

SUMMARY
Germline pathogenic mutations in BReast CAncer (BRCA1) genes are thought to drive normal fallopian tube epithelial (FTE) cell transformation to high-grade serous ovarian cancer. No human models capture the sequence of events for disease initiation and progression. Here, we generate induced pluripotent stem cells (iPSCs) from healthy individuals and young ovarian cancer patients with germline pathogenic BRCA1 mutations (BRCA1mut). Following differentiation into FTE organoids, BRCA1mut lines exhibit cellular abnormalities consistent with neoplastic transformation compared to controls. BRCA1mut organoids show an increased production of cancer-specific proteins and survival following transplantation into mice. Organoids from women with the most aggressive ovarian cancer show the greatest pathology, indicating the potential value to predict clinical severity prior to disease onset. These human FTE organoids from BRCA1mut carriers provide a faithful physiological in vitro model of FTE lesion generation and early carcinogenesis. This platform can be used for personalized mechanistic and drug screening studies.
In brief
Yucer et al. generate a human BRCA1 mutant iPSC-derived fallopian tube organoid model, which recapitulates BRCA1 mutant ovarian carcinogenesis in vitro and shows tumors in vivo. This model provides a biologically relevant platform to validate drugs and a basis for personalized early detection and preventative strategies for women carrying BRCA1 mutations.
Graphical abstract
INTRODUCTION
Ovarian cancer is the leading cause of gynecologic cancer death worldwide, with only slight improvements in overall survival rates over the last 40 years. Epithelial ovarian cancer represents a heterogeneous group of diseases with different cells of origin, histology, risk factors, and biologic behaviors. High-grade serous cancer (HGSC) is the most common and lethal subtype of ovarian cancer, with approximately 70% of newly diagnosed ovarian cancer cases classified as HGSC (Koshiyama et al., 2014, 2017; Kurman, 2013). Patients are typically diagnosed with late-stage (III/IV) clinical disease and present with wide-spread peritoneal metastases. The strongest genetic risk factor for developing HGSC is carrying a germline mutation in the BReast Cancer (BRCA1) gene (BRCA1mut). The risk of ovarian cancer in the general population is approximately 1.37% (Pearce et al., 2015), which jumps over 30-fold to 40%–59% among individuals that carry BRCA1mut (Kuchenbaecker et al., 2017). BRCA1 functions in the DNA break repair pathway through deficiencies in homologous recombination (HR). Nearly one-half of all HGSCs are HR deficient, most commonly from the loss of BRCA function due to germline or somatic mutations in the BRCA genes or loss of BRCA1 expression due to promotor methylation (Press et al., 2008).
Historically, ovarian cancer was believed to originate from the ovarian surface epithelium. Yet, recent studies have indicated that fallopian tube epithelial (FTE) cells are a common cell of origin for HGSC particularly in germline BRCA1mut carriers (Kurman, 2013; Labidi-Galy et al., 2017). The discovery that BRCA1mut patients undergoing risk-reducing surgery harbor serous tubal intraepithelial carcinoma (STIC) lesions in the FTE further supports the hypothesis that FTE is the predominant cell of origin of HGSC (Jones and Drapkin, 2013). As part of the STIC sequence, most pre-neoplastic lesions initially demonstrate nuclear accumulation of Tumor Protein p53 (TP53) and develop a “p53 signature,” prior to STIC and invasive serous carcinoma (Kuhn et al., 2012). Additional pathologic studies have supported STIC lesions as the precursor for HGSC in high-risk women carrying germline BRCA1 or BRCA2 mutations and in up to 60% of sporadic HGSC patients (Bolton et al., 2012; Labidi-Galy et al., 2017).
Immortalized cell lines and murine models have been used extensively to model the early-stage development of HGSCs from FTE cells, which includes the Brca;Tp53;Pten or Tp53(R172H) mutant with a Dicer/Pten double knockout mouse model that shows FTE transformation and HGSC initiation (Kim et al., 2015; Perets et al., 2013). But, few human-based in vitro models of FTE cells accurately recapitulate early-stage development of HGSC. The recent use of three-dimensional (3D) “organoid” models derived from human induced pluripotent stem cells (iPSCs) provides a powerful tool that can replicate physiologically relevant processes of disease in vitro (Liu et al., 2018; Sharma et al., 2020). This model system can self-renew and differentiate into multiple lineages and intrinsically self-organize to form 3D tissue architecture (Yamanaka, 2012). Importantly, iPSCs derived from patients with known genetic mutations will harbor not only the disease mutation but also the patient’s whole genetic background, which can help to elucidate how known and unknown mutations can together contribute to HGSC susceptibility, initiation, and progression (Curry et al., 2015). Several iPSC-derived disease models have been used to reproduce inherited high-risk cancers, such as an osteosarcoma model for Li-Fraumeni syndrome that recapitulated cancer pathology. (Crespo et al., 2017; Curry et al., 2015; Lee et al., 2015). BRCA1 patient-derived iPSCs have also been recently generated and provide an ideal model to study breast cancer events (Portier et al., 2021; Silva et al., 2021a, 2021b; Soyombo et al., 2013).
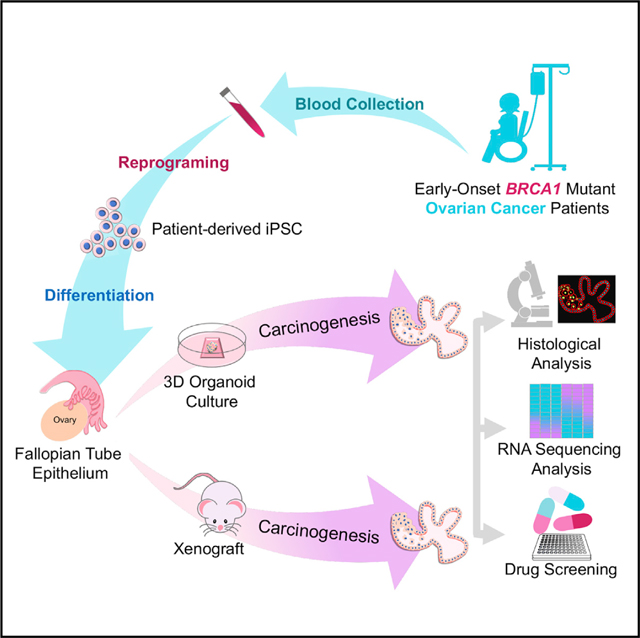
This study investigated early genomic alterations and carcinogenesis in patients with a BRCA1mut by using an iPSC-based disease model of FTE. Three different BRCA1mut-iPSC lines from patients with early-onset cancer were derived and 3D human BRCA1mut-FTE organoids were generated. Immunocytochemical staining of secretory and ciliated cellular components demonstrated that these organoid structures model human FTE. The BRCA1mut-FTE organoids showed structural abnormalities, including cellular crowding, loss of polarity, and severe atypia of the nuclei, which are signatures of the STIC. This finding indicates that BRCA1mut-FTE organoids recapitulate cancer-initiating events and create an in vitro system that can be used to model BRCA1mut-based ovarian cancer at early stages. Molecular signatures of this initial event in BRCA1mut-FTE organoids and in long-term cultures were characterized. Finally, the neoplastic transformation capacity and cancerous features of BRCA1mut-FTE organoids were explored using a mouse model. This human-derived FTE organoid model can be used to identify the critical sequence of genetic alterations involved in the carcinogenesis of HGSC and specific biomarkers of early FTE cell transformation.
RESULTS
BRCA1mut-iPSCs generated from early-onset ovarian cancer patients can be differentiated into FTE organoids
We previously developed a rapid and efficient method to create iPSC-derived 3D human FTE organoids that contained relevant cell types of the human FTE and a luminal architecture that closely reflects the organization of FTE tissues in vivo (Yucer et al., 2017). Briefly, modulation of BMP and WNT signaling (details in the STAR Methods section) was used to direct iPSC differentiation into Müllerian cells, and subsequent use of pro-Müllerian growth factors promoted differentiation into fallopian tube-like precursor cells. A 3D growth platform enabled the FTE organoid to self-organize into a convoluted luminal structure, permitting matured differentiation to a FTE lineage. Here, iPSCs were generated from three patients diagnosed with epithelial ovarian cancer, and all carried germline pathogenic mutations in the BRCA1 gene and three unaffected, non-BRCA1 mutation controls (Barrett et al., 2014). Genetic and clinical characteristics are provided in Table 1. Sequencing confirmed the presence of BRCA1 heterozygous mutations in iPSC lines (Figure 1A), with each patient having a different heterozygous mutation—79i-BRCA (IVGS5+1G > A, located at the junction between exon 5 and intron 6), 70i-BRCA (1048delA), and 08i-BRCA (3875del4, located in exon 11)—with all mutations predicted to be protein truncating and highly penetrant. qRT-PCR demonstrated that the BRCA1mut does not discernably alter BRCA1 transcript expression in iPSC lines from carriers versus non-carriers (Figure 1B). For western blot analysis, cultures were treated with the proteasomal inhibitor MG132 in order to facilitate the detection of BRCA1 that has low production under non-stress conditions. Although the 79i-BRCA line showed lower production, there was no significant difference overall between the BRCA1mut and control iPSC lines (Figures 1C and 1D). Both BRCA1mut and control iPSC lines displayed an embryonic stem cell-like morphology and exhibited pluripotent properties, including alkaline phosphatase activity, RNA expression of pluripotency factors (OCT4 and SOX2), and protein production of pluripotency markers (Figures 1E–1H). Clones of BRCA1mut and control iPSC lines exhibited normal karyotypes even after extensive culturing at passage 32 (Figure S1). Differentiation into three germ layers by using established protocols confirmed the differentiation capacity of BRCA1mut-iPSCs (Figure S2A; Chambers et al., 2009; Mae et al., 2013; Teo et al., 2014; Yucer et al., 2017). RNA expression analysis confirmed that control and BRCA1mut-iPSC lines had the capacity to differentiate into ectoderm-expressing PAX6, mesoderm-expressing BRACHYURY, and endoderm-expressing SOX17, with only slight variations in differentiation levels between lines (Figure S2A).
Table 1.
Clinical summary of controls and ovarian BRCA1mut cancer patients
| Cell ID | Parent tissue | Biopsy specimen | Sex | Karyotype | Mutation | Cancer stage | Age (years) |
|---|---|---|---|---|---|---|---|
| CS87i-CTR-n3 | Lymphoblastoid cells | Blood | Female | Normal | None | Unknown | |
| CS01iMEC-CTR-n4 | Mammary epithelial cells | Mammary Epithelial | Female | Normal | None | Unknown | |
| CS80i-CTR-Tn3 | PBMCs | Blood | Female | Normal | None | 48 | |
| CS08i-BRCA-n5 | Lymphoblastoid cells | Blood | Female | Normal | BRCA1: 3875del4 | IIC | 36.3 |
| CS70i-BRCA-n1 | Lymphoblastoid cells | Blood | Female | Normal | BRCA1: 1048delA | IIIC | 33.6 |
| CS79i-BRCA-n2 | Lymphoblastoid cells | Blood | Female | Normal | BRCA1: 1IVS5+1G > A | IIIC | 31.4 |
Figure 1. Characterization of BRCA1mut and control iPSC lines.

Lines 87i-CTR-n3, 80i-CTR-Tn3, 01iMEC-CTR-n4, 08i-BRCA-n5 and -n8, 70i-BRCA-n1 and -n2, and 79i-BRCA-n2 were characterized.
(A) Sequence analysis of heterozygous BRCA1mut in iPSC lines 79i-BRCA (IVGS5+1G > A located at the junction between exon 5 and intron 6), 70i-BRCA (1048delA), and 08i-BRCA (3875del4 located in exon 11). Heterozygous positions are indicated in comparison to 87i-CTR.
(B) BRCA1 gene expression in all controls and BRCA1mut-iPSC lines.
(C) Western blot for BRCA1, TP53, and Tubulin protein in iPSC lines after a 24-h treatment with the MG132 proteasomal inhibitor.
(D) BRCA1 protein normalized to Tubulin and quantified from multiple iPSC lines, with each point representing the band intensity from a separate line.
(E) Brightfield image of iPSC colonies for 87i-CTR, 08iBRCA, 70iBRCA, and 79iBRCA.
(F) Representative image of alkaline phosphatase (AP) staining of the 08i-BRCA line.
(G) Relative gene expression of OCT4 and SOX2 genes in all iPSC lines. Relative gene expression to 87i-CTR calculated using DDCt method and normalized to the endogenous GAPDH level.
(H) Immunocytochemistry of the 79i-BRCA iPSC line for pluripotent stem cell markers OCT4, NANOG, SOX2, TRA160, TRA181, and SSEA4. Scale bars, 25 μm. Error bars are standard error of the mean (SEM) (n = 3 independent biological experiments and each dot represents the average of 3 independent biological experiments per line).
In order to elucidate the role of BRCA1mut in ovarian cancer initiation in the FTE, we used our previously established protocol to generate iPSC-derived BRCA1mut-FTE organoids (Figure 2A; Yucer et al., 2017). The efficiency of FTE differentiation varied between lines, but all had the capacity to generate FTE cells. Quantifying the expression of key markers demonstrated differentiation into the mesoderm with BRACHYURY and MIXL1 and intermediate mesoderm with PAX2, GATA3, and OSR1 (Figures S2B and S2C). Consistent with our previous findings with control iPSC lines (Yucer et al., 2017), an increased expression of OVGP1 and WT1 over 8 days in culture confirmed that BRCA1mut-iPSC lines can produce a pre-Mullerian duct, indicating they model the precursors of FTE (Figure 2B). Lack of expression of the kidney marker SALL1 verified the specificity of Müllerian duct differentiation (Figure 2B). After 30 days in culture, BRCA1mut-FTE iPSCs formed 3D organoid structures with defined lumens lined by the cuboidal epithelial cells (Figures 2C and 2D). Importantly, these organoids expressed the epithelial marker CDH1 as well as FTE secretory and ciliated cell markers PAX8 and TUBB4A (Figure 2E; Figure S2D). TUBB4A production was restricted to the apical surface of columnar epithelial cells, consistent with the establishment of polarized epithelia.
Figure 2. BRCA1mut-iPSC-derived FTE organoids share characteristics of primary human FTE tissues.

(A) Timeline and factors for iPSC differentiation into FTE organoids. Control lines were reprogrammed from healthy female donor cells with peripheral blood mononuclear cells (PBMCs) used for CS80i-CTR-Tn3, lymphoblastoid cells (LCLs) used for CS87i-CTR-n3, and mammary epithelial cells used for CS01iMEC-CTR-n4. Lines CS08i-BRCA-n5, CS70i-BRCA-n1, and CS79i-BRCA-n2 were reprogrammed from LCLs from ovarian cancer patients with BRCA1mut.
(B) qRT-PCR quantification of gene expression for Müllerian duct markers, WT1 and OVGP1, and kidney markers SALL1 at day 8 of Müllerian duct differentiation.
(C and D) Brightfield image and H&E stain of BRCA1mut-FTE organoid (70i-BRCA line) at day 30 and high-magnification image. Scale bars, 50 mm and 10 mm.
(E) Immunocytochemistry for FTE markers PAX8 and TUBB4A and epithelial marker CDH1 in BRCA1mut-FTE organoids (70i-BRCA line) at day 30 in culture. Scale bar, 50 μm. Relative gene expression at iPSC stage (day 0) calculated using the ΔΔCt method and normalized to the endogenous GAPDH level for the 87i-CTR iPSC line. Error bars are SEM (n = 3 independent biological experiments). Student’s t test was used, *p ≤ 0.05.
BRCA1mut-FTE organoids exhibit precancerous pathological changes observed in pre-neoplastic ovarian cancer lesions
The pathological, phenotypic, and molecular properties of BRCA1mut-FTE organoids were compared to those of the control FTE organoids and human STICs, which are considered a precursor lesion of HGSC. A STIC lesion is defined based on distinct morphological and immunohistochemical features including severe nuclear atypia, architectural alterations, cellular crowding, loss of polarity, increased mitosis, and loss of ciliated cells (Vang et al., 2013; Voltaggio et al., 2016). Hematoxylin and eosin (H&E) staining was performed in order to determine whether BRCA1mut-FTE organoids cultured for 4 months develop STIC pathology. Compared to control organoids that exhibited a normal cuboidal cell morphology, BRCA1mut-FTE organoids displayed severe structural abnormalities with two distinct morphologies classified as Type I with a micropapillary-like structure or Type II with increased cell layers and nuclear stratification (Figures 3A and 3B). Organoids from all BRCA1mut-FTE lines had significantly varied tissue thickness distribution compared to controls, suggesting alteration in normal tissue architecture (Figure 3C; Figures S3A and S3B). To further analyze the Type I tissue architecture, an unbiased mathematical approach estimated cellular outgrowth incident or micropapillary-like structure formation based on the average surface roughness parameter (Figure S3C). Similar to the results from STIC histology, cellular outgrowths were significantly greater in organoids from all BRCA1mut-FTE lines than those in the controls, which is consistent with precancerous characteristics (Figure 3D; Figure S3D). We next investigated changes in nuclear atypia and structural abnormalities at the cellular level in 4-month cultures of BRCA1mut-FTE organoids. Calculating average nuclear area and surface of individual cells per organoid with 4’,6-diamidino-2-phenylindole (DAPI) and CDH1 staining showed that BRCA1mut-FTE organoids displayed cytological changes, including nuclear enlargement, cell surface distortion, and loss of columnar architecture (Figure 3E and 3F). Immunocytochemistry was used to characterize the pathology in 4-month-old BRCA1mut-FTE organoids. The production of TP53 and Ki67, which are hallmark proteins of STIC lesions that are expressed in dysplastic tissues and are used to confirm the preinvasive nature of cancer, indicates that BRCA1mut-FTE organoids exhibit precancerous characteristics similar to STIC in vitro (Figure 4A; Figure S4A). Glycoprotein CA125A (cancer antigen 125, also known as mucin 16 [MUC16]), which is overexpressed in most serous ovarian cancers and is used as a diagnostic serum biomarker, was also assessed (Felder et al., 2014). MUC16 protein promotes invasive behavior in preclinical models of cancer, both in vitro and in vivo (Rao et al., 2015). Remarkably, MUC16 production was considerably higher in BRCA1mut-FTE organoids and released into the lumen, suggesting that the BRCA1mut-FTE organoids have already acquired neoplastic features (Figure 4A). BRCA1mut and control FTE organoids have similar RNA expression levels for FTE secretory cell markers (PAX8, OVGP1, and TNFaIP2) and ciliated cell markers (FOXJ1 and TUBB4A), as well as the epithelial cell marker CDH1 (Figure 4B). Additionally, both BRCA1mut and control FTE organoids produced OVGP1 and TUBB4A, which were localized in the expected sub-compartments in control FTE organoids, with TUBB4A found along the lumen and OVGP1 located mostly along the basal region with a cohesive apical-to-basal polarity (Figure 4C; Figure S4B). Notably, control FTE organoids reached full maturation and formed clear cilia with the expression of its specific markers FOXJ1 and TUBB4A (Figures S4B and S4C). In contrast, proteins were distributed throughout the BRCA1mut-FTE organoids, indicating a loss of cellular identity and cohesive apical-to-basal polarity (Figure 4C; Figure S4D). RNA sequencing (RNA-seq) analysis of long-term cultures with gene set enrichment analysis (GSEA) and ingenuity pathways analysis (IPA) explored the molecular basis for STIC pathology in BRCA1mut-FTE organoids. Several ovarian cancer gene sets, including BUB1, CCNA2, CCNB2, CDC2, CDC20, CENPE, CENPF, ESPL1, HMMR, and HPN, as well as gene sets of the cell proliferation marker MKi76 and proliferating cell nuclear antigen (PCNA), were significantly upregulated in BRCA1mut-FTE organoids compared with those in controls (Figure 4D; Figure S5; Table S1; Adib et al., 2004; Buttermore et al., 2017; Cahill et al., 1998; Feng et al., 2019; Li et al., 2018; Sun et al., 2017). Similarly, IPA showed that cancer-related genes, including epithelial neoplasm and ovarian cancer, were enriched in BRCA1mut-FTE organoids (Table S2). Together, these data demonstrate that BRCA1mut-FTE organoids at 4 months displayed many features similar to STIC, suggesting that they recapitulate BRCA1mut-related ovarian cancer initiation.
Figure 3. BRCA1mut-FTE organoids exhibit histopathological features of STIC.

(A) Representative H&E stain of different histopathology for organoids at 4 months in culture, compared to 80i-CTR line. Type I obtained from the 08i-BRCA line and Type II obtained from the 79i-BRCA line, with images stitched using Stereo Investigator. Scale bar, 50 μm.
(B) Table for total organoids analyzed in each cell line at 4 months (n = 5 different biological replicates used per cell line).
(C) FTE organoid thickness distribution by individual lines.
(D) Cellular outgrowth measured as arithmetic average roughness by individual lines.
(E and F) Nuclei and cell surface distribution based on DAPI and CDH1 staining and corresponding quantification. Scale bars, 10 μM (E) 50 and 25 μm (F). (n = 3 different biological replicates used per organoid). Significance calculated using the Student’s t test and Welch ANOVA;*p < 0.05, ***p < 0.001, and ****p < 0.0001.
Figure 4. BRCA1mut-iPSC-derived FTE organoids recapitulate the molecular signature of STIC in vitro.

(A) Immunocytochemistry for TP53 and Ki67 and neoplastic marker MUC16 and quantification. Percentage of Ki67 and TP53/DAPI and area of MUC16 in lumen were calculated for 3 different control organoids and 3 different BRCA1 organoids (n = 2 different biological replicates for Ki67 and TP53, n = 3 different biological replicates for MUC16).
(B) Gene expression of FTE markers FOXJ1, PAX8, OVGP1, TUBB4A, TNFaIP2, and CDH1 at 4 months of culture. Color matrix of the heatmap shows TPM (transcripts per kilobase million).
(C) Immunocytochemistry for FTE secretory cell marker OVGP1 and ciliated cell marker TUBB4A in control and BRCA1mut-FTE organoids at 4 months.
(D) Differential gene expression and GSEA for n = 3 different control and n = 3 different BRCA1mut-FTE organoids at 4 months in culture. False discovery rate (FDR) value is <0.05. Z scores of the leading-edge genes were calculated and used to construct a heatmap. Scale bars, 50 mm. Significance was calculated using the Student’s t test; *p < 0.05 and ***p < 0.001.
BRCA1mut-FTE organoids recapitulate ovarian carcinogenesis
To determine whether the precancerous lesions progress to neoplastic transformation, in vitro cultures were maintained to 1 year and phenotypic and molecular characteristics were evaluated. The BRCA1mut-FTE organoids showed enhanced structural abnormalities with increased numbers of epithelial cells, which exhibited severe dysplastic phenotypes and evidence of transformation from a single epithelial layer to a disorganized multilayer epithelium that populated the luminal space (Figure 5A). Some cells in culture sloughed off from the surface of the lumen (Figure 5A, arrowhead), suggesting that the BRCA1mut epithelial cells may fail to undergo anoikis upon loss of contact with the basement membrane. Although the epithelial structures in control FTE organoids were very similar to normal human FTE tissues, BRCA1mut-FTE organoids lost normal epithelial structure and cellular identity (Figure 5B). Increased nuclear TP53 accumulation was observed in many epithelial cells from BRCA1mut-FTE organoids (Figure 5B). Collectively, these findings suggest that BRCA1mut-FTE organoids undergo neoplastic conversion over time.
Figure 5. BRCA1mut-iPSC-derived FTE organoids reveal ovarian cancer progression of carcinogenesis in vitro.

(A) H&E images for control (80i-CTR) and BRCA1mut-FTE organoids (79i-BRCA) at 1 year in culture.
(B) Immunocytochemistry of FTE markers OVGP1 and TUBB4A in human FTE tissue and control and BRCA1mut-FTE organoids. MUC16 and TP53 are shown as markers for neoplastic transformation in control and BRCA1mut-FTE organoids at 1 year in culture. Scale bars, 50 μm. Percentage of TP53 was calculated for n = 3 different controls and n = 3 different BRCA1mut-FTE organoids.
(C) Expression of BRCA1, ATM, ATR, and 53BP1 genes at 4 months and 1 year in culture in control and BRCA1mut-FTE organoids. Significance was calculated using Student’s t test; *p < 0.05.
(D) Differential gene expression and GSEA for control and BRCA1mut-FTE organoids at 1 year in culture. FDR value is <0.05. Z scores of the leading-edge genes were calculated and used to construct a heatmap.
BRCA1mut carriers often show a loss of the wild-type allele in the development of primary cancers, suggesting that the reduction to homozygosity and loss of function are prerequisites of cancer development (Cancer Genome Atlas Research Network, 2011; Esteller et al., 2000; Futreal et al., 1994; Russell et al., 2000). Reports suggest that the loss of heterozygosity (LOH) in the BRCA1 gene and DNA damage response (DDR) deficiency are strongly linked to the development of ovarian cancer (Bouwman et al., 2010; Gee et al., 2018; Weber and Ryan, 2015). To further evaluate the status of BRCA1 activity during neoplastic development in 1-year-old and 4-month-old BRCA1mut-FTE organoids, gene expression profiles were analyzed for BRCA1 and the key mediators of DDR including ATM (Ataxia Telangiectasia Mutated) kinase, ATR (ataxia-telangiectasia- and Rad3-related) kinase, and the nonhomologous end-joining factor 53BP1 (TP53 Binding Protein 1) (Bouwman et al., 2010; Hong et al., 2012; Weber and Ryan, 2015). In BRCA1mut-FTE organoids, but not control organoids, BRCA1 expression was significantly downregulated in 1-year cultures compared to 4-month cultures (Figure 5C), suggesting that there has been a loss of wild-type BRCA1 activity over time. Also, 53BP1 expression was significantly downregulated over time in BRCA1mut-FTE organoid cultures, and ATM and ATR kinases showed a trend toward downregulation (Figure 5C), suggesting that the functional loss or deregulation of these key proteins involved in the DDR may contribute to the progression of neoplastic transformation in BRCA1mut-FTE organoids.
Several mechanisms exist for the loss of function of the BRCA1 wild-type allele in heterozygous BRCA1mut carriers, including LOH, methylation, and somatic coding mutation. Loss of exon 11 is one of the common genomic alterations resulting in LOH in the BRCA1 gene. In order to evaluate whether the decrease in BRCA1 expression in 1-year organoids resulted from LOH, qRT-PCR analysis was performed to evaluate the allele-specific transcript expression in exon 11. Although full-length BRCA1 gene expression was significantly downregulated in BRCA1mut-FTE organoids at 1 year of culture, there was no evidence of changes in Delta Exon 11 expression (Figures S6A and S6B). The qRT-PCR data were confirmed with differential exon use analysis in bulk RNA-seq data, which showed no significant differential exon use at the BRCA1 locus detected between BRCA1mut and control organoids (Figure S6C). The presence of wild-type BRCA1 in BRCA1mut-FTE organoids suggests that downregulation of BRCA1 expression is not due to genomic deletion leading to changes in transcript expression.
RNA-seq and subsequent GSEA analysis showed that ovarian-cancer-related modules that promote cell migration, invasion, and malignant transformation including TPT1, NPM1, RAN, and predictive markers such as CK2β and PSME1 were significantly upregulated in BRCA1mut-FTE organoids, further suggesting neoplastic progression in vitro (Figure 5D; Figure S6D; Table S3; Chen et al., 2015; Liu et al., 2016; Londero et al., 2014; Ma et al., 2017; Wong et al., 2001; Zaoui et al., 2019). Thus, although 4-month organoid cultures exhibited a benign characteristic without evidence of malignant progression, the 1-year organoid cultures evolved into models that showed a malignant RNA profile. Strikingly, BRCA1mut-FTE organoids from the most aggressive case (79i-BRCA) exhibited upregulation of the MMP1 (Matrix Metallopeptidase 1) module, which may be critically involved in metastatic dissemination (Figure 5D; Behrens et al., 2001). Finally, IPA showed that cancer-related gene sets including epithelial neoplasm and malignant tumor were also highly enriched in 1-year cultures of BRCA1mut-FTE organoids compared to controls (Table S4).
Finally, in order to establish whether BRCA1mut-FTE organoids underwent neoplastic development in vivo, BRCA1mut and control-FTE organoids were differentiated in vitro for 1 month and implanted into NU/J athymic nude mouse mammary fad pad (MFP) at three different sites (Figure 6A; Lawson et al., 2015; Zhang et al., 2019). At 3 months post-transplantation, the fat pads were extracted for macroscopic histopathological examination. Both control and BRCA1mut-organoids remained structurally normal in the mammary gland (Figure S7A, red arrows). However, nodules were found outside of the mammary gland, and they exhibited severe cellular abnormalities reminiscent of precancerous and cancerous characteristics only in the BRCA1mut lines but not control lines (Figures S7B and S7C). H&E analysis suggested that these nodules contained both cancer and precancerous lesion formations (Figure S7C). Notably, immunohistochemical analysis demonstrated that unlike nodules from control-FTE organoids, BRCA1mut-FTE organoids showed substantial nuclear TP53 accumulation, which is the hallmark of an early time point in ovarian cancer development (Figure S7C). To further explore neoplastic transformation of BRCA1mut-FTE organoids in mice, the in vivo study was expanded to 6 months post-transplantation. Following transplantation of control and BRCA1mut-FTE organoids grown for 1 month in vitro, mice receiving BRCA1mut-FTE organoids developed several visible external tumors (Figure 6B). The relative tumor growth rate was determined and most visible BRCA1mut tumor lesions showed a significant growth rate compared to only one visible lesion in controls that did not show any growth (Figure 6C). To determine whether BRCA1mut-FTE organoid-derived tumor lesions have neoplastic transformation features, control and BRCA1mut tumor lesions were dissociated and primary cells were obtained. Primary cells from the control transplants did not survive (data not shown); however, primary cells from BRCA1mut tumor lesions have the ability to grow in a multilayer (Figure 6D) and exhibited clear production of FTE markers and cancer markers, including CDH1, PAX8, and Ki67, and increases in the frequency of double-stranded DNA break, shown with γH2AX accumulation and heterogenous TP53 staining, which is compatible with a wild-type pattern of TP53 expression (Figure 6E). Human nuclei marker production confirmed that primary cells from BRCA1mut tumor lesions were human epithelial cells (Figure 6E).
Figure 6. BRCA1mut-iPSC-derived FTE organoids show malignant characteristics of the BRCA1mut lesions in vivo.

(A) Schematic of organoid transplantation into mouse fat pads.
(B) Mice at 5 months post-transplantation of control-FTE organoids (87i-CTR line) and BRCA1mut-FTE organoids (79i-BRCA line).
(C) Control and BRCA1mut line tumor growth rates.
(D) Brightfield images of primary cells from BRCA1mut tumor lesions.
(E) Immunocytochemistry of primary cells from BRCA1mut tumor lesions shows cancer markers CDH1, PAX8, TP53, and Ki67; double-stranded DNA break marker γH2AX; and DAPI nuclei stain.
(F) Immunocytochemistry for TP53, Ki67, γH2AX, and cleaved Caspase 3 in primary cells from BRCA1mut tumor lesions treated for 72 h with 100 μM rucaparib, niraparib, olaparib, or DMSO, with DAPI nuclei stain.
(G) Cellular cytotoxicity assay (LDH assay) of primary cells from BRCA1mut tumor lesions over 72 h in response to 100 μM rucaparib, niraparib, or olaparib or DMSO.
(H) Brightfield images of primary cells from 8-months-old control and BRCA1mut-FTE organoids (87i-CTR, 08i-BRCA, 70i-BRCA, and 79i-BRCA).
(I–K) (I) Time course of LDH assay in response to 100 μM rucaparib treatment of primary cells from 8-monthsold control and BRCA1mut-FTE organoids (87i-CTR, 08i-BRCA, 70i-BRCA, and 79i-BRCA). PrestoBlue cell viability assay for primary cells from 8-monthold control and BRCA1mut-FTE organoids in response to 100 μM. (J) niraparib and (K) olaparib. Scale bars, 50 μm. Significance was calculated using 2-way ANOVA, Dunnett multiple comparisons;*p < 0.05, **p < 0.01, ***p < 0.001. Error bars are SD (n = 3 independent biological experiments).
To next explore BRCA1mut-related cancer characteristics, we investigated sensitivity to Poly (ADP-ribose) polymerase inhibitors (PARPis), which induce synthetic lethality in the context of HR deficiencies due to BRCA1mut (LaFargue et al., 2019; Lord and Ashworth, 2012, 2017). Federal Drug Administration-approved PARPi drugs used to treat epithelial ovarian cancer (olaparib, niraparib, and rucaparib) were evaluated in primary, monolayer cultures of 79i-BRCA cells derived from the mouse tumors. In response to rucaparib and niraparib treatment, primary cells showed significant DNA damage accumulation, with high γH2AX nuclear localization, compared to the control treatment (Figures 6F–6K), as well as elevated cell death demonstrated by TP53 and cleaved Caspase3 production and reduced levels of the cell proliferation marker Ki67 (Figure 6F; Figure S7D), and increased cellular cytotoxicity demonstrated by a lactate de-hydrogenase (LDH) assay (Figures 6G and S7E). Although olaparib did not appear to cause cellular cytotoxicity, it reduced cellular proliferation over time (Figures 6F and S7D).
To further explore the mechanism of PARPi actions on different BRCA1mut lines, primary cells dissociated from 8-month-old BRCA1mut-FTE and control organoids were treated with PARPi followed by cytotoxicity and cell viability assays (Figure 6H). Rucaparib treatment led to significant cell death in all BRCA1mut cells compared to the control treatment (Figure 6I). Niraparib and olaparib treatment reduced cellular viability at various levels, with a significant reduction in viability in the 08i-BRCA line, an initial significant change in the 70i-BRCA line, and no change in the 79i-BRCA line (Figures 6J and 6K). Collectively, these data show that BRCA1mut-FTE organoids exhibit severe cellular abnormalities, induced neoplastic transformation in vitro and in vivo, and recapitulated ovarian carcinogenesis. Importantly, this 3D human organoid model provides a platform to study cancer drug efficacy and specificity.
DISCUSSION
HGSC is the most common form of epithelial ovarian cancer, with a 5-year survival rate of ∼90% if detected at stage I. Unfortunately, no clinical biomarkers exist to reliably detect stage I/II HGSC, and there is a wide range of clinical presentation and behavior in BRCA1mut carriers and other high-risk populations. To address these clinical challenges, it is essential to better understand and model key molecular events involved in BRCA1-related ovarian cancer initiation and progression to improve predictive outcomes and interventions.
Current models that use established cell lines (normal and malignant) to study BRCA1 function are limited, as they do not reflect genetic characteristics of specific patients, nor do they recapitulate the complexity of the affected tissue or the dynamic changes that occur in disease initiation and progression. Recent studies have suggested that 3D cell culture systems better reflect the tissue and cell microenvironment than 2D models, and the behaviors of 3D-cultured cells are thought to recapitulate in vivo cellular responses. Several patient-biopsy-derived models and mouse 3D ovarian and other cancer models have been established and are shown to resemble the tumor structure in vitro (Crespo et al., 2017; Dumont et al., 2019; Kopper et al., 2019; Kurman, 2013; Lõhmussaar et al., 2020). Although biopsy-derived 3D organoids provide a drug screening platform and can be used for personalized therapy, this cell source cannot address how BRCA1mut leads to ovarian cancer initiation at the FTE and contributes to transformation over time. Finally, biopsy-derived organoids with introduced oncogenes and/or tumor suppressor mutations to induce cancer initiation fail to reveal critical molecular changes involved in BRCA1-related FTE transformation to ovarian cancer and cannot be used to improve early detection (Kopper et al., 2019; Lõhmussaar et al., 2020). Clearly, a better understanding of the initiation and early development of HGSC requires more relevant in vitro human models.
Here, we generated BRCA1mut iPSC lines that retained a pluripotent state, did not develop chromosomal abnormalities, and could be differentiated into the three germ layers, as shown in previous reports (Silva et al., 2021a; Soyombo et al., 2013). We differentiated control and BRCA1mut iPSCs into 3D FTE organoids that consisted of both the CDH1-positive epithelial layer and CDH1-negative fibroblast-like cells. We were surprised to find that iPSC-derived BRCA1mut lines, but not controls, generated FTE organoids with STIC histological characteristics such as cellular crowding, loss of polarity, and severe nuclear and cellular atypia. They also showed biomarker expression such as Ki67, TP53, and CA125/MUC16 that are common to ovarian cancer (Bast et al., 2005; Felder et al., 2014; Kuhn et al., 2012). Although BRCA1 expression was significantly downregulated in 1-year cultures compared to that of 4-month cultures, the data show that the mechanism for this ovarian cancer model is not based on LOH. Similarily, LOH was not observed following teratoma formation of BRCA1 iPSCs (Soyombo et al., 2013). Other mechanisms are currently being assessed with whole-genome and epigenetic sequencing.
Furthermore, the GSEA analysis indicated that BRCA1mut-FTE organoids cultured for 4 months retain highly similar key molecular signatures of ovarian cancer. For instance, hyaluronan-mediated motility receptor (HMMR), which is involved in microtubule spindle assembly and cell cycle progression, was one of the upregulated pathways (Buttermore et al., 2017; Maxwell et al., 2005, 2011). BRCA1 binds to HMMR and induces its degradation in normal cells and thus regulates apicobasal polarization of the breast luminal epithelium (Maxwell et al., 2011). Therefore, the loss of BRCA1 function linked to high HMMR expression can contribute to cancer progression (Anttila et al., 2000; Blanco et al., 2015; Maxwell et al., 2011). Similarly, elevated HMMR expression is associated with poor prognosis for BRCA1mut breast cancer (Blanco et al., 2015; Maxwell et al., 2011; Wang et al., 2014). Additionally, the expression of the potential PAX8 regulator lncRNA, namely PAX8-AS1 (PAX8 Antisense RNA 1), was significantly differentially expressed at the early time point for all the BRCA1mut lines (data not shown). Our data suggest that deregulation of PAX8 expression, which is the hallmark of ovarian cancer, is critical for ovarian cancer initiation and progression (Bowen et al., 2007). Therefore, BRCA1mut-FTE organoids at 4 months in culture recapitulate an early time point in ovarian cancer development that may reflect the neoplastic behavior of the original patient source and could provide a tool to identify genes for early detection. This platform can also benefit the development of new therapeutic approaches.
The FTE consists of two distinct cell populations, namely, ciliated columnar cells (FOXJ1+, TUBB4A+) and secretory cells (PAX8+, OVGP1+), and they share common progenitors. Balancing these cell populations along the FTE is critical for maintaining epithelial homeostasis. The FTE in HGSC shows reduced ciliated epithelial cells and increased PAX8-positive secretory cells, a phenotype that is considered a potential biomarker for the early stages of serous carcinogenesis (Coan et al., 2018; Tao et al., 2020; Wang et al., 2015). However, how these two cell populations shift during cancer initiation and progression is unknown. We demonstrated that control FTE cells consisted of distinct ciliated and secretory cell populations and reached full maturation by forming cilia, similar to normal FTE tissue. In contrast, early-stage BRCA1mut-FTE cells expressed both secretory cell markers (PAX8 and OVGP1) and TUBB4A in the same cells, which recapitulates the loss of cellular identity seen in HGSC fallopian tubes and suggests that this loss could be an early precancerous event.
We found that BRCA1mut-FTE organoids exhibited structural abnormalities with two distinct morphologies in early time points, which we considered as a precancerous time. We classified these abnormalities as Type I with a micropapillary-like structure or Type II with increased cell layers and nuclear stratification. STIC morphology displays the multilayered epithelium with stratification, loss of polarity, and absence of cilia, similar to Type II. Conversely, micropapillary structure formation is reported in HGSC, specifically in p53 wild-type cancer (Chui et al., 2021). Although we do not have further evidence that a primary structure predominates for neoplastic transformation, both types could result in neoplastic transformation.
Exploring carcinogenesis in this in vitro model revealed a strong correlation between disease severity in the BRCA patient and the phenotype in the organoids. Notably, organoids derived from the 79i-BRCA iPSC line (patient with stage IIIC ovarian cancer at age 31.4) exhibited a more aggressive molecular signature than organoids derived from the 08i-BRCA (patient with stage IIC ovarian cancer at age 36.3) or 70i-BRCA (patient with stage IIIC ovarian cancer at age 33.6) lines, suggesting that they faithfully recapitulated BRCA1mut-related phenotypes. Importantly, the metastatic feature of the BRCA1mut-FTE organoid was confirmed after transplantation into immunocompromised mice where further maturity of the cancer occurred, thus providing a physiologically relevant platform of induced cancer initiation in a short period. It appears that iPSC modeling predicts the severity of disease in these BRCA1 patients, although more cases are needed to confirm the phenotypic relationship between the severity of the organoid model and clinical outcomes.
PARPi therapy is as an exciting targeted strategy in BRCA-deficient ovarian cancers (Lord and Ashworth, 2012, 2017). PARPi inhibition induces DNA damage accumulation, activating DDR pathways that orchestrate cell fate decisions, including transient or prolonged cell proliferation arrest or apoptosis. In women with a BRCA germline and/or deficient and platinum-sensitive ovarian cancer, PARPi-targeted therapy increased rates of cancer-free survival. Unfortunately, not all patients respond to PARPi, and many of those patients who initially respond develop resistance. Here, we demonstrated BRCA1mut-dependent responses to different PARPi treatments. The efficacy and specificity of PARPi varied based on the BRCA1mut. Although rucaparib induced cell death specific to BRCA1mut cells, olparib and niraparib led to reduced cell proliferation at various levels across all lines. Our data suggest that this model may be used to study PARPi efficacy on different BRCA1mut with unique genetic backgrounds.
We established that BRCA1mut-FTE, but not control, organoids showed evidence of neoplastic development in vivo. Transplantation into mouse MFP has been shown to provide a similar hormonal microenvironment to the ovaries (Zhang et al., 2019). Interestingly, after engraftment into the MFP, BRCA1mut organoids formed subcutaneous nodules that exhibited cellular abnormalities with precancerous and cancerous characteristics. This finding was not observed with control organoids. It is unknown why the nodules formed outside the MFP. Although this result was unexpected, it was consistent throughout our studies and has been reported for xenografted human breast cancer cell lines that were found to be distinct from the mouse MFP (Samineni et al., 2011). Presumably subcutaneous regions provide a conducive microenvironment for neoplastic growth. MFP injection with a subsequent visualization of subcutaneous nodules permits tumor growth assessment over time in vivo, which could be key for drug screening.
The tumor microenvironment is composed of several cell types, including tumor-associated stromal cells, immune cells, and endothelial cells, as well as non-cellular components including the extracellular matrix (ECM) and other soluble factors (Cho et al., 2015; Jin and Jin, 2020). The role of the tumor microenvironment in facilitating cancer progression is gaining recognition (Jin and Jin, 2020), although a molecular understanding of the tumor microenvironment role in promoting neoplastic progression remains unclear. Our 3D model could provide a platform to study the contribution of the tumor microenvironment to neoplastic progression.
In summary, this 3D platform provides a more faithful physiological model that enables a deeper investigation into signaling pathways in early ovarian cancer initiation in both a tissue- and patient-specific context. This model offers a promising, biologically relevant platform to validate new drugs and targets for a cancer that is not well understood. By generation of 3D organoid models from various BRCA1mut patients, BRCA1mut-dependent responses to treatment can be elucidated and the information can help predict the drug sensitivity of individual patients based on their unique genetics. This iPSC-based BRCA model can provide a basis for personalized early detection and preventative strategies for women carrying BRCA1mut and potentially other high-risk mutations.
LIMITATIONS OF THE STUDY
This research is subject to some limitations. Although this iPSC-based BRCA model clearly shows abnormal cells in masses, it is still an in vitro model that does not fully recapitulate all in vivo histological features of HGSC (ovarian cancer) seen in patients. As an artificial culture environment does not include the hormonal environment and cell type heterogeneity of tissues in vivo, it may not reflect the entire biology of BRCA1-initiated cancer progression. Finally, although iPSC-derived FTE from BCRA1 patients clearly shows cancer initiation and early signs of progression, the validity of this model for later stages of cancer needs to be validated in further studies.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Clive Svendsen: Clive.Svendsen@cshs.org.
Materials availability
iPSC lines are available from Cedar-Sinai Biomanufacturing Center iPSC Core. Other materials generated in this study are available upon request.
Data and code availability
RNA-seq data have been deposited at GEO (GEO: GSE190134) and are publicly available as of publication date. Code used to generate data for this study will be available and shared by lead contact upon request. Original western blot images and microscopy data reported in this paper will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this paper is available and will be shared by the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
All iPSC lines were generated at the iPSC Core at Cedars-Sinai Medical Center. Parent cells were transfected with non-integrating episomal plasmid expressing 7 factors: OCT4, SOX2, KLF4, L-MYC, LIN28, SV40LT and p53 shRNA (pEP4 E02S ET2K, pCXLE-hOCT3/4-shp53-F, pCXLE-hUL, and pCXLE-hSK). Details regarding the disease status, sex, age at collection, and BRCA1 genotype for each iPSC line are provided in Table 1. All the cell lines and protocols in the present study were used in accordance with the guidelines approved by the stem cell research oversight committee (SCRO) and institutional review board (IRB) under the auspice IRB-SCRO Protocols Pro00032834 (iPSC Core Repository and Stem Cell Program) and Pro00021505 (Svendsen Stem Cell Program).
We selected 3 existing female control iPSC lines from the Cedars-Sinai Biomanufacturing Center iPSC Core. Control iPSC lines were reprogrammed from healthy female donor cells with peripheral blood mononuclear cells (PBMCs) used for CS80i-CTR-Tn3, lymphoblastoid cells (LCLs) used for CS87i-CTR-n3 and mammary epithelial cells used for CS01iMEC-CTR-n4. 3 female BRCA1mut patients’ LCLs were chosen from the Gilda Radner Hereditary Cancer Program repository and BRCA1mut iPSC lines (Lines CS08i-BRCA-n5, CS70i-BRCA-n1 and CS79i-BRCA-n2) were generated at Cedars-Sinai Biomanufacturing Center iPSC Core.
BRCA1mut iPSC lines were confirmed by the presence of BRCA1 heterozygous mutations based on DNA sequencing analysis. Results showed that each patient had a different heterozygous mutation - 79i-BRCA (IVGS5+1G > A, located at the junction between exon 5 and intron 6), 70i-BRCA (1048delA), and 08i-BRCA (3875del4, located in exon 11). These mutation matched to each patient’s clinical diagnosis.
Animals
Foxn1nu, formerly Hfh11nu (known as NU/J athymic nude) female mice at postnatal day 24 were purchased from the Jackson Laboratory and both control and BRCA1mut-FTE organoids were injected. All the animals maintained at vivarium facilities at the Cedars-Sinai Medical Center under standard conditions with the light on 12 hours and off 12 hours and ad libitum access to standard chow and water. All animal procedures were performed following the NIH Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (IACUC) of Cedars-Sinai Medical Center (protocol #005318).
METHOD DETAILS
Ethics Statement
All human samples were obtained using the approved IRB in accordance with relevant guidelines and regulations under IRB PRO00033469 protocol number. Informed consent was acquired from all human subjects and/or their legal guardians. Human B cells, mammary epithelial cells, and fibroblast cells were obtained from the Institute for Medical Research. The Cell Repository maintains the informed consent and privacy of the donor. All the cell lines and protocols in the present study were used in accordance with the guidelines approved by the stem cell research oversight committee (SCRO) and institutional review board (IRB) under the auspice IRB-SCRO Protocols Pro00032834 (iPSC Core Repository and Stem Cell Program) and Pro00021505 (Svendsen Stem Cell Program).
Generation and culture of iPSCs
Human iPSC lines were generated based on a previously published protocol (Barrett et al., 2014). The starting cell source for control lines was healthy donor female cells, with PBMCs used to generate control lines CS80i-CTR-Tn3, lymphoblastoid cells (LCLs) used to generate CS87i-CTR-n3 and mammary epithelial cells (from a mammary epithelium biopsy) used to generate CS01iMEC-CTR-n4. LCLs from ovarian cancer patients with BRCA1 mutations were used to generate lines CS08i-BRCA-n5, CS70i-BRCA-n1 and CS79i-BRCA-n2. Briefly, cells were cultured at 37°C and 5% CO2 in a humidified incubator, with LCLs maintained in RPMI 1640 (Life Technologies) media supplemented with 15% fetal bovine serum (FBS) and 2 mM L-glutamine, and mammary epithelial cells maintained in MammaryLife™ basal media (Life Line Cell Technology). LCLs and mammary epithelial cells were reprogrammed into virus-free iPSC lines with B cell Nucleofector Kit (VPA-1001, Lonza) using 1.5 mg of each episomal plasmid (Addgene) expressing 7 factors: OCT4, SOX2, KLF4, L-MYC, LIN28, SV40LT and p53 shRNA (pEP4 E02S ET2K, pCXLE-hOCT3/4-shp53-F, pCXLE-hUL, and pCXLE-hSK). Nucleofected cells were plated on feeder-independent BD Matrigel growth factor-reduced Matrix (Corning/BD Biosciences, #354230). All cultures were maintained at 20% O2 during the reprogramming process. Cells were initially cultured in their maintenance medium for 3 days post-nucleofection and gradually transitioned to reprogramming media (RM) by adding 1 mL RM to the original media daily for the next 3 days to aid in LCL attachment. Reprogramming media contains DMEM/F12, 1% NEAA, 1% GlutaMax, 1% N2, 2% B27, 0.5% Antibiotic-Antimycotic (GIBCO #15240–062), 0.1 μM β-mercaptoethanol, 100ng/ml bFGF (PeproTech), 1:1000 (∼1000 units) hLIF (Millipore, #LIF1010), 0.5 μM PD0325901 (Cayman Chemicals, #13034), 3 μM CHIR99021 (Tocris, #4423), 10 μM HA-100 (Santa Cruz Biotech, #203072), and 0.5 μM A-83–01 (Tocris, #2939). Cells were maintained in RM for 15 days with fresh media replenishment every other day. They were then gradually changed to chemically-defined mTeSR®1 medium (STEMCELL) between 17–20 days post-nucleofection. Colonies with iPSC-like morphology were mechanically isolated, transferred onto 12-well plates with fresh Matrigel Matrix, and maintained in mTeSR®1 medium on growth factor-reduced Matrigel Matrix (BD Biosciences)-coated plates at 37°C in a 5% CO2 incubator. For weekly passaging, colonies were lifted carefully with a cell scraper, removed using a 5 mL glass pipette, and replated at a 1:6 ratio.
Directed differentiation of iPSCs into 3D human FTE organoids
Human iPSCs were split onto Matrigel-coated plates and cultured in mTeSR®1 medium until 80% confluent. Cells were exposed to 100 ng/ml human recombinant activin A (Stemgent) and 3 μM CHIR99021 (Cayman Chemicals) to differentiate toward mesoderm, and cultured in DMEM/F12 (GIBCO) + Glutamax (Invitrogen) supplemented with 500 U/ml penicillin streptomycin (GIBCO) and 2% FBS with addition of 10 μM ROCK inhibitor Y-27632 (Stemgent) for 2 days. To differentiate cells toward intermeditae mesoderm (IM), media was changed to DMEM/F12 (GIBCO) + Glutamax (Invitrogen) supplemented with 0.1 mM non-essential amino acids (Invitrogen), 500 μ/ml penicillin/streptomycin (GIBCO), 0.55 mM 2-mercaptoethanol, 10% KOSR (Invitrogen), 100 ng/ml BMP4 (R&D Systems), 3 μCHIR99021 (Tocris), and 10 μM ROCK inhibitor Y-27632 (Stemgent) for 2 days. Spheroids were collected on day 4 from every well under a stereomicroscope using a 200 μL barrier pipette tip and pooled into a 1.5 mL microcentrifuge tube. Spheroids were then mixed with 50 μL Matrigel (BD Biosciences) and slowly pipetted into the middle of one well of a 24-well Nunclon Delta surface dish. The 3D droplet was allowed to solidify for 10–15 minutes in a tissue incubator, and Matrigel beads were then bathed in new fallopian tube media (nFTM) containing advanced Dulbecco’s modified Eagle medium/F12 supplemented with 12 mmol/L HEPES, 1 X Glutamax, 2% B27, 1% N2 (all from Life Technologies, Carlsbad, CA), and was supplemented with 10 ng/mL murine recombinant EGF (PeproTech), 10 ng/mL basic FGF, 30 ng/ml human Noggin (R&D system), 0.5 mM TGF-b R Kinase Inhibitor IV (SB431542, Calbiochem) and 1% penicillin/ streptomycin (Invitrogen). For FTE differentiation, 100 ng/ml human recombinant WNT4 (R&D Systems) with 10 μM ROCK inhibitor Y-27632 (Stemgent) were added for two days and followed by 20 ng/ml human recombinant Follistatin (PeproTech) with 10 μM ROCK inhibitor Y-27632 (Stemgent) for next two days. Media was replaced every 3–4 days as necessary, and cells were replated every two weeks. All the cultures were tested for mycoplasma contamination monthly.
Western blot analysis
iPSCs were treated with the proteasome inhibitor MG132 for 24 hours, and treated and nontreated cells were gently scraped off the plates, washed with phosphate buffered saline (PBS), and centrifuged at 15000 RPM for 1 min. Samples were lysed using 1X NETN buffer (20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 0.5 mM EDTA and 0.5% NP-40) supplemented with phosphatase/protease inhibitor cocktail (MS-SAFE, Sigma-Aldrich). Lysates were sonicated at a frequency of 20 kHz for 5 s (3times) on ice and incubated on ice for 20 min. Samples were centrifuged for 20 min at 4°C at 15000 RPM. Total soluble protein concentrations were measured using a Bradford assay (BIO-RAD). 4X Laemmli sample buffer (BIO-RAD 161–0774) was added to 100 μg of total protein extracts and samples were boiled for 5 min. Samples were run in 4%–20% Mini-PROTEAN TGX Precast gels (BIO-RAD, 456–1094) and transferred to nitro-cellulose membrane using 1X transfer buffer (25mM Tris, 190 mM glycine, 20% Methanol and 0.1% SDS) over night at 4°C. Membranes were blocked with Odyssey blocking buffer (LI-COR) and then incubated with primary BRCA1 antibody (Millipore, OP92), which is raised against the N-terminal region of BRCA1 protein, overnight at 4°C. Following incubation with dye-labeled mouse secondary antibody for 2 hours at room temperature, signals were visualized using an Odyssey Fc imaging system (LI-COR).
RNA Isolation and Real-time PCR Analysis
Total cellular RNA was isolated using QIAGEN RNeasy Mini kit following manufacturer recommendations (QIAGEN). RNeasy-treated total RNA (1 μg) was used for cDNA synthesis using the Quantitect Reverse Transcription Kit for cDNA synthesis for PCR (QIAGEN). Real-time PCR was performed using the SYBR Select Master Mix (Appliedbiosystem). The levels of expression of respective genes were normalized to corresponding GAPDH values and shown as fold change relative to the value of the control sample. All sample analyses were carried out in triplicate. List of primers used for real-time PCR experiments are listed in Table S5. At least one set of replicates was performed blindly.
Immunocytochemistry for Fallopian Tube Organoid
Organoids were fixed with 4% paraformaldehyde (PFA) in PBS for 20 minutes followed by three washes (3X) with PBS. The fixed organoids were then cryoprotected in 30% sucrose at 4°C overnight and embedded in OCT (Tissue-Tek). Frozen sections were collected at 12 μm using a cryostat onto glass slides and stored at −80°C. Each section was rehydrated with PBS for 5 min, permeabilized in PBS containing 2% Triton X-100 for 10 min at room temperature, blocked in a solution of 10% Normal Donkey Serum in PBS+0.05% Triton X-100 (PBS-T) for 1 hour at room temperature, followed by 2 hours incubation at room temperature in primary antibodies in blocking solution. The following primary antibodies were used at 1:200 dilution: PAX8 (Proteintech, 21384–1-AP), TUBB4A (Abcam, ab1315), POU5F1 (Stemgent, 09–0023), Nanog (Stemgent, 09–0020), SOX2 (Stemgent, 09–0024), TRA-1–60 (Stemgent, 09–0010), TRA-1–81 (Stemgent, 09–0011), SSEA4 (Stemgent, 09–0006), CDH1 (R&D System, AF648), OVGP1 (SIGMA, HPA062205), p53 (DO-1) (Santa Cruz Biotechnology, sc-126), Ki-67 (SP6) (Invitrogen, MA5–14520) MUC-16 (Sigma, HPA065600). The slides were then washed with PBS-T three times for 15 minutes each at room temperature and incubated with species-specific AF488 or AF594-conjugated secondary antibodies (Invitrogen) at 1:400 dilution, followed by 4’,6-diamidino-2-phenylindole (DAPI) (Molecular Probes, D3571) nuclear counterstain. Following three washes in PBS-T, the tissue was covered with a glass slide and imaged using Nikon/Leica microscopes. Each selected image is representative of a minimum of three independent experiments with at least two technical duplicates. At least one set of replicates was performed blindly.
Immunohistochemistry for TP53
Dissected tissues were fixed with 10% formalin (Fisher Chemical) for 1 hour at room temperature and then overnight at 4°C. The tissues were washed with PBS once and then were stored tissue in 70% EtOH overnight. Next day tissues were submitted to the Cedars-Sinai Pathology Core for paraffin embedding and then sectioning using a microtome. 5 mm slides were deparaffinized with xylene (3X) for 10 minutes each interval and then rehydrated in 100% EtOH (2X), 95% EtOH (2X), 75% EtOH, 50% EtOH at 5 minutes each. The slides were rinsed in tap water (2X) for 2 minutes intervals. The slides were microwaved 8 minutes at 100% power and 15 minutes at 30% power in Vector unmasking solution (Citric Acid Based, H-3300) for antigen retrieval and then cooled down to room temperature for 30 minutes. The slides were then washed with PBS and treated with 0.3% H2O2 in Methanol for peroxidase inactivation for 30 minutes at room temperature. The slides were washed with PBS (3X) with 2 minutes intervals, blocked in a solution of 3% BSA in PBS-T for 1 hour at room temperature, followed by overnight incubation at 4°C in TP53 primary antibody in 1:200 dilution in blocking solution. The slides were washed with PBS (3X) for 15 minutes each at room temperature, incubated with mouse-specific biotinylated secondary antibody for 2 hours at room temperature, washed with PBS (3X) for 15 minutes intervals. Then slides were treated with Avidin-Biotin Complex (ABC, VECTASTAIN® Elite® ABC-HRP Kit) for 30 minutes at room temperature and washed with PBS (3X) for 2 minutes intervals. The slides were then incubated with DAB solution (Vector, SK-4105) for 5 minutes and washed with distilled water (3X) for 5 minutes intervals. The slides were counterstained in hematoxylin (ImmunoMaster Hematoxylin, American MasterTech Scientific, Inc.) for 8 minutes, and then rinsed in running tap water for 5 minutes, dehydrated in 50% EtOH, 75% EtOH, 95% EtOH (2X), 100% EtOH (3X), at 1 minute each and Xylene (3X) at 5 minutes each. The tissues were covered with a glass cover slide using a mounting medium (Richard-Allan Scientific Mounting Medium).
H&E Staining
Organoid and fallopian tube tissues were fixed with 4% PFA in 1X PBS for 20 minutes, followed by PBS (3X) washes. The fixed organoids and fallopian tube tissues were cryopreserved in 30% sucrose at 4°C overnight and then embedded in OCT (Tissue-Tek). Frozen sections were collected at 12 μm using a cryostat onto glass slides and stored at −80°C. Each section was rehydrated with PBS for 5 minutes. The slides were stained in hematoxylin (ImmunoMaster Hematoxylin, American MasterTech Scientific, Inc.) for 8 minutes, then rinsed in running tap water for 5 minutes. The slides were then stained with eosin (Eosin Y Phloxine B, American MasterTech Scientific, Inc.) for 10 s. The slides were rinsed in tap water and dehydrated in 50% EtOH, 75% EtOH, 95% EtOH, 100% EtOH, and Xylene at 1 minute each. Tissues were covered with a glass coverslip using mounting medium (Richard-Allan Scientific Mounting Medium).
Organoid Thickness Measurement
Using Stereo Investigator 11.07 (MBF Bioscience), the “contour” tool was used to mark around FTE organoid sections on slides, which were then imaged at 20X magnification. The images were stitched together to yield one organoid section per file. The images were then imported into Neurolucdia 11.07 (MBF Bioscience) and FTE thickness measurements were taken using the “quick measure line” tool at increments of 200 μm along the epithelium of each imaged section for 228 organoids from 5 different biological replicates. Image analysis tool for three different control (87i-CTR, 80i-CTR, and 01iMEC-CTR) and three different BRCA1 mut (08i-BRCA,70i-BRCA and 79i-BRCA) organoids at 4-month culture, with 200 μm grids used as a reference. 10 to 20 different points based on the full size of organoids were used to calculate average thickness (Figure S5A) and cellular outgrowth was calculated using the surface roughness equation as follows: . Using the “ROI Manager” tool in ImageJ, individual cells stained with anti-Ecad were traced and analyzed to find the area of each FTE cell. Using ImageJ, the images of FTE sections were converted to binary and analyzed to find the area stained with DAPI. Using the DAPI count, the total area was divided to give us the nucleus size per FTE cell. At least one set of replicates was performed blindly.
Transcriptional Analysis of FTE Organoids
RNA integrity was determined on an Agilent 2100 Bioanalyzer and samples with RIN ≥ 9 were selected for cDNA library construction using Illumina TruSeq Stranded mRNA library preparation kit. Sample libraries were sequenced on an Illumina NextSeq 500 platform with 75bp single-end sequencing at the Cedars-Sinai Applied Genomics, Computation & Translational Core. Demultiplexing and conversion of raw sequencing data to FASTQ was performed with Illumina bcl2fastq software and reads were mapped to the GRCh38.p13 genome assembly (GENCODE 32) using Salmon version 1.1.0. Gene-level read counts were then quantified using the R/Bioconductor package tximport. Genes were filtered by an average read count of 3 per sample. Ensembl gene IDs without a matching HGNC symbol were also removed, resulting in 19,498 genes that met the filtering criteria. Raw counts were normalized using the variance stabilizing transformation of the DESeq2 package. Gene Set Enrichment Analysis (GSEA) was then performed using the filtered and normalized data to test enrichment in the C4: cancer gene neighborhood and cancer module gene sets from MSigDB (http://www.gsea-msigdb.org/gsea/index.jsp) using the default GSEA settings (Subramanian et al., 2005). Normalized expression values for the leading-edge subset of genes responsible for gene set enrichment were converted to Z-scores and plotted using the R/Bioconductor ComplexHeatmap package. A cutoff of FDR p value < 0.05 was used to determine which gene sets and pathways were significantly enriched in BRCA1mut FTE organoids.
For differential exon usage (DEU) analysis, RNaseq reads were aligned to GENCODE (version 38) primary genome assembly using STAR aligner (version 2.7.9a) with default settings. Aligned reads overlapping unique exon bins were quantified and visualized using DEXseq (version 1.38.0) (Anders et al., 2012) with false discovery rate set at 10%. Default settings were used for all DEXseq functions except for aggregate flag set to ‘-r no’ during annotation file preparation.
Mammary Fat Pad (MFP) Transplantation
All animal procedures were performed following the NIH Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (IACUC) of Cedars-Sinai Medical Center (protocol #005318). NU/J athymic nude female mice at postnatal day 24 were anesthetized by isoflurane, and surgery was performed. A 1.5 cm midline incision on the ventral surface of the skin was made beginning between the #4-mammary nipples and extending upward toward the thorax. Two contralateral incisions were prepared, beginning at the lower end of the previous incision, ending between the #4 and #5 mammary nipples, such that the incisions describe an inverted Y. The #4 nipples, the blood vessel near the inguinal lymph node in the #4 fat pad, and the blood vessel which courses between the #4 and #5 fat pads were cauterized. Since postnatal day 24, the #4 mammary gland has not grown beyond the lymph node, the triangular area described by the cautery points was surgically removed, resulting in the #4 mammary gland being “cleared.” The residual tissue called a “cleared” mammary fat pad was used to implant organoids. Meanwhile, organoids were collected in media and centrifuged, and the total packed volume of organoids was calculated. For 10 injections, 500 μL of total organoids were resuspended in 5 mL PBS, and 550 μL of organoids in PBS were relocated into the Eppendorf tube and then pelleted. The 1-month-old control and BRCA1mut-FTE organoids with similar sizes were resuspended in 20 μL Matrigel beads and then placed onto the cleared mammary fat pad using sterile micro-forceps. The surgery was carried out bilaterally, on both #4 mammary glands. After the surgery, the skin surrounding the incision was pinched and sutured with a silk suture, which was removed later. At 3 months and 6 months post-transplantation, animals were sacrificed, the brightfield images were taken, and nodules and fat pad where the organoids were transplanted were dissociated from the skin using scissors. At 5 months post-transplantation, tumor volume was recorded by caliper measurements using the formula (length [mm]) 3 (width [mm]) 3 (height [mm]) 3 (πX(4/3)). Relative tumor volume (RTV) was determined according to the formula RTV = Vn/V150 where V150 represents the tumor volume at day 150, which was the first measurement and Vn represents the tumor volume as measured after an interval of 10 days, respectively. Tissue was fixed in 10% formalin (Fisher Chemical) for 1 hour at room temperature and then overnight at 4°C, then dehydrated with 70% ethanol and paraffin-embedded according to standard protocol. 5 μm slides were cut and placed on glass slides for immunohistochemistry analysis for TP53 protein and H&E staining. Organoid injection, collection, and sectioning were performed blinded.
Tumor Cell Isolation and Characterization
Tumor tissues were removed from the animals and washed with 1X PBS. Tumors were minced with razor blades on an aseptic surface, transferred into a 15 mL canonical tube, and washed with PBS. Then cells were dissociated by incubating in nFTM media with 300 units/ml collagenase and 100 units/ml hyaluronidase (STEMCELL, #07912) and 2 units/ml DNaseI at 4°C overnight and centrifuged at 100 g for 10 minutes at 4°C to remove undigested tissue. Then, the supernatant was collected and centrifuged at 500 g for 10 minutes at 4°C and washed with PBS twice. Cells were resuspended in nFTM and passed through a 40 μm nylon mesh filter and then centrifuged at 500 g for 5 minutes at 4°C. Single cells were cultured on growth factor-reduced Matrigel Matrix (BD Biosciences)-coated plates at 37°C in a 5% CO2 incubator.
Single cells from tumors were plated on L-glass coverslips and grown as above. Cells were fixed in 4% PFA at room temperature for 10–15 minutes on day 5. Fixed coverslips were washed in PBS and permeabilized in PBS containing 2% Triton X-100 for 10 minutes at room temperature, then blocked in a solution of 10% Normal Donkey Serum in PBS-T for 1 hour at room temperature, followed by 2 hours incubation at room temperature in primary antibodies in blocking solution. The following primary antibodies were used at 1:200 dilution: PAX8 (Proteintech, 21384–1-AP), CDH1 (R&D System, AF648), Tp53 (DO-1) (Santa Cruz Biotechnology, sc-126), Ki-67 (SP6) (Invitrogen, MA5–14520), Anti-phospho-Histone H2A.X (Ser139) (Millipore, clone JBW301), Human Nuclei (MAB128–1). The coverslips were then washed with PBS-T (3X) for 15 minutes each at room temperature and incubated with species-specific AF488 or AF594-conjugated secondary antibodies (Invitrogen) at 1:400 dilution, followed by DAPI (Molecular Probes, D3571) counterstain. Coverslips were mounted on a glass slide, and images were acquired using a Nikon/Leica microscope.
PARPi Treatments
Cells from organoids or primary tumor cells were plated in flat-bottom 96-well plates at 1×104 per well and incubated for 3 days in FTE media. Rucaparib (AG-014699, Niraparib (Synonyms: MK-4827), and Olaparib (Synonyms: AZD2281; KU0059436) were prepared in 100 mM stock in DMSO and diluted according to treatments in the media. 100 μL of FTE media containing PARPi was add to the cells, and media was collected after 24, 48, and 72 hours for a LDH assay (CyQUANT, C20301), and cells were treated with the PrestoBlue (Invitrogen, 226392) reagent for cell viability assay.
LDH Assay
25 μL of media from PARPi treated cells were mixed with 25 μL of LDH substrate in a 96-well clear plate, and plates were incubated at room temperature in the dark for 30 minutes. 25 μL of LDH blocking solution were added to stop the reaction, and absorbance was measured at 490nm and 680nm. To determine LDH activity, the 680nm absorbances were subtracted from 490nm absorbance.
PrestoBlue Assays
After the PARPi treatments, 10 μL of PrestoBlue reagent was added into the well and incubated 1 hour at 37°C. Change in the fluorescent signal was recorded at 560/ 590 nm excitation/emission wavelengths.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed by using Prism software (GraphPad Software, La Jolla, California). All quantitative data were expressed as mean values ± Standard Error of the Mean (SEM) and analyzed by analysis of variance (ANOVA) with unequal variances known as Welch ANOVA or two-tailed paired t test followed by analysis of mean differences in three biological replicates. Differences were considered significant at *p ≤ 0.05, ** p ≤ 0.01, and *** p ≤ 0.001 and **** p ≤ 0.0001.
ADDITIONAL RESOURCES
Our study has not generated or contributed to a new website/forum, and it is not part of a clinical trial.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PAX8 | Proteintech | Cat#21384–1-AP |
| TUBB4A | Abcam | Cat# ab11315; RRID:AB_297919 |
| POU5F1 | Stemgent | Cat#09–0023; RRID:AB_2167689 |
| Nanog | Stemgent | Cat#09–0020; RRID:AB_2298294 |
| SOX2 | Stemgent, | Cat#09–0024: RRID:AB_2195775 |
| TRA-1−60 | Stemgent | Cat#09–0010; RRID:AB_1512170 |
| TRA-1−81 | Stemgent | Cat#09–0011; RRID:AB_1512171 |
| SSEA4 | Stemgent | Cat#09–0006; RRID:AB_1512169 |
| CDH1 | R&D System | Cat#AF648; RRID:AB_355504 |
| OVGP1 | SIGMA | Cat#HPA062205; RRID:AB_2684710 |
| p53 (DO-1) | Santa Cruz Biotechnology | Cat#sc-126; RRID:AB_628082 |
| Ki-67 (SP6) | Invitrogen | Cat#MA5–14520; RRID:AB_10979488 |
| MUC-16 | Sigma | Cat#HPA065600; RRID:AB_2732736 |
| Anti-phospho-Histone H2A.X (Ser139) | Millipore | Cat#16–202A; RRID:AB_568825 |
| Human Nuclei | Millipore | Cat#MAB128–1; RRID:AB_94090 |
| BRCA1 | Millipore | Cat#OP92; RRID:AB_2750876 |
| AF488 | Invitrogen | Cat#A32790; RRID:AB_2762833 and Cat# A32766; RRID:AB_2762823 |
| AF594 | Invitrogen | Cat#A32754; RRID:AB_2762827 and Cat# A32744; RRID:AB_2762826 |
| Chemicals, peptides, and recombinant proteins | ||
| Human recombinant activin A | Stemgent | Cat#03–0001 |
| Human recombinant BMP4 | R&D Systems | Cat#314-BP |
| Human Noggin | R&D system | Cat#6057-NG-01M |
| Murine recombinant EGF | PeproTech | Cat#AF-100–15 |
| Human recombinant basic FGF | PeproTech | Cat#100–18B |
| Human recombinant Follistatin | PeproTech | Cat#120–13 |
| Human recombinant WNT4 | R&D Systems | Cat#6076-WN-005 |
| SB431542 | Tocris | Cat#1614 |
| Glutamax | Life Technology | Cat#35050–061 |
| HEPES | Life Technology | Cat#15–630-106 |
| B27 supplement | GIBCO | Cat#17504044 |
| N2 supplement | GIBCO | Cat#17502048 |
| Matrigel | Corning/BD Biosciences | Cat#354230 |
| penicillin/ streptomycin | GIBCO | Cat#15140122 |
| Y-27632 | Stemgent | Cat#04–0012-02 |
| SYBR Select Master Mix | Appliedbiosystem | Cat#4472908 |
| OCT | Tissue-Tek | Cat#MPSMK-981385 |
| DAPI | Molecular Probes | Cat#D3571 |
| DAB solution | Vector | Cat#SK-4105 |
| Avidin-Biotin Complex | Vector | Cat#PK-4000 |
| Formalin | Fisher Chemical | Cat#23–245685 |
| ImmunoMaster Hematoxylin | American MasterTech Scientific | Cat#HXIMHPT |
| EOSIN WITH PHLOXINE B | American MasterTech Scientific | Cat#MPSM-STLB117 |
| Hyaluronidase | STEMCELL | Cat#07912 |
| Rucaparib (AG-G14699 | Chemietek | Cat#CT-AG01−10mg |
| OLAPARIB (AZD2281) | Chemietek | Cat#CT-A2281–100mg |
| NIRAPARIB (MK-4827) | Chemietek | Cat#CT-MK4827–10mg |
| Antibiotic-Antimycotic | GIBCO | Cat#15240-062 |
| hLIF | Millipore | Cat#LIF1010 |
| CHIR99021 | Tocris | Cat#4423 |
| HA-100 | Santa Cruz Biotech | Cat#203072 |
| A-83-10 | Tocris | Cat#2939 |
| mTeSR®1 medium | STEMCELL | Cat#85850 |
| KOSR | Invitrogen | Cat#10828028 |
| Advanced DMEM/F-12 | Life technologies | Cat#12634028 |
| Phosphatase/protease inhibitor cocktail | Sigma Aldrich | Cat#MSSAFE |
| Critical commercial assays | ||
| B cell Nucleofector Kit | Lonza | Cat#VPA-1001 |
| QIAGEN RNeasy Mini kit | QIAGEN | Cat#74004 |
| Quantitect Reverse Transcription Kit | QIAGEN | Cat#205311 |
| LDH assay | CyQUANT | Cat#C20301 |
| PrestoBlue assay | Invitrogen | Cat#226392 |
| 4X Laemmli sample buffer | BIO-RAD | Cat#161-0774 |
| Bradford assay | BIO-RAD | Cat#5000006 |
| Mini-PROTEAN TGX Precast gels | BIO-RAD | Cat#456–1094 |
| Odyssey blocking buffer | LI-COR | Cat#927–40000 |
| Deposited data | ||
| RNA Seq Data | This Paper | GEO: GSE190134 |
| Experimental models: Cell lines | ||
| CS87i-CTR-n3, female, age unknown, BRCA1 wildtype | Cedars-Sinai iPSC Core | CS87i-CTR |
| CS01iMEC-CTR, female, age unknown, BRCA1 wildtype | Cedars-Sinai iPSC Core | CS01iMEC-CTR |
| CS80i-CTR-Tn3, female, age 48, BRCA1 wildtype | Cedars-Sinai iPSC Core | CS80i-CTR |
| CS08i-BRCA-n5, female, age 36.3, BRCA1 mutant | Cedars-Sinai iPSC Core | CS08i-BRCA |
| CS70i-BRCA-n1, female, age 33.6, BRCA1 mutant | Cedars-Sinai iPSC Core | CS70i-BRCA |
| CS79i-BRCA-n2, female, age 31.4, BRCA1 mutant | Cedars-Sinai iPSC Core | CS79i-BRCA |
| Experimental models: Organisms/strains | ||
| Mouse: Foxn1nu,formerly Hfh11nu (known as NU/J athymic nude) | Jackson Laboratory | Cat#002019 |
| Oligonucleotides | ||
| Primers for Real-time PCR, see Table S5 | N/A | |
| Recombinant DNA | ||
| pEP4 E02S ET2K | Toombs et al., 2020 | Addgene 20927 |
| pCXLE-hOCT3/4-shp53-F | Toombs et al., 2020 | Addgene 27077 |
| pCXLEhUL | Toombs et al., 2020 | Addgene 27080 |
| pCXLE-hSK | Toombs et al., 2020 | Addgene 27078 |
| pCXWB-EBNA1 | Toombs et al., 2020 | Addgene 37624 |
| Software and algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Neurolucdia 11.07 | MBF Bioscience | https://www.mbfbioscience.com/ |
| Stereo Investigator 11.07 | MBF Bioscience | https://www.mbfbioscience.com/stereo-investigator |
| Prism software | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
Highlights.
BRCA1mut patient iPSC lines can be differentiated into fallopian tube epithelium
BRCA1mut fallopian tubes recapitulate ovarian carcinogenesis in vitro and in vivo
BRCA1mut fallopian tubes may provide a model to predict disease severity
BRCA1mut fallopian tube organoids provide a platform to study efficacy of cancer drugs
ACKNOWLEDGMENTS
This work was primarily funded by institutional support from the Regenerative Medicine Institute at Cedars-Sinai, with further support from the Ovarian Cancer Research Alliance Ann and Sol Schreiber Mentored Investigator Award, Veterans Administration Merit Award (VA-ORD BX004974), United States Department of Defense (DoD- W81XWH-17-BCRP), Breakthrough Award, Gilda Radner Hereditary Cancer Program, The Honorable Tina Brozman Foundation for Ovarian Cancer Research Rising Star Grant, and Precision Health Fund. We would like to express our gratitude to Dr. Soshana Svendsen for her critical review and editing of the manuscript and Kate Lawrenson for experiment suggestions.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing financial interests. Intellectual protection-patent pending for iPSC-derived human FTE organoid protocol has been initiated.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110146.
REFERENCES
- Adib TR, Henderson S, Perrett C, Hewitt D, Bourmpoulia D, Ledermann J, and Boshoff C (2004). Predicting biomarkers for ovarian cancer using gene-expression microarrays. Br. J. Cancer 90, 686–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Reyes A, and Huber W (2012). Detecting differential usage of exons from RNA-seq data. Genome Res. 22, 2008–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anttila MA, Tammi RH, Tammi MI, Syrjänen KJ, Saarikoski SV, and Kosma VM (2000). High levels of stromal hyaluronan predict poor disease outcome in epithelial ovarian cancer. Cancer Res. 60, 150–155. [PubMed] [Google Scholar]
- Barrett R, Ornelas L, Yeager N, Mandefro B, Sahabian A, Lenaeus L, Targan SR, Svendsen CN, and Sareen D (2014). Reliable generation of induced pluripotent stem cells from human lymphoblastoid cell lines. Stem Cells Transl. Med. 3, 1429–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bast RC Jr., Badgwell D, Lu Z, Marquez R, Rosen D, Liu J, Baggerly KA, Atkinson EN, Skates S, Zhang Z, et al. (2005). New tumor markers: CA125 and beyond. Int. J. Gynecol. Cancer 15, 274–281. [DOI] [PubMed] [Google Scholar]
- Behrens P, Rothe M, Florin A, Wellmann A, and Wernert N (2001). Invasive properties of serous human epithelial ovarian tumors are related to Ets-1, MMP-1 and MMP-9 expression. Int. J. Mol. Med. 8, 149–154. [DOI] [PubMed] [Google Scholar]
- Blanco I, Kuchenbaecker K, Cuadras D, Wang X, Barrowdale D, de Garibay GR, Librado P, Sánchez-Gracia A, Rozas J, Bonifaci N, et al. ; Teixeira; BCFR; SWE-BRCA; kConFab Investigators; GEMO (2015). Assessing associations between the AURKA-HMMR-TPX2-TUBG1 functional module and breast cancer risk in BRCA1/2 mutation carriers. PLoS One 10, e0120020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton KL, Chenevix-Trench G, Goh C, Sadetzki S, Ramus SJ, Karlan BY, Lambrechts D, Despierre E, Barrowdale D, McGuffog L, et al. ; EMBRACE; kConFab Investigators; Cancer Genome Atlas Research Network (2012). Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA 307, 382–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. (2010). 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 17, 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen NJ, Logani S, Dickerson EB, Kapa LB, Akhtar M, Benigno BB, and McDonald JF (2007). Emerging roles for PAX8 in ovarian cancer and endosalpingeal development. Gynecol. Oncol. 104, 331–337. [DOI] [PubMed] [Google Scholar]
- Buttermore ST, Hoffman MS, Kumar A, Champeaux A, Nicosia SV, and Kruk PA (2017). Increased RHAMM expression relates to ovarian cancer progression. J. Ovarian Res. 10, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, and Vogelstein B (1998). Mutations of mitotic checkpoint genes in human cancers. Nature 392, 300–303. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2011). Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, and Studer L (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Deng Y, Hua M, Xi Q, Liu R, Yang S, Liu J, Zhong J, Tang M, Lu S, et al. (2015). Expression and clinical role of TCTP in epithelial ovarian cancer. J. Mol. Histol. 46, 145–156. [DOI] [PubMed] [Google Scholar]
- Cho A, Howell VM, and Colvin EK (2015). The Extracellular Matrix in Epithelial Ovarian Cancer—A Piece of a Puzzle. Front. Oncol. 5, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chui MH, Momeni Boroujeni A, Mandelker D, Ladanyi M, and Soslow RA (2021). Characterization of TP53-wildtype tubo-ovarian high-grade serous carcinomas: rare exceptions to the binary classification of ovarian serous carcinoma. Mod. Pathol. 34, 490–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coan M, Rampioni Vinciguerra GL, Cesaratto L, Gardenal E, Bianchet R, Dassi E, Vecchione A, Baldassarre G, Spizzo R, and Nicoloso MS (2018). Exploring the Role of Fallopian Ciliated Cells in the Pathogenesis of High-Grade Serous Ovarian Cancer. Int. J. Mol. Sci. 19, 2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo M, Vilar E, Tsai S-Y, Chang K, Amin S, Srinivasan T, Zhang T, Pipalia NH, Chen HJ, Witherspoon M, et al. (2017). Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nat. Med. 23, 878–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curry EL, Moad M, Robson CN, and Heer R (2015). Using induced pluripotent stem cells as a tool for modelling carcinogenesis. World J. Stem Cells 7, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont S, Jan Z, Heremans R, Van Gorp T, Vergote I, and Timmerman D (2019). Organoids of epithelial ovarian cancer as an emerging preclinical in vitro tool: a review. J. Ovarian Res. 12, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Bussaglia E, Prat J, Harkes IC, Repasky EA, et al. (2000). Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl. Cancer Inst. 92, 564–569. [DOI] [PubMed] [Google Scholar]
- Felder M, Kapur A, Gonzalez-Bosquet J, Horibata S, Heintz J, Albrecht R, Fass L, Kaur J, Hu K, Shojaei H, et al. (2014). MUC16 (CA125): tumor biomarker to cancer therapy, a work in progress. Mol. Cancer 13, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Gu Z-Y, Li Q, Liu Q-H, Yang X-Y, and Zhang J-J (2019). Identification of significant genes with poor prognosis in ovarian cancer via bioinformatical analysis. J. Ovarian Res. 12, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal PA, Liu Q, Shattuck-Eidens D, Cochran C, Harshman K, Tavtigian S, Bennett LM, Haugen-Strano A, Swensen J, Miki Y, et al. (1994). BRCA1 mutations in primary breast and ovarian carcinomas. Science 266, 120–122. [DOI] [PubMed] [Google Scholar]
- Gee ME, Faraahi Z, McCormick A, and Edmondson RJ (2018). DNA damage repair in ovarian cancer: unlocking the heterogeneity. J. Ovarian Res. 11, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Li X, Zhao Y, Yang Q, and Kong B (2012). 53BP1 suppresses tumor growth and promotes susceptibility to apoptosis of ovarian cancer cells through modulation of the Akt pathway. Oncol. Rep. 27, 1251–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M-Z, and Jin W-L (2020). The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 5, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PM, and Drapkin R (2013). Modeling High-Grade Serous Carcinoma: How Converging Insights into Pathogenesis and Genetics are Driving Better Experimental Platforms. Front. Oncol. 3, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Coffey DM, Ma L, and Matzuk MM (2015). The ovary is an alternative site of origin for high-grade serous ovarian cancer in mice. Endocrinology 156, 1975–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopper O, de Witte CJ, Lõhmussaar K, Valle-Inclan JE, Hami N, Kester L, Balgobind AV, Korving J, Proost N, Begthel H, et al. (2019). An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat. Med. 25, 838–849. [DOI] [PubMed] [Google Scholar]
- Koshiyama M, Matsumura N, and Konishi I (2014). Recent concepts of ovarian carcinogenesis: type I and type II. BioMed Res. Int. 2014, 934261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiyama M, Matsumura N, and Konishi I (2017). Subtypes of ovarian cancer and ovarian cancer screening. Diagnostics (Basel) 7, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips K-A, Mooij TM, Roos-Blom M-J, Jervis S, van Leeuwen FE, Milne RL, Andrieu N, et al. ; BRCA1 and BRCA2 Cohort Consortium (2017). Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 317, 2402–2416. [DOI] [PubMed] [Google Scholar]
- Kuhn E, Kurman RJ, Vang R, Sehdev AS, Han G, Soslow R, Wang TL, and Shih IeM. (2012). TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma—evidence supporting the clonal relationship of the two lesions. J. Pathol. 226, 421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurman RJ (2013). Origin and molecular pathogenesis of ovarian high-grade serous carcinoma. Ann. Oncol. 24, x16–x21. [DOI] [PubMed] [Google Scholar]
- Labidi-Galy SI, Papp E, Hallberg D, Niknafs N, Adleff V, Noe M, Bhattacharya R, Novak M, Jones S, Phallen J, et al. (2017). High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 8, 1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFargue CJ, Dal Molin GZ, Sood AK, and Coleman RL (2019). Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol. 20, e15–e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson DA, Werb Z, Zong Y, and Goldstein AS (2015). The cleared mammary fat pad transplantation assay for mammary epithelial organogenesis. Cold Spring Harb. Protoc. 2015, pdb.prot078071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D-F, Su J, Kim HS, Chang B, Papatsenko D, Zhao R, Yuan Y, Gingold J, Xia W, Darr H, et al. (2015). Modeling familial cancer with induced pluripotent stem cells. Cell 161, 240–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Liu Z, Liang B, Chen S, Zhang X, Tong X, Lou W, Le L, Tang X, and Fu F (2018). Identification of core genes in ovarian cancer by an integrative meta-analysis. J. Ovarian Res. 11, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Lu S, Deng Y, Yang S, He S, Cai J, Qiang F, Chen C, Zhang W, Zhao S, et al. (2016). PSMB4 expression associates with epithelial ovarian cancer growth and poor prognosis. Arch. Gynecol. Obstet. 293, 1297–1307. [DOI] [PubMed] [Google Scholar]
- Liu C, Oikonomopoulos A, Sayed N, and Wu JC (2018). Modeling human diseases with induced pluripotent stem cells: from 2D to 3D and beyond. Development 145, dev156166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lõhmussaar K, Kopper O, Korving J, Begthel H, Vreuls CPH, van Es JH, and Clevers H (2020). Assessing the origin of high-grade serous ovarian cancer using CRISPR-modification of mouse organoids. Nat. Commun. 11, 2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Londero AP, Orsaria M, Tell G, Marzinotto S, Capodicasa V, Poletto M, Vascotto C, Sacco C, and Mariuzzi L (2014). Expression and prognostic significance of APE1/Ref-1 and NPM1 proteins in high-grade ovarian serous cancer. Am. J. Clin. Pathol. 141, 404–414. [DOI] [PubMed] [Google Scholar]
- Lord CJ, and Ashworth A (2012). The DNA damage response and cancer therapy. Nature 481, 287–294. [DOI] [PubMed] [Google Scholar]
- Lord CJ, and Ashworth A (2017). PARP inhibitors: Synthetic lethality in the clinic. Science 355, 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Wang X, He J, Xia J, and Li Y (2017). Increased expression of protein kinase CK2a correlates with poor patient prognosis in epithelial ovarian cancer. PLoS One 12, e0174037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mae S-I, Shono A, Shiota F, Yasuno T, Kajiwara M, Gotoda-Nishimura N, Arai S, Sato-Otubo A, Toyoda T, Takahashi K, et al. (2013). Monitoring and robust induction of nephrogenic intermediate mesoderm from human pluripotent stem cells. Nat. Commun. 4, 1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell CA, Keats JJ, Belch AR, Pilarski LM, and Reiman T (2005). Receptor for hyaluronan-mediated motility correlates with centrosome abnormalities in multiple myeloma and maintains mitotic integrity. Cancer Res. 65, 850–860. [PubMed] [Google Scholar]
- Maxwell CA, Benítez J, Gómez-Baldó L, Osorio A, Bonifaci N, Fernández-Ramires R, Costes SV, Guinó E, Chen H, Evans GJR, et al. ; HE-BON; EMBRACE; SWE-BRCA; BCFR; GEMO Study Collaborators; kConFab (2011). Interplay between BRCA1 and RHAMM regulates epithelial apicobasal polarization and may influence risk of breast cancer. PLoS Biol. 9, e1001199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce CL, Stram DO, Ness RB, Stram DA, Roman LD, Templeman C, Lee AW, Menon U, Fasching PA, McAlpine JN, et al. (2015). Population distribution of lifetime risk of ovarian cancer in the United States. Cancer Epidemiol. Biomarkers Prev. 24, 671–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perets R, Wyant GA, Muto KW, Bijron JG, Poole BB, Chin KT, Chen JYH, Ohman AW, Stepule CD, Kwak S, et al. (2013). Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell 24, 751–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portier L, Desterke C, Chaker D, Oudrhiri N, Asgarova A, Dkhissi F, Turhan AG, Bennaceur-Griscelli A, and Griscelli F (2021). iPSC-Derived Hereditary Breast Cancer Model Reveals the BRCA1-Deleted Tumor Niche as a New Culprit in Disease Progression. Int. J. Mol. Sci. 22, 1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press JZ, De Luca A, Boyd N, Young S, Troussard A, Ridge Y, Kaurah P, Kalloger SE, Blood KA, Smith M, et al. (2008). Ovarian carcinomas with genetic and epigenetic BRCA1 loss have distinct molecular abnormalities. BMC Cancer 8, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao TD, Tian H, Ma X, Yan X, Thapi S, Schultz N, Rosales N, Monette S, Wang A, Hyman DM, et al. (2015). Expression of the Carboxy-Terminal Portion of MUC16/CA125 Induces Transformation and Tumor Invasion. PLoS One 10, e0126633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell PA, Pharoah PD, De Foy K, Ramus SJ, Symmonds I, Wilson A, Scott I, Ponder BA, and Gayther SA (2000). Frequent loss of BRCA1 mRNA and protein expression in sporadic ovarian cancers. Int. J. Cancer 87, 317–321. [DOI] [PubMed] [Google Scholar]
- Samineni S, Glackin C, and Shively JE (2011). Role of CEACAM1, ECM, and mesenchymal stem cells in an orthotopic model of human breast cancer. Int. J. Breast Cancer 2011, 381080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Sances S, Workman MJ, and Svendsen CN (2020). Multi-lineage Human iPSC-Derived Platforms for Disease Modeling and Drug Discovery. Cell Stem Cell 26, 309–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva TP, Pereira CA, Oliveira AR, Raposo AC, Arez M, Cabral JMS, Milagre I, Carmo-Fonseca M, and da Rocha ST (2021a). Generation and characterization of induced pluripotent stem cells from a family carrying the BRCA1 mutation c.3612delA. Stem Cell Res. (Amst.) 52, 102242. [DOI] [PubMed] [Google Scholar]
- Silva TP, Pereira CA, Raposo AC, Oliveira AR, Arez M, Cabral JMS, Milagre I, Carmo-Fonseca M, and Rocha STD (2021b). Generation and characterization of induced pluripotent stem cells heterozygous for the Portuguese BRCA2 founder mutation. Stem Cell Res. (Amst.) 53, 102364. [DOI] [PubMed] [Google Scholar]
- Soyombo AA, Wu Y, Kolski L, Rios JJ, Rakheja D, Chen A, Kehler J, Hampel H, Coughran A, and Ross TS (2013). Analysis of induced pluripotent stem cells from a BRCA1 mutant family. Stem Cell Reports 1, 336–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Zhao H, Zhang C, Hu T, Wu J, Lin X, Luo D, Wang C, Meng L, Xi L, et al. (2017). Gene co-expression network reveals shared modules predictive of stage and grade in serous ovarian cancers. Oncotarget 8, 42983–42996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao T, Lin W, Wang Y, Zhang J, Chambers SK, Li B, Lea J, Wang Y, Wang Y, and Zheng W (2020). Loss of tubal ciliated cells as a risk for “ovarian” or pelvic serous carcinoma. Am. J. Cancer Res. 10, 3815–3827. [PMC free article] [PubMed] [Google Scholar]
- Teo AKK, Valdez IA, Dirice E, and Kulkarni RN (2014). Comparable generation of activin-induced definitive endoderm via additive Wnt or BMP signaling in absence of serum. Stem Cell Reports 3, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toombs J, Panther L, Ornelas L, Liu C, Gomez E, Martín-Ibáñez R, Cox SR, Ritchie SJ, Harris SE, Taylor A, et al. (2020). Generation of twenty four induced pluripotent stem cell lines from twenty four members of the Lothian Birth Cohort 1936. Stem Cell Res. (Amst.) 46, 101851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vang R, Shih IeM., and Kurman RJ (2013). Fallopian tube precursors of ovarian low- and high-grade serous neoplasms. Histopathology 62, 44–58. [DOI] [PubMed] [Google Scholar]
- Voltaggio L, Cimino-Mathews A, Bishop JA, Argani P, Cuda JD, Epstein JI, Hruban RH, Netto GJ, Stoler MH, Taube JM, et al. (2016). Current concepts in the diagnosis and pathobiology of intraepithelial neoplasia: A review by organ system. CA Cancer J. Clin. 66, 408–436. [DOI] [PubMed] [Google Scholar]
- Wang Z, Wu Y, Wang H, Zhang Y, Mei L, Fang X, Zhang X, Zhang F, Chen H, Liu Y, et al. (2014). Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc. Natl. Acad. Sci. USA 111, E89–E98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li L, Wang Y, Tang SN, and Zheng W (2015). Fallopian tube secretory cell expansion: a sensitive biomarker for ovarian serous carcinogenesis. Am. J. Transl. Res. 7, 2082–2090. [PMC free article] [PubMed] [Google Scholar]
- Weber AM, and Ryan AJ (2015). ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 149, 124–138. [DOI] [PubMed] [Google Scholar]
- Wong AS, Kim SO, Leung PC, Auersperg N, and Pelech SL (2001). Profiling of protein kinases in the neoplastic transformation of human ovarian surface epithelium. Gynecol. Oncol. 82, 305–311. [DOI] [PubMed] [Google Scholar]
- Yamanaka S (2012). Induced pluripotent stem cells: past, present, and future. Cell Stem Cell 10, 678–684. [DOI] [PubMed] [Google Scholar]
- Yucer N, Holzapfel M, Jenkins Vogel T, Lenaeus L, Ornelas L, Laury A, Sareen D, Barrett R, Karlan BY, and Svendsen CN (2017). Directed Differentiation of Human Induced Pluripotent Stem Cells into Fallopian Tube Epithelium. Sci. Rep. 7, 10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaoui K, Boudhraa Z, Khalifé P, Carmona E, Provencher D, and Mes-Masson A-M (2019). Ran promotes membrane targeting and stabilization of RhoA to orchestrate ovarian cancer cell invasion. Nat. Commun. 10, 2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Dolgalev I, Zhang T, Ran H, Levine DA, and Neel BG (2019). Both fallopian tube and ovarian surface epithelium are cells-of-origin for high-grade serous ovarian carcinoma. Nat. Commun. 10, 5367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data have been deposited at GEO (GEO: GSE190134) and are publicly available as of publication date. Code used to generate data for this study will be available and shared by lead contact upon request. Original western blot images and microscopy data reported in this paper will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this paper is available and will be shared by the lead contact upon request.