

Abstract
Neurodegenerative disorders and leukodystrophies are progressive neurologic conditions that can occur following the disruption of intricately coordinated patterns of gene expression. Exome sequencing has been adopted as an effective diagnostic tool for determining the underlying genetic etiology of Mendelian neurologic disorders, however genome sequencing offer advantages in its ability to identify and characterize copy number, structural, and sequence variants in noncoding regions. Genome sequencing from peripheral leukocytes was performed on two patients with progressive neurologic disease of unknown etiology following negative genetic investigations including exome sequencing. RNA sequencing from peripheral blood was performed to determine gene expression patterns in one of the patients. Potential causative variants were matched to the patients’ clinical presentation. The first proband was found to be heterozygous for a likely pathogenic missense variant in PLA2G6 (c.386T>C; p.Leu129Pro) and have an additional deep intronic variant in PLA2G6 (c.2035‐926G>A). RNA sequencing indicated this latter variant created a splice acceptor site leading to the incorporation of a pseudo‐exon introducing a premature termination codon. The second proband was heterozygous for a 261 kb deletion upstream of LMNB1 that included an enhancer region. Previous reports of copy number variants spanning this region of cis‐acting regulatory elements corroborated its pathogenicity. When combined with clinical presentations, these findings led to a definitive diagnosis of autosomal recessive infantile neuroaxonal dystrophy and autosomal dominant adult‐onset demyelinating leukodystrophy, respectively. In patients with progressive neurologic disease of unknown etiology, genome sequencing with the addition of RNA analysis where appropriate should be considered for the identification of causative noncoding pathogenic variants.
Keywords: PLA2G6, LMNB1, progressive neurologic disease, noncoding variants
Genome sequencing was performed on two probands with progressive neurologic disease of unknown etiology. The first proband was found to be heterozygous for a pathogenic missense variant in PLA2G6 (c.386T>C; p.Leu129Pro) and have an additional deep intronic variant in PLA2G6, (c.2035‐926G>A) creating a splice acceptor site detected with RNA sequencing. The second proband was heterozygous for a 261 kb deletion upstream of LMNB1 that included an enhancer region. These findings led to a definitive diagnosis of autosomal recessive infantile neuroaxonal dystrophy and autosomal dominant adult‐onset demyelinating leukodystrophy, respectively.
![]()
1. INTRODUCTION
Neurodegenerative disorders are a subset of progressive neurologic conditions characterized by dysfunction and death of select populations of neurons through proteotoxic stress and abnormalities in ubiquitin–proteasomal as well as autophagy‐lysosomal systems, oxidative stress, programmed cell death, and neuroinflammation (Dugger & Dickson, 2017). Similarly, leukodystrophies cause progressive neurologic disease through the degeneration of white matter, with cells involved in the axon–glia unit such as oligodendrocytes, astrocytes, ependymal cells, and microglia being specifically affected (Köhler et al., 2018).
The advent of next‐generation sequencing has allowed a comprehensive interrogation of relevant genes, revealing pathogenic variants in hundreds of genes that underly fundamental neuronal and white matter pathways. Exome sequencing performed in a large cohort across a range of clinical indications identified a definitive diagnosis for 31% for disorders of the central nervous system (Retterer et al., 2016). A more recent study reported a diagnostic yield of 40% among a diverse group of neurologic disorders when exome sequencing is undertaken (Córdoba et al., 2018). However, because this molecular testing focuses on the coding portion of the genome, it fails to detect noncoding variants beyond the canonical splice sites. Evidence has accumulated from genome and transcriptome sequencing that pathogenic mutations in over 75 disease‐associated genes occur in deep intronic regions, often due to pseudo‐exon inclusion or changes in regulatory element splicing (Vaz‐Drago et al., 2017).
Genome sequencing offers a greater breadth of coverage and thus is better equipped to identify and characterize copy number, structural, and sequence variants. In a study where participants with undiagnosed disease underwent exome sequencing followed by genome sequencing, 33% of diagnoses were missed by exome sequencing, and of these, 87% had copy number variants or noncoding variants identified by genome sequencing (Burdick et al., 2020).
Here, we present two study participants with progressive neurologic diseases of unknown etiology who were found to have novel pathogenic noncoding variants in the genes PLA2G6 and LMNB1, both of which were discovered through whole‐genome sequencing.
2. MATERIALS AND METHODS
2.1. Ethical compliance
This study was approved by the local Institutional Review Board at the University of Miami, USA. Clinical data and biological material were collected, stored, and used according to procedures in accordance with the ethical standards of the declaration of Helsinki protocols.
2.2. Participants
Participants were recruited for evaluation through the Undiagnosed Disease Network (UDN) at the University of Miami clinical site. Informed consent was obtained for NIH‐UDN protocol (15‐HG‐0130) from the affected individuals, or parents where appropriate.
2.3. Genome and RNA sequencing
Genome sequencing was performed using genomic DNA obtained from leukocytes. Trio‐genome analysis was performed for proband 1, and single‐genome testing was performed for proband 2. The sequencing was performed at Baylor Genetics as part of UDN workflow. Briefly, the library was prepared using the KAPA Hyper Prep kit and the Illumina NovaSeq 6000 platform for 150 bp paired‐end reads was used for sequence analysis. Average sequenced coverage over the genome was >40X, >97.5% target base was covered at >20X and SNP concordance to genotype array was >95%.
Genome sequencing data processing, sequence alignment to GRCh37/hg19, variant filtering and prioritization by allele frequency, predicted functional impact, and inheritance models were performed for both probands using Genesis 2.0 genome analysis software according to standard protocols (DePristo et al., 2011; McKenna et al., 2010). CNVnator was used for copy number variant detection. Candidate variants were prioritized based on the probands’ clinical phenotype and confirmed by Sanger sequencing.
RNA sequencing was performed for proband 1 at Baylor Genetics as previously described (Murdock et al., 2021). Briefly, a specimen of whole blood was processed using an Illumina stranded, polyA‐tailed kit then multiplexed and subjected to 150 bp paired‐end sequencing with approximately 30–50 million reads. Fastq files were aligned to the GRCh37/hg19 reference sequence using STAR‐2.6.1b in 2‐pass mode, and duplicates were marked with Picard. Gene count quantification for total RNA was performed using the GeneCounts function within STAR against the GENCODE v19 transcript file. RSEM was run with the STAR aligner to create fragments per kilobase of transcript per million mapped reads value (Bademci et al., 2020).
3. RESULTS
3.1. Clinical phenotype of proband 1
Proband 1 is a 5‐year‐old male with developmental delays beginning at 12 months of age. A neurologic assessment when he was 19 months old observed hypotonia in addition to worsening motor and speech delays. In‐home therapies failed to lead to sustained improvements over the ensuing months. He was also found to have monocular esotropia of the right eye and bilateral hyperopia upon ophthalmologic evaluation.
At 2 years of age, he had developmental regression as well as postural dystonia. He could no longer sit with assistance or scoot. He was also observed to have oropharyngeal dysphagia and failure to thrive. A gastrostomy tube was placed to encourage weight gain through nutritional supplementation.
Brain MRI demonstrated edema with restricted diffusion symmetrically in the bilateral caudate nuclei and putamen, and likely involving the bilateral thalami with moderate cerebellar hemisphere and vermian volume loss (Figure 1a). Electromyography with nerve conduction study demonstrated a primary axonal motor neuropathy with evidence of ongoing denervation.
FIGURE 1.
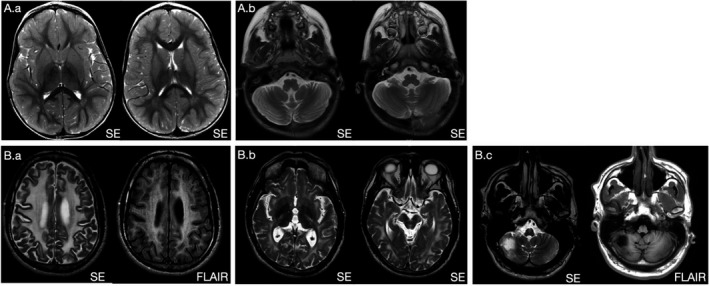
Brain MRI images of probands. Proband 1 (A.a) Axial T2W scans demonstrate diffuse increased signal abnormality within bilateral caudate and putamen and probable involvement of bilateral thalami with relative sparing of globus pallidus (A.b) Moderate cerebellar atrophy with prominence of cerebellar folia on axial T2W image through the level of posterior fossa. Proband 2 (B.a) Axial T2W and FLAIR images show extensive bilateral white matter signal hyperintensities. Basal ganglia were spared (not shown). (B.b) Axial T2W scans demonstrate increased signal within the bilateral internal capsules, and midbrain, including cerebral white matter. Middle cerebellar peduncles, pons, and medulla were also involved (not shown). (B.c) Axial T2W and FLAIR images reveal right inferior cerebellar encephalomalacia presumed to be due to prior vascular insult
The proband required the use of a wheelchair at 4 years of age. He could no longer support his head or use his hands. He had lost the ability to babble, and instead of using his eye gaze to make choices. He was also diagnosed with severe obstructive sleep apnea requiring treatment with bilevel positive airway pressure.
The etiology of his condition remained unknown despite biochemical and molecular genetics testing that was performed prior to his enrolling in the UDN and which included chromosomal microarray, fragile X analysis, as well as exome and mitochondrial sequencing, both in muscle and blood that was reported as normal.
3.2. Identification of novel deep intronic variant
Proband 1 was enrolled in the UDN at age 4. His evaluation consisted of reviewing past medical records, obtaining a complete medical and family history, performing a physical examination and trio genome and transcriptome sequencing. Trio genome sequencing of proband 1 revealed that both the proband and his unaffected mother were heterozygous for the PLA2G6 (NM_003560.4: c.386 T > C; p.Leu129Pro) variant in exon 3. This variant was found to have 6 prior entries in ClinVar with conflicting interpretations of pathogenicity. We interpret the variant as likely pathogenic based on ACMG criteria (Richards et al., 2015; Table S1). A second variant was found in the deep intronic region of PLA2G6 (NM_003560.4: c.2035‐926G>A) and interpreted as likely pathogenic (Table S2). Proband 1 and his unaffected father were both heterozygous for this variant, which is absent in the Genome Aggregation Database and predicted by multiple splice site prediction algorithms to create a splice acceptor site (Table S2 and S3).
RNA sequencing demonstrated the c.2035‐926G > A variant generates a 145 base pair pseudo‐exon with a premature stop codon within the pseudo‐exon (Figure 2a) and activates a cryptic donor gc splice site that is also used for alternative transcripts PLA2G6–236 and PLA2G‐237 (Figure 2b).
FIGURE 2.
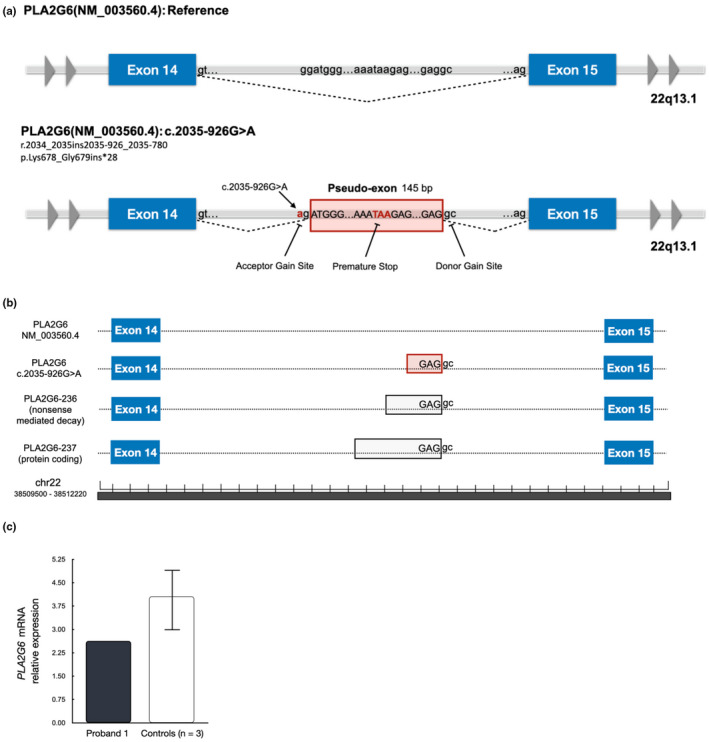
Molecular studies. (a) The PLA2G6(NM_003560.4): c.2035‐926G>A is a single nucleic acid substitution that creates a splice site acceptor gain site. As a consequence, a pseudo‐exon 145 base pairs in length is incorporated into the transcript. A premature stop codon within the pseudo‐exon is anticipated to produce nonsense‐mediated decay of the mRNA transcript. (b) Pseudo‐exon donor site found to be active in two other PLA2G6 transcripts based on information in Ensembl GRCh37/hg19. (c) Relative expression of PLA2G6 in whole blood of proband 1 found to be reduced when compared to controls as measured by PLA2G6 mRNA read counts normalized to total transcriptome exonic reads. Mean of three controls ± SD shown in graph
The presence of the premature stop codon within the pseudo‐exon is predicted to trigger nonsense‐mediated decay. Nevertheless, the transcript containing the pseudo‐exon was detected after performing RNA sequencing, though at reduced levels when compared to controls (Figure 2c). This suggests that the nonsense‐mediated decay occurred but was incomplete. On the basis of these data, the c.2035‐926G>A variant was re‐interpreted as likely pathogenic.
PLA2G6 encodes iPLA2B, a group of VIA calcium‐independent A2 phospholipases that are essential for maintaining cell membrane integrity. Pathogenic variants in PLA2G6 underlie infantile neuroaxonal dystrophy (INAD; OMIM #256600), which follows an autosomal recessive inheritance pattern and is characterized by progressive neurodegeneration starting in the first or second year of life that includes developmental delay, progressive motor, and intellectual deterioration, and marked hypotonia (Khateeb et al., 2006).
The identification of biallelic, likely pathogenic variants in PLA2G6 led the UDN to assign a definitive diagnosis of infantile neuroaxonal dystrophy to proband 1.
3.3. Clinical phenotype of proband 2
Proband 2 is a 47‐year‐old male who first presented with action tremors at age 42 that impaired his activities of daily living. He also demonstrated subtle evidence of cognitive decline. Over the ensuing 2 years, his walking speed decreased and showed signs of inattention and generalized apathy.
Brain MRI showed diffuse abnormal confluent white matter lesions involving the cerebrum as well as abnormal signal in the brainstem and cerebellar peduncles, thinning of corpus callosum, and right inferior cerebellar encephalomalacia, presumed to be due to an old vascular insult Figure 1b).
The patient’s cognitive decline progressively worsened, and he became unable to ambulate. He then developed pneumonia, which led to a prolonged hospitalization during which he was mechanically ventilated, however, he has since recovered and is at home.
He had extensive biochemical testing as well as a leukodystrophy gene panel, exome sequencing, and mitochondrial sequencing performed prior to his enrollment in the UDN. However, these results were non‐diagnostic and the cause of his condition remained unclear.
3.4. Identification of novel upstream deletion
Proband 2 enrolled in the UDN at age 46. His evaluation included the review of prior health records, obtaining a complete medical and family history, and singleton genome sequencing, as neither of his parents were available to participate.
A 261 kb heterozygous deletion was detected during CNV analysis that mapped to chr5:125844053–126,105,052 (GRCh37/hg19), which is upstream of LMNB1. The deletion was observed to overlap with previously reported deletions upstream of LMNB1 that involved the cis‐acting regulatory elements, in particular a putative enhancer region (Figure 3). In each of these published cases, deletions overlapping this LMNB1 enhancer region produced the phenotype associated with autosomal dominant adult‐onset demyelinating leukodystrophy with later‐onset autonomic dysfunction (Giorgio et al., 2015; Mezaki et al., 2018; Nmezi et al., 2019).
FIGURE 3.
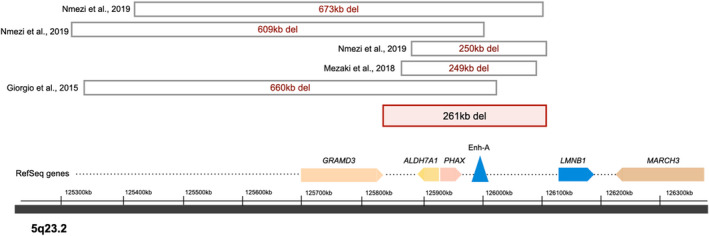
A 261 kb copy number variant was discovered at chr5:125844053‐126105052, upstream of LMNB1 as depicted. The deletion overlies a putative enhancer region for LMNB1 and five prior patients who are heterozygous for deletions overlying this area have been reported in the literature and share the autosomal dominant adult‐onset demyelinating leukodystrophy phenotype (this is based on information from the UCSC genome browser assembly GRCh37/hg19)
LMNB1 encodes lamin B, a component of the interphase nuclear lamina, which is required to maintain nuclear shape and mechanical integrity (Goldman et al., 2002). Pathogenic variants in LMNB1 have been shown to cause autosomal dominant adult‐onset demyelinating leukodystrophy (ADLD; OMIM #169500). This disorder is a slowly progressive leukoencephalopathy with an onset in the fourth and fifth decade of life that produces pyramidal and cerebellar dysfunction with symmetric demyelination of the central nervous system (Padiath et al., 2006).
Based on the identification of this heterozygous 261 kb deletion overlapping a minimum critical region in LMNB1, the UDN assigned a definitive diagnosis of adult‐onset demyelinating leukodystrophy to proband 2.
4. DISCUSSION
In this study, we identified novel noncoding variants in the genes PLA2G6 and LMNB1 that are associated with progressive neurologic disorders, namely infantile neuroaxonal dystrophy and autosomal dominant adult‐onset demyelinating leukodystrophy, in two previously undiagnosed individuals.
Proband 1 was found to be compound heterozygous for a missense variant and a pseudo‐exon splice site acceptor in PLA2G6 after performing genome sequencing followed by mRNA analysis. A very recently published study has established the potential pathogenicity of a similar deep intronic variant in PLA2G6 that alters the splice site acceptor leading to the inclusion of a pseudo‐exon in the transcript (Cavestro et al., 2021). Having identified a premature stop codon within the pseudo‐exon suggests that the transcript would undergo nonsense‐mediated decay. This is further supported by reduced read counts of the transcript containing the pseudo‐exon on RNA sequencing when compared to control samples and normalized for exonic reads (Figure 2c). However, the detection of the transcript also indicates that its nonsense‐mediated decay is incomplete.
The activation of a cryptic donor gc splice site instead of typical gt sequence has been well described in other genes (Burset et al., 2000), and in this case, was confirmed through mRNA analysis. Interestingly, two alternative PLA2G6 transcripts were found through Ensembl to use this donor gc splice site.
These sequencing results confirm the diagnosis of infantile neuroaxonal dystrophy, which is consistent with the patient’s presentation and disease progression. The findings also have prognostic value for the patient and may have future therapeutic implications as antisense oligonucleotide‐based treatments continue to be developed that are able to modulate pre‐mRNA splicing (Mezaki et al., 2021; Rigo et al., 2014).
Genome sequencing also revealed that Proband 2 was heterozygous for a deletion upstream of LMNB1 that overlaps a recently described minimum critical region of ~167 kb required for disease causation (Nmezi et al., 2019). Prior reports have shown that both duplications and deletions upstream of LMNB1 disrupt the function of the gene’s cis‐acting regulatory elements, leading to elevated mRNA expression of the gene and subsequent lamin B1 overproduction, demyelination, and the characteristic phenotype of a slowly progressive leukoencephalopathy with an onset in the fourth and fifth decade of life (Giorgio et al., 2015; Mezaki et al., 2018). The patient’s presentation closely matched the described disease course among individuals with upstream deletions as he exhibited autosomal dominant adult‐onset leukodystrophy with later‐onset dysautonomia.
Progressive neurologic disease involves genes with highly variable transcripts whose expression is tightly regulated based on developmental and tissue‐specific contexts. Alternative splicing has been shown to play a critical role for the central nervous system, which depends on a large repertoire of proteins to generate its complex neural circuits (Carvill & Mefford, 2020; Yan et al., 2015). Enhancers similarly regulate functional diversity across different classes of neuronal cells through intricate control of transcript expression (Carullo & Day, 2019). As such, noncoding variants beyond canonical splice sites can be expected to contribute significantly to the pathogenesis of neurologic disease.
Conventional stepwise strategies for genetic testing, such as chromosomal microarray followed by targeted next‐generation sequencing panels, are often costly, time‐consuming, and less likely to yield a molecular diagnosis when compared to exome sequencing. Recent longitudinal studies have further demonstrated that genome sequencing has a greater diagnostic yield than exome sequencing, as it can identify key types and regions of disease‐causing genomic variation such as copy number variants, structural variants, and intronic sequence variants (Burdick et al., 2020; Lionel et al., 2018).
The ACMG has recently released new guidelines that recommend exome or genome sequencing as a first‐ or second‐tier test for pediatric patients with congenital anomalies, developmental delays, and intellectual disabilities (Manickam et al., 2021). The authors note that the advantage of exome and genome sequencing with respect to cost‐effectiveness and clinical utility are greatest when ordered early in the diagnostic evaluation. We believe these patients with novel noncoding variants in PLA2G6 and LMNB1 illustrate the value of proceeding with genome sequencing, as well as RNA analysis when needed, to arrive at a definitive diagnosis.
5. CONCLUSION
In patients with progressive neurologic disease of unknown etiology, genome sequencing with the addition of RNA analysis where appropriate should be considered for the identification of causative noncoding pathogenic variants.
CONFLICT OF INTEREST
The authors report no conflicts of interest.
AUTHORS CONTRIBUTIONS
All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.
CONSORTIA (members of the Undiagnosed Diseases Network)
Maria T. Acosta, Margaret Adam, David R. Adams, Pankaj B. Agrawal, Mercedes E. Alejandro, Justin Alvey, Laura Amendola, Ashley Andrews, Euan A. Ashley, Mahshid S. Azamian, Carlos A. Bacino, Guney Bademci, Eva Baker, Ashok Balasubramanyam, Dustin Baldridge, Jim Bale, Michael Bamshad, Deborah Barbouth, Pinar Bayrak‐Toydemir, Anita Beck, Alan H. Beggs, Edward Behrens, Gill Bejerano, Jimmy Bennet, Beverly Berg‐Rood, Jonathan A. Bernstein, Gerard T. Berry, Anna Bican, Stephanie Bivona, Elizabeth Blue, John Bohnsack, Carsten Bonnenmann, Devon Bonner, Lorenzo Botto, Brenna Boyd, Lauren C. Briere, Elly Brokamp, Gabrielle Brown, Elizabeth A. Burke, Lindsay C. Burrage, Manish J. Butte, Peter Byers, William E. Byrd, John Carey, Olveen Carrasquillo, Ta Chen Peter Chang, Sirisak Chanprasert, Hsiao‐Tuan Chao, Gary D. Clark, Terra R. Coakley, Laurel A. Cobban, Joy D. Cogan, Matthew Coggins, F. Sessions Cole, Heather A. Colley, Cynthia M. Cooper, Heidi Cope, William J. Craigen, Andrew B. Crouse, Michael Cunningham, Precilla D'Souza, Hongzheng Dai, Surendra Dasari, Joie Davis, Jyoti G. Dayal, Matthew Deardorff, Esteban C. Dell'Angelica, Shweta U. Dhar, Katrina Dipple, Daniel Doherty, Naghmeh Dorrani, Argenia L. Doss, Emilie D. Douine, David D. Draper, Laura Duncan, Dawn Earl, David J. Eckstein, Lisa T. Emrick, Christine M. Eng, Cecilia Esteves, Marni Falk, Liliana Fernandez, Carlos Ferreira, Elizabeth L. Fieg, Laurie C. Findley, Paul G. Fisher, Brent L. Fogel, Irman Forghani, Laure Fresard, William A. Gahl, Ian Glass, Bernadette Gochuico, Rena A. Godfrey, Katie Golden‐Grant, Alica M. Goldman, Madison P. Goldrich, David B. Goldstein, Alana Grajewski, Catherine A. Groden, Irma Gutierrez, Sihoun Hahn, Rizwan Hamid, Neil A. Hanchard, Kelly Hassey, Nichole Hayes, Frances High, Anne Hing, Fuki M. Hisama, Ingrid A. Holm, Jason Hom, Martha Horike‐Pyne, Alden Huang, Yong Huang, Laryssa Huryn, Rosario Isasi, Fariha Jamal, Gail P. Jarvik, Jeffrey Jarvik, Suman Jayadev, Lefkothea Karaviti, Jennifer Kennedy, Dana Kiley, Shilpa N. Kobren, Isaac S. Kohane, Jennefer N. Kohler, Deborah Krakow, Donna M. Krasnewich, Elijah Kravets, Susan Korrick, Mary Koziura, Joel B. Krier, Seema R. Lalani, Byron Lam, Christina Lam, Grace L. LaMoure, Brendan C. Lanpher, Ian R. Lanza, Lea Latham, Kimberly LeBlanc, Brendan H. Lee, Hane Lee, Roy Levitt, Richard A. Lewis, Sharyn A. Lincoln, Pengfei Liu, Xue Zhong Liu, Nicola Longo, Sandra K. Loo, Joseph Loscalzo, Richard L. Maas, John MacDowall, Ellen F. Macnamara, Calum A. MacRae, Valerie V. Maduro, Marta M. Majcherska, Bryan C. Mak, May Christine V. Malicdan, Laura A. Mamounas, Teri A. Manolio, Rong Mao, Kenneth Maravilla, Thomas C. Markello, Ronit Marom, Gabor Marth, Beth A. Martin, Martin G. Martin, Julian A. Martínez‐Agosto, Shruti Marwaha, Jacob McCauley, Allyn McConkie‐Rosell, Colleen E. McCormack, Alexa T. McCray, Elisabeth McGee, Heather Mefford, J. Lawrence Merritt, Matthew Might, Ghayda Mirzaa, Eva Morava, Paolo M. Moretti, Deborah Mosbrook‐Davis, John J. Mulvihill, David R. Murdock, Anna Nagy, Mariko Nakano‐Okuno, Avi Nath, Stan F. Nelson, John H. Newman, Sarah K. Nicholas, Deborah Nickerson, Shirley Nieves‐Rodriguez, Donna Novacic, Devin Oglesbee, James P. Orengo, Laura Pace, Stephen C. Pak, J. Carl Pallais, Christina GS. Palmer, Jeanette C. Papp, Neil H. Parker, John A. Phillips III, Jennifer E. Posey, Lorraine Potocki, Bradley Power, Barbara N. Pusey, Aaron Quinlan, Wendy Raskind, Archana N. Raja, Deepak A. Rao, Genecee Renteria, Chloe M. Reuter, Lynette Rives, Amy K. Robertson, Lance H. Rodan, Jill A. Rosenfeld, Natalie Rosenwasser, Francis Rossignol, Maura Ruzhnikov, Ralph Sacco, Jacinda B. Sampson, Susan L. Samson, Mario Saporta, C. Ron Scott, Judy Schaechter, Timothy Schedl, Kelly Schoch, Daryl A. Scott, Vandana Shashi, Jimann Shin, Rebecca Signer, Edwin K. Silverman, Janet S. Sinsheimer, Kathy Sisco, Edward C. Smith, Kevin S. Smith, Emily Solem, Lilianna Solnica‐Krezel, Ben Solomon, Rebecca C. Spillmann, Joan M. Stoler, Jennifer A. Sullivan, Kathleen Sullivan, Angela Sun, Shirley Sutton, David A. Sweetser, Virginia Sybert, Holly K. Tabor, Amelia L. M. Tan, Queenie K.‐G. Tan, Mustafa Tekin, Fred Telischi, Willa Thorson, Audrey Thurm, Cynthia J. Tifft, Camilo Toro, Alyssa A. Tran, Brianna M. Tucker, Tiina K. Urv, Adeline Vanderver, Matt Velinder, Dave Viskochil, Tiphanie P. Vogel, Colleen E. Wahl, Stephanie Wallace, Nicole M. Walley, Chris A. Walsh, Melissa Walker, Jennifer Wambach, Jijun Wan, Lee‐kai Wang, Michael F. Wangler, Patricia A. Ward, Daniel Wegner, Mark Wener, Tara Wenger, Katherine Wesseling Perry, Monte Westerfield, Matthew T. Wheeler, Jordan Whitlock, Lynne A. Wolfe, Jeremy D. Woods, Shinya Yamamoto, John Yang, Muhammad Yousef, Diane B. Zastrow, Wadih Zein, Chunli Zhao, Stephan Zuchner.
Supporting information
Table S1
Table S2
Table S3
Figure S1
ACKNOWLEDGMENTS
We are grateful to our patients, their families, and the staff at the UDN and University of Miami for participating. We also thank Suat Fitoz from Ankara University for his help interpreting images.
Borja, N. , Bivona, S. , Peart, L. S. , Johnson, B. , Gonzalez, J. , Barbouth, D. , Moore, H. , Guo, S. , Undiagnosed Disease Network , Bademci, G. & Tekin, M. (2022). Genome sequencing reveals novel noncoding variants in PLA2G6 and LMNB1 causing progressive neurologic disease. Molecular Genetics & Genomic Medicine, 10, e1892. 10.1002/mgg3.1892
†A complete members of the Undiagnosed Diseases Network can be found in the Appendix at the end of the manuscript.
Funding informationResearch reported in this manuscript was supported by the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under award number [1U01HG010230]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Contributor Information
Mustafa Tekin, Email: mtekin@med.miami.edu.
Undiagnosed Disease Network:
Maria T. Acosta, Margaret Adam, David R. Adams, Pankaj B. Agrawal, Mercedes E. Alejandro, Justin Alvey, Laura Amendola, Ashley Andrews, Euan A. Ashley, Mahshid S. Azamian, Carlos A. Bacino, Guney Bademci, Eva Baker, Ashok Balasubramanyam, Dustin Baldridge, Jim Bale, Michael Bamshad, Deborah Barbouth, Pinar Bayrak‐Toydemir, Anita Beck, Alan H. Beggs, Edward Behrens, Gill Bejerano, Jimmy Bennet, Beverly Berg‐Rood, Jonathan A. Bernstein, Gerard T. Berry, Anna Bican, Stephanie Bivona, Elizabeth Blue, John Bohnsack, Carsten Bonnenmann, Devon Bonner, Lorenzo Botto, Brenna Boyd, Lauren C. Briere, Elly Brokamp, Gabrielle Brown, Elizabeth A. Burke, Lindsay C. Burrage, Manish J. Butte, Peter Byers, William E. Byrd, John Carey, Olveen Carrasquillo, Ta Chen Peter Chang, Sirisak Chanprasert, Hsiao‐Tuan Chao, Gary D. Clark, Terra R. Coakley, Laurel A. Cobban, Joy D. Cogan, Matthew Coggins, F. Sessions Cole, Heather A. Colley, Cynthia M. Cooper, Heidi Cope, William J. Craigen, Andrew B. Crouse, Michael Cunningham, Precilla D’Souza, Hongzheng Dai, Surendra Dasari, Joie Davis, Jyoti G. Dayal, Matthew Deardorff, Esteban C. Dell’Angelica, Shweta U. Dhar, Katrina Dipple, Daniel Doherty, Naghmeh Dorrani, Argenia L. Doss, Emilie D. Douine, David D. Draper, Laura Duncan, Dawn Earl, David J. Eckstein, Lisa T. Emrick, Christine M. Eng, Cecilia Esteves, Marni Falk, Liliana Fernandez, Carlos Ferreira, Elizabeth L. Fieg, Laurie C. Findley, Paul G. Fisher, Brent L. Fogel, Irman Forghani, Laure Fresard, William A. Gahl, Ian Glass, Bernadette Gochuico, Rena A. Godfrey, Katie Golden‐Grant, Alica M. Goldman, Madison P. Goldrich, David B. Goldstein, Alana Grajewski, Catherine A. Groden, Irma Gutierrez, Sihoun Hahn, Rizwan Hamid, Neil A. Hanchard, Kelly Hassey, Nichole Hayes, Frances High, Anne Hing, Fuki M. Hisama, Ingrid A. Holm, Jason Hom, Martha Horike‐Pyne, Alden Huang, Yong Huang, Laryssa Huryn, Rosario Isasi, Fariha Jamal, Gail P. Jarvik, Jeffrey Jarvik, Suman Jayadev, Lefkothea Karaviti, Jennifer Kennedy, Dana Kiley, Shilpa N. Kobren, Isaac S. Kohane, Jennefer N. Kohler, Deborah Krakow, Donna M. Krasnewich, Elijah Kravets, Susan Korrick, Mary Koziura, Joel B. Krier, Seema R. Lalani, Byron Lam, Christina Lam, Grace L. LaMoure, Brendan C. Lanpher, Ian R. Lanza, Lea Latham, Kimberly LeBlanc, Brendan H. Lee, Hane Lee, Roy Levitt, Richard A. Lewis, Sharyn A. Lincoln, Pengfei Liu, Xue Zhong Liu, Nicola Longo, Sandra K. Loo, Joseph Loscalzo, Richard L. Maas, John MacDowall, Ellen F. Macnamara, Calum A. MacRae, Valerie V. Maduro, Marta M. Majcherska, Bryan C. Mak, May Christine V. Malicdan, Laura A. Mamounas, Teri A. Manolio, Rong Mao, Kenneth Maravilla, Thomas C. Markello, Ronit Marom, Gabor Marth, Beth A. Martin, Martin G. Martin, Julian A. Martínez‐Agosto, Shruti Marwaha, Jacob McCauley, Allyn McConkie‐Rosell, Colleen E. McCormack, Alexa T. McCray, Elisabeth McGee, Heather Mefford, J. Lawrence Merritt, Matthew Might, Ghayda Mirzaa, Eva Morava, Paolo M. Moretti, Deborah Mosbrook‐Davis, John J. Mulvihill, David R. Murdock, Anna Nagy, Mariko Nakano‐Okuno, Avi Nath, Stan F. Nelson, John H. Newman, Sarah K. Nicholas, Deborah Nickerson, Shirley Nieves‐Rodriguez, Donna Novacic, Devin Oglesbee, James P. Orengo, Laura Pace, Stephen C. Pak, J. Carl Pallais, Christina G. S. Palmer, Jeanette C. Papp, Neil H. Parker, John A. Phillips, Jennifer E. Posey, Lorraine Potocki, Bradley Power, Barbara N. Pusey, Aaron Quinlan, Wendy Raskind, Archana N. Raja, Deepak A. Rao, Genecee Renteria, Chloe M. Reuter, Lynette Rives, Amy K. Robertson, Lance H. Rodan, Jill A. Rosenfeld, Natalie Rosenwasser, Francis Rossignol, Maura Ruzhnikov, Ralph Sacco, Jacinda B. Sampson, Susan L. Samson, Mario Saporta, C. Ron Scott, Judy Schaechter, Timothy Schedl, Kelly Schoch, Daryl A. Scott, Vandana Shashi, Jimann Shin, Rebecca Signer, Edwin K. Silverman, Janet S. Sinsheimer, Kathy Sisco, Edward C. Smith, Kevin S. Smith, Emily Solem, Lilianna Solnica‐Krezel, Ben Solomon, Rebecca C. Spillmann, Joan M. Stoler, Jennifer A. Sullivan, Kathleen Sullivan, Angela Sun, Shirley Sutton, David A. Sweetser, Virginia Sybert, Holly K. Tabor, Amelia L. M. Tan, Queenie K.‐G. Tan, Mustafa Tekin, Fred Telischi, Willa Thorson, Audrey Thurm, Cynthia J. Tifft, Camilo Toro, Alyssa A. Tran, Brianna M. Tucker, Tiina K. Urv, Adeline Vanderver, Matt Velinder, Dave Viskochil, Tiphanie P. Vogel, Colleen E. Wahl, Stephanie Wallace, Nicole M. Walley, Chris A. Walsh, Melissa Walker, Jennifer Wambach, Jijun Wan, Lee‐kai Wang, Michael F. Wangler, Patricia A. Ward, Daniel Wegner, Mark Wener, Tara Wenger, Katherine Wesseling Perry, Monte Westerfield, Matthew T. Wheeler, Jordan Whitlock, Lynne A. Wolfe, Jeremy D. Woods, Shinya Yamamoto, John Yang, Muhammad Yousef, Diane B. Zastrow, Wadih Zein, Chunli Zhao, and Stephan Zuchner
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Bademci, G. , Abad, C. , Cengiz, F. B. , Seyhan, S. , Incesulu, A. , Guo, S. , Fitoz, S. , Atli, E. I. , Gosstola, N. C. , Demir, S. , Colbert, B. M. , Seyhan, G. C. , Sineni, C. J. , Duman, D. , Gurkan, H. , Morton, C. C. , Dykxhoorn, D. M. , Walz, K. , & Tekin, M. (2020). Long‐range cis‐regulatory elements controlling GDF6 expression are essential for ear development. The Journal of clinical investigation, 130(8), 4213–4217. 10.1172/JCI136951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdick, K. J. , Cogan, J. D. , Rives, L. C. , Robertson, A. K. , Koziura, M. E. , Brokamp, E. , Duncan, L. , Hannig, V. , Pfotenhauer, J. , Vanzo, R. , Paul, M. S. , Bican, A. , Morgan, T. , Duis, J. , Newman, J. H. , Hamid, R. , Phillips, J. A., 3rd , & Network, U. D. (2020). Limitations of exome sequencing in detecting rare and undiagnosed diseases. American Journal of Medical genetics Part A, 182(6), 1400–1406. 10.1002/ajmg.a.61558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burset, M. , Seledtsov, I. A. , & Solovyev, V. V. (2000). Analysis of canonical and non‐canonical splice sites in mammalian genomes. Nucleic Acids research, 28(21), 4364–4375. 10.1093/nar/28.21.4364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carullo, N. , & Day, J. J. (2019). Genomic enhancers in brain health and disease. Genes, 10(1), 43. 10.3390/genes10010043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvill, G. L. , & Mefford, H. C. (2020). Poison exons in neurodevelopment and disease. Current Opinion in Genetics & Development, 65, 98–102. 10.1016/j.gde.2020.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavestro, C. , Panteghini, C. , Reale, C. , Nasca, A. , Fenu, S. , Salsano, E. , Chiapparini, L. , Garavaglia, B. , Pareyson, D. , Di Meo, I. , & Tiranti, V. (2021). Novel deep intronic mutation in PLA2G6 causing early‐onset Parkinson's disease with brain iron accumulation through pseudo‐exon activation. Neurogenetics, 22(4), 347–351. 10.1007/s10048-021-00667-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Córdoba, M. , Rodriguez‐Quiroga, S. A. , Vega, P. A. , Salinas, V. , Perez‐Maturo, J. , Amartino, H. , Vásquez‐Dusefante, C. , Medina, N. , González‐Morón, D. , & Kauffman, M. A. (2018). Whole exome sequencing in neurogenetic odysseys: An effective, cost‐ and time‐saving diagnostic approach. PloS one, 13(2), e0191228. 10.1371/journal.pone.0191228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo, M. A. , Banks, E. , Poplin, R. , Garimella, K. V. , Maguire, J. R. , Hartl, C. , Philippakis, A. A. , del Angel, G. , Rivas, M. A. , Hanna, M. , McKenna, A. , Fennell, T. J. , Kernytsky, A. M. , Sivachenko, A. Y. , Cibulskis, K. , Gabriel, S. B. , Altshuler, D. , & Daly, M. J. (2011). A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nature Genetics, 43(5), 491–498. 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugger, B. N. , & Dickson, D. W. (2017). Pathology of neurodegenerative diseases. Cold Spring Harbor Perspectives in Biology, 9(7), a028035. 10.1101/cshperspect.a028035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio, E. , Robyr, D. , Spielmann, M. , Ferrero, E. , Di Gregorio, E. , Imperiale, D. , Vaula, G. , Stamoulis, G. , Santoni, F. , Atzori, C. , Gasparini, L. , Ferrera, D. , Canale, C. , Guipponi, M. , Pennacchio, L. A. , Antonarakis, S. E. , Brussino, A. , & Brusco, A. (2015). A large genomic deletion leads to enhancer adoption by the lamin B1 gene: a second path to autosomal dominant adult‐onset demyelinating leukodystrophy (ADLD). Human Molecular Genetics, 24(11), 3143–3154. 10.1093/hmg/ddv065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman, R. D. , Gruenbaum, Y. , Moir, R. D. , Shumaker, D. K. , & Spann, T. P. (2002). Nuclear lamins: building blocks of nuclear architecture. Genes & Development, 16(5), 533–547. 10.1101/gad.960502 [DOI] [PubMed] [Google Scholar]
- Khateeb, S. , Flusser, H. , Ofir, R. , Shelef, I. , Narkis, G. , Vardi, G. , Shorer, Z. , Levy, R. , Galil, A. , Elbedour, K. , & Birk, O. S. (2006). PLA2G6 mutation underlies infantile neuroaxonal dystrophy. American Journal of Human Genetics, 79(5), 942–948. 10.1086/508572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler, W. , Curiel, J. , & Vanderver, A. (2018). Adulthood leukodystrophies. Nature Reviews Neurology, 14(2), 94–105. 10.1038/nrneurol.2017.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionel, A. C. , Costain, G. , Monfared, N. , Walker, S. , Reuter, M. S. , Hosseini, S. M. , Thiruvahindrapuram, B. , Merico, D. , Jobling, R. , Nalpathamkalam, T. , Pellecchia, G. , Sung, W. , Wang, Z. , Bikangaga, P. , Boelman, C. , Carter, M. T. , Cordeiro, D. , Cytrynbaum, C. , Dell, S. D. , … Marshall, C. R. (2018). Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole‐genome sequencing as a first‐tier genetic test. Genetics in Medicine, 20(4), 435–443. 10.1038/gim.2017.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manickam, K. , McClain, M. R. , Demmer, L. A. , Biswas, S. , Kearney, H. M. , Malinowski, J. , Massingham, L. J. , Miller, D. , Yu, T. W. , Hisama, F. M. , & ACMG Board of Directors . (2021). Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence‐based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genetics in Medicine, 23(11), 2029–2037. 10.1038/s41436-021-01242-6 [DOI] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. , Altshuler, D. , Gabriel, S. , Daly, M. , & DePristo, M. A. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20(9), 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezaki, N. , Miura, T. , Ogaki, K. , Eriguchi, M. , Mizuno, Y. , Komatsu, K. , Yamazaki, H. , Suetsugi, N. , Kawajiri, S. , Yamasaki, R. , Ishiguro, T. , Konno, T. , Nozaki, H. , Kasuga, K. , Okuma, Y. , Kira, J. I. , Hara, H. , Onodera, O. , & Ikeuchi, T. (2018). Duplication and deletion upstream of LMNB1 in autosomal dominant adult‐onset leukodystrophy. Neurology. Genetics, 4(6), e292. 10.1212/NXG.0000000000000292 [DOI] [PMC free article] [PubMed]
- Mezaki, N. , Miura, T. , Ogaki, K. , Eriguchi, M. , Mizuno, Y. , Komatsu, K. , Yamazaki, H. , Suetsugi, N. , Kawajiri, S. , Yamasaki, R. , Ishiguro, T. , Konno, T. , Nozaki, H. , Kasuga, K. , Okuma, Y. , Kira, J. I. , Hara, H. , Onodera, O. , Aziz, M. C. , … Carvill, G. L. (2021). Targeting Poison Exons to Treat Developmental and Epileptic Encephalopathy. Developmental neuroscience, 43(3‐4), 241–246. 10.1159/000516143 [DOI] [PubMed] [Google Scholar]
- Murdock, D. R. , Dai, H. , Burrage, L. C. , Rosenfeld, J. A. , Ketkar, S. , Müller, M. F. , Yépez, V. A. , Gagneur, J. , Liu, P. , Chen, S. , Jain, M. , Zapata, G. , Bacino, C. A. , Chao, H. T. , Moretti, P. , Craigen, W. J. , Hanchard, N. A. , Network, U. D. , & Lee, B. (2021). Transcriptome‐directed analysis for Mendelian disease diagnosis overcomes limitations of conventional genomic testing. The Journal of Clinical Investigation, 131(1), e141500. 10.1172/JCI141500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nmezi, B. , Giorgio, E. , Raininko, R. , Lehman, A. , Spielmann, M. , Koenig, M. K. , Adejumo, R. , Knight, M. , Gavrilova, R. , Alturkustani, M. , Sharma, M. , Hammond, R. , Gahl, W. A. , Toro, C. , Brusco, A. , & Padiath, Q. S. (2019). Genomic deletions upstream of lamin B1 lead to atypical autosomal dominant leukodystrophy. Neurology. Genetics, 5(1), e305. 10.1212/NXG.0000000000000305 [DOI] [PMC free article] [PubMed]
- Retterer, K. , Juusola, J. , Cho, M. T. , Vitazka, P. , Millan, F. , Gibellini, F. , Vertino‐Bell, A. , Smaoui, N. , Neidich, J. , Monaghan, K. G. , McKnight, D. , Bai, R. , Suchy, S. , Friedman, B. , Tahiliani, J. , Pineda‐Alvarez, D. , Richard, G. , Brandt, T. , Haverfield, E. , … Bale, S. (2016). Clinical application of whole‐exome sequencing across clinical indications. Genetics in Medicine, 18(7), 696–704. 10.1038/gim.2015.148 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigo, F. , Seth, P. P. , & Bennett, C. F. (2014). Antisense oligonucleotide‐based therapies for diseases caused by pre‐mRNA processing defects. Advances in Experimental Medicine and Biology, 825, 303–352. 10.1007/978-1-4939-1221-6_9 [DOI] [PubMed] [Google Scholar]
- Padiath, Q. S. , Saigoh, K. , Schiffmann, R. , Asahara, H. , Yamada, T. , Koeppen, A. , Hogan, K. , Ptácek, L. J. , & Fu, Y. H. (2006). Lamin B1 duplications cause autosomal dominant leukodystrophy. Nature genetics, 38(10), 1114–1123. 10.1038/ng1872 [DOI] [PubMed] [Google Scholar]
- Vaz‐Drago, R. , Custódio, N. , & Carmo‐Fonseca, M. (2017). Deep intronic mutations and human disease. Human Genetics, 136(9), 1093–1111 10.1007/s00439-017-1809-4 [DOI] [PubMed] [Google Scholar]
- Yan, Q. , Weyn‐Vanhentenryck, S. M. , Wu, J. , Sloan, S. A. , Zhang, Y. , Chen, K. , Wu, J. Q. , Barres, B. A. , & Zhang, C. (2015). Systematic discovery of regulated and conserved alternative exons in the mammalian brain reveals NMD modulating chromatin regulators. Proceedings of the National Academy of Sciences of the United States of America, 112(11), 3445–3450 10.1073/pnas.1502849112 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2
Table S3
Figure S1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.