

SUMMARY
Macrophage adhesion and stretching have been shown to induce interleukin (IL)-1β production, but the mechanism of this mechanotransduction remains unclear. Here we specify the molecular link between mechanical tension on tissue-resident macrophages and activation of the NLRP3 inflammasome, which governs IL-1β production. NLRP3 activation enhances antimicrobial defense, but excessive NLRP3 activity causes inflammatory tissue damage in conditions such as pulmonary fibrosis and acute respiratory distress syndrome. We find that the actin-bundling protein L-plastin (LPL) significantly enhances NLRP3 assembly. Specifically, LPL enables apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC) oligomerization during NLRP3 assembly by stabilizing ASC interactions with the kinase Pyk2, a component of cell-surface adhesive structures called podosomes. Upon treatment with exogenous NLRP3 activators, lung-resident alveolar macrophages (AMs) lacking LPL exhibit reduced caspase-1 activity, IL-1β cleavage, and gasdermin-D processing. LPL−/− mice display resistance to bleomycin-induced lung injury and fibrosis. These findings identify the LPL-Pyk2-ASC pathway as a target for modulation in NLRP3-mediated inflammatory conditions.
Graphical Abstract
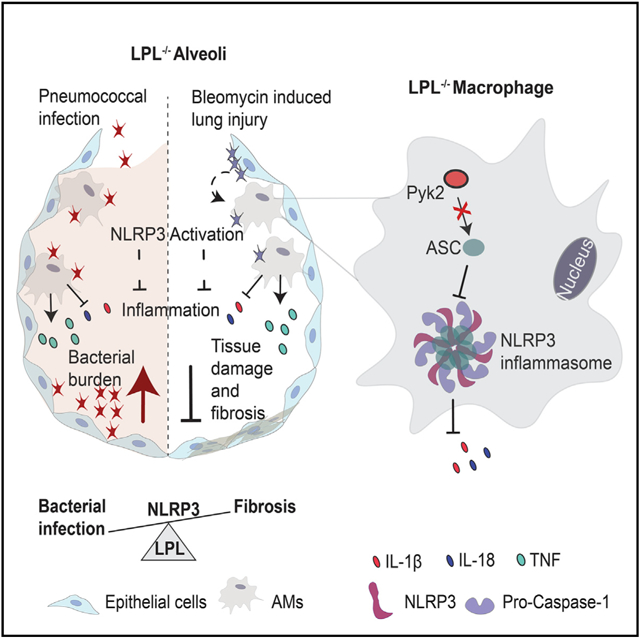
In brief
In this study, Joshi et al. identify a crucial modulator, L-plastin, in lung inflammation. L-plastin supports the macrophage inflammatory response to enhance lung fibrosis during lung injury by connecting inflammation and mechanical stimuli in a process called mechanotransduction. The findings from this study will help determine efficient targets for diagnosis and treatment of lung inflammatory diseases.
INTRODUCTION
Macrophage adhesion and stretching induce the pro-inflammatory mediator interleukin (IL)-1β. The process by which adhesion or mechanical stress is sensed by a cell and converted to a chemical signal is called mechanotransduction. Multiple inflammatory disease states, such as bacterial pneumonia, lung fibrosis, and acute respiratory distress syndrome, are characterized by changes in tissue compliance (i.e., stiffness). Understanding how macrophages convert mechanical signals to IL-1β production may illuminate the pathogenesis of multiple human disease states.
Here we identify a previously unrecognized mediator, L-plastin (LPL), in adhesion-regulated IL-1β production in macrophages via the nucleotide-binding oligomerization domain (NOD), leucine-rich repeat (LRR)-containing proteins (NLR) family protein 3 (NLRP3) inflammasome pathway. NLRP3 activation drives essential anti-pathogen responses by producing pro-inflammatory cytokines, such as IL-1β and IL-18, whereas overabundant NLRP3 activation can trigger autoinflammatory diseases. NLRP3 activation relies on signaling through two distinct pathways. First, a priming signal is provided via Toll-like receptor (TLR) activation of nuclear factor κB (NF-κB) (Joshi et al., 2021) to upregulate synthesis of pro-IL-1β and inflammasome components; in addition, priming is sufficient for production of certain pro-inflammatory mediators, such as tumor necrosis factor alpha (TNF-α) and IL-6 (Petrilli et al., 2005; Hirano et al., 2017; John et al., 2019). Second, an activation (assembly) signal induces oligomerization of inflammasome components, resulting in auto-activation of caspase-1, which then cleaves pro-IL-1β to its bioactive form (Martinon et al., 2002; Petrilli et al., 2005). NLRP3-activating signals are initiated by pathogens or host-derived damage-associated molecular patterns (DAMPs), such as uric acid (Martinon et al., 2006) or ATP (Le Feuvre et al., 2002). NLRP3 engages with the inactive pro-form of caspase-1 through an adaptor called ASC (apoptosis-associated speck-like protein containing a caspase activation and recruitment domain [CARD]) (Srinivasula et al., 2002).
Previously, macrophages deficient in the actin-bundling protein LPL have been noted to exhibit diminished IL-1β production upon adhesion to tissue culture plates (Chen et al., 2003). LPL, also called leukocyte cytosolic protein-1 (LCP-1), is conserved in humans and mice (Shinomiya et al., 1994) and is normally expressed only in hematopoietic cells (Goldstein et al., 1985; Lin et al., 1993). LPL is also aberrantly expressed in non-immune malignant cells (Goldstein et al., 1985; Lin et al., 1993). Ectopic LPL expression is found in many invasive metastatic human cancers, such as melanoma (Klemke et al., 2007), breast cancer (Tiedemann et al., 2019), colorectal cancer (Otsuka et al., 2001), and prostate cancer (Chen et al., 2017), and is considered a diagnostic marker in many solid tumors (Strickler et al., 2014).
LPL supports formation of dynamic F-actin structures, such as podosomes, invadopodia, and filopodia, during cell adhesion, migration, invasion, and metastasis (Van Audenhove et al., 2016). We also observed reduced IL-1β production accompanied by increased bacterial loads and mortality in LPL−/− mice during pneumococcal lung infection (Deady et al., 2014). Although LPL−/− mice harbor numerically fewer alveolar macrophages (AMs) compared with wild-type (WT) mice (Todd et al., 2016), we now identify a second, functional defect explaining the susceptibility of LPL−/− mice to pneumococcal pneumonia. We find that LPL is required, downstream of the assembly signal, for maximal NLRP3 activation in macrophages. Under conditions of equivalent priming, LPL deficiency reduces ASC multimerization and subsequent activation of caspase-1, impairing NLRP3 assembly, as measured by release of IL-1β and IL-18 and gasdermin-D cleavage. Mechanistically, loss of LPL disrupts the association of the kinase Pyk2, a major component of macrophage podosomes, with the NLRP3 adaptor molecule ASC. These data define a complete signaling pathway from macrophage podosomes to NLRP3 assembly, providing a molecular explanation for how tensile stress on macrophages regulates their IL-1β production. Broadening the translational importance of this pathway, we found that LPL−/− mice were also resistant to NLRP3-dependent inflammatory diseases; namely, bleomycin-induced lung fibrosis and monosodium urate (MSU)-induced peritonitis. These findings suggest an essential role of LPL in augmenting NLRP3 assembly in macrophages for effective host defense against bacterial pathogens and supporting auto-inflammatory diseases. Our findings reveal an LPL-dependent molecular basis for mechanosensitivity of NLRP3 activation in macrophages and illuminate potential avenues for targeted therapy in infectious and inflammatory diseases.
RESULTS
NLRP3-mediated production of IL-1β and IL-18 is diminished in LPL-deficient AMs
LPL supports podosome formation in macrophages, and macrophages lacking LPL produce less IL-1β upon adhesion to culture surfaces (Chen et al., 2003). NLRP3-induced IL-1β production is modulated by mechanical stimuli, such as cyclic stretching during tendinitis or stress exerted by mechanical ventilation during lung inflammation/injury and fibrosis (Kuipers et al., 2011; Wu et al., 2013; Chen et al., 2018). Because LPL supports podosome formation, and because podosomes “sense” adhesion-induced mechanical forces, we hypothesized that LPL may contribute to IL-1β through mechanotransduction.
To test this hypothesis, we measured NLRP3-mediated IL-1β release from cells cultured on surfaces of varying compliance (measure in kilopascal elastic modulus). AMs were cultured on collagen-coated silica gels of low compliance (64 kPa) and high compliance (0.2 kPa) corresponding to hard tissue (bone) and soft tissue (lung) elastic modulus, respectively. With increased compliance (decreased stiffness), AMs from WT mice exhibited increased IL-1β production. Significantly less IL-1β was produced by LPL-deficient AMs, and LPL-deficient AMs were unresponsive to changes in the compliance of culture surfaces (Figure 1A). Importantly, LPL-deficiency did not ablate IL-1β production but removed the modulation by mechanosensation and reduced the total amount released.
Figure 1. Macrophages from LPL−/− mice exhibit reduced IL-1β production after NLRP3 activation.
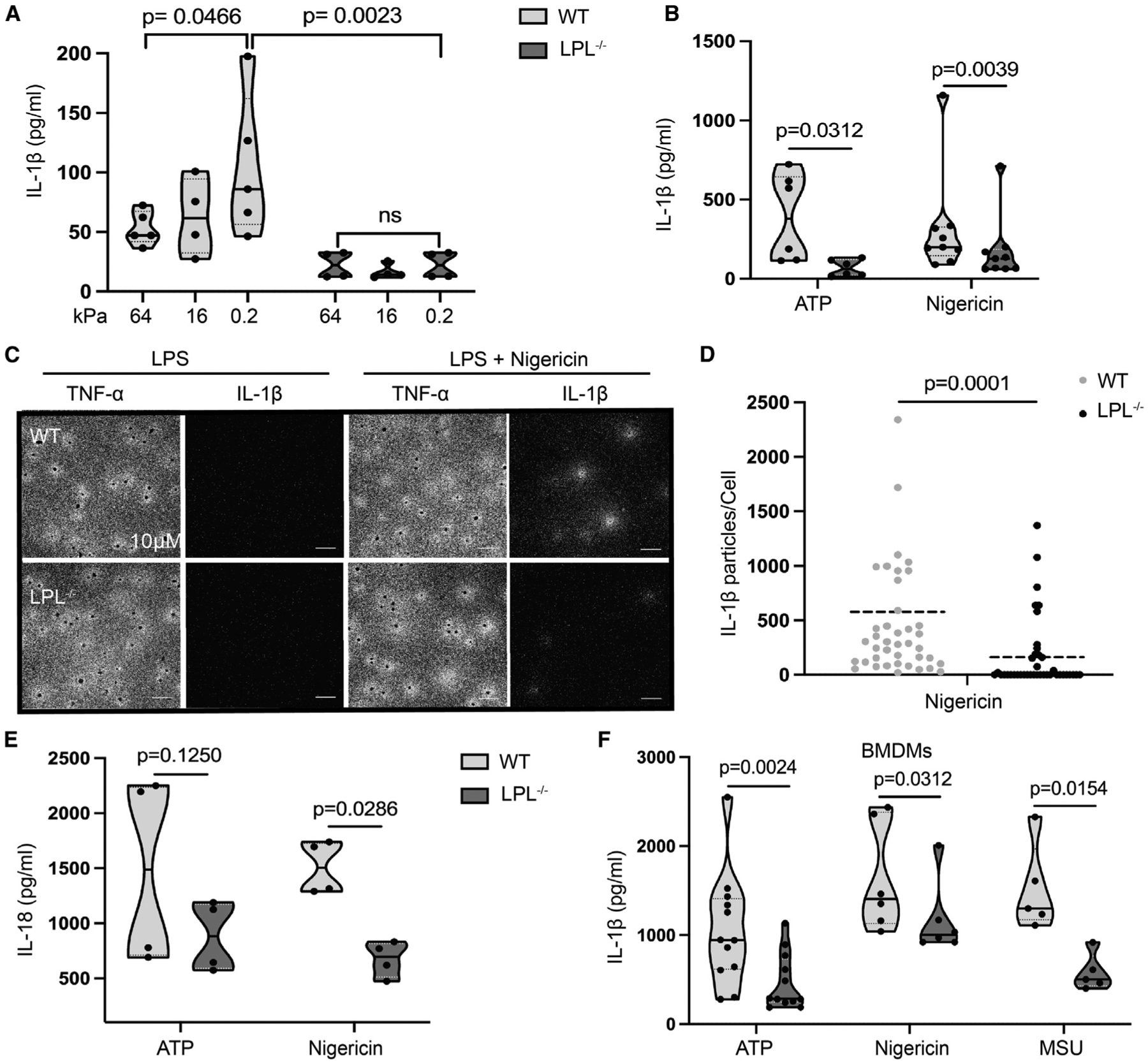
(A and B) AMs obtained from naive WT (light gray) and LPL−/− (dark gray) mice were cultured on collagen-coated silica gel with corresponding stiffness (shown in kilopascal) (A) or plastic culture plates (B). Cells were primed with LPS and then stimulated with ATP; IL-1β in culture supernatant was quantified by ELISA.
(A) Data points, n = 4 (WT, 16 kPa; LPL−/−, 0.2, 16, and 64 kPa) or n = 5 (WT, 0.2 kPa and 64 kPa) from 3 independent experiments are shown.
(B) n = 6 from 4 independent experiments (ATP) and n = 9 from 5 independent experiments (nigericin).
(C) AMs were incubated on a culture surface coated with anti-IL-1β or anti-TNF-α antibodies as indicated, primed with LPS, and activated with nigericin. Locally released IL-1β and TNF-α were detected by plasmonic-fluor nanoprobe-conjugated anti-IL-1β and TNF-α antibodies. Microscopy images at 20× magnification show IL-1β as white clusters around individual cells. Each cell was visualized by DAPI (seen as black dots) staining; scale bars, 10μm. Representative results from 3 independent experiments are shown.
(D) Quantification of numbers of IL-1β particles released around AMs from microscopy analysis of plasmonic-fluor images. Representative results from 3 biological repeats are shown (n = 40 data points of independent cells).
(E) AMs were primed with LPS and then activated with ATP or nigericin. Secreted IL-18 in culture supernatants was quantified by ELISA. Results representing 4 independent experiments (n = 4 data points) are shown.
(F) BMDMs were primed with LPS and then activated with ATP, nigericin, and MSU. IL-1β in culture supernatants was quantified using ELISA. Represented results are showing data points n = 12 (ATP) from 7 independent experiments and n = 6 (nigericin) and n = 5 (MSU) from 3 independent experiments.
In graphs, each symbol represents an individual data point, the solid line shows the median, and the dashed line shows the interquartile range. The p values were determined by ANOVA (A), Wilcoxon test (B, E, and F), and Mann-Whitney test (C).
We have previously noted reduced IL-1β in bronchoalveolar lavage (BAL) fluid of LPL−/− mice after pneumococcal challenge (Deady et al., 2014). Pneumococcal infection activates NLRP3 and induces IL-1β, which promotes antimicrobial defense (Witzenrath et al., 2011; Cho et al., 2018). AM expression of LPL promotes efficient clearance of pulmonary pneumococcal infections (Deady et al., 2014; Todd et al., 2016); reduced IL-1β production in response to NLRP3 activation is consistent with the increased susceptibility of LPL−/− mice to pneumococcal infection.
To further evaluate a cell-intrinsic role of LPL in NLRP3 activation, we measured IL-1β production by AMs isolated from WT and LPL−/− mice (Figure 1B) after lipopolysaccharide (LPS) priming and exposure to other NLRP3 activators. We noted variability in the amount of IL-1β produced by WT cells across multiple independent experiments. The variability in results is due to the biological variability expected when analyzing primary cells (instead of cell lines) as well as the technical variability inherent in performing independent experiments over an extended time period, using different lots of chemical activators and quantifying with different lots of ELISA reagents. Despite the biological and technical variability, we detected significantly less IL-1β in culture supernatants of LPL-deficient AMs (analyzed concurrently with WT AMs) primed with LPS and then treated with the NLRP3 activators ATP or nigericin (Figure 1B). To determine whether LPL deficiency affected inflammasome priming, assembly, or both, we compared TNF-α and IL-1β production using highly sensitive, ultrabright fluorescent nanoprobes (plasmonic-fluor) that capture cytokines as they are released (Luan et al., 2020). LPS (priming) is sufficient to induce TNF-α, but nigericin (activation signal) is required to induce IL-1β secretion. Microscopic analysis showed that a lower proportion of LPL-deficient AMs released IL-1β after nigericin treatment, consistent with reduced NLRP3 assembly; however, TNF-α production remained similar in both groups, suggesting that priming was intact in LPL-deficient AMs (Figure 1C). Quantification of nanoprobe fluorescence confirmed that significantly fewer IL-1β particles associated with LPL-deficient AMs (Figure 1D).
The inflammatory cytokine IL-18 is also produced after NLRP3 activation. Because macrophages constitutively express pro-IL-18 (de Zoete et al., 2014), release of IL-18 depends primarily on NLRP3 assembly. Primed LPL-deficient AMs also secreted significantly less IL-18 after challenge with ATP and nigericin (Figure 1E). Reduced IL-18 release, concordant with reduced IL-1β production, suggests that LPL is required for signaling to assemble NLRP3. To evaluate a separate macrophage lineage and support further biochemical analysis, we next tested ex-vivo-differentiated bone marrow-derived macrophages (BMDMs). LPL-deficient BMDMs also displayed reduced IL-1β production following NLRP3 activation using specific activators such as ATP, nigericin, and MSU (Figure 1F). Equivalent expression of the receptor NLRP3 and the adaptor ASC in multiple macrophage lineages (lung, peritoneum, and bone marrow derived) from WT and LPL−/− mice was revealed by immunoblotting (Figure S1A), indicating that impaired IL-1β production by LPL-deficient macrophages was not due to loss of essential inflammasome components. Impaired IL-1β production by BMDMs derived from LPL−/− mice also indicates that the observed defect in AMs is not merely due to impaired AM development. Finally, priming was equivalent among WT and LPL−/− macrophages, as shown by LPS-induced pro-IL-1β expression (Figure S1B). These results reveal an essential role of LPL in NLRP3 assembly and the resulting IL-1β and IL-18 production.
LPL promotes caspase-1 activation
Inflammasome assembly activates the effector caspase-1, which catalyzes maturation of IL-1β by cleaving pro-IL-1β (Black et al., 1989). Thus, we hypothesized that LPL would impair assembly signal transduction upstream of caspase-1 activation. We measured caspase-1 activity through flow cytometry analysis of AMs labeled with a fluorochrome-conjugated, caspase-1-specific molecular inhibitor (carboxyfluorescein [FAM]-Tyr-Val-Ala-Asp [YVAD]-fluoromethyl ketone [FMK]-fluorochrome inhibitor of caspases [FLICA]). Analysis of activated AMs demonstrated a reduced proportion of LPL-deficient AMs with active caspase-1 (Figures 2A, S1C, and S1D). Quantification of the median fluorescence intensity (MFI) of FAM-FLICA confirmed significantly reduced caspase-1 activity in LPS-primed AMs lacking LPL upon treatment with nigericin or ATP (Figure 2B).
Figure 2. NLRP3-induced caspase-1 activation and processing of IL-1β and gasdermin-D are reduced in LPL-deficient macrophages.
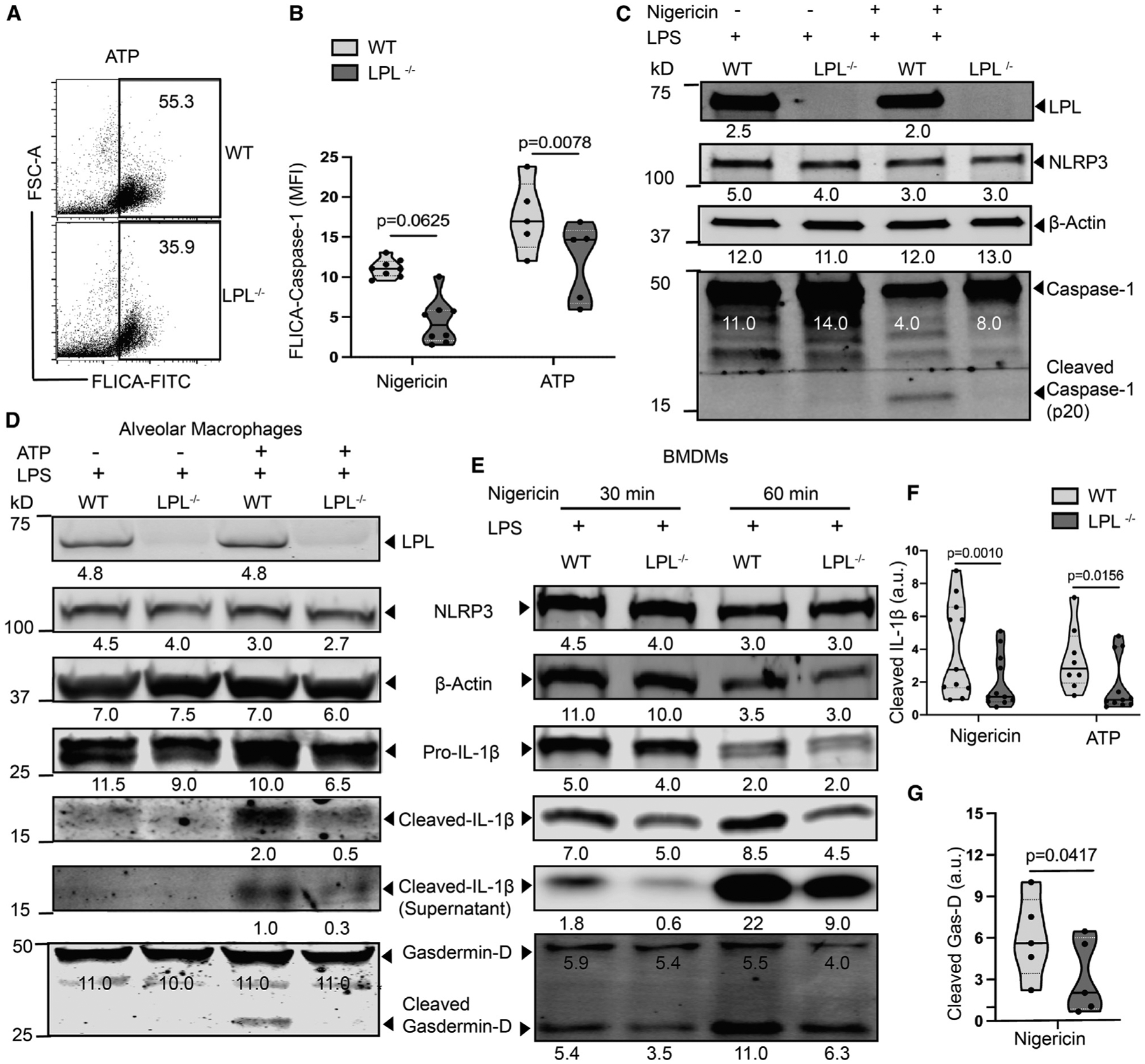
(A and B) AMs were primed with LPS, activated with nigericin or ATP, and then labeled with FAM-FLICA to detect active caspase-1.
(A) Representative fluorescence-activated cell sorting (FACS) analysis with FAM-FLICA-labeled cells in response to ATP activation. Representative results from 3 independent experiments are shown.
(B) Quantified median fluorescence intensity (MFI) of FAM-FLICA fluorescence. Results representing n = 8 from 4 independent experiments (nigericin) and n = 5 from 3 independent experiments (ATP) are shown.
(C) Immunoblot of cell lysates from BMDMs primed with LPS with or without subsequent nigericin activation. Full-length and cleaved caspase-1 were detected using specific antibodies, as indicated. Density is shown below each protein band. A representative result from 3 independent experiments is shown.
(D) Immunoblot of cell lysates of AMs (primed with LPS and activated with ATP) probed for cleaved IL-1β. Culture supernatants were used to detect extracellular cleaved IL-1β. Density is shown below each protein band. Here, NLRP3 expression serves as an LPS priming control and β-actin as total cell control. Representative results from 3 independent experiments are shown.
(E) Immunoblot of BMDMs (primed with LPS and activated with nigericin) probed for IL-1β (culture supernatant and cell lysates) and cellular gasdermin-D cleavage (shown from a replicated immunoblot). Density is shown below each protein band. A representative result from 4 independent experiments is shown.
(F and G) Densitometry quantification from multiple immunoblots of cleaved IL-1β after 30 M nigericin and ATP treatment(F) and gasdermin-D after 30 M nigericin treatment (G). Here, β-actin is used as the loading control.
(F) Representative result showing n = 11 from 5 independent experiments (nigericin) and n = 7 from 4 independent experiments (ATP).
(G) n = 5 from 3 independent experiments are shown.
In (B), (F), and (G), each symbol represents a value from an independent data point, with the solid line at the median and the dashed line showing the interquartile range. The p values were determined with a Wilcoxon test. See also Figures S1C and S1D.
Caspase-1 activation is characterized by self-cleavage (Martinon et al., 2002). Therefore, we also performed immunoblotting to evaluate caspase-1 cleavage after NLRP3 assembly (Figure 2C). We detected the cleaved form of caspase-1 (p20 fragment) in WT cells but observed very little p20 in LPL-deficient cells. Equivalent levels of pro-caspase-1 and NLRP3 in WT and LPL-deficient cells indicated intact priming, again suggesting that transduction of the assembly signal to caspase-1 cleavage was impaired by LPL deficiency (Figure 2C).
Caspase-1 is the terminal effector of the inflammasome activation pathway and cleaves pro-IL-1β and -IL-18 to bioactive forms. In addition to IL-1β and IL-18 processing, caspase-1 triggers pyroptosis by maturing the mediator gasdermin-D (Shi et al., 2015). Cleaved gasdermin-D oligomerizes in cell membranes to form pores, inducing cell death and facilitating release of mature IL-1β and IL-18 (Martinon et al., 2002; Shi et al., 2015; He et al., 2015). To confirm diminished caspase-1 activity in LPL-deficient macrophages after NLRP3 assembly, we examined IL-1β and gasdermin-D cleavage. Immunoblotting of cell lysates and culture supernatants revealed significantly reduced cleavage products of IL-1β and gasdermin-D in LPL-deficient AMs activated with ATP (Figure 2D) despite equivalent levels of pro-IL-1β and full-length gasdermin-D. Similarly, nigericin-induced IL-1β maturation was reduced in LPL-deficient AMs (Figure S1E).
Parallel analysis of BMDMs yielded similar results; LPL-deficient cells exhibited reduced processing of IL-1β and gasdermin-D following nigericin exposure, whereas LPS-induced priming (as assessed by NLRP3 expression) was comparable in WT and LPL-deficient BMDMs (Figure 2E). LPL-deficient BMDMs also displayed significantly reduced IL-1β cleavage (Figure 2F) and gasdermin-D processing (Figure 2G) in response to nigericin and ATP. Our findings of reduced processing of IL-1β and gasdermin-D in the absence of LPL corroborate disruption of the assembly signal in the process of NLRP3 activation.
LPL enhances ASC oligomerization
NLRP3 assembly is initiated by oligomerization of the adaptor protein ASC (Hoss et al., 2017), which also recruits pro-caspase-1 to the assembling NLRP3 complex. Oligomerized ASC complexes, visualized microscopically as “ASC specks,” indicate active inflammasome formation (Stutz et al., 2013). To define how LPL promotes NLRP3 assembly, we evaluated ASC speck formation using ASC-Citrine reporter mice (Tzeng et al., 2016). ASC specks were readily visible in WT AMs after NLRP3 activation but ASC specks formed in LPL-deficient cells ~50% less frequently (Figures 3A and 3B). We next assessed NLRP3-induced ASC oligomers using immunoblotting. In primed cells, ASC exists as a monomer. In response to the assembly signal, ASC multimerizes and translocates to the detergent-insoluble fraction. Upon treatment with NLRP3 activators, reduced translocation of ASC oligomers to the detergent-insoluble fraction was seen in LPL-deficient cells (Figure 3C). Whole-cell lysates showed comparable monomeric ASC levels, indicating that diminished oligomerization was not due to reduced availability of the ASC monomer. Confirmatory immunoblotting revealed reduced IL-1β cleavage product in LPL-deficient cells (Figure 3C, bottom panel). These observations suggest that LPL supports ASC oligomerization to enhance NLRP3 assembly.
Figure 3. LPL-deficient macrophages exhibit less ASC speck formation and reduced oligomerization upon NLRP3 activation.
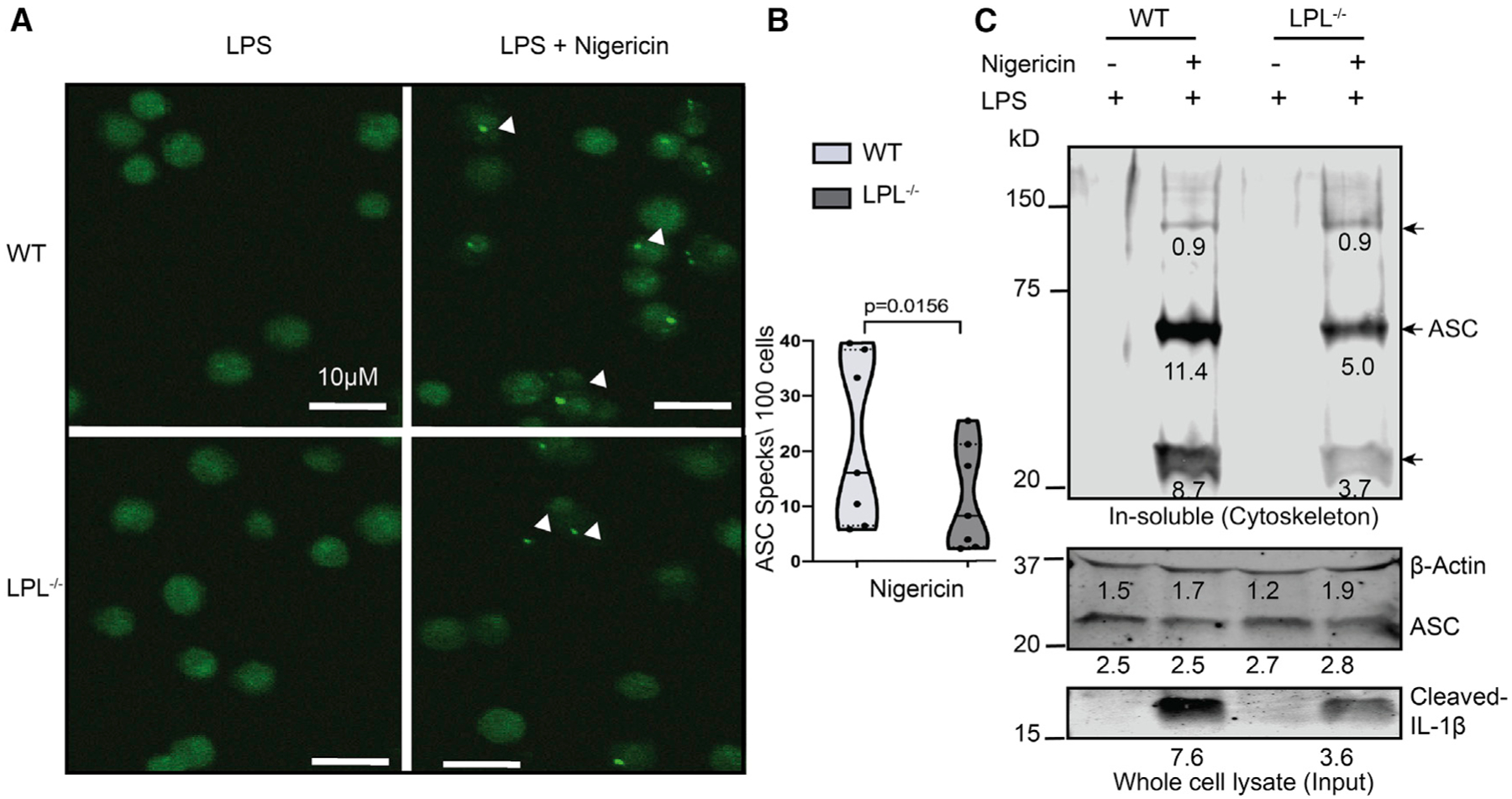
(A) AMs from ASC reporter WT and LPL−/− mice were isolated, LPS primed, and treated with nigericin. A microscopy image at 20× magnification shows ASC specks, visible as bright green fluorescent points (indicated by arrowheads). Scale bars, 10μm.
(B) Quantification of ASC speck count per 100 cells. In the graph, the solid line is at the median and the dashed line shows the interquartile range. The p values were obtained using a Wilcoxon test..
(A and B) Results represent 7 data points from 4 independent experiments.
(C) BMDMs were primed with LPS, activated with nigericin, and then lysed with Triton X-100. The Triton X-100-insoluble portion was crosslinked with disuccinimidyl suberate (DSS). The immunoblot of the detergent-insoluble fraction (cytoskeleton portion) was probed for oligomerized ASC. Triton X-100-lysed whole-cell lysate was used as input control for total ASC availability. Densitometry analysis values are shown below each band. Here, β-actin was used as a loading control, and cleaved IL-1β confirmed NLRP3 activation. Representative results from 2 independent experiments are shown.
Although NLRP3 requires ASC for activation (Elliott et al., 2018), other inflammasomes, such as NLRC4, do not because NLRC4 directly interacts with caspase-1 via CARD-CARD interactions (Nambayan et al., 2019; Dick et al., 2016; Vajjhala et al., 2012). We therefore hypothesized that, if LPL enhances inflammasome assembly by increasing ASC recruitment, then NLRC4 activation would be unaffected by loss of LPL. We tested NLRC4 activation in BMDMs after transfection with its activator flagellin. NLRC4-induced IL-1β production was equivalent in WT and LPL-deficient BMDMs (Figure S2A), and immunoblots showed comparable IL-1β cleavage in WT and LPL-deficient macrophages (Figure S2B). The LPL independence of NLRC4 activation is consistent with an ASC-dependent function of LPL in NLRP3 assembly.
LPL supports Pyk2:ASC interaction
We next considered possible molecular links between LPL and ASC. LPL is essential for formation and stability of macrophage podosomes, which are integrin-stimulated, F-actin-linked multimolecular complexes supporting myeloid cell adhesion and migration. Podosomes recruit Src family, Syk family, and focal adhesion kinase (FAK)/Pyk2 kinases, which organize as a “ring” around an F-actin “core.” We have previously demonstrated specific disruption of Pyk2 signaling in LPL-deficient B cells (Todd et al., 2013). Pyk2 also sensitizes ASC to oligomerization via direct interaction and phosphorylation of ASC tyrosine residue 146 (Tyr146); increased Pyk2 signaling augments ASC multimerization after NLRP3 activation, enhancing IL-1β output (Chung et al., 2016). Therefore, we tested whether LPL is required for Pyk2-ASC interactions during NLRP3 assembly by immunoprecipitating Pyk2 from BMDMs after LPS priming and/or NLRP3 activation and immunoblotting for ASC. As expected, there was no ASC associated with Pyk2 after LPS priming only (Figure 4A). In WT BMDMs, NLRP3 activation induced robust association of ASC with Pyk2; however, in LPL-deficient BMDMs, Pyk2-associated ASC was detected only weakly (Figure 4A). Similarly, co-immunoprecipitation of Pyk2-associated NLRP3 was reduced in LPL-deficient cells compared with the WT (Figure 4A). LPL was not detectable with anti-Pyk2 or anti-ASC immunoprecipitation (data not shown).
Figure 4. LPL stabilizes Pyk2-ASC interaction by regulating Pyk2 localization.
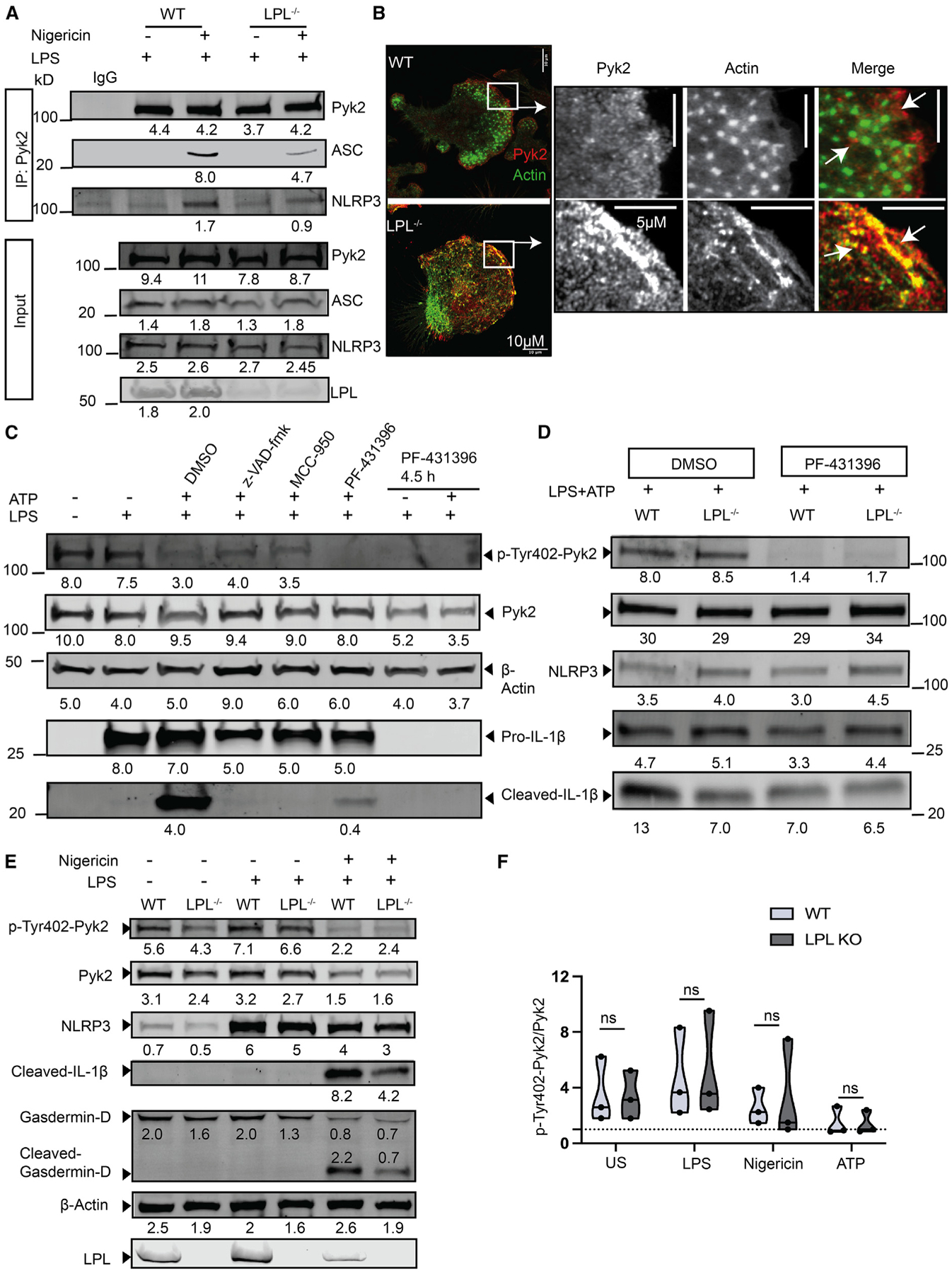
(A) LPS-primed or nigericin-activated NLRP3-stimulated BMDM cell lysates were subjected to Pyk2 immunoprecipitation (IP) using a specific antibody. Pulled down Pyk2 and co-precipitated ASC and NLRP3 were probed by immunoblotting. Here, initial cell lysate is used as an input control, and immunoglobulin G (IgG) isotype is used as a negative control for anti-Pyk2 IP. Results representing 2 independent experiments are shown..
(B and C) BMDMs were cultured on cover glass in serum-free medium for 1.5 h prior to serum addition to induce podosome formation (B, green dots). Cells were stained for F-actin with phalloidin-AF488 (green) and anti-Pyk2-IgG-DyLight594 (red). A 60× image (scale bars, 10μm) is shown here, with the insets showing podosomes (arrows; scale bars, 5 μm). Results represent 2 independent experiments. In (C), WT BMDMs were primed with LPS and stimulated for NLRP3 activation by ATP (30 min) in the absence and presence of z-VAD-fmk (50 μM, 1 h), MCC-950 (50 μM, 1 h), and PF-431396 (25 μM) for 1 h or 4.5 h. Cell lysates were probed for Pyk2 phosphorylation and IL-1β products by immunoblotting. The density of each band is shown below. Here, β-actin is the total cell control..
(D) WT and LPL−/− BMDMs were LPS primed and activated with ATP (30 min) in the presence of PF-431396 (25 μM, 1 h), and cell lysates were analyzed by immunoblotting for Pyk2 phosphorylation and IL-1β processing. Here, NLRP3 is the priming control, and density is shown below each band. (C and D) The results represent 2 independent experiments..
(E) Unstimulated (US), LPS-primed, and NLRP3-activated (nigericin) WT and LPL−/− BMDM cell lysates were analyzed for phosphorylated (Tyr-402) Pyk2 and processed gasdermin-D and IL-1β (cleaved). Here, NLRP3 expression, showing priming signal and β-actin, serves as loading control. Densitometry values are shown below each protein band. The control immunoblot for LPL is derived from a separate immunoblot run (not normalized) of the same lysates. A representative immunoblot from 3 independent experiments is shown..
(F) The ratio of phosphorylated Pyk2 (Tyr-402) to total Pyk2 is measured by densitometry from multiple experiments. Conditions include US, LPS priming, and after NLRP3 activation (nigericin or ATP). Here, each dot represents an individual data point, showing median (solid line) and interquartile range (dashed line). Data points (n = 3) are from Figures 4D, 4E, and S2C and replicate independent experiments (data not shown). Statistical significance was measured by Mann-Whitney test.
We then tested whether LPL is required for correct Pyk2 localization. When viewing podosomes en face by super-resolution microscopy, the ultrastructural ring-and-core appearance can be appreciated. The podosome core is formed by tightly cross-linked F-actin filaments, whereas integrin-associated proteins and signaling molecules, such as vinculin or Pyk2, surround the core in a ring-like structure. We have found previously that LPL stabilizes the F-actin cores (Zhou et al., 2016). This ring-and-core ultrastructure cannot always be resolved using traditional confocal microscopy, and then all podosome components appear to co-localize (Zhou et al., 2016). We therefore employed super-resolution microscopy to view podosome formation in BMDMs derived from WT and LPL−/− mice, after podosome formation was induced by serum stimulation (Zhou et al., 2016). In WT cells, Pyk2 localized to the ring of podosomes surrounding F-actin cores, as expected; however, in LPL-deficient cells, podosomes were organized poorly, with irregular and dense F-actin co-localizing with Pyk2 (Figure 4B). LPL is thus required for correct cellular localization of Pyk2, and absence of LPL reduces Pyk2 interaction with ASC.
Inhibition of Pyk2 activity, using the specific inhibitor PF-431396 added at the time of NLRP3 activation (Kedan et al., 2018), reduced IL-1β cleavage in cells treated with NLRP3 activators, in agreement with prior reports (Chung et al., 2016); in comparison, IL-1β cleavage was completely abolished by inhibiting caspase-1 (N-Benzyloxycarbonyl-Val-Ala-Asp(O-Me) fluoromethyl ketone [z-VAD-fmk]) or NLRP3 (MCC-950) (Figure 4C). Because Pyk2 also regulates MyD88 signaling downstream of TLR4 engagement (Xi et al., 2010), early inhibition of Pyk2 during LPS priming (4.5-h incubation) abrogated pro-IL-1β expression (Figure 4C), preventing further analysis of NLRP3 assembly. We confirmed equivalent inhibition of Pyk2 Tyr-402 phosphorylation by PF-431396 in WT and LPL−/− cells. However, although PF-431396 impaired IL-1β cleavage in WT cells, it did not further reduce the already impaired IL-1β processing in LPL−/− cells (Figure 4D). The failure to further impair IL-1β production in macrophages lacking LPL by pharmacological inhibition of Pyk2 activity suggests that LPL and Pyk2 function in the same molecular pathway during NLRP3 assembly.
We did not find reduced Pyk2 phosphorylation (Tyr-402) in macrophages lacking LPL at steady state, and Pyk2 phosphorylation was increased comparably upon LPS priming. Pyk2 phosphorylation was reduced equivalently after NLRP3 activation (Figure 4E), regardless of the NLRP3 activator used (Figures S2C and 4F). Thus, LPL deficiency disrupts the interaction of Pyk2 with ASC through a mechanism independent of Tyr402 phosphorylation, possibly through mislocalization (Figure 4B).
Although Pyk2-mediated phosphorylation of ASC promotes NLRP3 assembly, assembly of the ASC-dependent inflammasome AIM2 rests on FAK-mediated phosphorylation of ASC at a different tyrosine residue (Chung et al., 2016). We therefore tested whether AIM2 inflammasome activation depends on LPL. We found no significant differences in IL-1β production after stimulating WT and LPL-deficient BMDMs with poly(dA:dT) (Figure S2D). Equivalent AIM2 activation in WT and LPL-deficient macrophages is concordant with the dependence of FAK-mediated (and Pyk2-independent) regulation of ASC during AIM2 activation (Chung et al., 2016). The dependence of NLRP3 but not AIM2 activation on LPL is consistent with a model of LPL-mediated, Pyk2-specific regulation of NLRP3.
Impaired pneumococcal clearance in LPL−/− mice is NLRP3 driven
Pulmonary production of IL-1β enhances clearance of various infections, including Streptococcus pneumoniae (Lemon et al., 2015). The pneumococcal toxin pneumolysin is a major virulence factor and activates NLRP3 (Garcia-Suarez Mdel et al., 2007; McNeela et al., 2010). Deficient IL-1β production by LPL-deficient AMs suggests a second mechanism (beyond reduced AM numbers) for impaired clearance of pneumococci in LPL−/− mice (Todd et al., 2016). We confirmed defective IL-1β and IL-18 production by LPL-deficient AMs in response to pneumolysin (Figures 5A and 5B). Caspase-1 activity was also reduced significantly in LPL-deficient AMs after pneumolysin exposure (Figures 5C and S2E). We then evaluated clearance of pneumococcal isolates (D39; Streptococcus pneumoniae serotype 2) that did (D39) or did not (D39Δply) express pneumolysin and, therefore, did or did not activate NLRP3 (Garcia-Suarez Mdel et al., 2007; McNeela et al., 2010; Lemon et al., 2015; Figure 5D). As seen before, LPL−/− mice demonstrated impaired clearance of WT pneumococci (Figure 5E); however, clearance of D39Δply was unchanged by LPL deficiency (Figure 5F). Correlating with the bacterial loads, D39 infection induced significant weight loss in LPL−/− mice but infection with D39Δply did not (Figures 5D and 5G). The dependence of defective bacterial clearance in LPL−/− mice on the presence of pneumolysin, a potent NLRP3 activator, suggests that a functional defect in NLRP3 activation also enhances pneumococcal susceptibility in LPL−/− mice.
Figure 5. LPL−/− mouse lungs sufficiently cleared pneumococcal infection.
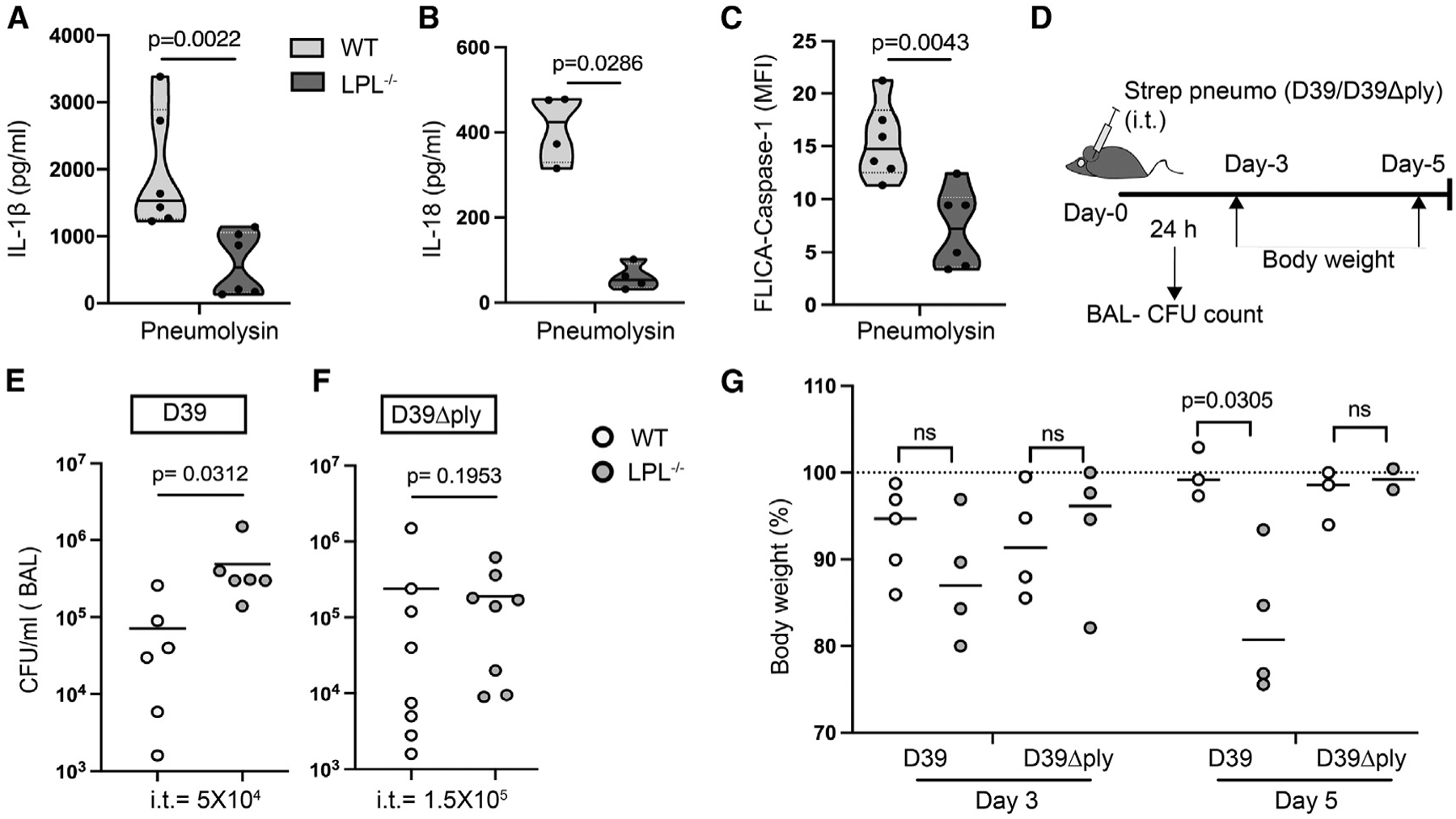
(A–C) AMs obtained from naive WT (light gray) and LPL−/− (dark gray) mice were primed with LPS and stimulated with pneumolysin, and then IL-1β (A) and IL-18 (B) in culture supernatant was quantified by ELISA. Represented results are from 3 independent experiments showing data points; n = 6 (A) and n = 4 (B). In (C), AMs were primed with LPS, activated with pneumolysin, and then labeled with FAM-FLICA to detect active caspase-1. Represented results n = 6 from 3 independent experiments are shown. Here, each dot represents an individual data point, showing median (solid line) and interquartile range (dashed line)..
(D) Schematic showing time points of analysis after pneumococcal infection, such as BAL colony-forming unit (CFU) count and body weight monitoring.
(E and F) Mice were injected intra-tracheal (i.t.) with the indicated dose of Streptococcus pneumoniae (D39 or D39Δply), and after 24 h, CFUs were counted from BAL fluid. Represented results show 3 independent experiments from 6 (E) and 8 (F) mice..
(G) WT and LPL−/− mice were administered (i.t.) Streptococcus pneumoniae (D39 or D39Δply). Body weight loss after infection is presented here on the indicated days, with a dashed line showing initial weight. Data represent 2 independent experiments..
In graphs, each dot represents an individual mouse (E–G) with median (solid lines) and standard deviation (error bars). The p values were calculated with a Mann Whitney test (A–C), Wilcoxon test (E), and ANOVA (G).
Using a new mouse model in which the endogenous LPL locus was modified to substitute the non-phosphorylatable alanine for a serine residue at position 5 (S5A), we recently showed that Ser-5 phosphorylation of LPL is required for pneumococcal clearance by splenic macrophages. However, homozygous S5A mice were able to clear pneumococci from the lungs (Anaya et al., 2021). To evaluate whether S5 phosphorylation of LPL regulates NLRP3 activation, we evaluated IL-1β production in BMDMs derived from WT or S5A mice after NLRP3 activation (Figure S2F). BMDMs from S5A mice exhibited NLRP3-induced IL-1β production similar to WT cells after ATP stimulation (Figure S2G). NLRP3-activated gasdermin-D and IL-1β processing after nigericin or ATP treatment in cells from S5A mice was also equivalent to the WT (Figure S2F). These results are in agreement with our previous observation that S5A mice showed no defect in pneumococcal clearance in our lung infection model (Anaya et al., 2021).
LPL−/− mice are resistant to bleomycin-induced lung injury and fibrosis
NLRP3 drives multiple inflammatory diseases characterized by ongoing tissue injury (Kuipers et al., 2011; Nystrom and Bruckner-Tuderman, 2018). NLRP3 activation and subsequent IL-1β release have been implicated in acute lung injury (ALI), which results in inflammation and fibrosis. Resident AMs are critical for NLRP3-induced ALI and fibrosis (Aran et al., 2019; dos Santos et al., 2015). We therefore investigated whether LPL deficiency would ameliorate pulmonary fibrosis in the well-characterized, NLRP3-dependent, bleomycin-induced ALI model (Izbicki et al., 2002; Moore and Hogaboam, 2008). Bleomycin directly injures lung epithelium, promoting uric acid production and triggering NLRP3 assembly; the resulting IL-1β production initiates an inflammatory response that further damages the tissue (Wilson et al., 2010). Subsequent tissue repair and remodeling feature fibroblast proliferation and enhanced collagen deposition, which are hallmarks of fibrosis (Gasse et al., 2009).
To test the hypothesis that reduced NLRP3 activation in LPL−/− mice would increase resistance to bleomycin-induced fibrosis, we challenged WT and LPL−/− mice with a single dose of intranasal bleomycin (Izbicki et al., 2002) and weighed the mice weekly (Figure 6A). Bleomycin challenge caused an ~10% decrease in body weight in WT mice after 1 week, whereas LPL−/− mice had not lost weight at this time point (Figure 6B). Three weeks after bleomycin treatment, H&E staining of lung sections revealed massively damaged lungs in WT mice, with inflammatory infiltrates. However, lung sections obtained from bleomycin-treated LPL−/− mice revealed tissue architecture similar to PBS-treated mice (Figure 6C). Compared with that of WT mice, BAL fluid from bleomycin-challenged LPL−/− mice had significantly reduced hydroxyproline content, indicating lower collagen accumulation (Figure 6D). Masson trichrome staining, used to highlight collagen deposition, revealed extensive fibrotic areas in the lungs of bleomycin-treated WT mice; in comparison, very little fibrosis was evident in lungs from bleomycin-treated LPL−/− mice (Figure 6E). Additionally, bleomycin-treated LPL−/− lungs had lower levels of cleaved IL-1β in BAL fluid in comparison with WT mice (Figure S3A). To assess cellular inflammatory responses, we analyzed BAL fluid 24 h after bleomycin treatment for the influx of polymorphonuclear leukocytes (i.e., neutrophils) and depletion of AMs (Reader et al., 2020; Misharin et al., 2013). LPL−/− mice exhibited significantly fewer CD11b+Ly6G+ neutrophils in the BAL fluid (Figure S3B) and did not lose as many CD11c+SiglecF+ AMs (Figure S3C) compared with WT mice. We similarly confirmed that LPL deficiency ameliorated another NLRP3-mediated inflammatory disease, MSU-induced peritonitis (Figure S4). LPL−/− mice exhibited attenuated recruitment of inflammatory cells (e.g., neutrophils and monocytes) upon MSU treatment. Protection from NLRP3-dependent, bleomycin-induced lung fibrosis in LPL−/− mice confirms the physiological relevance of LPL in activation of NLRP3.
Figure 6. LPL−/− mice are resistant to bleomycin-induced lung fibrosis.
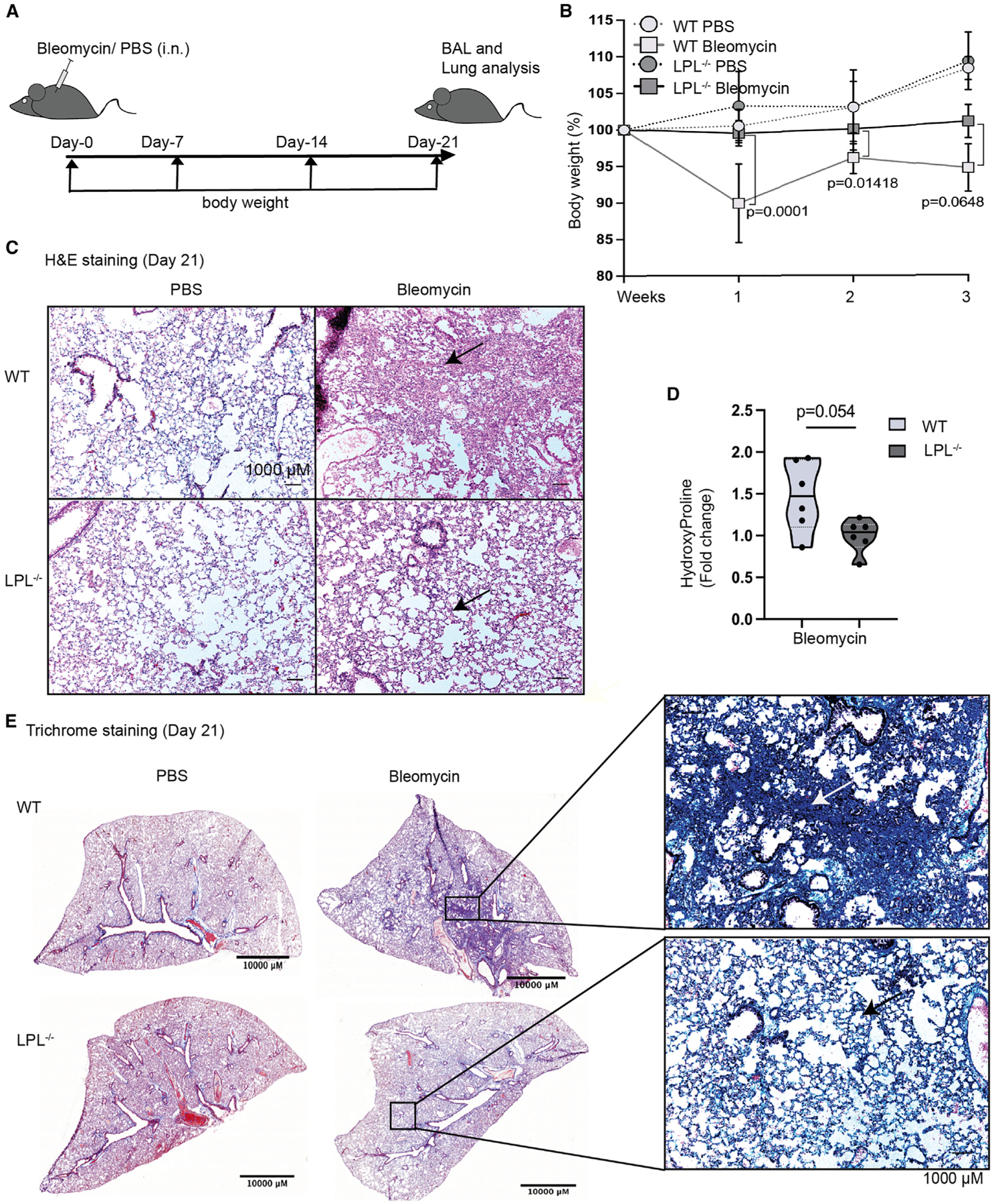
(A) To evaluate lung fibrosis in mice, a single dose of bleomycin (2 mg/kg) was administered intranasally (i.n.). Body weight was measured weekly, and BAL and lungs were obtained 21 days after challenge.
(B) Change in body weight plotted from PBS-treated (control) and bleomycin-treated mice.
(C) Sections of lungs obtained from PBS- and bleomycin-treated mice were analyzed for histological changes using H&E staining. Representative images at 10× magnification (scale bars, 1,000 μm) show lung sections with normal alveoli (empty holes) and damaged tissue (pink) (indicated by arrows).
(D) Three weeks after bleomycin challenge, accumulated hydroxyproline was quantified. Fold increase in hydroxyproline over PBS control mice is shown, with each symbol representing a single mouse, solid line at median, and dashed lines at interquartile range.
(E) Lung slices were stained with Masson trichrome to illuminate collagen deposition (blue, arrows), shown in representative section images (10× magnification); scale bars, 10,000 μ and 1,000 μm (inset).
Data were combined from 2 independent experiments, using 3 mice in each bleomycin group and 2 mice in the PBS control group in each experiment. The p values were determined by ANOVA (B) and Mann-Whitney test (D).
DISCUSSION
The NLRP3 inflammasome drives significant acute inflammation and tissue damage through IL-1β and other downstream mediators. Although actin cytoskeleton elements have been shown previously to regulate NLRP3 priming and the resulting NF-κB activation (Kustermans et al., 2008; Xi et al., 2010), evidence showing that actin-binding proteins participate in transducing the NLRP3 assembly signal has been lacking. Here we demonstrate a clear requirement for LPL in enhancing NLRP3 assembly and IL-1β production, independent of priming. Without LPL, ASC oligomerization and NLRP3 assembly were reduced following exposure to various NLRP3 activators, resulting in reduced caspase-1 activation and reduced cleavage of pro-IL-1β, pro-IL-18, and gasdermin-D. The molecular link from LPL to ASC is the kinase Pyk2, which has been shown previously to sensitize ASC to oligomerization (Vajjhala et al., 2012). In LPL-deficient cells, association of Pyk2 with ASC and NLRP3 was reduced, indicating disruption of the Pyk2:ASC signaling axis. We further showed that LPL balances effective host defense against autoinflammation; LPL−/− mice were more susceptible to pneumococcal infection (where NLRP3 was required for defense) but more resistant to bleomycin-induced pulmonary fibrosis.
LPL deficiency disrupts multiple immune functions, predominantly by disrupting processes relying on adhesion-based signaling. For instance, LPL is required for full T cell activation and normal T cell motility (Morley, 2013) and supports B cell motility and marginal-zone B cell development (Todd et al., 2011). Among myeloid cells, LPL is required for normal macrophage and monocyte migration under restrictive conditions (Zhou et al., 2016) but is dispensable for neutrophil migration (Chen et al., 2003). LPL is also required for normal maturation of AMs, and we found that LPL−/− mice possess about 50% fewer AMs than their WT counterparts (Todd et al., 2016). Although numerical deficiency of AMs in LPL−/− mice may be sufficient to increase susceptibility to pneumococcal infection (Deady et al., 2014), we now show that these mice have a second, functional defect that also enhances pneumococcal susceptibility because full NLRP3 assembly and IL-1β production are needed for pathogen clearance (Witzenrath et al., 2011; McNeela et al., 2010). Although a maturation defect in AMs from LPL-deficient animals could theoretically lead to subsequent activation impairment, we also found impaired NLRP3 activation in BMDMs derived from LPL−/− mice. Impaired NLRP3 activation in two separate macrophage lineages, one of which does not exhibit maturation defects, implies that diminished NLRP3 activation in AMs is not due to a developmental defect.
Identification of LPL as a modulator of NLRP3-mediated inflammatory signaling has significant implications for multiple human inflammatory and fibrotic diseases. LPL is a well-established, critical component of macrophage podosomes, multimolecular signaling complexes that support macrophage adhesion, migration, and mechanotransduction (Zhou et al., 2016); mechanotransduction is the translation of cell adhesion or stretching into intracellular signaling (van den Dries et al., 2019). Macrophage podosomes require LPL for stabilization (Zhou et al., 2016). Using super-resolution microscopy, the ring-and-core ultrastructure of podosomes becomes apparent. The core of podosomes is formed by tightly cross-linked F-actin, whereas integrin-associated and signaling proteins localize to a ring surrounding the F-actin core. Loss of LPL disrupts podosome organization and displaces the kinase Pyk2 from the ring, although phosphorylation of Pyk2 remains intact. Intact Pyk2 phosphorylation suggests that recruitment and activation of upstream kinases, such as the tyrosine kinase Syk2, are independent of LPL in this signaling pathway. In contrast, Syk2 phosphorylation downstream of β2 integrin signaling in neutrophils was disrupted by LPL deficiency (Chen et al., 2003). Pyk2 phosphorylation was also disrupted downstream of CXCL12 signaling in LPL-deficient B cells (Todd et al., 2011). Thus, the requirement for LPL in modulating Pyk2 phosphorylation appears to vary by cell type and cell stimulus. We did not identify a direct interaction between LPL and Pyk2, possibly because of technical limitations associated with co-immunoprecipitation analyses (for instance, if LPL and Pyk2 are co-localized to the detergent-insoluble cytoskeletal fraction of cell lysates, then co-immunoprecipitation would be impossible). However, we do find cellular mislocalization of Pyk2 in LPL-deficient macrophages, which correlates with a reduction in direct association of Pyk2 with ASC. Direct interaction of Pyk2 and ASC results in tyrosine phosphorylation of ASC, which enhances subsequent assembly of the NLRP3 inflammasome. However, Pyk2-mediated phosphorylation of ASC is dispensable for recruitment of ASC into the AIM2 inflammasome, and phosphorylation of ASC has not been shown to be required for NLRC4 activation (Chung et al., 2016). Thus, the specific function of Pyk2 in accentuating NLRP3 assembly is consistent with our observation that LPL deficiency impairs NLRP3 but not AIM2 or NLRC4 activation. The kinase Pyk2 participates in mechanotransduction (Duong and Rodan, 2000), and we show that loss of LPL removes the mechanosensitivity of NLRP3 activation (Figure 1A). With identification of a pathway linking podosomes through Pyk2 to NLRP3 assembly, we provide the first molecular explanation for the previously observed phenomenon that macrophage adhesion or stretching induces IL-1β (Chen et al., 2003; Kuipers et al., 2011; Wu et al., 2013). We propose that long-term changes in tissue compliance during chronic fibrotic processes could exert positive feedback signals on inflammatory processes through this pathway.
The N-terminal domain of LPL encompasses several regulatory features, including serine phosphorylation sites at Ser5 and Ser7, calmodulin-binding domains, and a threonine phosphorylation site at Thr89 (Janji et al., 2006; Morley, 2012). Because Ser5 is heavily phosphorylated in multiple immune cell lineages after stimulation (Wang et al., 2010; Hagi et al., 2006; Freeley et al., 2012; De Clercq et al., 2013), most work has focused on Ser5 phosphorylation of LPL in regulating cell activation and motility. We reported recently that Ser5 phosphorylation is required for efficient splenic clearance of blood-stream infection with Streptococcus pneumoniae, myeloid cell phagocytosis, and generation of splenic marginal-zone macrophages (Anaya et al., 2021). However, we have not noted defects in macrophage podosome formation or NLRP3 inflammasome activation in macrophages from S5A mice (Anaya et al., 2021). Further work using these mice will illuminate the specific requirements for Ser5 phosphorylation and how modification at other sites may compensate for or coordinate with this regulatory feature.
In contrast to its disease-promoting role in pulmonary fibrosis, LPL enhances survival after pneumococcal lung infection (Deady et al., 2014). The benefit of LPL in infection, in contrast to its pathologic contribution to fibrosis, can be explained by its requirement for full NLRP3 activation. Enhanced NLRP3 assembly and increased IL-1β production promote pathogen clearance during infection (Witzenrath et al., 2011; McNeela et al., 2010). Identification of LPL as a modulator of NLRP3 in infection and fibrosis may illuminate certain poorly understood disease processes, such as aging-associated inflammation, during which ongoing NLRP3 activation induces progression of pulmonary fibrosis (Latz and Duewell, 2018). In some clinical situations, aging fibrotic lungs exhibit increased susceptibility to pneumococcal infection (Stout-Delgado et al., 2016), which, in turn, accelerates fibrogenesis via pneumolysin-mediated NLRP3 activation (Knippenberg et al., 2015). Further elucidation of LPL-supported enhancement of NLRP3 assembly may clarify pathophysiological mechanisms common to infection and chronic inflammatory diseases, such as cryopyrin-associated periodic syndrome (CAPS), familial Mediterranean fever (FMF), and pyogenic arthritis (Mortimer et al., 2016; Carta et al., 2015; Wilson and Cassel, 2010).
Mechanical signals that modulate inflammatory responses have been correlated with integrin-induced F-actin rearrangement in a variety of cell types, such as chondrocytes, dendritic cells, and macrophages (McWhorter et al., 2013; Chen et al., 2014; Mennens et al., 2017). Recently, in vitro work observed that decreased tissue compliance (increased substrate stiffness) downregulated macrophage NLRP3 activity (Escolano et al., 2021). However, a molecular mechanism linking mechanosensation to NLRP3 activation has been elusive (Carpentier et al., 2019; Burger et al., 2016; Li et al., 2008; Joshi and Morley, 2019). Direct binding of F-actin with ASC and NLRP3 inhibited NLRP3 activation, with depolymerization of F-actin by Latrunculin enhancing NRLP3 assembly (Burger et al., 2016; Li et al., 2008). The F-actin severing protein Flightless-l (Flil), together with LRR Flightless-1-interacting protein 2 (LRRFIP2), also inhibits NLRP3 activation via direct binding (Burger et al., 2016). Flil severs F-actin filaments, generating free G-actin and limiting F-actin polymerization. Additionally, Flil can directly bind to caspase-1 to regulate its intracellular localization to inhibit NLRP3 assembly (Li et al., 2008). Flil and the LRRFIP3 regulator B cell adaptor for phosphoinositide 3-kinase (BCAP) also regulate NLRP3 activation. BCAP interacts directly with Flil and LRRFIP3, and loss of BCAP results in enhanced NLRP3 activation (Carpentier et al., 2019). Interestingly, LPL and multiple other actin-binding proteins, such as Myosin, gelsolin, and Arp2/3, also interact with BCAP, but the molecular mechanism of this complex is not yet known (Carpentier et al., 2019). In LPL S5A mutant macrophages, intact podosomes correlate with uninhibited NLRP3 inflammasome assembly, consistent with intact integrin-induced, F-actin-mediated mechanotransduction to NLRP3 assembly. Our results showing an LPL requirement for mechanical signaling because of substrate stiffness further confirm LPL-supported mechanotransduction in NLRP3 assembly. Targeting NLRP3 inflammasomes and mechanotransduction could open new paradigms for treating such inflammatory diseases. The NLRP3-specific inhibitor MCC-950 (Coll et al., 2015) controls liver inflammation and fibrosis in a murine model (Mridha et al., 2017). Further interrogation of this molecular signaling pathway transducing the NLRP3 assembly signal will drive increased understanding of the pathophysiology and options for modulation of infectious, inflammatory, and fibrotic conditions.
Limitations of the study
We note several limitations of our current study. First, we used small-molecule inhibition of Pyk2, but not genetic ablation, to test the requirement of Pyk2 in NLRP3 activation. Genetic ablation could not be performed in our system because priming of NLRP3 was also dependent on Pyk2. Second, the observation that phosphorylation of Pyk2 was unaffected in LPL-deficient macrophages contrasts with results from our prior work in B cells; delineating the differential requirements for Pyk2 phosphorylation in various immune cell lineages and downstream of different cell stimuli will be important in future work. Third, analyses of NLRP3-mediated inflammatory diseases (pneumococcal lung infection, MSU-induced peritonitis, and bleomycin-induced lung fibrosis) were performed in LPL−/− mice, in which all immune cells lack LPL. Future work performed in mice expressing cell-specific deletions of LPL will identify additional lineages critical for the observed phenotypes. Finally, all work was performed in mouse models, and translation to human systems remains a future goal.
We identified LPL as an enhancer of NLRP3 inflammasome activation and illuminated a molecular pathway from the macrophage podosome to IL-1β production. Delineating an adhesion-based pathway that modulates pro-inflammatory signals may provide new therapeutic targets for diseases that are difficult to treat, like idiopathic pulmonary fibrosis (IPF).
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Any additional information and inquiries regarding reagent and experimental resources should be directed to the lead contact, Sharon Celeste Morley (morleys@wustl.edu).
Materials availability
This study did not generate new unique reagents.
There are restrictions on the availability of bacterial strains, due to a material transfer agreement (MTA) requirement.
Data and code availability
No standardized data types are reported in this study. All data reported in this paper will be shared by lead contact upon request.
This study does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal procedures are conducted in accordance with the National Institute of Health (NIH) Guide for the care and Use of Laboratory animals and approved by the Institutional Animal Care and Use Committee (IACUC), Washington University, St. Louis. Animals were housed at the Animal Resource Center (CU-AMC) and were routinely monitored for general health, cage changes, feeding and overcrowding.
Mice were age (8–10 weeks) and gender matched. Wild-type (WT) and LPL−/− mice with C57BL/6 background were used at 6–10 weeks of age (Chen et al., 2003). ASC-Citrine reporter mice (B.6 Gt (ROSA)26Sortm1.1(CAG-Pycard/mCitrine*,−CD2*)Dtg) were purchased from The Jackson Laboratory. LPL S5A mice with C57BL/6 background were generated with use of designed guide RNA targeting the change by using the CRISPR/Cas9 system provided by Genome Engineering & iPSC Center at the Washington University School of Medicine (WUSM), St. Louis, MO and used with CRISPR control WT mice (Anaya et al., 2021). Used mouse model and bacterial strain are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ASC/TMS1 | Cell signaling Technology | AB_2799736 |
| NLRP3 | Cell signaling Technology | AB_2722591 |
| IL-1β | R and D Systems | AB_416684 |
| Caspase-1 | Millipore | AB_2068894 |
| β-Actin | Cell signaling Technology | AB_2797972 |
| GAPDH-HRP | Sigma-Aldrich | AB_1078992 |
| Cleaved IL-1β | Cell signaling Technology | AB_2799421 |
| L-Plastin (LPL) | Wang et al., 2010 | N/A |
| Pyk2 antibody mouse | Cell signaling Technology | AB_2174093 |
| Anti-Rabbit-IgG-AF680 | Thermo Fisher Scientific | AB_2535758 |
| anti-Rabbit IgG DyLight™ 800 | Rockland | AB_2610841 |
| anti-mouse IgG-IRDye800 | Rockland | AB_220125 |
| anti-mouse IgG-AF680 | Thermo Fisher Scientific | AB_2535724 |
| DyLight™ 594 AffiniPure Rabbit | Jackson ImmunoResearch | AB_2340204 |
| Anti-Mouse IgG + IgM (H+L) | Laboratories | |
| Phospho-Pyk2 (Tyr402) Antibody #3291 | Cell signaling Technology | AB_2300530 |
| CD11b-PE-Cy7 | BioLegend | AB_312799 |
| CD45-BV510 | BioLegend | AB_2563378 |
| F4/80-APC | BioLegend | AB_893493 |
| Ly6G-PE | BioLegend | AB_1186104 |
| Ly6C-PercP-Cy5.5 | BioLegend | AB_1659242 |
| Bacterial and virus strains | ||
| Streptococcus pneumoniae D39 | Dr. J. Rosch Rowe; (Rowe et al., 2019) | NCBI:txid373153 |
| Streptococcus pneumoniae D39Δply | Dr. Elaine I. Tuomanen; (Orihuela et al., 2004) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Lipopolysaccharide (LPS) | Invivogen | Cat# tlrl-3pelps |
| Nigericin | Sigma Aldrich | Cat# N7143 |
| Adenosine triphosphate (ATP) | Sigma Aldrich | Cat# 10127523001 |
| Flagellin protein | Invivogen | Cat# tlrl-stfla |
| Monosodium urate (MSU) | AdipoGen Life Science | Cat# AG-CR1-3950-2002 |
| Z-vad-fmk | Enzo LifeSciences | Cat# BML-P416-000 |
| MCC-950 | Selleckchem | Cat# S8930 |
| PF-431396 | Sigma-Aldrich | Cat# PZ0185 |
| Collagen-1 | Corning | Cat# 354236 |
| Streptavidin Cy5-plasmonic-fluors | Luan et al., 2020 | N/A |
| Phalloidin-AlexaFluor-488 | Invitrogen | Cat# A12379 |
| DOTAP liposomal transfection reagent | Sigma Aldrich | Cat# 11202375001 |
| Bleomycin sulfate | Fisher Scientific | Cat# 50148547 |
| p-dimethyl amino benzaldehyde (p-DMAB | Sigma Aldrich | Cat# 156477 |
| Critical commercial assays | ||
| Mouse IL-1β ELISA assay kit | Thermo Fisher Scientific | AB_2574946 |
| Mouse IL-18 ELISA assay kit | Thermo Fisher Scientific | AB_2575692 |
| Mouse IL-6 ELISA assay kit | Thermo Fisher Scientific | AB_2574989 |
| Mouse TNF-α ELISA assay kit | Thermo Fisher Scientific | AB 2575080 |
| FAM-FLICA® caspase-1 assay Kit | Immunochemistry | Cat# M0815 |
| Experimental models: Organisms/strains | ||
| C57BL/6 mice | N/A | |
| B6.Cg-Gt(ROSA)26Sortm1(CAG-Pycard/mCitrine*,- CD2*)Dtg/J Mus musculus (ASC-Citrine reporter mice) | The Jackson Laboratory | IMSR_JAX:030743 |
| C57BL/6 LPL-S5A mice | Anaya et al., 2021 | N/A |
| C57BL/6 LPL−/− mice | Chen et al., 2003 | N/A |
| Software and algorithms | ||
| Prism9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Image Studio lite | LI-COR Biosciences | N/A |
| FlowJo | BD Biosciences | https://www.flowjo.com/solutions/flowjo |
| Other | ||
| Silica gel containing glass bottom 96 well plates | Advanced BioMatrix | Cat# 5255; #5261 |
| StrataClean resin | Agilent Technologies | Cat# 400714 |
| Anti-mouse IgG-conjugated beads | Rockland | Cat# 00-8811-25 |
Pneumococcal infections
Streptococcus pneumoniae (D39; serotype 2) (Rowe et al., 2019) or its pneumolysin-deficient mutant (D39Δply) (Orihuela et al., 2004) were grown overnight on plates containing trypticase soy agar with 5% sheep blood. Mice were challenged with ~5 × 104 colony forming units (cfu) in 20 μL of Dulbecco’s PBS [DPBS]) via intra-tracheal instillation (Deady et al., 2014). Bronchoalveolar lavage (BAL) fluid was obtained 24 h after challenge and bacterial burden quantified by serial dilution. Infected mice were monitored once daily for change in body weight for 5 d. For body weight loss experiment, infected mice were monitored once daily for change in body weight for 5 days and animals that lost >20% of starting weight were euthanized. WT D39 bacteria was kindly provided by Dr. J. Rosch (Rowe et al., 2019) and pneumolysin deficient (D39Δply) strain was generous gift from Dr. Elaine I. Tuomanen (Orihuela et al., 2004).
MSU-induced peritonitis
Monosodium urate (MSU; 250 μg in 100 μL PBS) (AdipoGen Life Science; Cat#AG-CR1-3950-2002) or PBS was administered via i. p. injection. Mice were sacrificed 6 h post-challenge. Peritoneal exudate was analyzed by flow cytometry for infiltrating immune cells (BD LSRFortessa™). Directly fluorescently labeled antibodies used at 1:200 dilution for flow cytometry staining are, CD11b- PE-Cy7, CD45- BV510, F4/80-APC, Ly6G-PE, Ly6C-PercP-Cy5.5. Flow cytometry data was analyzed using FlowJo (BD) software. For antibodies details refer to key resources table.
Bleomycin-induced lung fibrosis
Mice received a single intranasal dose of bleomycin sulfate (2 mg/kg) (Fisher Scientific; Cat#50148547). Body weights were obtained weekly and mice sacrificed 21 d after inoculation. BAL fluid was analyzed for IL-1β via immunoblot and for hydroxyproline content by a colorimetric assay using Ehrlich’s reagent (p-dimethyl amino benzaldehyde (p-DMAB)) (Van Dyken et al., 2017). Lungs were fixed in 4% paraformaldehyde and 8-μm sections generated. Lung sections underwent staining with hematoxylin and eosin or Masson trichrome, and images were acquired by the core facility of the Division of Comparative Medicine at Washington University.
In vitro macrophage stimulation
To obtain alveolar macrophages (AMs) bronchoalveolar lavage (BAL) was harvested from naïve mice using DPBS (gibco; Cat#14190-136) containing 1% fetal bovine serum (FBS) (Sigma-Aldrich; Cat#F2442) and 0.6mM EDTA. BAL was resuspended in complete DMEM (gibco; Cat#11965-084) and after 1 hour of culture, non-adherent cells were removed via washing with fresh culture media. Adherent AMs were used for further stimulations (Zhou et al., 2016).
For bone marrow derived macrophages (BMDMs), mice femurs and tibias were harvested and bone marrow was flushed with DPBS containing 1% FBS. Collected bone marrow cells were treated with BD lysis buffer (BD; Cat#555899) for lysing red blood cells. To induce differentiation to macrophages, bone marrow cells cultured in DMEM supplemented with 10% L929 conditioned media (containing macrophage colony stimulating factor (M-CSF) and 10% FBS, 1% penicillin/streptomycin (Lonza; Cat# BW17602E). BMDMs were collected using cellstripper™ buffer (Corning; Cat#25-056-CI) after 7 days of culture and then further used for stimulations.
For invitro activation assays, macrophages were maintained in Dulbecco’s modified essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (dos Santos et al., 2015). For NLRP3 activation, priming ‘signal-1’ was provided by TLR4 activation using LPS (500 ng/ml for BMDMs and 1 μg/ml for AMs) (InvivoGen; Cat#tlrl-3pelps) treatment for 4 hrs. After that, specific NLRP3 inflammasome activators such as nigericin (20μM; Sigma Aldrich; Cat#N7143; 30 m), ATP (5mM; Sigma (Roche); Cat#10127523001; 30 m) or pneumolysin toxin (1 μg; 1 h) were added. (Other concentrations or durations are specified in the figure legends). Purified pneumolysin toxin was received as a kind gift from Dr. Elaine I. Tuomanen, St. Jude Children’s Research Hospital, TN, USA. For NLRC4 inflammasome stimulation, LPS primed (500 ng/ml for 4 h) macrophages were transfected with purified flagellin protein (1 μg/ml for 1 h; S. typhimurium; InvivoGen; Cat#tlrl-stfla) using DOTAP liposomal transfection reagent (Sigma (Roche); Cat#11202375001) for 2 h in OptiMEM media (gibco; Cat#31985070) (Miao et al., 2006). Pharmacological inhibitors e.g., Pan-caspase-1 inhibitor; Z-vad-fmk (Enzo LifeSciences; Cat# BML-P416-000), NLRP3 inhibitor; MCC-950 (Selleckchem; Cat#S8930) and Pyk2 inhibitor; PF-431396 (Sigma-Aldrich; Cat#PZ0185) were added 30 min before NLRP3 activation. To induce mechanotransduction AMs were cultured on Collagen-1 (Corning; Cat#354236, 100μg/ml) coated silica gel containing glass bottom 96 well plates (Advanced BioMatrix; Cat#5255 and #5261). For reagents details refer to key resources table.
In all experiments primary cells were harvested from sex and age matched mice. Data represented in the study are from sex-randomized experiments.
METHOD DETAILS
ELISA
Culture supernatants (fresh or stored at −80°C) from stimulated macrophages were determined by using ELISA detection kits to measure secreted IL-1β (AB_2574946) and IL-18 (AB_2575692) as per manufacturer instructions. In parallel, for LPS priming signal, TNF-α (AB_2575080) and IL-6 (AB_2574989) were detected from culture medium using ELISA technique as per manufacturer instructions. IL-1β quantification was normalized to corresponding levels of TNF-α (in AMs) and IL-6 (in BMDMs), to standardize for priming across multiple experiments. For reagent details refer to key resources table.
Confocal and super-resolution microscopy
Fluoro-dot assay
To detect localized cytokine secretion from AMs, novel plasmon-enhanced fluoro-dot assay was used (Luan et al., 2020). AMs were seeded at density of 5,000 cells/well on glass-bottom 96-well black plates (Cellvis; Cat#P96-1.5H-N) coated with ELISA capture antibody (2 μg/ml in PBS). After NLRP3 activation, cells were fixed using 4% neutral buffered formalin (NBF) and incubated with ELISA biotinylated detection antibodies (anti-TNF-α and anti-IL-1β) followed by streptavidin Cy5-plasmonic-fluors. Nuclei were visualized with DAPI. Images were acquired using a Nikon TsR2 epifluorescence illumination microscope with 20× air objective, 0.75 numerical aperture for secreted cytokines. For image processing and quantification of IL-1β particles from each cell/cluster, ImageJ was used.
ASC speck detection.
AMs isolated from WT and LPL−/− ASC-citrine reporter mice (Tzeng et al., 2016) were cultured on 8 chamber cover glass (Eppendorf; Cat#0030742036). For NLRP3 activation nigericin (20 μM for 30 m) was exposed after LPS (1 μg/ml for 4 h) priming. For microscopic preparation, cells were fixed in 2% paraformaldehyde (PFA) and mounted on slides using DAPI containing mounting medium (Vectashield; Cat#H-1200-10). Images were acquired using Olympus FLUOVIEW FV1000, software version FV10-ASW 2.0.0.7, with a 20× air objective and 0.75 numerical aperture. ASC specks were quantified in cells by a blinded observer using ImageJ.
Podosome imaging.
Synchronous podosome formation in BMDMs was induced by exposure to FBS-containing DMEM for 30 min after 1.5-h serum starvation (Zhou et al., 2016). Cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton™-x-100, blocked in 5% normal goat serum, then stained with Phalloidin-AlexaFluor-488 (Invitrogen; Cat#A12379) (for F-actin) and anti-Pyk2 antibody (Cell Signaling; cat#3480S). Anti-Pyk2 antibody was detected via secondary antibody conjugated to DyLight-594 (Jackson ImmunoResearch Laboratories, West Grove, PA). Nuclei were visualized with DAPI. Images were acquired using a Zeiss LSM 880 II with Airyscan FAST confocal microscope with a 60× oil objective and numerical aperture 1.40.
Measurement of caspase-1 activation
To determine activated caspase-1 upon NLRP3 activation, AMs were stimulated with various stimulants as described earlier. Activated cells were stained using FAM-FLICA® caspase-1 assay Kit (Immunochemistry; Cat#M0815) as per manufacturer instructions. FAM-FLICA® stained AMs were subjected to flow cytometry (BD LSRFortessa™) for assessing caspase-1 activity at FITC fluorochrome excitation. To obtain NLRP3 induced MFI, autofluorescence from unstained cells was subtracted from NLRP3 activated cells.
Immunoblotting
Cell lysates were prepared using RIPA lysis buffer containing protease and phosphatase Inhibitors (Sigma Aldrich: Cat#PPC1010) and further processed in Laemmli buffer and stored in −20°C. Cell lysates were resolved using SDS-PAGE gels and proteins were transferred on PVDF-membranes (Bio-Rad; Cat#165800). For detecting proteins, membranes were incubated with specific primary antibodies overnight at 4°C. Used primary and secondary antibodies are listed in key resources table. Primary antibodies diluted in TBST (5% milk or 3% BSA), ASC/TMS1 (1:500), NLRP3 (1:500), IL-1β/IL-1F2 (1:1000), caspase-1 (1:1000), β-Actin (1:4000), GAPDH-HRP (1:2000), Cleaved IL-1β (1:500), I2A2 (m-LPL) (1:500), anti-Pyk2 antibody mouse (1:1000), and anti-Phosphorylated (Tyr-402)-Pyk2 antibody mouse (1:500) for application. Secondary antibody directed to primary antibody used at dilution of 1:10000; anti-rabbit IgG-AF680, anti-Rabbit IgG DyLight 800, anti-mouse IgG-IRDye800, anti-mouse IgG-AF680. For detecting IL-1β, supernatants were incubated with StrataClean resin (Agilent Technologies; Cat#400714) overnight. Beads bound proteins were eluted by boiling with Laemmli buffer and used for immunoblotting. For imaging, membranes were scanned in LI-COR-Odyssey imager. Densitometry analysis was performed using ImageStudio™ (LI-COR) software. In figures, corresponding molecular weights are indicated in kD units with shown protein bands.
ASC oligomerization assay
NLRP3 activated BMDMs cells were lysed in wells using 1% Triton™ x-100 (Sigma-Aldrich) containing protease and phosphatase inhibitors (Sigma Aldrich; Cat#PPC1010). For cross-linking oligomerized ASC, Triton™ x-100 insoluble fraction of cell lysate was subjected to 2 mM disuccinimidyl suberate (DSS) (Thermofisher; Cat#A39267) treatment for 30 m at room temperature. Pellets were then dissolved by boiling in Laemmli buffer and resolved in SDS-PAGE together with input control (Triton™ x-100 soluble fraction of cell lysate).
Co-immunoprecipitation assay
After NLRP3 activation (LPS + nigericin), BMDMs (1 × 107 cells per sample) were lysed in immunoprecipitation (IP) buffer (Pierce; Cat#87788). Lysates were incubated overnight with anti-Pyk2 antibody. Complexes were precipitated using anti-mouse IgG-conjugated beads (ROCKLAND; Cat#00-8811-25) as per manufacturer’s instructions. Samples were immunoblotted for co-precipitated ASC and/or NLRP3 protein. Mouse-specific IgG isotype was used for IP negative control, and whole cell lysate was used as input control.
QUANTIFICATION AND STATISTICAL ANALYSIS
Immunoblots were visualized using a LI-COR Odyssey imager. Densitometry analysis was performed using ImageStudio™ (LI-COR) software. Microscopic images were analyzed using ImageJ software (NIH). Flowcytometry results calculated (frequency and median fluorescent intensity (MFI)) by FLOWJO (BD) software. ELISA results were quantified using spectrophotometer at 450 nm absorbance (BioTek).
Quantified data were graphed and analyzed using Prism (GraphPad Software, La Jolla, CA). Non-parametric Mann-Whitney, Wilcoxon or ANOVA tests were employed, as indicated in figure legends, and p values ≤ 0.05 were considered significant. In figure legends, n = is number of data points (from ELISA and immunoblot densitometry analysis) collected from parallel stimulations (technical repeats) within independent experiments (biological replicates) or otherwise mentioned in figure legends.
Supplementary Material
Highlights.
Macrophages require L-plastin for optimal NLRP3 inflammasome activation
LPL stabilizes Pyk2 kinase interaction with ASC in NLRP3 assembly to link podosomes
Podosome-mediated mechanotransduction in inflammasome signaling requires L-plastin
L-plastin supports bleomycin-induced lung fibrosis via NLRP3 pathway
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants R01-AI104732, R21EB030171, R01 AI118719, HL148033, R01AI139540, and R56 AI104732 (to S.C.M.), National Science Foundation grants CBET-1900277 and CMMI1548571 (to S.C.M.), American Lung Association grant ETRA 736343 (to S.C.M.), Washington University in Saint Louis Children’s Discovery Institute grants CDI-CORE-2019-813 and CDI-CORE-2015-505 (to S.C.M.), Foundation for Barnes-Jewish Hospital grants 3770 and 4642 (to S.C.M.), American Association of Immunologists AAI Careers in immunology fellowship (to S.C.M.), National Institutes of Health grant R21EB030171 (to S.S.), National Science Foundation CBET-1900277 (to S.S.), National Institutes of Health grant R01 AI118719 (to F.S.), and National Institutes of Health grant HL148033 (to S.J.V.D.). The content of this study is the authors’ sole responsibility and does not necessarily represent official NIH views.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110507.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Anaya EP, Lin X, Todd EM, Szasz TP, and Morley SC (2021). Novel mouse model reveals that serine phosphorylation of L-plastin is essential for effective splenic clearance of pneumococcus. J. Immunol 206, 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, et al. (2019). Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol 20, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Audenhove I, Denert M, Boucherie C, Pieters L, Cornelissen M, and Gettemans J (2016). Fascin rigidity and L-plastin flexibility cooperate in cancer cell invadopodia and filopodia. J. Biol. Chem 291, 9148–9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black RA, Kronheim SR, Merriam JE, March CJ, and Hopp TP (1989). A pre-aspartate-specific protease from human leukocytes that cleaves pro-interleukin-1 beta. J. Biol. Chem 264, 5323–5326. [PubMed] [Google Scholar]
- Burger D, Fickentscher C, De Moerloose P, and Brandt KJ (2016). F-actin dampens NLRP3 inflammasome activity via Flightless-I and LRRFIP2. Sci. Rep 6, 29834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier SJ, Ni M, Duggan JM, James RG, Cookson BT, and Hamerman JA (2019). The signaling adaptor BCAP inhibits NLRP3 and NLRC4 inflammasome activation in macrophages through interactions with Flightless-1. Sci. Signal 12, eaau0615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta S, Penco F, Lavieri R, Martini A, Dinarello CA, Gattorno M, and Rubartelli A (2015). Cell stress increases ATP release in NLRP3 inflammasome-mediated autoinflammatory diseases, resulting in cytokine imbalance. Proc. Natl. Acad. Sci. U S A 112, 2835–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Mocsai A, Zhang H, Ding RX, Morisaki JH, White M, Roth-fork JM, Heiser P, Colucci-Guyon E, Lowell CA, et al. (2003). Role for plastin in host defense distinguishes integrin signaling from cell adhesion and spreading. Immunity 19, 95–104. [DOI] [PubMed] [Google Scholar]
- Chen C, Xie J, Deng L, and Yang L (2014). Substrate stiffness together with soluble factors affects chondrocyte mechanoresponses. ACS Appl. Mater. Inter 6, 16106–16116. [DOI] [PubMed] [Google Scholar]
- Chen C, Cai Q, He W, Lam TB, Lin J, Zhao Y, Chen X, Gu P, Huang H, Xue M, et al. (2017). AP4 modulated by the PI3K/AKT pathway promotes prostate cancer proliferation and metastasis of prostate cancer via upregulating L-plastin. Cell Death Dis 8, e3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Zhou J, Zhang B, Chen Z, Luo Q, and Song G (2018). Cyclic stretching exacerbates tendinitis by enhancing NLRP3 inflammasome activity via F-actin depolymerization. Inflammation 41, 1731–1743. [DOI] [PubMed] [Google Scholar]
- Cho SJ, Rooney K, Choi AMK, and Stout-Delgado HW (2018). NLRP3 inflammasome activation in aged macrophages is diminished during Streptococcus pneumoniae infection. Am. J. Physiol. Lung Cell Mol. Physiol 314, L372–L387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung IC, Ouyang CN, Yuan SN, Li HP, Chen JT, Shieh HR, Chen YJ, Ojcius DM, Chu CL, Yu JS, et al. (2016). Pyk2 activates the NLRP3 inflammasome by directly phosphorylating ASC and contributes to inflammasome-dependent peritonitis. Sci. Rep 6, 36214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq S, Zwaenepoel O, Martens E, Vandekerckhove J, Guillabert A, and Gettemans J (2013). Nanobody-induced perturbation of LFA-1/L-plastin phosphorylation impairs MTOC docking, immune synapse formation and T cell activation. Cell Mol. Life Sci 70, 909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, et al. (2015). A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med 21, 248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deady LE, Todd EM, Davis CG, Zhou JY, Topcagic N, Edelson BT, Ferkol TW, Cooper MA, Muenzer JT, and Morley SC (2014). L-plastin is essential for alveolar macrophage production and control of pulmonary pneumococcal infection. Infect. Immun 82, 1982–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick MS, Sborgi L, Ruhl S, Hiller S, and Broz P (2016). ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat. Commun 7, 11929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Dries K, Linder S, Maridonneau-Parini I, and Poincloux R (2019). Probing the mechanical landscape - new insights into podosome architecture and mechanics. J. Cell Sci 132, jcs236828. [DOI] [PubMed] [Google Scholar]
- Duong L,T, and Rodan GA (2000). PYK2 is an adhesion kinase in macrophages, localized in podosomes and activated by beta(2)-integrin ligation. Cell Motil. Cytoskeleton 47, 174–188. [DOI] [PubMed] [Google Scholar]
- Van Dyken SJ, Liang HE, Naikawadi RP, Woodruff PG, Wolters PJ, Erle DJ, and Locksley RM (2017). Spontaneous chitin accumulation in airways and age-related fibrotic lung disease. Cell 169, 497–509.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott EI, Miller AN, Banoth B, Iyer SS, Stotland A, Weiss JP, Gottlieb RA, Sutterwala FS, and Cassel SL (2018). Cutting edge: mitochondrial assembly of the NLRP3 inflammasome complex is initiated at priming. J. Immunol 200, 3047–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escolano JC, Taubenberger AV, Abuhattum S, Schweitzer C, Farrukh A, Del Campo A, Bryant CE, and Guck J (2021). Compliant substrates enhance macrophage cytokine release and NLRP3 inflammasome formation during their pro-inflammatory response. Front. Cell Dev. Biol 9, 639815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Feuvre RA, Brough D, Iwakura Y, Takeda K, and Rothwell NJ (2002). Priming of macrophages with lipopolysaccharide potentiates P2X7-mediated cell death via a caspase-1-dependent mechanism, independently of cytokine production. J. Biol. Chem 277, 3210–3218. [DOI] [PubMed] [Google Scholar]
- Freeley M, O’Dowd F, Paul T, Kashanin D, Davies A, Kelleher D, and Long A (2012). L-plastin regulates polarization and migration in chemokine-stimulated human T lymphocytes. J. Immunol 188, 6357–6370. [DOI] [PubMed] [Google Scholar]
- Garcia-Suarez Mdel M, Florez N, Astudillo A, Vazquez F, Villaverde R, Fabrizio K, Pirofski LA, and Mendez FJ (2007). The role of pneumolysin in mediating lung damage in a lethal pneumococcal pneumonia murine model. Respir. Res 8, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasse P, Riteau N, Charron S, Girre S, Fick L, Petrilli V, Tschopp J, Lagente V, Quesniaux VF, Ryffel B, and Couillin I (2009). Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am. J. Respir. Crit. Care Med 179, 903–913. [DOI] [PubMed] [Google Scholar]
- Goldstein D, Djeu J, Latter G, Burbeck S, and Leavitt J (1985). Abundant synthesis of the transformation-induced protein of neoplastic human fibroblasts, plastin, in normal lymphocytes. Cancer Res 45, 5643–5647. [PubMed] [Google Scholar]
- Hagi A, Hirata H, and Shinomiya H (2006). Analysis of a bacterial lipopolysaccharide-activated serine kinase that phosphorylates p65/L-plastin in macrophages. Microbiol. Immunol 50, 331–335. [DOI] [PubMed] [Google Scholar]
- He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, and Han J (2015). Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res 25, 1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano S, Zhou Q, Furuyama A, and Kanno S (2017). Differential regulation of IL-1β and IL-6 release in murine macrophages. Inflammation 40, 1933–1943. [DOI] [PubMed] [Google Scholar]
- Hoss F, Rodriguez-Alcazar JF, and Latz E (2017). Assembly and regulation of ASC specks. Cell Mol. Life Sci 74, 1211–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izbicki G, Segel MJ, Christensen TG, Conner MW, and Breuer R (2002). Time course of bleomycin-induced lung fibrosis. Int. J. Exp. Pathol 83, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janji B, Giganti A, De Corte V, Catillon M, Bruyneel E, Lentz D, Plastino J, Gettemans J, and Friederich E (2006). Phosphorylation on Ser5 increases the F-actin-binding activity of L-plastin and promotes its targeting to sites of actin assembly in cells. J. Cell Sci 119, 1947–1960. [DOI] [PubMed] [Google Scholar]
- John V, Kotze LA, Ribechini E, Walzl G, Du Plessis N, and Lutz MB (2019). Caveolin-1 controls vesicular TLR2 expression, p38 signaling and T cell suppression in BCG infected murine monocytic myeloid-derived suppressor cells. Front. Immunol 10, 2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi H, and Morley SC (2019). Cells under stress: the mechanical environment shapes inflammasome responses to danger signals. J. Leukoc. Biol 106, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi H, Lunz B, Peters A, Zölch M, Berberich I, and Berberich-Siebelt F (2021). The extreme C-terminus of IRAK2 assures full TRAF6 ubiquitination and optimal TLR signaling. Mol. Immunol 134, 172–182. [DOI] [PubMed] [Google Scholar]
- Kedan A, Verma N, Saroha A, Shreberk-Shaked M, Müller AK, Nair NU, and Lev S (2018). PYK2 negatively regulates the Hippo pathway in TNBC by stabilizing TAZ protein. Cell Death Dis 9, 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemke M, Rafael MT, Wabnitz GH, Weschenfelder T, Konstandin MH, Garbi N, Autschbach F, Hartschuh W, and Samstag Y (2007). Phosphorylation of ectopically expressed L-plastin enhances invasiveness of human melanoma cells. Int. J. Cancer 120, 2590–2599. [DOI] [PubMed] [Google Scholar]
- Knippenberg S, Ueberberg B, Maus R, Bohling J, Ding N, Tort Tarres M, Hoymann HG, Jonigk D, Izykowski N, Paton JC, et al. (2015). Streptococcus pneumoniae triggers progression of pulmonary fibrosis through pneumolysin. Thorax 70, 636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuipers MT, Aslami H, Janczy JR, Sluijs KFVD, Vlaar APJ, Wolthuis EK, Choi G, Roelofs JJTH, Flavell RA, Sutterwala FS, et al. (2011). Ventilator-induced lung injury is mediated by the NLRP3 inflammasome. Anesthesiology 116, 11. [DOI] [PubMed] [Google Scholar]
- Kustermans G, El Mjiyad N, Horion J, Jacobs N, Piette J, and LegrandPoels S (2008). Actin cytoskeleton differentially modulates NF-kappaB-mediated IL-8 expression in myelomonocytic cells. Biochem. Pharmacol 76, 1214–1228. [DOI] [PubMed] [Google Scholar]
- Latz E, and Duewell P (2018). NLRP3 inflammasome activation in inflammaging. Semin. Immunol 40, 61–73. [DOI] [PubMed] [Google Scholar]
- Lemon JK, Miller MR, and Weiser JN (2015). Sensing of interleukin-1 cytokines during Streptococcus pneumoniae colonization contributes to macrophage recruitment and bacterial clearance. Infect. Immun 83, 3204–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yin HL, and Yuan J (2008). Flightless-I regulates proinflammatory caspases by selectively modulating intracellular localization and caspase activity. J. Cell Biol 181, 321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CS, Park T, Chen ZP, and Leavitt J (1993). Human plastin genes. Comparative gene structure, chromosome location, and differential expression in normal and neoplastic cells. J. Biol. Chem 268, 2781–2792. [PubMed] [Google Scholar]
- Luan J, Seth A, Gupta R, Wang Z, Rathi P, CAO S, Gholami Derami H, Tang R, Xu B, Achilefu S, et al. (2020). Ultrabright fluorescent nanoscale labels for the femtomolar detection of analytes with standard bioassays. Nat. Biomed. Eng 4, 518–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Burns K, and Tschopp J (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426. [DOI] [PubMed] [Google Scholar]
- Martinon F, Petrilli V, Mayor A, Tardivel A, and Tschopp J (2006). Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241. [DOI] [PubMed] [Google Scholar]
- McNeela EA, Burke A, Neill DR, Baxter C, Fernandes VE, Ferreira D, Smeaton S, El-Rachkidy R, Mcloughlin RM, Mori A, et al. (2010). Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog 6, e1001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhorter FY, Wang T, Nguyen P, Chung T, and Liu WF (2013). Modulation of macrophage phenotype by cell shape. Proc. Natl. Acad. Sci. U S A 110, 17253–17258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennens SFB, Bolomini-Vittori M, Weiden J, Joosten B, Cambi A, and Van Den Dries K (2017). Substrate stiffness influences phenotype and function of human antigen-presenting dendritic cells. Sci. Rep 7, 17511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, and Aderem A (2006). Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol 7, 569–575. [DOI] [PubMed] [Google Scholar]
- Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, and Perlman H (2013). Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol 49, 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore BB, and Hogaboam CM (2008). Murine models of pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol 294, L152–L160. [DOI] [PubMed] [Google Scholar]
- Morley SC (2012). The actin-bundling protein L-plastin: a critical regulator of immune cell function. Int. J. Cell Biol 2012, 935173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley SC (2013). The actin-bundling protein L-plastin supports T-cell motility and activation. Immunol. Rev 256, 48–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer L, Moreau F, Macdonald JA, and Chadee K (2016). NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat. Immunol 17, 1176–1186. [DOI] [PubMed] [Google Scholar]
- Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, Haczeyni F, Teoh NC, Savard C, Ioannou GN, et al. (2017). NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol 66, 1037–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambayan RJT, Sandin SI, Quint DA, Satyadi DM, and De Alba E (2019). The inflammasome adapter ASC assembles into filaments with integral participation of its two Death Domains, PYD and CARD. J. Biol. Chem 294, 439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystrom A, and Bruckner-Tuderman L (2018). Injury- and inflammation-driven skin fibrosis: the paradigm of epidermolysis bullosa. Matrix Biol 68–69, 547–560. [DOI] [PubMed] [Google Scholar]
- Orihuela CJ, Gao G, Francis KP, Yu J, and Tuomanen EI (2004). Tissue-specific contributions of pneumococcal virulence factors to pathogenesis. J. Infect. Dis 190, 1661–1669. [DOI] [PubMed] [Google Scholar]
- Otsuka M, Kato M, Yoshikawa T, Chen H, Brown EJ, Masuho Y, Omata M, and Seki N (2001). Differential expression of the L-plastin gene in human colorectal cancer progression and metastasis. Biochem. Biophys. Res. Commun 289, 876–881. [DOI] [PubMed] [Google Scholar]
- Petrilli V, Papin S, and Tschopp J (2005). The inflammasome. Curr. Biol 15, R581. [DOI] [PubMed] [Google Scholar]
- Reader BF, Sethuraman S, Hay BR, Thomas Becket RV, Karpurapu M, Chung S, Lee YG, Christman JW, and Ballinger MN (2020). IRAK-M regulates monocyte trafficking to the lungs in response to bleomycin challenge. J. Immunol 204, 2661–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe HM, Karlsson E, Echlin H, Chang TC, Wang L, Van Opijnen T, Pounds SB, Schultz-Cherry S, and Rosch JW (2019). Bacterial factors required for transmission of Streptococcus pneumoniae in mammalian hosts. Cell Host Microbe 25, 884–891.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Santos G, Rogel MR, Baker MA, Troken JR, Urich D, Morales-Nebreda L, Sennello JA, Kutuzov MA, Sitikov A, Davis JM, et al. (2015). Vimentin regulates activation of the NLRP3 inflammasome. Nat. Commun 6, 6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. [DOI] [PubMed] [Google Scholar]
- Shinomiya H, Hirata H, Saito S, Yagisawa H, and Nakano M (1994). Identification of the 65-kDa phosphoprotein in murine macrophages as a novel protein: homology with human L-plastin. Biochem. Biophys. Res. Commun 202, 1631–1638. [DOI] [PubMed] [Google Scholar]
- Srinivasula SM, Poyet JL, Razmara M, Datta P, Zhang Z, and Alnemri ES (2002). The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J. Biol. Chem 277, 21119–21122. [DOI] [PubMed] [Google Scholar]
- Stout-Delgado HW, Cho SJ, Chu SG, Mitzel DN, Villalba J, El-Chemaly S, Ryter SW, Choi AM, and Rosas IO (2016). Age-dependent susceptibility to pulmonary fibrosis is associated with NLRP3 inflammasome activation. Am. J. Respir. Cell Mol. Biol 55, 252–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickler AG, Vasquez JG, Yates N, and Ho J (2014). Potential diagnostic significance of HSP90, ACS/TMS1, and L-plastin in the identification of melanoma. Melanoma Res 24, 535–544. [DOI] [PubMed] [Google Scholar]
- Stutz A, Horvath GL, Monks BG, and Latz E (2013). ASC speck formation as a readout for inflammasome activation. Methods Mol. Biol 1040, 91–101. [DOI] [PubMed] [Google Scholar]
- Tiedemann K, Sadvakassova G, Mikolajewicz N, Juhas M, Sabirova Z, Tabaries S, Gettemans J, Siegel PM, and Komarova SV (2019). Exosomal release of L-plastin by breast cancer cells facilitates metastatic bone osteolysis. Transl Oncol 12, 462–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd EM, Deady LE, and Morley SC (2011). The actin-bundling protein L-plastin is essential for marginal zone B cell development. J. Immunol 187, 3015–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd EM, Deady LE, and Morley SC (2013). Intrinsic T- and B-cell defects impair T-cell-dependent antibody responses in mice lacking the actin-bundling protein L-plastin. Eur. J. Immunol 43, 1735–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd EM, Zhou JY, Szasz TP, Deady LE, D’Angelo JA, Cheung MD, Kim AH, and Morley SC (2016). Alveolar macrophage development in mice requires L-plastin for cellular localization in alveoli. Blood 128, 2785–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng TC, Schattgen S, Monks B, Wang D, Cerny A, Latz E, Fitzgerald K, and Golenbock DT (2016). A fluorescent reporter mouse for inflammasome assembly demonstrates an important role for cell-bound and free ASC specks during in vivo infection. Cell Rep 16, 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vajjhala PR, Mirams RE, and Hill JM (2012). Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. J. Biol. Chem 287, 41732–41743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Morley SC, Donermeyer D, Peng I, Lee WP, Devoss J, Danilenko DM, Lin Z, Zhang J, Zhou J, et al. (2010). Actin-bundling protein L-plastin regulates T cell activation. J. Immunol 185, 7487–7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SP, and Cassel SL (2010). Inflammasome-mediated autoinflammatory disorders. Postgrad. Med 122, 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, and Wynn TA (2010). Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J. Exp. Med 207, 535–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, Reppe K, Meixenberger K, Dorhoi A, Ma J, et al. (2011). The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J. Immunol 187, 434–440. [DOI] [PubMed] [Google Scholar]
- Wu J, Yan Z, Schwartz DE, Yu J, Malik AB, and Hu G (2013). Activation of NLRP3 inflammasome in alveolar macrophages contributes to mechanical stretch-induced lung inflammation and injury. J. Immunol 190, 3590–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi CX, Xiong F, Zhou Z, Mei L, and Xiong WC (2010). PYK2 interacts with MyD88 and regulates MyD88-mediated NF-kappaB activation in macrophages. J. Leukoc. Biol 87, 415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JY, Szasz TP, Stewart-Hutchinson PJ, Sivapalan J, Todd EM, Deady LE, Cooper JA, Onken MD, and Morley SC (2016). L-Plastin promotes podosome longevity and supports macrophage motility. Mol. Immunol 78, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Zoete MR, Palm NW, Zhu S, and Flavell RA (2014). Inflammasomes. Cold Spring Harb. Perspect. Biol 6, a016287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No standardized data types are reported in this study. All data reported in this paper will be shared by lead contact upon request.
This study does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.