

Abstract
There is growing interest in the potential use of phosphodiesterase (PDE) inhibitors for colorectal cancer (CRC) prevention and treatment. The present study has tested the idea that PDE inhibitors inhibit growth and viability of CRC cell lines by increasing cyclic guanosine monophosphate (cGMP) and activating cGMP-dependent protein kinase (PKG). Colon cancer cell lines and those with ectopic PKG2 expression were treated with membrane-permeable 8Br-cGMP or inhibitors of PDE5, PDE9, and PDE10a. Levels of cGMP capable of activating PKG were measured by immunoblotting for phosphorylation of vasodilator-stimulated phosphoprotein (VASP). The effects of treatment on CRC cell proliferation and death were measured using hemocytometry with trypan blue. Treatment with 8Br-cGMP had no effect on CRC cell proliferation or death. Endogenous PKG activity was undetectable in any of the CRC cells, but expression of ectopic PKG2 conferred modest inhibition of proliferation but did not affect cell death. Extremely high concentrations of all the PDE inhibitors reduced proliferation in CRC cell lines, but none of them increased cGMP levels, and the effect was independent of PKG expression. The inability of the PDE inhibitors to increase cGMP was due to the lack of endogenous cGMP generating machinery. In conclusion, PDE inhibitors that target cGMP only reduce CRC growth at clinically unachievable concentrations, and do so independent of cGMP signaling through PKG.
SIGNIFICANCE STATEMENT
A large number of in vitro studies have reported that PDE inhibitors block growth of colon cancer cells by activating cGMP signaling, and that these drugs might be useful for cancer treatment. Our results show that these drugs do not activate cGMP signaling in colon cancer cells due to a lack of endogenous guanylyl cyclase activity, and that growth inhibition is due to toxic effects of clinically unobtainable drug concentrations.
Introduction
Cancers of the colon and rectum (CRC) are some of the most commonly diagnosed, and are responsible for 9% of all cancer deaths due to the late stage of diagnosis, when treatments are largely ineffective. It is therefore widely recognized that both primary prevention and the development of new therapeutics to treat advanced CRC are top priorities to reduce the health burden. Phosphodiesterases (PDEs) comprise a large family of enzymes that degrade cyclic nucleotides. They are also established therapeutic targets owing to diverse expression and functions in a variety of biologic processes (Lugnier, 2006; Maurice et al., 2014). A large and growing body of literature supports the idea that PDE inhibitors have antitumor effects in the gastrointestinal tract.
Some of the earliest studies demonstrated that a weak PDE5 inhibitor called sulindac sulphone (exisulind) could cause regression of colorectal polyps in human patients (Stoner et al., 1999; van Stolk et al., 2000). While exisulind was not approved for use in patients owing to toxicity at levels required for therapeutic effects (Stoner et al., 1999; van Stolk et al., 2000), efforts continued to understand the antitumor mechanism. Exisulind was reported to inhibit growth and induce apoptosis of colon cancer cell lines grown in vitro by increasing cGMP and activating type-1 cGMP-dependent protein kinase (PKG1) (Thompson et al., 2000; Joe et al., 2003). PKG1-dependent suppression of Wnt/β-catenin signaling and activation of cJun-N-terminal kinase were shown to mediate the proapoptotic effects of PDE5 inhibition (Soh et al., 2000; Thompson et al., 2000; Whitt et al., 2012; Li et al., 2013). While PDE5 is specific to cGMP, more recently, inhibition of the dual-specificity enzyme PDE10A has also been reported to inhibit colon cancer cell growth by a cGMP-dependent inhibition of β-catenin/T cell factor signaling (Li et al., 2015a; Li et al., 2015b; Lee et al., 2016).
Observations made by several independent groups are incongruent with the idea that activation of cGMP/PKG signaling by PDE inhibitors can inhibit colon cancer cell growth and viability. It has been suggested that the anticancer effect of exisulind is independent of cGMP signaling (Pitari et al., 2006). Others have shown that PDE5 inhibitors can synergize with the cytotoxic effects of chemotherapeutic drugs, but do not affect growth of colon cancer cell lines when used as a single agent (Booth et al., 2014; Booth et al., 2016; Booth et al., 2017). The importance of PKG downstream of PDE5 inhibitors has also been questioned, since both isoforms (PKG1 and PKG2) have been shown to be downregulated in colon cancer tissue compared with normal colon epithelium and are undetectable in colon cancer cell lines (Hou et al., 2006; Wang et al., 2012). In normal colon epithelium, cGMP is produced upon activation of the receptor guanylyl-cyclase (GC-C) by the peptide hormones guanylin and uroguanylin (Forte, 2004). Epithelial PDE5 antagonizes the effects of GC-C agonists by degrading cGMP (Bakre et al., 2000). Treatment of mice with PDE5 inhibitors has been shown to increase epithelial cGMP levels and suppress the proliferation in the colon crypt in a PKG2-dependent manner (Wang et al., 2014). In addition, it was recently reported that treatment with the PDE5 inhibitor sildenafil can inhibit carcinogenesis in two different models of colon cancer in mice (Islam et al., 2017; Sharman et al., 2018). In contrast to in vitro studies with colon cancer cell lines, the in vivo inhibition of carcinogenesis was shown to result from suppression of polyp initiation, and that sildenafil treatment did not affect the growth of existing tumors (Islam and Browning, 2018).
The present study has tested the idea that treatment of colon cancer cells with PDE inhibitors in vitro can inhibit growth by increasing cGMP and activating PKG signaling. PDE inhibitors were shown to reduce proliferation of colon cancer cell lines at extremely high concentrations that are clinically irrelevant. However, even at the high levels of PDE inhibitors, treatment did not activate cGMP signaling because the colon cancer cell lines lack endogenous cGMP-generating machinery. Taken together, it is concluded physiologic doses of PDE inhibitors do not activate cGMP signaling or affect the proliferation of colon cancer cell lines grown in vitro.
Materials and Methods
Tissue Culture and Reagents
The colon cancer cell lines LS174T, Caco2, HCT116, HT29, and SW480 were obtained from the American Type Culture Collection (ATCC); maintained in 5% CO2 in RMPI-1640 medium containing 10% FBS; and supplemented with 2 mM L-glutamine, 10 IU/ml penicillin, and 10 µg/ml streptomycin. ATCC-derived colon cancer cell lines were made inducible for type 2 PKG expression using a second-generation tetracycline-inducible system based on the Tet-On-Advanced and pLVX-puro vector systems (Takara Bio, Mountain View, CA). Packaging was with pLP1, pLP2, and pLP/VSVG vectors, and virus production using 293FT cells was carried out as detailed in the manufacturer instructions (ViraPower; Thermo Fisher Scientific). High titers of both viruses were mixed and used to transduce cell lines using polybrene, after which populations of lentiviral-infected cells were selected for using cell-optimized G418 concentrations and 2 µg/ml puromycin. Parallel transductions using GFP-expressing viruses indicated close to 100% expression in all cell lines. Inducible cell lines were subsequently cultured using_maintenance medium supplemented with 200 µg/ml of G418 and 0.2 µg/ml of puromycin. Before experimentation, the cells were grown up for one passage in the absence of antibiotics.
The doxycycline, puromycin, G418, and 8Br-cGMP were from Sigma Chemical (St. Louis, MO) and all other chemicals were from Thermo Fisher Scientific. The sildenafil citrate (Revatio) and Linaclotide (Linzess) were extracted from pharmacy-grade pills/capsules in water and stored as aliquots at −80°C. TAK-063 (PDE-10a inhibitor) and PF-04447943 (PDE-9 inhibitor) were from MedChemExpress (Monmouth Junction, NJ).
Cell Proliferation and Viability Assays
To measure cell growth, triplicate wells of 12-well plates were seeded with 5 × 104 cells, and the next day a baseline cell number (day 0) and viability were determined using a TC-20 cell counter with trypan blue (BioRad, Hercules, CA). For all cell lines, the density was estimated to be less than 10% confluent on day 0 and subconfluent on day 3. To quantitate the cells, the medium was removed, and after 5 minutes in trypsin the medium and any floating cells were added back to the well. The cells in the mixture were then pelleted by centrifugation and resuspended in PBS. Drugs to be tested were added to the wells on day 0 and cell counting was carried out on day 3. For quantitation of cell lines with inducible PKG2, doxycycline (0.2 µg/ml) was added prior to seeding and was replaced on day 0. Each experiment containing triplicate wells for each condition was repeated at least three times. Cell numbers measured at day 3 were normalized as a percentage of the mean control value obtained for untreated cells in each replicate. This was done to minimize the variation in cell counts at day 0, which is at the lower range for the TC-20 hemocytometer.
Western Blotting
Cells were lysed by incubation in ice-cold lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 0.25% deoxycholate) supplemented with a protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Clarified protein extracts were quantified using a bicinchoninic acid protein quantification kit (Thermo Fisher Scientific). Proteins were then separated on 10% polyacrylamide minigels (BioRad, Hercules, CA), transferred to nitrocellulose membranes and blocked with 5% bovine serum albumin in PBS containing 0.025% Tween 20. Antibodies used for immunoblotting: β-actin (1:2000, Sigma), PKG2 (mAb E7; 1:200, Santa Cruz Biotechnology), β-catenin C-terminus (BD Biosciences, San Jose, CA). Phospho-VASP (Ser239), VASP, ERK-P, extracellular signal-regulated kinase (ERK), AKT-P, and AKT were all from Cell Signaling (1:1000; Danvers, MA). Blots were visualized on film using Pierce-ECL substrate (Thermo Fisher Scientific). Select film exposures were scanned and bands on the grayscale images were quantitated using ImageJ software. For semiquantitative analysis, the band intensities were normalized to β-actin (β-catenin) or to the total protein level (ERK-P and AKT-P) for each treatment.
Statistical Analysis
The cell proliferation data were calculated as a percentage of the control (untreated) cells for each experiment, and are expressed as mean ± S.D. All cell proliferation and death data were analyzed for significance using a one-sided ANOVA followed by Dunnett’s multiple comparisons test comparing to the untreated control. The differences were considered to be significant when p < 0.05.
Results
Intracellular cGMP Does Not Inhibit Colon Cancer Cell Proliferation in the Absence of Ectopic PKG2 Expression
Several studies have reported that PDE inhibitors inhibit colon cancer cell proliferation, with the implication that elevated cGMP levels and activation of PKG mediate the effect. To directly interrogate the effects of increased intracellular cGMP on colon cancer cell growth, the membrane-permeable, PDE-resistant analog 8Br-cGMP was used. Treatment of parental LS174T colon cancer cells with 8Br-cGMP did not cause phosphorylation of VASP at the PKG-preferred site, Ser239, which demonstrates the absence of endogenous PKG. Expression of ectopic PKG2 in these cells (LS174T-PKG2) conferred a dose-dependent phosphorylation of VASP in response to 8Br-cGMP (Fig. 1A). The lower mobility band observed at higher concentrations of 8Br-cGMP represents additional phosphorylation of Ser157 that causes a change in mobility on SDS-PAGE gels (Smolenski et al., 1998). Treatment with 8Br-cGMP did not affect proliferation or viability of the parental LS174T cells. However, 8Br-cGMP dose-dependently inhibited proliferation of the LS174T-PKG2 cells, with a maximum effect of 30% at 10 µM, but did not affect cell death (Fig. 1B, C). This effect was not unique to LS174T cells as similar studies using Caco2 and HCT116 cells were also unable to detect PKG activity, and there was no observed effect of cGMP on proliferation or cell death in the parental cell lines (Fig. 2). Ectopic expression of PKG2 conferred reduced proliferation of HCT116 cells and viability in Caco2 cells in response to 8Br-cGMP treatment, but these effects were modest.
Fig. 1.
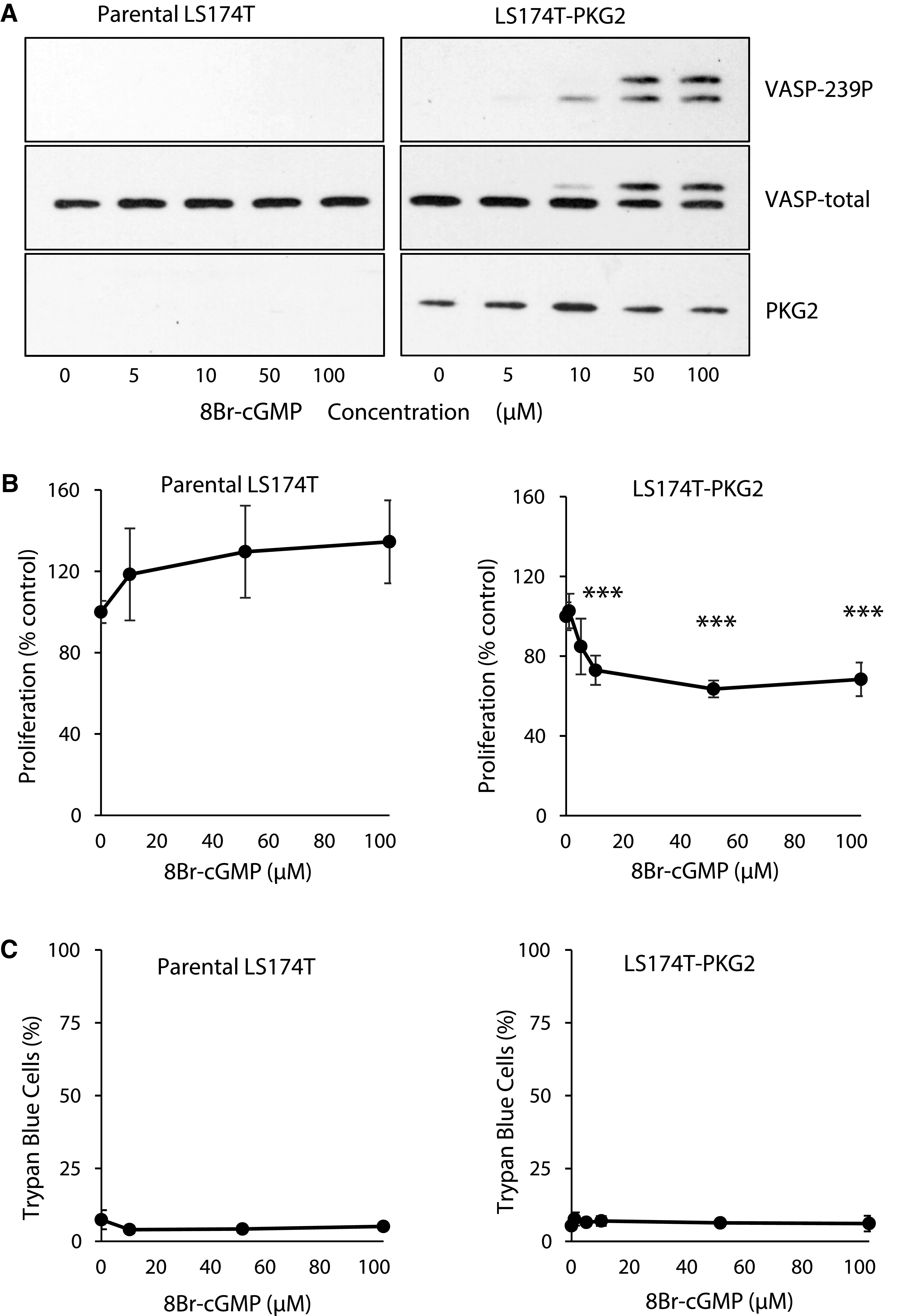
Ectopic PKG2 is required for cGMP to inhibit the growth of LS174T colon cancer cells. Parental LS174T colon cancer cells and those engineered to express ectopic PKG2 were treated with 8Br-cGMP at the indicated concentrations. (A) The cGMP signaling was measured by immunoblotting for VASP phosphorylation at Ser239 (VASP-239P). The effect of 8Br-cGMP of parental and PKG2-expressing LS174T cells on (B) cell proliferation and (C) cell viability. Data show means, and error bars are S.D. ***P < 0.0001.
Fig. 2.
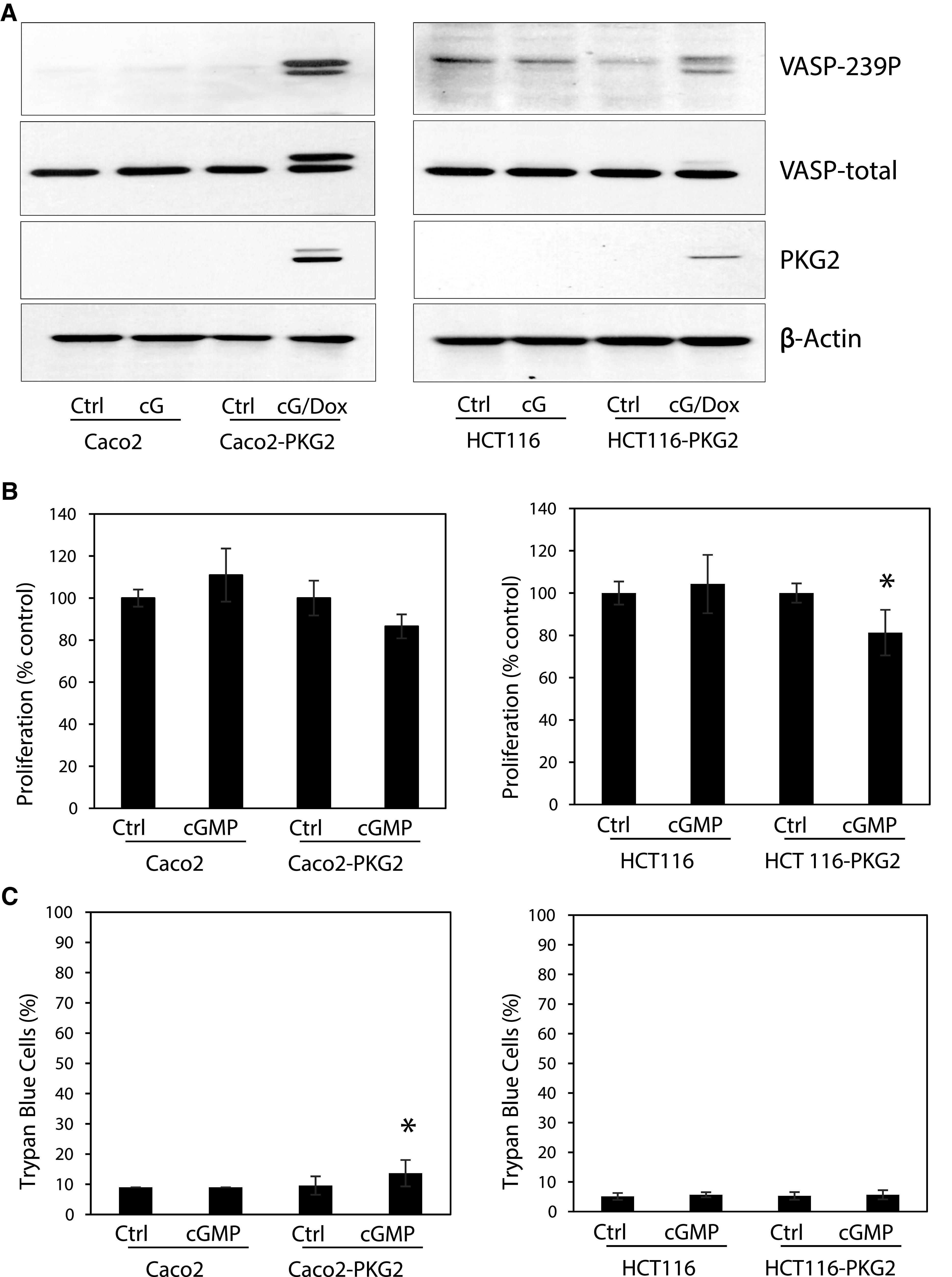
The inability of cGMP to affect proliferation is common among colon cancer cell lines. (A) Parental Caco2 and HCT116 colon cancer cells and those with inducible PKG2 expression were treated with 8Br-cGMP (100 µM) and doxycycline (Dox) to induce PKG2 expression as indicated. The cGMP signaling was measured by immunoblotting for VASP phosphorylation at Ser239 (VASP-239P). The effect of 8Br-cGMP treatment on (B) cell proliferation (hemocytometer), and (C) cell viability (trypan blue exclusion) is shown. Data show means, and error bars are S.D. *P < 0.05.
PDE Inhibitors Suppress Colon Cancer Cell Growth Independent of cGMP/PKG2
The inability of high levels of intracellular cGMP to significantly affect proliferation or viability in several colon cancer cell lines is incongruent with published reports describing the growth-inhibitory effects of cGMP-elevating PDE inhibitors (Thompson et al., 2000; Li et al., 2013; Li et al., 2015b; Lee et al., 2016). To investigate this discrepancy, inhibitors of cGMP-specific PDE5 (sildenafil) and PDE9 (PF-04447943), as well as dual specificity PDE10A (TAK-063) were used to treat parental and PKG-expressing LS174T cells (Fig. 3). Surprisingly, all three inhibitors inhibited cell proliferation at concentrations higher than 10 µM in both parental and PKG2-expressing cells (Fig. 3A). At the highest dose (200 µM), the inhibition of proliferation in the parental LS174T cells was least for sildenafil (50%) and maximal for Pf-04447943 (80%), with TAK-063 in between (65%). Results were similar in the LS174T-PKG2 cells, except that sildenafil was less effective (25% inhibition). Trypan blue staining showed that these drugs had marginal effects on cell viability that were typically less than 20% at all concentrations, except for the highest concentration of TAK-063, which was toxic (Fig. 3B). The ability of the PDE inhibitors to suppress proliferation in the parental cells contrasted with the effect of 8Br-cGMP, which did not affect parental cells, and only moderately inhibited growth in PKG2-expressing cells. These observations indicated that the antiproliferative effect of PDE inhibitors might be independent of cGMP and PKG. To test this, the ability of the PDE inhibitors to activate PKG in LS174T cells was determined using VASP phosphorylation as a readout (Fig. 3C). While 8Br-cGMP treatment caused robust VASP phosphorylation in the LS174T-PKG2 cells, none of the PDE inhibitors were effective except for 50 µM TAK-063, which caused a faint band in some experiments. This effect was not unique to LS174T cells, as similar results were obtained using Caco2 and HCT116 cells (Fig. 4). Treatment of HCT116 or Caco2 with 50 µM of the PDE inhibitors reduced proliferation to varying degrees in both parental and PKG2-expressing cells, with TAK-063 as the most effective in both cell lines. However, none of the PDE inhibitors caused VASP phosphorylation, indicating that the antiproliferative effect was independent of both cGMP and PKG.
Fig. 3.
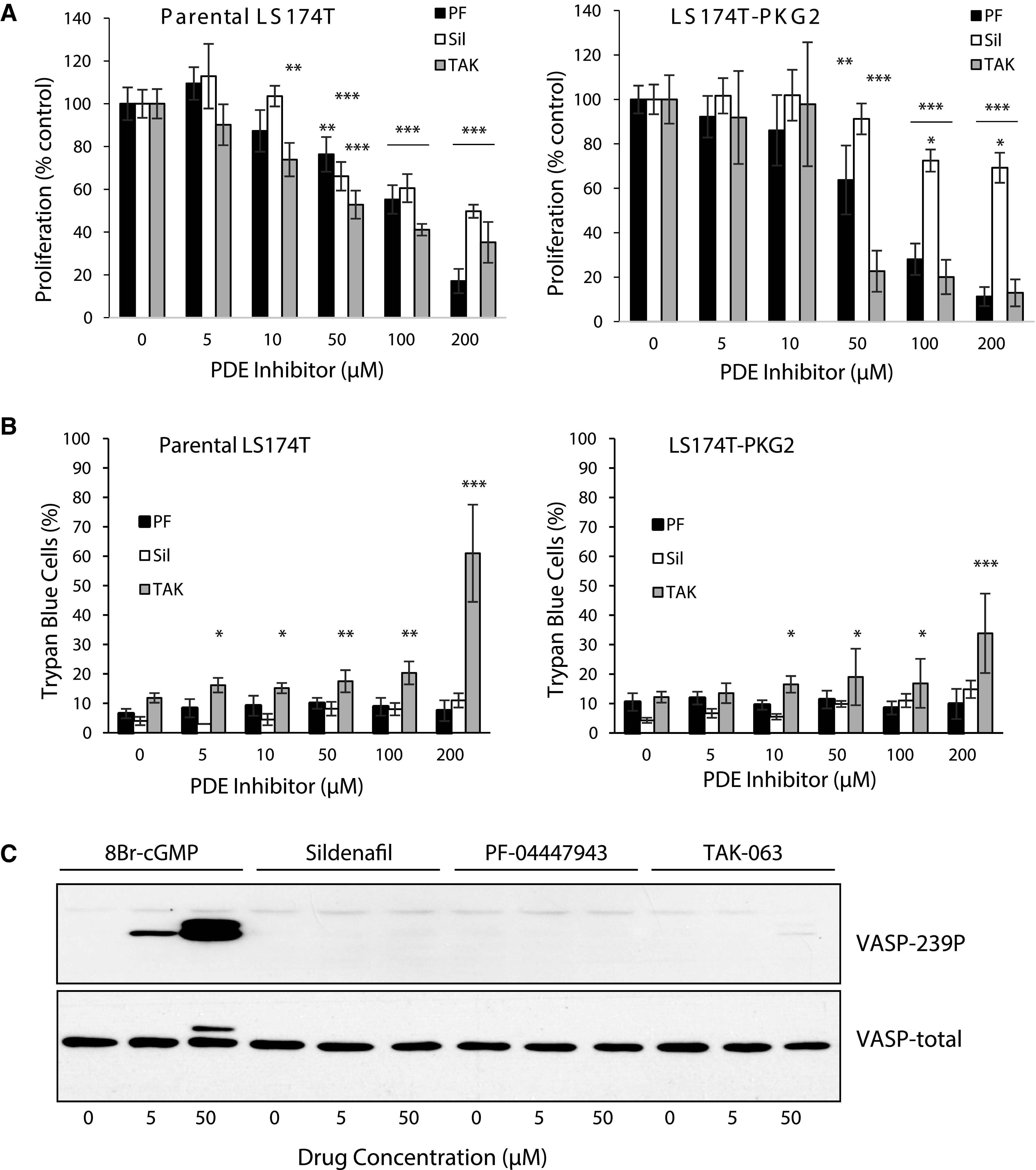
High doses of cGMP-PDE inhibitors suppress growth of LS174T colon cancer cells by a cGMP-independent mechanism. Parental and PKG2-expressing LS174T colon cancer cells were treated for 3 days with different doses of PDE inhibitors. Sildenafil (Sil)-PDE5, PF-04447943 (PF)-PDE9, and TAK-063 (TAK)-PDE10a. The effect of PDE inhibitor treatment on (A) cell proliferation (hemocytometer), and (B) cell viability (trypan blue exclusion). (C) The effect of PDE inhibitors on cGMP levels in LS-174T-PKG2 cells was measured by immunoblotting for phosphorylation of VASP (VASP-239P). Data show means, and error bars are S.D. ***P < 0.0001, **P < 0.005, *P < 0.05.
Fig. 4.
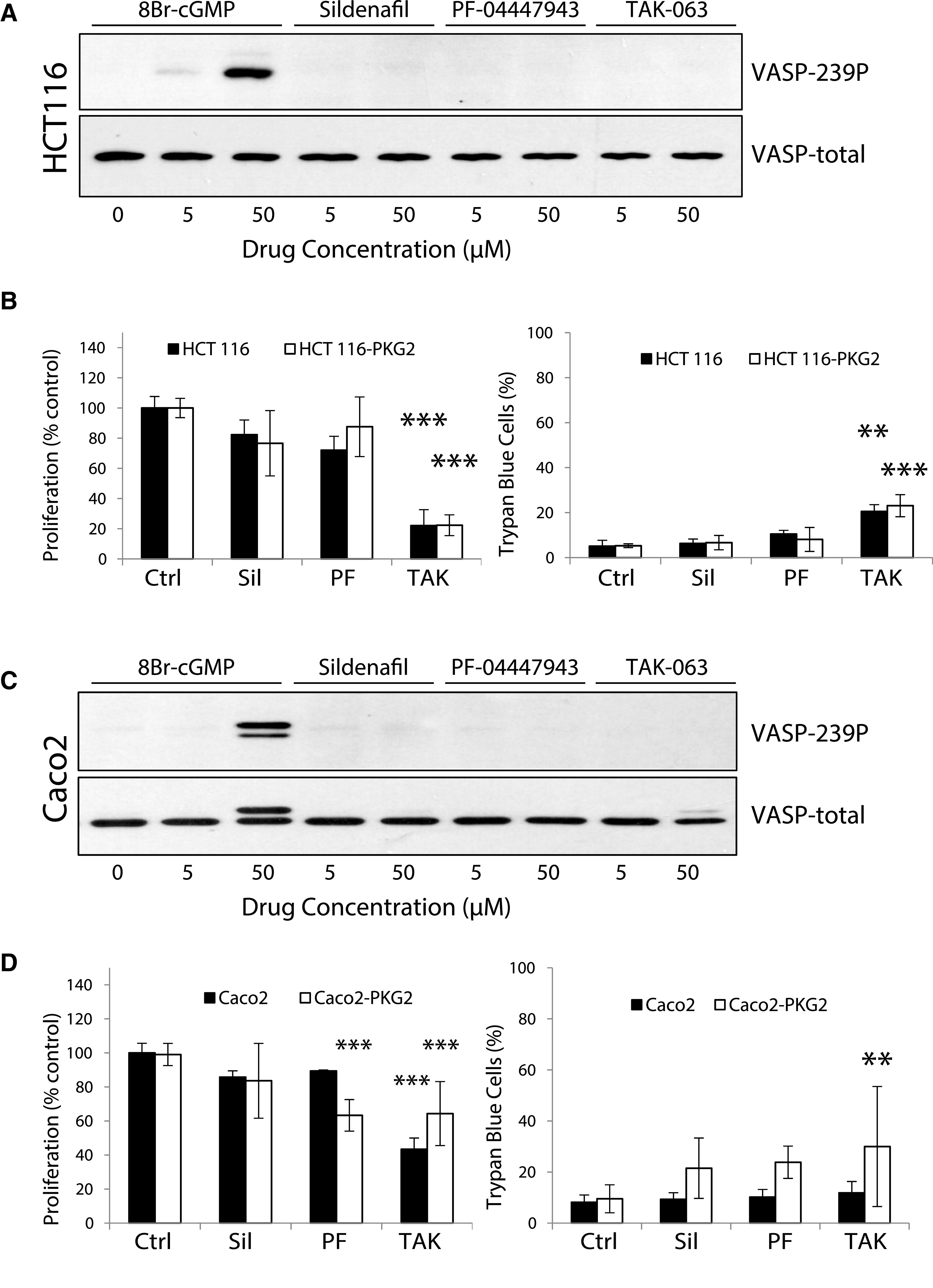
The cGMP/PKG-independent inhibition of proliferation by PDE-inhibitors is a common feature of colon cancer cell lines. HCT116 and Caco2 colon cancer cells were either untreated or treated with the PDE5 inhibitor sildenafil (Sil), the PDE9 inhibitor PF-04447943 (PF), and the PDE10a inhibitor TAK-063 (TAK) as indicated. The ability of the drugs to increase cGMP in (A) HCT116-PKG2 and (C) Caco2-PKG2 was measured by immunoblotting for VASP phosphorylation at Ser239 (VASP-239P) using 8Br-cGMP as a positive control. The effect of 50 µM of the PDE inhibitors on proliferation and viability was measured in both parental and PKG2-expressing (B) HCT116 and (D) Caco2 colon cancer cells. Cells were treated with 50 µM of Sil, PF, or TAK for 3 days. Cell death was measured using trypan blue exclusion. Data show means, and error bars are S.D. ***P < 0.0001, **P < 0.005.
Colon Cancer Cell Lines Lack Endogenous cGMP-Generating Machinery
The inability of the PDE inhibitors to elicit a functional cGMP response as measured by PKG-dependent phosphorylation of VASP was unanticipated based upon previous studies (Thompson et al., 2000; Li et al., 2013; Lee et al., 2016). Phosphodiesterases antagonize guanylyl-cyclases by hydrolytically cleaving the cGMP to produce GMP. The source of cGMP in the colon epithelium is membrane-bound GC-C following activation by guanylin (Forte, 2004). GC-C is maintained in colon tumors (Carrithers et al., 1996; Cagir et al., 1999; Birbe et al., 2005), but expression is more variable in colon cancer cell lines (Waldman et al., 1998). However, the GC-C agonist guanylin is rapidly downregulated during tumorigenesis (Cohen et al., 1998; Steinbrecher et al., 2000; Wilson et al., 2014). To determine whether functional GC-C is expressed in colon cancer cell lines, we measured VASP phosphorylation following treatment with the GC-C agonist linaclotide in combination with the PDE5 inhibitor sildenafil in different colon cancer cell lines that we engineered to express ectopic PKG2 (Fig. 5A). Out of four cell lines that were used in previous studies of PDE inhibitors, only LS174T and Caco2 produced phospho-VASP bands that were comparable to positive controls treated with 8Br-cGMP. LS174T cells expressing ectopic PKG2 were subsequently treated with different doses of linaclotide to identify the lowest concentration capable of activating cGMP/PKG signaling (Fig. 5B). The cells were then treated with the PDE inhibitors, together with the low dose of linaclotide (10 nM), to determine whether these drugs could amplify the basal cGMP production (Fig. 5C). Sildenafil produced a robust VASP phosphorylation at both 1 and 10 µM, whereas the PF-04447943 and TAK-063 were much less effective, with weak bands appearing only at the higher 10 μM dose. Taken together, these results strongly support the idea that the growth-inhibitory effects of PDE inhibitors in colon cancer cells does not involve activation of cGMP signaling, and is more likely to result from off-target effects at higher doses.
Fig. 5.
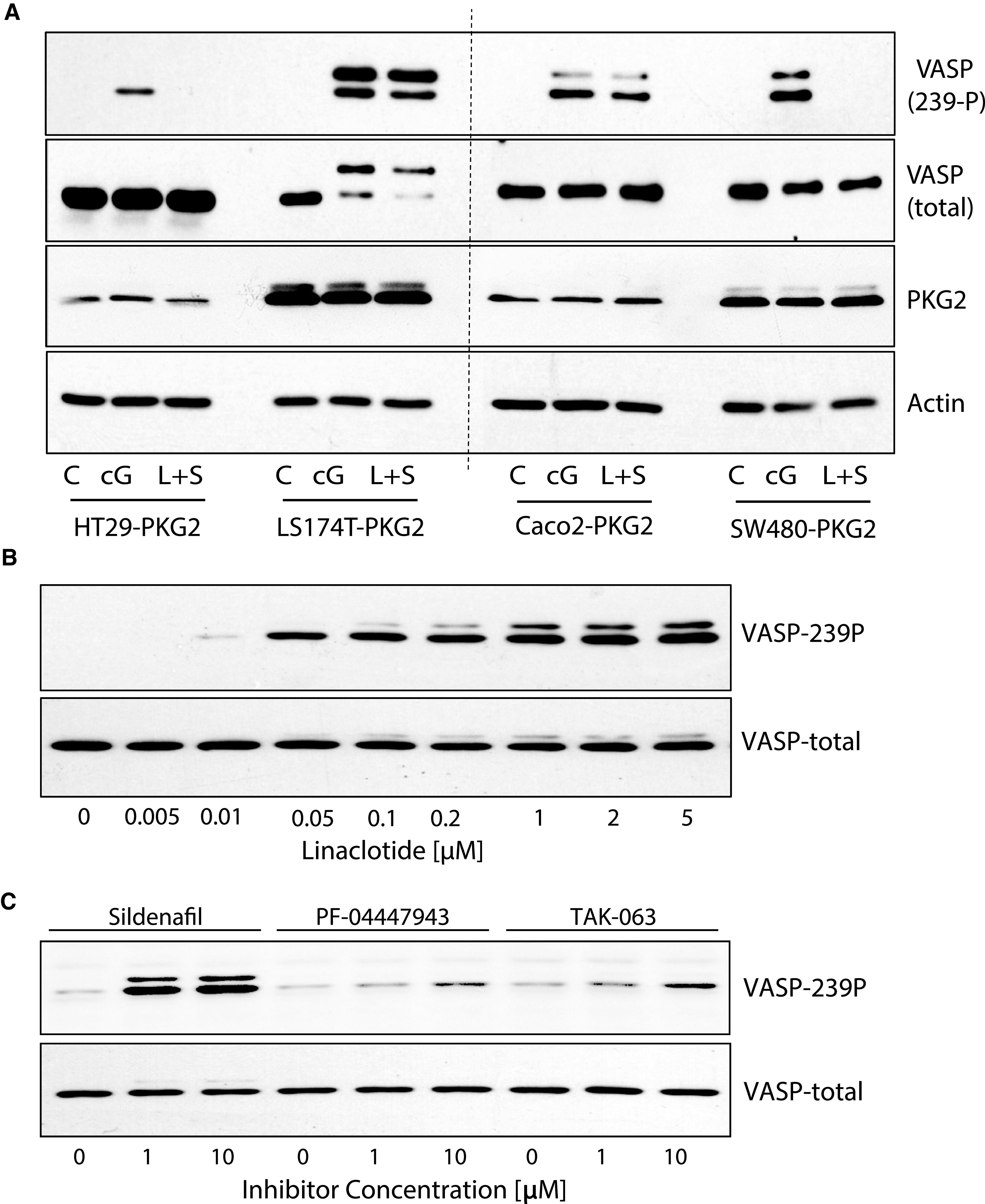
PDE inhibitors do not increase cGMP in colon cancer cell lines due to a lack of endogenous cGMP production. The expression of functional guanylyl-cyclase-C (GC-C) in colon cancer cells engineered to express ectopic PKG2 was determined by measuring VASP phosphorylation in response to treatment with the GC-C agonist linaclotide. (A) Different cell lines induced to express PKG2 as indicated below, were either untreated (control, C), treated with 8Br-cGMP (cG), or with the combination of linaclotide and sildenafil (L+S). Western blots were probed for β-actin as a loading control for PKG2 expression. (B) Dose-response study of linaclotide stimulation of LS174T-PKG2 cells to determine the dose required to generate minimal cGMP/PKG2 activity as measured by VASP phosphorylation. (C) LS174T-PKG2 cells were treated with PDE inhibitors as indicated, in the presence of 10 nM linaclotide. Relative cGMP levels were then measured by immunoblotting for VASP-239P. All immunoblots were reprobed with antitotal VASP to indicate relative phosphorylation.
The Effect of PDE Inhibitors on Survival Signaling in Colon Cancer Cells
The mechanisms that have been proposed to mediate the antiproliferative effects of PDE inhibitors and cGMP in colon cancer cells have centered upon growth and survival pathways downstream of receptor-tyrosine kinases, and Wnt signaling. The effect of PDE inhibitors on these pathways was examined in the GC-C–expressing LS174T and Caco2 cell lines (Fig. 6). These studies showed that phospho-ERK levels were transiently suppressed by treating cells with 200 µM of Sildenafil or 100 µM of the PDE9, 10 inhibitors in both cell lines. This inhibition was more pronounced in Caco2 cells treated with PF-04447943 and TAK-063 (40%–70% inhibition), compared with sildenafil (10% inhibition), but in all cases the phospho-ERK normalized after 24 hours. Phospho-AKT and total β-catenin levels were not inhibited in either cell line by any of the drugs.
Fig. 6.
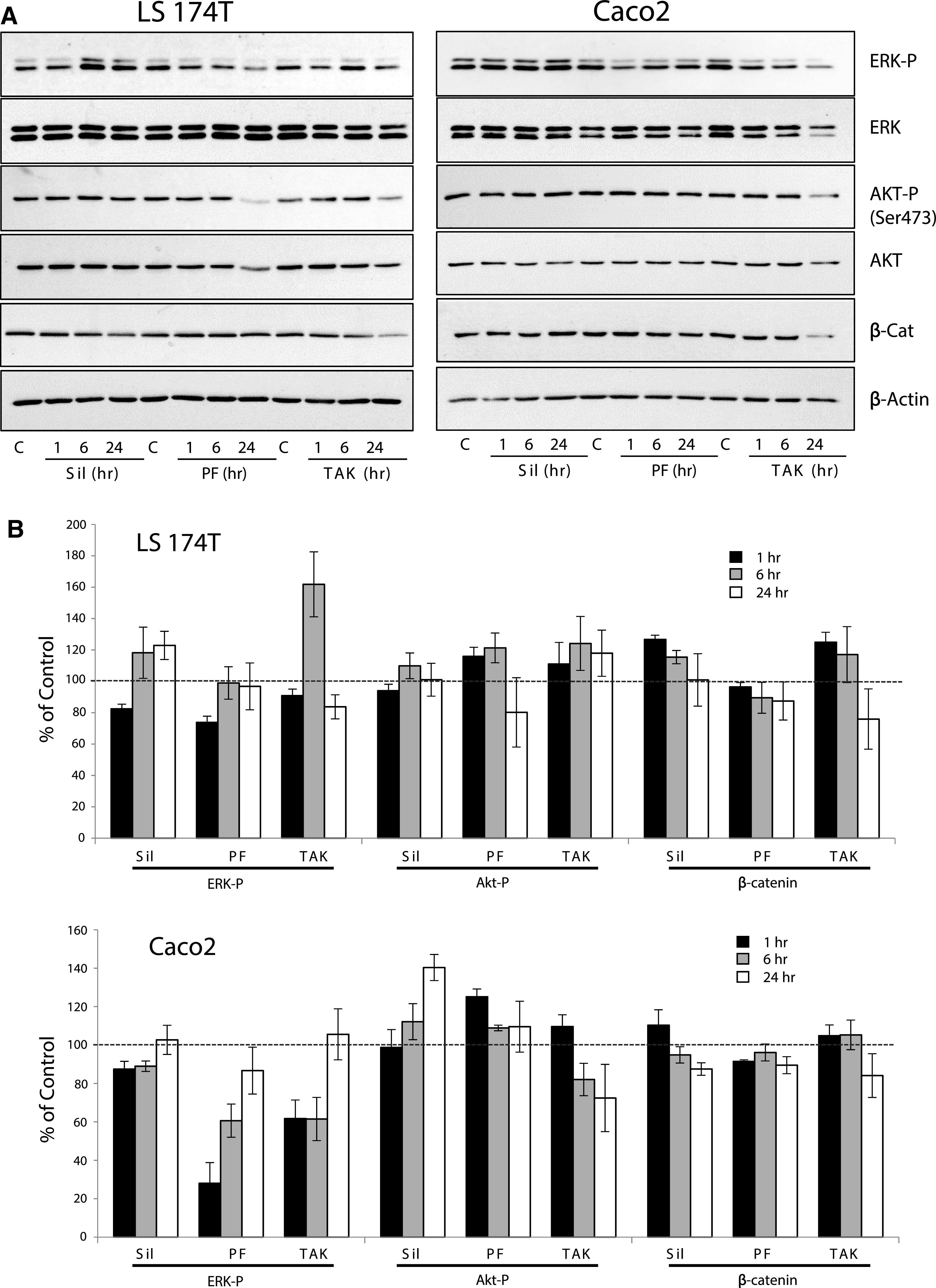
The effect of high-dose PDE inhibitors on proliferation/survival signaling in colon cancer cell lines. (A) LS174T and Caco2 colon cancer cells were treated with 200 µM Sildenafil (Sil), 100 µM PF-04447943 (PF), or 100 µM TAK-063 (TAK) for different times as indicated. The effects of treatment on ERK, AKT, and β-catenin were determined by immunoblotting. (B) Densitometric analysis of blots, where ratios of band intensities for ERK-P/ERK, AKT-P/AKT, and β-catenin/β-actin were calculated and expressed as a percent of the basal level (0 hour time point; dashed line).
Discussion
A large body of work spanning two decades has highlighted cGMP-elevating PDE inhibitors for the prevention and treatment of colorectal cancer. Building on this foundation, recent large-cohort retrospective epidemiologic studies support their efficacy in humans and provide the impetus for prospective clinical studies to interrogate their utility in patients (Huang et al., 2019; Sutton et al., 2020). However, there are inconsistencies in the literature regarding the potential utility of cGMP-PDE inhibitors as a primary prevention modality or as chemotherapy for cancer treatment. The present study provides new information that impacts the interpretation of foundational in vitro studies of PDE inhibitors in colon cancer cell lines.
A key observation shown here is that high levels of intracellular cGMP do not inhibit proliferation or affect the viability of ATCC-derived colon cancer cell lines. In addition, the colon cancer cell lines do not express detectable PKG activity that has been reported to mediate antiproliferative signaling. Ectopic expression of PKG2 in colon cancer cells conferred a modest antiproliferative response to cGMP treatment in some colon cancer cell lines, but did not affect viability. While the number of cell lines used in these studies is not exhaustive, the results were similar between cells of different genetic etiology and are supported by observations from several independent groups using diverse gastrointestinal cancer cell lines (Wang et al., 2012; Wu et al., 2013; Booth et al., 2014).
Another key observation from the present study is that inhibitors of PDE5, PDE9, and PDE10a all suppressed colon cancer cell proliferation, but only at concentrations exceeding 10 µM. These results are consistent with observations made by several independent laboratories, which have reported EC50 values for inhibiting colon cancer cell growth ranging from 20 µM to over 500 µM for different PDE inhibitors (Table 1). Notably, these concentrations are between 10,000 to almost 100,000 times the published IC50 for the target PDE enzyme. This massive disconnect in effective concentration suggests that the inhibitory mechanism is due to off-target effects. Most importantly, the EC50 values for inhibiting colon cancer cell growth in vitro are almost 50 times the published peak plasma concentrations (Cmax) of these drugs when administered at high doses to human patients. The results shown here are consistent with the antiproliferative effects of PDE5 and PDE10a inhibitors on colon cancer cell lines as published by other laboratories, but the doses required are clinically unachievable.
Table 1.
Effective doses of cGMP-PDE inhibitors
| Target | Drug | IC50 | EC50a | Human Plasma [Cmax] |
|---|---|---|---|---|
| PDE5 | Sildenafil | 3.9 nM(Gupta et al., 2005) | 66-271 µM (Li et al., 2013; Mei et al., 2015) | 1.2 µM(Gupta et al., 2005) (100 mg dose) |
| Tadalafil | 1-5 nM(Gupta et al., 2005; Ahmed, 2018) | 38 µM(Li et al., 2013) | 0.87 µM(Gupta et al., 2005) (20 mg dose) | |
| Exisulind | 113 µM(Soh et al., 2000) | 165-557 µM(Thompson et al., 2000) | 13-27 µM(van Stolk et al., 2000) (40-200 mg dose) | |
| PDE9 | Pf-04447943 | 12 nM(Charnigo et al., 2019) | 90 µMb | 0.12-0.63 µM (Charnigo et al., 2019) |
| BI-409306 | 52 nM(Rosenbrock et al., 2019) | ND | 1.37 µM(Moschetti et al., 2016) (100 mg dose) | |
| PDE10 | TAK-063 | 0.3 nM(Harada et al., 2015) | 22 µMb | 0.57 µM(Tsai et al., 2016) (1000 mg dose) |
| Pf-2545920 | 0.16 nM(Lee et al., 2016) | 14-20 µM(Lee et al., 2016) | 0.1-0.52 µM(Delnomdedieu et al., 2017) (10-30 mg dose) |
aInhibition of colon cancer cell growth.
bFrom the present study. ND: Not determined.
The notion that the antiproliferative effect of the cGMP-PDE inhibitors is due to off-target effects is supported by the lack of effect of 8Br-cGMP on colon cancer cell growth. The present study measured activation of PKG as a functional readout for cGMP because the biologic significance of absolute cGMP levels is unclear. A surprising observation was that none of the PDE inhibitors studied were able to increase cGMP levels to activate PKG in the colon cancer cell lines. The underlying reason was clearly shown to be due to the lack of endogenous cGMP-generating machinery. Without a basal rate of cGMP production to provide a substrate for PDEs, their inhibition had no effect. In agreement with previous studies, it was shown here that functional GC-C was only expressed in 2 of the cell lines tested (Waldman et al., 1998). GC-C is the main source of cGMP in the colon epithelium, which is activated by peptide hormone guanylin, which is universally lost in colon cancer (Wilson et al., 2014). In the presence of GC-C and the synthetic agonist linaclotide, a physiologic dose (1 µM) of the PDE5 inhibitor sildenafil potently activated cGMP signaling as measured by VASP-phosphorylation. The lack of effect of PDE9 and PDE10a inhibitors in the linaclotide-treated cells demonstrated that PDE5 is the central enzyme controlling cGMP levels in colon cancer cells, as has been reported by others (Bakre et al., 2000; Piazza et al., 2000; Mei et al., 2015). The slight increase in cGMP detected at very high concentrations of the PDE9 and PDE10a inhibitors in the present study likely reflects crossover inhibition of PDE5.
The antiproliferative effect of high doses of PDE inhibitors was shown here to be an off-target effect of these drugs, but specific signaling pathways have previously been implicated as important. The most prevalent mechanism proposed is the cGMP/PKG-mediated downregulation of β-catenin protein levels, leading to suppression of TCF-target gene expression (Thompson et al., 2000; Whitt et al., 2012; Li et al., 2015b). However, inhibition of AKT and ERK pathways in cells expressing ectopic PKG2 has also been reported (Wu et al., 2012; Wu et al., 2013; Wang et al., 2017). There was no obvious effect on any of these signaling pathways that was common to the inhibitors, or across cell lines with any single inhibitor. A recent study showed that treating SW480 and HCT116 colon cancer cells with 300 µM sildenafil for 48 hours inhibited proliferation, which was associated with reduced β-catenin levels (Mei et al., 2015). The mechanism was shown to involve increased redox stress and G1 arrest, but no specific pathway was indicated. It is possible that higher inhibitor concentrations over a longer period might have resulted in additional toxicity, but our results demonstrate that direct targeting of specific proliferative signaling pathways is unlikely to have a role.
An important conclusion from the present work is that the effective concentration of cGMP-PDE inhibitors used in colon cancer studies must be a critical factor when evaluating the significance of in vitro observations. The present study did not address the potential benefits of using PDE5 inhibitors in combination with traditional chemotherapy (Booth et al., 2014; Booth et al., 2017) or as immunomodulatory agents (Serafini et al., 2006; Lin et al., 2017). However, the concentrations of cGMP-PDE inhibitors that are needed to directly suppress colon cancer cell growth are clinically unachievable. Studies using knockout mice with defective cGMP signaling have shown that cGMP activation of PKG2 regulates intestinal homeostasis by slowing epithelial turnover (Steinbrecher et al., 2002; Li et al., 2007; Wang et al., 2012). Increasing cGMP using physiologic doses of PDE5 inhibitors or GC-C agonists, inhibits colon carcinogenesis in mice (Chang et al., 2017; Islam et al., 2017; Sharman et al., 2018). The mechanism was proposed to involve the reducing tumor initiation rather than inhibiting the growth of existing tumors, suggesting that PDE5 inhibitors might be useful for colon cancer prevention (Islam and Browning, 2018; Browning, 2019).
In summary, the present results demonstrate that proliferation and viability of colon cancer cell lines are not affected by cGMP or physiologic doses of cGMP-elevating PDE inhibitors. Extremely high doses of PDE inhibitors can inhibit colon cancer cell proliferation using an off-target mechanism that does not involve cGMP/PKG signaling.
Abbreviations
- ATCC
American Type Culture Collection
- CRC
colorectal cancer
- ERK
extracellular signal-regulated kinase
- GC-C
guanylyl cyclase C
- PDE
phosphodiesterase
- PKG
cGMP-dependent protein kinase
- VASP
vasodilator-stimulated phosphoprotein
Authorship Contributions
Participated in research design: Hou, Wren, D. Browning.
Conducted experiments: Hou, Wren, Mylarapu, K. Browning, Islam, D. Browning.
Contributed new reagents or analytic tools: Vega, D. Browning.
Performed data analysis: Hou, Wren, D. Browning.
Wrote or contributed to the writing of the manuscript: Wang, Vega, D. Browning.
Footnotes
This work was supported by a grant from the National Institutes of Health [Grant CA172627‐01A1] (to D.D.B.), and by a grant from the Georgia Research Alliance [GRA.VL21.E1] (to D.D.B.).
No author has an actual or perceived conflict of interest with the contents of this article.
References
- Ahmed NS (2018) Tadalafil: 15 years’ journey in male erectile dysfunction and beyond. Drug Dev Res 80:683–701. [DOI] [PubMed] [Google Scholar]
- Bakre MM, Sopory S, Visweswariah SS (2000) Expression and regulation of the cGMP-binding, cGMP-specific phosphodiesterase (PDE5) in human colonic epithelial cells: role in the induction of cellular refractoriness to the heat-stable enterotoxin peptide. J Cell Biochem 77:159–167. [PubMed] [Google Scholar]
- Birbe R, Palazzo JP, Walters R, Weinberg D, Schulz S, Waldman SA (2005) Guanylyl cyclase C is a marker of intestinal metaplasia, dysplasia, and adenocarcinoma of the gastrointestinal tract. Hum Pathol 36:170–179. [DOI] [PubMed] [Google Scholar]
- Booth L, Albers T, Roberts JL, Tavallai M, Poklepovic A, Lebedyeva IO, Dent P (2016) Multi-kinase inhibitors interact with sildenafil and ERBB1/2/4 inhibitors to kill tumor cells in vitro and in vivo. Oncotarget 7:40398–40417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth L, Roberts JL, Cruickshanks N, Conley A, Durrant DE, Das A, Fisher PB, Kukreja RC, Grant S, Poklepovic A,, et al. (2014) Phosphodiesterase 5 inhibitors enhance chemotherapy killing in gastrointestinal/genitourinary cancer cells. Mol Pharmacol 85:408–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth L, Roberts JL, Poklepovic A, Gordon S, Dent P (2017) PDE5 inhibitors enhance the lethality of pemetrexed through inhibition of multiple chaperone proteins and via the actions of cyclic GMP and nitric oxide. Oncotarget 8:1449–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning DD(2019) The enduring promise of phosphodiesterase 5 inhibitors for colon cancer prevention. Transl Gastroenterol Hepatol 4:83–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagir B, Gelmann A, Park J, Fava T, Tankelevitch A, Bittner EW, Weaver EJ, Palazzo JP, Weinberg D, Fry RD,, et al. (1999) Guanylyl cyclase C messenger RNA is a biomarker for recurrent stage II colorectal cancer. Ann Intern Med 131:805–812. [DOI] [PubMed] [Google Scholar]
- Carrithers SL, Parkinson SJ, Goldstein SD, Park PK, Urbanski RW, Waldman SA (1996) Escherichia coli heat-stable enterotoxin receptors. A novel marker for colorectal tumors. Dis Colon Rectum 39:171–181. [DOI] [PubMed] [Google Scholar]
- Chang WL, Masih S, Thadi A, Patwa V, Joshi A, Cooper HS, Palejwala VA, Clapper ML, Shailubhai K (2017) Plecanatide-mediated activation of guanylate cyclase-C suppresses inflammation-induced colorectal carcinogenesis in Apc+/Min-FCCC mice. World J Gastrointest Pharmacol Ther 8:47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charnigo RJ, Beidler D, Rybin D, Pittman DD, Tan B, Howard J, Michelson AD, Frelinger AL III, Clarke N (2019) PF-04447943, a Phosphodiesterase 9A Inhibitor, in Stable Sickle Cell Disease Patients: A Phase Ib Randomized, Placebo-Controlled Study. Clin Transl Sci 12:180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MB, Hawkins JA, Witte DP (1998) Guanylin mRNA expression in human intestine and colorectal adenocarcinoma. Lab Invest 78:101–108. [PubMed] [Google Scholar]
- Delnomdedieu M, Forsberg A, Ogden A, Fazio P, Yu CR, Stenkrona P, Duvvuri S, David W, Al-Tawil N, Vitolo OV,, et al. (2017) In vivo measurement of PDE10A enzyme occupancy by positron emission tomography (PET) following single oral dose administration of PF-02545920 in healthy male subjects. Neuropharmacology 117:171–181. [DOI] [PubMed] [Google Scholar]
- Forte LR Jr (2004) Uroguanylin and guanylin peptides: pharmacology and experimental therapeutics. Pharmacol Ther 104:137–162. [DOI] [PubMed] [Google Scholar]
- Gupta M, Kovar A, Meibohm B (2005) The clinical pharmacokinetics of phosphodiesterase-5 inhibitors for erectile dysfunction. J Clin Pharmacol 45:987–1003. [DOI] [PubMed] [Google Scholar]
- Harada A, Suzuki K, Kamiguchi N, Miyamoto M, Tohyama K, Nakashima K, Taniguchi T, Kimura H (2015) Characterization of binding and inhibitory properties of TAK-063, a novel phosphodiesterase 10A inhibitor. PLoS One 10:e0122197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Gupta N, Schoenlein P, Wong E, Martindale R, Ganapathy V, Browning D (2006) An anti-tumor role for cGMP-dependent protein kinase. Cancer Lett 240:60–68. [DOI] [PubMed] [Google Scholar]
- Huang W, Sundquist J, Sundquist K, Ji J (2019) Use of Phosphodiesterase 5 Inhibitors Is Associated With Lower Risk of Colorectal Cancer in Men With Benign Colorectal Neoplasms. Gastroenterology 157:672–681.e4. [DOI] [PubMed] [Google Scholar]
- Islam BN, Browning DD (2018) Phosphodiesterase-5 inhibitors for colon cancer chemoprevention. Aging (Albany NY) 10:2216–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam BN, Sharman SK, Hou Y, Bridges AE, Singh N, Kim S, Kolhe R, Trillo-Tinoco J, Rodriguez PC, Berger FG,, et al. (2017) Sildenafil Suppresses Inflammation-Driven Colorectal Cancer in Mice. Cancer Prev Res (Phila) 10:377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joe AK, Liu H, Xiao D, Soh JW, Pinto JT, Beer DG, Piazza GA, Thompson WJ, Weinstein IB (2003) Exisulind and CP248 induce growth inhibition and apoptosis in human esophageal adenocarcinoma and squamous carcinoma cells. J Exp Ther Oncol 3:83–94. [DOI] [PubMed] [Google Scholar]
- Lee K, Lindsey AS, Li N, Gary B, Andrews J, Keeton AB, Piazza GA (2016) β-catenin nuclear translocation in colorectal cancer cells is suppressed by PDE10A inhibition, cGMP elevation, and activation of PKG. Oncotarget 7:5353–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Chen X, Zhu B, Ramírez-Alcántara V, Canzoneri JC, Lee K, Sigler S, Gary B, Li Y, Zhang W,, et al. (2015a) Suppression of β-catenin/TCF transcriptional activity and colon tumor cell growth by dual inhibition of PDE5 and 10. Oncotarget 6:27403–27415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Lee K, Xi Y, Zhu B, Gary BD, Ramírez-Alcántara V, Gurpinar E, Canzoneri JC, Fajardo A, Sigler S,, et al. (2015b) Phosphodiesterase 10A: a novel target for selective inhibition of colon tumor cell growth and β-catenin-dependent TCF transcriptional activity. Oncogene 34:1499–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Xi Y, Tinsley HN, Gurpinar E, Gary BD, Zhu B, Li Y, Chen X, Keeton AB, Abadi AH,, et al. (2013) Sulindac selectively inhibits colon tumor cell growth by activating the cGMP/PKG pathway to suppress Wnt/β-catenin signaling. Mol Cancer Ther 12:1848–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Lin JE, Chervoneva I, Schulz S, Waldman SA, Pitari GM (2007) Homeostatic control of the crypt-villus axis by the bacterial enterotoxin receptor guanylyl cyclase C restricts the proliferating compartment in intestine. Am J Pathol 171:1847–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Wang J, Wang L, Wen J, Guo Y, Qiao W, Zhou J, Xu G, Zhi F (2017) Phosphodiesterase-5 inhibition suppresses colonic inflammation-induced tumorigenesis via blocking the recruitment of MDSC. Am J Cancer Res 7:41–52. [PMC free article] [PubMed] [Google Scholar]
- Lugnier C (2006) Cyclic nucleotide phosphodiesterase (PDE) superfamily: a new target for the development of specific therapeutic agents. Pharmacol Ther 109:366–398. [DOI] [PubMed] [Google Scholar]
- Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, Manganiello VC (2014) Advances in targeting cyclic nucleotide phosphodiesterases. Nat Rev Drug Discov 13:290–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei XL, Yang Y, Zhang YJ, Li Y, Zhao JM, Qiu JG, Zhang WJ, Jiang QW, Xue YQ, Zheng DW,, et al. (2015) Sildenafil inhibits the growth of human colorectal cancer in vitro and in vivo. Am J Cancer Res 5:3311–3324. [PMC free article] [PubMed] [Google Scholar]
- Moschetti V, Boland K, Feifel U, Hoch A, Zimdahl-Gelling H, Sand M (2016) First-in-human study assessing safety, tolerability and pharmacokinetics of BI 409306, a selective phosphodiesterase 9A inhibitor, in healthy males. Br J Clin Pharmacol 82:1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza GA, Xu S, Klein-Szanto A, Ahnen DJ, Li H, Liu L, David M, Pamukcu R, Thompson WJ (2000) Overexpression of CGMP phosphodiesterase (cG PDE) in colonic neoplasias compared to normal mucosa. Gastroenterology 118:A282. [Google Scholar]
- Pitari GM, Li T, Baksh RI, Waldman SA (2006) Exisulind and guanylyl cyclase C induce distinct antineoplastic signaling mechanisms in human colon cancer cells. Mol Cancer Ther 5:1190–1196. [DOI] [PubMed] [Google Scholar]
- Rosenbrock H, Giovannini R, Schänzle G, Koros E, Runge F, Fuchs H, Marti A, Reymann KG, Schröder UH, Fedele E,, et al. (2019) The Novel Phosphodiesterase 9A Inhibitor BI 409306 Increases Cyclic Guanosine Monophosphate Levels in the Brain, Promotes Synaptic Plasticity, and Enhances Memory Function in Rodents. J Pharmacol Exp Ther 371:633–641. [DOI] [PubMed] [Google Scholar]
- Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, Dolcetti L, Bronte V, Borrello I (2006) Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med 203:2691–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharman SK, Islam BN, Hou Y, Singh N, Berger FG, Sridhar S, Yoo W, Browning DD (2018) Cyclic-GMP-Elevating Agents Suppress Polyposis in ApcMin mice by Targeting the Preneoplastic Epithelium. Cancer Prev Res (Phila) 11:81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolenski A, Bachmann C, Reinhard K, Hönig-Liedl P, Jarchau T, Hoschuetzky H, Walter U (1998) Analysis and regulation of vasodilator-stimulated phosphoprotein serine 239 phosphorylation in vitro and in intact cells using a phosphospecific monoclonal antibody. J Biol Chem 273:20029–20035. [DOI] [PubMed] [Google Scholar]
- Soh JW, Mao Y, Kim MG, Pamukcu R, Li H, Piazza GA, Thompson WJ, Weinstein IB (2000) Cyclic GMP mediates apoptosis induced by sulindac derivatives via activation of c-Jun NH2-terminal kinase 1. Clin Cancer Res 6:4136–4141. [PubMed] [Google Scholar]
- Steinbrecher KA, Tuohy TM, Heppner Goss K, Scott MC, Witte DP, Groden J, Cohen MB (2000) Expression of guanylin is downregulated in mouse and human intestinal adenomas. Biochem Biophys Res Commun 273:225–230. [DOI] [PubMed] [Google Scholar]
- Steinbrecher KA, Wowk SA, Rudolph JA, Witte DP, Cohen MB (2002) Targeted inactivation of the mouse guanylin gene results in altered dynamics of colonic epithelial proliferation. Am J Pathol 161:2169–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoner GD, Budd GT, Ganapathi R, DeYoung B, Kresty LA, Nitert M, Fryer B, Church JM, Provencher K, Pamukcu R,, et al. (1999) Sulindac sulfone induced regression of rectal polyps in patients with familial adenomatous polyposis. Adv Exp Med Biol 470:45–53. [DOI] [PubMed] [Google Scholar]
- Sutton SS, Magagnoli J, Cummings TH, Hardin JW (2020) The Association Between Phosphodiesterase-5 Inhibitors and Colorectal Cancer in a National Cohort of Patients. Clin Transl Gastroenterol 11:e00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson WJ, Piazza GA, Li H, Liu L, Fetter J, Zhu B, Sperl G, Ahnen D, Pamukcu R (2000) Exisulind induction of apoptosis involves guanosine 3′,5′-cyclic monophosphate phosphodiesterase inhibition, protein kinase G activation, and attenuated beta-catenin. Cancer Res 60:3338–3342. [PubMed] [Google Scholar]
- Tsai M, Chrones L, Xie J, Gevorkyan H, Macek TA (2016) A phase 1 study of the safety, tolerability, pharmacokinetics, and pharmacodynamics of TAK-063, a selective PDE10A inhibitor. Psychopharmacology (Berl) 233:3787–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Stolk R, Stoner G, Hayton WL, Chan K, DeYoung B, Kresty L, Kemmenoe BH, Elson P, Rybicki L, Church J,, et al. (2000) Phase I trial of exisulind (sulindac sulfone, FGN-1) as a chemopreventive agent in patients with familial adenomatous polyposis. Clin Cancer Res 6:78–89. [PubMed] [Google Scholar]
- Waldman SA, Barber M, Pearlman J, Park J, George R, Parkinson SJ (1998) Heterogeneity of guanylyl cyclase C expressed by human colorectal cancer cell lines in vitro. Cancer Epidemiol Biomarkers Prev 7:505–514. [PubMed] [Google Scholar]
- Wang R, Islam BN, Bridges A, Sharman SK, Hu M, Hou Y, Somanath PR, Venable L, Singh N, Kim S,, et al. (2017) cGMP Signaling Increases Antioxidant Gene Expression by Activating Forkhead Box O3A in the Colon Epithelium. Am J Pathol 187:377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Kwon IK, Singh N, Islam B, Liu K, Sridhar S, Hofmann F, Browning DD (2014) Type 2 cGMP-dependent protein kinase regulates homeostasis by blocking c-Jun N-terminal kinase in the colon epithelium. Cell Death Differ 21:427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Kwon IK, Thangaraju M, Singh N, Liu K, Jay P, Hofmann F, Ganapathy V, Browning DD (2012) Type 2 cGMP-dependent protein kinase regulates proliferation and differentiation in the colonic mucosa. Am J Physiol Gastrointest Liver Physiol 303:G209–G219. [DOI] [PubMed] [Google Scholar]
- Whitt JD, Li N, Tinsley HN, Chen X, Zhang W, Li Y, Gary BD, Keeton AB, Xi Y, Abadi AH,, et al. (2012) A novel sulindac derivative that potently suppresses colon tumor cell growth by inhibiting cGMP phosphodiesterase and β-catenin transcriptional activity. Cancer Prev Res (Phila) 5:822–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C, Lin JE, Li P, Snook AE, Gong J, Sato T, Liu C, Girondo MA, Rui H, Hyslop T,, et al. (2014) The paracrine hormone for the GUCY2C tumor suppressor, guanylin, is universally lost in colorectal cancer. Cancer Epidemiol Biomarkers Prev 23:2328–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Chen Y, Jiang L, Li Y, Lan T, Wang Y, Qian H (2013) Type II cGMP-dependent protein kinase inhibits epidermal growth factor-induced phosphatidylinositol-3-kinase/Akt signal transduction in gastric cancer cells. Oncol Lett 6:1723–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Chen Y, Qu R, Lan T, Sang J (2012) Type II cGMP-dependent protein kinase inhibits EGF-triggered signal transduction of the MAPK/ERK-mediated pathway in gastric cancer cells. Oncol Rep 27:553–558. [DOI] [PubMed] [Google Scholar]