| SRR10397096 | SRR6384965 | 7 | Georgia | Azerbaijan | 2.2.1 | MDR | MDR | 1.20 | 1.17 | 3.17 |
| SRR1159053 | SRR5153222 | 7 | Belarus | Georgia | 2.2.1 | XDR | MDR | 2.16 | 2.11 | 5.71 |
| SRR1159053 | SRR5153279 | 7 | Belarus | Georgia | 2.2.1 | XDR | MDR | 2.39 | 2.34 | 6.33 |
| SRR5153830 | SRR7516307 | 7 | Moldova | Georgia | 2.2.1 | MDR | MDR | 0.51 | 0.50 | 1.35 |
| SRR10379966 | SRR10397218 | 7 | Moldova | Georgia | 4.2.1 | MDR | MDR | 0.48 | 0.17 | 0.31 |
| SRR7516307 | SRR10380089 | 7 | Georgia | Moldova | 2.2.1 | MDR | MDR | 1.96 | 1.92 | 5.18 |
| SRR10380072 | SRR9738492 | 7 | Moldova | Romania | 4.2.1 | MDR | MDR | 4.77 | 1.69 | 3.08 |
| SRR3743405 | SRR9738492 | 7 | Moldova | Romania | 4.2.1 | MDR | MDR | 1.05 | 0.37 | 0.68 |
| SRR1159053 | SRR6356966 | 8 | Belarus | Azerbaijan | 2.2.1 | XDR | MDR | 2.36 | 2.30 | 6.23 |
| SRR1163177 | SRR7655469 | 8 | Belarus | Azerbaijan | 2.2.1 | MDR | MDR | 6.59 | 6.45 | 17.43 |
| SRR5153718 | SRR5153279 | 8 | Azerbaijan | Georgia | 2.2.1 | MDR | MDR | 0.15 | 0.14 | 0.38 |
| SRR5153279 | SRR6384965 | 8 | Georgia | Azerbaijan | 2.2.1 | MDR | MDR | 2.11 | 2.06 | 5.57 |
| SRR6356966 | SRR11033734 | 8 | Azerbaijan | Georgia | 2.2.1 | MDR | Poly DR | 2.47 | 2.42 | 6.53 |
| SRR6807742 | SRR9738492 | 8 | Moldova | Romania | 4.2.1 | MDR | MDR | 3.83 | 1.36 | 2.47 |
| SRR1163140 | SRR5153707 | 9 | Belarus | Azerbaijan | 4.3.3 | XDR | MDR | 2.57 | 0.91 | 1.66 |
| SRR1159053 | SRR5153718 | 9 | Belarus | Azerbaijan | 2.2.1 | XDR | MDR | 2.25 | 2.20 | 5.94 |
| SRR1163140 | SRR7655952 | 9 | Belarus | Azerbaijan | 4.3.3 | XDR | MDR | 4.97 | 1.76 | 3.21 |
| SRR1163140 | SRR7657747 | 9 | Belarus | Azerbaijan | 4.3.3 | XDR | MDR | 4.97 | 1.76 | 3.20 |
| SRR5153329 | SRR6356966 | 9 | Georgia | Azerbaijan | 2.2.1 | XDR | MDR | 0.51 | 0.50 | 1.35 |
| SRR5153619 | SRR6356977 | 9 | Georgia | Azerbaijan | 2.2.1 | XDR | XDR | 0.38 | 0.37 | 1.01 |
| SRR7516458 | SRR6357007 | 9 | Georgia | Azerbaijan | 2.2.1 | XDR | MDR | 0.48 | 0.47 | 1.28 |
| SRR6384965 | SRR10397106 | 9 | Azerbaijan | Georgia | 2.2.1 | MDR | MDR | 0.23 | 0.23 | 0.62 |
| SRR10380139 | SRR10397218 | 9 | Moldova | Georgia | 4.2.1 | MDR | MDR | 0.71 | 0.25 |
0.46 |
| SRR11033734 | SRR10380214 | 9 | Georgia | Moldova | 2.2.1 | Poly DR | MDR | 0.54 | 0.53 | 1.43 |
| SRR10380234 | SRR6384965 | 9 | Moldova | Azerbaijan | 2.2.1 | MDR | MDR | 2.82 | 2.75 | 7.44 |
| SRR5153877 | SRR9738492 | 9 | Moldova | Romania | 4.2.1 | MDR | MDR | 1.15 | 0.41 | 0.74 |
| SRR6807719 | SRR9738492 | 9 | Moldova | Romania | 4.2.1 | MDR | MDR | 2.98 | 1.06 | 1.92 |
| SRR1159002 | SRR7655952 | 10 | Belarus | Azerbaijan | 4.3.3 | MDR | MDR | 5.90 | 2.09 | 3.81 |
| SRR1159290 | SRR7655952 | 10 | Belarus | Azerbaijan | 4.3.3 | MDR | MDR | 4.71 | 1.67 | 3.04 |
| SRR1159002 | SRR7657747 | 10 | Belarus | Azerbaijan | 4.3.3 | MDR | MDR | 5.90 | 2.09 | 3.80 |
| SRR1159290 | SRR7657747 | 10 | Belarus | Azerbaijan | 4.3.3 | MDR | MDR | 4.71 | 1.67 | 3.04 |
| SRR7516382 | SRR6356982 | 10 | Georgia | Azerbaijan | 2.2.1 | Sensitive | Sensitive | 0.59 | 0.58 | 1.57 |
| SRR6356988 | SRR7516382 | 10 | Azerbaijan | Georgia | 2.2.1 | MDR | Sensitive | 2.63 | 2.58 | 6.96 |
| SRR7516399 | SRR6356977 | 10 | Georgia | Azerbaijan | 2.2.1 | MDR | XDR | 0.46 | 0.45 | 1.22 |
| SRR5153718 | SRR10397096 | 10 | Azerbaijan | Georgia | 2.2.1 | MDR | MDR | 1.05 | 1.03 | 2.79 |
| SRR6356966 | SRR11033716 | 10 | Azerbaijan | Georgia | 2.2.1 | MDR | MDR | 2.80 | 2.74 | 7.39 |
| SRR6807730 | SRR10397096 | 10 | Moldova | Georgia | 2.2.1 | Sensitive | MDR | 3.29 | 3.22 | 8.70 |
| SRR10380234 | SRR10397106 | 10 | Moldova | Georgia | 2.2.1 | MDR | MDR | 3.05 | 2.98 | 8.06 |
| SRR5153860 | SRR10397218 | 10 | Moldova | Georgia | 4.2.1 | MDR | MDR | 2.18 | 0.77 | 1.40 |
| SRR10379908 | SRR10397218 | 10 | Moldova | Georgia | 4.2.1 | MDR | MDR | 1.73 | 0.61 | 1.11 |
| SRR10380080 | SRR10397218 | 10 | Moldova | Georgia | 4.2.1 | MDR | MDR | 4.01 | 1.42 | 2.59 |
| SRR10380120 | SRR10397218 | 10 | Moldova | Georgia | 4.2.1 | MDR | MDR | 2.90 | 1.03 | 1.87 |
| SRR11033734 | SRR10380199 | 10 | Georgia | Moldova | 2.2.1 | Poly DR | Mono DR | 0.58 | 0.57 | 1.54 |
| SRR7516428 | SRR10379886 | 10 | Georgia | Moldova | 2.2.1 | Sensitive | MDR | 1.67 | 1.64 | 4.42 |
| SRR9738492 | SRR10379966 | 10 | Romania | Moldova | 4.2.1 | MDR | MDR | 0.70 | 0.25 | 0.45 |
| SRR10380160 | SRR9738492 | 10 | Moldova | Romania | 4.2.1 | MDR | MDR | 0.51 | 0.18 | 0.33 |
| SRR10380245 | SRR9738492 | 10 | Moldova | Romania | 4.2.1 | XDR | MDR | 4.83 | 1.71 | 3.12 |
The asterisk (*) indicates the sample with a possible mixed infection.
Closer inspection of these four pairs of samples from Table 1 found that they also had very similar DR SNP panels. Two of the pairs had identical SNP panels. The sample pair from Azerbaijan and Kazakhstan (SRR7655790 and SRR10303352, respectively) was separated by less than one year (288 days) and had identical DR SNPs. The remaining three pairs consisted of two older samples from Belarus (SRR1159053 and SRR1163177) with one newer sample from Georgia (SRR10397096) and two from Azerbaijan (SRR6384965 and SRR7655469). The older sample from Belarus that was very similar to a single sample from Azerbaijan (SRR1163177 and SRR7655469, respectively) was separated by 6.6 years but still had an identical panel of DR SNPs. The other older sample from Belarus (SRR1159053) was very similar to two samples from Georgia (SRR10397096) and Azerbaijan (SRR6384965). The elapsed time between this Belarus sample and the Georgia sample was 3.3 years, while the Azerbaijan sample was collected 4.5 years after the Belarus sample. This Belarus sample had the katG S315T and rpoB S450L DR SNPs. The newer Azerbaijan sample additionally had the gyrA D94G fluoroquinalone DR SNP. The Georgia sample had a complex genotype. It had multiple DR SNPs including the katG S315T and rpoB S450L SNPs but all the DR SNPs in this sample were present at intermediate frequencies (55.2%−60.5% of the reads). This pattern of nearly equal frequencies of reads across DR SNP loci in the sample is consistent with this sample having a mixed infection. Without further sequencing efforts it is impossible to determine the exact DR SNP status of the individual strains present in this sample. It should also be mentioned that if there were extraneous factors that could have increased substitution rates (such as hitchhiking of genomic SNPs with DR SNPs under selection due to therapy (18)) then there could be additional candidates for direct transmission across international borders.
Overall, we have found that magnitudes of pairwise distance among Mtb genomes are due to differences among lineages. Interestingly, some sublineages within lineage 4 are as divergent from each other as they are from lineage 2. There are also some rare cases where individual isolates are extremely divergent from members of their same phylogenomic lineage. The use of full genome sequence data with curated isolate metadata, particularly sample collection date, allow the identification of isolates from different countries that are closely linked genealogically. This is strong evidence for international transmission of DR TB. These relationships would be difficult to ascertain from data collected under a more limited geographic, genealogical, or temporal scope.
4. Discussion
The samples deposited in the TB Portals database provide researchers the basic resources to investigate broad patterns of pathogen genetic variation and how it correlates with clinical and phenotypic aspects of these samples. Since one aspect of these samples provided by individual countries is their retrospective analysis as part of local research initiatives the samples will reflect individual donors’ research foci. This can lead to subtle biases in the data, such as the presence of multiple samples from individual patients when the time-course of infection is of interest. Factors such as this must be taken into account when performing broader analyses of TB Portals data.
For these types of analyses multiple instances of serial samples from single patients could influence the results and must be accommodated. As an example, for our analysis of the pairwise distances this bias should skew the distribution of pairwise differences, inflating the peak at lower values. Even after controlling for relapse cases (removing one or more samples from a single patient if the number of genomic SNP differences was < 10 SNPs) large peaks in the frequency distribution at small distances were still present. These peaks are due to pairwise relationships among samples within the same phylogenomic lineage, especially lineage 2. Within the TB Portals samples many of these close relationships are due to samples consistent with high levels of person-to-person transmission in individual countries. Previous phylogenomic analyses of the Belarus and Moldova samples found closely related clusters which are consistent with this phenomenon (4,5). Interestingly, evaluation of samples across the entire database found pairs of samples that were consistent with close transmission but were collected in different countries. Therefore, our genomic analysis identified several cases of transmission of specific drug-resistant TB lineages across international borders. In spite of the potential bias in TB Portals samples due to emphasis on local DR outbreaks we still see obvious peaks around 1500 SNPs difference because these depositions still contain samples across divergent lineages (lineage 2 and lineage 4), and this is reflected in all of the single-country plots (Supplemental Figure 1). The presence of specific biases in the samples from individual collaborators did not negate other general patterns that were present more broadly through the cohort.
The Mtb genome is notorious for having a class of loci for which aligning typical short-read genomic data are difficult. These loci, termed PE/PPE loci, encode for proteins carrying proline-glutamate (PE) and proline-proline-glutamate (PPE) repeat motifs at their N terminus and they make up approximately 10% of the Mtb genome(19). The combination of repetitive elements, sequence conservation, and high GC content makes mapping of reads to these regions problematic. Typically, these loci are masked from Mtb genomic analyses due to these complications. This filtering step could potentially bias any results based on genomic distances by excluding the variation at these loci. In our pairwise distance analysis we did not exclude these regions so the distances reported include their variation. However, the problematic alignment of short reads at many of these loci could lead to inaccuracies in the estimation of SNP distances due to read misalignments(20). Our experience has been that the inaccuracies in pairwise distances due to misalignment of reads at PE/PPE loci is small with respect to the additional information gained by including these loci. Recent advances in sequencing technology, especially with regarding long-read sequencing, show promise in resolving many of these issues.
Another aspect of the pairwise distance analysis is the presence of ambiguous nucleotide calls in the data. Since the Mtb genome is haploid, these ambiguities will be the result of sequencing errors or pathogen population structure in the sample. Sequencing errors are controlled by the quality assurance steps of the data processing pipeline so are considered to be of minimal impact. The quasispecies nature of infections is the major source of ambiguities in these data (21). In one case, one of the samples identified as being closely related to samples from other countries was found to have majority nucleotide calls at DR loci on the order of 50–60% of the reads at these sites. This can be an issue depending on how the distance calculation algorithm deals with ambiguity codes in the nucleotide sequences.
5. Conclusions
Bryant, et al. state that the “establishment of whole-genome databases will further enhance the possibility to compare samples to exclude or propose transmission.”(22) Tuberculosis is one of many diseases that are exacerbated by the increasing threat of drug-resistant pathogens. Understanding and documenting the molecular basis of drug-resistance on a broad scale would allow the fine tuning of diagnostics and treatment of evolving pathogens. Continuous full-genome sequencing to monitor for new variants of pathogens is essential for development of new drugs, vaccines and diagnostics. The TB Portals is an important resource for the investigation of M. tuberculosis molecular evolution as it connects a curated database of clinical and image data with publicly available whole-genome sequence records. The TB Portals does not store fully assembled M. tuberculosis genomes, we instead compute and make available all DR SNP data, complementing the submitted DST tests results with genomic-wide analysis. For all TB Portals sequencing projects, the raw genome sequence data files are deposited in the publicly accessible SRA database. The breadth of the samples in the TB Portals database allows the discovery of heretofore unrecognized genomic relationships among isolates across geographic regions (the breadth of which will increase with future depositions). The availability of complete genomes with associated sample collection dates allows for the inference of genealogical connections among the samples deposited in the TB Portals database. The collection of DR SNP data, in conjunction with clinical and other metadata curated in the TB Portals, provides the research community with an extraordinary resource that can be used for retrospective pathogen genomic analysis across broad geographic, temporal, and evolutionary perspectives. Large, broadly sampled databases of pathogen genomic sequence data, when combined with other dimensions of these samples such as clinical and/or image data, facilitate the investigation of epidemiological hypotheses.
Supplementary Material
Acknowledgement
We would like to thank Madeline Galac, Hernan Lorenzi, David Stern, Mariam Quiñones and an anonymous reviewer for providing valuable suggestions that greatly improved this manuscript.
Funding
KW and BJ are supported with Federal funds from the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health, Department of Health and Human Services under BCBB Support Services Contract HHSN316201300006W/HHSN27200002 to MSC, Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Data
Supplementary data are available at online.
Conflict of interest
The authors report no financial interests or connections, direct or indirect, or other situations that might raise the question of bias in the work reported in this manuscript.
Data Availability
The main TB Portals website is freely available at https://tbportals.niaid.nih.gov/. Multi-domain data analysis can be performed through the TB Portals DEPOT at https://depot.tbportals.niaid.nih.gov/. Genomic SNP analysis can be performed through the TB Portals GAP at https://gap.tbportals.niaid.nih.gov/#!/dashboard/home.
References
- 1.Rosenthal A, Gabrielian A, Engle E, Hurt DE, Alexandru S, Crudu V, Sergueev E, Kirichenko V, Lapitskii V, Snezhko E et al. (2017) The TB Portals: an Open-Access, Web-Based Platform for Global Drug-Resistant-Tuberculosis Data Sharing and Analysis. J Clin Microbiol, 55, 3267–3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(2021) Global Tuberculosis Report. World Health Organization, Geneva. [Google Scholar]
- 3.Skrahina A, Hurevich H, Zalutskaya A, Sahalchyk E, Astrauko A, Hoffner S, Rusovich V, Dadu A, de Colombani P, Dara M et al. (2013) Multidrug-resistant tuberculosis in Belarus: the size of the problem and associated risk factors. Bull World Health Organ, 91, 36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wollenberg KR, Desjardins CA, Zalutskaya A, Slodovnikova V, Oler AJ, Quinones M, Abeel T, Chapman SB, Tartakovsky M, Gabrielian A et al. (2017) Whole-Genome Sequencing of Mycobacterium tuberculosis Provides Insight into the Evolution and Genetic Composition of Drug-Resistant Tuberculosis in Belarus. J Clin Microbiol, 55, 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wollenberg K, Harris M, Gabrielian A, Ciobanu N, Chesov D, Long A, Taaffe J, Hurt D, Rosenthal A, Tartakovsky M et al. (2020) A retrospective genomic analysis of drug-resistant strains of M. tuberculosis in a high-burden setting, with an emphasis on comparative diagnostics and reactivation and reinfection status. BMC Infect Dis, 20, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moreno-Molina M, Shubladze N, Khurtsilava I, Avaliani Z, Bablishvili N, Torres-Puente M, Villamayor L, Gabrielian A, Rosenthal A, Vilaplana C et al. (2021) Genomic analyses of Mycobacterium tuberculosis from human lung resections reveal a high frequency of polyclonal infections. Nat Commun, 12, 2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenfeld G, Gabrielian A, Wang Q, Gu J, Hurt DE, Long A and Rosenthal A (2021) Radiologist observations of computed tomography (CT) images predict treatment outcome in TB Portals, a real-world database of tuberculosis (TB) cases. PLoS One, 16, e0247906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang F, Yu H, Kantipudi K, Karki M, Kassim YM, Rosenthal A, Hurt DE, Yaniv Z and Jaeger S (2021) Differentiating between drug-sensitive and drug-resistant tuberculosis with machine learning for clinical and radiological features. Quant Imag Med Surg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Molder F, Jablonski KP, Letcher B, Hall MB, Tomkins-Tinch CH, Sochat V, Forster J, Lee S, Twardziok SO, Kanitz A et al. (2021) Sustainable data analysis with Snakemake. F1000Res, 10, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H and Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R and Genome Project Data Processing, S. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK et al. (2014) Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One, 9, e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X and Ruden DM (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin), 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM and Lee JJ (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience, 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M and Chen WM (2010) Robust relationship inference in genome-wide association studies. Bioinformatics, 26, 2867–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Menardo F, Duchene S, Brites D and Gagneux S (2019) The molecular clock of Mycobacterium tuberculosis. PLoS Pathog, 15, e1008067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coll F, McNerney R, Guerra-Assuncao JA, Glynn JR, Perdigao J, Viveiros M, Portugal I, Pain A, Martin N and Clark TG (2014) A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat Commun, 5, 4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eldholm V, Norheim G, von der Lippe B, Kinander W, Dahle UR, Caugant DA, Mannsaker T, Mengshoel AT, Dyrhol-Riise AM and Balloux F (2014) Evolution of extensively drug-resistant Mycobacterium tuberculosis from a susceptible ancestor in a single patient. Genome Biol, 15, 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ates LS (2020) New insights into the mycobacterial PE and PPE proteins provide a framework for future research. Mol Microbiol, 113, 4–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elghraoui A, Modlin SJ and Valafar F (2017) SMRT genome assembly corrects reference errors, resolving the genetic basis of virulence in Mycobacterium tuberculosis. BMC Genomics, 18, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schurch AC, Kremer K, Kiers A, Daviena O, Boeree MJ, Siezen RJ, Smith NH and van Soolingen D (2010) The tempo and mode of molecular evolution of Mycobacterium tuberculosis at patient-to-patient scale. Infect Genet Evol, 10, 108–114. [DOI] [PubMed] [Google Scholar]
- 22.Bryant JM, Schurch AC, van Deutekom H, Harris SR, de Beer JL, de Jager V, Kremer K, van Hijum SA, Siezen RJ, Borgdorff M et al. (2013) Inferring patient to patient transmission of Mycobacterium tuberculosis from whole genome sequencing data. BMC Infect Dis, 13, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The main TB Portals website is freely available at https://tbportals.niaid.nih.gov/. Multi-domain data analysis can be performed through the TB Portals DEPOT at https://depot.tbportals.niaid.nih.gov/. Genomic SNP analysis can be performed through the TB Portals GAP at https://gap.tbportals.niaid.nih.gov/#!/dashboard/home.