

Abstract
Background
De novo variants are a common cause to rare intellectual disability syndromes, associated with low recurrence risk. However, when such variants occur pre‐zygotically in parental germ cells, the recurrence risk might be higher. Still, the recurrence risk estimates are mainly based on empirical data and the prevalence of germline mosaicism is often unknown.
Methods
To establish the prevalence of mosaicism in parents of children with intellectual disability syndromes caused by de novo variants, we performed droplet digital PCR on DNA extracted from blood (43 trios), and sperm (31 fathers).
Results
We detected low‐level mosaicism in sperm‐derived DNA but not in blood in the father of a child with Kleefstra syndrome caused by an EHMT1 variant. Additionally, we found a higher level of paternal mosaicism in sperm compared to blood in the father of a child with Gillespie syndrome caused by an ITPR1 variant.
Conclusion
By employing droplet digital PCR, we detected paternal germline mosaicism in two intellectual disability syndromes. In both cases, the mosaicism level was higher in sperm than blood, indicating that analysis of blood alone may underestimate germline mosaicism. Therefore, sperm analysis can be clinically useful to establish the recurrence risk for parents and improve genetic counselling.
Keywords: de novo variant, droplet digital PCR (ddPCR), germline mosaicism, intellectual disability, sperm (semen)
By employing droplet digital PCR, we detected paternal germline mosaicism in two intellectual disability syndromes. In both cases, the mosaicism level was higher in sperm than blood, indicating that analysis of blood alone may underestimate germline mosaicism. Therefore, sperm analysis can be clinically useful to establish the recurrence risk for parents and improve genetic counselling.
1. BACKGROUND
A human genome varies, on average, at 4–5 million sites compared to the human reference genome (Pasmant & Pacot, 2020). Massively parallel sequencing (MPS) studies have estimated the de novo rate of single nucleotide variants (SNVs), to be approximately 10−8 per generation (Kong et al., 2012; Michaelson et al., 2012; Pasmant & Pacot, 2020; Rahbari et al., 2016; Roach et al., 2010). Therefore, a newborn child is estimated to harbour, on average, 40–80 de novo SNVs, with 1–2 affecting the coding sequences with the potential to result in a disease (Breuss et al., 2020; Jónsson et al., 2018; Kong et al., 2012; Pasmant & Pacot, 2020). Such variants are usually thought to originate pre‐zygotically, in the parental germ cells, or post‐zygotically, in an early mitotic division in the developing embryo (Acuna‐Hidalgo et al., 2016). De novo variants occur three to four times more often in paternal germ cells than in maternal germ cells and are more prevalent with increasing paternal age (Acuna‐Hidalgo et al., 2016; Kong et al., 2012; Michaelson et al., 2012; Pasmant & Pacot, 2020; Rahbari et al., 2016). In the last decade, the increased use of trio exome and genome sequencing has established that de novo variants are a common cause behind genetic diseases, underlying approximately 30% of all diagnosed cases (Acuna‐Hidalgo et al., 2016; Rauch et al., 2012; Stefanski et al., 2021; Stranneheim et al., 2021).
During genetic counselling of parents to children with disease‐causing de novo variants, clinical geneticists rely on empirical data regarding the overall recurrence risk in future pregnancies. Parental investigations most often depend on genetic analysis of blood‐derived DNA. Currently, a recurrence risk of 1% is commonly used during counselling (Campbell, Stewart, et al., 2014; de Lange et al., 2019; Myers et al., 2018; Röthlisberger & Kotzot, 2007). However, the recurrence risk may be higher or lower depending on if one of the parents is a germline mosaic or not. Investigation of germline mosaicism in females is not as straightforward as in males due to the invasive procedure required when collecting oocytes (Breuss et al., 2020; Møller et al., 2019). Unless multiple children are born with the same de novo variant, germline mosaicism is generally not investigated, leaving the recurrence risk unknown for most families at risk (Wilbe et al., 2017).
Understanding underlying genetic mechanisms behind a disorder is of essence to accurately assess recurrence risk and evaluate options for prenatal or preimplantation genetic testing for future pregnancies. The increased availability of MPS technology has improved the chance to detect parental mosaicism even in blood‐derived DNA, since several MPS applications have enhanced sensitivity than older methods such as Sanger sequencing (Campbell, Yuan, et al., 2014; Cao et al., 2019; Gambin et al., 2020; Jónsson et al., 2018; Krupp et al., 2017; Rahbari et al., 2016; Wright et al., 2019). This has led to improved detection of parental mosaicism, with current data suggesting that the frequency of parental mosaicism might be much higher than 1% for some genetic disorders, for example, some genetic forms of epilepsy (Møller et al., 2019; Myers et al., 2018; Nakayama et al., 2018; Xu et al., 2015; Yang et al., 2017, 2019). Yet, few studies have looked at the occurrence of mosaicism in sperm samples. In this study, we aim to investigate the prevalence and level of germline and somatic mosaicism in different tissues in parents of children with intellectual disability syndromes caused by de novo SNVs, by analysing parental blood and paternal sperm samples with droplet digital PCR (ddPCR).
2. MATERIAL AND METHODS
2.1. Ethical compliance
This study was performed in accordance with the Declaration of Helsinki, and the local ethical board approved the study. Informed consents were obtained from each participating individual or their legal guardians prior to their inclusion in the study according to local ethical guidelines.
2.2. Study subjects
All included probands with de novo disease‐causing variants (n = 44) were initially referred for clinical diagnostic testing with trio whole‐exome sequencing (WES) or whole‐genome sequencing (WGS) at the Department of Clinical Genetics at Karolinska University Hospital, Stockholm, Sweden, between the years 2011 and 2019. DNA was extracted from peripheral blood of mothers (n = 43), fathers (n = 44), probands (n = 44) and from sperm samples of fathers (n = 31) (Figure 1). In one proband‐father duo, paternal mosaicism in blood was diagnosed by clinical routine parental testing (Figure 1).
FIGURE 1.
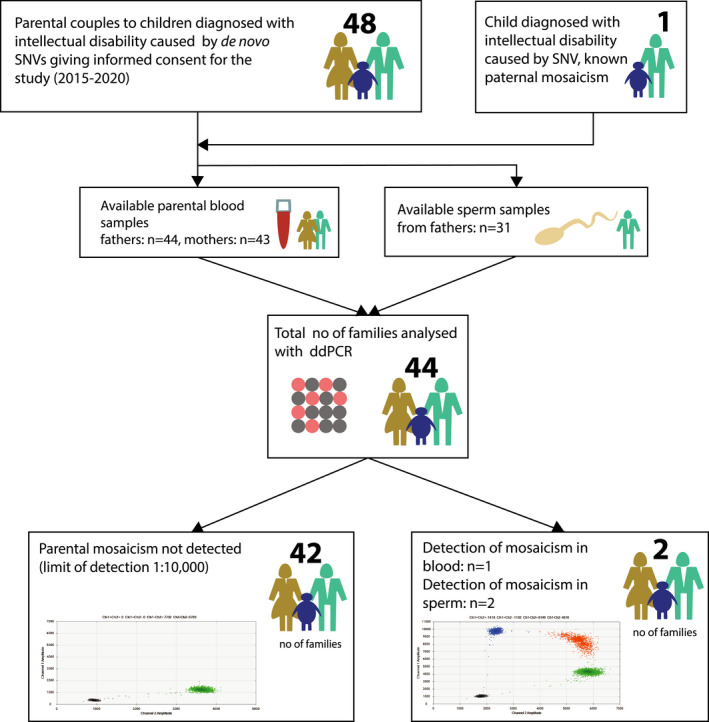
Overview of the study. We offered parents to children diagnosed with a genetic syndrome including intellectual disability due to de novo disease‐causing SNVs for participation in this cohort. All patients were initially referred for clinical diagnostic testing with trio whole‐genome sequencing/whole‐exome sequencing at the Department of Clinical Genetics of Karolinska university hospital, Stockholm, Sweden. After signing written consent, a test kit was sent home to fathers to provide sperm sample. Blood‐derived DNA was available prior to the study at the Karolinska university laboratory. Four families were excluded due to not sending in sperm sample (n = 2), lack of availability to parental blood samples and/or positive control in the family (n = 2). ddPCR, droplet digital PCR; SNV, single nucleotide variant
2.3. DNA isolation and massively parallel sequencing
As part of routine clinical diagnostics, DNA extraction from peripheral blood had been performed prior to this study at the Department of Clinical Genetics at the Karolinska University Hospital following standard procedures. Whole‐exome sequencing or WGS had been performed at Clinical Genomics at Science for Life Laboratory using either HiSeq X (Illumina Inc, San Diego, CA, USA) or NovaSeq 6000 (Illumina Inc, San Diego, CA, USA) aiming at 100x or 30x median read depth respectively (Stranneheim et al., 2021). All probands included in this cohort had been diagnosed with genetic variants classified as ‘pathogenic’ or ‘likely pathogenic’ according to the American College of Medical Genetics and Genomics (ACMG) guidelines for interpretation of genetic variants (Richards et al., 2015).
2.4. DNA isolation from sperm samples
Sperm samples (n = 31) were collected in 15 ml Falcon tubes between the years 2017 and 2020 and were stored at −18 °C until DNA extraction. DNA was isolated using the Qiagen Mini Amp Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions for tissue isolation. The concentrations were determined by Qubit™ dsDNA Broad Range Assay Kit in Qubit™ 3.0 fluorometer (Life Technologies, Carlsbad, CA, USA).
2.5. Droplet digital PCR
For each mutation in this cohort, a unique TaqMan® assay was designed by and ordered from ThermoFisher Scientific (Waltham, Massachusetts, USA). Probes detecting the mutant allele were labelled with FAM fluorophore and wild‐type allele with VIC fluorophore. Sixty‐six nanograms of genomic DNA was mixed with ddPCR Supermix for Probes (No dUTP) (Bio‐Rad, Hercules, CA, USA) according to the manufacturer's protocol. The droplets were generated using QX200 Droplet Generator (Bio‐Rad, Hercules, CA, USA) according to the manufacturer's instructions. After the amplification was performed in CFX96 Real‐Time Thermal Cycler (Bio‐Rad, Hercules, CA, USA) according to the manufacturer's protocol with adjustments of annealing temperature (58.8–60.5°C) for each probe, the droplets were scanned using QX200 QuantaSoft Droplet Reader (Bio‐Rad, Hercules, CA, USA). QuantaSoft Analysis Pro Software (Bio‐Rad, Hercules, CA, USA) was used to analyse ddPCR data through a Poisson distribution. After analysis, data were manually inspected and when needed, the droplets were further grouped using the manual selection tools in the software. Each sample was run in triplicates. When needed, the number of replicates were increased up to 36 wells.
3. RESULTS
In total, we analysed 44 unique variants with ddPCR located in 32 different genes (Table 1). Six genes (ARID1B, ANKRD11, GRIN2B, SYNGAP, PURA, EFTUD2) were mutated in more than one patient in the cohort (Figure 2a). ARID1B was the most commonly mutated gene (n = 5), followed by ANKRD11 and GRIN2B (n = 4) (Figure 2a). Missense variants accounted for the majority of variants (37%) (Figure 2b). The mean maternal age at conception for participating mothers was 33.2 years and ranged from 21 to 45 years of age at conception (Figure 2c). The mean paternal age for participating fathers was 37.7 years and ranged from 25 to 59 years of age at conception (Figure 2c). In families with more than one child, no recurrence of disease was reported.
TABLE 1.
All analysed variants including clinical diagnosis and parental ages at conception
| Gene | Diagnosis | Reference‐ID | Variant | Amino acid change | Paternal age | Maternal age |
|---|---|---|---|---|---|---|
| ACTG1 | Baraitser–Winter syndrome 2 | NM_001199954.1 | c.439C > T | p.Arg147Cys | 34 | 32 |
| ANKRD11 | KBG syndrome | NM_013275.5 | c.5663del | p.Ala1888Glufs*75 | 36 | 35 |
| ANKRD11 | KBG syndrome | NM_001256182.1 | c.6513dup | p.Gly2172Argfs*14 | 25 | 21 |
| ANKRD11 | KBG syndrome | NM_013275.5 | c.3770_3771del | p.Lys1257ARGfs*25 | 39 | 35 |
| ANKRD11 | KBG syndrome | NM_013275.5 | c.1903_1907del | p.Lys635Glnfs*26 | 41 | 38 |
| ARID1B | Coffin–Siris syndrome 1 | NM_020732.3 | c.1876C > T | p.Gln626* | 26 | 26 |
| ARID1B | Coffin–Siris syndrome 1 | NM_020732.3 | c.5404C > T | p.Arg1802* | 32 | 32 |
| ARID1B | Coffin–Siris syndrome 1 | NM_020732.3 | c.5023C > T | p.Gln1675* | 39 | 37 |
| ARID1B | Coffin–Siris syndrome 1 | NM_020732.3 | c.4466_4466dup | p.Tyr1490Leufs20* | 38 | 33 |
| ARID1B | Coffin–Siris syndrome 1 | NM_020732.3 | c.5267_5270del | p.Glu1756Alafs*9 | 39 | 36 |
| ASXL1 | Bohring–Opitz syndrome | NM_015338.5 | c.4189_4190del | p.Gly1397Serfs*26 | 52 | 36 |
| EFTUD2 | Mandibulofacial dysostosis | NM_004247 | c.1705C > T | p.Arg569* | 53 | 42 |
| EFTUD2 | Mandibulofacial dysostosis | NM_001258353.1 | c.427_427del | p.Thr143Hisfs*7 | 46 | 35 |
| EHMT1 | Kleefstra syndrome | NM_024757.4 | c.2986C > T | p.Gln996* | 33 | 30 |
| FOXP1 | Mental retardation with language impairment with or without autistic features | NM_032682.5 | c.1062G > T | p. Gln354His | 31 | 33 |
| GRIN2B | Mental retardation, autosomal dominant 6 | NM_000834.3 | c.2539C > T | p.Arg847* | 38 | 33 |
| GRIN2B | Mental retardation, autosomal dominant 6 | NM_000834.3 | c.2189 T > C | p.Ile730Thr | 26 | 27 |
| GRIN2B | Mental retardation, autosomal dominant 6 | NM_000834.3 | c.2086C > A | p.Arg696Ser | 44 | 36 |
| GRIN2B | Mental retardation, autosomal dominant 6 | NM_000834.3 | c.1652 T > C | p.Leu551Ser | 28 | 30 |
| HDAC8 | Cornelia de Lange syndrome 5 | NM_018486.2 | c.913G > A | p.Gly305Ser | 38 | 29 |
| IQSEC2 | Mental retardation, X‐linked 1/78 | NM_001111125 | c.2984G > A | p.Arg995Gln | 41 | 39 |
| ITPR1 | Gillespie syndrome | NM_002222.5 | c.7642_7644del | p.Lys2548del | 44 | 37 |
| KAT6B | SBBYSS syndrome | NM_012330.3 | c.3147G > A | p= | 37 | 34 |
| KCNQ2 | Developmental and epileptic encephalopathy 7 | NM_172107.3 | c.1057C > G | p.Arg353Gly | 37 | 30 |
| KMT2D | Kabuki syndrome | NM_003482.3 | c.8141delT | p.Val2714Glyfs*19 | 33 | 34 |
| MECP2 | Rett syndrome | NM_004992 | c.808C > T | p.Arg270* | 45 | 33 |
| MYH3 | Arthrogryposis, distal, type 2B (Sheldon‐Hall) | NM_002470.3 | c.4256A > T | p.Lys1419Met | 37 | 32 |
| NF1A | Brain malformations and urinary defects | NM_005595 | c.946 + 1G > A | 35 | 33 | |
| PHF6 | Borjeson–Forssman–Lehmann syndrome | NM_001015877 | c.966C > A | p.Tyr322* | 41 | 38 |
| POGZ | White–Sutton syndrome | NM_207171.2 | c.3541C > T | p.His1181Tyr | 59 | 32 |
| PURA | Mental retardation, autosomal dominant 31 | NM_005859.4 | c.692 T > G | p.Phe231Cys | 44 | 38 |
| PURA | Mental retardation, autosomal dominant 31 | NM_005859.4 | c.487C > T | Gln163* | 52 | 42 |
| RIT1 | Noonan syndrome 8 | NM_006912.5 | c.270G > C | p.Met90Ile | 35 | 28 |
| SATB2 | Glass syndrome | NM_015265.3 | c.1148_1148del | p.Ala383GLufs*30 | 32 | 30 |
| SRCAP | Floating–Harbour syndrome | NM_006662.2 | c.7330C > T | p.Arg2444* | 33 | 35 |
| STXBP1 | Developmental and epileptic encephalopathy 4 | NM_003165.3 | c.1439C > T | p.Pro480Leu | 40 | 32 |
| SYNGAP1 | Mental retardation, autosomal dominant 5 | NM_006772.2 | c.3415C > T | p.Gln1139* | 30 | 30 |
| SYNGAP1 | Mental retardation, autosomal dominant 5 | NM_006772.2 | c.1783_1783del | p.Leu595Cys | 46 | 45 |
| TCOF1 | Treacher–Collins syndrome | NM_001008656.2 | c.11622G > A | p.Trp541* | 37 | 30 |
| TUBB3 | Cortical dysplasia type 1 | NM_006086.2 | c.785G > A | p.Arg262His | 30 | 30 |
| TUBG1 | Cortical dysplasia type 4 | NM_001070.4 | c.776C > T | p.Ser259Leu | 35 | 26 |
| USP9X | Mental retardation, X‐linked 99 | NM_001039590 | c.2554C > T | p.Arg852* | 31 | 31 |
| WAC | Desanto–Shinawi syndrome | NM_016628 | c.1537C > T | p.Arg468* | 34 | 33 |
| ZEB2 | Mowat–Wilson syndrome | NM_014795 | c.1106del | p.Leu369* | 35 | 34 |
| ZIC2 | Holoprosencephaly 5 | NM_007129.3 | c.1225C > T | p.Arg409Trp | 36 | 34 |
FIGURE 2.
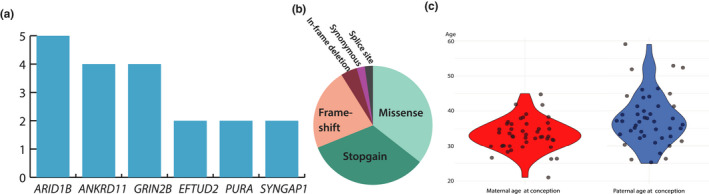
Summary of cohort data. Genes mutated more than one time in the cohort (a). ARID1B was the most prevalent mutated gene in our cohort (n = 5), followed by ANKRD11 and GRIN2B (n = 4) other genes appearing more than one time were EFTUD2, PURA and SYNGAP1. Variant type distribution (b). Missense variants accounted for the majority (37%), followed by stopgain variants (33%), frameshift variants (23%). Less prevalent were inframe‐deletions (4%), synonymous and splice site variants (2%). Violin plot of paternal and maternal ages at conception of all analysed parents (c). The mean maternal age at conception was 33.2 years and ranged from 21 to 45 years of age. The mean paternal age was 37.7 years and ranged from 25 to 59 years of age at conception. There was one outlier; 59 years of age at conception of one father
In the 30 sperm samples without previously detected mosaicism in blood, ddPCR discovered germline mosaicism in one healthy father (Figure 1, Figure 3h). His child presented with intellectual disability, autism, seizures, hypoplastic midface, hypertelorism, synophrys, sparse teeth and was diagnosed with Kleefstra syndrome (MIM # 610253) due to a missense variant in the EHMT1 gene (NM_024757.4: c.2986C > T, p.[Gln996*]). We detected the EMHT1 variant in the father at a level of 1.1% in sperm (Table 2, Figure 3g,h, Figure 4a). To ensure higher sensitivity, the ddPCR assay for blood‐derived paternal DNA was performed on approximately 600,000 droplets with an estimated sensitivity of 1:100,000. Even though we detected 105 positive signals out of approximately 600,000 droplets, we concluded that these were false positive as they did not show any statistical significance compared to negative control.
FIGURE 3.
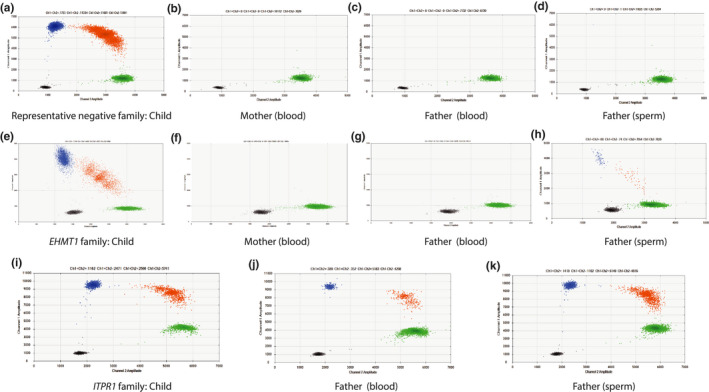
2D plot of ddPCR data. Blue cluster (FAM) shows signals from mutant allele, green signals (VIC) from reference allele and red signals from both mutant and reference alleles. Black cluster represents the wells where no amplification signal was detected. Axes show channel 1 (y) and channel 2 (x) amplitudes. The index patient is used as a positive control since he/she is a known heterozygous carrier of the variant. (a)–(d) shows representative negative ddPCR data from a family with blood‐derived DNA from the index patient/child (a), mother (b), father (c) and paternal sperm‐derived DNA (d). In this representative family, a ddPCR assay for the variant c.2539C > T in the GRIN2B gene was designed. The plots show that only the child in this assay displays mutant signals. (e)–(h) shows 2D plots of ddPCR data in the EHMT1 family with blood‐derived DNA from the index patient/child (e), mother (f), father (g) and paternal sperm‐derived DNA (h) in the index patient (e) and paternal sperm (h) FAM‐positive droplets (blue and red) were detected, signalling mutant alleles in those two individuals. (i)–(k) shows 2D plot of ddPCR data in the ITPR1 family with blood‐derived DNA from the index patient/child (i), father (j) and paternal sperm‐derived DNA (k) FAM‐positive droplets (blue and red) were seen in all three samples. Maternal DNA was not available at the time of the ddPCR assay
TABLE 2.
Summary of ddPCR data in families where parental mosaicism was detected
| ddPCR results | EHMT1 family | ITPR1 family |
|---|---|---|
| Gene | EHMT1 | ITPR1 |
| Variant | c.2986C > T | c.7642_7644del |
| Amino acid change | p.Gln996* | p.Lys2548del |
| Transcript | NM_024757.4 | NM_002222.5 |
| ACMG Class | Likely pathogenic (4) | Pathogenic (5) |
| Parent with findings | Father | Father |
| VAF (%) | ||
| Blood | – | 9.28 |
| Sperm | 1.11 | 20.24 |
| Number of droplets | ||
| Father (blood), in total | 586,705 | 35,553 |
| VIC positive | 325,202 | 19,323 |
| FAM positive | – | 2741 |
| Father (sperm), in total | 284,766 | 37,380 |
| VIC positive | 145,311 | 20,416 |
| FAM positive | 2202 | 6789 |
| Reached sensitivity | 1:100,000 | 1:10,000 |
Note: Variants were classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines for interpretation of genetic variants.
Abbreviation: VAF, variant allelic fraction.
FIGURE 4.
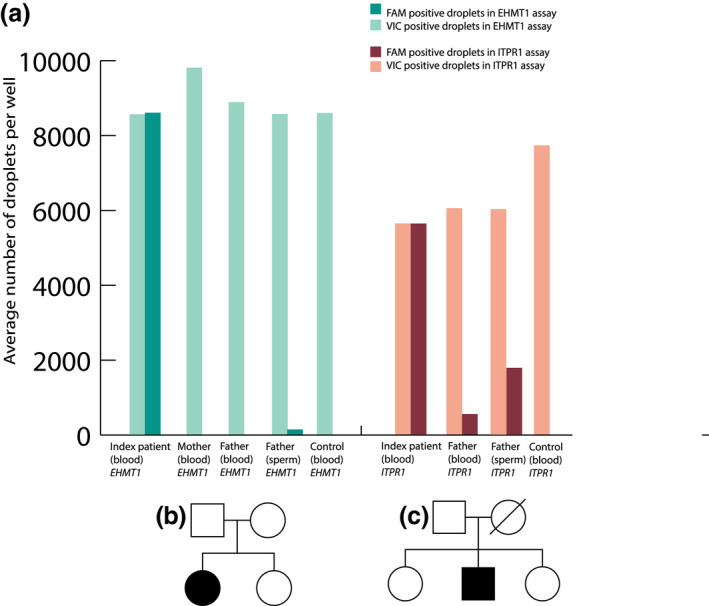
Summary of results in families where parental mosaicism was detected (a). For the EHMT1 family (in green), the heterozygote child (index patient) had a total of 49.9% of mutation‐positive droplets (dark green; FAM positive) and 50.1% wild‐type‐positive droplets (light green; VIC positive). The blood samples from the mother and father were negative, with 0% mutated droplets identified. Sperm sample from the father identified 1.1% mutated droplets (dark green). For the ITPR1 family (in red), the index patient had a total of 50.0% of mutation‐positive droplets (dark red; FAM positive) and 50.0% wild‐type‐positive droplets (light red; VIC positive). Blood and sperm from the father had 9.3% and 20.2%, respectively, positive droplets. Since the mother had passed away, DNA was not available in the ITPR1 family. All samples were run at the least in triplicates and each assay with positive findings was replicated with similar results. Pedigrees of EHMT1 family (b) and ITPR1 family (c)
In addition, a father with previously known blood mosaicism of an ITPR1 pathogenic variant (NM_002222.5: c.7642_7644del, p.[Lys2548del]) was included in this study for the investigation of mosaicism in sperm‐derived DNA using ddPCR (Figure 3k, Figure 4a). We detected 20.3% mosaicism of the variant in sperm compared to 9.3% in blood (Table 2). His child presented with iris hypoplasia, ataxia, moderate intellectual disability, autism and was diagnosed with Gillespie syndrome (MIM # 206700). Even though we found varying levels of mosaicism in the investigated tissues, he is healthy and has two other healthy children (Figure 4c).
In total, we detected germline mosaicism in approximately 3% (1 of 30) of the fathers whose child received a molecular diagnosis of an intellectual disability syndrome caused by a de novo SNV without previous findings in parents. We did not detect any somatic mosaicism in any maternal blood samples (n = 43), nor in paternal blood samples (n = 43) in the parents with previously unknown mosaic status. Additionally, in a father with previously known mosaicism in blood, we detected a higher level of mosaicism in sperm (20%) compared to blood (9%).
4. DISCUSSION
Mosaicism in sperm has not been widely studied and comparison of mosaicism levels between germline and somatic cells are limited to few studies (Table 3). In our relatively large cohort, we detected paternal mosaicism in approximately 3% of tested sperm samples and observed a consistently higher level of mosaicism in sperm compared to blood.
TABLE 3.
Cohorts of parental mosaicism
| Study | Number of families/trios analysed for parental mosaicism | Diagnosis | Tissue analysed | Ratio of families with parental mosaicism |
|---|---|---|---|---|
| Campbell, Yuan et al. (2014) | 100 | Mixed deletion syndromes | Blood | 4% |
| Xu et al. (2015) | 174 | Dravet syndrome |
Blood Hair follicles (2), buccal mucosa (1), saliva (2), urine (2), sperm (1) |
8.6% |
| Zillhardt et al. (2016) | 18 | Malformations of cortical development | Blood | 22.2% |
| Yang et al. (2017) | 112 (blood)/56 (sperm) | Dravet syndrome | Blood, sperm | 17.86% (in sperm) |
| Krupp et al. (2017) | 2264 | Autism spectrum disorder | Blood | 6.8% |
| Myers et al. (2018) | 120 | Epileptic encephalopaties | Blood, saliva | 8.3% |
| Hu (2019) | 19 | Holoprosencephaly | Blood | 26% |
| Legrand et al. (2019) | 36 | COL3A1‐related vascular Ehlers–Danlos syndrome | Blood | 2–3% |
| Møller et al. (2019) | 75 | Epilepsia |
Blood In 26 parents: additional oral mucosa and urothelium |
6.6% |
| de Lange et al. (2019) | 80 | Dravet syndrome | Blood | 6.25% |
| Yang et al. (2019) | 80 (blood)/51 (sperm) | ATP1A3‐related alternating hemiplegia | Blood, sperm | 4% (in sperm) |
| Cao et al. (2019) |
2373 (trios) 9619 (proband‐only) |
Mixed | Blood | 0.3% |
| Wright et al. (2019) | 420 | Mixed developmental disorders | Blood, saliva | 0.5% |
| Gambin et al. (2020) |
768 (BHCMG cohort) 46 (BG cohort) |
Mixed |
Blood In three parents: additional hair follicles, saliva, buccal cells, urine |
2% (BHCMG 16/768) 23% (BG cohort 11/46) |
| Breuss et al. (2020) | 14 | Autism spectrum disorder | Sperm | 21.4% |
| Shu et al. (2021) | 237 | Mixed neurodevelopmental diseases | Blood | 3.0% |
| Our study (2021) | 43 (blood)/31 (sperm) | Mixed ID syndromes | Blood, sperm | 3% (in sperm) |
Abbreviation: ID, intellectual disability.
Here, we report two families with germline mosaicism. One index patient has Kleefstra syndrome, caused by a paternally inherited pathogenic missense variant (c.2986C > T, p.[Gln996*]) in the EHMT1 gene. A mosaic splice site EHMT1 variant has previously been reported in a mother of a child with Kleefstra syndrome (Rump et al., 2013). To the best of our knowledge, this is the first reported paternally inherited mosaic SNV in Kleefstra syndrome (de Boer et al., 2018; Hervé et al., 2015; Rump et al., 2013; Willemsen et al., 2011). Both paternal and maternal mosaic structural aberrations have been described in five families with Kleefstra syndrome caused by a 9q34.3 deletion (de Boer et al., 2018; Hervé et al., 2015; Willemsen et al., 2011). In one study, autism‐spectrum disorder was reported in three mosaic parents (de Boer et al., 2018). Mosaic levels ranged from 40% to 80% in the parents, and mosaicism was detected in other tissues than the gonads. Unlike the previously reported cases, no autistic features are noted in the father of the EHMT1 family in our cohort, which can possibly be explained by low‐level mosaicism (1%) confined to the germ cells.
The ITPR1 variant (c.7642_7644del, p.[Lys2548del]) in the second family has previously been reported as de novo in six other patients with iris hypoplasia and cerebellar ataxia, some of the patients had global delay or mild to moderate intellectual disability (Farwell et al., 2015; Gerber et al., 2016; McEntagart et al., 2016; Synofzik et al., 2018). However, this is the first time this variant is described as mosaic in a father of a child affected by Gillespie syndrome. The father is healthy and has a normal level of intellectual function.
Considering the level of sperm mosaicism in the father with the ITPR1 variant (20%), compared to the father with the EHMT1 variant (1%), the recurrence risk appears higher in the ITPR1 family. Yet, the recurrence risk to future offspring depends on several factors, such as the timing of mutation and mutation type (Breuss et al., 2020, 2021; Jónsson et al., 2018). Breuss et al. (2021) divide sperm mosaicism into different types depending on timing of the mutations, each associated with a different recurrence risk. According to this classification, we hypothesize that the ITPR1 variant in the father in our cohort is a type IIIa mutation, since it was found in blood as well sperm and therefore might have originated during embryogenesis. We hypothesize that the EHMT1 variant in the father in our cohort is possibly a type IIb mutation; a mutation with relatively low allelic fraction compared to type III mutations but with proliferation advantages in the spermatocyte.
Previous cohort studies of de novo variants have reported varying prevalence of parental mosaicism (Table 3) (Breuss et al., 2020; Campbell, Yuan, et al., 2014; de Lange et al., 2019; Hu et al., 2019; Jónsson et al., 2018; Legrand et al., 2019; Møller et al., 2019; Myers et al., 2018; Nakayama et al., 2018; Xu et al., 2015; Yang et al., 2017, 2019). In contrast to parental mosaicism studies on peripheral blood, sperm has only been investigated in three cohorts previously (Breuss et al., 2020; Yang et al., 2017, 2019). Those studies found a higher prevalence of paternal mosaicism in sperm than we did. All the probands reported by Yang et al. (2017) had Dravet syndrome caused by pathogenic SCN1A variants and 10 of the 56 fathers carried the disease‐causing variant in sperm (18%). The study for ATP1A3 mosaicism found 4% (2/51) mosaicism in the analysed sperm samples (Yang et al., 2019). We investigated sperm in a genetically heterogeneous cohort and found one parent (3%) with isolated germline mosaicism in sperm. Susceptibility to mutagenesis during the spermatogenesis as well as timing of mutations during paternal embryogenesis may affect the abundance of mutations in germline and somatic tissues, and the overall recurrence risk (Breuss et al., 2020, 2021; Jónsson et al., 2018). Larger studies of sperm mosaicism in different genetic disorders need to be performed to delineate the recurrence risk in specific conditions.
Varying levels of mosaicism between diverse tissues have also been observed in other studies (Breuss et al., 2020; Møller et al., 2019; Pasmant & Pacot, 2020; Wilbe et al., 2017; Xu et al., 2015; Yang et al., 2017). In line with our observation, Yang et al. (2017) also found that the level of mosaicism in sperm of the fathers were higher than in their blood. Further support for differences in mosaicism levels between tissues is provided in a study by Moller et al., that used targeted MPS with a minimum read depth of 850x on 75 parental couples to patients with epilepsy due to alleged de novo variants (Møller et al., 2019). Similar to our findings of higher allelic fractions in sperm than in blood, these studies support the notion that results from blood cannot be automatically translated to recurrence risk.
Our findings also demonstrate the importance of using sensitive methods in routine genetic parental testing, in agreement with several previous studies (Campbell, Yuan, et al., 2014; de Lange et al., 2019; Liu et al., 2020; Wilbe et al., 2017; Xu et al., 2015; Yang et al., 2017). Here we used TaqMan assays specifically designed and optimized for ddPCR analysis of unique targets in each family. Although this approach may seem costly and laborious, it gives a more accurate level of parental mosaicism. However, with reduced cost of MPS, deep sequencing might be an attractive option instead of designing family specific assays. Still, even though WES/WGS might be used for identification of parental mosaicism in trio analysis, there is a risk that mosaic variants in parents may hinder the identification of potentially disease‐causing variants in the children, as those variants might be filtered out due to their detection in one of the parental blood samples. Therefore, it could be of importance to analyse the MPS data as a singleton or one parent at‐a‐time approach.
Due to technical difficulties of obtaining oocytes for analysis, we are limited to study germline mosaicism in males only, making the actual rate of germline mosaicism and true recurrence risk difficult to conclude. Nevertheless, sperm analysis adds important information regarding parental mosaicism since 80% of de novo variants arise on the paternal haplotype (Acuna‐Hidalgo et al., 2016; Breuss et al., 2021; Kong et al., 2012; Rahbari et al., 2016). For future studies, it would also be interesting to collect samples from tissues representing all the three different germ layers during embryonic development, in order to understand when the mutational event has occurred and further deciphering the mechanisms of mosaicism.
In conclusion, we report upon two unaffected fathers with germline mosaicism, showing higher level of mosaicism in sperm than in blood, which is important to consider for genetic counselling. Consequently, mosaic variants in parents might be missed by routine clinical blood‐derived DNA analysis. Therefore, after genetic counselling, sperm analysis in fathers can be offered as part of the routine testing of parents to children diagnosed with de novo variants causing rare diseases.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Ann Nordgren and Fulya Taylan designed the study. Sofia Frisk, Helena Malmgren, Kristina Lagerstedt‐Robinson, Alexandra Wachtmeister and Nina Jäntti contributed to data collection. Sofia Frisk, Nina Jäntti and Alexandra Wachtmeister did the laboratory work. Sofia Frisk and Fulya Taylan prepared the Figures. Sofia Frisk, Bianca Tesi, Anna Lindstrand, Tobias Laurell, Ann Nordgren and Fulya Taylan contributed to data interpretation. Sofia Frisk, Ann Nordgren and Fulya Taylan wrote the manuscript. All authors revised the manuscript and approved the final version.
ETHICS APPROVAL
The local Ethics Committee at the Karolinska Institute approved the study (Dnr 2012–2106‐31/4), which followed the tenets of the Declaration of Helsinki. Informed consents were obtained from each participating individual or their legal guardians prior to their inclusion in the study.
PARENTAL/GUARDIAN CONSENT OBTAINED
Yes.
ACKNOWLEDGMENTS
This work was supported by grants from the Swedish Research Council, the Region Stockholm (combined residency and PhD training programme), Karolinska Institutet, the Swedish Brain Foundation, the Swedish Rare Diseases Research Foundation (Sällsyntafonden) and The Hållsten Research Foundation.
Frisk, S. , Wachtmeister, A. , Laurell, T. , Lindstrand, A. , Jäntti, N. , Malmgren, H. , Lagerstedt‐Robinson, K. , Tesi, B. , Taylan, F. & Nordgren, A. (2022). Detection of germline mosaicism in fathers of children with intellectual disability syndromes caused by de novo variants. Molecular Genetics & Genomic Medicine, 10, e1880. 10.1002/mgg3.1880
Fulya Taylan and Ann Nordgren shared senior authorships.
DATA AVAILABILITY STATEMENT
All ddPCR data and sequences for Taqman Assays are available upon reasonable request.
REFERENCES
- Acuna‐Hidalgo, R. , Veltman, J. A. , & Hoischen, A. (2016). New insights into the generation and role of de novo mutations in health and disease. Genome Biology, 17(1), 241. 10.1186/s13059-016-1110-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuss, M. W. , Antaki, D. , George, R. D. , Kleiber, M. , James, K. N. , Ball, L. L. , Hong, O. , Mitra, I. , Yang, X. , Wirth, S. A. , Gu, J. , Garcia, C. A. B. , Gujral, M. , Brandler, W. M. , Musaev, D. , Nguyen, A. , McEvoy‐Venneri, J. , Knox, R. , Sticca, E. , … Gleeson, J. G. (2020). Autism risk in offspring can be assessed through quantification of male sperm mosaicism. Nature Medicine, 26(1), 143–150. 10.1038/s41591-019-0711-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuss, M. W. , Yang, X. , & Gleeson, J. G. (2021). Sperm mosaicism: Implications for genomic diversity and disease. Trends in Genetics, 37, 890–902. 10.1016/j.tig.2021.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, I. M. , Stewart, J. R. , James, R. A. , Lupski, J. R. , Stankiewicz, P. , Olofsson, P. , & Shaw, C. A. (2014). Parent of origin, mosaicism, and recurrence risk: Probabilistic modeling explains the broken symmetry of transmission genetics. American Journal of Human Genetics, 95(4), 345–359. 10.1016/j.ajhg.2014.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, I. M. , Yuan, B. , Robberecht, C. , Pfundt, R. , Szafranski, P. , McEntagart, M. E. , Nagamani, S. C. , Erez, A. , Bartnik, M. , Wiśniowiecka‐Kowalnik, B. , Plunkett, K. S. , Pursley, A. N. , Kang, S. H. , Bi, W. , Lalani, S. R. , Bacino, C. A. , Vast, M. , Marks, K. , Patton, M. , … Stankiewicz, P. (2014). Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. American Journal of Human Genetics, 95(2), 173–182. 10.1016/j.ajhg.2014.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, Y. , Tokita, M. J. , Chen, E. S. , Ghosh, R. , Chen, T. , Feng, Y. , Gorman, E. , Gibellini, F. , Ward, P. A. , Braxton, A. , Wang, X. , Meng, L. , Xiao, R. , Bi, W. , Xia, F. , Eng, C. M. , Yang, Y. , Gambin, T. , Shaw, C. , … Stankiewicz, P. (2019). A clinical survey of mosaic single nucleotide variants in disease‐causing genes detected by exome sequencing. Genome Medicine, 11(1), 48. 10.1186/s13073-019-0658-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer, A. , Vermeulen, K. , Egger, J. I. M. , Janzing, J. G. E. , de Leeuw, N. , Veenstra‐Knol, H. E. , den Hollander, N. , van Bokhoven, H. , Staal, W. , & Kleefstra, T. (2018). EHMT1 mosaicism in apparently unaffected parents is associated with autism spectrum disorder and neurocognitive dysfunction. Molecular Autism, 9, 5. 10.1186/s13229-018-0193-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange, I. M. , Koudijs, M. J. , van 't Slot, R. , Sonsma, A. C. M. , Mulder, F. , Carbo, E. C. , van Kempen, M. , Nijman, I. J. , Ernst, R. F. , Savelberg, S. M. C. , Knoers, N. V. A. M. , Brilstra, E. H. , & Koeleman, B. P. C. (2019). Assessment of parental mosaicism in SCN1A‐related epilepsy by single‐molecule molecular inversion probes and next‐generation sequencing. Journal of Medical Genetics, 56(2), 75–80. 10.1136/jmedgenet-2018-105672 [DOI] [PubMed] [Google Scholar]
- Farwell, K. D. , Shahmirzadi, L. , el‐Khechen, D. , Powis, Z. , Chao, E. C. , Tippin Davis, B. , Baxter, R. M. , Zeng, W. , Mroske, C. , Parra, M. C. , Gandomi, S. K. , Lu, I. , Li, X. , Lu, H. , Lu, H. M. , Salvador, D. , Ruble, D. , Lao, M. , Fischbach, S. , … Tang, S. (2015). Enhanced utility of family‐centered diagnostic exome sequencing with inheritance model‐based analysis: Results from 500 unselected families with undiagnosed genetic conditions. Genetics in Medicine, 17(7), 578–586. 10.1038/gim.2014.154 [DOI] [PubMed] [Google Scholar]
- Gambin, T. , Liu, Q. , Karolak, J. A. , Grochowski, C. M. , Xie, N. G. , Wu, L. R. , Yan, Y. H. , Cao, Y. , Coban Akdemir, Z. H. , Wilson, T. A. , Jhangiani, S. N. , Chen, E. , Eng, C. M. , Muzny, D. , Posey, J. E. , Yang, Y. , Zhang, D. Y. , Shaw, C. , Liu, P. , … Stankiewicz, P. (2020). Low‐level parental somatic mosaic SNVs in exomes from a large cohort of trios with diverse suspected mendelian conditions. Genetics in Medicine, 22, 1768–1776. 10.1038/s41436-020-0897-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber, S. , Alzayady, K. J. , Burglen, L. , Brémond‐Gignac, D. , Marchesin, V. , Roche, O. , Rio, M. , Funalot, B. , Calmon, R. , Durr, A. , Gil‐da‐Silva‐Lopes, V. L. , Ribeiro Bittar, M. F. , Orssaud, C. , Héron, B. , Ayoub, E. , Berquin, P. , Bahi‐Buisson, N. , Bole, C. , Masson, C. , … Fares Taie, L. (2016). Recessive and dominant de novo ITPR1 mutations cause Gillespie syndrome. American Journal of Human Genetics, 98(5), 971–980. 10.1016/j.ajhg.2016.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervé, B. , Roume, J. , Cognard, S. , Fauvert, D. , Molina‐Gomes, D. , & Vialard, F. (2015). Low‐level mosaicism of a de novo derivative chromosome 9 from a t(5,9)(q35.1;q34.3) has a major phenotypic impact. European Journal of Medical Genetics, 58(6–7), 346–350. 10.1016/j.ejmg.2015.04.005 [DOI] [PubMed] [Google Scholar]
- Hu, P. , Martinez, A. F. , Kruszka, P. , Berger, S. , Roessler, E. , & Muenke, M. (2019). Low‐level parental mosaicism affects the recurrence risk of holoprosencephaly. Genetics in Medicine, 21, 1015–1020. 10.1038/s41436-018-0261-8 [DOI] [PubMed] [Google Scholar]
- Jónsson, H. , Sulem, P. , Arnadottir, G. A. , Pálsson, G. , Eggertsson, H. P. , Kristmundsdottir, S. , Zink, F. , Kehr, B. , Hjorleifsson, K. E. , Jensson, B. Ö. , Jonsdottir, I. , Marelsson, S. E. , Gudjonsson, S. A. , Gylfason, A. , Jonasdottir, A. , Jonasdottir, A. , Stacey, S. N. , Magnusson, O. T. , Thorsteinsdottir, U. , … Stefansson, K. (2018). Multiple transmissions of de novo mutations in families. Nature Genetics, 50(12), 1674–1680. 10.1038/s41588-018-0259-9 [DOI] [PubMed] [Google Scholar]
- Kong, A. , Frigge, M. L. , Masson, G. , Besenbacher, S. , Sulem, P. , Magnusson, G. , Gudjonsson, S. A. , Sigurdsson, A. , Jonasdottir, A. , Jonasdottir, A. , Wong, W. S. , Sigurdsson, G. , Walters, G. B. , Steinberg, S. , Helgason, H. , Thorleifsson, G. , Gudbjartsson, D. F. , Helgason, A. , Magnusson, O. T. , … Stefansson, K. (2012). Rate of de novo mutations and the importance of father's age to disease risk. Nature, 488(7412), 471–475. 10.1038/nature11396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupp, D. R. , Barnard, R. A. , Duffourd, Y. , Evans, S. A. , Mulqueen, R. M. , Bernier, R. , Rivière, J. B. , Fombonne, E. , & O'Roak, B. J. (2017). Exonic mosaic mutations contribute risk for autism Spectrum disorder. American Journal of Human Genetics, 101(3), 369–390. 10.1016/j.ajhg.2017.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legrand, A. , Devriese, M. , Dupuis‐Girod, S. , Simian, C. , Venisse, A. , Mazzella, J. M. , Auribault, K. , Adham, S. , Frank, M. , Albuisson, J. , & Jeunemaitre, X. (2019). Frequency of de novo variants and parental mosaicism in vascular Ehlers‐Danlos syndrome. Genetics in Medicine, 21(7), 1568–1575. 10.1038/s41436-018-0356-2 [DOI] [PubMed] [Google Scholar]
- Liu, Q. , Karolak, J. A. , Grochowski, C. M. , Wilson, T. A. , Rosenfeld, J. A. , Bacino, C. A. , Lalani, S. R. , Patel, A. , Breman, A. , Smith, J. L. , Cheung, S. W. , Lupski, J. R. , Bi, W. , & Stankiewicz, P. (2020). Parental somatic mosaicism for CNV deletions ‐ A need for more sensitive and precise detection methods in clinical diagnostics settings. Genomics, 112(5), 2937–2941. 10.1016/j.ygeno.2020.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEntagart, M. , Williamson, K. A. , Rainger, J. K. , Wheeler, A. , Seawright, A. , de Baere, E. , Verdin, H. , Bergendahl, L. T. , Quigley, A. , Rainger, J. , Dixit, A. , Sarkar, A. , López Laso, E. , Sanchez‐Carpintero, R. , Barrio, J. , Bitoun, P. , Prescott, T. , Riise, R. , McKee, S. , … FitzPatrick, D. R. (2016). A restricted repertoire of de novo mutations in ITPR1 cause Gillespie syndrome with evidence for dominant‐negative effect. American Journal of Human Genetics, 98(5), 981–992. 10.1016/j.ajhg.2016.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson, J. J. , Shi, Y. , Gujral, M. , Zheng, H. , Malhotra, D. , Jin, X. , Jian, M. , Liu, G. , Greer, D. , Bhandari, A. , Wu, W. , Corominas, R. , Peoples, A. , Koren, A. , Gore, A. , Kang, S. , Lin, G. N. , Estabillo, J. , Gadomski, T. , … Sebat, J. (2012). Whole‐genome sequencing in autism identifies hot spots for de novo germline mutation. Cell, 151(7), 1431–1442. 10.1016/j.cell.2012.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller, R. S. , Liebmann, N. , Larsen, L. H. G. , Stiller, M. , Hentschel, J. , Kako, N. , Abdin, D. , di Donato, N. , Pal, D. K. , Zacher, P. , Syrbe, S. , Dahl, H. A. , & Lemke, J. R. (2019). Parental mosaicism in epilepsies due to alleged de novo variants. Epilepsia, 60(6), e63–e66. 10.1111/epi.15187 [DOI] [PubMed] [Google Scholar]
- Myers, C. T. , Hollingsworth, G. , Muir, A. M. , Schneider, A. L. , Thuesmunn, Z. , Knupp, A. , King, C. , Lacroix, A. , Mehaffey, M. G. , Berkovic, S. F. , Carvill, G. L. , Sadleir, L. G. , Scheffer, I. E. , & Mefford, H. C. (2018). Parental mosaicism in “de novo” epileptic encephalopathies. New England Journal of Medicine, 378(17), 1646–1648. 10.1056/NEJMc1714579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama, T. , Ishii, A. , Yoshida, T. , Nasu, H. , Shimojima, K. , Yamamoto, T. , Kure, S. , & Hirose, S. (2018). Somatic mosaic deletions involving SCN1A cause Dravet syndrome. American Journal of Medical Genetics. Part A, 176(3), 657–662. 10.1002/ajmg.a.38596 [DOI] [PubMed] [Google Scholar]
- Pasmant, E. , & Pacot, L. (2020). Should we genotype the sperm of fathers from patients with ‘de novo’ mutations? European Journal of Endocrinology, 182(1), C1–c3. 10.1530/eje-19-0759 [DOI] [PubMed] [Google Scholar]
- Rahbari, R. , Wuster, A. , Lindsay, S. J. , Hardwick, R. J. , Alexandrov, L. B. , Turki, S. A. , Dominiczak, A. , Morris, A. , Porteous, D. , Smith, B. , Stratton, M. R. , UK10K Consortium , & Hurles, M. E. (2016). Timing, rates and spectra of human germline mutation. Nature Genetics, 48(2), 126–133. 10.1038/ng.3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch, A. , Wieczorek, D. , Graf, E. , Wieland, T. , Endele, S. , Schwarzmayr, T. , Albrecht, B. , Bartholdi, D. , Beygo, J. , di Donato, N. , Dufke, A. , Cremer, K. , Hempel, M. , Horn, D. , Hoyer, J. , Joset, P. , Röpke, A. , Moog, U. , Riess, A. , … Strom, T. M. (2012). Range of genetic mutations associated with severe non‐syndromic sporadic intellectual disability: An exome sequencing study. Lancet, 380(9854), 1674–1682. 10.1016/s0140-6736(12)61480-9 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach, J. C. , Glusman, G. , Smit, A. F. , Huff, C. D. , Hubley, R. , Shannon, P. T. , Rowen, L. , Pant, K. P. , Goodman, N. , Bamshad, M. , Shendure, J. , Drmanac, R. , Jorde, L. B. , Hood, L. , & Galas, D. J. (2010). Analysis of genetic inheritance in a family quartet by whole‐genome sequencing. Science, 328(5978), 636–639. 10.1126/science.1186802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röthlisberger, B. , & Kotzot, D. (2007). Recurrence risk in de novo structural chromosomal rearrangements. American Journal of Medical Genetics. Part A, 143a(15), 1708–1714. 10.1002/ajmg.a.31826 [DOI] [PubMed] [Google Scholar]
- Rump, A. , Hildebrand, L. , Tzschach, A. , Ullmann, R. , Schrock, E. , & Mitter, D. (2013). A mosaic maternal splice donor mutation in the EHMT1 gene leads to aberrant transcripts and to Kleefstra syndrome in the offspring. European Journal of Human Genetics, 21(8), 887–890. 10.1038/ejhg.2012.267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu, L. , Zhang, Q. , Tian, Q. , Yang, S. , Peng, X. , Mao, X. , Yang, L. , Du, J. , & Wang, H. (2021). Parental mosaicism in de novo neurodevelopmental diseases. American Journal of Medical Genetics, 185(7), 2119–2125. 10.1002/ajmg.a.62174 [DOI] [PubMed] [Google Scholar]
- Stefanski, A. , Calle‐López, Y. , Leu, C. , Pérez‐Palma, E. , Pestana‐Knight, E. , & Lal, D. (2021). Clinical sequencing yield in epilepsy, autism spectrum disorder, and intellectual disability: A systematic review and meta‐analysis. Epilepsia, 62(1), 143–151. 10.1111/epi.16755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranneheim, H. , Lagerstedt‐Robinson, K. , Magnusson, M. , Kvarnung, M. , Nilsson, D. , Lesko, N. , Engvall, M. , Anderlid, B. M. , Arnell, H. , Johansson, C. B. , Barbaro, M. , Björck, E. , Bruhn, H. , Eisfeldt, J. , Freyer, C. , Grigelioniene, G. , Gustavsson, P. , Hammarsjö, A. , Hellström‐Pigg, M. , … Wedell, A. (2021). Integration of whole genome sequencing into a healthcare setting: High diagnostic rates across multiple clinical entities in 3219 rare disease patients. Genome Medicine, 13(1), 40. 10.1186/s13073-021-00855-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synofzik, M. , Helbig, K. L. , Harmuth, F. , Deconinck, T. , Tanpaiboon, P. , Sun, B. , Guo, W. , Wang, R. , Palmaer, E. , Tang, S. , Schaefer, G. B. , Gburek‐Augustat, J. , Züchner, S. , Krägeloh‐Mann, I. , Baets, J. , de Jonghe, P. , Bauer, P. , Chen, S. R. W. , Schöls, L. , & Schüle, R. (2018). De novo ITPR1 variants are a recurrent cause of early‐onset ataxia, acting via loss of channel function. European Journal of Human Genetics, 26(11), 1623–1634. 10.1038/s41431-018-0206-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilbe, M. , Gudmundsson, S. , Johansson, J. , Ameur, A. , Stattin, E. L. , Annerén, G. , Malmgren, H. , Frykholm, C. , & Bondeson, M. L. (2017). A novel approach using long‐read sequencing and ddPCR to investigate gonadal mosaicism and estimate recurrence risk in two families with developmental disorders. Prenatal Diagnosis, 37(11), 1146–1154. 10.1002/pd.5156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen, M. H. , Beunders, G. , Callaghan, M. , de Leeuw, N. , Nillesen, W. M. , Yntema, H. G. , van Hagen, J. , Nieuwint, A. W. , Morrison, N. , Keijzers‐Vloet, S. T. , Hoischen, A. , Brunner, H. G. , Tolmie, J. , & Kleefstra, T. (2011). Familial Kleefstra syndrome due to maternal somatic mosaicism for interstitial 9q34.3 microdeletions. Clinical Genetics, 80(1), 31–38. 10.1111/j.1399-0004.2010.01607.x [DOI] [PubMed] [Google Scholar]
- Wright, C. F. , Prigmore, E. , Rajan, D. , Handsaker, J. , McRae, J. , Kaplanis, J. , Fitzgerald, T. W. , FitzPatrick, D. , Firth, H. V. , & Hurles, M. E. (2019). Clinically‐relevant postzygotic mosaicism in parents and children with developmental disorders in trio exome sequencing data. Nature Communications, 10(1), 2985. 10.1038/s41467-019-11059-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X. , Yang, X. , Wu, Q. , Liu, A. , Yang, X. , Ye, A. Y. , Huang, A. Y. , Li, J. , Wang, M. , Yu, Z. , Wang, S. , Zhang, Z. , Wu, X. , Wei, L. , & Zhang, Y. (2015). Amplicon resequencing identified parental mosaicism for approximately 10% of “de novo” SCN1A mutations in children with Dravet syndrome. Human Mutation, 36(9), 861–872. 10.1002/humu.22819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X. , Liu, A. , Xu, X. , Yang, X. , Zeng, Q. , Ye, A. Y. , Yu, Z. , Wang, S. , Huang, A. Y. , Wu, X. , Wu, Q. , Wei, L. , & Zhang, Y. (2017). Genomic mosaicism in paternal sperm and multiple parental tissues in a Dravet syndrome cohort. Scientific Reports, 7(1), 15677. 10.1038/s41598-017-15814-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X. , Yang, X. , Chen, J. , Li, S. , Zeng, Q. , Huang, A. Y. , Ye, A. Y. , Yu, Z. , Wang, S. , Jiang, Y. , Wu, X. , Wu, Q. , Wei, L. , & Zhang, Y. (2019). ATP1A3 mosaicism in families with alternating hemiplegia of childhood. Clinical Genetics, 96(1), 43–52. 10.1111/cge.13539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zillhardt, J. L. , Poirier, K. , Broix, L. , Lebrun, N. , Elmorjani, A. , Martinovic, J. , Saillour, Y. , Muraca, G. , Nectoux, J. , Bessieres, B. , Fallet‐Bianco, C. , Lyonnet, S. , Dulac, O. , Odent, S. , Rejeb, I. , Ben Jemaa, L. , Rivier, F. , Pinson, L. , Geneviève, D. , …. Chelly, J. (2016). Mosaic parental germline mutations causing recurrent forms of malformations of cortical development. European Journal of Human Genetics, 24(4), 611–614. 10.1038/ejhg.2015.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All ddPCR data and sequences for Taqman Assays are available upon reasonable request.