

Abstract
The unicellular ciliate Paramecium contains a large vegetative macronucleus with several unusual characteristics, including an extremely high coding density and high polyploidy. As macronculear chromatin is devoid of heterochromatin, our study characterizes the functional epigenomic organization necessary for gene regulation and proper Pol II activity. Histone marks (H3K4me3, H3K9ac, H3K27me3) reveal no narrow peaks but broad domains along gene bodies, whereas intergenic regions are devoid of nucleosomes. Our data implicate H3K4me3 levels inside ORFs to be the main factor associated with gene expression, and H3K27me3 appears in association with H3K4me3 in plastic genes. Silent and lowly expressed genes show low nucleosome occupancy, suggesting that gene inactivation does not involve increased nucleosome occupancy and chromatin condensation. Because of a high occupancy of Pol II along highly expressed ORFs, transcriptional elongation appears to be quite different from that of other species. This is supported by missing heptameric repeats in the C-terminal domain of Pol II and a divergent elongation system. Our data imply that unoccupied DNA is the default state, whereas gene activation requires nucleosome recruitment together with broad domains of H3K4me3. In summary, gene activation and silencing in Paramecium run counter to the current understanding of chromatin biology.
The degree of epigenetic differentiation and the organization of eukaryotic genomes are usually adapted to the complexity of an organism: Chromatin serves as an additional layer of information, either for manifestation of gene expression patterns, for the cyclic condensation of chromosomes, or for microtubule-assisted separation of DNA in mitotic divisions. Chromatin further influences the proper processing of functional mRNAs as histone modifications influence Pol II dynamics and its interaction with RNA modifying components, such as the capping enzyme or the spliceosome.
Paramecium tetraurelia is a unicellular organism belonging to the SAR clade (including Stramenophiles, alveolates, and Rhizaria), which is as distant to plants, fungi, and animals. Paramecium is a ciliate, a phylum of Alveolata, and shows an unusual nuclear feature: Although unicellular, these cells already differentiate between germline and soma by germline micronuclei (MICs) and somatic macronuclei (MACs). Both differ in structural and functional aspects. MICs are small (1–2 µm) and transcriptionally inactive during vegetative growth, because the large (∼30-µm) MACs transcribe all necessary genes to allow for cell proliferation (Bétermier and Duharcourt 2014). During sexual reproduction, haploid meiotic nuclei are reciprocally exchanged and fuse to a zygote nucleus: This creates new MICs and MACs, whereas the new developing MAC (anlagen) already transcribes some genes involved in development (Furrer et al. 2017; Rzeszutek et al. 2020).
The genomic structures between MICs and MACs are quite different. MICs contain thousands of short transposon remnants (internal eliminated sequences [IESs]), which become deleted by a RNAi-related mechanism during macronuclear development (Allen and Nowacki 2020). The MAC differs from the MICs by the absence of IESs and transposons (Guérin et al. 2017). In addition, MAC chromosomes are tiny in size, usually <1 Mb, because MIC chromosomes are fragmented into many (about 200) different MAC chromosomes. These are amplified then to about 800 copies each, resulting in a massive polyploidy. The separation of that many DNA molecules, approximately 200 MAC chromosomes × 800n, is realized by amitotic divisions of the MAC: Replicated DNA becomes distributed to daughter nuclei without chromosome condensation and a typical mitotic spindle. The latter would be useless as the absence of centromeres (Lhuillier-Akakpo et al. 2016) and, consequently, kinetochores would not allow for attachment of microtubules.
In 2006, the Paramecium macronuclear genome project revealed two highly unexpected findings: (1) an exceptionally high number of genes (about 40,000), most of them resulting from three successive whole-genome duplications, and (2) an exceptionally high coding density of 78%. The latter is owing to tiny introns, predominantly 25 bp in length, and small intergenic regions (352 bp on average) (Aury et al. 2006).
Chromatin during the amitotic M phase remains uncondensed, suggesting that the MAC does not harbor the full genetic requirements to create highly condensed chromatin. In addition, interphase chromatin was reported to show several unusual features compared with other species based on chromatin spread preparations, for instance, the finding of several unusual filament types and the appearance of a low level of polyteny between individual transcription nodes (Samuel et al. 1981). Classical heterochromatin is believed to be absent from the MAC, although a deeper biochemical insight in the MAC chromatin organization is still missing. The same holds true for the presence of classical repressive histone marks in the vegetative MAC, raising the question of how gene repression is regulated. Another epigenetic mark, 5-methylcytosine, is known to be involved in the negative regulation of gene expression in many eukaryotes. However, 5-methylcytosine is reportedly absent in MAC DNA (Singh et al. 2018).
Hence, the contribution of dynamic MAC chromatin modifications to the regulation of gene expression remains poorly understood in ciliates. We know from other organisms that chromatin marks have functions in RNA processing and active elongation of transcription. Current studies of mammalian chromatin report functions for well-positioned nucleosomes in the context of Pol II phosphorylation and interaction with RNA modifying enzymes. This raises the question of how such a regulation is realized in ciliates, specifically in Paramecium.
+1 nucleosome positioning, for instance, was indicated to correlate with Pol II pausing and increased recruitment of negative elongation factor (NELF) (Jimeno-González and Reyes 2016). Whereas initiation of transcription is accompanied by phosphorylation of serine 5, P-TEFb was shown to mediate the conversion of the Pol II complex from its initiation to the processive elongation form, which includes phosphorylation of serine 2 (Egloff and Murphy 2008; Buratowski 2009). Promoter proximal pausing is known to be controlled by the negative regulators NELF and DSIF, whereas the C-terminal domain (CTD) of Pol II interacts with the capping components for 5′-capping of the nascent mRNA. Similarly, polyadenylation and splicing are controlled by both the CTD of Pol II and correctly positioned nucleosomes (Böhm and Östlund Farrants 2011). Especially for the latter aspect, alternative splicing has been implicated to be regulated by alternative CTD phosphorylation regulated by the SWI/SNF chromatin remodeling complex (Batsché et al. 2006), rich heptad repeat. Although we do not know much about these mechanisms in ciliates, we suspect them to differ from the above-described CTD regulation and interaction with additional components in metazoans. This suspicion arises from the missing Pol II heptameric repeats in Paramecium, which likely also affect the interacting complexes owing to a coevolutionary effect. One of those complexes involved in transcription coactivation and elongation, the Mediator complex, for instance, significantly differs from Tetrahymena to other species (Zhao and Liu 2019). As a consequence, we currently do not understand the role of the ciliate epigenome architecture concerning Pol II activity in terms of initiation, elongation, pausing, and interaction with complexes. In this work, we aim to understand the epigenomic organization of the polyploid vegetative MAC of P. tetraurelia. These cells contain two diploid and transcriptionally silent micronuclei, which divide by classical mitosis during cellular fission, whereas the MAC divides amitotically: Stretching and outlining results in uncontrolled separation of uncondensed chromosomes (Fig. 1A). The interpretation of any MAC epigenome data requires a look for the genomic structure of the chromosomes. During their processing from MIC chromosomes after sexual recombination, heterochromatic regions such as telomeres, centromeres, satellites, and transposons become eliminated in addition to about 45,000 transposon remnants called IES elements (Fig. 1B). Fragments undergo de novo telomere addition, resulting in small acentromeric chromosomes with a size of <1 Mb. These chromosomes exist at varying lengths owing to imprecise eliminations of repeated sequences (Duret et al. 2008). Compared with other species, even the related ciliate Tetrahymena, the Paramecium MAC genome shows an extremely high coding density of ∼80%, with small intergenic regions and tiny introns of 25 nt on average (Aury et al. 2006).
Figure 1.
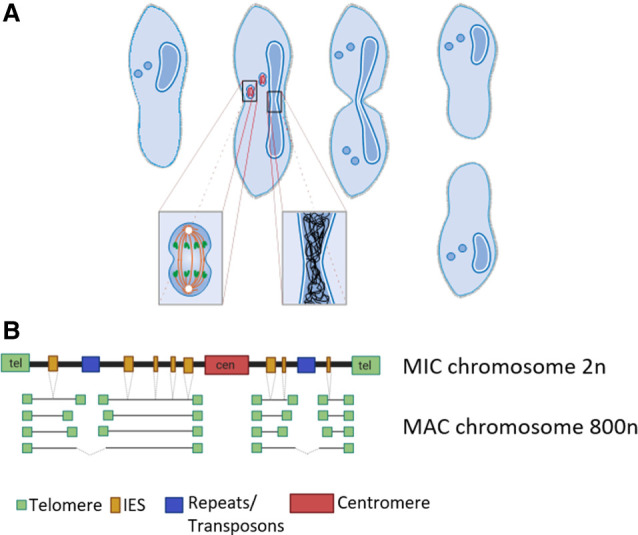
Paramecium vegetative cell divisions and chromosomal structure of MIC and MAC. (A) Paramecium tetraurelia showing two generative MICs and one vegetative MAC. Cell division involves mitotic separation of condensed MIC chromosomes and amitotic separation of uncondensed MAC chromosomes. While MICs and MAC divide, the nuclear envelope remains at both nuclei. (Figure courtesy of Jens Boenigk and Martin Simon.) (B) Chromosomes of the diploid MIC are large and contain centromeres and telomeres similar to canonical eukaryotic chromosomes. In addition, they consist of about 45,000 internal eliminated sequence (IES) elements and repeats (transposons, minisatellites). During macronuclear development after sexual reproduction (not shown here), telomeres, centromeres, repeats, and IESs become eliminated by different mechanisms. Although IESs are precisely excised, elimination of repeats and, presumably, centromeres is imprecise, resulting in fragmentation into heterogenous macronuclear chromosomes (with rare fusion of fragments). All macronuclear fragments show de novo telomere addition and amplification to 800n (created with BioRender [https://biorender.com]).
Results
Unusual properties of the macronuclear genome
The mechanisms of DNA elimination described above during development of the Paramecium MAC result in a highly compact genome with striking differences in comparison to Schizosaccharomyces pombe and individual metazoans (Fig. 2A,B).
Figure 2.

Features of the Paramecium genome in comparison to other organisms. (A) Comparisons of distribution of genes (green arrows) along the chromosomes of selected organisms to highlight the variation in coding density (P. tetraurelia, Tetrahymena thermophila, Schizosaccharomyces pombe, Drosophila menlanogaster, Homo sapiens). A window of 300 kb is shown for each chromosome in a genome browser. (B) Summary of genomic features of the same organisms named in A. For details on collected data, see Methods. (C) Detection of histone modifications in vegetative Paramecium nuclei by immunofluorescence staining. DNA in the nuclei is stained with DAPI (blue), and antibodies directed against the three indicated modifications (H3K4me3, H3K9ac, H3K27me3) were labeled with a secondary Alexa Fluor 594 conjugated antibody (red). Arrowheads point at micronuclei; arrows indicate position of the macronucleus. Other panels show brightfield and overlay of signals. Representative overlays of Z-stacks of magnified views are shown. Scale bar, 10 µm.
To quantify global epigenome organization in Paramecium, we first investigated the distribution of histone H3 modifications in the vegetative MAC, because histone modifications are major contributors to chromatin architecture. Immunofluorescence analysis with histone H3–specific antibodies shows H3K4me3 and H3K27me3 occurring in both MICs and the MAC, whereas H3K9ac is present in the MAC only (Fig. 2C). The MAC H3K27me3 signal is usually weak in immunofluorescence, similar to earlier reports (Ignarski et al. 2014), and shows slight unspecific staining of extranuclear structures as the oral apparatus. To test the specificity of the antibodies for their respective target, competition assays using dot-blots were performed and are shown in Supplemental Figure S1.
Low nucleosome occupancy in intergenic regions and silent genes
To characterize nucleosome positioning, mononucleosomal DNA was isolated after digestion of MAC chromatin with micrococcal nuclease (MNase). Reads were mapped to the genome and normalized against a digest of naked DNA, resulting in discrete peaks for both setups using 10 or 128 U MNase (Fig. 3A), corresponding to light and heavy digestion. As the figure suggests that intergenic regions show low nucleosome occupancy, we separately analyzed coding genes and intergenic regions, the latter being defined as the region in between the transcription start site (TSS)/transcription termination site (TTS) of the gene of interest and the TSS/TTS (depends on the orientation) of the upstream gene. Figure 3B shows that genes show increased nucleosome occupancy in the 5′- and 3′-coding regions associated with drops in occupancy in flanking noncoding regions. The latter indeed show general low occupancy (Fig. 3C). For further quantification, we dissected genes by their expression levels (Fig. 3D) and calculated the associated nucleosome occupancy. Figure 3E shows the MNase signals quantified in intergenic regions and quantiles of genes. Intergenic regions show the lowest nucleosome occupancy. Please note that these values are not normalized for the individual gene length of groups, given in Supplemental Figure S2A. In support of these analyses, Supplemental Figure S2B shows the occupancy only of the most prominent nucleosome (+1) in these gene groups. Genes show increasing nucleosome occupancy with increasing gene expression levels. This is an unexpected result, as unoccupied DNA is believed to be highly accessible for Pol II and therefore usually defined as active chromatin. Our results here suggest that this is the opposite in the Paramecium MAC.
Figure 3.

MNase-seq results reveal well-positioned +1 nucleosomes. (A) Exemplary view of nucleosome distribution along the MAC scaffolds of Paramecium. Top panel shows the peak distribution in a 12-kb window, and the bottom panel shows the magnified view on one gene. For both panels, the top row shows the coverage track from poly(A) mRNA-seq followed by the tracks for nucleosome occupancy obtained by light (10 U) and heavy (128 U) MNase digestion of Paramecium nuclei. Coverage tracks were visualized using the Integrative Genomics Viewer (IGV) browser (Robinson et al. 2011). (B) Profile plot of nucleosome distribution at the transcription start site (TSS; left) and transcription termination site (TTS; right) for genes >1 kb and digestion conditions as in A. The plot organization resembles the nucleosome profile along the gene body/intergenic region with dotted lines indicating excluded regions in the center of both plots. (C) Same plot as in B, but for intergenic regions >1 kb. Horizontal line is drawn to aid comparison between B and C. (D) Ranking of genes by their mRNA expression values from low to high (Q1–Q5) and (E) total sum occupancy for the genes in each expression quantile and the intergenic regions. Occupancy values are shown for mild and heavy digest side by side.
Prominent +1 nucleosomes mark actively transcribed genes
We aim to analyze the nucleosome positioning and occupancy in genes more in detail. Genomic analysis of MNase data revealed well-positioned +1 and −1 nucleosomes at the TSS (Fig. 4A). Especially the presence of −1 nucleosomes differs from analog analyses of MNase data from Tetrahymena, S. pombe, Drosophila melanogaster, but they are apparent in humans (Supplemental Fig. S3). As such, their presence in Paramecium is surprising and requires additional analysis. In addition, the comparison to other species shows that downstream nucleosomes (downstream from +1) in Paramecium are apparently much less pronounced; already, the +2 nucleosome signal is roughly background, which is in contrast to Tetrahymena, S. pombe, and Drosophila showing slightly decreasing peak values inside the gene bodies (Supplemental Fig. S3). The recent paper of Gnan et al. (2022) did not identify these putative −1 nucleosomes in Paramecium. This difference is not owing to the bioinformatics pipelines, because Supplemental Figure S3 shows still the absence of putative −1 nucleosomes when our MNase pipeline is applied on the data of Gnan et al. (2022). We therefore conclude that the difference is owing to the MNase conditions. We used formaldehyde-fixed material in contrast to fresh chromatin. It seems suitable that our MNase digests are weaker compared with the relatively harsh conditions on native chromatin. Lighter MNase digests can obtain signals of nucleosomes, which are otherwise hidden: For example, in Tetrahymena, light MNase digests indeed show a weak −1 signal, which was similar to our data Xiong et al. (2016). We added the MNase profiles of the latter data of Tetrahymena, analyzed with our MNase pipeline to Supplemental Figure S3. As a result, one indeed needs to take the MNase conditions into account. We cannot exclude that other MNase conditions applied to the analyses of yeast, flies, and human chromatin (Supplemental Fig. S3) could produce alternative patterns. In the following, we aimed to see whether the positioning of −1 nucleosomes could be owing to short intergenic regions. We therefore dissected the Paramecium genes owing to two parameters: intergenic distance and orientation of genes. We considered bidirectional promoter genes, in which the two start sites of both genes are adjacent (start–start [SS]), or unidirectional genes, in which one start site is paired with the end of the other gene (start–end [SE]) (Supplemental Fig. S4A). These two categories were additionally classified into four groups based on their intergenic distance. The number of genes in each category is given in Figure 4B. Figure 4C shows nucleosome positioning of these categories at the TSS and the TTS. Most apparent, putative −1 nucleosomes are much more pronounced in genes with short 5′-intergenic regions >142 bp, and this is true for the SE and the SS configuration. In addition, TTSs also show well-positioned nucleosomes at the ultimate 3′-end of ORFs, which are more pronounced in the SE configuration. The absence of −1 nucleosomes in genes with a longer intergenic region let us conclude that these putative −1 nucleosomes are either +1 or TTS nucleosomes of upstream genes, but no true −1 nucleosomes. They could, however, have a function in regulation of both genes, being “coincidental” −1 nucleosomes in point of view of our analysis.
Figure 4.

Positioning of nucleosomes in relation to gene expression. (A) Profile plot for nucleosome distribution relative to the transcription start site (TSS) for all analyzed Paramecium genes. Signal for 1000 bp upstream of and downstream from the TSS is shown. For comparison, MNase-seq data from T. thermophila were plotted in the same manner. (B) Dissection of neighboring Paramecium genes based on their configuration and intergenic distance (ID). Table shows separation of genes by configuration and ID, ranked from short distances (G1) to long distances (G4). The last two columns indicate numbers of genes in each configuration and ID group. (C) Nucleosome profiles in a 2-kb window centered at the TSS (left) or the TTS (right) for neighboring genes in SS and SE configuration are shown. Genes were additionally separated by the length of intergenic distances; see color-coding in B. The nucleosome profiles in relation to their distance (x-axis) to TSS (D), TTS (E), and intron–exon junction (F) are shown for gene categories based on their expression levels. (G) Box plots showing the mRNA expression (y-axis; log10 TPM + 1) of genes with different intron frequency groups (number of introns per 100 bp; x-axis). A Kruskal–Wallis test showed that the expression distribution between all pairs of intron frequency groups is significantly different (P < 2.2 × 10−16).
We consequently asked for a potential coregulation of genes at bidirectional promoters. Correlation analysis of neighboring genes suggests a high degree of coregulation of all neighbor genes regardless of the configuration (Supplemental Fig. S4A,B). However, Supplemental Figure S4C shows that we cannot identify a higher degree of coregulation in genes under the same bidirectional promoter, suggesting that even short intergenic distances are sufficient to control regulation of gene expression independently of the neighbor gene. However, our data indicate that genes with bidirectional promoters tend to have a longer intergenic distance (Supplemental Fig. S4D), suggesting that selection pressure acts on these regions to separate bidirectional genes from each other. Gene length itself seems not to have a strong effect on TSS and TTS nucleosome positioning (Supplemental Fig. S5).
We sought to investigate whether nucleosome positioning is changed with differences in gene expression levels (Fig. 4D,E). At both ends of a gene, TSS and TTS, well-positioned nucleosomes can be found in highly expressed genes only. In contrast, these regions and also gene bodies of silent genes appear to be almost devoid of well-positioned nucleosomes.
We can detect well-positioned di-nucleosomes around introns (Fig. 4F). As mentioned, the 25-nt introns are among the shortest reported in eukaryotes (Russell et al. 1994). Intron splicing appears to result from efficient intron definition, rather than exon definition as in multicellular species, although only 3 nt define the 5′- and 3′-splice sites (Jaillon et al. 2008). Our data do not reveal any associations of intron nucleosomes with intron length (Supplemental Fig. S6A). As our MNase data suggest a general low occupancy of nucleosomes in gene bodies, intron-associated di-nucleosomes could be an exception to this. We correlated the intron frequency (number of introns per 100 bp) with gene expression levels (Fig. 4G) and found increasing mRNA levels with increasing intron frequency, an effect independent of the gene length (Supplemental Fig. S6B). Thus, introns in Paramecium may be involved in transcriptional regulation by recruitment of nucleosomes to gene bodies.
Broad histone mark domains in gene bodies
To extend the chromatin analysis to histone modifications, chromatin immunoprecipitation followed by sequencing (ChIP-seq) was performed from vegetative cells. We used the NEXSON procedure (Arrigoni et al. 2016) involving isolation of intact MACs without MICs. Another advantage of this procedure was that we were able to use the very same MAC preparations for both MNase- and ChIP-seq. We used antibodies for the activation-associated marks H3K9ac and H3K4me3, as well as an antibody for the repressive mark H3K27me3. It is necessary to note here that H3 variants have been described in P. tetraurelia (Lhuillier-Akakpo et al. 2016): Divergent and putative development-associated H3 variants cannot be detected with the antibodies used here; it is not likely that these antibodies can dissect the five H3 variants expressed during vegetative growth, which means that ChIP should detect all of these variants, as well as the putative H3.3. The observed ChIP-seq signatures of these three marks showed rather broad signals, which were not comparable to sharp peaks of metazoan ChIP-seq signals. Thus, we refrained from a peak-calling approach and used ChromHMM (Ernst and Kellis 2012) to segment the entire MAC genome into 200-bp bins, representing approximately the resolution of a nucleosome including a spacer region, for de novo determination of reoccurring combinatorial and spatial signal patterns. We found that five different chromatin states (CSs) could be observed (trying to increase the number of states resulted in highly similar states, and we continued all further analyses with five states). Heatmaps in Figure 5A show the contribution of the individual signals to each CS and, on the right, the quantitative assignment of each CS to different regions of the genome. We abbreviate all five CSs as CS1 to CS5.
Figure 5.

Segmentation analysis using ChromHMM. (A) The chromatin state (CS) assignments are shown as a heatmap of emission parameters from a five-state ChromHMM model (left). Each row corresponds to a ChromHMM state, and each column represents a different epigenetic mark. The darker the color of an epigenetic mark for a state, the higher the probability of observing that epigenetic mark in that state. Heatmap showing the overlap fold enrichment of each ChromHMM state (row) in different genomic annotations (columns; right). Enrichment values are obtained from the overlap enrichment functionality of ChromHMM with a column-specific color scale. (B) The fold enrichment of each state in 200-bp bins within a 2-kb window around the TSS is shown. Enrichment values are obtained from the neighborhood enrichment functionality of ChromHMM with a uniform color scale. (C) Box plots show mRNA expression (y-axis; 10 TPM + 1) of genes whose loci overlap at least 80% with a respective state (right). Additionally, genes were separated by their assigned state in 300 bp upstream of the TSS (N − 300) and the first 300 bp of the gene body (N + 300), and mRNA expression values of these genes are plotted (left, middle). Sketch on top of the plots visualizes the arrangement of the three analyzed regions.
One major finding of the segmentation is represented in CS4. ChromHMM defines this state as being almost free of any signal; this state is moreover attributed to the highest percentage of the genome (Fig. 5A, right). This may support our previous assumption that a high amount of MAC DNA is free of nucleosomes and therefore also of transcription-altering histone marks. In contrast, MNase and histone mark signals can be found in CS1–CS3 and CS5. Their ChromHMM signature shows dynamic combinations between the three investigated histone marks, and the occurrence of these states also varies in different genomic areas. Focusing on histone marks around the TSS, CS1 and CS2, both enriched in H3K9ac and H3K4me3, show strong accumulation at the +1 nucleosome (Fig. 5B). All other CSs show depletion at +1, especially CS3, which suggests that especially H3K27me3 is depleted at these gene loci.
To go deeper into the role of the individual marks and states in association with gene expression, we dissected genes into categories overlapping with a CS (1) for >80% of the entire gene body, (2) with first 300 bp of the ORF, or (3) with 300 bp of the noncoding upstream region. We consequently correlated this with the gene expression level of these genes (Fig. 5C). Genes with high levels of H3K9ac and H3K4me3 (CS1) are highly expressed. Focusing to the role of H3K27me3, its high abundance in CS2, associated genes showing the highest expression level, is an argument against a repressive function of this histone mark. Only few genes (91) can be attributed to CS3, the only state in which the H3K27me3 signal dominates over H3K4me3 and H3K9ac; although the genes appear to be quite lowly expressed, the small number of genes does not allow for a conclusion about a possible repressive function of H3K27me3.
Genes associated with CS5 show low levels of H3K4me3 and H3K9ac with the absence of H3K27me3, and these genes show an intermediate gene expression level. CS4 shows the lowest gene expression level and, in agreement with the quantitative analysis, the highest number of genes. We conclude that gene silencing in the MAC is associated with genomic loci that consist predominantly of free and accessible DNA. Comparing the 80% gene overlap category to the upstream and the 5′-coding region, our analysis indicates that the upstream region contributes less to gene regulation. Mainly the 5′-CDS and the ORF appear to be involved in gene regulation, which fits to our conclusions from MNase data. We can therefore conclude that gene transcription is mainly associated with high levels of H3K9ac and H3K4me3 at the +1 nucleosome. We do not see direct evidence for a repressive function of H3K27me3. These results now raise several questions, especially about the role of the prominent +1 nucleosome in transcriptional activation: Could this be a place for RNA Polymerase II pausing in order to regulate gene expression?
Pol II occupancy correlates with gene expression levels
To characterize Pol II occupancy and activity, it is important to note that Paramecium Pol II diverges from conserved metazoan and most unicellular Pol II. In Paramecium, as well in Tetrahymena, the consensus serine-rich repeats are missing, but the CTD shows overall a high percentage of serines (Fig. 6A). As commercial Pol II antibodies target the heptamers in the CTD, we had to produce an antibody of our own against the P. tetraurelia CTD of RPB1. After affinity purification and specificity checks by IF and western blots of cellular fractions (Supplemental Fig. S7), ChIP was performed as described. Figure 6B shows high Pol II occupancy of genes showing high expression and vice versa. Here, the analysis of all genes of the genome results in a quite equal distribution of Pol II along the ORF.
Figure 6.

Analysis of RNA polymerase II pausing. (A) Multiple sequence alignment of the RNA polymerase II enzyme's RPB1 subunit in different organisms is shown. The C-terminal end of RPB1 is zoomed in to show the difference in conserved regions of some ciliates to other organisms. For details, see Supplemental Methods. (B, left) Box plots of gene expression (y-axis; log10 TPM) split in 10 quantiles are shown; higher quantiles mean higher expression. (Right) Pol II enrichment (y-axis) profiles of genes in respective quantiles are shown. Distance shown on the x-axis is scaled; that is, all genes (TSS–TTS) are either stretched or shrunken to a length of 1500 bp. A 500-bp window upstream of and downstream from the gene loci is included. Enrichment profiles were plotted using deepTools2. (C) A graphical representation of the regions included in polymerase pausing index (PI) calculation is shown. We categorized a gene as paused if the PI ≥ 1.5. The table summarizes numbers of paused/not paused genes for selected organisms (Supplemental Table S1 contains details on Pol II data sets). (D) Same as the Pol II enrichment profiles in B, but genes are split based on the status of Pol II pausing.
We consequently asked whether Pol II pausing at the +1 nucleosome can be observed, and we calculated a pausing index (PI) by dividing the Pol II coverage of the TSS by the coverage of the gene body (Fig. 6C). Dissecting paused and nonpaused genes by a threshold of PI larger than 1.5, we compared Pol II occupancy of Paramecium to other species. Figure 6D shows that Paramecium is the only species with similar occupancy of paused and nonpaused genes. The overall distribution of Paramecium Pol II is highly different to other species. In humans, S. pombe, and Tetrahymena, nonpaused genes show increasing coverage along the ORF (for detailed heatmaps, see Supplemental Fig. S8A). This is different in Paramecium, in which nonpaused genes show in general higher occupancy and less decrease along the ORF. Considering the huge differences in gene length distribution for the different species, we additionally analyzed subsets of genes with approximately the same length (Supplemental Fig. S8B) and still observed the similar Pol II distribution as shown in Figure 6D. The pattern of Paramecium appears different to other species, suggesting that regulated pausing at the +1 nucleosome occurs only rarely. This is to some extent also true for Tetrahymena and yeast with the difference that paused genes here show a clearer peak at the TSS along with a strong decrease along the ORF. Such patterns cannot be identified in Paramecium. Paramecium in contrast shows a clear drop in Pol II occupancy before the TSS and at the TTS: This seems in agreement with our hypothesis that regulation of gene expression occurs mainly inside ORFs. We further analyzed whether pausing is associated with reduced full-length mRNA production. Supplemental Figure S8C shows that we see a significantly lower expression of paused genes in Tetrahymena and S. pombe; only in humans do paused genes show higher mRNA levels. Thus, Pol II pausing may indeed be a mechanism of gene regulation, but used in a different manner. Especially in Paramecium, the mRNA levels between paused and nonpaused genes show the smallest differences, although significant, suggesting that pausing is more involved in fine-tuning transcription rather than on/off switching.
H3K4me3 is the most important predictor of gene expression
Integrating all the data generated, we started by characterizing their distribution over all genes categorized by two factors, namely, gene expression and gene length. Figure 7A shows the input normalized profiles of different epigenetic marks and GC content based on the gene expression groups. Genes in heatmaps are sorted by gene length. MNase, Pol II, H3K4me3, and H3K9ac show accumulation in the 5′-CDS in expressed genes with decreasing intensity along the ORF. However, most signals are still high and correlate to gene expression level in the 3′-CDS. The 5′-accumulation is not that pronounced in H3K27me3, which shows more equal distribution along the ORF. Hence, we further investigated how the epigenetic marks are distributed along the gene structure, based on their length. MNase signals show a strongly phased pattern in all categories of gene expression, which becomes apparent when genes are sorted by length. Supplemental Figure S10A shows a strong positive correlation of exon length and nucleosome counts in exons. Similarly, nucleosome occupancy is positively correlated with gene expression (Fig. 7A). Similar to the strongly phased signals of MNase, we observe that Pol II signals are also phased and show positive association with gene expression.
Figure 7.

Distribution of epigenetic marks. (A) Distribution of epigenetic marks in different transcriptomic groups. Heatmaps show the input normalized enrichment values for different epigenetic marks. Genes (rows) are split into three categories based on gene expression—high (TPM > 2), low (0 < TPM < 2), and silent (TPM = 0)—and are sorted by decreasing order of gene length in each, which is visualized by the length distribution graph on the left. Distance shown on the x-axis is scaled; that is, all genes (TSS–TTS) are either stretched or shrunken to a length of 1500 bp, adding 1000 bp upstream of and downstream from the gene. Heatmaps were plotted using deepTools2; black lines in intergenic regions reflect missing data at this position. (B) Distribution of epigenetic marks for a subset of 4000 genes with discrete length of ∼1.2 kb. Plots show the signal in the upstream intergenic region, the TSS and the TTS of genes belonging to the similar expression categories as in B.
All epigenetic marks are consistently low at 5′- and 3′-noncoding regions, showing a clear gap in all analyses and thus fostering the assumption that intergenic regions hardly contribute to gene regulation. All silent genes have very faint signals of all epigenetic marks, supporting our conclusion that lowly occupied nearly naked DNA is a hallmark of gene inactivation in Paramecium.
The visualization in the heatmaps in Figure 7A reveals a phasing pattern for almost all marks, as genes are ordered by gene length in each expression group. This means that nucleosomes are indeed well positioned in all ORFs and along the entire length, but with varying intensity, owing to differences in gene expression. As one will have assumed then that the histone marks need to follow the nucleosome pattern, this follows also the GC content oscillations in position and quantity. As such, this cis-factor likely contributes nucleosome positioning and, consequently, gene expression. We investigated the effects of gene length and mRNA levels and observed that shorter genes show higher mRNA levels (Supplemental Fig. S9), and as such, gene length itself appears to be a factor limiting transcriptional efficiency. We observe the phasing pattern also for Pol II occupancy. This would suggest that Pol II shows association with nucleosomes along the entire ORF, and the higher Pol II occupancy in highly expressed genes does not indicate that this association is a mechanism of transcriptional inhibition. In agreement with the conclusion from the PI analyses, this Pol II nucleosome association appears to be a mark of highly expressed genes, although one could get the impression that Pol II stops at every single nucleosome, which could also be an argument for inefficient elongation. Figure 7B shows the signals of the epigenetic marks in a subset of genes with similar gene length (∼1200 kb), thus avoiding the projection of small and large genes. As we observed some intriguing patterns of histone marks, especially of H3K27me3, which is abundant in highly expressed genes, we checked the correlation of all epigenetic marks with each other with mRNA (Supplemental Fig. S10A). We observed that all epigenetic marks are positively correlated (Pearson's correlation > 0.6) with each other, and with mRNA (Pearson's correlation > 0.30). We wondered about the individual contribution of gene characteristics and epigenomic marks to gene expression. Thus, we constructed a machine learning classifier to predict genes as highly or lowly expressed using epigenetic features and genic features (see Methods). Our model is based on a random forest algorithm, which accurately predicts gene expression with an average area under the precision-recall curve (PR-AUC) of 0.74 and 0.76 for genic or epigenetic features, respectively. The model combining all information performed best (PR-AUC of 0.82) (Fig. 8A). These differences were statistically significant (Supplemental Fig. S10B). The experiments in Figure 8A were performed using histone marks in the complete gene body. When quantification is restricted to the proximal TSS region (TSS + 300 bp), performance decreased (Supplemental Fig. S10C), supporting a role of those marks throughout the gene body.
Figure 8.

Prediction of gene expression by epigenetic marks and genomic features. (A) Results of classifying low and high gene groups using different data (genic: related to gene structure; epigenetic: using abundance of histone marks and MNase; both: genic and epigenetic). PrecisionRecall curve with average values from a 40-fold cross-validation with random forests indicating features by different colors. (B) Analysis of feature importance using both genic and epigenetic features (underlining color indicates type on y-axis; see legend for A). Features are listed in decreasing order of classification importance from top to bottom. The importance (SHAP value; x-axis) of a feature for each gene illustrates its contribution to classification as high or low, with positive and negative SHAP values, respectively. The color gradient depicts the feature value in scale from low to high, for example, the length of a gene (third row). For example, long genes strongly contribute to the prediction of lowly expressed genes. (C) Genes were separated into four groups by their plasticity, which is defined by a large variation in gene expression among different conditions. The box plot shows the distribution of classifier performance values for genes with different plasticity (50-fold CV-based PR-AUC) for the same three feature sets as A. The number of genes in each plastic gene group was randomly subsampled to have equal number of genes in high and low expressed category. (D) Distribution of CSs among plastic gene groups. We only included genes with a ChromHMM state overlap of at least 80% (see Fig. 5). Additionally, partial correlation values for H3K4me3-H3K9ac (cross) and H3K4me3-H3K27me3 (circle) are red for each group.
Further, we interrogated the best-performing model on the importance of each feature in obtaining the classification (Fig. 8B). According to the feature importance values calculated on our best-performing model, H3K4me3, intron frequency, and gene length are the top three features required to classify gene expression. Intergenic length and H3K27me3 are among the least important features for our model. The presence of H3K27me3 in the whole gene body, with its high correlation to other histone marks and highly expressed genes, does raise the question of the role of H3K27me3 in MAC nucleosomes of Paramecium.
H3K4me3 and H3K27me3 co-occur at plastic genes
We consequently asked for the contribution of individual features to gene regulation. We used RNA-seq data from environmental states that include four different serotypes at different temperatures, starvation, heat shock, and cultivation at 4°C (Cheaib et al. 2015). Using those data, we dissected genes showing large expression variations (high plasticity) during the vegetative growth in different environments to identify dynamically regulated genes from housekeeping genes (see Methods) (Supplemental Fig. S11). We defined four classes of plasticity (G1–G4), in which G4 genes showed the largest variation. We again used the random forest algorithm to analyze whether genic/epigenetic factors contribute to the accuracy of gene expression prediction for each gene plasticity group. The performance of expression prediction decreased for genes with higher plasticity (Fig. 8C). Thus, plasticity of gene expression seems to be accompanied with additional and unknown features contributing to gene regulation.
To get further insights, we checked the CSs based on our ChromHMM segmentation of the four categories of plastic genes (Fig. 8D). These show gradual differences, with most apparent increase that of CS4 and decrease that of CS2. This suggests that epigenetic marks are used not only for control of gene expression but moreover for gene regulation. We studied the differences of histone marks of these categories in more detail and calculated the partial correlation between different modifications (see Methods). Figure 8D shows an increase in partial correlation of H3K4me3/H3K27me3 for the most plastic genes only, suggesting that the interplay between histone marks varies in the four considered groups.
Discussion
Genomic and epigenomic paradoxes
At first glance, the genomic structure of the Paramecium MAC seems paradoxical. Although Paramecium is extremely gene-rich, with approximately 40,000 genes (Aury et al. 2006), the size limitations of intergenic regions and introns provide only restricted capacity for differential gene regulation. This is different compared with genomic/epigenomic features in metazoans, because unicellular organisms do not need to differentiate into distinct tissues with all the known epigenetic manifestations to guarantee for cell type–specific gene expression patterns. However, the Paramecium epigenome still needs to manage dynamic regulation of gene expression and proper transcription of mRNA. We know that histone marks do not just control condensation and transcriptional on/off switches but interact with capping enzymes, splicing factors, and elongation factors to guarantee mature mRNA synthesis (Jimeno-González and Reyes 2016).
Thus, we aimed to answer the question in which manner the MAC epigenome signature is associated with transcriptional regulation in this ciliate. Its nucleosomes appear to hold some secrets as recent results show that the nucleosome repeat length is only ∼151 bp, which means that the linker DNA between nucleosomes is only few base pairs long (Gnan et al. 2022). Our data show that nucleosome occupancy appears to be associated in general with active transcription, because segmentation of MNase and ChIP data shows a large number of genes where our setup detects only low or no signals (CS4 in Fig. 5). Correlation of this CS with gene expression indicates that low nucleosome occupancy, regardless of the histone marks, is associated with silent or lowly expressed genes. One could therefore interpret naked or lowly occupied DNA as a default state, which needs to be occupied with nucleosomes first to become transcribed into mRNA. As such, the epigenome of Paramecium appears paradoxical as well, as gene inactivation becomes realized by low nucleosome occupancy, and this is contrary to the classical models.
Textbooks describe gene inactivation by a hierarchical chromatin folding from open 10-nm fibers to condensed and higher occupied 30-nm filaments. Active transcription accompanied by open, accessible chromatin in mammals was highly supported in the last years by many studies of DNA accessibility using ATAC, NOMe, DNase-seq, or methods free of enzymatic steps like sedimentation velocity centrifugation (Klemm et al. 2019; Nordström et al. 2019; Ishihara et al. 2021). Our data do not support this model for Paramecium MAC chromatin, suggesting a different chromatin-associated mechanism of gene inactivation. This raises many more questions about how, in particular, spurious and aberrant transcription of Pol II in open regions is inhibited or whether this could be tolerated to some extent. In most species, condensation of chromatin is accompanied with linker histone H1 recruitment and studies on Drosophila chromatin show H1 occurring exclusively at closed heterochromatic loci (Nalabothula et al. 2014). We are not able to identify a MAC histone H1 variant in Paramecium, supporting the idea of condensation-free gene inactivation. To be precise, we have to distinguish MAC and MIC linker histones in ciliates. Tetrahymena has distinct MAC- and MIC-specific H1 histones, where the MAC version (Hho1) is nonessential (Schulman et al. 1987). HHO1 knockouts show an overall decondensation of MAC chromatin (Huang et al. 1999). Indeed, a Hho1 homolog is not present in Paramecium, or it may be more divergent to identify. However, the recent findings of Gnan et al. (2022) showed an extremely short linker DNA length between Paramecium nucleosomes compared with other species, and the investigators speculate that this could correlate with the absence of a canonical H1 ortholog.
Bistable H3K4/K27me3 as a mark of poised genes?
Another question we followed is whether the H3K27me3 could be involved in gene inactivation. Our ChIP data do not suggest that H3K27me3 is associated exclusively with silent or lowly expressed genes. When we asked for the function of this modification in the vegetative MAC, its role is unlikely the condensation of chromatin, and the segmentation shows H3K27me3 co-occurring in varying ratios with the H3K9ac and H3K4me3. Our data suggest that genes with high regulation dynamics show an increasing correlation for H3K27me3 and H3K4me3. This is one of the best-studied bivalent domains for poised chromatin, where chromatin is placed into a waiting state for future activation, and this was described to occur particularly in embryonic stem cells (Pan et al. 2007; Zhao et al. 2007). There is an ongoing debate whether poised chromatin is bistable or bivalent, the latter representing a background population of fragments with active and silent marks, whereas bistability means the frequent switching between monostable active and silent states (Sneppen and Ringrose 2019). The polyploidy of the Paramecium MAC introduces here an additional layer of complexity. Similar to ChIPs of different cell states from a culture of metazoan cell cultures, which cannot dissect different cell states of a mixture from a real bivalent domain, we cannot be sure that the 800 copies of a gene in the MAC are coregulated.
If Paramecium, for instance, would use gene dosage to regulate gene expression level, one would expect different ratios of marks: some copies silent, some copies active. This is what we can observe to some extent, because the random forest analysis suggests that the K4/K27me3 ratio explains gene expression levels better than the H3K27me3 alone. In a previous study, increased H3K27me3 levels in association with decreased levels of H3K4me3 at an endogenous reporter gene have been shown to go along with siRNA mediated silencing (Götz et al. 2016), which supports the K4/K27me3 ratio hypothesis for controlling gene expression levels. In addition, the finding that we see increasing partial correlation values of K4/K27me3 in genes that show high regulation dynamics could be called poised as such. This suggests that the bivalency of K4/K27me3 in chromatin poising could be an ancient and general mechanism rather than an invention of metazoans.
In Paramecium, the Polycomb group methyltransferease Ezl1 was shown to mediate both H3K9me3 and H3K27me3 during development: Loss of these marks is accompanied by loss of transposon repression and elimination and, in addition, a transcriptional up-regulation of early developmental genes (Frapporti et al. 2019). As Ezl1 shows also low expression during vegetative growth, it remains to be elaborated whether Ezl1 or another SET-domain-containing enzyme catalyzes the replicative maintenance of H3K27me3 during vegetative cell divisions. In addition, it remains to note that a putative repressive function of H3K27me3 could, in principle, be blocked by a phospho-switch by a neighboring serine-residue as this was initially shown for loss of binding of HP1 to H3K9me9 in context with H3K10 serine phosphorylation (Fischle et al. 2005). However, this is unlikely for Paramecium H3K27me3 as all H3 variants miss the conserved serine 28 in Paramecium (Supplemental Fig. S1; Lhuillier-Akakpo et al. 2016).
From an evolutionary point of view, this could imply that although Paramecium is unicellular, the epigenomic repertoire already has the capacity to manifest vegetative gene expression regulation during development, meaning to place histone marks for poising genes. The inheritance of gene expression pattern was previously shown also for the multigene family of surface antigen genes as transcription of a single gene follows the expression pattern of its cytoplasmic parent (Baranasic et al. 2014; Simon and Plattner 2014), but we would need to analyze the genome-wide extent of such an inheritance and/or whether such a mechanism is coupled with other genomic parameters like, for instance, subtelomeric localization of the respective genes.
ChIP-seq reveals broad domains instead of narrow peaks
When looking for the distribution of marks along genes, the absence of narrow peaks becomes apparent as all histone mark distributions are more comparable to broad domains instead of local and narrow peaks, which explains the failure of peak calling. Broad domains were also found in mammals. For instance, H3K27me3 was shown in mammalian chromatin to be distributed along ORFs (Zhou et al. 2011). Also in mammals, broad H3K4me3 was shown for tumor-suppressor genes with exceptionally high expression, where this mark has also been attributed to transcriptional elongation (Chen et al. 2015). In addition to tumor-suppressors, broad H3K4me3 domains have been implicated with genes for cellular identity and transcriptional consistency; as the broadest domains show increased Pol II pausing, the investigators suggest the broad mark as a buffer domain to ensure the robustness of the transcriptional output (Benayoun et al. 2014). This model could also fit to our observations, which suggest not only that H3K4me3 is the key regulator of transcription but that H3K4me3 appears in broad domains along ORFs highly covered with Pol II. Concerning the different patterns of Pol II along ORFs compared with other species, either for poised or nonpoised genes, the buffer domain model could hold true for the majority of Paramecium genes.
Nucleosome positioning and GC content
Paramecium has an exceptional genome composition with an average GC content of 28%, including the even more AT-rich intergenic regions. It is known that GC content favors nucleosome positioning (Tillo and Hughes 2009). Our data show that nucleosome occupancy is mostly restricted to ORFs, which would correlate to increased GC levels but also correlated to gene expression levels as higher expressed genes show higher occupancy of promoter proximal- and intron-associated nucleosomes. It is difficult to reason how much the sequence content of the Paramecium genome itself encodes the deposition of nucleosomes from our data. There is ample discussion about the DNA sequence preferences of nucleosomes (Meyer and Liu 2014), and also MNase-seq can generate a signature of higher occupancy at GC-rich regions on naked as well as occupied DNA (Chung et al. 2011). One may conclude that this bias explains the large drop of MNase-seq read occupancy at intergenic regions. However, analysis of ChIP-seq data shows a similar drop at intergenic regions and similar phasing patterns in our data, and Supplemental Figure S12 suggests that our procedure and the applied PCR amplification have minimized GC biases. We argue that it is unlikely to observe these trends exclusively owing to methodological biases in AT content.
Our results of nucleosome positioning fit to observations in Tetrahymena, where well-positioned nucleosomes in the MAC match GC oscillations but are also affected by trans-factors, for example, the transcriptional landscape (Xiong et al. 2016). In addition, studies in Tetrahymena revealed that N6-methyladenine (6mA) is preferentially found at the AT-rich linker DNA of well-positioned nucleosomes of Pol II transcribed genes (Wang et al. 2017; Luo et al. 2018). Also, in Paramecium, 6mA sites enriched between well-positioned nucleosomes are positively correlated with gene expression (Hardy et al. 2021). The latter finding would fit our observations: The more nucleosomes, the more 6mA, the more transcription.
Qualitative aspects of gene expression
To understand the relation between epigenomic data and gene expression, throughout this study we categorized genes based on their expression levels (high, low, silent). Although this categorization helps, it should be treated with a grain of salt as the cut-offs are rather arbitrary. Another aspect that requires cautious interpretation is the analyses presented in Figure 7. Specifically, Figure 7A shows the linear relation between epigenetic signals and mRNA expression in a qualitative manner. The random forests analysis, presented in Figure 7, B and C, reveals both the linear and nonlinear relationships inherent in the epigenetic data while calculating the probabilities to predict/classify a gene as highly or lowly expressed. For example, we can observe that H3K9ac is directly proportional to the different expression groups in Figure 7A. However, Figure 7C suggests genes with low H3K9ac are associated with high expression. Although this may seem counter-intuitive, both results are correct owing to the high colinearity of epigenetic marks (Supplemental Fig. S10). Hence, the random forests model relies on the H3K9ac signal only when the H3K4me3 signal is not sufficient to increase the probability of predicting a gene as highly expressed.
A divergent mechanism of transcriptional elongation
How can the highly regulated CTD phosphorylation and interaction with the different RNA modification and elongation complexes of metazoans be compared to our data? Paramecium Pol II does not show the serine-rich heptamer repeats. Thus, it would be surprising if a regulated and patterned phosphorylation of individual serines would be possible. As the Paramecium CTD is still rich in serines, although not organized in a repeat structure, it still seems likely that phosphorylation could be an activating mark. This needs to be discovered, and we need to note here that our polyclonal serum against one peptide, including unphosphorylated serines, could miss CTD variants being phosphorylated. An argument against this would be that we can detect Pol II, for example, in the center and 3′-regions of genes, where most serines are phosphorylated in mammalian CTDs. It seems quite tempting to speculate that Pol II of Paramecium does not need to be that highly regulated compared with mammals. First of all, alternative splicing is extremely limited; no single example of exon skipping has been reported (Jaillon et al. 2008); and, therefore, the well-positioned nucleosomes do not need to control this. In addition, the data of Gnan et al. (2022) support the idea that the GC content, not nucleosome positioning, contributes to splice efficiency. Introns are recognized by intron definition, and even artificially introduced introns in GFP are efficiently spliced (Jaillon et al. 2008). Our data suggest that introns serve in nucleosome positioning that may permit more intron accumulation in genes, increasing transcription. This would be supported by our data showing that genes with higher intron frequency show higher transcript levels.
Concerning the issues of pausing and elongation, our data suggest pausing to occur, but the pattern is different to other species because we find high levels of Pol II associated with nucleosomes along the entire ORF not only restricted to +1 nucleosomes. Given the fact that +1 nucleosomes are quite prominent, the question raises whether the stops of Pol II at +1 nucleosomes are mechanistically different from stops at all nucleosomes inside the ORF or whether this is a general phenomenon of Paramecium Pol II to stop at nucleosomes, maybe by less efficient elongation. For instance, the tiny introns of Paramecium do not contribute to a significant enlargement of transcriptional units compared with other species with introns, which are often much larger than the exons. It is therefore the question whether Pol II elongation has the need to be highly supported. Paramecium and Tetrahymena miss homologs of NELF, and two recent studies showed the mediator complex, a key regulator of Pol II interaction with transcription and elongation factors, to be highly divergent in Tetrahymena (Garg et al. 2019; Tian et al. 2019). Additionally, in Paramecium we cannot identify all components of the Paf complex regulating elongation, 3′-end processing, and histone modification (Jaehning 2010). Especially, the subunit Paf1, involved in serine phosphorylation of the CTD of Pol II, is missing, which fits to the missing serine repeats of the CTD. Because of the lack of canonical elongation systems going along with a lack of conserved serine residues, we conclude that transcriptional elongation in Paramecium is regulated differently. As discussed above, broad H3K4me3 going along with increased occupancy of Pol II in ORFs might be an alternative control of transcription by buffer domains. It seems tempting to speculate this strange form of Pol II buffering represents an alternative or maybe an ancient form of elongation control.
This is the first description of the Paramecium vegetative chromatin landscape, which appears to be quite different to that of other unicellular eukaryotes and multicellular species. Broad domains along the gene bodies regulate transcription, whereas the noncoding and nonexpressed regions are devoid of epigenetic information. Paradoxically, our data also indicate silent genes to be devoid of epigenetic information, and it has to be clarified if and how the cell prevents spurious Pol II activity at these unoccupied regions. The Pol II distribution we observe is also quite different to other species; the process of transcriptional initiation and elongation appears to be controlled without sophisticated control of CTD phosphorylation and canonical complexes, like NELF, Paf, and Mediator, that assist Pol II in generating mature mRNA. However, this work here attributes to the vegetative nucleus only. We have to keep in mind that the transcriptional machinery needs to switch its mode of action to lncRNA transcription from the meiotic micronuclei during development. As such, functional and temporal dynamics require more alterations of the polymerase complex than in other species. There are plenty of challenges left, especially about the control of Pol II without or with limited CTD phosphorylation. Our study shows the unusual pattern of Pol II in expressed genes and in the light of so many missing interaction partners of Pol II; it is not a surprise that the epigenome looks different from other species in addition to the fact that no mitotic condensation is necessary in the MAC. Concerning Pol II interaction complexes, future studies will need to show whether some components are absent or whether they are too divergent such that reverse genetics cannot identify them. Their identification and contribution to Pol II activity and modulation will shed light on the mechanisms controlling mRNA and lncRNA transcription and the epigenetic marks in support of them. The comparison of the divergent mRNA transcription in Paramecium might unravel new basic principles of how, for example, a gene can be silenced in absence of repressive marks, and these principles might be applicable to understand the regulation of individual genes in other species.
Methods
Cell culture and RNA isolation
P. tetraurelia cells (strain 51) of serotype A were cultured as described before using Klebsiella planticola for regular food in wheat grass powder (WGP) (Simon et al. 2006). All cultures for this study were grown at 31°C. To ensure the vegetative state of the MAC, cells were stained with DAPI.
Genomic annotations
The genomic features shown in Figure 2B are captured from the annotations of the respective organisms, namely, from ParameciumDB (strain 51, version 2), Tetrahymena Genome Database (version 2014) (Stover et al. 2006), PomBase (version 2020) (The Gene Ontology Consortium 2019), and the ensemble database for D. melanogaster (release 98), and Homo sapiens (release 100) (Yates et al. 2020).
Antibodies
ChIP-seq-grade antibodies directed against histone modifications were purchased from Diagenode: H3K9ac C15410004, H3K27me3 C15410195, and H3K4me3 C15410003. For the antibody against P. tetraurelia RPB1, the peptide SPHYTSHTNSPSPSYRSS-C was used for immunization. Purification and testing of specificity by western blots and immunostaining were performed as described recently (Drews et al. 2021). Because there are some amino acid differences in the N-terminal tail of the Paramecium H3P1 to Human H3 (Supplemental Fig. S1A), the peptide PtH3K27me3 TKAARK(me3)TAPAVG was synthesized, and binding affinity of the purchased H3K27me3 antibody to the PtH3k27me3 peptide was verified by dot-blots and competition assays. For details, see Supplemental Methods.
Fixation of cells
Isolation of intact MACs from fixed cells was performed using an adapted NEXSON protocol (Arrigoni et al. 2016). Two to 3 million cells were washed twice in Volvic and starved for 20 min at 31°C. After harvesting (2500 rpm, 2 min), the cell pellet without remaining media was resuspended in 2 mL fixative solution (20 mM Tris-HCL at pH 8, 0.5 mM EGTA, 1 mM EDTA, 10 mM NaCl, 1% methanol-free formaldehyde). After incubation (15 min, room temperature), the reaction was quenched by adding glycine to a final concentration of 125 mM. Cells were centrifuged (3300g, 3 min, 4°C), and the supernatant was discarded. The pellet was washed once in ice-cold PBS buffer and once in PBS buffer supplemented with cOmplete protease inhibitor cocktail, EDTA-free (PIC; Roche 11873580001). Cell suspension was split in half and centrifuged (3300g, 5 min, 4°C), and cell pellets were flash-frozen in liquid nitrogen.
MNase-seq
One aliquot was thawed on ice, resuspended in 2 mL Farnham laboratory buffer (5 mM PIPES at pH 8, 85 mM KCl, 0.5% NP-40), and evenly split into precooled 1.5-mL Bioruptor tubes (Diagenode). After sonication (15 sec on/30 sec off, five cycles, 4°C) using Bioruptor 300 (Diagenode) 5 µL was stained with DAPI to verify isolation of intact MACs. Cell suspension was centrifuged twice (3000g, 5 min, 4°C) with washing of the pellet in Farnham laboratory buffer in between. The following isolation of DNA covered by mononucleosomes was isolated as described previously (Xiong et al. 2016). One aliquot of isolated nuclei was resuspended in 1× MNase buffer (50 mM Tris-HCL at pH 8.0, 5 mM CaCl2) and split into portions of 20,000 nuclei per reaction. After centrifugation (3000g, 5 min, 4°C) nuclei pellets were resupended in 500 µL MNase reaction buffer (50 mM Tris-HCl at pH 8.0, 5 mM CaCl2, 10 mM β-mercaptoethanol, 1% NP-40, 500 ng BSA). To each reaction, 10 or 128 U of MNase (NEB M0247S) was added, and after incubation (10 min, 37°C, 450 rpm), the reaction was stopped (10 mM EGTA, 1 mM EDTA, 5 min, 450 rpm). DNA corresponding to the size of mononucleosomes (100–200 bp) was isolated from a 3% agarose gel using a MinElute gel extraction kit (Qiagen 28604). As input, nuclei were treated with Proteinase K, extracted as described, and treated with 0.1 U or 1.5 U MNase (5 min, 28°C) and extracted again. DNA library preparation was performed using NEBNext Ultra DNA library prep kit for Illumina (NEB E7370) with 10 ng input, 11 PCR cycles, and KAPA Taq HotStart DNA polymerase (Kapa Biosystems KK1512). The MNase-seq read count correlation of four independent replicates, each, used for subsequent analyses can be found in Supplemental Figure S13.
ChIP-seq
Nuclei pellets were resuspended in shearing buffer (10 mM Tris-HCl at pH 8, 0.1% SDS, 1 mM EDTA) and transferred in fresh, precooled Bioruptor tubes. The suspension was sonicated (30 sec on/30 sec off, five cycles, 4°C). After centrifugation (16,000g, 10 min, 4°C), the supernatant was aliquoted in 100-µL portions and stored at −80°C. To control shearing efficiency, 50 µL was decrosslinked using Proteinase K (20 mg/mL), followed by phenol/chloroform/isoamylalcohol extraction, which was repeated after RNase A (10 mg/mL) digestion. Aliquots of 2 µg were run on a 1.5% agarose gel. Eight micrograms of adequately sheared chromatin was subjected to immunoprecipitation using an iDeal ChIP-seq kit for histones (Diagenode C01010050) with 2 µg of antibodies against histone modifications or 10 µg of custom RPB1 antibody. Input was generated by putting 1 µL of chromatin aside without mixing to antibodies. After overnight IP and elution from the magnetic beads, precipitated chromatin and the input kept aside were decrosslinked, RNase A–treated, and extracted as described above. DNA library preparation was performed using a NEBNext Ultra DNA Library Prep Kit for Illumina for serine-rich heptad repeats (NEB E7370) with 10 ng input, 11 PCR cycles, and KAPA Taq HotStart DNA polymerase (Kapa Biosystems KK1512). Precipitated DNA and input DNA were equally handled. ChIP-seq read count correlation of four independent replicates of H3K4me3, H3K27me3, and H3K9ac IP each, used for subsequent analyses, can be found in Supplemental Figure S14.
Sequencing and preprocessing
DNA libraries resulting from MNase digestion and ChIP were sequenced on an Illumina HiSeq 2500 in high-output run mode, and reads were adapter and quality trimmed. For details, see Supplemental Methods. All MNase, Pol II, and histone ChIP-seq reads were aligned to the MAC genome P. tetraurelia (strain 51, version 2) (Arnaiz et al. 2012) after quality control. For details, see Supplemental Methods. We used deepTools2 (Ramírez et al. 2016) to investigate the quality of replicates (multiBamSummary, plotFingerprint, and plotCorrelation tools) with subsequent down-sampling of some histone ChIP replicates, which had rather high coverage (see Supplemental Table S1). We used the DANPOS2 (Chen et al. 2013) software for position or peak calling with default parameters. We used the dpos functionality to call the positions of MNase and Pol II peaks and the dpeak functionality for histone ChIP peak calling. MNase-seq data were normalized to naked DNA inputs, whereas ChIP-seq data were normalized to the respective input files listed in Supplemental Table S1. Further, we made use of the profile functionality of DANPOS2 to visualize how a chromatin feature is distributed in a genomic annotation of interest (see Figs. 3, 4).
Segmentation analysis of chromatin marks
We used ChromHMM (Ernst and Kellis 2012) to perform genome-wide segmentation using the histone marks (H3K27me3, H3K4me3, H3K9ac) and MNase data. The genome was binarized into 200-bp bins based on a Poisson background model using the BinarizeBam function. This was used to learn a CS model with five states using the LearnModel function. We used the plotProfile and plotHeatmap functionality of deepTools2 to create scaled enrichment plots of different chromatin features.
Gene expression and intron data
We used the mRNA expression data of strain 51 wild-type serotype A from our previous work (European Nucleotide Archive [ENA, http://www.ebi.ac.uk/ena] accession number PRJEB9464) (Cheaib et al. 2015). We quantified the expression using Salmon (v0.8.2) (Patro et al. 2017) default parameters for all replicates and used the mean of replicates in all downstream analyses. We used the transcript annotation from the MAC genome of P. tetraurelia (version 2; strain 51) (Arnaiz et al. 2017). For intron profiles, we created a 20-bp window centered on the first and last intron base of the 5′-exon–intron junction and the 3′-intron–exon junction. We plotted the nucleosome profile for 1500 bp around this window with the center of x-axis representing the junctions (see Fig. 4F).
Comparative Pol II analysis and PI
We used the data sets mentioned in Supplemental Table S1 for the comparative Pol II analysis of different organisms shown in Figure 6. We calculated the PI, after applying a threshold on the number of reads in the TSS region of genes (see Supplemental Fig. S8), depending on the distribution of read counts of individual data sets. The thresholds are mentioned in Figure 6C. mRNA quantification was performed using the default parameters of Salmon with transcripts obtained from the respective genomic annotations mentioned above (mean of replicates). We defined a region starting at 30 bp upstream of the TSS until 300 bp downstream from the TSS as the TSS region, and a region starting at 300 bp downstream from the TSS until the TTS as the gene body. The PI is calculated as a ratio of reads (in TPM) in the TSS region compared with reads in the gene body. Genes with a PI greater than 1.5 were considered as paused.
Classification of gene expression using random forests
After removing 1369 silent genes (TPM = 0), we split the remaining genes into 19,090 high (TPM > 2) and 20,001 low expressed genes (TPM > 2). Cut-offs were determined using the first quartile of the distribution of wild-type 51A serotype mRNA expression. For these gene sets, gene body normalized read counts were calculated for H3K27me3, H3K4me3, H3K9ac, Pol II, and MNase, as well as the ratio of H3K4me3 and H3K27me3. We also obtained three genetic features: gene length, intron frequency, and intergenic length. We built a random forests classifier in Python (version 3) using the default parameters available with the scikit-learn package (Pedregosa et al. 2011). We used all available data to train the model using a 40-fold cross-validation (CV) method, and the CV-based PR-AUC was used to evaluate the performance of different models. A PR-AUC of one would represent a perfect model, which 100% of the time would correctly predict whether a gene is highly or lowly expressed. Further, we used the shap package (Lundberg et al. 2020) to calculate the global and local feature importance.
Partial correlation networks
We investigated the partial correlation of any two epigenetic marks of interest after removing the effects of other measured epigenetic marks by using the sparse partial correlation networks method (Lasserre et al. 2013). We used the gene body normalized signals of all the epigenetic marks in this study and the mRNA expression for this analysis.
Analyses of gene expression plasticity
The mean TPM for each gene over different conditions (expression data from serotype A, B, D, and H as well as heat shock conditions) (Cheaib et al. 2015) was calculated. The absolute deviation from the mean for each gene was calculated. We refer to genes with a large fluctuation as plastic genes. For the random forests analysis of plastic genes, we grouped all genes in four groups of roughly similar gene numbers. We performed random down-sampling (five times) of highly or lowly expressed genes such that there is an equal number of genes in both groups for classification.
Data access
All raw read data generated in this study have been submitted to the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena/browser/home) under accession number PRJEB46233.
Supplementary Material
Acknowledgments
We thank Karl Nordstöm for the help with the initial analyses and Salmon DNA sequencing and Laura Arrigoni and Ulrike Boenisch for support with NEXSON and ChIP-seq. We thank Sandra Duharcourt and Melody Matelot for sharing unpublished MNase data sets. This work was supported by grants from the German Research Council (DFG) to M.S. (SI1379/3-1), M.H.S. (3140/1-1), and M.J. (CRC894). A.S. was supported by the German Federal Ministry of Research and Education grant for de.NBI (031L0101D). We acknowledge the support of the Freiburg Galaxy Team, University of Freiburg (Germany) funded by the Collaborative Research Centre 992 Medical Epigenetics (DFG grant SFB 992/1 2012) and the German Federal Ministry of Education and Research BMBF grant 031 A538A de.NBI-RBC.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276126.121.
Competing interest statement
The authors declare no competing interests.
References
- Allen SE, Nowacki M. 2020. Roles of noncoding RNAs in ciliate genome architecture. J Mol Biol 432: 4186–4198. 10.1016/j.jmb.2019.12.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaiz O, Mathy N, Baudry C, Malinsky S, Aury J-M, Wilkes CD, Garnier O, Labadie K, Lauderdale BE, Le Mouäl A, et al. 2012. The Paramecium germline genome provides a niche for intragenic parasitic DNA: evolutionary dynamics of internal eliminated sequences. PLoS Genet 8: e1002984. 10.1371/journal.pgen.1002984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaiz O, Van Dijk E, Bétermier M, Lhuillier-Akakpo M, de Vanssay A, Duharcourt S, Sallet E, Gouzy J, Sperling L. 2017. Improved methods and resources for paramecium genomics: transcription units, gene annotation and gene expression. BMC Genomics 18: 483. 10.1186/s12864-017-3887-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigoni L, Richter AS, Betancourt E, Bruder K, Diehl S, Manke T, Bönisch U. 2016. Standardizing chromatin research: a simple and universal method for ChIP-seq. Nucleic Acids Res 44: e67. 10.1093/nar/gkv1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aury J-M, Jaillon O, Duret L, Noel B, Jubin C, Porcel BM, Ségurens B, Daubin V, Anthouard V, Aiach N, et al. 2006. Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature 444: 171–178. 10.1038/nature05230 [DOI] [PubMed] [Google Scholar]
- Baranasic D, Oppermann T, Cheaib M, Cullum J, Schmidt H, Simon M. 2014. Genomic characterization of variable surface antigens reveals a telomere position effect as a prerequisite for RNA interference-mediated silencing in Paramecium tetraurelia. mBio 5: e01328. 10.1128/mBio.01328-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batsché E, Yaniv M, Muchardt C. 2006. The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nat Struct Mol Biol 13: 22–29. 10.1038/nsmb1030 [DOI] [PubMed] [Google Scholar]
- Benayoun BA, Pollina EA, Ucar D, Mahmoudi S, Karra K, Wong ED, Devarajan K, Daugherty AC, Kundaje AB, Mancini E, et al. 2014. H3k4me3 breadth is linked to cell identity and transcriptional consistency. Cell 158: 673–688. 10.1016/j.cell.2014.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bétermier M, Duharcourt S. 2014. Programmed rearrangement in ciliates: Paramecium. Microbiol Spectr 2: MDNA3-0035-2014. 10.1128/microbiolspec.MDNA3-0035-2014 [DOI] [PubMed] [Google Scholar]
- Böhm S, Östlund Farrants A. 2011. Chromatin remodelling and RNA processing. In RNA processing (ed. Grabowski P), p. 1. IntechOpen, London. 10.5772/20998 [DOI] [Google Scholar]
- Buratowski S. 2009. Progression through the RNA polymerase II CTD cycle. Mol Cell 36: 541–546. 10.1016/j.molcel.2009.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheaib M, Dehghani Amirabad A, Nordström KJ, Schulz MH, Simon M. 2015. Epigenetic regulation of serotype expression antagonizes transcriptome dynamics in Paramecium tetraurelia. DNA Res 22: 293–305. 10.1093/dnares/dsv014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Xi Y, Pan X, Li Z, Kaestner K, Tyler J, Dent S, He X, Li W. 2013. DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res 23: 341–351. 10.1101/gr.142067.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Chen Z, Wu D, Zhang L, Lin X, Su J, Rodriguez B, Xi Y, Xia Z, Chen X, et al. 2015. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat Genet 47: 1149–1157. 10.1038/ng.3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H-R, Dunkel I, Heise F, Linke C, Krobitsch S, Ehrenhofer-Murray AE, Sperling SR, Vingron M. 2011. The effect of micrococcal nuclease digestion on nucleosome positioning data. PLoS One 5: e15754. 10.1371/journal.pone.0015754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drews F, Karunanithi S, Götz U, Marker S, deWijn R, Pirritano M, Rodrigues-Viana AM, Jung M, Gasparoni G, Schulz MH, et al. 2021. Two Piwis with Ago-like functions silence somatic genes at the chromatin level. RNA Biol 18: 757–769. 10.1080/15476286.2021.1991114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Cohen J, Jubin C, Dessen P, Goût J-F, Mousset S, Aury J-M, Jaillon O, Nöel B, Arnaiz O, et al. 2008. Analysis of sequence variability in the macronuclear DNA of Paramecium tetraurelia: a somatic view of the germline. Genome Res 18: 585–596. 10.1101/gr.074534.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff S, Murphy S. 2008. Cracking the RNA polymerase II CTD code. Trends Genet 24: 280–288. 10.1016/j.tig.2008.03.008 [DOI] [PubMed] [Google Scholar]
- Ernst J, Kellis M. 2012. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods 9: 215–216. 10.1038/nmeth.1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, Allis CD. 2005. Regulation of hp1–chromatin binding by histone h3 methylation and phosphorylation. Nature 438: 1116–1122. 10.1038/nature04219 [DOI] [PubMed] [Google Scholar]
- Frapporti A, Pina CM, Arnaiz O, Holoch D, Kawaguchi T, Humbert A, Eleftheriou E, Lombard B, Loew D, Sperling L, et al. 2019. The Polycomb protein Ezl1 mediates H3K9 and H3K27 methylation to repress transposable elements in Paramecium. Nat Commun 10: 2710. 10.1038/s41467-019-10648-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furrer DI, Swart EC, Kraft MF, Sandoval PY, Nowacki M. 2017. Two sets of Piwi proteins are involved in distinct sRNA pathways leading to elimination of germline-specific DNA. Cell Rep 20: 505–520. 10.1016/j.celrep.2017.06.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg J, Saettone A, Nabeel-Shah S, Cadorin M, Ponce M, Marquez S, Pu S, Greenblatt J, Lambert J-P, Pearlman RE, et al. 2019. The Med31 conserved component of the divergent mediator complex in Tetrahymena thermophila participates in developmental regulation. Curr Biol 29: 2371–2379.e6. 10.1016/j.cub.2019.06.052 [DOI] [PubMed] [Google Scholar]
- The Gene Ontology Consortium. 2019. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res 47: D330–D338. 10.1093/nar/gky1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnan S, Matelot M, Weiman M, Arnaiz O, Guérin F, Sperling L, Bétermier M, Thermes C, Chen C-L, Duharcourt S. 2022. GC content but not nucleosome positioning directly contributes to intron-splicing efficiency in Paramecium. Genome Res (this issue) 32: 699–709. 10.1101/gr.276125.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz U, Marker S, Cheaib M, Andresen K, Shrestha S, Durai DA, Nordström KJ, Schulz MH, Simon M. 2016. Two sets of RNAi components are required for heterochromatin formation in trans triggered by truncated transgenes. Nucleic Acids Res 44: 5908–5923. 10.1093/nar/gkw267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guérin F, Arnaiz O, Boggetto N, Wilkes CD, Meyer E, Sperling L, Duharcourt S. 2017. Flow cytometry sorting of nuclei enables the first global characterization of Paramecium germline DNA and transposable elements. BMC Genomics 18: 327. 10.1186/s12864-017-3713-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy A, Matelot M, Touzeau A, Klopp C, Lopez-Roques C, Duharcourt S, Defrance M. 2021. DNAModAnnot: a R toolbox for DNA modification filtering and annotation. Bioinformatics 37: 2738–2740. 10.1093/bioinformatics/btab032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Smothers JF, Wiley EA, Allis CD. 1999. A nonessential HP1-like protein affects starvation-induced assembly of condensed chromatin and gene expression in macronuclei of Tetrahymena thermophila. Mol Cell Biol 19: 3624–3634. 10.1128/MCB.19.5.3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignarski M, Singh A, Swart EC, Arambasic M, Sandoval PY, Nowacki M. 2014. Paramecium tetraurelia chromatin assembly factor-1-like protein PtCAF-1 is involved in RNA-mediated control of DNA elimination. Nucleic Acids Res 42: 11952–11964. 10.1093/nar/gku874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara S, Sasagawa Y, Kameda T, Yamashita H, Umeda M, Kotomura N, Abe M, Shimono Y, Nikaido I. 2021. Local states of chromatin compaction at transcription start sites control transcription levels. Nucleic Acids Res 49: 8007–8023. 10.1093/nar/gkab587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaehning JA. 2010. The Paf1 complex: platform or player in RNA polymerase II transcription? Biochim Biophys Acta 1799: 379–388. 10.1016/j.bbagrm.2010.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon O, Bouhouche K, Gout J-F, Aury J-M, Noel B, Saudemont B, Nowacki M, Serrano V, Porcel BM, Ségurens B, et al. 2008. Translational control of intron splicing in eukaryotes. Nature 451: 359–362. 10.1038/nature06495 [DOI] [PubMed] [Google Scholar]
- Jimeno-González S, Reyes JC. 2016. Chromatin structure and pre-mRNA processing work together. Transcription 7: 63–68. 10.1080/21541264.2016.1168507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm SL, Shipony Z, Greenleaf WJ. 2019. Chromatin accessibility and the regulatory epigenome. Nat Rev Genet 20: 207–220. 10.1038/s41576-018-0089-8 [DOI] [PubMed] [Google Scholar]
- Lasserre J, Chung H-R, Vingron M. 2013. Finding associations among histone modifications using sparse partial correlation networks. PLoS Comput Biol 9: e1003168. 10.1371/journal.pcbi.1003168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lhuillier-Akakpo M, Guérin F, Frapporti A, Duharcourt S. 2016. DNA deletion as a mechanism for developmentally programmed centromere loss. Nucleic Acids Res 44: 1553–1565. 10.1093/nar/gkv1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg SM, Erion G, Chen H, DeGrave A, Prutkin JM, Nair B, Katz R, Himmelfarb J, Bansal N, Lee S-I. 2020. From local explanations to global understanding with explainable AI for trees. Nat Mach Intell 2: 56–67. 10.1038/s42256-019-0138-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo G-Z, Hao Z, Luo L, Shen M, Sparvoli D, Zheng Y, Zhang Z, Weng X, Chen K, Cui Q, et al. 2018. N6-Methyldeoxyadenosine directs nucleosome positioning in Tetrahymena DNA. Genome Biol 19: 200. 10.1186/s13059-017-1381-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer CA, Liu XS. 2014. Identifying and mitigating bias in next-generation sequencing methods for chromatin biology. Nat Rev Genet 15: 709–721. 10.1038/nrg3788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalabothula N, McVicker G, Maiorano J, Martin R, Pritchard JK, Fondufe-Mittendorf YN. 2014. The chromatin architectural proteins HMGD1 and H1 bind reciprocally and have opposite effects on chromatin structure and gene regulation. BMC Genomics 15: 92. 10.1186/1471-2164-15-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordström KJ, Schmidt F, Gasparoni N, Salhab A, Gasparoni G, Kattler K, Müller F, Ebert P, Costa IG, DEEP consortium, et al. 2019. Unique and assay specific features of NOMe-, ATAC- and DNase I-seq data. Nucleic Acids Res 47: 10580–10596. 10.1093/nar/gkz799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan G, Tian S, Nie J, Yang C, Ruotti V, Wei H, Jonsdottir GA, Stewart R, Thomson JA. 2007. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell 1: 299–312. 10.1016/j.stem.2007.08.003 [DOI] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. 2017. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14: 417–419. 10.1038/nmeth.4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. 2011. Scikit-learn: machine learning in Python. J Mach Learn Res 12: 2825–2830. [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T. 2016. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44: W160–W165. 10.1093/nar/gkw257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell CB, Fraga D, Hinrichsen RD. 1994. Extremely short 20–33 nucleotide introns are the standard length in Paramecium tetraurelia. Nucleic Acids Res 22: 1221–1225. 10.1093/nar/22.7.1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzeszutek I, Maurer-Alcala X, Nowacki M. 2020. Programmed genome rearrangements in ciliates. Cell Mol Life Sci 77: 4615–4629. 10.1007/s00018-020-03555-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel C, Mackie J, Sommerville J. 1981. Macronuclear chromatin organization in Paramecium primaurelia. Chromosoma 83: 481–492. 10.1007/BF00328274 [DOI] [PubMed] [Google Scholar]
- Schulman IG, Cook RG, Richman R, Allis CD. 1987. Tetrahymena contain two distinct and unusual high mobility group (HMG)-like proteins. J Cell Biol 104: 1485–1494. 10.1083/jcb.104.6.1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon M, Plattner H. 2014. Unicellular eukaryotes as models in cell and molecular biology: critical appraisal of their past and future value. Int Rev Cell Mol Biol 309: 141–198. 10.1016/B978-0-12-800255-1.00003-X [DOI] [PubMed] [Google Scholar]
- Simon MC, Marker S, Schmidt HJ. 2006. Posttranscriptional control is a strong factor enabling exclusive expression of surface antigens in Paramecium tetraurelia. Gene Expr 13: 167–178. 10.3727/000000006783991809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Vancura A, Woycicki RK, Hogan DJ, Hendrick AG, Nowacki M. 2018. Determination of the presence of 5-methylcytosine in Paramecium tetraurelia. PLoS One 13: e0206667. 10.1371/journal.pone.0206667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneppen K, Ringrose L. 2019. Theoretical analysis of Polycomb-Trithorax systems predicts that poised chromatin is bistable and not bivalent. Nat Commun 10: 2133. 10.1038/s41467-019-10130-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover NA, Krieger CJ, Binkley G, Dong Q, Fisk DG, Nash R, Sethuraman A, Weng S, Cherry JM. 2006. Tetrahymena genome database (TGD): a new genomic resource for Tetrahymena thermophila research. Nucleic Acids Res 34: D500–D503. 10.1093/nar/gkj054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian M, Mochizuki K, Loidl J. 2019. Non-coding RNA transcription in Tetrahymena meiotic nuclei requires dedicated mediator complex-associated proteins. Curr Biol 29: 2359–2370.e5. 10.1016/j.cub.2019.05.038 [DOI] [PubMed] [Google Scholar]
- Tillo D, Hughes TR. 2009. G+C content dominates intrinsic nucleosome occupancy. BMC Bioinformatics 10: 442. 10.1186/1471-2105-10-442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chen X, Sheng Y, Liu Y, Gao S. 2017. N6-Adenine DNA methylation is associated with the linker DNA of H2A.Z-containing well-positioned nucleosomes in Pol II-transcribed genes in Tetrahymena. Nucleic Acids Res 45: 11594–11606. 10.1093/nar/gkx883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong J, Gao S, Dui W, Yang W, Chen X, Taverna SD, Pearlman RE, Ashlock W, Miao W, Liu Y. 2016. Dissecting relative contributions of cis- and trans-determinants to nucleosome distribution by comparing Tetrahymena macronuclear and micronuclear chromatin. Nucleic Acids Res 44: 10091–10105. 10.1093/nar/gkw684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates AD, Achuthan P, Akanni W, Allen J, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Azov AG, Bennett R, et al. 2020. Ensembl 2020. Nucleic Acids Res 48: D682–D688. 10.1093/nar/gkz966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Liu Y. 2019. Transcription regulation: tales of a divergent mediator. Curr Biol 29: R685–R688. 10.1016/j.cub.2019.06.033 [DOI] [PubMed] [Google Scholar]
- Zhao XD, Han X, Chew JL, Liu J, Chiu KP, Choo A, Orlov YL, Sung W-K, Shahab A, Kuznetsov VA, et al. 2007. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell 1: 286–298. 10.1016/j.stem.2007.08.004 [DOI] [PubMed] [Google Scholar]
- Zhou VW, Goren A, Bernstein BE. 2011. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Gene 12: 7–18. 10.1038/nrg2905 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.