

Abstract
The use of Disease progression models (DPMs) in Drug Development has been widely adopted across therapeutic areas as a method for integrating previously obtained disease knowledge to elucidate the impact of novel therapeutics or vaccines on disease course, thus quantifying the potential clinical benefit at different stages of drug development programs. This paper provides a brief overview of DPMs and the evolution in data types, analytic methods, and applications that have occurred in their use by Quantitive Clinical Pharmacologists. It also provides examples of how these models have informed decisions and clinical trial design across several therapeutic areas and at various stages of development. It briefly describes potential new applications of DPMs utilizing emerging data sources, and utilizing new analytic techniques, and discuss new challenges faced such as requiring description of multiple endpoints, rapid model development, application of machine learning-based analytics, and use of high dimensional and real-world data. Considerations for the continued evolution future of DPMs to serve as community-maintained expert systems are also provided.
KEY WORDS: decision-making, disease progression model, drug development, MIDD
Introduction
A fundamental tenet of model-informed drug development (MIDD) is incorporation of available information to inform the development of a new medicine [1]. For many diseases, understanding the course of disease as a function of time, disease severity, and impact of treatment can aid in answering questions related to impact of a new medicine, and how it can be of greatest value to the patient. A model-informed approach offers an ideal solution to incorporate all the types of information available to the researcher across disparate sources. It serves as the framework to integrate knowledge, and to build and grow an expert understanding of both disease and treatment impacts that can inform the development and use of medicines.
Disease progression modeling (DPM) integrates mathematical functions and underlying scientific pathophysiologic principles to quantitatively describe the time course of disease progression. The key concepts and developments leading to their increased use have been well-described previously [2, 3]. Historically, these models have been empiric, but more recent examples include semi-mechanistic, systems biology, and systems pharmacology approaches [4].
Over time, the complexity of data types used, analytic approaches, and potential applications have continued to grow. However, irrespective of the underlying modeling techniques, these models normally contain three elements essential for use in drug development (see Fig. 1). The three components of DPMs allow for use across a variety of applications at various stages in drug development.
Fig. 1.
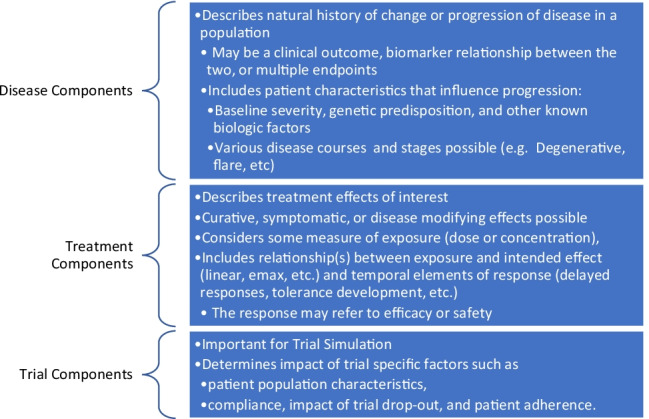
Components of a Disease Progression Model for Use in Drug Development.
DPM Applications
During development, MIDD strategies encompass a variety of model types as tools to inform different decision points and problems, ideally in an integrated and complimentary manner. Each model type offers a different approach and may be constructed on different data. They can inform different problems or may be used together to offer fidelity to a particular decision (assuming they make the same recommendation). Often, model types evolve in parallel and inform each other, and may be used interchangeably. DPMs have become an important tool in the Quantitive Clinical pharmacologist’s toolkit, and the application of DPMs to inform decision making has grown over time with the realization of their potential. They have become one of the most common applications for models in drug development.
The utility of connecting disease progression models with clinical trial simulations was identified early, dating back to the pivotal work of Nick Holford and others in Parkinson’s and Alzheimer’s Disease [5–7]. These early examples illustrated the approach and benefit to understanding disease progression linked to clinical outcomes and illustrated the potential of data sharing and meta-analyses. These analyses provided insights int the nature of the drug effects within the clinical trials used as a data source. Since then, a diverse array of similar examples across multiple therapeutic areas have been completed and made available in the public domain [8–10].
An important learning from these initial clinical trial-informed efforts was that data pooled solely from one or a few completed clinical trials rarely contained sufficient information to fully characterize disease progression (e.g., insufficient duration of observation). As investigators explored other applications of DPMs, it became necessary to utilize a broader array of existing and emerging data sources, especially if the intent was to fully characterize and link progression attributes and biomarkers to long-term disease outcomes or if accounting for real-world experience.
Over time, there have been significant advances in linking observed outcomes back to the underlying mechanisms of drug action, and to related biomarkers. Mechanistic-based models inform decisions related to pathway and target selection, candidate selection, biomarker strategy, patient selection and optimal study design for early signals of efficacy [2]. Mechanistic DPMs may identify patient populations most likely to respond to therapy. They can identify patient-responder phenotypes that inform enrollment criteria and that aid in assessment of commercial value by determining the prevalence of the proposed target indication. They can answer the longitudinal design-related clinical study questions (duration of study, optimal timing of assessments). DPMs can assess impact of drug combinations, especially when combined with a Quantitive Systems Pharmacology (QSP) model. In more recent examples, QSP models form the basis for disease progression models themselves [11].
A common application of DPMs in drug development is use of QSP-linked DPMs to design and interpret clinical Proof of Concept, guide portfolio decisions including franchise ranking and DPM-linked clinical trial simulation models to evaluate design scenarios and judge the probability of technical success (PTS) across various design options [1, 12]. The trial simulation application is particularly important given the stage and cost of development at this juncture (typically phase 2 or 3 but can also reflect post-marketing efforts). Beyond clinical trial simulation, there are also applications in health economic and clinical outcomes research supported by both academic and governmental agencies that could inform both funding allocation and policy decisions respectively [13, 14] and amongst the provider and payer communities to support formulary and reimbursement policy and decisions [15].
At later stages of development this type of model framework can be used to evaluate clinical study designs including endpoints, substrata, and sample size as well as clinical operations including patient and site selection.
Data Sources and Requirements for DPMs
The extent to which modeling efforts are successful often depends upon the availability and appropriateness of the data used to construct them, development of analysis plans apriori, and alignment of MIDD deliverables with timing of decision points with the team responsible. Irrespective of the planned application, and given the increasing diversity of DPM application [2], early investment and planning in data requirements is warranted to maximize a DPM’s value. Early development and planning also allows for model enhancement over time, and to add data as it is generated and to answer emerging questions in a timely manner.
A fundamental concept in MIDD is that the systematic collection and quantification of results from all available sources is required to best inform decision making at every stage [1]. As such, data requirements for a DPM model may vary based on the intended context of use (COU). As model requirements change with stage of development, different data may be required to inform new COU as the utility of the model is challenged by later-stage development questions. An early-stage model may be informed by natural history data, patient registry data and preclinical data that describe mechanistic underpinnings of the relevant disease biology (like QSP model requirements). In later stages, the model may be augmented by earlier patient studies within the program, advancements in understanding of the disease, or from patient data from previous programs in the same population. While providing flexibility in the types of information that can be incorporated, these disparate data types (large, survey-based, and often unstructured data with small, structured data or simply parameter estimates with distributional data or assumptions) can also create a challenge for assumptions around suitability for data integration and model definition.
In general, the data used to inform DPMs has evolved with the growing complexity of data sources available (see Fig. 2). Early DPM examples primarily were constructed using individual level data taken from within clinical trials and natural history studies used to inform the model. These individual level DPMs were based on clinical endpoints, incorporated linear progression of disease and potential symptomatic or disease modifying treatment effects [5, 6], but the data itself was often not available outside of the organizations that generated it. When published, this information was typically aggregated to a set of summary statistics. As individual level data was not readily available to the larger group of quantitative scientists, aggregate literature data was used to characterize disease progression in longitudinal model based meta-analyses (MBMA) [7, 8]. These MBMA were able to incorporate more information across industrial and academic sources which were previously unavailable. Further evolution of DPM branched into combined individual and aggregate data derived longitudinal MBMA [7, 40].
Fig. 2.

Flow of Information from Data Source to DPM Development Utilization*. *Information Propagates from Data Sources (light green nodes) to Utilization (brown, tan, lavender, and pink nodes) through Data Types (red nodes) and Model Types (light red, purple, and light purple nodes). The Evolution of DPMs is Depicted by Linkage Color. The light blue linkages are the earliest, which expanded to include natural history data sources (dark blue) and Aggregate Level Data Types (green). The Current State Includes Non-clinical Data Sources and Mechanistic DPM (peach). Real World Evidence (orange linkages) is Depicted as Potential Future State.
As biomarker data (e.g., imaging, HbA1c concentrations, etc.) became more prevalent in clinical trials, their use in DPMs also increased leading to more complex, semi-mechanistic models. Non-clinical sources were utilized, leading to sets of physiological parameters which furthered the development of mechanistic DPM type models. In our current state, real world evidence is also being incorporated into different data types.
Information flows from source, or observation, to developmental utility (Fig. 2). Differing data sources (Clinical Trials, Natural History studies, non-clinical experiments, and real-world evidence) provide the basis for data types (individual clinical endpoint, aggregated clinical endpoints, clinical biomarkers, [16–18] physiological parameters). DPM types (individual, aggregate, and mechanistic) characterizing these data types, or combinations of such, are utilized for a range of development decisions. The DPM transforms the information into quantitative knowledge which is actionable. DPMs may incorporate information from different sources, and data types, and are constructed using a range of methodologies. Though different, an overlap in utility is apparent in which different types of DPM can address the same development question (eg – the time course of a novel clinical trial).
The progression of a model can be thought of as part of a model development lifecycle (MDLC) in which model structures are evolved in an iterative fashion, each building on the previous work. Implicit to the concept of a MDLC is the addition of more information, thereby increasing precision of parameter estimates, or improved characterization of clinical endpoints or trajectory of disease [18]. A future state may be the merging of empirical and mechanistic approaches into holistic DPMs describing both pathophysiology of the disease and the distribution of clinical endpoints supporting development utility ranging from the selection of mechanism to providing a Bayesian prior for more efficient study design.
The past decade has also seen significant advances in the regulatory science real-world data (RWD) and Real-World Evidence (RWE) framework. These advancements along with regulatory guidance [19] have driven the increased use of from electronic health records and claims databases to provide the basis for evidence in support of drug effectiveness (RWE). The use of RWD has now also become an important data source for DPMs. RWD sources include electronic health records (EHRs), claims and billing activities, product and disease registries, patient-generated data including in home-use settings, and data gathered from other sources that can inform on health status, such as mobile devices. The addition of various RWD sources may also be relevant to incorporate the clinical signs and symptoms of clinical care into a DPM. Such data allows the evaluation of the existing standard of care and the performance of existing treatments to be considered and can be useful if the DPM is coupled with a clinical trial simulation model [20].
Analytic Methodologies
Initial approaches
Cook and Bies [2] describe three broad classes of DPMs: empirical, semi-mechanistic, and systems biology DPMs, with their application and subsequent appearance in the literature occurring in that order. With advancements in the types of DPM models used, and increased complexity of data types, there has also been an evolution in analytic methodologies that have developed. Initial empiric models describing subjective scoring utilized linear and non-linear mixed effects models. Subsequently more complex models such as asymptotic progress, physiological turnover, and growth and decay models have been utilized and have been well described [21]. The next section focuses on more recent advances in analytic techniques.
Latent Variable Disease Progression Models
In many cases, clinical endpoints are composites of prespecified observations (or assessments) which are combined as a single measure of disease state. Due to the way in which they are defined, these endpoints may be bounded at one or both ends which may cause complications when modeling near the boundaries. Additionally, each of the assessments, or subscales, can contribute different amount of information to the underlying understanding of disease state depending on severity. Empirical model approaches have been developed which characterize the progression of disease as a latent variable. In this methodology the disease is indirectly characterized based on information from a series of clinical endpoints. Applications range in complexity from a logit transformation in which the probability of an endpoint is characterized, to models utilizing item response which characterize the probability for each of the endpoint components (subscales).
Latent variable disease progression modeling, charactering primary clinical endpoints, has been employed in myriad of indications. Selected examples can be seen in rheumatoid arthritis [22–25], psoriasis [26, 27], ulcerative colitis [24], and Alzheimer’s disease [28, 29] Applications of item response disease progression modeling have been applied to Alzheimer’s disease [30, 31], Parkinson’s disease [31, 32], and multiple sclerosis [33]. In these examples the bounded nature of the clinical endpoint has been appropriately described enabling the interpretation of underlying disease progression, and in some examples a treatment effect, to be established. This type of modeling has also shown utility in being able to simulate different response rates through clinical trial simulation. The item response methodology, characterizing the information for each of the subcomponents of a composite scale, enables clinical trial simulation into the subscales. A further benefit of the item response model is that it can be used to integrate different variants of a clinical scale [32] and potentially future applications integrating multiple clinical scales which describe different levels of disease severity (ex—CDR-SOB, ADAS-COG, NPI, SIB).
Natural language Processing and Machine Learning
There has been significant progress in the availability of knowledge and data across biological and human scales, facilitated by advances in omics technologies, digital data platforms integrating clinical trial and patient registry data, and hospital or primary care data such as EHR, claims. For the Life Sciences industry, the question has become where these data can be systematically leveraged for advancing knowledge on human disease, and for developing innovations in medical treatments or vaccines for disease eradication or management. This exponential increase in data generation and availability is met with commensurate improvements in computer technology and advancement in computational models geared for handling big data science.
Take for example the problem of researching the literature for novel or historical insights on disease or drug mechanisms. With the increasing size of the PubMed corpus (several million full text articles), it is increasingly difficult to research specific topics comprehensively without additional search tools and capabilities. Natural language processing (NLP) has been an evolving discipline, with deep roots in computer science and discrete mathematical modeling among others, that is progressed to be an important tool for assimilating large amounts of knowledge, example from PubMed, and building knowledge graphs representing relationships across fields or variables of interest to the researcher [34]. These graph models can be quantitatively mined for specific information and surfacing the relevant data or knowledge the research is looking for in a systematic and data driven way. Given the extent and evolution of data and knowledge that DPMs rely on for both mechanistic and patient level information, NLP models can be an invaluable tool for integrating and assimilating the relevant knowledge and data from respective knowledge sources.
Advances in measurement technologies have also facilitated the interrogation of broad metabolic or proteomic scale characteristics of disease and drug action. These have over time played important roles in advancing our understanding of disease [35, 36] One notable and timely example is host-virus interactions and how DNA or RNA based viruses can evade the immune system, or hijack cellular machinery to reproduce and spread. Although these data have the potential to elucidate disease mechanisms or drug action, they also have brought forward computational and technical challenges due to the size of the data and complexity of potential interactions. Advancement in machine learning (ML) based models coupled with computing technology have allowed us to tackle increasingly larger data sets and derive biological or mechanistic while maintaining statistical rigor and soundness. As we apply ML models to big data derived from mechanistic data sets (e.g., omics data), we can advance specific biological mechanisms or biomarker strategies that can subsequently be represented in DPMs [37]. With larger scale data, and applications where causality or inference is less important, for example, medical outcome of an imaging-based diagnostic, Artificial Intelligence based models, grounded in deep learning models such as large neural networks, have been the tool of choice. These AI based approaches have successfully alleviated the burden of manual readouts where machine readouts based on AI technology is validated and are also increasingly utilized as a platform [38] in early drug discovery to advance potential novel targets forward based on volumes of discovery biology and chemical libraries.
Regulatory Considerations for DPM use
Often, DPMs are developed de novo for use within a development program and used to describe the data contained within an individual submission. In some cases, the model is subsequently published. This approach is inefficient and limits the potential for models to evolve.
Regulatory Path to model qualification and COU
A longstanding interest of the global regulatory community as potentially enabling significant progress in drug development has been the application of scientific advances as new tools to aid the development process. Such tools have been shown to speed up the availability of new products that may be safer and more effective. The Center for Drug Evaluation and Research (CDER) of the US FDA has undertaken multiple initiatives to support the development of new drug development tools (DDTs). Among these efforts has been the creation of a formal qualification process, described in a formal guidance [9] that CDER can use when working with submitters of DDTs to guide development as submitters refine the tools and rigorously evaluate them for use in the regulatory process. The DDT qualification process is intended to expedite development of publicly available DDTs that can be widely employed. Drug developers can use a DDT that has been qualified within a specific context of use (COU) [FDA Guidance 2020] for the qualified purpose during drug development if: [1] The study is conducted properly, [2] the DDT is used for the qualified purpose and [3] at the time of qualification, there is no new information that conflicts with the basis for qualification. Once a DDT has been qualified, CDER reviewers feel more confident in the application of the DDT within the qualified COU and do not have to re-confirm DDT utility.
Qualification is an expectation that within the stated COU, the DDT can be relied on to have a specific interpretation and application in drug development and regulatory review. The COU describes the way the DDT is to be used and the purpose of the use. A complete COU statement describes the circumstances under which the DDT is qualified and the boundaries within which the available data adequately support use of the DDT. Once a DDT has been qualified for a specific COU in drug development, it can be used to produce analytically valid measurements that can be relied on to have a specific use and interpretable meaning. The DDT can then be used by drug developers for the qualified context in IND, NDA, and BLA submissions without the relevant CDER review group reconsidering and reconfirming suitability.
The process for DDT qualification provides a framework for interactions between CDER and DDT submitters to guide the collection of data to support a DDT’s prospectively specified COU. The qualification process consists of three stages: [1] an initiation stage, [2] a consultation and advice stage, and [3] a review stage for the qualification determination. The appropriate review offices will participate in the entire qualification process for the DDT. The goal of the process is to reach a determination about the adequacy of the submitted data to support DDT qualification within a COU. An important future goal of the COU process would be the establishment of more formalized planning regarding governance and provenance of disease progression models and the underlying code. Provenance is an important aspect given that the COU should carry some version of an “expiration date” as the demands and utility of models in this category evolves with data, knowledge of disease biology and the pool of agents and procedures used to treat target diseases. Caretakers of the various models should represent the mutual desires of regulators and the scientific community.
Illustrative Example Applications
Evolution of DPMS in Neurodegenerative Disorders (Alzheimer’s)
The evolution of DPMs in medicines development for Alzheimer’s disease (AD) over the last three decades illustrates how DPMs have evolved with increased treatment options, understanding of disease, improved analytic techniques, emergence of new data types, and increased trial design complexity (see Table I). It also highlights how investigators build on previous knowledge to continue to evolve models and to develop them into expert systems for each disease.
Table I.
Some Illustrative Disease Progression Models across Therapeutic Areas.
| Model Reference | Therapeutic Area and Class | Data Informing Model | Model Structure and Type | Application |
|---|---|---|---|---|
| Holford et al. 1992 [5] | Alzheimer’s disease | Alzheimer’s patient data (placebo, tacrine 40 mg, tacrine 80 mg) for 10 weeks | Nonlinear mixed effects | Description of symptomatic effects of tacrine in Alzheimer’s patients from a single trial |
| Barendregt et al. 2003 [13] | Asthma/epidemiology | Victorian Burden of Disease Study/Australian Bureau of Statistics | Multi-state life table | A generic model for the assessment of disease epidemiology |
| Kowalski et al. 2008 [49] | Surgery Pain | Data from a parallel-group, placebo-controlled study evaluating single oral doses of placebo, 3, 10, and 60 mg SC-75416 capsules, and 50 mg rofecoxib with 50 patients per group | Nonlinear logistic-normal | Dose selection and clinical development |
| Hutmacher et al. 2008 [22] | Rheumatoid Arthritis (RA) | Dose ranging in 264 RA patients | Introduction of indirect latent response variable | Dose Selection |
| Ito et al. 2010 [7] | Alzheimer’s disease | 576 mean ADAS-cog changes from baseline data points of 52 trials in the literature, representing data from approximately 19,972 patients and more than 84,000 individual observations | Nonlinear mixed effects | Trial duration determination for disease modifying agents |
| Ito et al. 2011 [39] | Alzheimer’s disease | 817 mild to moderate patients in a Natural history study | Nonlinear mixed effects | Incorporation and use of biomarkers as predictive markers of disease progression |
| Hu et al. 2011 [26] | Psoriasis | Two Phase III studies in severe Psoriasis | Informative dropout/Longitudinal Mixed Effects logistic regression | Support of dose regimen and support for alternative regimens |
| Hu et al. 2011 [50] | Psoriasis | Two Phase III studies in severe Psoriasis | Latent variable with standard logit transofrmation to bound tails | Improvement to previous model by bounding outcomes |
| Rogers et al. 2012 [40] | Alzheimer’s disease | Literature and Patient-level data from Open registry | Beta-regression | First model approved in the FDA Fit for Purpose Pathway. Shared model |
| Ueckert et al. 2014 [30] | Alzheimer’s disease | 2744 patients, ADAS-Cog | Item response Theory | Early example of application of IRT to DPMs |
| Conrado et al. 2014 [28] | Alzheimer’s disease | 4495 patients ADAS-COG11 from Coalition Against Major Disease Database (15 RCTs) | Beta regression | Model refinement and verification rates of progression of earlier publications |
| Hu et al. 2014 [51] | Methodology | NA | Latent variable indirect response | New Method Description |
| Novakovic et al. 2017 [33] | Multiple sclerosis (RRMS) | 1319 multiple sclerosis patients | Item response theory | Estimation of drug disease modifying effect |
| Karelina et al. 2017 [52] | Alzheimer’s | TG576 mouse data, longitudinal soluble and insoluble CSF and plasma A-beta healthy and patient data, Pib-PET data (healthy subjects) | Mechanisitic (QSP) | Estimation of required trial duration for observation of disease modifying effects in AD |
| Kaddi et al. 2018 [4] | Acid Sphingomyelinase Deficiency | Literature (enzyme kinetics) preclinical studies, natural History Study, Phase I patient studies, | Mechanistic (QSP) with 4 sub-models (pk, molecular, cellular, organ) | Assessment of systemic pharmacological effects in adult and pediatric patients, variability within and across these patient populations, and extrapolation of treatment response from adults to pediatrics |
| Gottipati et al. 2019 [32] | Parkinson’s disease | 554 patients UPDRS and MDS-UPDRS | Item response | |
| Louis et al. 2019 [48] | None | Riemannian Geometry Learning for Disease Progression Modelling | Riemannian manifold learning | Method development |
| Kim et al., 2020 [53] | Alzheimer’s disease | 645 patients (129 SCI, 270 AMCI, and 246 ADD) followed up more than three times to obtain CDR-SB scores at the Samsung Medical Center from Jan. 2003 to Dec. 2015. Data shared by author upon request | Mixed-effect predictive model expressed in SAS | Understanding of the effect of education on cognitive trajectories – no described regulatory use |
| Fourage et al., 2021 [54] | Centronuclear myopathy | 59 patients, 15 with DNM2 mutation and 44 patients with mutation in the MTM1 gene. Patients had been evaluated every 3 months under 2 years of age, every 6 months between 2 and 6 years of age, and, for patients older than 6, at 6 months and 12 months after enrolment and then once a year | Bayesian predictive model—Proc MCMC in SAS 9.4 | Model adequately predicted the natural evolution of patients over the duration of the study and will facilitate future trial designs that can cope with disease rarity |
| Rao et al. 2021 [11] | COVID-19 | Literature (in vitro mechanistic data), observational and RCT trial summary data across multiple studies | Integrated multi-endpoint QSP model of within-host SARS-CoV-2 viral dynamics and the immune response | Utilized for duration Selection for Paxlovid™ |
| Wang et al. 2022 [55] | COPD | real-world longitudinal Electronic Medical Record (EMR) database of over 300,000 patients | Unsupervised learning/Markov Model | no |
AD DPMs were some of the first reported in the literature, and were used to describe the symptomatic effects of cholinesterase inhibitors in AD patients by Holford and Peace [5] and Ito et. al. [7] utilized summary level literature data from 52 studies representing nearly 20,000 patients to describe impact of disease severity and age on yearly progression of the most commonly used clinical outcome measure, and to further describe treatment effect. The model was used to describe both symptomatic and disease modifying effects, and to determine expected differences in highlighted their use to describe effects observed in clinical trials.
With advancements in understanding of disease biology, and incorporation of specific biomarkers and genetic tests in studies, DPMs were able to further characterize factors impacting progression of AD. Ito et al. [39] published further work based on a natural history study that incorporated imaging data, biomarkers, and genetic information. Subsequent work was undertaken by Rogers et. al. in collaboration with Ito et al., the Critical Path Institute and FDA to utilize both patient level and summary level data that were available [40]. A beta-regression approach was used that allowed for both data types to inform the model. This model also formed the basis for a fit-for-purpose pathway for drug development and was the first tool deemed suitable under that regulatory program. The model and supporting materials were made available as open source for community use.
With increasing understanding that late-stage patients may have progressed too far to respond to disease modifying agents, a shift to testing disease modifying agents at the earlier stages of AD with resultant slower rates of progression, there was a need to understand whether existing elements of the ADASD-cog were more sensitive to detecting treatment effect and/or if new endpoints would be needed in patients with mild cognitive impairment. Ueckert et al. [30] applied Item Response Theory (IRT) to determine which items within the ADAS-cog provided the most information by stage of disease.
In parallel to advances in DPMs, both systems biology and systems pharmacology models also advanced and provided even further insights into relationship between the emerging imaging, genetic and protein biomarkers, and trial outcomes. Karelina et al. [52] looked at how mechanistic translational models can allow for prediction of long-term clinical trials at various stages of disease. Systems biology approaches capture the disease in the broader context of CNS neurodegeneration and help provide insights into potential targets and pathways for exploration [41].
Inflammation and Immunology
MIDD has been applied in the inflammation and immunology areas to characterize disease progression and to provide dosing rationale for a myriad of indications such as ulcerative colitis, psoriasis, and rheumatoid arthritis [22]. In these indications the progression of disease has been implemented through placebo (or standard of care) and active treatment indirect response functions in which the clinical endpoints have been described using bounded outcome methodologies [22]. While many of these examples are applied to the observations from a single study or combined studies for a single novel therapeutic, the expectation of a mature disease progression model is to synthesize information across clinical studies and new molecular entities (NMEs). Hu et al. [51] applied information from multiple studies, and NMEs, utilizing an empirical model describing the expectation of disease and standard of care to provide a phase 2 dose regimen decision for a novel therapeutic in psoriasis. The Immunology and Inflammation therapeutic area has a wealth of information and has provided the opportunity for a holistic model describing the behavior of disease and standard of care in a clinical trial. Such models provide a basis for trial design quantifying the duration of treatment needed to observe an effect. As an informative prior they enable a reduction in the number of patients needed to demonstrate the effect of a novel treatment. In both cases the application of DPMs would accelerate development decision making while maintaining scientific rigor.
Rare Disease: Bronchopulmonary Dysplasia
Comprising approximately 8000 diverse disease states linked only by their “rare” prevalence designation (disease or condition that affects less than 200,000 people in the United States by the US FDA) [42] is a broad array of conditions that often begins at birth or soon thereafter sometimes with very short life expectancy and rarely in a manageable condition in adulthood [42]. It is only through the Orphan Drug Act of 1983 that this therapeutic area has been properly incentivized for financial motivation to spur private sector R&D to make inroads to the myriad of diseases in this class.
A recently supported effort of the FDA, Critical Path Institute, and International Neonatal Consortium (INC) has promoted the execution of pilot projects that generate RWE to support regulatory decision making in neonatal drug development. One such pilot is focused on developing a validated definition of Bronchopulmonary dysplasia (BPD). In addition to the definition, the BPD pilot will also assess the extent to which a large, multisource aggregation of RWD will allow identification of validated risk factors for, and surrogate endpoints representing, BPD, and the inclusion of these in clinical trial simulations that help identify risk factors and surrogate that are fit-for-purpose for hypothetical studies aiming to prevent or treat BPD and its related long-term complications. The backbone of the proposed trial simulations will be a qualified, fit-for-purpose disease progression model (and likely other models).
While BPD is described as a disease, in fact its better classified as a syndrome – a condition of a premature neonate requiring “oxygen supplementation at a particular level and for a particular duration in the postnatal period” [57]. The probability of a BPD diagnosis depends mainly on gestational age and birth weight. Babies born after only 22–24 weeks of development have an 80% chance of being diagnosed with BPD [58]. At this age, lungs are just starting to develop alveoli and the premature exposure to air breathing disrupts this process. While knowledge of the many factors involved in alveologenesis is steadily accumulating, specific endotypes remain to be defined with the quantitative detail needed for both QSP and disease progression models. A major caveat is that this knowledge is obtained primarily in a range of model systems and using a variety of manipulations to induce BPD-like lungs. Longitudinal data reflecting disease progression are very limited, both in humans and model animals. In humans, longitudinal data in neonates are limited for obvious reasons. Besides records of oxygen supplementation, there is some longitudinal data on the efficiency of gas exchange, showing that premature neonates with BPD are less efficient compared to neonates without BPD and that both improve over time [59]. Ultrasound imaging also appears promising as a source of data easily obtained from neonates [60, 61]. At present, a landscaping exercise is in progress to assemble credible mechanistic and RWD sources that would be the foundation of both QSP and disease progression models for BPD. Figure 3 illustrates the nature of the repeated measure data of current clinical and convenience sampling and the gaps particularly at the onset of disease progression where sampling is limited or nonexistent. It is hoped that this effort even if it does not support the full COU desired will be a starting point for future models and encourage more informative BPD trials with sampling that compliments current data and knowledge gaps. It is also the hope and intent that the landscaping and integration of mechanistic, RWD and disease progression data, and incorporation into a coupled disease-progression and QSP platform can facilitate the discovery and translation of novel targets and identify optimal timepoints for therapeutic intervention.
Fig. 3.

Schematic of BPD Disease Progression with Variables of Clinical Interest Linked to Stage of Progression.
Neuroscience
Evolving Approaches (Systems Biology/Data Science)
Mechanism-driven DPMs (mDPMs) describe the time evolution of disease characteristics with fit for purpose mechanistic description and can often be tied to QSP models to provide a more comprehensive representation of underlying biology for the respective mechanisms. mDMPs provide decision value across the discovery and translational medicine continuum, such informing the design and interpretation of POC/POM clinical studies, and informing the biomarker strategy, as tied to a disease or to a therapeutic MOA. This begs the question – how does one inform mechanisms that can be incorporated into mDPMs Various data sources are often relied upon when interrogating disease mechanisms or drug MOA, such as non-clinical in-vitro or in-vivo models. Although these efforts and tools generate important data, but not sufficient by themselves. Systems biology has deep academic roots and has over time extended its reach from basic science application, elucidating system wide etiology of disease and drug action, into more recently having increasingly direct influence on key drug discovery and development milestones. e.g., identifying MOA of a compound, or facilitating translation into the clinic. The advantage and promise of systems biology as a discipline is the data driven, scientifically objective approach to discovery and elucidation of disease mechanisms and drug action. One data source we have discussed earlier here is the importance of disease registries for the assembly and engineering of DPMs. These same registries can be additionally utilized as an important source of identification of underlying biology implicated along the time evolution of disease progression. Big data approaches such as metabolomics and proteomics (derived from patient samples from these registries) have been invaluable tools in discovery of novel mechanisms implicated in disease. When coupled with advanced data science approaches e.g., machine learning, they represent an innovative and data driven pipeline for discovery of disease mechanisms, and incorporation into mDPMs or mDPM-QSP platforms.
Multi-endpoint QSP Models: COVID-19
A recent example for a novel oral treatment for COVID-19 illustrates the flexibility of a DPM to integrate information from disparate sources and to build on existing models by incorporating rapidly emerging data to quickly answer important questions regarding drug development [11]. The model provided understanding across several different biomarker endpoints, and clinical outcomes, and was used to inform study design (specifically treatment duration).
In this example, a QSP model of the pathogenesis and treatment of SARS-CoV-2 infection streamlined and accelerated the development and Emergency Use Authorization of a novel medicine to treat COVID-19. Utilizing an updated version of a previously published preliminary model of the immune response to SARS-CoV-2 infection (significantly updated with emerging data from a curated dataset spanning viral load and immune responses in plasma and lung) allowed for in silico exploration of the uncertainties of clinical trial design to rapidly inform development decisions for upcoming clinical trial duration. The authors identified a population of parameter sets to generate heterogeneity in pathophysiology and treatment and tested this model against published reports from interventional SARS-CoV-2 targeting Ab and anti-viral trials. Upon generation and selection of a virtual population, they matched both the placebo and treated responses in viral load in these trials. They extended the model to predict the rate of hospitalization or death within a population. To validate this approach, they showed the model matched a published subgroup analysis of patients treated with neutralizing Abs. By simulating intervention at different timepoints post infection, the model predicted efficacy is not sensitive to interventions within five days of symptom onset, but efficacy is dramatically reduced if more than five days pass post-symptom onset prior to treatment, as borne out in the clinical trial [43].
Challenges and Opportunities
Challenges exist for routine, standardized approaches for the development and use of well-characterized and robust DPMs. These include but are not limited to the following:
more consistent evaluation and regulatory feedback regarding the construction and utility of DPMs
- more diverse and collaborative drug development culture which embraces the contributions of a truly multidisciplinary community that is required to develop such models as opposed to other model types in the MIDD toolbox
-
○a more collaborative environment for the sharing of data, code, and models.
-
○a more collaborative and neutral governance and provenance environment that is both efficient and comprehensive.
-
○
Despite these challenges, progress has been made in all these areas. Recent publications and FDA public meetings [2, 9] provides initial thoughts on best practices for DPMs. As the field is still evolving and the COU for the various DPM types can be very varied, a heavily prescriptive approach is unwarranted though these initial thoughts form the basis of what will surely evolve as a meaningful guide. Collaboration happens of course but the more consistent engagement of academic thought leaders particularly those in the disease biology arena of targeted therapeutic areas should be more commonly expected. Notions of model ownership need also to be examined and resolved so more can contribute and benefit from such collaborations.
An important requirement for the future success of DPM is the collaborative spirit and effort that must guide the next generation of models and ultimately enhance their utility beyond drug development purposes. As alluded to previously, this will require a relaxed view of model ownership and a broader adoption of open science principles. As some have pointed out [44], despite the increasing availability of Open Science (OS) infrastructure and the rise in policies to change behavior, OS practices are not yet the norm. The benefits are clear it would seem—less error-prone and more visible models, not only to peers from the same and other scientific disciplines but also greater penetration to the public, who can appreciate the economic benefits of knowledge dissemination. Moreover, engaging in OS practices facilitates the sharing and reuse of data, materials, and code in the scientific community [45, 46], contributes to enriched scholarly output and literacy, and increases trust in the process [47] The obstacles to meaningful OS adoption are typically grounded in financial concerns over intellectual property (IP) and heavily constrained by past legal practice. An important evolution for this collaboration and more consistent OS engagement will require legal agreements and data use agreements more focused on shared IP where financial incentives are agreed upon without constraining the creative process and the OS approach.
Conclusions and Path Forward
Despite past challenges, the use of DPMs to inform drug development is becoming routine. DPMs flexibility in allowing integration of information from various sources in a quantitive manner make them indispensable for use in informing trial design and improving confidence in decision making during all stages of drug development. They are now routinely accepted and used in support of drug submissions worldwide. The key questions surrounding DPMs are no longer whether they have validity, do they add value and where they can be applied, but rather how their use can be expanded to incorporate emerging complex data types, to answer more and more complex development questions stemming from new modalities and emerging health risks, and how to do so quickly and efficiently so they accelerate development of new medicines.
There is a growing need for disease models to inform multiple safety, biomarker, and efficacy endpoints simultaneously, a requirement that may not be suited for classical empiric DPM approaches. While ML approaches are used to recognize patterns in large data, and complex statistical methodologies [48] have been proposed, they lack the underlying ability to integrate basic pharmacologic principles and drug-specific information that Quantitive clinical pharmacology “expert-systems” like QSP models afford, and that can allow for hypothesis generation (i.e. for identification of new targets or pathways). In addition, QSP approaches, based in fundamental principles of pharmacology, allow for models to build. grow and evolve as new information emerges. They can be shared and maintained by a community of users [45].
Finally, going forward, DPMs may be combined with other emerging tools and technologies to decrease patient burden. While randomized controlled trials have been considered the standard for demonstration of efficacy, there has been a significant drive for increased patient inclusivity and use of patient centric designs to minimize patient burden and to provide maximal benefit to patients seeking clinical trials as a care option. Hybrid study designs that include features of RCTs with use of RWD can combine the advantages of both [62]. A potential synergy is DPMS utilizing RWD as an informative Bayesian prior to augment control arms of a study. An appropriate drug-disease-trial model could significantly minimize the number of patients needed in the control arm, improving likelihood that a patient receives active therapy. This could be of particular benefit for populations that are not part of initial approvals and that are typically included in post-approval commitments, such as pediatrics by minimizing the number of patients needed in the control arms and maximizing the likelihood of being randomized to active treatment.
Declarations
Conflicts of interests/Competing interests
The authors received salary support from the Critical Path Institute (JB), Pfizer Pharmaceuticals (BC and TN) and Axcella Therapeutics (KA). Other than salary support, the authors did not receive support from any organization for the submitted work.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jeffrey S. Barrett, Email: jbarrett@c-path.org
Tim Nicholas, Email: timothy.nicholas@pfizer.com.
Karim Azer, Email: kazer@axcellatx.com.
Brian W. Corrigan, Email: brian.corrigan@pfizer.com
REFERENCES
- 1.Lalonde RL, Kowalski KG, Hutmacher MM, Ewy W, Nichols DJ, Milligan PA, et al. Model-based drug development. Clin Pharmacol Ther. 2007;82(1):21–32. doi: 10.1038/sj.clpt.6100235. [DOI] [PubMed] [Google Scholar]
- 2.Cook SF, Bies RR. Disease Progression Modeling: Key Concepts and Recent Developments. Curr Pharmacol Rep. 2016;2(5):221–230. doi: 10.1007/s40495-016-0066-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mould DR. Models for Disease Progression: New Approaches and Uses (vol 92, pg 125, 2012) Clinical Pharmacology & Therapeutics. 2012;92(5):666. doi: 10.1038/clpt.2012.155. [DOI] [PubMed] [Google Scholar]
- 4.Kaddi CD, Niesner B, Baek R, Jasper P, Pappas J, Tolsma J, et al. Quantitative Systems Pharmacology Modeling of Acid Sphingomyelinase Deficiency and the Enzyme Replacement Therapy Olipudase Alfa Is an Innovative Tool for Linking Pathophysiology and Pharmacology. CPT Pharmacometrics Syst Pharmacol. 2018;7(7):442–452. doi: 10.1002/psp4.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holford NH, Peace KE. Methodologic aspects of a population pharmacodynamic model for cognitive effects in Alzheimer patients treated with tacrine. Proc Natl Acad Sci U S A. 1992;89(23):11466–11470. doi: 10.1073/pnas.89.23.11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parkinson Study G Effect of deprenyl on the progression of disability in early Parkinson's disease. N Engl J Med. 1989;321(20):1364–1371. doi: 10.1056/NEJM198911163212004. [DOI] [PubMed] [Google Scholar]
- 7.Ito K, Ahadieh S, Corrigan B, French J, Fullerton T, Tensfeldt T, et al. Disease progression meta-analysis model in Alzheimer's disease. Alzheimers Dement. 2010;6(1):39–53. doi: 10.1016/j.jalz.2009.05.665. [DOI] [PubMed] [Google Scholar]
- 8.Raket LL. Statistical Disease Progression Modeling in Alzheimer Disease. Front Big Data. 2020;3:24. doi: 10.3389/fdata.2020.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Best Practices for Development and Application of Disease Progression Models https://www.fda.gov/drugs/news-events-human-drugs/best-practices-development-and-application-disease-progression-models-11192021-11192021: FDA; 2021 [cited 2022 January 23, 2022]. Available from: https://www.fda.gov/drugs/news-events-human-drugs/best-practices-development-and-application-disease-progression-models-11192021-11192021. Accessed 22 Dec 2021.
- 10.Iwasa S, Kudo T, Takahari D, Hara H, Kato K, Satoh T. Practical guidance for the evaluation of disease progression and the decision to change treatment in patients with advanced gastric cancer receiving chemotherapy. Int J Clin Oncol. 2020;25(7):1223–1232. doi: 10.1007/s10147-020-01684-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dai W, Rao R, Sher A, Tania N, Musante CJ, Allen R. A Prototype QSP Model of the Immune Response to SARS-CoV-2 for Community Development. CPT Pharmacometrics Syst Pharmacol. 2021;10(1):18–29. doi: 10.1002/psp4.12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gobburu JV, Lesko LJ. Quantitative disease, drug, and trial models. Annu Rev Pharmacol Toxicol. 2009;49:291–301. doi: 10.1146/annurev.pharmtox.011008.145613. [DOI] [PubMed] [Google Scholar]
- 13.Barendregt JJ, Van Oortmarssen GJ, Vos T, Murray CJ. A generic model for the assessment of disease epidemiology: the computational basis of DisMod II. Popul Health Metr. 2003;1(1):4. doi: 10.1186/1478-7954-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johns B, Baltussen R, Hutubessy R. Programme costs in the economic evaluation of health interventions. Cost Eff Resour Alloc. 2003;1(1):1. doi: 10.1186/1478-7547-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seigfried RJ, Corbo T, Saltzberg MT, Reitz J, Bennett DA. Deciding which drugs get onto the formulary: a value-based approach. Value Health. 2013;16(5):901–906. doi: 10.1016/j.jval.2013.03.1623. [DOI] [PubMed] [Google Scholar]
- 16.de Winter W, DeJongh J, Post T, Ploeger B, Urquhart R, Moules I, et al. A mechanism-based disease progression model for comparison of long-term effects of pioglitazone, metformin and gliclazide on disease processes underlying Type 2 Diabetes Mellitus. J Pharmacokinet Pharmacodyn. 2006;33(3):313–343. doi: 10.1007/s10928-006-9008-2. [DOI] [PubMed] [Google Scholar]
- 17.Choy S, Kjellsson MC, Karlsson MO, de Winter W. Weight-HbA1c-insulin-glucose model for describing disease progression of type 2 diabetes. CPT Pharmacometrics Syst Pharmacol. 2016;5(1):11–19. doi: 10.1002/psp4.12051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conrado DJ, Nicholas T, Tsai K, Macha S, Sinha V, Stone J, et al. Dopamine Transporter Neuroimaging as an Enrichment Biomarker in Early Parkinson's Disease Clinical Trials: A Disease Progression Modeling Analysis. Clin Transl Sci. 2018;11(1):63–70. doi: 10.1111/cts.12492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.FDA. Considerations for the Use of Real-World Data and Real-World Evidence To Support Regulatory Decision-Making for Drug and Biological Products FDA: FDA; 2021 [Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-use-real-world-data-and-real-world-evidence-support-regulatory-decision-making-drug. Accessed 22 Dec 2021.
- 20.Dagenais S, Russo L, Madsen A, Webster J, Becnel L. Use of Real-World Evidence to Drive Drug Development Strategy and Inform Clinical Trial Design. Clin Pharmacol Ther. 2022;111(1):77–89. doi: 10.1002/cpt.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mould DR. Models for disease progression: new approaches and uses. Clin Pharmacol Ther. 2012;92(1):125–131. doi: 10.1038/clpt.2012.53. [DOI] [PubMed] [Google Scholar]
- 22.Hutmacher MM, Krishnaswami S, Kowalski KG. Exposure-response modeling using latent variables for the efficacy of a JAK3 inhibitor administered to rheumatoid arthritis patients. J Pharmacokinet Pharmacodyn. 2008;35(2):139–157. doi: 10.1007/s10928-007-9080-2. [DOI] [PubMed] [Google Scholar]
- 23.Hu C, Xu Z, Rahman MU, Davis HM, Zhou H. A latent variable approach for modeling categorical endpoints among patients with rheumatoid arthritis treated with golimumab plus methotrexate. J Pharmacokinet Pharmacodyn. 2010;37(4):309–321. doi: 10.1007/s10928-010-9162-4. [DOI] [PubMed] [Google Scholar]
- 24.Hu C, Adedokun OJ, Zhang L, Sharma A, Zhou H. Modeling near-continuous clinical endpoint as categorical: application to longitudinal exposure-response modeling of Mayo scores for golimumab in patients with ulcerative colitis. J Pharmacokinet Pharmacodyn. 2018;45(6):803–816. doi: 10.1007/s10928-018-9610-0. [DOI] [PubMed] [Google Scholar]
- 25.Li L, Zhang Y, Ma L, Ji P, Yim S, Chowdhury B, et al. Exposure-Response Modeling and Power Analysis of Components of ACR Response Criteria in Rheumatoid Arthritis (Part 1: Binary Model) J Clin Pharmacol. 2017;57(9):1097–1106. doi: 10.1002/jcph.891. [DOI] [PubMed] [Google Scholar]
- 26.Hu C, Szapary PO, Yeilding N, Zhou H. Informative dropout modeling of longitudinal ordered categorical data and model validation: application to exposure-response modeling of physician's global assessment score for ustekinumab in patients with psoriasis. J Pharmacokinet Pharmacodyn. 2011;38(2):237–260. doi: 10.1007/s10928-011-9191-7. [DOI] [PubMed] [Google Scholar]
- 27.Hu C, Randazzo B, Sharma A, Zhou H. Improvement in latent variable indirect response modeling of multiple categorical clinical endpoints: application to modeling of guselkumab treatment effects in psoriatic patients. J Pharmacokinet Pharmacodyn. 2017;44(5):437–448. doi: 10.1007/s10928-017-9531-3. [DOI] [PubMed] [Google Scholar]
- 28.Conrado DJ, Denney WS, Chen D, Ito K. An updated Alzheimer's disease progression model: incorporating non-linearity, beta regression, and a third-level random effect in NONMEM. J Pharmacokinet Pharmacodyn. 2014;41(6):581–598. doi: 10.1007/s10928-014-9375-z. [DOI] [PubMed] [Google Scholar]
- 29.Samtani MN, Xu SX, Russu A, Adedokun OJ, Lu M, Ito K, et al. Alzheimer's disease assessment scale-cognitive 11-item progression model in mild-to-moderate Alzheimer's disease trials of bapineuzumab. Alzheimers Dement (N Y) 2015;1(3):157–169. doi: 10.1016/j.trci.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ueckert S, Plan EL, Ito K, Karlsson MO, Corrigan B, Hooker AC, et al. Improved utilization of ADAS-cog assessment data through item response theory based pharmacometric modeling. Pharm Res. 2014;31(8):2152–2165. doi: 10.1007/s11095-014-1315-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gottipati G, Karlsson MO, Plan EL. Modeling a Composite Score in Parkinson's Disease Using Item Response Theory. AAPS J. 2017;19(3):837–845. doi: 10.1208/s12248-017-0058-8. [DOI] [PubMed] [Google Scholar]
- 32.Gottipati G, Berges AC, Yang S, Chen C, Karlsson MO, Plan EL. Item Response Model Adaptation for Analyzing Data from Different Versions of Parkinson's Disease Rating Scales. Pharm Res. 2019;36(9):135. doi: 10.1007/s11095-019-2668-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Novakovic AM, Krekels EH, Munafo A, Ueckert S, Karlsson MO. Application of Item Response Theory to Modeling of Expanded Disability Status Scale in Multiple Sclerosis. AAPS J. 2017;19(1):172–179. doi: 10.1208/s12248-016-9977-z. [DOI] [PubMed] [Google Scholar]
- 34.Ramponi A, Giampiccolo S, Tomasoni D, Priami C, Lombardo R. High-Precision Biomedical Relation Extraction for Reducing Human Curation Efforts in Industrial Applications. Ieee Access. 2020;8:150999–151011. doi: 10.1109/ACCESS.2020.3014862. [DOI] [Google Scholar]
- 35.Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, et al. The Human Cell Atlas. Elife. 2017;6. [DOI] [PMC free article] [PubMed]
- 36.Reiss DJ, Baliga NS, Bonneau R. Integrated biclustering of heterogeneous genome-wide datasets for the inference of global regulatory networks. BMC Bioinformatics. 2006;7:280. doi: 10.1186/1471-2105-7-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peterson EJR, Ma S, Sherman DR, Baliga NS. Network analysis identifies Rv0324 and Rv0880 as regulators of bedaquiline tolerance in Mycobacterium tuberculosis. Nat Microbiol. 2016;1(8):16078. doi: 10.1038/nmicrobiol.2016.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tse EG, Aithani L, Anderson M, Cardoso-Silva J, Cincilla G, Conduit GJ, et al. An Open Drug Discovery Competition: Experimental Validation of Predictive Models in a Series of Novel Antimalarials. J Med Chem. 2021;64(22):16450–16463. doi: 10.1021/acs.jmedchem.1c00313. [DOI] [PubMed] [Google Scholar]
- 39.Ito K, Corrigan B, Zhao Q, French J, Miller R, Soares H, et al. Disease progression model for cognitive deterioration from Alzheimer's Disease Neuroimaging Initiative database. Alzheimers Dement. 2011;7(2):151–160. doi: 10.1016/j.jalz.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 40.Rogers JA, Polhamus D, Gillespie WR, Ito K, Romero K, Qiu R, et al. Combining patient-level and summary-level data for Alzheimer's disease modeling and simulation: a beta regression meta-analysis. J Pharmacokinet Pharmacodyn. 2012;39(5):479–498. doi: 10.1007/s10928-012-9263-3. [DOI] [PubMed] [Google Scholar]
- 41.Lloret-Villas A, Varusai TM, Juty N, Laibe C, Le NovEre N, Hermjakob H, et al. The Impact of Mathematical Modeling in Understanding the Mechanisms Underlying Neurodegeneration: Evolving Dimensions and Future Directions. CPT Pharmacometrics Syst Pharmacol. 2017;6(2):73–86. doi: 10.1002/psp4.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miller KL, Fermaglich LJ, Maynard J. Using four decades of FDA orphan drug designations to describe trends in rare disease drug development: substantial growth seen in development of drugs for rare oncologic, neurologic, and pediatric-onset diseases. Orphanet J Rare Dis. 2021;16(1). [DOI] [PMC free article] [PubMed]
- 43.EPIC-HR: Study of Oral PF-07321332/Ritonavir Compared With Placebo in Nonhospitalized High Risk Adults With COVID-19 [Internet]. 2021 [cited February 13, 2022]. Available from: https://clinicaltrials.gov/ct2/show/NCT04960202. Accessed 22 Dec 2021.
- 44.Armeni K, Brinkman L, Carlsson R, Eerland A, Fijten R, Fondberg R, et al. Towards wide-scale adoption of open science practices: The role of open science communities. Sci Publ Policy. 2021;48(5):605–611. doi: 10.1093/scipol/scab039. [DOI] [Google Scholar]
- 45.Allen C, Mehler DMA. Open science challenges, benefits and tips in early career and beyond. PLoS Biol. 2019;17(5):e3000246. doi: 10.1371/journal.pbio.3000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Milham MP, Craddock RC, Son JJ, Fleischmann M, Clucas J, Xu H, et al. Assessment of the impact of shared brain imaging data on the scientific literature. Nat Commun. 2018;9(1):2818. doi: 10.1038/s41467-018-04976-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tennant JP, Waldner F, Jacques DC, Masuzzo P, Collister LB, Hartgerink CH. The academic, economic and societal impacts of Open Access: an evidence-based review. F1000Res. 2016;5:632. [DOI] [PMC free article] [PubMed]
- 48.Louis M, Couronne R, Koval I, Charlier B, Durrleman S. Riemannian Geometry Learning for Disease Progression Modelling. Lect Notes Comput Sc. 2019;11492:542–553. doi: 10.1007/978-3-030-20351-1_42. [DOI] [Google Scholar]
- 49.Kowalski KG, Olson S, Remmers AE, Hutmacher MM. Modeling and simulation to support dose selection and clinical development of SC-75416, a selective COX-2 inhibitor for the treatment of acute and chronic pain. Clin Pharmacol Ther. 2008;83(6):857–866. doi: 10.1038/sj.clpt.6100374. [DOI] [PubMed] [Google Scholar]
- 50.Hu C, Yeilding N, Davis HM, Zhou H. Bounded outcome score modeling: application to treating psoriasis with ustekinumab. J Pharmacokinet Pharmacodyn. 2011;38(4):497–517. doi: 10.1007/s10928-011-9205-5. [DOI] [PubMed] [Google Scholar]
- 51.Hu C. Exposure-response modeling of clinical end points using latent variable indirect response models. CPT Pharmacometrics Syst Pharmacol. 2014;3:e117. doi: 10.1038/psp.2014.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karelina T, Demin O, Jr, Demin O, Duvvuri S, Nicholas T. Studying the Progression of Amyloid Pathology and Its Therapy Using Translational Longitudinal Model of Accumulation and Distribution of Amyloid Beta. CPT Pharmacometrics Syst Pharmacol. 2017;6(10):676–685. doi: 10.1002/psp4.12249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim KW, Woo SY, Kim S, Jang H, Kim Y, Cho SH, et al. Disease progression modeling of Alzheimer's disease according to education level. Sci Rep. 2020;10(1):16808. doi: 10.1038/s41598-020-73911-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fouarge E, Monseur A, Boulanger B, Annoussamy M, Seferian AM, De Lucia S, et al. Hierarchical Bayesian modelling of disease progression to inform clinical trial design in centronuclear myopathy. Orphanet J Rare Dis. 2021;16(1):3. doi: 10.1186/s13023-020-01663-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang XS, D. Wang, F. . Unsupervised Learning of Disease Progression Models. Available from: https://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.434.8794&rep=rep1&type=pdf. Accessed 22 Dec 2021.
- 56."Orphan Drug Act of 1983". US Food and Drug Administration. 4 January 1983. Retrieved 26 December 2021. https://www.govinfo.gov/content/pkg/STATUTE-96/pdf/STATUTE-96-Pg2049.pdf. Accessed 22 Dec 2021.
- 57.SurateSolaligue DE, Rodríguez-Castillo JA, Ahlbrecht K, Morty RE. Recent advances in our understanding of the mechanisms of late lung development and bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2017;313(6):L1101–L1153. doi: 10.1152/ajplung.00343.2017. [DOI] [PubMed] [Google Scholar]
- 58.Thébaud B, Goss KN, Laughon M, Whitsett JA, Abman SH, Steinhorn RH, Aschner JL, Davis PG, McGrath-Morrow SA, Soll RF, Jobe AH. Bronchopulmonary dysplasia Nat Rev Dis Primers. 2019;5(1):78. doi: 10.1038/s41572-019-0127-7.PMID:31727986;PMCID:PMC6986462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Choi YJ, Stoecklin B, Hemy NR, Hall GL, Doherty DA, Simpson SJ, Pillow JJ. Pulmonary Gas Exchange Improves over the First Year in Preterm Infants with and without Bronchopulmonary Dysplasia. Neonatology. 2021;118(1):98–105. doi: 10.1159/000513357. [DOI] [PubMed] [Google Scholar]
- 60.Hoshino, Y., Arai, J., Hirono, K., Maruo, K., Kajikawa, D., Yukitake, Y., Hinata, A., & Miura, R. (2021). Gravity-induced loss of aeration and atelectasis development in the preterm lung: a serial sonographic assessment. Journal of perinatology : official journal of the California Perinatal Association, 1–6. [DOI] [PMC free article] [PubMed]
- 61.Oulego-Erroz I, Alonso-Quintela P, Terroba-Seara S, et al. Early assessment of lung aeration using an ultrasound score as a biomarker of developing bronchopulmonary dysplasia: a prospective observational study. J Perinatol. 2021;41:62–68. doi: 10.1038/s41372-020-0724-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baumfeld Andre E, Reynolds R, Caubel P, Azoulay L, Dreyer NA. Trial designs using real-world data: The changing landscape of the regulatory approval process. Pharmacoepidemiol Drug Saf. 2020;29(10):1201–1212. doi: 10.1002/pds.4932. [DOI] [PMC free article] [PubMed] [Google Scholar]