

Abstract
Oxidative stress (OS) refers to the enhancement of oxidation and the decreased of related antioxidant enzymes activity under pathological conditions, resulting in relatively excess reactive oxygen species (ROS), causing cytotoxicity, which leads to tissue damage and is linked to neurodegenerative diseases, cardiovascular diseases, diabetes, cancers, and many other pathologies. As an important intracellular signaling molecule, ROS can regulate numerous physiological actions, such as vascular reactivity and neuronal function. According to several studies, the uncontrolled production of ROS is related to vascular injury. The growing evidence revealing how traditional risk factors translate into ROS and lead to vasculitis and other vascular diseases. In this review, we sought to mainly discuss the role of ROS and antioxidant mechanisms in vascular-related diseases, especially cardiovascular and common macrovascular diseases.
1. Introduction
Oxidative stress (OS) refers to the imbalance of oxidative metabolism in vivo, mainly due to the enhancement of oxidation, which promotes inflammatory responses and the production of large amounts of oxidative intermediate products, such as free radicals, including superoxide, hydroxyl and nitric oxide, and other ROS, including hydrogen peroxide [1]. ROS formation primarily occurs in the mitochondria and endoplasmic reticulum of eukaryotic cells [2]. Oxidative stress induced by the production of excess ROS has been a pivotal mechanism in cardiovascular diseases [3]. Research shows that ROS can promote the generation of inflammatory cytokines, causing inflammation and impairing the functions of vascular cells by activating transcription factors, upregulating adhesion molecules, stimulating chemokine production, and recruiting inflammatory cells [4, 5]. Based on accumulating evidence, excessive ROS-induced altered vascular functions have been demonstrated, including endothelial cell (ECs) damage, hyperplasia of vascular smooth muscle cells (VSMCs), and structural remodeling [6]. Oxidative stress also plays an important role in vascular diseases by regulating the release of both vasoconstricting and vasodilating factors of ECs and autophagy [7, 8] (Figure 1(e)).
Figure 1.
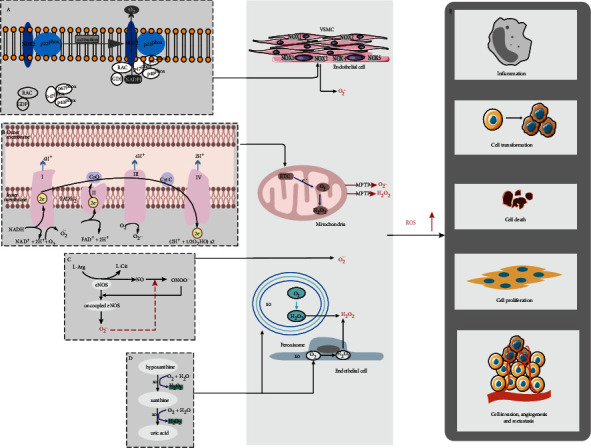
Sources of ROS production. (a) ROS from the NOX2. (b) Electron transfer and ROS generation in ETC. (c) Uncoupled eNOS produced superoxide. (d) OX produced superoxide and hydrogen peroxide by degrading of purines and conversion of hypoxanthine.
Growing evidences revealing how traditional risk factors translate into ROS and contribute to heart and vascular diseases [9, 10]. Uncontrolled production of ROS is interrelated with vascular injury. We opted to discuss the important intermediate role of oxidative stress in vascular-related diseases, particularly cardiovascular and common macrovascular diseases.
First, we detailed the production of ROS in the vessels and their physiological functions. Second, we investigated the implications of ROS in cardiovascular and common macrovascular diseases. For ECs and VSMCs, oxidative stress can be particularly significant as they can activate multiple pathways to induce either cell proliferation, or migration and death [11, 12]. Considering the importance of oxidative stress in the pathogenesis of vascular dysfunction, in the last part of this review, we explored the potential therapeutic strategies of antioxidant therapy in vascular diseases.
2. Sources of Intracellular ROS Production
ROS can play an important role in many intracellular responses. ROS-dependent production in a positive feedback way is the key to continued oxidative stress, which has been linked to interactions between different ROS-producing oxidases systems, and ultimately alters many functions of the vascular cells [13]. The intracellular production of ROS often occurs in the endoplasmic reticulum and mitochondria [14]. As highly reactive molecules, ROS mainly include typical free radicals which contains at least one unpaired electron such as hydroxyl radical (·OH), superoxide (O2−·), and hydrogen peroxides (H2O2), and meanwhile, there are adducts such as hypochlorous acid and nitrogen-containing species [15, 16]. O2−·is the most basic form of ROS produced by cells among them, and others can be produced through O2−· metabolism. Under normal circumstances, a handful of ROS are sustainedly produced in cells. However, excessive production of ROS can be caused by the stimulation of pathological factors. Further, failure of the ROS scavenging system is the major cause of oxidative stress. There are multiple sources of ROS, such as NADPH oxidases (NOXs), the mitochondrial electron transport chain (ETC), uncoupled endothelial nitric oxide synthase (eNOS), and xanthine oxidase (XO). ROS overload often occurs due to the intricate relationships between different oxidase components [17–19] (Figure 1).
2.1. ROS from the NADPH Oxidase (NOX) Activity
NOX activity is the primary ROS resource [6, 20]. The NOX family consists of seven catalytic subunits, termed NOX1-NOX5 and DUOX1-DUOX2. NOXs are multitransmembrane proteins, and DUOXs are seven-transmembrane proteins which C-terminus exposed to the cytosol. Whereas the function of NOX-derived ROS in vasculature also depends on the cell type. Of these, NOX1 and NOX4 are present in VSMCs, while NOX2 and NOX4 are primarily expressed in ECs [21]. The NOX2 protein is stable when constitutively associated with p22phox [22]. NOX2 can be activated to generate superoxide (O2−) if the cytosolic components p40phox, p47phox, and p67phox transferred to the NOX2/p22phox complex, causing the endothelial dysfunction [23–25]. The functional studies of NOX4 have shown that ROS generated by NOX4 is dependent on the p22phox protein, and NOX4 is a high degree of homology to NOX2 [26] (Figure 1(a)).
A portion of NOX-derived ROS are released into the extracellular space which can affect the extracellular matrix (ECM) [27], which another important step in angiogenic response, and participate normal angiogenesis and the development of aortic disease [20, 28].
2.2. ROS from the Mitochondrial Electron Transport Chain (ETC)
ROS are produced as by-products of mitochondrial aerobic metabolism, therefore, a mass of ROS are mainly produced via the mitochondrial ETC during normal aerobic metabolism [29]. Based on evidences, approximately 10-fold higher ROS are present in the mitochondrial than in the nucleus and other organelles, and this unmasking explained why mitochondria are primary targets for ROS-induced damage [30].
Complex I is the main intracellular sites of ROS production in the mitochondria and complex III followed. The essential function of complex I is to transfer 2 electrons from reduced NADH to FMN and then to ubiquinone (CoQ) through a series of iron-sulfur centers, where electrons can be generated at both the FMN and CoQ binding sites [31] (Figure 1(b)). As the only reducing electron carrier in complex III, ubisemiquinone can move freely and leak an electron to O2, thereby reducing O2 to ROS [32, 33].
2.3. Xanthine Oxidase (XO)
XO is another major intracellular oxidase. The main functions of XO include the degradation of purines and conversion of hypoxanthine to xanthine which is further converted to uric acid [34]. XOs are found in ECs and plasma and produce superoxide and H2O2 by donating electrons to molecular oxygen [35–37] (Figure 1(d)).
2.4. Uncoupled Endothelial Nitric Oxide Synthase (eNOS)
Studies have shown that eNOS can be activated to produce vascular protective medium, nitric oxide (NO) [5]. NO oxidative inactivation is rapidly induced by excess superoxide, and persistent oxidative stress induces eNOS uncoupling, producing superoxide at the expense of NO. Whereas the decreased NO is a well-established cardiovascular risk factor [35, 38] (Figure 1(c)).
3. Oxidative Stress in Common Vascular Diseases
3.1. Oxidative Stress in Atherosclerosis
Atherosclerosis (AS) is regarded as a chronic immunoinflammatory disease [39], which is closely related to oxidative stress caused by the production of excess ROS [40]. A generally accepted statement about AS is damage-response theories. Endothelial injury is caused by various stimuli, which causes plasma components such as low-density lipoprotein cholesterol (LDL) to infiltrate and accumulate in the intima, forms atherosclerotic plaques, and leads to narrowing of the vascular lumen [41].
ROS exhibit different mechanisms in the occurrence and development of atherosclerosis. Nonoxidized LDLs by themselves do not risk factors for AS due to its lower affinity for macrophages. When oxidized, LDL becomes oxidized LDL (ox-LDL) in response to ROS [42]. Ox-LDL can stimulate ECs to secrete a variety of inflammatory factors, while promoting vascular cells to secrete ROS by activating NOXs. In particular, ox-LDL increases the expression of vascular endothelial growth factor (VEGF) in macrophages and induces HIF-1α, significantly increasing lumen formation in ECs [43, 44]. Consequently, oxidative stress plays a pathogenic role in promoting AS by inducing macrophage infiltration and ox-LDLs to thicken the blood vessel walls [17]. Several decades ago, researchers discovered that ROS are involved in the development of atherosclerotic plaques by oxidizing unsaturated fatty acids in membrane lipids [1, 3]. Foam cells are the result of binding and uptake of ox-LDL by scavenger receptors on the surface of macrophages. In addition, ROS can promote the expression of scavenger receptors in SMCs and their transformation into foam cells [45]. A recent study showed that autophagy deficiency in VSMCs can promote AS by regulating the inflammatory response [46]. These figures suggest that various factors affect the synthesis of ROS directly or indirectly, leading to the occurrence of AS.
Iron is considered as a risk factor for cardiovascular system owing to its ability to catalyze ROS formation [47]. Vinchi and his team were the first to prove the connection between nontransferrin bound serum iron (NTBI) and AS by investigating a mouse model (ApoE-/- FPNwt/C326S), they observed that iron is heavily deposited in the arterial media layer in the presence of elevated NTBI, induces ROS production, apoptosis of SMCs and ECs, and stimulates a large number of MCP-1-mediated monocyte recruitment, formation of unstable plaques, and eventually aggravates atherosclerosis [48, 49]. As a novel pathway of regulated cell death (RCD), the role of ferroptosis in AS is unclear but there is no doubt that ferroptosis is closely related to iron metabolism and promotes oxidative stress leading to cell death [50, 51].
Prolonged estrogen deficiency is an important risk factor for AS in older women [52]. Estrogen plays a direct protective role on the blood vessels by regulating gene expression and function in ECs [53]. Yang et al. successfully induced postmenopausal AS in mouse by feeding the ovariectomized mice a high-fat diet. It has also been confirmed that estrogenic hormones such as dioscin can play an antioxidant role through activating the PGC-1α/ERα pathway to prevent postmenopausal AS [54]. Notably, chronic infectious disease-associated pathogens have been detected in atherosclerotic plaques, such as pathogenic bacteria including the periodontitis-associated Porphyromonas gingivalis (P. gingivalis). Further, Xie et al. revealed that BMAL1-downregulation and its associated circadian clock disturbance aggravate P. gingivalis-induced atherosclerosis in ApoE-/- mice by elevating oxidative stress formation [55].
3.2. Oxidative Stress in Aortic Dissection (AD)
The term “aortic dissection (AD)” refers to the process of intimal tearing within the aortic wall, causing a false lumen and rapid expansion in the aorta. AD is a common clinical type of acute aortic syndrome (AAS), with high mortality and poor prognosis. Hypertension and intimal injury are two important factors in the formation of AD, and atherosclerotic degeneration of aorta is a common precipitating factor for intimal injury, which often leads to intimal tears and the destruction of vascular wall structure [56]. VSMCs and ECs are two main cell types in the vascular system. Therefore, dysfunction of VSMCs and ECs is a key factor in the formation of AD. In pathophysiology, AD is characterized by excessive VSMC loss, ECM degradation, and inflammation. The intima of the aorta consists of collagen, elastin fibers, and a monolayer of ECs. As a result, the aorta is prone to intimal damage by inflammation or physical stimulation, eventually leading to intimal tears [6, 57, 58].
ROS result in ECM remodeling and SMCs apoptosis by activating multiple hypertrophy-signaling kinases and transcription factors and inducing the release of matrix metalloproteinases (MMPs), which leads to ECM remodeling and induces smooth muscle cell apoptosis [59, 60]. Studies have also shown that SMC apoptosis is related to the oxidative stress caused by ribosome biogenesis. Previously, Wu et al. found decreased ribosome biogenesis in aortic-SMCs, which results in apoptosis associated with p53-dependent proliferative inhibition [61]. Based on emerging evidence, autophagy is connected with inflammation, and excessive autophagy leads to the death of ECs, which can result in intimal tear [62]. Consequently, improving endothelial dysfunction, ECM degradation, and SMCs death, all of which can be potential targets for the non-surgical treatment of AD.
Liu and Desai previously revealed that transforming growth factor-beta (TGF-β) induces myofibroblast differentiation by activating NOX4 to promote ROS production [63]. Meanwhile, TGF-β1 induces hydrogen peroxide-inducible clone 5 (Hic-5) expression in a delayed manner, which can bind to heat shock protein HSP27 to counter these effects [64, 65]. Zou et al. showed that HSP27 produced by VSMCs was higher in the aortic wall of AD patients and verified that HSP27 plays a positive role in preventing AD by promoting cell proliferation and inhibiting apoptosis [66]. Accordingly, through further research, Desai et al. discovered that TGF-β1-induced Hic-5 degrades NOX4 through a ubiquitination pathway, limiting the senescence of myofibroblasts by a negative feedback mechanism [67]. This is the first study to certify the posttranslational regulation of NOX4 (Figure 2).
Figure 2.
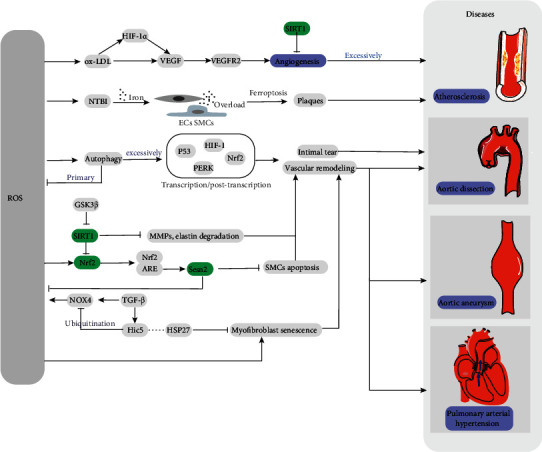
The related mechanism of ROS and diseases.
Sestrin2 (Sesn2) is an antioxidant protein involved in diseases by regulating levels of oxidative stress [68]. The upregulation of Sesn2 is reported to decrease ox-LDL-induced ROS production and alleviate the apoptosis of macrophage. Sesn2 is also found to be increased in both the aortic tissues and plasma of AD patients. Therefore, through further studies, Xiao et al. found that Sesn2 attenuates Ang II-induced SMCs apoptosis and prevents AD through Nrf2-ARE pathway [69–71].
Potente and his team showed that SIRT1, which belongs to the Sirtuin protein family, is a pivotal protein that regulates angiogenesis. Functional defects of SIRT1 can lead to downregulation of genes involved in vascular development and remodeling, resulting in the vascular-related diseases [72]. Melatonin, one of the hormones secreted by the pineal gland, has been shown to regulate circadian rhythms and play an important role in antioxidative stress and downregulation of MMPs [73]. Xia et al. demonstrated in mouse experiments that melatonin reduces oxidative stress and VSMCs loss by activating SIRT1 signaling in a receptor-dependent manner, which can be a potential therapeutic target in AD [74].
In recent years, researchers have shown that autophagy is important in the pathogenesis of AD. As a common mode of regulated cell death, autophagy is responsible for the degradation of damaged organelles and some proteins which cannot be degraded by proteasomes [75]. Metabolic stress or oxidative stress induces autophagy to degrade potentially harmful ROS-producing organelles, such as the mitochondria. Therefore, autophagy is inherently cytoprotective [76, 77]. Based on emerging evidence, excessive stimulation of autophagy can lead to the death of ECs, which can result in aberrant vascular remodeling or intimal tear. Therefore, whether autophagy is protective or deleterious in AD depends on whether it limits or increases abnormal endothelial proliferation, or whether it prevents normal angiogenesis [50, 78]. Besides of this, autophagy has been confirmed in the lungs of humans with pulmonary arterial hypertension (PAH) and may have a protective effect on endothelial injury that initiates vascular remodeling in PAH [79].
Resveratrol (RES), as a phytoestrogen, is widely found in red wine and grapes and has been proven to have antioxidant properties [80]. Van Andel et al. found that RES can effectively combat oxidative stress by inhibiting NOX production of mitochondrial superoxide, which has a protective effect on the aorta [81].
3.3. Oxidative Stress in Aortic Aneurysm (AA)
AA refers to permanent abnormal expansion or localized bulging caused by local pathological weakness of the aortic wall and extension after hypotonia, without intimal tearing. SMCs are the major components of the aortic wall, and their loss through apoptosis or necrosis is a major defining feature of both AA and AD [82]. AA is also characterized by abnormalities in the ECM that compromise the structural integrity of the aorta, and MMPs play a key role in ECM remodeling. Inflammation and ROS are cardinal features of AA. However, ROS in the pathogenesis of AA and AD has received less attention than AS [83–85].
Superoxide dismutase (SOD) is an effective antioxidant enzyme. In fact, zinc can combine with SOD3 as a cofactor to alleviate oxidative stress [86]. Therefore, Zn deficiency plays a crucial role in oxidative damage and inflammatory responses, such as wound healing and homeostasis [87, 88]. Socha et al. determined the mineral content in aorta tissue samples from 108 patients with AA and found that zinc concentration in AA patients was significantly lower than that in healthy subjects [89]. Accordingly, supplementation with antioxidant trace elements in patients with aortic aneurysm may improve the prognosis and mortality of patients.
Nrf2 and its main intracellular regulator, Keap1, function in a pervasive intracellular defense mechanisms to against oxidative stress [90]. Nrf2 can protect cells and restore redox homeostasis by scavenging excessive ROS levels, so it has been proved to have a function of preventing diseases [91, 92]. Kopacz et al. found that inhibiting Nrf2 transcriptional activity in mouse helped AA formation, which can be prevented by simvastatin [93]. Interestingly, inhibition of Nrf2 expression has previously been found to increase the activation of the RAC1-dependent nuclear factor-κB (NF-κB), which means that Nrf2 also plays a critical role in counteracting NF-κβ-driven inflammatory responses [94]. Lithium chloride (LiCl) is a compound which can effectively reduce inflammatory response. Xu and his team provided the first evidence that LiCl inhibits ROS production, facilitates the synthesis of elastin, and maintains the stability of aorta structure by regulating the GSK3β/SIRT1/NF-κB cascade, ultimately preventing the development of AA [95]. Additionally, LiCl was found to significantly induce SIRT1 expression, which can reduce oxidative stress and VSMC loss to prevent AA [72]. Based on recent evidence, the deficiency of kappa B kinase epsilon (IKK-epsilon) inhibitors also effectively inhibits the generation of ROS, macrophage infiltration, and VSMC apoptosis [96]. Such finding highlights a novel therapeutic strategy for the prevention and treatment of AA by targeting IKK-epsilon.
Aldehyde dehydrogenase 2 (ALDH2) is an enzyme associated with aldehyde metabolism that is mainly located in mitochondria and is closely related to oxidative metabolism of aldehydes produced by lipid peroxidation. Studies have found that lack of ALDH2 can lead to the imbalance between antioxidant defense activity and ROS production, aggravating oxidative stress. Therefore, as an antioxidant gene, ALDH2 activator plays a potential therapeutic role in AA [97, 98].
Heme oxygenase-1 (HO-1) is a cell-protective heme degrading enzyme, which regulates inflammatory responses by limiting intracellular levels of oxyhemoglobin [99]. HO-1 has been reported to attenuate the development of AA in mouse models induced by Ang-II. However, Kopacz et al. revealed the dual role of HO-1 in AA formation, which can prevent the development of AA while simultaneously exacerbating the formation of AA states and reducing the risk of rupture [100, 101].
3.4. Oxidative Stress in Pulmonary Arterial Hypertension (PAH)
PAH is a complex degenerative disease and refers to a resting elevated mean pulmonary arterial pressure (≥25 mmHg) and normal wedge pressure (<15 mmHg) [102, 103]. The pathogenesis of PAH is still unclear and is characterized by excessive proliferation of vascular cells and abnormal vascular remodeling. EC dysfunction has been found to produce a variety of endothelial vasoactive mediators, which in turn promote proliferation and migration of VSMCs. And ultimately, hyperproliferation of disordered ECs and VSMCs can induce remodeling of small pulmonary arteries [104–106].
In recent decades, with the development of molecular genetics, the genetics of PAH has evolved rapidly as a research hotspot. Loss of bone morphogenetic protein (BMP) signaling was found to be a major risk factor for PAH development, especially BMPR2 [106]. BMPR2 activates serine/threonine kinases by encoding TGF-β II receptors, resulting in transcriptional regulation of phosphorylated Smads. Studies have proved that PAH is caused by mutations in BMPR2 affecting the TGFβ/Smads signaling pathway [107]. The findings of several recent studies have shown increased DNA damage in lung vascular cells from PAH patients. Federici et al. verified this phenomenon through measurements and found that DNA damage is prevalent in PAHs, which may be genetically related. This study is the first to directly link oxidative stress-induced DNA damage to ROS overload in PAH cells [108].
In the pathogenesis of PAH, the dysfunction of ECs causes homeostasis imbalance of endothelium-derived vasodilator and constrictor factors [105]. Several studies have shown that the generation of endothelium-derived nitric oxide (EDNO) is essential for maintaining vascular homeostasis [5]. Substantial evidence supports that excessive production of ROS directly inactivates EDNO, acts as cell signaling molecules, and promotes protein dysfunction [109]. Phosphodiesterases-5 (PDE-5), which is highly expressed in lung tissue and significant upregulated in PAH, can lead to endothelial dysfunction by inactivating cGMP affecting the vasodilator-NO action. Therefore, the use of PDE inhibitors is of great significance in the treatment of PAH [110, 111].
4. Oxidative Stress in Angiogenesis
Although high concentrations of ROS have vascular-damaging effects, oxidative stress has been proved to take an active role in angiogenesis [112]. Typically, ROS derived from NOXs and mitochondrial can induce angiogenesis directly or indirectly [113]. These mechanisms often involve hypoxia-inducible factor/vascular endothelial growth factor (VEGF) signaling, which is essential for normal vascular development and has great potential in wound repair and the treatment of ischemic diseases [114, 115].
5. New Advances in Antioxidant Therapy
In summary, excessive generation of ROS or failure of the ROS clearance system is the main factor causing ROS accumulation. Consequently, inhibiting the generation of ROS and promoting the metabolism of ROS are essential for the antioxidative stress response. Xie et al. revealed that CoQ10 can affect mitochondrial function, inhibit ETC-derived ROS production, and control oxidative stress responses by activating the AMPK/YAP/OPA1 pathway [116]. It has been established that the antioxidant system plays a key role in the process of antioxidative stress (Table 1).
Table 1.
Role of antioxidants on vascular diseases.
| Antioxidants | Effect on antioxidant system | Reference |
|---|---|---|
| CoQ10 | Activating AMPK-YAP-OPA1 pathway, decrease mitochondrial superoxide | 116 |
| Resveratrol (RES) | As a phytoestrogen, decrease NOX, mitochondrial superoxide | 54,81 |
| Zn | Combines with SOD3 as a cofactor | 86-88 |
| Catalase | Detoxify hydrogen peroxide into water | 117 |
| GSH-Px | Reducing properties | 117 |
| Statins | Decrease NOX, prevent eNOs uncoupling | 93 |
| Lithium chloride (LiCl) | Regulation GSK3β/SIRT1/NF-κB cascade, decrease inflammation, MMPs, and superoxide | 72,95 |
| β-Carotene | β-Carotene | 117 |
| Vitamin C | Water-soluble antioxidant | 119 |
| Vitamin E | Activated to α-tocopherol, preventing lipid per-oxidation | 117 |
| Melatonin | Via activation of SIRT1 signaling in a receptor-dependent manner. | 69 |
The antioxidant system can protect tissues from ROS toxicity, which includes antioxidant enzymes such as superoxide dismutase (SODs), catalase and glutathione peroxidase (GSH-Px), and nonenzymatic antioxidants such as bilirubin, α-tocopherol, and β-carotene [117].
Among them, SODs are the most potent antioxidant enzymes and are metalloproteins which catalyze the conversion of superoxide anions (•O2−) into H2O2. Three isoforms of SOD required different cofactors to assist their activities: copper (Cu) and zinc (Zn) are cofactors for SOD1 and SOD3, whereas the SOD2 tetramer requires manganese (Mn) as a cofactor [118]. The average concentrations of Zn and Cu in the aorta of patients with AA were significantly lower than those in the aorta samples of healthy individuals [89]. Therefore, supplementation of micronutrients with antioxidant properties such as Zn and Cu in the perioperative period can improve the prognosis of surgery to a certain extent.
Catalase and Gpx convert hydrogen peroxide into water, and Gpx has reducing properties. Vitamin C, which is known as ascorbic acid, is also recognized to be the most effective antioxidant in plasma [119]. As a lipophilic vitamin, vitamin E exerts a powerful antioxidant effect by preventing lipid peroxidation.
6. Conclusion and Perspectives
In this review, we introduce the mechanism of ROS production and analyze the role of oxidative stress responses in the pathogenesis of AS, AD, AA, and PAH in detail. As mentioned previously, the imbalance between prooxidant and antioxidant systems results in the excessive production of ROS, which affects the inflammatory response and cell proliferation, death and transformation through various pathways, leading to the occurrence of a variety of common diseases (Figure 1). Aortic diseases are life-threatening conditions with no effective medicines for the treatment, which mainly include aortic open surgery or endovascular replacement.
At present, studies have confirmed that ROS can regulate the proliferation, apoptosis, extracellular matrix remodeling, and intima damage of vascular cells through a variety of classical molecules and pathways, such as Nrf2, TGF-β, and the novel pathways of regulated cell death such as autophagy and ferroptosis also play an important role, thus causing the occurrence of a variety of vascular diseases (Figure 2). As biomarkers of these diseases, well-controlled ROS may be beneficial for preventing cardiovascular disease and promoting angiogenesis during tissue repair. Therefore, our research is mainly devoted to the prevention, improvement of the prognosis, and effective nonsurgical therapies for such diseases via the identification of targets for antioxidant drugs.
As we all know, as the most common dietary antioxidant compounds, vitamin A, C, E, and other trace elements, phenolic compounds, and carotenoids are abundantly found in plants and served as a safe way to prevent diseases. In addition, as effective antioxidants, antioxidant enzymes and their cofactors, such as zinc and copper, have also been extensively studied and play a great potential in the prevention and treatment of diseases.
Acknowledgments
This work was supported by Department of Human Resource and Social Security of Shanxi province China (grant number 2019-29) and Science and Technology Department of Shanxi Province (grant number 201701D121152).
Conflicts of Interest
The authors declare that they have no competing interests.
References
- 1.Halliwell B. The role of oxygen radicals in human disease, with particular reference to the vascular system. Haemostasis . 1993;23(Supplement 1):118–126. doi: 10.1159/000216921. [DOI] [PubMed] [Google Scholar]
- 2.Kaludercic N., Di Lisa F. Mitochondrial ROS formation in the pathogenesis of diabetic cardiomyopathy. Frontiers in Cardiovascular Medicine . 2020;7:p. 12. doi: 10.3389/fcvm.2020.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kattoor A. J., Pothineni N. V. K., Palagiri D., Mehta J. L. Oxidative stress in atherosclerosis. Current Atherosclerosis Reports . 2017;19(11):p. 42. doi: 10.1007/s11883-017-0678-6. [DOI] [PubMed] [Google Scholar]
- 4.Ruan Y., Jiang S., Musayeva A., Gericke A. Oxidative stress and vascular dysfunction in the retina: therapeutic strategies. Antioxidants . 2020;9(8) doi: 10.3390/antiox9080761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas S. R., Witting P. K., Drummond G. R. Redox control of endothelial function and dysfunction: molecular mechanisms and therapeutic opportunities. Antioxidants & Redox Signaling . 2008;10(10):1713–1766. doi: 10.1089/ars.2008.2027. [DOI] [PubMed] [Google Scholar]
- 6.Xu T., Ding W., Ji X., et al. Oxidative stress in cell death and cardiovascular diseases. Oxidative Medicine and Cellular Longevity . 2019;2019 doi: 10.1155/2019/9030563.9030563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao S., Cheng C. K., Zhang C. L., Huang Y. Interplay between oxidative stress, cyclooxygenases, and prostanoids in cardiovascular diseases. Antioxidants & Redox Signaling . 2021;34(10):784–799. doi: 10.1089/ars.2020.8105. [DOI] [PubMed] [Google Scholar]
- 8.Scherz-Shouval R., Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends in Biochemical Sciences . 2011;36(1):30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 9.Münzel T., Camici G. G., Maack C., Bonetti N. R., Fuster V., Kovacic J. C. Impact of oxidative stress on the heart and vasculature: part 2 of a 3-part series. Journal of the American College of Cardiology . 2017;70(2):212–229. doi: 10.1016/j.jacc.2017.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H., Horke S., Forstermann U. Oxidative stress in vascular disease and its pharmacological prevention. Trends in Pharmacological Sciences . 2013;34(6):313–319. doi: 10.1016/j.tips.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 11.Bir S. C., Kolluru G. K., Fang K., Kevil C. G. Redox balance dynamically regulates vascular growth and remodeling. Seminars in Cell & Developmental Biology . 2012;23(7):745–757. doi: 10.1016/j.semcdb.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reuter S., Gupta S. C., Chaturvedi M. M., Aggarwal B. B. Oxidative stress, inflammation, and cancer: how are they linked? Free Radical Biology & Medicine . 2010;49(11):1603–1616. doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai H., Harrison D. G. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circulation Research . 2000;87(10):840–844. doi: 10.1161/01.RES.87.10.840. [DOI] [PubMed] [Google Scholar]
- 14.Cao S. S., Kaufman R. J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxidants & Redox Signaling . 2014;21(3):396–413. doi: 10.1089/ars.2014.5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winterbourn C. C. The biological chemistry of hydrogen peroxide. Methods in Enzymology . 2013;528:3–25. doi: 10.1016/B978-0-12-405881-1.00001-X. [DOI] [PubMed] [Google Scholar]
- 16.Knaus U. G. Oxidants in physiological processes. Handbook of Experimental Pharmacology . 2021;264:27–47. doi: 10.1007/164_2020_380. [DOI] [PubMed] [Google Scholar]
- 17.Fukai T., Ushio-Fukai M. Cross-talk between NADPH oxidase and mitochondria: role in ROS signaling and angiogenesis. Cell . 2020;9(8):p. 1849. doi: 10.3390/cells9081849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Awad M. A., Aldosari S. R., Abid M. R. Genetic alterations in oxidant and anti-oxidant enzymes in the vascular system. Frontiers in cardiovascular . 2018;5:p. 107. doi: 10.3389/fcvm.2018.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Portelli S. S., Hambly B. D., Jeremy R. W., Robertson E. N. Oxidative stress in genetically triggered thoracic aortic aneurysm: role in pathogenesis and therapeutic opportunities. Redox Report . 2021;26(1):45–52. doi: 10.1080/13510002.2021.1899473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lassegue B., San Martin A., Griendling K. K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circulation Research . 2012;110(10):1364–1390. doi: 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drummond G. R., Selemidis S., Griendling K. K., Sobey C. G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nature Reviews. Drug Discovery . 2011;10(6):453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sumimoto H., Hata K., Mizuki K., et al. Assembly and activation of the phagocyte NADPH oxidase. Specific interaction of the N-terminal Src homology 3 domain of p47phox with p22phox is required for activation of the NADPH oxidase. The Journal of Biological Chemistry . 1996;271(36):22152–22158. doi: 10.1074/jbc.271.36.22152. [DOI] [PubMed] [Google Scholar]
- 23.Vermot A., Petit-Härtlein I., Smith S. M. E., Fieschi F. NADPH oxidases (NOX): an overview from discovery, molecular mechanisms to physiology and pathology. Antioxidants . 2021;10(6):p. 890. doi: 10.3390/antiox10060890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Groemping Y., Rittinger K. Activation and assembly of the NADPH oxidase: a structural perspective. The Biochemical Journal . 2005;386(3):401–416. doi: 10.1042/BJ20041835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bedard K., Krause K. H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological Reviews . 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 26.Martyn K. D., Frederick L. M., von Loehneysen K., Dinauer M. C., Knaus U. G. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cellular Signalling . 2006;18(1):69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 27.Mital S., Liao J. K. Statins and the myocardium. Seminars in Vascular Medicine . 2004;4(4):377–384. doi: 10.1055/s-2004-869594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y., Murugesan P., Huang K., Cai H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: novel therapeutic targets. Nature Reviews. Cardiology . 2020;17(3):170–194. doi: 10.1038/s41569-019-0260-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dubois-Deruy E., Peugnet V., Turkieh A., Pinet F. Oxidative stress in cardiovascular diseases. Antioxidants . 2020;9(9) doi: 10.3390/antiox9090864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo J., Mills K., le Cessie S., Noordam R., van Heemst D. Ageing, age-related diseases and oxidative stress: what to do next? Ageing Research Reviews . 2020;57, article 100982 doi: 10.1016/j.arr.2019.100982. [DOI] [PubMed] [Google Scholar]
- 31.Zhao R. Z., Jiang S., Zhang L., Yu Z. B. Mitochondrial electron transport chain, ROS generation and uncoupling (review) International Journal of Molecular Medicine . 2019;44(1):3–15. doi: 10.3892/ijmm.2019.4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turrens J. F., Alexandre A., Lehninger A. L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Archives of Biochemistry and Biophysics . 1985;237(2):408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 33.Villalpando-Rodriguez G. E., Gibson S. B. Reactive oxygen species (ROS) regulates different types of cell death by acting as a rheostat. Oxidative Medicine and Cellular Longevity . 2021;2021:17. doi: 10.1155/2021/9912436.9912436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montezano A. C., Dulak-Lis M., Tsiropoulou S., Harvey A., Briones A. M., Touyz R. M. Oxidative stress and human hypertension: vascular mechanisms, biomarkers, and novel therapies. The Canadian Journal of Cardiology . 2015;31(5):631–641. doi: 10.1016/j.cjca.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 35.Forstermann U. Oxidative stress in vascular disease: causes, defense mechanisms and potential therapies. Nature Clinical Practice. Cardiovascular Medicine . 2008;5(6):338–349. doi: 10.1038/ncpcardio1211. [DOI] [PubMed] [Google Scholar]
- 36.Borges F., Fernandes E., Roleira F. Progress towards the discovery of xanthine oxidase inhibitors. Current Medicinal Chemistry . 2002;9(2):195–217. doi: 10.2174/0929867023371229. [DOI] [PubMed] [Google Scholar]
- 37.George J., Struthers A. D. Role of urate, xanthine oxidase and the effects of allopurinol in vascular oxidative stress. Vascular Health and Risk Management . 2009;5(1):265–272. doi: 10.2147/VHRM.S4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Forstermann U., Xia N., Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circulation Research . 2017;120(4):713–735. doi: 10.1161/CIRCRESAHA.116.309326. [DOI] [PubMed] [Google Scholar]
- 39.Hansson G. K., Hermansson A. The immune system in atherosclerosis. Nature Immunology . 2011;12(3):204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 40.Zhang D. X., Gutterman D. D. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. American Journal of Physiology. Heart and Circulatory Physiology . 2007;292(5):H2023–H2031. doi: 10.1152/ajpheart.01283.2006. [DOI] [PubMed] [Google Scholar]
- 41.Ketelhuth D. F., Hansson G. K. Cellular immunity, low-density lipoprotein and atherosclerosis: break of tolerance in the artery wall. Thrombosis and Haemostasis . 2011;106(11):779–786. doi: 10.1160/TH11-05-0321. [DOI] [PubMed] [Google Scholar]
- 42.Marchio P., Guerra-Ojeda S., Vila J. M., Aldasoro M., Victor V. M., Mauricio M. D. Targeting early atherosclerosis: a focus on oxidative stress and inflammation. Oxidative Medicine and Cellular Longevity . 2019;2019:32. doi: 10.1155/2019/8563845.8563845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salomonsson L., Pettersson S., Englund M. C. O., Wiklund O., Ohlsson B. G. Post-transcriptional regulation of VEGF expression by oxidised LDL in human macrophages. European Journal of Clinical Investigation . 2002;32(10):767–774. doi: 10.1046/j.1365-2362.2002.01072.x. [DOI] [PubMed] [Google Scholar]
- 44.Hutter R., Speidl W. S., Valdiviezo C., et al. Macrophages transmit potent proangiogenic effects of oxLDL in vitro and in vivo involving HIF-1α activation: a novel aspect of angiogenesis in atherosclerosis. Journal of Cardiovascular Translational Research . 2013;6(4):558–569. doi: 10.1007/s12265-013-9469-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chistiakov D. A., Melnichenko A. A., Myasoedova V. A., Grechko A. V., Orekhov A. N. Mechanisms of foam cell formation in atherosclerosis. Journal of Molecular Medicine (Berlin, Germany) . 2017;95(11):1153–1165. doi: 10.1007/s00109-017-1575-8. [DOI] [PubMed] [Google Scholar]
- 46.Osonoi Y., Mita T., Azuma K., et al. Defective autophagy in vascular smooth muscle cells enhances cell death and atherosclerosis. Autophagy . 2018;14(11):1991–2006. doi: 10.1080/15548627.2018.1501132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vinchi F., Muckenthaler M. U., Da Silva M. C., Balla G., Balla J., Jeney V. Atherogenesis and iron: from epidemiology to cellular level. Frontiers in Pharmacology . 2014;5 doi: 10.3389/fphar.2014.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Araujo J. A., Romano E. L., Brito B. E., et al. Iron overload augments the development of atherosclerotic lesions in rabbits. Arteriosclerosis, Thrombosis, and Vascular Biology . 1995;15(8):1172–1180. doi: 10.1161/01.ATV.15.8.1172. [DOI] [PubMed] [Google Scholar]
- 49.Vinchi F., Porto G., Simmelbauer A., et al. Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. European Heart Journal . 2020;41(28):2681–2695. doi: 10.1093/eurheartj/ehz112. [DOI] [PubMed] [Google Scholar]
- 50.Chen Y., He Y., Wei X., Jiang D. S. Targeting regulated cell death in aortic aneurysm and dissection therapy. Pharmacological Research . 2021;176, article 106048 doi: 10.1016/j.phrs.2021.106048. [DOI] [PubMed] [Google Scholar]
- 51.Wei X., Yi X., Zhu X.-H., Jiang D.-S. Posttranslational modifications in ferroptosis. Oxidative Medicine and Cellular Longevity . 2020;2020:12. doi: 10.1155/2020/8832043.8832043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sample S. J., Racette M. A., Hao Z., Thomas C. F., Behan M., Muir P. Functional adaptation in female rats: the role of estrogen signaling. PLoS One . 2012;7(9, article e43215) doi: 10.1371/journal.pone.0043215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lamas A. Z., Caliman I. F., Dalpiaz P. L. M., et al. Comparative effects of estrogen, raloxifene and tamoxifen on endothelial dysfunction, inflammatory markers and oxidative stress in ovariectomized rats. Life Sciences . 2015;124:101–109. doi: 10.1016/j.lfs.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 54.Yang Q., Wang C., Jin Y., et al. Disocin prevents postmenopausal atherosclerosis in ovariectomized LDLR-/- mice through a PGC-1α/ERα pathway leading to promotion of autophagy and inhibition of oxidative stress, inflammation and apoptosis. Pharmacological Research . 2019;148, article 104414 doi: 10.1016/j.phrs.2019.104414. [DOI] [PubMed] [Google Scholar]
- 55.Xie M., Tang Q., Nie J., et al. BMAL1-downregulation aggravates <i>Porphyromonas gingivalis</i>-induced atherosclerosis by encouraging oxidative stress. Circulation Research . 2020;126(6):e15–e29. doi: 10.1161/CIRCRESAHA.119.315502. [DOI] [PubMed] [Google Scholar]
- 56.Tchana-Sato V., Sakalihasan N., Defraigne J. O. Aortic dissection. Revue Médicale de Liège . 2018;73(5-6):290–295. [PubMed] [Google Scholar]
- 57.Goldfinger J. Z., Halperin J. L., Marin M. L., Stewart A. S., Eagle K. A., Fuster V. Thoracic aortic aneurysm and dissection. Journal of the American College of Cardiology . 2014;64(16):1725–1739. doi: 10.1016/j.jacc.2014.08.025. [DOI] [PubMed] [Google Scholar]
- 58.Sun Y., Xiao Y., Sun H., et al. miR-27a regulates vascular remodeling by targeting endothelial cells' apoptosis and interaction with vascular smooth muscle cells in aortic dissection. Theranostics . 2019;9(25):7961–7975. doi: 10.7150/thno.35737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsutsui H., Kinugawa S., Matsushima S. Oxidative stress and heart failure. American Journal of Physiology. Heart and Circulatory Physiology . 2011;301(6):H2181–H2190. doi: 10.1152/ajpheart.00554.2011. [DOI] [PubMed] [Google Scholar]
- 60.Liu W., Wang B., Wang T., et al. Ursodeoxycholic acid attenuates acute aortic dissection formation in angiotensin II-infused apolipoprotein E-deficient mice associated with reduced ROS and increased Nrf2 levels. Cellular Physiology and Biochemistry . 2016;38(4):1391–1405. doi: 10.1159/000443082. [DOI] [PubMed] [Google Scholar]
- 61.Wu Q., Hong J., Wang Z., et al. Abnormal ribosome biogenesis partly induced p53-dependent aortic medial smooth muscle cell apoptosis and oxidative stress. Oxidative Medicine and Cellular Longevity . 2019;2019:19. doi: 10.1155/2019/7064319.7064319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qin Y., Zheng B., Yang G. S., et al. _Salvia miltiorrhiza-_ derived Sal-miR-58 induces autophagy and attenuates inflammation in vascular smooth muscle cells. Mol Ther Nucleic Acids . 2020;21:492–511. doi: 10.1016/j.omtn.2020.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu R. M., Desai L. P. Reciprocal regulation of TGF-β and reactive oxygen species: a perverse cycle for fibrosis. Redox Biology . 2015;6:565–577. doi: 10.1016/j.redox.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dabiri G., Tumbarello D. A., Turner C. E., van de Water L. TGF- _β_ 1 slows the growth of pathogenic myofibroblasts through a mechanism requiring the focal adhesion protein, Hic-5. The Journal of Investigative Dermatology . 2008;128(2):280–291. doi: 10.1038/sj.jid.5700975. [DOI] [PubMed] [Google Scholar]
- 65.Jia Y., Ransom R. F., Shibanuma M., Liu C., Welsh M. J., Smoyer W. E. Identification and characterization of hic-5/ARA55 as an hsp27 binding protein∗. The Journal of Biological Chemistry . 2001;276(43):39911–39918. doi: 10.1074/jbc.M103510200. [DOI] [PubMed] [Google Scholar]
- 66.Zou S., Liao M., Yang J., et al. Heat shock protein 27 plays a protective role in thoracic aortic dissection by promoting cell proliferation and inhibiting apoptosis. Cellular & Molecular Biology Letters . 2017;22(1):p. 24. doi: 10.1186/s11658-017-0056-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Desai L. P., Zhou Y., Estrada A. V., et al. Negative regulation of NADPH oxidase 4 by hydrogen peroxide-inducible clone 5 (Hic-5) protein∗. The Journal of Biological Chemistry . 2014;289(26):18270–18278. doi: 10.1074/jbc.M114.562249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pasha M., Eid A. H., Eid A. A., Gorin Y., Munusamy S. Sestrin2 as a novel biomarker and therapeutic target for various diseases. Oxidative Medicine and Cellular Longevity . 2017;2017:10. doi: 10.1155/2017/3296294.3296294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xiao T., Zhang L., Huang Y., et al. Sestrin2 increases in aortas and plasma from aortic dissection patients and alleviates angiotensin II-induced smooth muscle cell apoptosis via the Nrf2 pathway. Life Sciences . 2019;218:132–138. doi: 10.1016/j.lfs.2018.12.043. [DOI] [PubMed] [Google Scholar]
- 70.Hu H. J., Shi Z. Y., Lin X. L., Chen S. M., Wang Q. Y., Tang S. Y. Upregulation of Sestrin2 expression protects against macrophage apoptosis induced by oxidized low-density lipoprotein. DNA and Cell Biology . 2015;34(4):296–302. doi: 10.1089/dna.2014.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Park S. J., Cho S. S., Kim K. M., et al. Protective effect of sestrin2 against iron overload and ferroptosis-induced liver injury. Toxicology and Applied Pharmacology . 2019;379, article 114665 doi: 10.1016/j.taap.2019.114665. [DOI] [PubMed] [Google Scholar]
- 72.Potente M., Ghaeni L., Baldessari D., et al. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes & Development . 2007;21(20):2644–2658. doi: 10.1101/gad.435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reiter R. J., Tan D., Rosales-Corral S., Galano A., Zhou X., Xu B. Mitochondria: central organelles for melatonin's antioxidant and anti-aging actions. Molecules . 2018;23(2):p. 509. doi: 10.3390/molecules23020509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xia L., Sun C., Zhu H., et al. Melatonin protects against thoracic aortic aneurysm and dissection through SIRT1-dependent regulation of oxidative stress and vascular smooth muscle cell loss. Journal of Pineal Research . 2020;69(1, article e12661) doi: 10.1111/jpi.12661. [DOI] [PubMed] [Google Scholar]
- 75.Racanelli A. C., Kikkers S. A., Choi A. M. K., Cloonan S. M. Autophagy and inflammation in chronic respiratory disease. Autophagy . 2018;14(2):221–232. doi: 10.1080/15548627.2017.1389823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schaaf M. B., Houbaert D., Meçe O., Agostinis P. Autophagy in endothelial cells and tumor angiogenesis. Cell Death and Differentiation . 2019;26(4):665–679. doi: 10.1038/s41418-019-0287-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li L., Tan J., Miao Y., Lei P., Zhang Q. ROS and autophagy: interactions and molecular regulatory mechanisms. Cellular and Molecular Neurobiology . 2015;35(5):615–621. doi: 10.1007/s10571-015-0166-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chichger H., Rounds S., Harrington E. O. Endosomes and autophagy: regulators of pulmonary endothelial cell homeostasis in health and disease. Antioxidants & Redox Signaling . 2019;31(13):994–1008. doi: 10.1089/ars.2019.7817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang C. F., Zhao F. Y., Xu S. L., Liu J., Xing X. Q., Yang J. Autophagy in pulmonary hypertension: emerging roles and therapeutic implications. Journal of Cellular Physiology . 2019;234(10):16755–16767. doi: 10.1002/jcp.28531. [DOI] [PubMed] [Google Scholar]
- 80.Bonnefont-Rousselot D. Resveratrol and cardiovascular diseases. Nutrients . 2016;8(5):p. 250. doi: 10.3390/nu8050250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van Andel M. M., Groenink M., Zwinderman A., Mulder B., de Waard V. The potential beneficial effects of resveratrol on cardiovascular complications in Marfan syndrome patients(-)insights from rodent-based animal studies. International Journal of Molecular Sciences . 2019;20(5):p. 1122. doi: 10.3390/ijms20051122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lai C. H., Chang C. W., Lee F. T., et al. Targeting vascular smooth muscle cell dysfunction with xanthine derivative KMUP-3 inhibits abdominal aortic aneurysm in mice. Atherosclerosis . 2020;297:16–24. doi: 10.1016/j.atherosclerosis.2020.01.029. [DOI] [PubMed] [Google Scholar]
- 83.Quintana R. A., Taylor W. R. Cellular mechanisms of aortic aneurysm formation. Circulation Research . 2019;124(4):607–618. doi: 10.1161/CIRCRESAHA.118.313187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gurung R., Choong A. M., Woo C. C., Foo R., Sorokin V. Genetic and epigenetic mechanisms underlying vascular smooth muscle cell phenotypic modulation in abdominal aortic aneurysm. International Journal of Molecular Sciences . 2020;21(17):p. 6334. doi: 10.3390/ijms21176334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li Z., Kong W. Cellular signaling in abdominal aortic aneurysm. Cellular Signalling . 2020;70, article 109575 doi: 10.1016/j.cellsig.2020.109575. [DOI] [PubMed] [Google Scholar]
- 86.Lewandowski L., Kepinska M., Milnerowicz H. The copper-zinc superoxide dismutase activity in selected diseases. European Journal of Clinical Investigation . 2019;49(1, article e13036) doi: 10.1111/eci.13036. [DOI] [PubMed] [Google Scholar]
- 87.Guo C. H., Wang C. L. Effects of zinc supplementation on plasma copper/zinc ratios, oxidative stress, and immunological status in hemodialysis patients. International Journal of Medical Sciences . 2013;10(1):79–89. doi: 10.7150/ijms.5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rink L., Gabriel P. Zinc and the immune system. The Proceedings of the Nutrition Society . 2000;59(4):541–552. doi: 10.1017/S0029665100000781. [DOI] [PubMed] [Google Scholar]
- 89.Socha K., Karwowska A., Kurianiuk A., et al. Estimation of selected minerals in aortic aneurysms-impaired ratio of zinc to lead may predispose? Biological Trace Element Research . 2021;199(8):2811–2818. doi: 10.1007/s12011-020-02410-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Itoh K., Ishii T., Wakabayashi N., Yamamoto M. Regulatory mechanisms of cellular response to oxidative stress. Free Radical Research . 1999;31(4):319–324. doi: 10.1080/10715769900300881. [DOI] [PubMed] [Google Scholar]
- 91.Bellezza I., Giambanco I., Minelli A., Donato R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim Biophys Acta Mol Cell Res . 2018;1865(5):721–733. doi: 10.1016/j.bbamcr.2018.02.010. [DOI] [PubMed] [Google Scholar]
- 92.Hybertson B. M., Gao B., Bose S. K., McCord J. M. Oxidative stress in health and disease: the therapeutic potential of Nrf2 activation. Molecular Aspects of Medicine . 2011;32(4-6):234–246. doi: 10.1016/j.mam.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 93.Kopacz A., Werner E., Grochot-Przęczek A., et al. Simvastatin attenuates abdominal aortic aneurysm formation favoured by lack of Nrf2 transcriptional activity. Oxidative Medicine and Cellular Longevity . 2020;2020 doi: 10.1155/2020/6340190.6340190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bellezza I., Mierla A. L., Minelli A. Nrf2 and NF-κB and their concerted modulation in cancer pathogenesis and progression. Cancers . 2010;2(2):483–497. doi: 10.3390/cancers2020483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Xu T., Wang S., Li X., et al. Lithium chloride represses abdominal aortic aneurysm via regulating GSK3β/SIRT1/NF-κB signaling pathway. Free Radical Biology & Medicine . 2021;166:1–10. doi: 10.1016/j.freeradbiomed.2021.02.007. [DOI] [PubMed] [Google Scholar]
- 96.Chai H., Tao Z., Qi Y., et al. IKK epsilon deficiency attenuates angiotensin II-induced abdominal aortic aneurysm formation in mice by inhibiting inflammation, oxidative stress, and apoptosis. Oxidative Medicine and Cellular Longevity . 2020;2020 doi: 10.1155/2020/3602824.3602824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Choi H., Tostes R. C., Webb R. C. Mitochondrial aldehyde dehydrogenase prevents ROS-induced vascular contraction in angiotensin-II hypertensive mice. Journal of the American Society of Hypertension . 2011;5(3):154–160. doi: 10.1016/j.jash.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tsai S. H., Hsu L. A., Tsai H. Y., et al. Aldehyde dehydrogenase 2 protects against abdominal aortic aneurysm formation by reducing reactive oxygen species, vascular inflammation, and apoptosis of vascular smooth muscle cells. The FASEB Journal . 2020;34(7):9498–9511. doi: 10.1096/fj.201902550RRR. [DOI] [PubMed] [Google Scholar]
- 99.Ryter S. W., Choi A. M. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Translational Research . 2016;167(1):7–34. doi: 10.1016/j.trsl.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kopacz A., Klóska D., Werner E., et al. A dual role of heme oxygenase-1 in angiotensin II-induced abdominal aortic aneurysm in the normolipidemic mice. Cell . 2021;10(1):p. 163. doi: 10.3390/cells10010163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jiang W. C., Chen C. M., Hamdin C. D., et al. Therapeutic potential of heme oxygenase-1 in aneurysmal diseases. Antioxidants . 2020;9(11):p. 1150. doi: 10.3390/antiox9111150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mandras S., Kovacs G., Olschewski H., et al. Combination therapy in pulmonary arterial hypertension-targeting the nitric oxide and prostacyclin pathways. Journal of Cardiovascular Pharmacology and Therapeutics . 2021;26(5):453–462. doi: 10.1177/10742484211006531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hoeper M. M., Bogaard H. J., Condliffe R., et al. Definitions and diagnosis of pulmonary hypertension. Journal of the American College of Cardiology . 2013;62(25):D42–D50. doi: 10.1016/j.jacc.2013.10.032. [DOI] [PubMed] [Google Scholar]
- 104.Sakao S., Tatsumi K., Voelkel N. F. Endothelial cells and pulmonary arterial hypertension: apoptosis, proliferation, interaction and transdifferentiation. Respiratory Research . 2009;10(1):p. 95. doi: 10.1186/1465-9921-10-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Thenappan T., Ormiston M. L., Ryan J. J., Archer S. L. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ . 2018;360, article j5492 doi: 10.1136/bmj.j5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Southgate L., Machado R. D., Gräf S., Morrell N. W. Molecular genetic framework underlying pulmonary arterial hypertension. Nature Reviews. Cardiology . 2020;17(2):85–95. doi: 10.1038/s41569-019-0242-x. [DOI] [PubMed] [Google Scholar]
- 107.Lane K. B., Machado R. D., Pauciulo M. W., et al. Heterozygous germline mutations in _BMPR2_ , encoding a TGF-β receptor, cause familial primary pulmonary hypertension. Nature Genetics . 2000;26(1):81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 108.Federici C., Drake K. M., Rigelsky C. M., et al. Increased mutagen sensitivity and DNA damage in pulmonary arterial hypertension. American Journal of Respiratory and Critical Care Medicine . 2015;192(2):219–228. doi: 10.1164/rccm.201411-2128OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sharma A., Bernatchez P. N., de Haan J. B. Targeting endothelial dysfunction in vascular complications associated with diabetes. International journal of vascular medicine . 2012;2012 doi: 10.1155/2012/750126.750126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Montani D., Chaumais M. C., Guignabert C., et al. Targeted therapies in pulmonary arterial hypertension. Pharmacology & Therapeutics . 2014;141(2):172–191. doi: 10.1016/j.pharmthera.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 111.Rybalkin S. D., Yan C., Bornfeldt K. E., Beavo J. A. Cyclic GMP phosphodiesterases and regulation of smooth muscle function. Circulation Research . 2003;93(4):280–291. doi: 10.1161/01.RES.0000087541.15600.2B. [DOI] [PubMed] [Google Scholar]
- 112.Kim Y. W., Byzova T. V. Oxidative stress in angiogenesis and vascular disease. Blood . 2014;123(5):625–631. doi: 10.1182/blood-2013-09-512749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yun J., Rocic P., Pung Y. F., et al. Redox-dependent mechanisms in coronary collateral growth: the "redox window" hypothesis. Antioxidants & Redox Signaling . 2009;11(8):1961–1974. doi: 10.1089/ars.2009.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kim Y. M., Kim S. J., Tatsunami R., Yamamura H., Fukai T., Ushio-Fukai M. ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. American Journal of Physiology. Cell Physiology . 2017;312(6):C749–C764. doi: 10.1152/ajpcell.00346.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Huang Y. J., Nan G. X. Oxidative stress-induced angiogenesis. Journal of Clinical Neuroscience . 2019;63:13–16. doi: 10.1016/j.jocn.2019.02.019. [DOI] [PubMed] [Google Scholar]
- 116.Xie T., Wang C., Jin Y., et al. CoenzymeQ10-induced activation of AMPK-YAP-OPA1 pathway alleviates atherosclerosis by improving mitochondrial function, inhibiting oxidative stress and promoting energy metabolism. Frontiers in Pharmacology . 2020;11:p. 1034. doi: 10.3389/fphar.2020.01034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liguori I., Russo G., Curcio F., et al. Oxidative stress, aging, and diseases. Clinical Interventions in Aging . 2018;Volume 13:757–772. doi: 10.2147/CIA.S158513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sharifi-Rad M., Anil Kumar N. V., Zucca P., et al. Lifestyle, oxidative stress, and antioxidants: back and forth in the pathophysiology of chronic diseases. Frontiers in Physiology . 2020;11:p. 694. doi: 10.3389/fphys.2020.00694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kalyanaraman B. Teaching the basics of redox biology to medical and graduate students: oxidants, antioxidants and disease mechanisms. Redox Biology . 2013;1(1):244–257. doi: 10.1016/j.redox.2013.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]