

Abstract
Background
Hypomagnesemia, seizures, and mental retardation (HSMR) syndrome is a rare genetic disease. Presently, only 24 cases have been reported and the clinical features of the disease are yet to be fully described, thereby making diagnosis challenging.
Methods
Trio‐whole‐exome sequencing was used for the patient and her parents, and the structure of the variant protein was analyzed by molecular dynamics. Finally, the characteristics of HSMR were summarized by reviewing the previous literature.
Results
The main disease manifestations in the patient were seizures, liver function damage, hypomagnesemia, atrial septal defect, and sinus arrhythmia. A novel mutation in CNNM2 (c.566A>G/p.Tyr189Cys) was identified by genetic detection. The parents were wild type, and the mutation was rated as pathogenic by American College of Medical Genetics and Genomics guidelines. Ab initio modeling and molecular dynamics simulation show that the mutation destroys the surrounding hydrogen bonds, which may reduce the local stability of the protein structure. In the previous literature, only 24 children with HSMR have been reported, mainly manifested as hypomagnesemia, mental retardation, seizures, and language and motor impairment.
Conclusion
We have reported the second case of HSMR in the Chinese population, which further expands the phenotypic spectrum of congenital heart disease and the variation spectrum of CNNM2.
Keywords: CHD, CNNM2, HSMR, hypomagnesemia, mental retardation, seizures
Hypomagnesemia, seizures, and mental retardation (HSMR) syndrome is a rare genetic disease. Presently, only 24 cases have been reported and the clinical features of the disease have not been fully described, thereby making diagnosis challenging.

1. INTRODUCTION
The cystathionine‐β‐synthase (CBS)‐pair domain divalent metal cation transport mediator (CNNM) family includes four proteins (CNNM1–4), which maintain Mg2+ homeostasis in vivo. These proteins are mainly expressed in the basolateral membrane of the loop of Henle and other tissues such as the brain, lung, liver, spleen, and heart (Chen et al., 2020; Li et al., 2021; Stuiver et al., 2011). CNNM2 heterozygous mutations lead to hypomagnesemia, seizures, and mental retardation syndrome (HSMR, OMIM#616418), first reported in 2011 (Stuiver et al., 2011). To date, only 24 cases of HSMR caused by CNNM2 mutations have been reported worldwide; therefore, the phenotypic characteristics of the syndrome are still unclear (Accogli et al., 2019; Arjona et al., 2014; Bamhraz et al., 2021; Franken, Müller, et al., 2021; García‐Castaño et al., 2020; Stuiver et al., 2011). We have identified a novel heterozygous variant of CNNM2, c.566A>G/p.Tyr189Cys, resulting in hypomagnesemia, hypocalcemia, seizures, mental retardation, and other typical features of HSMR accompanied by congenital heart disease (CHD) and liver enzyme abnormalities in a Chinese child. This is the second report of an association between a CNNM2 variant and HSMR in the Chinese population. Revealing variant associations with the syndrome may lead to improved differential diagnoses.
2. MATERIALS AND METHODS
2.1. Case presentation
The patient was a 4‐month and 9‐day‐old female infant who was delivered naturally and had normal physical development. She experienced one episode of uninduced focal epilepsy at 4 months of age, during which she held her hands tightly. The upper limb tremor was relieved after several seconds. Biochemical examination indicated abnormal serum magnesium and calcium levels, alanine, and aspartate aminotransferases, suggesting aberrant liver function. A cardiac function examination revealed high levels of creatine kinase. While a Doppler ultrasound showed characteristics of CHD, including atrial septal defect (ASD), an electrocardiogram (ECG) suggested sinus arrhythmia (Table 1). Magnetic resonance imaging (MRI) and electroencephalography revealed no abnormalities. The parents were healthy and denied the marriage of close relatives. There was no family history of genetic disorders.
TABLE 1.
Clinical characteristics of the patient with CNNM2 variant c.566A>G/p.Tyr189Cys
| Essential information | Patient | Reference values |
|---|---|---|
| Gender | Female | – |
| Age of onset | 4 months | – |
| Age of diagnosis | 6 months | – |
| Seizure type | Focal | – |
| Motor development | Unstable vertical head | – |
| Intellectual development | Mild mental retardation | – |
| Language development | No abnormality | – |
| Biochemical examination | ||
| Serum K+ (mmol/L) | 4.59 | 3.50–5.30 |
| Serum Na+ (mmol/L) | 138.59 | 137–147.00 |
| Serum Mg2+ (mmol/L) | 0.62↓ | 0.75–1.02 |
| Serum Ca2+ (mmol/L) | 1.92↓ | 2.11–2.52 |
| Serum P (mmol/L) | 1.88↑ | 0.85–1.51 |
| Urea (mmol/L) | 1.65↓ | 3.10–8.00 |
| Glucose (mmol/L) | 5.99 | 3.89–6.11 |
| Alanine transaminase (U/L) | 99.50↑ | 9.00–50.00 |
| Aspartate aminotransferase (U/L) | 138.5↑ | 15.0–40.0 |
| Alkaline phosphatase (U/L) | 201.7↑ | 45.0–125.0 |
| Creatine kinase isoenzyme MB (U/L) | 56↑ | 0–24 |
| Creatine kinase (U/L) | 197 | 50–310 |
| Lactate dehydrogenase (U/L) | 471.00↑ | 120.00–250.00 |
| Lactic acid (mmol/L) | 7.8↑ | 0.4–1.8 |
| Ceruloplasmin (g/L) | 0.19↓ | 0.30–0.65 |
| Imaging examination | ||
| EEG | No abnormality | – |
| Head MRI | No abnormality | – |
| Echocardiography | Atrial septal defect | – |
| ECG | Sinus arrhythmia, P–P interval difference > 0.12 s | – |
Abbreviations: ECG, electrocardiogram; EEG, electroencephalogram; MRI, magnetic resonance imaging.
2.2. Genetic testing and variant analysis (including ethical considerations)
The work described in this case report has been carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans (http://www.wma.net/e/policy/b3.htm). The Hospital Ethics Committee approved the study and publication of the report (PJ2020‐08‐21). After obtaining informed consent from the parents, the patient and parents underwent trio‐whole‐exome sequencing (trio‐WES). Peripheral blood (3 ml, in an ethylenediaminetetraacetic acid‐coated tube to prevent coagulation) was collected, and the white blood cell DNA was extracted as per the manufacturer’s instructions for the genomic extraction kit (CWBIO, Beijing, China). After constructing the library, the sequence was captured using an Illumina Noveseq 6000 high‐throughput sequencer (Illumina, San Diego, CA, USA). Normal population distribution databases, including dbSNP, ExAC, and 1000 Genomes, were screened for suspicious variants, and bioinformatic prediction analysis (SIFT, Polyphen2, and MutationTaster) was carried out. The amino acid sequencing homology was analyzed using Mega7.0, and their pathogenicity was evaluated based on the American College of Medical Genetics and Genomics (ACMG) guidelines.
2.3. Molecular dynamics (MD) simulation
Using the QUARK ab initio calculation (1–430aa is ab initio construction, 430–823aa is the crystal structure, PDB Code, 6N7E) (QUARK, https://zhanglab.ccmb.med.umich.edu/QUARK/), a three‐dimensional (3D) model of the CNNM2 protein was constructed. Using Modeller10.1 (https://salilab.org/modeller/) multi‐template modeling, model_multi.py, and other scripts for multi‐template modeling and 3D structure simulation, the tertiary structure of the full‐length protein was obtained (Supporting Information 1). The entire simulation process included system construction, energy minimization, NVT and NPT ensemble simulation, and finished product simulation. Finally, the interaction between the variant and the surrounding residues was analyzed with Chimera 1.15 (http://www.cgl.ucsf.edu/chimera/).
3. RESULTS
3.1. Genetic testing
A CNNM2 heterozygous variant (NM_017649: c.566A>G/p.Tyr189Cys) was detected in the patient but not in the parents or the normal population database. A search of the ClinVar database and Human Gene Mutation Database (HGMD) revealed the variant to be a novel CNNM2 mutation, not yet reported. The results of the SIFT, Polyphen2, and MutationTaster analyses were 0.01 (damaging), 0.99 (probably damaging), and 0.99999 (disease‐causing), respectively. The variant was rated as pathogenic (PS2 + PM1 + PM2 + PP2 + PP3) as per the ACMG guidelines (Richards et al., 2015). Homology analysis found Tyr189 to be highly conserved among the different species (Figure 1).
FIGURE 1.
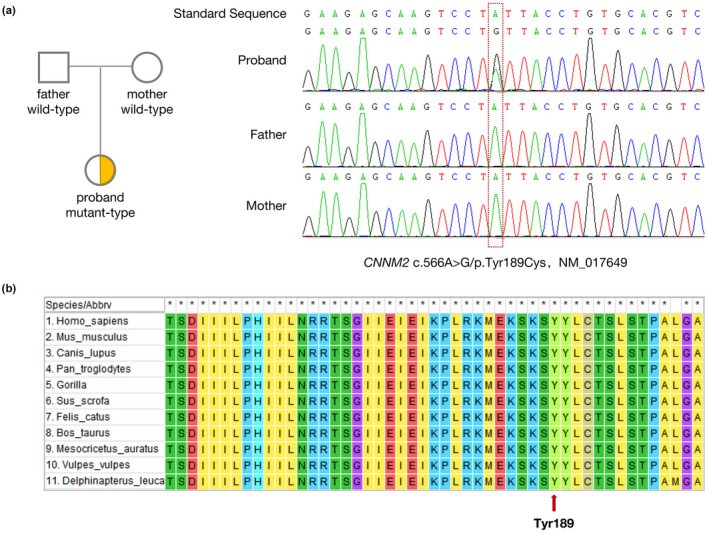
(a) Trio‐whole exome sequencing (trio‐WES) identified that the CNNM2 gene of the infant had heterozygous variation (c.566A>G/p.Tyr189Cys), and the parents were wild‐type at the ectopic site of the variant. Therefore, this indicates a denovo mutation of CNNM2 gene. Sanger sequencing confirmed the existence of the variant. (b) Tyr189 is highly conserved among the different species studied
3.2. MD results
The root mean square deviation (RMSD) results showed that the mutant and wild type were balanced at around 2000 ps, and their conformational overlap map suggested that the variant may not affect the secondary protein structure. However, further interaction analysis indicated that the wild‐type Tyr189 formed a hydrogen bond with Asn83 and a water bridge with Glu637 and Ser205, whereas the Cys189 in the mutant had no polar interaction with the surrounding residues due to its small side chain. As a result, compared with Tyr189, Cys189 had weaker interactions with the surrounding residues (Figure 2).
FIGURE 2.
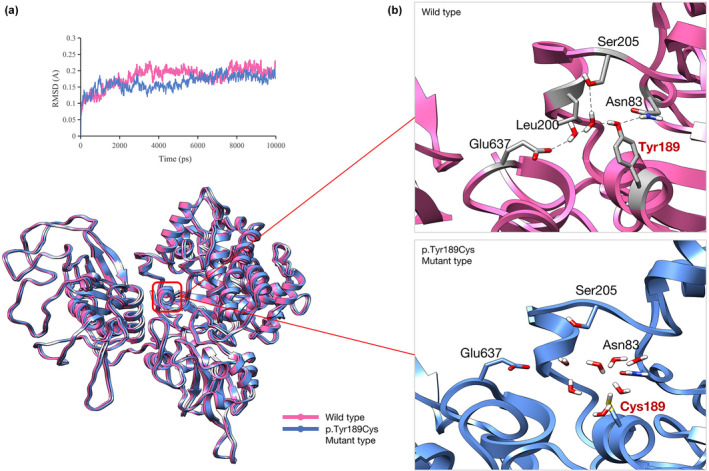
Molecular dynamics (MD) results. (a) The root mean square deviation (RMSD) analysis shows the mutant and wild‐type balanced at about 2000 ps, and their conformational overlap diagram shows no effect on the secondary structure of CNNM2. (b) An in‐depth analysis of the local interaction shows that Tyr189 forms a hydrogen bond with Asn83 and a water bridge with Glu637 and Ser205 in the wild‐type structure. However, in the mutant, due to the small side chain of Cys189, all the water molecules fill the pockets, precluding polar interaction with the surrounding residues, thus reducing the local stability
4. DISCUSSION
HSMR is a rare genetic disease characterized by hypomagnesemia, seizures, and intellectual disability due to renal Mg2+ consumption. In some cases, additional symptoms, such as obesity or autism spectrum disorder, have been reported (Arjona et al., 2014). To date, only 24 HSMR cases have been reported worldwide and the characteristic clinical spectrum has not been well described, making its differential diagnosis challenging (Accogli et al., 2019; Arjona et al., 2014; Bamhraz et al., 2021; Franken, Müller, et al., 2021; García‐Castaño et al., 2020; Stuiver et al., 2011). We retrospectively analyzed the reported cases and found that in addition to the HSMR triad, language retardation, motor development disorder, and obesity also occur in the high incidence phenotypes of the syndrome. A few patients will also have ASD, microcephaly, and hypocalciuria (Figure 3a). The infant in this study had typical HSMR features, such as seizures, hypomagnesemia, and mild intellectual impairment at 4 months of age. It is worth noting that the infant also showed evidence of CHD with elevated myocardial enzymes and atrial septal defect. ECG showed sinus arrhythmia; however, few previously reported HSMR cases have shown congenital heart disease symptoms. Accogli et al. (2019) reported that a fourth male newborn in a family member of an HSMR patient showed severe phenotypes, including severe cardiac structural abnormalities, ECG abnormalities, and craniofacial malformations, and died of cardiogenic shock on the third day after birth. Arrhythmia in HSMR patients may be caused by early afterdepolarization. Since Mg2+ affects myocardial contraction mainly by antagonizing Ca2+ regulation, hypomagnesemia can increase the risk of arrhythmia (Tangvoraphonkchai & Davenport, 2018). Although aberrant CNNM2 activity generally leads to hypomagnesemia, reports of arrhythmia are rare, which could be due to the lack of relevant cohort studies and case reports. In addition, a direct correlation between cardiac structural abnormalities and CNNM2 function is not yet clear. However, in a mouse model of magnesium deficiency, various cardiovascular structural abnormalities, including aortic wall thinning, tissue disorder, elastic fiber rupture, and collagen abnormality, were observed. This collection of symptoms may be related to increased matrix metalloproteinase (MMP)‐2 and MMP‐9 activities (Pagès et al., 2003). Therefore, CNNM2 defects may indirectly affect the cardiovascular system.
FIGURE 3.
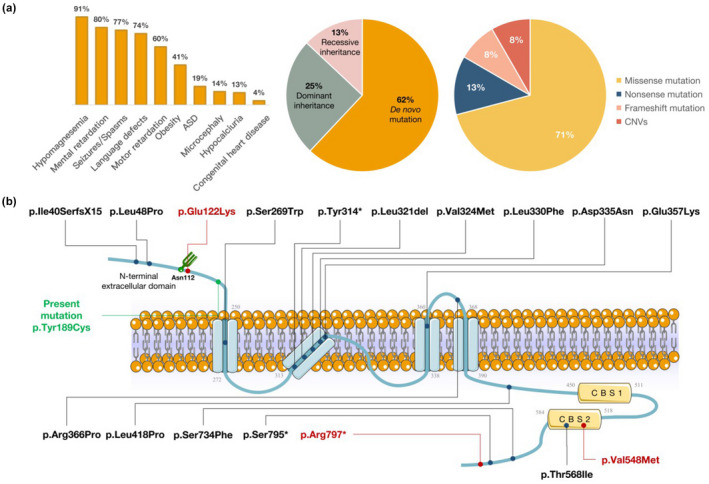
Literature review of hypomagnesemia, seizures, and mental retardation (HSMR) syndrome caused by a CNNM2 gene mutation. (a) According to the phenotypic analysis of 24 reported HSMR cases, we found that in addition to the known triad (hypomagnesemia, seizures, and mental retardation), other high‐frequency phenotypes include language retardation, motor development disorder, and obesity. Furthermore, a few patients will have atrial septal defect (ASD), microcephaly, and other phenotypes. CNNM2 gene mutations causing HSMR are mainly de novo, followed by both dominant and recessive inheritance patterns. The main type of variation found in the literature review was missense mutations. Nonsense mutation, frameshift mutation, and copy number variations (CNVs) were also reported. (b) The protein encoded by CNNM2 contains an N‐terminal extracellular domain, four transmembrane α‐helix transmembrane domains, cystathionine β‐synthase (CBS‐par) domain, and the cyclic nucleotide binding homology (CNBH) domain. Homozygous mutation of key amino acids or domains of proteins may lead to more serious clinical manifestations. For example, the homozygous mutation adjacent to Asn112 can lead to a severe nervous system phenotype (Arjona et al., 2014). A missense mutation of the CBS2 domain can also lead to a severe nervous system phenotype and even death (Accogli et al., 2019). Red indicates severe phenotype, black indicates reported loci, and green indicates variant loci of our patient
The majority of CNNM2 mutations leading to HSMR are de novo mutations, followed by dominant inheritance, and a few patients exist with recessive inheritance. Additionally, in previously reported cases, CNNM2 variations tend to be missense mutations. Other types, such as nonsense mutations, frameshift mutations, and copy number variations, have also been reported (Figure 3a). The severity of the HSMR phenotype may be related to the mutation site and the genetic pattern of the CNNM2 gene mutation. If the pathogenic mutation occurs at a key site and is homozygous, it may have a more serious nervous system phenotype. CNNM2 is located on chromosome 10q24.32 and encodes a protein of 875 amino acids, which features an N‐terminal extracellular domain, four transmembrane α‐helix transmembrane domains, and the cyclic nucleotide‐binding homology (CNBH) domain (Chen et al., 2020). There is a glycosylation site (Asn112) in the N‐terminal extracellular domain, which can recruit signal recognition particles to perform the signal peptide cleavage process and plays a role in key functions such as membrane stability and protein localization (de Baaij et al., 2012; Vagin et al., 2009). A mutation near the locus can result in severe nervous system‐related phenotypes, such as intractable epilepsy, microcephaly, severe intellectual impairment, and abnormal brain MRI (Arjona et al., 2014). Additionally, CBS‐pair dimerization (CBS1, CBS2) mediated by the CNBH domain has important biological functions. This domain significantly affects the physiological role of Mg2+ efflux (Hardy et al., 2019). Patients with CNNM2 mutations affecting the CBS2 structure can also have severe nervous system‐related phenotypes such as refractory epilepsy, severe intellectual impairment, and even death (Accogli et al., 2019) (Figure 3b). The novel mutation (c.566A>G/p.Tyr189Cys) in this case appears in the N‐terminal extracellular domain of the CNNM2 protein. Through ab initio modeling and molecular dynamic simulation, we determined that the mutation may not affect the secondary structure of the protein. However, the ectopic point destroys the polar bond of the surrounding residues, which may affect the local stability of the N‐terminal structure.
The mechanism by which the loss of function of CNNM2 leads to abnormal Mg2+ metabolism and affects the function of the nervous system remains controversial, but studies have shown that the gene is located in the midbrain–hindbrain boundary, which can affect the development and differentiation of the central nervous system (CNS). Therefore, the CNNM2 protein function is very important for the development of CNS constituent regions (Arjona et al., 2014; Franken, Seker, et al., 2021). In the study of model organisms, both Cnnm2 −/− mouse and zebrafish models showed the absence of lethal brain structure in the early stage of the embryo. For example, around 36% of Cnnm2 −/− mouse embryos had anencephaly, showing that neural tube development defects led to a brain phenotype (Franken, Seker, et al., 2021). In the phenotype of an electrolyte disorder, the Cnnm2 +/− mouse model is consistent with the observations of the human phenotype. Both Cnnm2 +/− mouse models show that the level of Mg2+ in hemorrhage is reduced; however, the excretion of Mg2+ in the urine is not significantly increased. Mice exposed to a low‐magnesium diet may even show a reduction of Mg2+ excretion in the urine, which suggests the ability to compensate for Mg2+ reabsorption. However, hypomagnesemia and decreased urinary Mg2+ excretion can be explained as renal magnesium loss, indicating that Cnnm2 can regulate renal Mg2+ (Franken, Seker, et al., 2021; Funato et al., 2017).
At present, there are no relevant treatment guidelines for HSMR. Clinically, treatment has generally been symptomatic. For example, most cases of hypomagnesemia are treated with oral magnesium supplements, yet serum magnesium content is not effectively increased after treatment. For antiepileptic treatment, two or more antiepileptic drugs (AEDs) are usually used. However, according to the reported literature, more than 50% of patients still do not show complete alleviation of epilepsy symptoms after AED treatment. The low control rate of AEDs may be related to the use of different drugs among research teams (Arjona et al., 2014; Franken, Seker, et al., 2021). In the current case, treatment with reduced glutathione for liver protection and an oral calcium/magnesium supplement was used. Blood magnesium content increased but was still lower than the normal range. As of the 3‐month follow‐up, there had been no other seizure activity. Therefore, increasing the serum magnesium content may be a promoting factor in the alleviation of cardiovascular problems, inflammation, and seizures caused by HSMR (Rodelo‐Haad et al., 2020). Since the role of the CNNM2 protein in renal magnesium ion transport is still controversial, understanding this may be the key to finding effective therapeutic targets for the treatment of related complications caused by hypomagnesemia in HSMR (Arjona & de Baaij, 2018).
5. CONCLUSION
In conclusion, we performed a genetic analysis and provided a clinical case description of a Chinese infant with HSMR through trio‐WES. The infant showed epilepsy and mild intellectual impairment without inducement. Biochemical examination showed electrolyte disorders such as hypomagnesemia and hypocalcemia. In addition, the infant had rare HSMR phenotypes such as abnormal myocardial enzymes caused by CHD and abnormal liver enzymes caused by impaired liver function. However, there are still some limitations to this study. For example, there is a lack of long‐term follow‐up records and in‐depth analysis of treatment for HSMR patients. In conclusion, this report enriches the knowledge of the phenotypic and variation spectrum of HSMR. Clinicians should consider the possibility of CNNM2 mutations in infants with hypomagnesemia, seizures, mental retardation, and CHD symptoms. The early identification of HSMR is of positive significance for clinical hierarchical management of the disease.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The study was approved by the ethics committee of the First Affiliated Hospital of Anhui Medical University (PJ2020‐08‐21). Written informed consent was provided by the participant.
Supporting information
Supinfo
ACKNOWLEDGMENT
We would like to thank the family of the reported case for their cooperation in this study.
Xu, X. , Hou, S. , Sun, W. , Zhu, J. , Yuan, J. , Cui, Z. , Wu, D. & Tang, J. (2022). Rare hypomagnesemia, seizures, and mental retardation in a 4‐month‐old patient caused by novel CNNM2 mutation Tyr189Cys: Genetic analysis and review. Molecular Genetics & Genomic Medicine, 10, e1898. 10.1002/mgg3.1898
Xiaoyan Xu and Shu Hou contributed equally to this work.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Accogli, A. , Scala, M. , Calcagno, A. , Napoli, F. , Di Iorgi, N. , Arrigo, S. , Mancardi, M. M. , Prato, G. , Pisciotta, L. , Nagel, M. , Severino, M. , & Capra, V. (2019). CNNM2 homozygous mutations cause severe refractory hypomagnesemia, epileptic encephalopathy and brain malformations. European Journal of Medical Genetics, 62(3), 198–203. 10.1016/j.ejmg.2018.07.014 [DOI] [PubMed] [Google Scholar]
- Arjona, F. J. , de Baaij, J. H. , Schlingmann, K. P. , Lameris, A. L. , van Wijk, E. , Flik, G. , Regele, S. , Korenke, G. C. , Neophytou, B. , Rust, S. , Reintjes, N. , Konrad, M. , Bindels, R. J. , & Hoenderop, J. G. (2014). CNNM2 mutations cause impaired brain development and seizures in patients with hypomagnesemia. PLoS Genetics, 10(4), e1004267. 10.1371/journal.pgen.1004267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arjona, F. J. , & de Baaij, J. H. F. (2018). CrossTalk opposing view: CNNM proteins are not Na+ /Mg2+ exchangers but Mg2+ transport regulators playing a central role in transepithelial Mg2+ (re)absorption. The Journal of Physiology, 596(5), 747–750. 10.1113/JP275249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamhraz, A. A. , Franken, G. A. C. , de Baaij, J. H. F. , Rodrigues, A. , Grady, R. , Deveau, S. , & Chanchlani, R. (2021). Diagnostic dilemma in an adolescent girl with an eating disorder, intellectual disability, and hypomagnesemia. Nephron, 19, 1–4. 10.1159/000518173 [DOI] [PubMed] [Google Scholar]
- Chen, Y. S. , Kozlov, G. , Fakih, R. , Yang, M. , Zhang, Z. , Kovrigin, E. L. , & Gehring, K. (2020). Mg2+‐ATP sensing in CNNM, a putative magnesium transporter. Structure, 28(3), 324–335.e4. 10.1016/j.str.2019.11.016 [DOI] [PubMed] [Google Scholar]
- de Baaij, J. H. , Stuiver, M. , Meij, I. C. , Lainez, S. , Kopplin, K. , Venselaar, H. , Müller, D. , Bindels, R. J. , & Hoenderop, J. G. (2012). Membrane topology and intracellular processing of cyclin M2 (CNNM2). The Journal of Biological Chemistry, 287(17), 13644–13655. 10.1074/jbc.M112.342204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franken, G. A. C. , Müller, D. , Mignot, C. , Keren, B. , Lévy, J. , Tabet, A. C. , Germanaud, D. , Tejada, M. I. , Kroes, H. Y. , Nievelstein, R. A. J. , Brimble, E. , Ruzhnikov, M. , Claverie‐Martin, F. , Szczepańska, M. , Ćuk, M. , Latta, F. , Konrad, M. , Martínez‐Cruz, L. A. , Bindels, R. J. M. , … de Baaij, J. H. F. (2021). The phenotypic and genetic spectrum of patients with heterozygous mutations in cyclin M2 (CNNM2). Human Mutation, 42(4), 473–486. 10.1002/humu.24182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franken, G. A. C. , Seker, M. , Bos, C. , Siemons, L. A. H. , van der Eerden, B. C. J. , Christ, A. , Hoenderop, J. G. J. , Bindels, R. J. M. , Müller, D. , Breiderhoff, T. , & de Baaij, J. H. F. (2021). Cyclin M2 (CNNM2) knockout mice show mild hypomagnesaemia and developmental defects. Scientific Reports, 11(1), 8217. 10.1038/s41598-021-87548-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato, Y. , Yamazaki, D. , & Miki, H. (2017). Renal function of cyclin M2 Mg2+ transporter maintains blood pressure. Journal of Hypertension, 35(3), 585–592. 10.1097/HJH.0000000000001211 [DOI] [PubMed] [Google Scholar]
- García‐Castaño, A. , Madariaga, L. , Antón‐Gamero, M. , Mejia, N. , Ponce, J. , Gómez‐Conde, S. , Pérez de Nanclares, G. , De la Hoz, A. B. , Martínez, R. , Saso, L. , Martínez de LaPiscina, I. , Urrutia, I. , Velasco, O. , Aguayo, A. , Castaño, L. , & Gaztambide, S. (2020). Novel variant in the CNNM2 gene associated with dominant hypomagnesemia. PLoS One, 15(9), e0239965. 10.1371/journal.pone.0239965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy, S. , Kostantin, E. , Wang, S. J. , Hristova, T. , Galicia‐Vázquez, G. , Baranov, P. V. , Pelletier, J. , & Tremblay, M. L. (2019). Magnesium‐sensitive upstream ORF controls PRL phosphatase expression to mediate energy metabolism. Proceedings of the National Academy of Sciences of the United States of America, 116(8), 2925–2934. 10.1073/pnas.1815361116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , Bao, S. , Wang, W. , Shi, X. , Hu, Y. , Li, F. , Zhao, Q. , Zheng, F. , & Lin, Z. (2021). Case report: CNNM2 mutations cause damaged brain development and intractable epilepsy in a patient without hypomagnesemia. Frontiers in Genetics, 12, 705734. 10.3389/fgene.2021.705734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagès, N. , Gogly, B. , Godeau, G. , Igondjo‐Tchen, S. , Maurois, P. , Durlach, J. , & Bac, P. (2003). Structural alterations of the vascular wall in magnesium‐deficient mice. A possible role of gelatinases a (MMP‐2) and B (MMP‐9). Magnesium Research, 16(1), 43–48. [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodelo‐Haad, C. , Pendón‐Ruiz de Mier, M. V. , Díaz‐Tocados, J. M. , Martin‐Malo, A. , Santamaria, R. , Muñoz‐Castañeda, J. R. , & Rodríguez, M. (2020). The role of disturbed mg homeostasis in chronic kidney disease comorbidities. Frontiers in Cell and Developmental Biology, 8, 543099. 10.3389/fcell.2020.543099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuiver, M. , Lainez, S. , Will, C. , Terryn, S. , Günzel, D. , Debaix, H. , Sommer, K. , Kopplin, K. , Thumfart, J. , Kampik, N. B. , Querfeld, U. , Willnow, T. E. , Němec, V. , Wagner, C. A. , Hoenderop, J. G. , Devuyst, O. , Knoers, N. V. , Bindels, R. J. , Meij, I. C. , & Müller, D. (2011). CNNM2, encoding a basolateral protein required for renal Mg2+ handling, is mutated in dominant hypomagnesemia. American Journal of Human Genetics, 88(3), 333–343. 10.1016/j.ajhg.2011.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tangvoraphonkchai, K. , & Davenport, A. (2018). Magnesium and cardiovascular disease. Advances in Chronic Kidney Disease, 25(3), 251–260. 10.1053/j.ackd.2018.02.010 [DOI] [PubMed] [Google Scholar]
- Vagin, O. , Kraut, J. A. , & Sachs, G. (2009). Role of N‐glycosylation in trafficking of apical membrane proteins in epithelia. American Journal of Physiology. Renal Physiology, 296(3), F459–F469. 10.1152/ajprenal.90340.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supinfo
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.