

Abstract
Tumour cells are characterized by having lost their differentiation state. They constitutively secrete small extracellular vesicles (sEV) called exosomes when they come from late endosomes. Dendrogenin A (DDA) is an endogenous tumour suppressor cholesterol‐derived metabolite. It is a new class of ligand of the nuclear Liver X receptors (LXR) which regulate cholesterol homeostasis and immunity. We hypothesized that DDA, which induces tumour cell differentiation, inhibition of tumour growth and immune cell infiltration into tumours, could functionally modify sEV secreted by tumour cells. Here, we have shown that DDA differentiates tumour cells by acting on the LXRβ. This results in an increased production of sEV (DDA‐sEV) which includes exosomes. The DDA‐sEV secreted from DDA‐treated cells were characterized for their content and activity in comparison to sEV secreted from control cells (C‐sEV). DDA‐sEV were enriched, relatively to C‐sEV, in several proteins and lipids such as differentiation antigens, “eat‐me” signals, lipidated LC3 and the endosomal phospholipid bis(monoacylglycero)phosphate, which stimulates dendritic cell maturation and a Th1 T lymphocyte polarization. Moreover, DDA‐sEV inhibited the growth of tumours implanted into immunocompetent mice compared to control conditions. This study reveals a pharmacological control through a nuclear receptor of exosome‐enriched tumour sEV secretion, composition and immune function. Targeting the LXR may be a novel way to reprogram tumour cells and sEV to stimulate immunity against cancer.
Keywords: bis(monoacylglycero)phosphate, cancer, cholesterol, dendrogenin A, differentiation, exosomes, Lysobisphosphatidic acid, nuclear receptor
1. INTRODUCTION
Dendrogenin A (DDA) is a naturally occurring cholesterol‐derived metabolite discovered in mammals, including humans, which displays tumour suppressor properties. It is a new class of ligand and modulator of the Liver X receptor (LXR), a nuclear receptor and a ligand‐dependent transcription factor (Bauriaud‐Mallet et al., 2019; Segala et al., 2017). LXRs control the lipid metabolism, tumour cell proliferation as well as immune responses (Lin & Gustafsson, 2015; Pontini & Marinozzi, 2021; Silvente‐Poirot et al., 2018). At low concentrations, DDA induces tumour cell redifferentiation and cell‐cycle arrest in various mouse and human tumour cells (Bauriaud‐Mallet et al., 2019; de Medina et al., 2013; de Medina et al., 2009). In melanoma and breast cancer tumour cells, DDA activates melanogenesis and characteristics of lactation, respectively (Bauriaud‐Mallet et al., 2019; de Medina et al., 2009; de Medina et al., 2013). The study of some differentiation characteristics in mammary and thyroid tumour cells indicates a LXRβ‐dependent mechanism (Bauriaud‐Mallet et al., 2019). At higher concentrations, DDA triggers autophagic tumour cell death by acting via the LXRβ (Mouchel et al., 2020; Segala et al., 2017; Serhan et al., 2020). Interestingly, DDA‐induced tumour cell differentiation was associated with tumour growth inhibition as well as increased animal survival and immune cell infiltration into melanoma and mammary tumours (de Medina et al., 2013).
Tumour cells, which have evolved toward a dedifferentiated state, constitutively produce small extracellular vesicles (sEV) of 30–150 nm in diameter. sEV is also produced by a large variety of immune and nonimmune cells. They facilitate intercellular communication by allowing exchange of materials between cells (Mathieu et al., 2019; Record et al., 2011). sEV originates from the intracellular endosome compartment named multivesicular bodies (MVB), and the intraluminal vesicles (ILV) inside MVB are named “exosomes” when secreted. sEV are also released from the shedding of the peripheral plasma membrane and termed “ectosomes” (Kalluri & Lebleu, 2020; Record et al., 2018). sEV contain different types of cargos (proteins, lipids, nucleic acids) representative of the parental cells and their content results from the selective sorting processes operating in the parental cells. They can induce biological functions in target cells following their internalization. sEV are important components of the immune system (Pitt et al., 2014). Antigen‐presenting‐cell‐derived sEV have been reported to activate T lymphocytes and immune responses (Wolfers et al., 2001) and to induce antitumour effects and long‐term survival in animals when loaded with tumour peptides (Zitvogel et al., 1998). sEV derived from dendritic cells (DC) have been used in clinical trials as a potential antitumour vaccine with some clinical benefits (Dai et al., 2008; Escudier et al., 2005).
In contrast, tumour‐sEV intervenes in cancer cell plasticity, resistance to therapy and cancer progression facilitating metastatic dissemination and has inhibitory effects on cells of the immune system that might contribute to tumour‐induced immune evasion (Hoshino et al., 2015; Kalluri & Lebleu, 2020; Li & Nabet, 2019; Ortiz et al., 2019; Peinado et al., 2012; Robbins & Morelli, 2014; Whiteside, 2016).
In the present study, we hypothesized that DDA, which stimulates tumour differentiation and immune cell infiltration into tumours, could modify the content and activity of sEV secreted by tumour cells. The present study highlights a pharmacological control by DDA, via its target receptor, of tumour sEV production, composition and immunogenicity.
2. MATERIALS AND METHODS
2.1. Materials
The B16F10 cell line is a spontaneous mouse melanoma of the C57Bl/6 (H‐2b) strain expressing low levels of MHC class I molecules, BRAF wild type, and the SKMEL28 cell line is a malignant melanoma of human origin, BRAF V600E mutated, obtained from (LGC standards, France). E0771 cells are from a murine breast adenocarcinoma from Tebu‐bio, France. Mouse embryonic fibroblasts (MEF) were derived from 13.5 dpc wild‐type or Lxrβ−/− embryos, as previously described (Segala et al., 2017). DDA was from Affichem, France. 22(R)HC was from Sigma/Merck, France. Standards of BMP for mass spectrometry quantification of the compound were from Avanti Polar lipids (USA/COGER France). The fluorescent lipid probe Bodipy–Ceramide was from Invitrogen–Molecular Probes. L‐α‐Phosphatidylethanolamine‐N‐(lissamine rhodamine B sulfonyl) (Ammonium Salt) [Egg Liss Rhod PE #810146] was supplied by Avanti Polar Lipids. Primary antibodies: Antibodies against CD81 (clone MT81) was a generous gift from Dr Rubinstein (INSERM‐UMR 1004, Villefuif, France). Anti‐LBPA (BMP) (clone 6C4) was also obtained from TEBU, France (ref 117ZPLBPA) and Sigma/Merck, France (ref MABT837). Anti‐LC3 was from Novus biological (NB100‐2331) and Merck/Sigma (L8918). Other antibodies were from: Abcam: antiannexin A1 [EPR19432], ab214486; anti‐Flotilin Ab 133497; antityrosinase (EP1577Y); anticalreticulin ab92516 and ab2907; anti‐Melan A ab199935; anti‐CD63 (abcam 217345); BD Bioscience: anti‐CD63 FITC (# 555288); anti‐CD9 FITC (#555371); Santa‐Cruz (TEBU France, Cliniscience France): anti‐CD9 (clone KMC8.8: sc‐18869), anti‐TRP2 (C9) sc‐74439 [previously (D18) sc‐10451]; anti‐Melan A (A103) sc‐20032; anti‐ALIX (C11) sc‐271975 and anti‐Rab27b, sc‐517602; Sigma: anti‐Rab27a (HPA 001333) and Rab27b (HPA 019849); anti‐Tsg101 (HPA 006161). Merck: antiactin mab1501. Secondary antibodies and isotypes : FITC‐conjugated secondary antibodies (antimouse:sc‐2010, antirabbit:sc‐2359) were from Santa‐Cruz /TEBU/Cliniscience, France, or from BD Bioscience (antirat #554016), eBioscience (antimouse #11‐4011‐85), Invitrogen (antirabbit #F2765). Control isotypes: FITC‐conjugated antimouse IgGK (#555748) and IgG2a (#555573) and PE‐conjugated antimouse (#559940) were from BD Bioscience. AF488‐conjugated goat antimouse and AF594‐conjugated goat antimouse (A11005) antibodies were from Life Technology. AF488 antirabbit (4412S) was from Cell Signaling. LC3I and LC3 II enriched cell fractions from PC3 cells were from Nanotools (1041/PC3‐LC3I and 1042/PC3‐LC3II) and were used as positive controls.
2.1.1. Cell culture
Cells were grown at 37°C in a humidified atmosphere with 5% CO2. All cell lines were tested once a month for mycoplasma contamination using Mycoalert Detection kit (Lonza, France). All culture reagents were from Gibco, France except foetal bovine serum (FBS) which was from Dutscher, France. B16F10 cells were grown in DMEM 4 g/L sucrose medium (ref 41966) supplemented with 2 mM L‐glutamine, 10 % FBS, penicillin and streptomycin (50 units/ml). SKMEL28 cells were grown in RPMI1640 (ref 21875) supplemented with 2 mM L‐glutamine, 10% FBS previously heat‐inactivated at 56°C for 1 h and penicillin and streptomycin (50 units/ml). E0771 were grown in RPMI 1640 (ref 21875) supplemented with 10% FBS, penicillin and streptomycin (50 units/ml) and 10 mM HEPES.
2.1.2. Cell cycle and apoptosis analysis
Experiments were performed as described previously (Segala et al., 2017). Briefly, cells were treated with DDA at the indicated concentrations or the solvent vehicle for 24 h. Cell cycle analysis was performed by flow cytometry after treating with RNase and staining with propidium iodide (PI) and the fraction of cells in the different phases of the cycle was calculated using flowJo software (Ashland, Oregon). Cell apoptosis was analysed by flow cytometry using the apoptosis detection kit (Sigma, France, ref APOAF). The percentage of Annexin‐V–FITC/ PI‐positive cells was determined using CellQuest software (BD Biosciences).
2.1.3. Cell transmission electron microscopy
Experiments were performed as described previously (Segala et al., 2017). Briefly, cells seeded onto 6‐well culture plates were treated for 24 h with the solvent vehicle or DDA at the indicated concentrations and then fixed on the culture plates with 2% glutaraldehyde in cacodylate buffer (0.1 M sodium cacodylate, 0.012 M calcium chloride, pH 7.4) for 4 h and washed with 0.2 M sodium cacodylate with 0.006 M calcium chloride for 12 h. Then, cells were postfixed with 1% OsO4 in cacodylate buffer with 1.5% K3Fe(CN)6 for 1 h, washed twice with distilled water, and prestained with an aqueous solution of uranyl acetate at 2% for 12 h. Samples were dehydrated in an ascending ethanol series and embedded in epoxy resin (Epon 812). Ultrathin sections (50 nm) were mounted on 150 mesh collodion‐coated copper grids and poststained with 3% uranyl acetate in 50% ethanol and with 8.5% lead citrate before being examined with an H300 Hitachi electron microscope at an accelerating voltage of 75 kV.
2.1.4. sEV negative stain electron microscopy
sEV were fixed by 2% glutaraldehyde. Collodion/carbon coated copper grids, with discharged, placed onto a drop of 30 microliters of sEV solution. After 1 min, grid was then stained for 10 s by inverting onto a drop of uranyl acetate aqueous solution. The grids were blotted using filter paper after staining step. The samples were visualized by Hitachi HU12A at 80 kV.
2.1.5. sEV cryomicroscopy examination
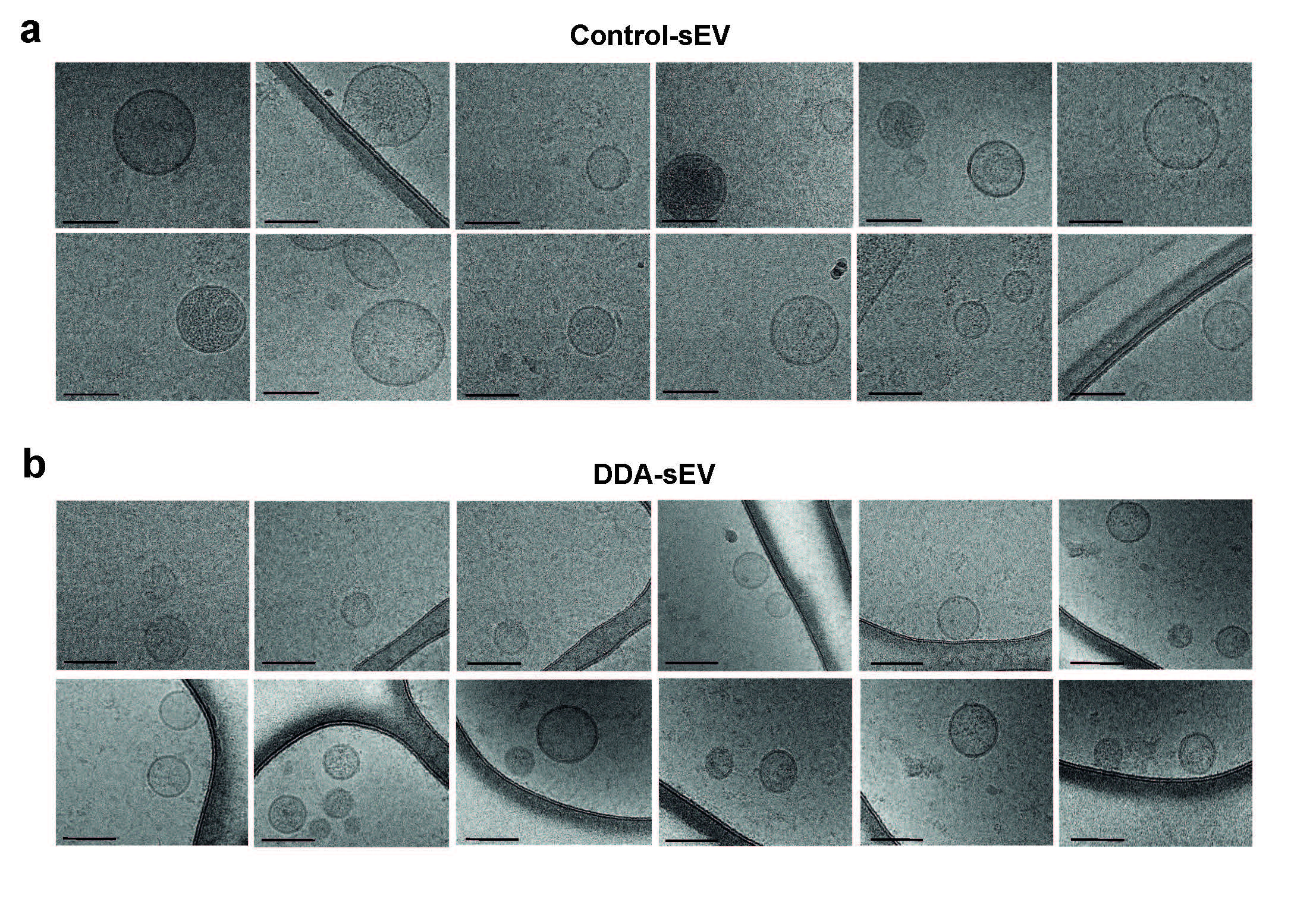
sEV (10 μg) were fixed with ethane and examined by high contrast transmission electronic microscopy with a JEM1400 or 2100 equipment at the METI platform IBCG Toulouse III University. Data are representative of measurements from 400 vesicles in each condition.
2.1.6. Cell immunogold labelling for transmission electron microscopy
Cells, treated as indicated, were pelleted and embedded at ‐30°C in HM20, acrylic resin and 80 nm sections on membrane nickel grid were performed. The immunogold labelling was automated with a Leica EM IGL. First, aldehyde groups were inactivated with 50 mM glycine in PBS, for 15 min and nonspecific antigenic sites were blocked with 0.8% BSA, 0.1% Cold Water Fish Skin gelatin in PBS, for 15 min. The sections were incubated with the primary antibody, purified mouse anti‐LBPA (BMP), Echelon biosciences Z‐PLBPA (TEBU, France) or Rabbit anti‐LC3, Novus biological NB100‐2331, diluted 1/25 in the blocking solution for 2 h at room temperature then washed with PBS (six times 5 min) and incubated with the secondary antibody GAM IgG ultrasmall, Aurion 800.177, for BMP or GAR ultrasmall, Aurion 800.011, for LC3, diluted 1/100 in BSA‐PBS solution, for 90 min at room temperature. The sections were washed with PBS (six times 5 min), postfixed with 2.0% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4), for 5 min and then washed with water. The sections were then subjected to silver enhancement for 25 min, washed with water and then contrast stained with 2% uranyl acetate and observed with a Hitachi HT 7700 transmission electron microscope.
2.1.7. Cell labelling with N‐Rh‐PE
As described in (Vidal et al., 1997), cells were incubated in suspension with 3 μM L‐α‐Phosphatidylethanolamine‐N‐(lissamine rhodamine B sulfonyl) or (N‐Rh‐PE) in PBS for 60 min at 4°C to allow N‐Rh‐PE to bind to the plasma membrane while blocking endocytosis. Cells were then pelleted and washed with PBS to remove excess of N‐Rh‐PE. Cells were transferred into 6‐well plates containing cover‐slips and treated with 1 μM DDA or vehicle overnight at 37°C to allow endocytosis. After this time, culture medium was removed and cells were washed with 3% w/v BSA in PBS to remove N‐Rh‐PE remaining on the cell periphery in order to focus labelling on newly formed endosomes. Cover‐slips were recovered, cells were fixed with 3.7% PFA, labelled with DAPI, and cover‐slips were mounted with mowiol and cells were examined by wide‐angle fluorescent microscopy.
2.1.8. Cell immunostaining
The indicated tumour cells were seeded on coverslip and treated for 24 h with solvent vehicle, DDA or 22(R)HC or their combination, as indicated and at the indicated concentrations. Cells were then washed and fixed with 4% PFA for 15 min at 25°C and permeabilized by 0.05% saponin or triton X100/1% BSA in PBS for 15 min at 25°c. Cells were washed and then incubated with the indicated primary antibody: anti‐BMP mouse antibody, Merck/Sigma Aldrich MAB T837 clone 6C4, 1/100 or anti‐LC3 rabbit antibody, Novusbio #NB100‐2331, 1/200 or with anti‐CD63 rabbit antibody (Abcam 217345), 1/100; for 24 h at 4°C or with anti‐BMP plus anti‐LC3 or with BMP plus CD63 for colocalization experiments. Cells were washed twice in 1% BSA‐0.1% or TritonX100 PBS and incubated with a AF594‐conjugated goat antimouse (A11005) (1/500) or with an AF488‐conjugated antirabbit (4412S, Cell signalling) 1/500, respectively, for 1 h at 25°C, then washed three times with 1% BSA‐0.1% TritonX100 PBS. Coverlips were mounted with 10 μl of a ProLong Gold Antifade Mountant with DAPI solution and cells were examined with a confocal LSM 880 FAS equipment (Figure 2) or with the confocal Leica LSM 780 (Figure S11) or with a fluorescence wide‐angle microscope (Figure 4, 5).
FIGURE 2.
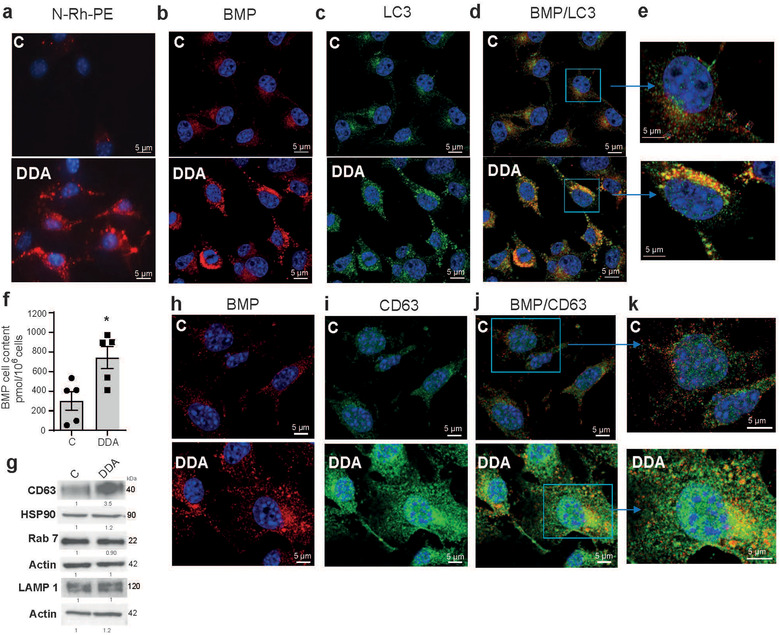
Characterization of endocytic vesicles in control‐ or DDA‐treated B16F10 cells. B16F10 tumour cells were treated with the solvent vehicle (control) or 1 μM DDA for 24 h and analysed as indicated. (a) Representative pictures of cells labeled with N‐Rh‐PE (red labelling) and observed by fluorescence microscopy, n = 3. (b) Representative confocal pictures of BMP immunostaining in cells (red labelling), n = 3. (c) Representative confocal pictures of LC3 immunostaining in cells (green labelling), n = 3. (d‐e) Representative confocal pictures of BMP (red labelling) and LC3 (green labelling) coimmunostaining in cells and magnification of cells showing BMP/LC3 colocalization in DDA‐treated cells (yellow labelling), n = 3. (f) Quantification of BMP in cells by HILIC/MS analysis (n = 5). Data are the means ± SEM of five independent experiments performed in triplicate (*P < 0.05, t test, two‐tailed). g) Representative immunoblot analysis of the expression of endosomal proteins related to different stages of the endocytic pathway (CD63, LAMP1, Rab7 and control (HSP90 or actin), (n = 3). h) Representative confocal pictures of BMP immunostaining in cells (red labelling), n = 3. (i) Representative confocal pictures of CD63 immunostaining in cells (green labelling), n = 3. (j‐k) Representative confocal pictures of BMP (red labelling) and CD63 (green labelling) coimmunostaining in cells and magnification of cells showing BMP/CD63 colocalization in DDA‐treated cells (yellow labelling), n = 3. In IF experiments, nuclei in (a‐e) were stained with DAPI. In immunoblot experiments, densitometry values indicate changes in protein expression relative to control treated cells and normalized to HSP90 or actin as indicated
FIGURE 4.
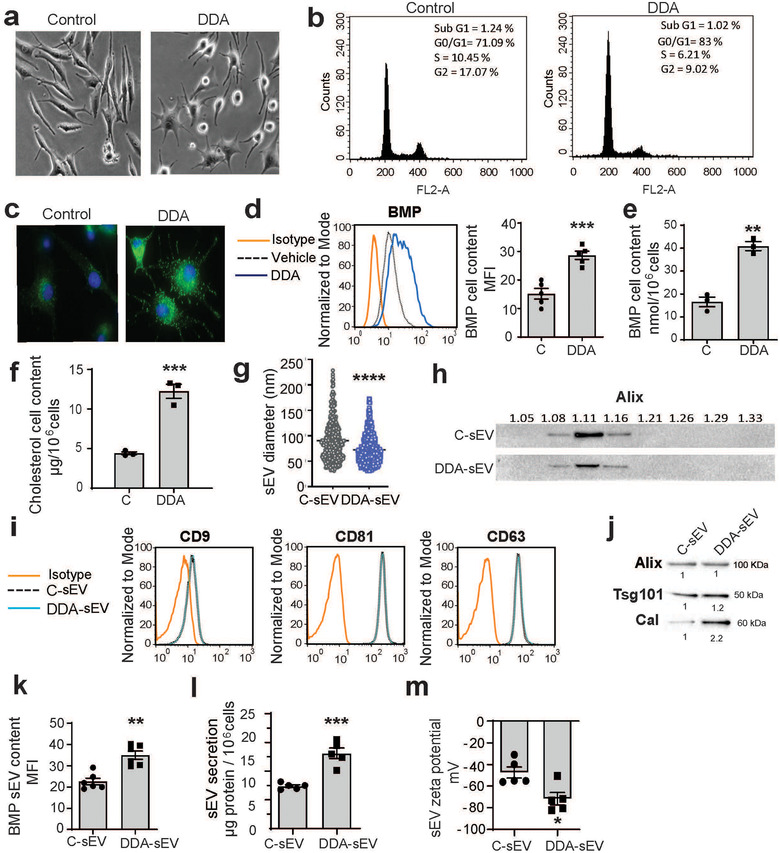
Characterization of sEV secreted from control‐ or DDA‐treated SKMEL28 cells. SKMEL28 tumour cells were treated with the solvent vehicle (control) or 2 μM DDA for 24 h and analysed as indicated. (a) Cells were observed by light microscopy (×40), (n = 3). (b) Representative flow cytometry analysis of cell cycle distribution, (n = 3). (c) Representative picture of BMP immunostaining in cells observed by fluorescence microscopy (x 63), (n = 3). (d) Representative flow cytometry analysis of BMP quantification in cells. The plot indicates the median fluorescence intensity (MFI) of BMP content in cells (n = 5). (e) BMP cell content quantified by HILIC/MS analysis, (n = 3). (f) Cholesterol cell content quantified by GC/MS analysis, (n = 3). (g) Size distribution analysis of C‐sEV and DDA‐sEV obtained from cryo‐microscopy examination of 400 vesicles in each condition. (h) Buoyant density of sEV monitored by Alix content, (n = 2). (i) Tetraspanin content of sEV analysed by flow cytometry, (n = 3). (j) Immuno‐blot analysis of Alix, Tsg101 and calreticulin content in sEV, (n = 3), with densitometry values showing changes in protein expression relative to C‐sEV and normalized to Alix. (k) BMP content of sEV analysed by flow cytometry (n = 6). (l) sEV production recovered from 106 cells was analysed by measuring protein content (n = 5). (m) Zeta potential was measured using the Zeta View equipment from Particle Metrix GmbH (Formulaction, Toulouse, France) to monitor sEV surface charge (n = 5). Data in d, e, f, g, k, l, m, are the means ± SEM of 3–5 independent experiments (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, t test, two‐tailed)
FIGURE 5.

DDA differentiates melanoma cells by acting through the LXRβ. (a,b) SKMEL28 proficient (shC) or deficient (sh3LXRβ and sh4LXRβ) for the expression of the LXRβ, were treated or not, for 24 h, with 2 μM DDA. (a) Representative immunoblot analysis of the expression of proteins related to melanogenesis differentiation (Tyrosinase (Tyr), TRP2, Melan A), the ESCRT pathway (Alix, Tsg101) and sEV secretion (Rab27a and Rab27b), (n = 3), with densitometry values showing changes in protein expression relative to control treated cells and normalized to actin or HSP90 as indicated. (b) Analysis of BMP cell content by HILIC/MS, (n = 3), (*P < 0.05, ns: not significant, t test, two‐tailed). (c) Representative pictures of BMP immunostaining in cells observed by fluorescence microscopy (magnification x 63) of SKMEL28 cells treated or not, for 24 h, with 2 μM DDA or 5μM 22R‐HC or 5 μM 22R‐HC and 2 μM DDA, n = 3. (d) Representative immunoblot analysis of the expression of the endosomal protein CD63, (n = 3), with densitometry values showing changes in protein expression relative to control treated cells and normalized to HSP90
2.1.9. sEV preparation
Cells were seeded in 175 cm2 culture flasks (Falcon) at 1.8 × 106 cells for SKMEL28 and 1.2 × 106 for B16F10 in their complete culture medium. The medium was removed 24 h later and replaced by the same medium prepared with EV‐free FBS obtained by overnight ultracentrifugation at 110 000 g at 4°c and passed through a 0.2 μm filter. B16F10 cells were treated with 1 μM DDA or 5 μM 22(R)HC or the solvent vehicle for 24 h. SKMEL28 cells were treated with 2 μM DDA or solvent vehicle for 24 h. sEV preparation were carried out under sterile conditions using sterilizable ultracentrifuge tubes (Beckman Coulter). sEV were recovered from the culture medium which was processed by differential centrifugations as described previously (Subra et al., 2010). Briefly, cell culture medium was sequentially centrifuged at 4°c at 1200 x g for 5 min and 10 000 x g for 30 min. The supernatant was recovered and sEV were then pelleted at 110 000 x g for 70 min, resuspended in 20 ml PBS and centrifuged again at 110 000 x g for 70 min. Finally, sEV pellet was diluted in calcium free‐PBS. sEV were endotoxin‐free as checked using a LAL chromo endotoxin quantitation kit (Thermo‐Fisher Scientific, France).
2.1.10. sEV quantification
Different methods were used to quantify sEV. (1) Protein content in sEV were quantified by the colorimetric method of Lowry in the presence of 0.1% w/v SDS to disrupt membranes as previously reported (Subra et al., 2010). (2) The fluorescent lipidic probe Bodipy–Ceramide was also used to quantify sEV secreted by increasing concentrations of DDA (Laulagnier et al., 2004). sEV were recovered by cut‐off filtration using Vivaspin filters (Sartorius) (Gastpar et al., 2005). In this experiment, culture medium was harvested and first centrifuged at 3200 x g for 15 min through a 0.2 μm Vivaspin filter, then through a 1000 kDa Vivaspin filter by centrifugation at 3200 x g for 15 min. sEV recovered from the filter were then labelled with the fluorescent lipid Bodipy‐FL‐C5‐ceramide (Invitrogen–Molecular Probes, D3521) for 1 h at 37°C. Excess of Bodipy‐ceramide was removed by filtration and washings through the 1000 KDa Vivaspin filter. sEV recovered from the filter were then bound on 4 μm latex beads and fluorescence was quantified by flow cytometry, and (3) sEV number was measured with the NanoSight equipment (Malvern Instruments, France) or with the qNano equipment from Izon (Oxford, UK). The Zeta View equipment from Particle Metrix GmbH (Formulation, Toulouse, France) was used to quantify sEV released by the MEF cells.
2.1.11. Zeta potential measure
The Zeta View equipment from Particle Metrix GmbH (Formulation, Toulouse, France) was also used to measure the Zeta potential of sEV which monitors the peripheral electric charge of sEV (Gordillo‐Galeano & Mora‐Huertas, 2018). Acquisitions were based on 2.5–3.5 × 1013 vesicles injected into the equipment.
2.1.12. Characterization of sEV markers by flow cytometry analysis
sEV (5 μg) were bound onto 5 μl of 4 μm aldehyde‐sulphate latex beads (Invitrogen, #A37304; Thermo‐Fisher Scientific) in 200 μl PBS for 1 h at 25°C with gentle periodical shaking. Free sites on latex beads were next saturated with 100 μl vesicle‐free FBS for 30 min at 25°C. Beads with bound sEV were centrifuged for 5 min at 5000 rpm, washed in 200 μl PBS, and diluted in 100 μl FACS buffer. Specific primary antibody or control isotype (1:50) were added and incubated at 25°c for 30 min. After centrifugation and washing, secondary antibody (1:100) was added and incubated for 30 min at 25 °c. Beads with bound antibody‐labelled sEV were diluted in 1 ml FACS buffer and analysed by flow cytometry (FACScalibur or LSRII, Becton‐Dickinson).
2.1.13. Characterization of sEV by immunocapture and flow cytometry analysis
Inmunocapture of sEV followed by bound antibody‐labelled sEV was evaluated according to (Ostrowski et al., 2010). Briefly, 3 μg of antimouse CD63 (Santa‐Cruz, sc5275) or 2 μg anti‐BMP (Millipore MAB T837 clone 6C4), were coupled to 4‐μm aldehyde‐sulphate latex beads (Invitrogen #A37307) for 24 h at 4°C under shaking at 300 rpm. Beads with bound antibody were washed twice with PBS and were centrifuged for 5 min at 5000 rpm. Free sites on latex beads were next saturated with 200 μl solution PBS ‐ BSA 4% for 30 min at 25°C with shaking. Then, 2 μl of aldehyde‐sulfate latex beads bound with the antibody of interest were coupled to 5 μg of sEV in 100 μl of PBS for 24 h at 4°C under shaking at 300 rpm. After two centrifugations for 5 min at 5000 rpm and two washes, free sites on latex beads +sEV were next saturated with 200 μl solution PBS ‐ BSA 4% for 1 h at 25°C under shaking. The captured sEV was then incubated with the indicated specific primary antibody for detection: anti‐BMP (Millipore, MAB T837 clone 6C4) 1/50 or anti‐LC3 (Sigma/Merck, L8918), 1/20 or anti‐CD63 (Santa‐Cruz, sc5275), 1/20) at 25°C for 2 h. After two centrifugations for 5 min at 5000 rpm and two washes, the secondary antibody (1/50) was incubated with captured sEV for 1 h at 4°C. Beads with bound antibody‐labelled sEV were diluted in 200 μl of PBS‐BSA 4%. 50000 events were analysed by MACSQuant‐10 flow cytometer (Miltenyi Biotec) and further analyses were performed using FlowJo Software. Data are shown as percentage of positive beads.
2.1.14. Characterization of sEV markers by immuno‐blot analysis
5 to 20 μg sEV (based on protein content) were diluted in sample buffer and denaturated by heating at 60°C for 10 min. Identical amounts of proteins were deposited in each well and proteins were resolved in SDS‐PAGE and transferred onto PVDF membranes, saturated with 5% w/v nonfat milk in TBS‐Tween 0.1%. Antibodies were added in 1% w/v nonfat milk in TBS‐Tween 0.1% at the indicated dilutions according to the manufacturer recommendations. For LC3 expression, LC3I and LC3 II enriched cell fractions from PC3 cells were used as positive controls according to the manufacturer recommendations. Revelation from immunoblotting was performed by enhanced chemiluminescence and analysed by ChemiDoc imager (BioRad).
2.1.15. sEV density analysis
sEV density was measured through a sucrose gradient. 50 μg sEV in 100 μl PBS was deposited on top of a discontinuous gradient constituted by nine layers of increasing sucrose concentration from 0.25 M to 2.25 M and a cushion of 2.5 M sucrose, and centrifuged at 160 000 g for 16 h in swinging buckets. Fractions of 1 ml were harvested, diluted in 10 ml PBS and centrifuged for 2 h at 110 000 g. Pellets were recovered in Laemli buffer and their protein content resolved through SDS‐PAGE, then probed for the expression of Alix or CD9.
2.1.16. Lipid analysis
Cholesterol in cells (5‐10106 ×) and sEV (50‐100 μg) was quantified by GC‐MS at the lipid analysis facility of the Faculté de Médecine Saint–Antoine as reported in (Serhan et al., 2020). BMP quantification was performed from lipid extracts of cells (10 to 20 × 106cells) or sEV (100‐150 μg proteins) by hydrophilic interaction liquid chromatography‐tandem mass spectrometry (HILIC‐MS/MS) according to (Scherer et al., 2010) at the lipid analysis facility Saint–Antoine, Paris, France. BMP was also quantified by the mean fluorescence intensity (MFI) (Chapuy‐Regaud et al., 2013) measured by flow cytometry following intracellular staining with the 6C4 monoclonal anti‐BMP antibody, and in sEV bound on latex beads following their staining with the antibody. Analysis was performed with FACScalibur or LSRII, Becton–Dickinson equipments.
2.1.17. Filipin staining
Cells grown on glass coverslips were treated with the solvent vehicle or DDA for 24 h and then fixed with 3.7% paraformaldehyde for 15 min at 25°c followed by washing twice with PBS. Cells were then incubated with filipin at 50 mg/ml, (Sigma/Merck, France) for 75 min at 25°c followed by washing twice with PBS. Cells were analysed using a Nikon eclipse 90i microscope.
2.1.18. Tumour growth analysis in vivo
C57BL/6 female mice (6 weeks old) were purchased from Janvier Labs (France). All mice were maintained in specific pathogen‐free conditions. All of the animal procedures for the care and use of laboratory animals were conducted according to the guidelines of our institution and followed the general regulations governing animal experimentation. Exponentially growing cells were harvested, washed twice in PBS and suspended in PBS (106 cells/ml). B16F10 cells (105 cells in 100 μl PBS) were subcutaneously injected into the right flank of the mice. On the day of cell transplantation at day 0 and at day 7, animals were treated intradermal in the opposite flank with 2 μg of sterile DDA‐sEV or C‐sEV in 100 μl PBS or with 100 μl PBS. The tumour size was measured every 2–3 days with a caliper and the volume was calculated using the formula [width2 × length]/2.
2.1.19. Stable ShRNA transfection
ShLXRβ SKMEL28 and shC SKMEL28 cells were obtained as described in (Segala et al., 2017). E0771 cells were transfected with puromycin resistant shRNA plasmids specific to the LXRβ gene or with a nonspecific shRNA plasmid (Psi‐nH1shLXRbeta (geneCopoeia, Tebu‐Bio, France), using the NEON transfection system (Invitrogen) and according to the supplier's instructions. Selection of stable clones was obtained after growing the cells for 1 month in the presence of puromycin (0.5 μg/ml).
2.1.20. RNA isolation and analysis
Total RNA was extracted from E0771 cells using trizol and was quantified with NanoDrop. Complementary DNA (cDNA) synthesis was performed with 1 μg RNA iScript Reverse Transcriptase (Bio‐Rad). 25 ng cDNA was amplified using SyBR Supermix (Bio‐Rad). The forward and reverse primers used were: 5′‐AACCTGCCAGATGGATGCCTTC‐3′ and 5′‐GCTGCTGCTGTTGCTGCTT‐3′ for LXRβ and 5′‐ACTTCGTGCAAGAAATGCTGAA‐3′ and 5′‐CAGTTGTCCGTGGCTCTCT‐3′ for Tata Box binding protein (TBP). Amplification was carried out as follows: 95°C for 3 min, followed by 40 cycles at 95°C for 15 s, 60°C for 1 min (iCycler, Bio‐Rad). The levels of LXRβ in shC E0771 and shLXRβ E0771 cells were normalized to TBP.
2.1.21. Human DC maturation and mixed lymphocyte reaction
Human DC generation and treatment were performed as previously reported (Perrin‐Cocon et al., 2017; Perrin‐Cocon et al., 2018). Peripheral blood mononuclear cells were isolated from human peripheral blood of healthy donors.
Monocytes (106 cells /ml; purity > 90%) were differentiated to immature DC (iDC), cultured in RPMI 1640 with 2 mM glutamine, 10 mM Hepes, 40 ng/ml gentamycin (Life Technologies), 10% FCS for 7 days with 100 ng/ml human rGM‐CSF and rIL‐4 (Human DC cytokine package, Peprotech). Cells were treated at day 6 for the last 24 h with 20 μg/ml C‐sEV or DDA‐sEV or with 0,1 μg/ml ultrapure LPS (from Escherichia coli 0111:B4; InvivoGen, Toulouse, France) as a positive control of DC maturation. All cells and supernatants were collected at day 7. All DC were more than 95% pure CD14 – CD1a + .
2.1.22. Phenotyping
Phenotype was analysed on a FACSCanto (BD Biosciences, Le Pont de Claix, France) using FITC‐conjugated anti‐CD14, anti‐CD1a, HLA‐DR, ‐CD80, ‐CD54, and PE‐conjugated ‐CD86, ‐CD83 and ‐CD40 (Beckman Coulter). Cytokines IL12, IL6 and TNFα were assayed using a cytokine specific Cytometric Bead Array (CBA) Flex Sets (BD Biosciences).
2.1.23. Mixed Lymphocyte Reaction (MLR)
T lymphocytes were purified from peripheral blood leukocytes (PBL) and were more than 95% pure as assessed by CD3 labelling. Primary MLR were conducted in 96 well flat‐bottom culture plates. Washed DC were cocultured in triplicates with 2 × 105 allogeneic T cells in 200 μl complete culture medium at 1/10, 1/20 or 1/40 DC/T cell ratio. After 5 days, 100 μl of the culture supernatant was analysed for cytokine secretion assay by CBA (Agaugué et al., 2006).
2.1.24. Statistical analyses
Statistical analyses were carried out as specified in the figure legends using Graph Pad Prism software (v7 and v8). Statistical difference was considered when P ≤ 0.05.
3. RESULTS
3.1. DDA increases the formation of endocytic vesicles in tumour cells and secretes sEV including exosomes
Using transmission electron microscopy technique (Figure 1a), we observed that mouse B16F10 melanoma cells treated with concentrations of DDA (1 or 2.5 μM) inducing cell differentiation (de Medina et al., 2013), cell cycle arrest but not apoptosis (Figure S1a‐b), contained numerous vesicles compared to control cells (Figure 1 b‐c). These vesicles displayed a morphology similar to that of MVB (Figure 1c‐e) and secreted small vesicles (Figure 1c circle,d,g‐h) whose size (30–100 nm) fitted with that of sEV that we isolated by ultracentifugation (Figure 1f). As shown in Figure 1g–h, sEV released under DDA treatment includes exosomes which are formed and secreted extracellularly from MVB. To confirm the endocytic nature of DDA‐induced vesicles, B16F10 cells were labelled with either N‐(lissamine rhodamine B sulfonyl) phosphatidylethanolamine (N‐Rh‐PE) or with an antibody directed against bis(monoacylglycero)phosphate (BMP), formerly called lysobisphosphatidic acid (LBPA). These two lipids accumulate in late endosomes (Kobayashi et al., 1998; Vidal et al., 1997). In addition, BMP is enriched in the internal membrane of endosomes and MVB (Kobayashi et al., 2002; Kobayashi et al., 2001) and is involved in ILV biogenesis (Matsuo et al., 2004). DDA‐treated tumour cells showed an enhanced N‐Rh‐PE staining (Figures 2a and S1c) and anti‐BMP immunostaining (Figures 2b and S1d) compared to control cells, suggesting that DDA increased endosomes and MVB in these cells. Since the lipidated form of LC3 (LC3II) was recently reported to control sEV loading and secretion from MVB (Leidal et al., 2020), we measured LC3‐II puncta accumulation that was increased by DDA via the LXRβ during lethal autophagy (Segala et al., 2017) and determined whether BMP and LC3 colocalized. DDA‐treated cells showed a marked increase in LC3 puncta immunostaining compared to control cells (Figure 2c) and a colocalization of LC3 with BMP in puncta structures (Figures 2d‐e and S2). Immunogold analysis confirmed that BMP and LC3 are found in MVB (Figure S3a‐b). Quantification of BMP in tumour cells by liquid chromatography coupled to mass spectrometry (LC/MS) indicated a 1.9‐fold increase in BMP level in DDA‐treated cells compared to control cells (Figure 2f). Cell cholesterol, that is present in MVB and in the intraluminal vesicles of MVB (Möbius et al., 2003), was measured by gas chromatography analysis coupled to mass spectrometry (GC/MS). As shown in Figure S3c, cholesterol was increased by 1.3‐fold in DDA‐treated B16F10 cells compared to control cells. Consistent with this data, filipin staining confirmed that DDA induced the accumulation of puncta of free sterols in cells (Figure S3d).
FIGURE 1.

DDA increases the formation of endocytic vesicles in tumour cells and secretes sEV including exosomes. (a‐e, g‐h) B16F10 tumour cells were treated with the solvent vehicle (control) or 1 μM DDA for 24 h and prepared and analysed by TEM as indicated in (a). (b‐e, g‐h) Representative pictures of cells examined by transmission electron microscopy, n = 3. DDA‐treated cells showed numerous MVB close to the plasma membrane (c‐e, g‐h) compared to control cells (b). The extracellular release of sEV called exosomes can be observed from a MVB fused to the plasma membrane (g‐h). (f) Representative pictures of DDA‐sEV secreted from B16F10 cells treated with 1 μM DDA for 24 h and isolated by ultracentrifugation, n = 3
We then determined whether endosomal proteins such as CD63, LAMP1 and Rab7, which label endosomes at different stages of the endocytic pathway (Mathieu et al., 2021), were increased in DDA‐treated cells compared to control‐treated cells by immunoblot analysis. The expression of CD63, that is more specific of the MVB endosome subset, was increased by 3.5‐fold in DDA‐treated B16F10 cells compared to control cells while Rab7 and LAMP1 had an expression mostly similar to that of control cells (Figures 2g and S3e). These data indicates that DDA increases selectively CD63 in addition to BMP in cells. We next investigated the subcellular distribution of CD63 and BMP. Immunofluorescence analysis indicated an increased content in both endosomal markers in DDA‐treated cells compared to control cells (Figure 2h‐i). Noteworthy, a colocalization between CD63 and BMP was evidenced in some puncta structures (Figures 2 j‐k and S4a), which indicated the presence of CD63 and BMP in MVB.
Together these results suggest an accumulation of late endosomes in DDA‐treated cells compared to control cells, associated with the secretion of sEV, some of which being exosomes (Figure 1g,h).
3.2. DDA increases the secretion and modifies the content of tumour sEV with anti‐tumour activity
To confirm vesicle secretion, sEV was isolated from culture media of B16F10 tumour cells treated with 1 μM DDA (named DDA‐sEV) or with the solvent vehicle (named C‐sEV). DDA‐sEV and C‐sEV content were analysed and compared for their density and the presence of markers found in exosomes or ectosomes as well as markers of DDA activity. The density of both types of vesicles was similar and ranged between 1.10‐1.17 (Figure S4b). The tetraspanins CD9, CD63, CD81 (Figure 3a), the ESCRT‐associated proteins Alix and Tsg 101 (Figures 3b and S4c‐d) were found in equal amounts in both DDA‐sEV and C‐sEV while Flotillin was decreased by half in DDA‐sEV (Figure 3b and S4e). DDA‐sEV was enriched by approximately 1.5‐fold in tyrosinase (Tyr) compared to C‐sEV (Figures 3b and S4f) in agreement with the fact that DDA induced melanoma B16F10 cells differentiation into a melanocytic phenotype associated with an increased melanin accumulation and tyrosinase activity (de Medina et al., 2013). In addition, Annexin A1 was increased by 2‐fold and its cleaved form by 3‐fold in DDA‐sEV (Figures 3b and S4g) and in calreticulin by 1.2‐fold (Figure 3c) compared to C‐sEV. Moreover, DDA‐sEV was enriched in the lipidated form of LC3 (LC3 II) by 4‐fold (Figures 3b and S5a). Immunocapture experiments showed that around 88 % of BMP‐positive DDA‐sEV contained LC3 indicating a MVB origin for this population of DDA‐sEV while only 57% of BMP‐positive C‐sEV were positive for LC3 (Figure S5b).
FIGURE 3.
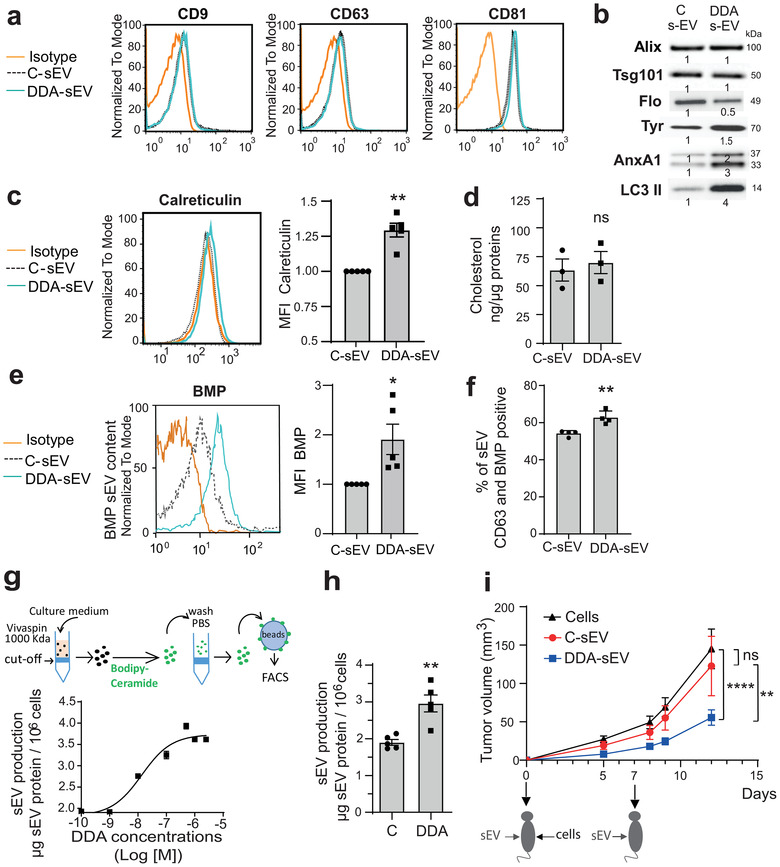
Characterization of sEV secreted from control‐ or DDA‐treated B16F10 cells. Analysis of sEV isolated from the culture media of B16F10 cells treated with the solvent vehicle (C‐sEV) or DDA (DDA‐sEV) for 24 h. (a) Representative flow cytometry analysis of sEV tetraspanin content (n = 3). (b) Representative immunoblot analysis of markers of sEV (Alix, Tsg101, Flotilin (Flo), LC3 II, Annexin A1 (AnxA1) and the melanocytic marker tyrosinase (Tyr) (n = 3) with densitometry values showing changes in protein expression relative to C‐sEV and normalized to Alix. (c) Representative flow cytometry analysis of sEV calreticulin content (n = 5). (d) GC/MS analysis of cholesterol content in sEV (n = 3). (e) Representative flow cytometry analysis of BMP quantification in sEV (n = 5). (f) Immunocapture of CD63‐positive sEV with an anti‐CD63 antibody fixed to beads and analysis of the presence of BMP in CD63‐positive sEV by flow cytometry analysis (n = 4). Data is expressed as percentage of positive beads and are the mean ± SEM of four independent experiments (*P < 0.05, t test, two‐tailed). (g) Quantification of sEV production from B16F10 cells, treated with increasing concentrations of DDA, monitored after sEV labelling with the lipid membrane probe bodipy‐ceramide as described in the schematic diagram, (n = 3). (h) sEV production recovered from 106 cells was measured by measuring protein content (n = 5). (i) Mice (15/group) were implanted with B16F10 tumour cells in the right flank and treated two times, at day 0 and day 7, in the contralateral flank with PBS or with 2 μg of C‐sEV or DDA‐sEV. Mean tumour volumes (± SEM) are shown, two‐way ANOVA, **P < 0.01, ****P < 0.0001. ns: not significant. Data are representative of three independent experiments. The plots in (c) and (e) show the median fluorescence intensity (MFI) of DDA‐ sEV relative to C‐sEV. Data in c, d, e, g, h are the means ± SEM of 3–5 independent experiments (*P < 0.05 and **P < 0.01, t test, two‐tailed; ns: not significant)
Similar level of cholesterol was found in DDA‐sEV and C‐sEV (Figure 3d) while DDA‐sEV was enriched in BMP by approximately 2‐fold (Figure 3e). Since the expression of CD63, that is more specific of MVB and ILV, was increased in DDA‐treated cells relative to control cells (Figures 2g and S3e), we analysed by immunocapture experiments the colocalization of CD63 with BMP in C‐sEV and DDA‐sEV. As shown in Figures 3f and S5c‐d, 54% of CD63‐positive C‐sEV were positive for BMP, and this amount significantly increased to 63 % for DDA‐sEV positive for CD63 and BMP. These data show that BMP and CD63 colocalize in a large part of DDA‐sEV subpopulations. In summary, immunocapture experiments confirm, at the level of sEV, the colocalization observed in parental cells of BMP with LC3 and BMP with CD63.
DDA stimulated a concentration‐dependent increase in DDA‐sEV secretion, monitored by fluorescent labelling of the vesicles (Laulagnier et al., 2004), that was maximum at 1 μM DDA and reached 1.9‐fold that of C‐sEV (Figure 3g). The half‐maximal effective concentration (EC50) of DDA was 13.4 ± 0.21 nM. The measure of sEV protein content (Figure 3h) or vesicle number by Nano Tracking Analysis (NTA) technology (Figure 6a) confirmed that DDA‐sEV secretion was increased compared to C‐sEV, by 1.6‐fold and 2‐fold, respectively. The activity of C‐sEV and DDA‐sEV was then tested on the growth of B16F10 tumours implanted into syngenic mice compared to vehicle treatment. As shown in Figures 3i and S6a‐e, intradermal injection, at day 0 and 7, of 2 μg DDA‐sEV secreted from B16F10 cells significantly inhibited the growth of B16F10 tumours by approximately 50 % compared to similar treatments with C‐sEV or with the solvent vehicle, indicating that DDA‐sEV display antitumour activity.
FIGURE 6.
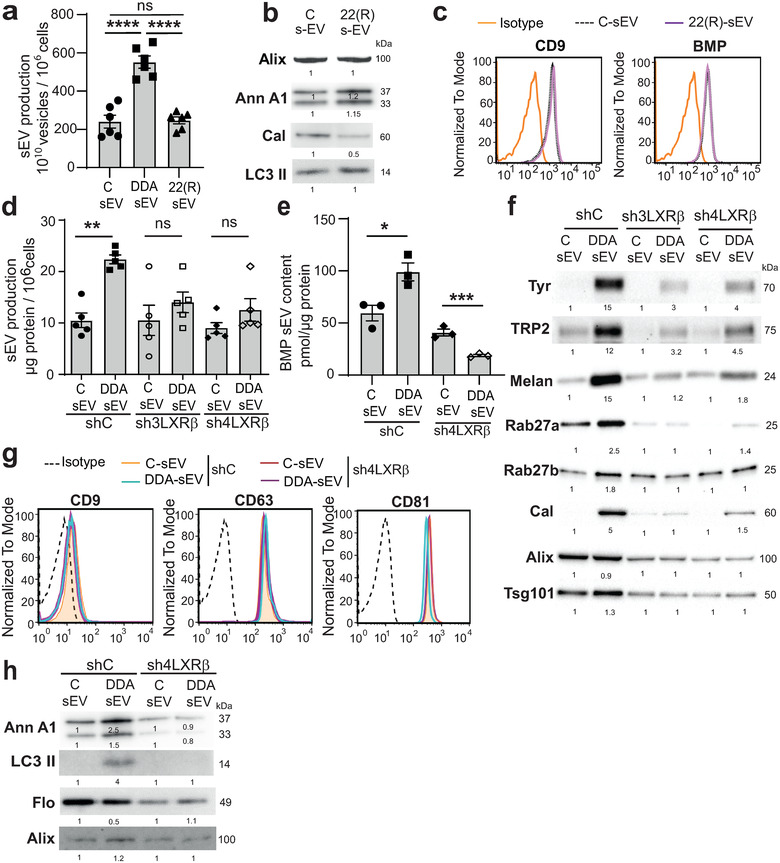
LXRβ regulates secretion and composition of DDA‐sEV from mouse and human melanoma cells. (a‐c) sEV were isolated from the media of either mouse B16F10 cells treated or not, for 24 h, with DDA 1 μM or 22(R) 5 μM, or (d‐h) of human melanoma cells SKMEL28 proficient (shC) or deficient (sh3LXRβ and sh4LXRβ) for the expression of the LXRβ, treated or not, for 24 h, with DDA 2 μM and sEV were analysed as indicated. (a) B16F10 sEV production recovered from 106 cells treated as indicated, was measured by counting secreted vesicles using the Nanosight equipment (Malvern Instrument, France) (n = 3). (b) Representative immunoblots analysis of Alix, Annexin A1 (AnxA1), Calreticulin (Cal) and LC3‐II content in B16F10 sEV, n = 3. (c) Representative cytometry analysis of CD9 and BMP content in B16F10 sEV treated or not with 22(R), n = 3. (d) SKMEL28 sEV production recovered from 106 cells was analysed by measuring sEV protein content (n = 5). (e) Analysis of BMP content in SKMEL28 sEV by HILIC/MS, (n = 3). (f) Representative immunoblots analysis of Tyrosinase (Tyr), TRP2, Melan A, Rab27a and b, calreticulin (Cal), Alix and Tsg101 content in SKMEL28 sEV, n = 3. (g) Representative cytometry analysis of tetraspanin content in SKMEL28 sEV, (n = 3). (h) Representative immunoblots analysis of Annexin A1 (AnxA1) and LC3‐II content in SKMEL28 sEV, n = 3. Data in a, d, e, are the means ± SEM of 3–5 independent experiments. (a :****P < 0.0001, one‐way ANOVA ; d,e : *P < 0.05, **P < 0.01, t test, two‐tailed). (b, f, h) densitometry values show changes in protein expression relative to C‐sEV and normalized to Alix
We then characterized human melanoma SKMEL28 cells (BRAF mutated) upon treatment with a concentration of DDA inducing cell differentiation and cell cycle arrest (Figure 4a‐b). Immunofluorescence studies with an anti‐BMP antibody revealed an increased labelling in DDA‐treated SKMEL28 cells compared to control cells (Figure 4c) and an increase of around 2‐fold was measured by flow cytometry analysis (Figure 4d). In addition, LC/MS analysis indicated a 2.5‐fold increase in cell BMP content (Figure 4e). GC/MS analysis indicated that cholesterol content was 2.8‐fold higher in DDA‐treated cells than in control cells (Figure 4f).
In these conditions, we then analysed the ability of DDA to produce modified sEV from SKMEL‐28 cells. DDA and vehicle cell treatment triggered the release of sEV with a mean diameter measured by cryo‐microscopy of 72.5 ± 1.3 nm for DDA‐sEV and of 90.4 ± 1.9 for C‐sEV (Figures 4g and S7a‐b) and a density that ranged between 1.08 and 1.16 (Figures 4h and S8a). Their characterization revealed the presence of CD9, CD81, CD63 (Figure 4i), Alix and Tsg101 (Figures 4j and S8b) that were present in similar levels in C‐sEV and DDA‐sEV. DDA‐sEV also featured a higher content in calreticulin (2.2‐fold) (Figures 4j and S8c) and BMP (1.6‐fold) (Figure 4k) compared to C‐sEV. Moreover, DDA treatment of SKMEL28 cells increased the amount of DDA‐sEV released by 1.8‐fold compared to control‐treated cells (Figure 4l). The measure of the zeta potential indicated a significant difference between DDA‐sEV and C‐sEV with a higher electronegativity for DDA‐sEV (Figure 4 m). Together these data indicated that DDA modified B16F10 and SKMEL28 tumour cells to produce higher amounts of DDA‐sEV with modified content, size and charge compared to C‐sEV.
3.3. DDA differentiates melanoma cells by acting on the LXRβ
Since DDA mediates characteristics of cell differentiation in mammary and thyroid tumours by acting through the LXRβ (Bauriaud‐Mallet et al., 2019), we determined whether changes in melanoma parental cells and sEV induced by concentrations of DDA inducing cell differentiation were regulated by this receptor. We used SKMEL28 cells knocked down for the expression of the LXRβ (clones Sh3LXRβ or sh4LXRβ) and control cells (clone ShC) (Segala et al., 2017). We characterized these cells for the expression of melanocytic differentiation markers as well as markers that are important for sEV formation and production (Colombo et al., 2014; Yamaguchi et al., 2007). DDA‐mediated melanoma cell differentiation was associated in shC cells with a higher expression of proteins involved in the melanogenesis pathway such as tyrosinase (Tyr) (2.5‐fold), TRP2 (2.6‐fold) and melan A (1.7‐fold) (Figures 5a and S9a‐b and S10b). The silencing of LXRβ expression completely inhibited the increased expression of Tyr and TRP2 but did not affect that of melan A (Figures 5a and S9a‐b and S10b). Moreover, the expression of Rab27a was 1.65 higher in shC cells upon DDA treatment compared to control treatment and its expression was decreased in shLXRβ cells treated with DDA relative to control treatment (Figures 5a and S9c and S10c). The DDA/LXRβ complex had an opposite effect on Rab27b expression and decreased the expression of Rab27b in shC cells while this effect was reversed in shLXRβ cells (Figures 5a and S9d and S10c). In contrast, Alix and Tsg101 were not regulated by DDA in shC and shLXRβ cells compared to control treatment, indicating that these proteins are not under the control of the DDA/LXRβ complex (Figures 5a and S10a and S10c). Quantification of BMP, by LC/MS, in shC and shLXRβ, indicated a significant increase of 1.9‐fold in DDA‐treated shC cells compared to control treatment, while no significant BMP increase was measured in shLXRβ cells (Figure 5b), indicating a LXR‐dependent mechanism regulating BMP metabolism. We then evaluated the effect of DDA on BMP melanoma cell content in comparison with 22(R)‐hydroxycholesterol (22(R)HC), a canonical endogenous ligand of the LXR, by immunofluorescence studies. As shown in Figure 5c, DDA‐treated cells showed an increased punctate BMP staining pattern compared to control‐treated cells, while 22(R)HC‐treated cells did not. In addition, in cells cotreated with 22(R)HC and DDA, BMP‐labelling induced by DDA was completely inhibited. Similar results were obtained in B16F10 cells (Figure S11a). These data indicate that DDA increases BMP content in SKMEL28 and B16F10 cells through the LXRβ, while 22(R)HC has an opposite effect and inhibits DDA‐induced BMP accumulation. Since CD63 expression increased in DDA‐treated B16F10 cells (Figure 2), we evaluated whether the content of CD63 was increased in DDA‐treated SKMEL28 cells and whether LXRβ mediated this effect. As shown in Figures 5d and S10d, CD63 expression was increased under DDA‐treatment in shC cells compared to control‐treated cells while in shLXRβ cells this effect was abolished, indicating that the DDA/LXRβ complex increased the expression of CD63.Together these data indicate that DDA increases the cell levels of Tyrosinase, TRP2, Rab27a, BMP and CD63 and decreases that of Rab27b by acting through the LXRβ.
3.4. DDA increases the secretion of modified tumour‐sEV through an LXRβ‐dependent mechanism
Regarding the opposite effect of 22(R)HC on BMP biogenesis in tumour cells compared to DDA, we evaluated its impact on the secretion of sEV. Treatment of B16F10 cells with 22(R)HC did not significantly increase sEV secretion compared to vehicle treatment as indicated by the measure of the number of vesicles by NTA analysis (NanoSight), while DDA increased sEV secretion by 2.3‐fold (Figure 6a). Characterization of the content of 22(R)HC‐sEV indicated that they contain Alix, lipidated LC3, CD9 and BMP in equal amounts compared to C‐sEV (Figures 6b‐c and S11b‐c). In addition, they were not enriched in annexin A1 and the level of calreticulin was decreased compared to C‐sEV (Figures 6b and S11d‐e).
We then tested DDA on a tumour cell line of a different origin and the implication of the LXRβ on DDA‐sEV secretion and content. Mouse mammary E0771 tumour cells were knocked‐down for the expression of LXRβ by using shRNA specific of the LXRβ (shLXRβ cells) in comparison to control shRNA (shC cells) (Figure S12a). As shown in Figure S12b, DDA treatment of shC E0771 cells increased by two times the production of DDA‐sEV compared to C‐sEV while this effect was abolished in DDA‐treated shLXRβ E0771 cells, indicating that DDA‐sEV secretion is controlled by the LXRβ. sEV characterization indicated that DDA‐sEV and C‐sEV secreted from shC E0771 cells contained CD9 in equal amounts but DDA‐sEV was enriched in calreticulin and BMP by 1.6 and 2.3‐fold, respectively, compared to C‐sEV (Figure S12c). In contrast, DDA‐sEV purified from shLXRβ mammary tumour cells were not enriched in these markers (Figure S12c), indicating a LXRβ‐dependent mechanism driving their content in DDA‐sEV. The use of mouse embryonic fibroblasts (MEF) isolated from LXRβ knock‐out mice (Segala et al., 2017) confirmed that the DDA/LXRβ complex mediates sEV secretion (Figure S12d).
To further assess the role of this receptor in secretion and DDA‐sEV characteristics, the impact of knocking‐down LXRβ in SKMEL28 parental cells on DDA‐induced sEV secretion was investigated. As shown in Figure 6d, DDA induced a 1.8‐fold increase in DDA‐sEV secretion from shC cells compared to C‐sEV treatment. In contrast, no significant increase in DDA‐sEV secretion was measured upon DDA‐treatment of sh3LXRβ or sh4LXRβ cells, indicating that DDA acts through the LXRβ to promote sEV secretion. The measure of the vesicle size generated by control or DDA treatment of shC and shLXRβ cells by NTA analysis indicated that around 93 % of the vesicles obtained in each of these conditions had a size < 200 nm (Figure S13a). Vesicles < 50 nm accounted for less than 0.5%. Treatment of shC cells with DDA increased the secretion of DDA‐sEV that ranged between 70 nm and 90 nm compared to vehicle, but not from shLXRβ cells, indicating that DDA triggers the generation of smaller vesicles in a LXRβ‐dependent manner (Figure S13a).
We then characterized the sEV produced by shC and shLXRβ SKMEL28 cells. sEV released upon DDA or control treatment from shC or shLXRβ parental cells indicated that all sEV types had a similar density ranging between 1.11 and 1.16 (Figure S13b). The evaluation of different markers important for sEV biogenesis and secretion or modulated by DDA in parental cells were evaluated. DDA significantly increased the content of BMP in DDA‐sEV secreted from shC cells analysed by LC/MS (Figure 6e) or flow cytometry (Figure S13c). In contrast, no increase in BMP was measured in DDA‐sEV released from shLXRβ cells (Figures 6e and S13c), indicating that DDA/LXRβ complex regulates the content of this lipid in DDA‐sEV. Moreover, DDA increased the content of the proteins tyrosinase, Trp2, Melan A, Rab27a, Rab27b and calreticulin in DDA‐sEV (Figures 6f and S14a‐b) by acting through the LXRβ since their enrichment was decreased in DDA‐sEV recovered from shLXRβ cells compared to C‐sEV. In contrast, DDA did not modify the amount of Alix in DDA‐sEV secreted from shC or shLXRβ cells compared to C‐sEV (Figures 6f and S15a) nor that of CD9, CD63 and CD81 (Figure 6g) and slightly increased Tsg101 in DDA‐sEV secreted from shC cells (Figures 6f and S15b). DDA also increased the annexin A1‐level by 2.5‐fold and its cleaved form by 1.5‐fold in DDA‐sEV secreted from shC cells compared to C‐sEV, while no enrichment of this protein was observed in DDA‐sEV recovered from DDA‐treated‐shLXRβ cells, indicating an involvement of the LXRβ in the enrichment of this protein (Figures 6h and S15c). Moreover, DDA treatment of shC cells increased the content of LC3 II in DDA‐sEV by 4‐fold compared to C‐sEV (Figures 6h and S15d), while decreasing that of flotillin by half (Figures 6h and S15e) and both effects were LXRβ‐dependent.
3.5. DDA stimulates the secretion of immunogenic sEV from human tumour cells through a LXRβ‐dependent mechanism
Dendritic cells (DC) are potent activators of naïve T cells, inducing different functional T‐cell responses (priming or tolerance) according to their maturation state. Since sEV is efficiently internalized by DC and may have different impact according to the parental cells producing these sEV (Groot Kormelink et al., 2018; Valenti et al., 2006), we evaluated the function of DDA‐sEV and C‐sEV recovered from shC or shLXRβ SKMEL28 cells on the maturation of human monocyte‐derived DC and T lymphocyte polarization. As observed in Figure 7a, (left bars) and Figure S16, the surface expression of MHC class II molecules (HLA‐DR), the costimulatory molecules B7‐1 (CD80) and B7‐2 (CD86), the intercellular adhesion molecule ICAM‐1 (CD54) and CD40 involved in T cell activation was increased between 2.5 times (for CD54) to 11.6 times (for HLA‐DR) in DC challenged with DDA‐sEV compared to DC challenged with C‐sEV. The expression of the late DC maturation marker, CD83, was also enhanced, by 6‐ and 8‐fold, in DC challenged with DDA‐sEV. This phenotypic maturation was accompanied by a functional maturation evidenced by the secretion of cytokines representative of mature DC such as IL12p40, IL6 and TNFα (Figure 7b, left bars). These data indicate that DDA‐sEV recovered from shC cells promoted the functional maturation of human DC. In contrast, when similar experiments were performed with DDA‐sEV or C‐sEV recovered from shLXRβ cells, DC remain mainly immature according to the expression of phenotypic markers (Figure 7a, right bars and Figure S16) and low cytokine secretion, especially of IL‐12 (Figure 7b, right bars).
FIGURE 7.
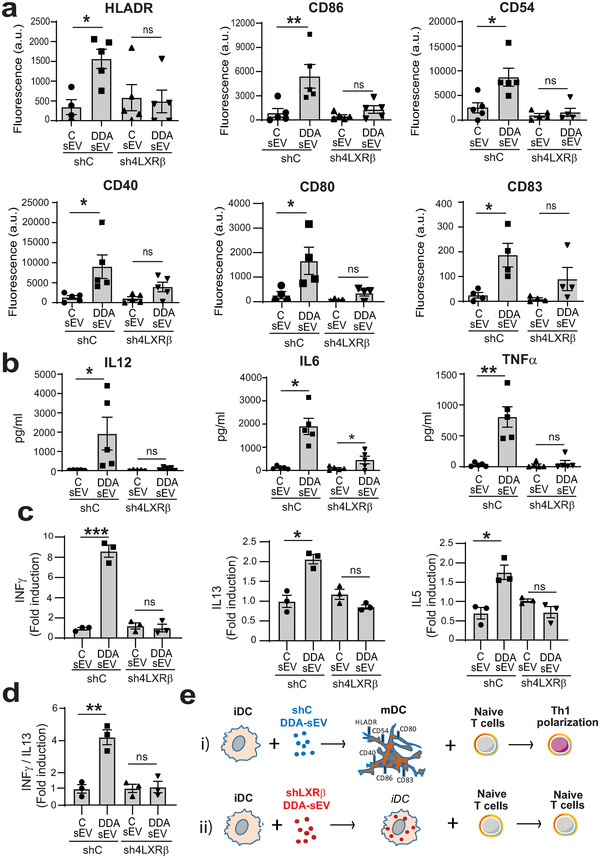
The DDA/ LXRβ complex induces the secretion of DDA‐sEV from human SKMEL28 cells, which triggers human DC maturation and Th1 T lymphocyte polarization. sEV were isolated from the media of human melanoma cells SKMEL28 proficient (shC, black bars) or deficient (sh4LXRβ, gray bars) for the expression of the LXRβ, treated or not, for 24 h, with DDA 2 μM and sEV were analysed for their immunogenicity. (a‐b) Human DC, incubated for 24 h with 20 μg/ml sEV recovered from the indicated SKMEL28 cells and treatments, were analysed for: (a) the expression of markers of DC maturation by flow cytometry. The plots show the mean fluorescence intensity (MFI) of the indicated marker, (n = 5‐4) and (b) for cytokine released in the media measured by CBA assay, (n = 5). (c) Mixed Lymphocyte Reaction (MLR) between DC maturated with sEV recovered from the indicated cells (as indicated in a,b) and naïve T cells (1/10 DC/LT cell ratio) was performed and culture supernatant was analysed for cytokine secretion by CBA assay, (n = 3). Data are normalized to control conditions (DCs without sEV) in the same experiment. d) IFNγ/IL‐13 ratio was calculated for each biological replicate to determine the Th1 (above 1) versus Th2 (below 1) balance of T cell activation. (a‐d) Data are the means ± SEM of 3–5 independent experiments (*P < 0.05, **P < 0.01, ***P < 0.001, t test, two‐tailed). (e) Diagram of the results obtained in these different experiments showing that i) DDA‐sEV, isolated from the medium of DDA‐treated‐shC SKMEL28 cells proficient for the LXRβ expression, transform immature DC (iDC) into functional mature DC (mDC) that are able to promote T cell activation with a Th1 polarization; ii) DDA‐sEV, isolated from the medium of DDA‐treated‐shLXRβ SKMEL28 cells deficient for LXRβ expression, mainly remain immature DC (iDC) and these iDC do not activate naïve T cells. Thus, DDA, by a LXRβ‐dependent mechanism, induces the secretion from SKMEL28 cells of DDA‐sEV promoting human DC maturation that acquire the ability to activate T cells
The quality of the functional maturation of DC triggered by DDA‐sEV from shC or shLXRβ cells was then investigated by analysing their ability to activate T lymphocytes in Mixed Leukocyte Reaction (MLR). As shown in Figure 7c‐d, DC maturated by DDA‐sEV issued from shC cells (left bars), strongly induced the secretion of IFNγ by T lymphocytes (ranges of 388–488 pg/ml) and weakly increased IL‐13 (ranges of 99–120 pg/ml) and IL‐5 secretion (ranges 21–29 pg/ml), compared to control DC challenged by C‐sEV issue from shC cells (ranges of 54–84, 39–64 and 5–11 pg/ml for INFγ and IL13 and IL5, respectively). As previously shown by Tkach et al (Tkach et al., 2017), we used the ratio of secreted IFNγ versus IL‐13 to determine whether DDA‐sEV activated a Th1 polarization. Indeed, the 4‐fold IFNγ/IL‐13 secretion ratio induced by treatment with DDA‐sEV issue from shC cells was representative of a Th1 polarization (above 1) while that induced by treatment with C‐sEV issue from shC cells was around 1 (Figure 7d, left bars). In contrast, no stimulation of T lymphocytes was observed when DC, challenged with DDA‐sEV issued from shLXRβ cells, were used in the DC/T lymphocyte alloreaction compared to the control conditions (Figure 7c, right bars) and the IFNγ/IL‐13 secretion ratio was about 1. In summary, the DDA/LXRβ complex in SKMEL28 cells induces the secretion of DDA‐sEV which promote human DC phenotypic and functional maturation and T cell activation (Figure 7e).
4. DISCUSSION
This study highlights a pharmacological control by DDA, through the LXRβ, of characteristics of tumour cell differentiation and sEV production, composition and function. The sEV secretion response to DDA treatment, measured by different techniques, is important in the tumour cell lines tested and corresponds to approximately 1.5 to 2‐fold that of C‐sEV and increases in a DDA concentration‐dependent manner, in agreement with a receptor‐mediated mechanism. In contrast, the canonical LXR ligand, 22(R)HC, does not increase sEV secretion. The DDA/LXRβ complex increases the tumour cell expression of Rab27a while decreasing the level of Rab27b and increases the sorting of both proteins toward DDA‐sEV. In the ventral prostate of mice lacking LXRα and LXRβ gene expression, Rab27b mRNA expression is upregulated, indicating a transcriptional control of the expression of this gene by the LXR (Viennois et al., 2012). These data are consistent with the fact that DDA regulates the expression of this protein via the LXRβ and, here, our results extend this control to Rab27a. Rab27a and Rab27b were described to promote the secretion of sEV and MVB docking to the plasma membrane (Ostrowski et al., 2010). It was also observed that these proteins have different subcellular distributions with Rab27a being preferentially associated with CD63 positive‐MVB, while Rab27b was predominantly present in the transgolgi network area (Ostrowski et al., 2010). The increased expression of Rab27a in tumour cells by the DDA/LXRβ complex indicates that at least part of DDA‐sEV have an MVB origin and is in agreement with cell electronic microscopy analysis showing the release of DDA‐exosomes from MVB and with the fact that the DDA/LXRβ complex increases BMP content in tumour cells and in DDA‐sEV. Indeed, BMP accumulates in the internal membranes of late endosomes, in MVB as well as in ILV, but is not detected at the plasma membrane (Kobayashi et al., 1998) (Möbius et al., 2003). BMP is a fusogenic lipid at the acidic pH of endosomes (Kobayashi et al., 2002), which promotes the formation of ILV when added in liposomes (Matsuo et al., 2004). Therefore, the increase in BMP levels in cells and sEV mediated by the DDA/LXRβ complex is consistent with the higher secretion of DDA‐sEV. Moreover, the canonical LXR agonist, 22(R)HC, which does not increase BMP levels in tumour cells and sEV and inhibits DDA‐induced BMP accumulation in cells, does not increase sEV secretion. Together these results highlight, for the first time, that the BMP metabolism is controlled by a nuclear receptor, namely, the LXRβ, and reveal the opposite action of DDA and 22R(R)HC on this event and on sEV secretion, as already reported for autophagy when DDA was compared to 22(R)HC or other LXR ligands (Segala et al., 2017).
Consistent with these data, the expression of CD63, which is specific of MVB and ILV as BMP, was increased by the DDA/LXRβ complex upon differentiation of tumour cells. The study of the subcellular distribution of BMP and CD63 indicated an increased content in both endosomal markers in DDA‐treated cells compared to control cells and a colocalization between CD63 and BMP in puncta structures, which indicated the presence of CD63 and BMP in MVB in DDA‐treated cells. A sorting process of both CD63 and BMP then occurred between MVB membrane and intraluminal vesicles as assessed by immunocapture experiments performed on sEV, showing that a major part of DDA‐sEV are BMP and CD63 positive.
DDA‐sEV contains different markers found in sEV, such as CD63 (specific of exosome), CD9, CD81, Alix and TSG101, at similar levels to those found in C‐sEV. These data suggest that the DDA/LXRβ complex promotes an enhanced biogenesis process independent of the ESCRT pathway since Alix and TSG101 are known to regulate the ESCRT biogenesis of sEV (Colombo et al., 2013). Interestingly, DDA‐sEV is enriched in LC3 II, the lipidated form of LC3, compared to C‐sEV, while sEV generated by 22(R)HC is not. LC3 II is well described as an autophagic protein and autophagy is well described for its degradative role. However, recently, part of the autophagy machinery has been implicated in cellular secretion and an LC3 II‐dependent EV loading and secretion, arising mostly from MVB, have been described (Leidal et al., 2020). Here, immunogold labelling indicated that MVB in DDA‐treated B16F10 cells contain LC3 II and BMP and immunocapture experiments confirmed that LC3 and BMP colocalize in subpopulations of DDA‐sEV and C‐sEV and showed that BMP‐positive DDA‐sEV are enriched in LC3 compared to C‐sEV. Moreover, we showed that LC3 II enrichment into DDA‐sEV is mediated by the DDA/LXRβ complex. Together these results reveal a new mechanism in sEV secretion driven by the LXR and indicate that sEV secretion may be pharmacologically controlled by an oxygenated cholesterol metabolite via its target receptor. Importantly, this LXRβ‐mediated mechanism is not limited to melanoma but is also involved in mammary tumour cells and mouse embryo fibroblasts.
In addition, melanoma cell differentiation induced by DDA was associated with an increased expression of the key enzymes involved in melanin biosynthesis such as Tyrosinase and Trp2 (Yamaguchi et al., 2007) and this effect was mediated by the LXRβ. These data are in agreement with the fact that DDA increases melanin production and tyrosinase activity in melanoma cells (de Medina et al., 2009; de Medina et al., 2013) and that the LXRβ regulates melanogenesis in human primary melanocytes and melanoma cells (Lee et al., 2013). Indeed, LXR ligands, TO901317, 22(R)HC and GW3965, suppress the expression of the melanogenic enzymes, Tyrosinase, Trp1 and Trp2 and inhibit melanin synthesis stimulated by the α‐melanocyte stimulating hormone (Lee et al., 2013). The increased expression by DDA of Melan A, which is a protein involved in the structural maturation of melanosomes (Yamaguchi et al., 2007), seems to not be affected by the silencing of the LXRβ in melanoma cells. Moreover, the DDA/LXRβ complex increases the sorting of Tyrosinase, Trp2 as well as Melan A in DDA‐sEV, indicating that DDA‐sEV are enriched in several melanocytic antigens of differentiation. In addition, we showed that calreticulin is enriched in DDA‐sEV and its sorting in DDA‐sEV is driven by the LXRβ. This protein is found in the endoplasmic reticulum (ER) and is frequently used as a ER marker; however, other localizations of calreticulin within cells have been reported, such as the cytoplasm, nucleus and plasma membrane (Krause & Michalak, 1997). Calreticulin is a danger‐associated molecular pattern (DAMP) which acts as an eat‐me signal to recruit DC, when exposed at the surface of tumour cells treated with some chemotherapies and contributes to immunogenic cell death (ICD) (Kepp et al., 2014; Obeid et al., 2007). Moreover, induction of calreticulin expression in DC promotes their maturation in vitro (Liu et al., 2016). In the present study, DDA‐sEV enriched in calreticulin could represent a subset of sEV coming from the plasma membrane of DDA‐treated cells. However, here, no expression of calreticulin was measured at the plasma membrane in control and DDA‐treated cells (Figure S3d), suggesting that they may arise from MVB. DDA‐sEV are also enriched in annexin A1, which is found in sEV coming from the plasma membrane and/or MVB (Jeppesen et al., 2019; Leoni et al., 2015), and its enrichment in DDA‐sEV is mediated by the LXRβ. The release of annexin A1 into the extracellular space is also a feature of ICD and is required for calreticulin exposure to the tumour cell membrane (Baracco et al., 2019).
We previously reported that DDA is a molecule of the self, not present in tumours, which induces tumour redifferentiation and inhibition of tumour growth in mice (de Medina et al., 2013). Here, we show that tumour cell redifferentiation induced by DDA is mediated by the LXRβ. This results in the production of DDA‐sEV which decreases tumour growth compared to C‐sEV or vehicle treatments, indicating that DDA‐sEV contributes to DDA antitumour response. In addition, the DDA/LXRβ complex in human melanoma cells promotes the secretion of DDA‐sEV which induce human DC maturation promoting a Th1 polarization of T cells. These data indicate that the DDA/LXRβ complex controls the loading of key immunostimulatory factors in DDA‐sEV. Thus, this work reveals the possibility of a pharmacological control of tumour sEV production and functions. More generally, our data suggest that DDA may have a physiological function in maintaining cell integrity and differentiation associated with immune system surveillance by acting on the LXRβ and by producing sEV.
In conclusion, our results indicate that targeting the LXRβ with DDA in tumour cells may be an original strategy to increase the immune response against melanoma and possibly others tumours. Our study highlights a novel pathway controlling tumour sEV secretion, content and immune function with an unexpected role for the LXRβ activated by a new class of ligand.
ACKOWLEDGEMENTS
We acknowledge the contribution of the Etablissement Français du Sang Auvergne ‐ Rhône‐Alpes. We acknowledge Laetitia Ligat at the CRCT technical facility for her technical support and helpful advices. Mehdi Attia was supported by La Fondation Toulouse Cancer Santé. Julio Bunay was supported by the Institut National du Cancer (INCA), grant PLBio 20–160. This work was supported by recurrent grants from INSERM and Toulouse University, from the Fondation Toulouse Cancer Santé and the Institut National du Cancer (INCA), grants PLBio 12–130 and PLBio 20–160.
AUTHOR CONTRIBUTIONS
Michel Record: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Supervision; Validation; Visualization; Writing – original draft; Writing – review & editing. Mehdi Attia: Formal analysis; Investigation; Methodology; Validation. Kevin Carayon: Formal analysis; Investigation; Methodology; Validation; Visualization. Laly Pucheu: Investigation; Methodology; Validation; Visualization. Julio Bunay: Formal analysis; Investigation; Methodology; Validation; Visualization. Régis Soulès: Formal analysis; Investigation; Methodology; Validation; Visualization. Silia Ayadi: Investigation; Methodology. Laure Perrin‐Cocon: Formal analysis; Investigation; Methodology; Supervision; Validation; Visualization; Writing – original draft. Florence Bourgailh: Formal analysis; Investigation; Methodology; Validation; Visualization. Vincent Lotteau: Formal analysis; Investigation; Supervision; Validation; Visualization. Marc Poirot: Conceptualization; Formal analysis; Funding acquisition; Investigation; Methodology; Supervision; Validation; Visualization; Writing – original draft. Sandrine Silvente‐Poirot: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Supervision; Validation; Visualization; Writing – original draft; Writing – review & editing. Philippe de Medina: Conceptualization; Formal analysis; Investigation; Methodology; Project administration; Supervision; Validation; Visualization; Writing – original draft
CONFLICT OF INTEREST
All the authors disclose no competing interests in this work.
PERMISSION TO REPRODUCE MATERIAL FROM OTHER SOURCES
We did not reproduce material from other sources.
ETHICAL STATEMENT
All of the animal procedures for the care and use of laboratory animals were conducted according to the guidelines of our institutions and followed the general regulations governing animal experimentation.
Supporting information
Supplementary information
Supplementary information
{kind=link}
Supplementary information
{kind=link}
Supplementary information
{kind=link}
Supplementary information
Supplementary information
Supplementary information
{kind=link}
Supplementary information
{kind=link}
Supplementary information
{kind=link}
Supplementary information
{kind=link}
Supplementary information
Supplementary information
Supplementary information
Supplementary information
{kind=link}
Supplementary information
Supplementary information
{kind=link}
Supplementary information
Record, M. , Attia, M. , Carayon, K. , Pucheu, L. , Bunay, J. , Soulès, R. , Ayadi, S. , Payré, B. , Perrin‐Cocon, L. , Bourgailh, F. , Lamazière, A. , Lotteau, V. , Poirot, M. , Silvente‐Poirot, S. , & de Medina, P. (2022). Targeting the liver X receptor with dendrogenin A differentiates tumour cells to secrete immunogenic exosome‐enriched vesicles. Journal of Extracellular Vesicles, 11,e12211. 10.1002/jev2.12211
Contributor Information
Michel Record, Email: michel.record@inserm.fr.
Marc Poirot, Email: marc.poirot@inserm.fr.
Sandrine Silvente‐Poirot, Email: sandrine.poirot@inserm.fr.
Philippe de Medina, Email: philippe.de-medina@inserm.fr.
DATA AVAILABILITY STATEMENT
The data for the study are available from the corresponding authors upon request.
REFERENCES
- Agaugué, S. , Perrin‐Cocon, L. , Coutant, F. , André, P. , & Lotteau, V. (2006). 1‐Methyl‐tryptophan can interfere with TLR signaling in dendritic cells independently of IDO activity. Journal of Immunology 177, 2061–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baracco, E. E. , Petrazzuolo, A. , & Kroemer, G. (2019).Assessment of annexin A1 release during immunogenic cell death. Methods in Enzymology 629, 71–79. [DOI] [PubMed] [Google Scholar]
- Bauriaud‐Mallet, M. , Vija‐Racaru, L. , Brillouet, S. , Mallinger, A. , de Medina, P. , Rives, A. , Payre, B. , Poirot, M. , Courbon, F. , & Silvente‐Poirot, S. (2019). The cholesterol‐derived metabolite dendrogenin A functionally reprograms breast adenocarcinoma and undifferentiated thyroid cancer cells. Journal of Steroid Biochemistry and Molecular Biology 192, 105390. [DOI] [PubMed] [Google Scholar]
- Chapuy‐Regaud, S. , Subra, C. , Requena, M. , de Medina, P. , Amara, S. , Delton‐Vandenbroucke, I. , Payre, B. , Cazabat, M. , Carriere, F. , Izopet, J. , Poirot, M. , & Record, M. (2013). Progesterone and a phospholipase inhibitor increase the endosomal bis(monoacylglycero)phosphate content and block HIV viral particle intercellular transmission. Biochimie 95, 1677–1688. [DOI] [PubMed] [Google Scholar]
- Colombo, M. , Moita, C. , van Niel, G. , Kowal, J. , Vigneron, J. , Benaroch, P. , Manel, N. , Moita, L. F. , Théry, C. , & Raposo, G. (2013). Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. Journal of Cell Science 126, 5553–5565. [DOI] [PubMed] [Google Scholar]
- Colombo, M. , Raposo, G. , & Théry, C. (2014). Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annual Review of Cell and Developmental Biology 30, 255–289. [DOI] [PubMed] [Google Scholar]
- Dai, S. , Wei, D. , Wu, Z. , Zhou, X. , Wei, X. , Huang, H. , & Li, G. (2008). Phase I clinical trial of autologous ascites‐derived exosomes combined with GM‐CSF for colorectal cancer. Molecular Therapy 16, 782–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Medina, P. , Paillasse, M. R. , Payré, B. , Silvente‐Poirot, S. , & Poirot, M. (2009). Synthesis of new alkylaminooxysterols with potent cell differentiating activities: Identification of leads for the treatment of cancer and neurodegenerative diseases. Journal of Medicinal Chemistry 52, 7765–7777. [DOI] [PubMed] [Google Scholar]
- de Medina, P. , Paillasse, M. R. , Segala, G. , Voisin, M. , Mhamdi, L. , Dalenc, F. , Lacroix‐Triki, M. , Filleron, T. , Pont, F. , Saati, T. A. l. , Morisseau, C. , Hammock, B. D. , Silvente‐Poirot, S. , & Poirot, M. (2013). Dendrogenin A arises from cholesterol and histamine metabolism and shows cell differentiation and anti‐tumour properties. Nature Communication 4, 1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escudier, B. , Dorval, T. , Chaput, N. , André, F. , Caby, M.‐P. , Novault, S. , Flament, C. , Leboulaire, C. , Borg, C. , Amigorena, S. , Boccaccio, C. , Bonnerot, C. , Dhellin, O. , Movassagh, M. , Piperno, S. , Robert, C. , Serra, V. , Valente, N. , Le Pecq, J.‐B. , … Zitvogel, L. (2005). Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived‐exosomes: Results of thefirst phase I clinical trial. Journal of Translational Medicine 3, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastpar, R. , Gehrmann, M. , Bausero, M. A. , Asea, A. , Gross, C. , Schroeder, J. A. , & Multhoff, G. (2005). Heat shock protein 70 surface‐positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Research 65, 5238–5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordillo‐Galeano, A. , & Mora‐Huertas, C. E. (2018). Solid lipid nanoparticles and nanostructured lipid carriers: A review emphasizing on particle structure and drug release. European Journal of Pharmaceutics and Biopharmaceutics 133, 285–308. [DOI] [PubMed] [Google Scholar]
- Groot Kormelink, T. , Mol, S. , de Jong, E. C. , & Wauben, M. H. M. (2018). The role of extracellular vesicles when innate meets adaptive. Seminars in Immunopathology 40, 439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino, A. , Costa‐Silva, B. , Shen, T.‐L. , Rodrigues, G. , Hashimoto, A. , Tesic Mark, M. , Molina, H. , Kohsaka, S. , Di Giannatale, A. , Ceder, S. , Singh, S. , Williams, C. , Soplop, N. , Uryu, K. , Pharmer, L. , King, T. , Bojmar, L. , Davies, A. E. , Ararso, Y. , … Lyden, D. (2015). Tumour exosome integrins determine organotropic metastasis. Nature 527, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen, D. K. , Fenix, A. M. , Franklin, J. L. , Higginbotham, J. N. , Zhang, Q. , Zimmerman, L. J. , Liebler, D. C. , Ping, J. , Liu, Q. , Evans, R. , Fissell, W. H. , Patton, J. G. , Rome, L. H. , Burnette, D. T. , & Coffey, R. J. (2019). Reassessment of exosome composition. Cell 177, 428–445 e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri, R. , Lebleu, V. S. , The biology, function, and biomedical applications of exosomes. Science 367 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepp, O. , Senovilla, L. , Vitale, I. , Vacchelli, E. , Adjemian, S. , Agostinis, P. , Apetoh, L. , Aranda, F. , Barnaba, V. , Bloy, N. , Bracci, L. , Breckpot, K. , Brough, D. , Buqué, A. , Castro, M. G. , Cirone, M. , Colombo, M. I. , Cremer, I. , Demaria, S. , … Galluzzi, L. (2014). Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 3, e955691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, T. , Beuchat, M.‐H. , Chevallier, J. , Makino, A. , Mayran, N. , Escola, J.‐M. , Lebrand, C. , Cosson, P. , Kobayashi, T. , & Gruenberg, J. (2002). Separation and characterization of late endosomal membrane domains. Journal of Biological Chemistry 277, 32157–32164. [DOI] [PubMed] [Google Scholar]
- Kobayashi, T. , Stang, E. , Fang, K. S. , de Moerloose, P. , Parton, R. G. , & Gruenberg, J. (1998). A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature 392, 193–197. [DOI] [PubMed] [Google Scholar]
- Kobayashi, T. , Startchev, K. , Whitney, A. J. , & Gruenberg, J. (2001). Localization of lysobisphosphatidic acid‐rich membrane domains in late endosomes. Biological Chemistry 382, 483–485. [DOI] [PubMed] [Google Scholar]
- Krause, K.‐H. , & Michalak, M. (1997). Calreticulin. Calreticulin. Cell 88, 439–443. [DOI] [PubMed] [Google Scholar]
- Laulagnier, K. , Grand, D. , Dujardin, A. , Hamdi, S. , Vincent‐Schneider, H. , Lankar, D. , Salles, J.‐P. , Bonnerot, C. , Perret, B. , & Record, M. (2004). PLD2 is enriched on exosomes and its activity is correlated to the release of exosomes. Febs Letters 572, 11–14. [DOI] [PubMed] [Google Scholar]
- Lee, C. S. , Park, M. , Han, J. , Lee, J.‐H. , Bae, I.‐H. , Choi, H. , Son, E. D. , Park, Y‐H. , & Lim, K.‐M. (2013). Liver X receptor activation inhibits melanogenesis through the acceleration of ERK‐mediated MITF degradation. Journal of Investigative Dermatology 133, 1063–1071. [DOI] [PubMed] [Google Scholar]
- Leidal, A. M. , Huang, H. H. , Marsh, T. , Solvik, T. , Zhang, D. , Ye, J. , Kai, F. , Goldsmith, J. , Liu, J. Y. , Huang, Y.‐H. , Monkkonen, T. , Vlahakis, A. , Huang, E. J. , Goodarzi, H. , Yu, L. , Wiita, A. P. , & Debnath, J. (2020). The LC3‐conjugation machinery specifies the loading of RNA‐binding proteins into extracellular vesicles. Nature Cell Biology 22, 187–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoni, G. , Neumann, P.‐A. , Kamaly, N. , Quiros, M. , Nishio, H. , Jones, H. R. , Sumagin, R. , Hilgarth, R. S. , Alam, A. , Fredman, G. , Argyris, I. , Rijcken, E. , Kusters, D. , Reutelingsperger, C. , Perretti, M. , Parkos, C. A. , Farokhzad, O. C. , Neish, A. S. , Nusrat, A. , Annexin A1‐containing extracellular vesicles and polymeric nanoparticles promote epithelial wound repair. Journal of Clinical Investigation 125, 1215–1227 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, I. , & Nabet, B. Y. (2019). Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Molecular cancer 18, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C.‐Y. , & Gustafsson, J.‐Å. (2015). Targeting liver X receptors in cancer therapeutics. Nature Reviews Cancer 15, 216–224. [DOI] [PubMed] [Google Scholar]
- Liu, X. , Li, J. , Liu, Y. , Ding, J. , Tong, Z. , Liu, Y. , Zhou, Y. , & Liu, Y. (2016). Calreticulin acts as an adjuvant to promote dendritic cell maturation and enhances antigen‐specific cytotoxic T lymphocyte responses against non‐small cell lung cancer cells. Cellular Immunology 300, 46–53. [DOI] [PubMed] [Google Scholar]
- Mathieu, M. , Martin‐Jaular, L. , Lavieu, G. , & Théry, C. (2019). Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell‐to‐cell communication. Nature Cell Biology 21, 9–17. [DOI] [PubMed] [Google Scholar]
- Mathieu, M. , Névo, N. , Jouve, M. , Valenzuela, J. I. , Maurin, M. , Verweij, F. J. , Palmulli, R. , Lankar, D. , Dingli, F. , Loew, D. , Rubinstein, E. , Boncompain, G. , Perez, F. , & Théry, C. (2021). Specificities of exosome versus small ectosome secretion revealed by live intracellular tracking of CD63 and CD9. Nature Communication 12, 4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo, H. , Chevallier, J. , Mayran, N. , Le Blanc, I. , Ferguson, C. , Fauré, J. , Blanc, N. S. , Matile, S. , Dubochet, J. , Sadoul, R. , Parton, R. G. , Vilbois, F. , & Gruenberg, J. (2004). Role of LBPA and Alix in multivesicular liposome formation and endosome organization. Science 303, 531–534. [DOI] [PubMed] [Google Scholar]
- Möbius, W. , Van Donselaar, E. , Ohno‐Iwashita, Y. , Shimada, Y. , Heijnen, H. F. G. , Slot, J. W. , & Geuze, H. J. (2003). Recycling compartments and the internal vesicles of multivesicular bodies harbor most of the cholesterol found in the endocytic pathway. Traffic (Copenhagen, Denmark) 4, 222–231. [DOI] [PubMed] [Google Scholar]
- Mouchel, P. L. , Serhan, N. , Betous, R. , Farge, T. , Saland, E. , de Medina, P. , Hoffmann, J. S. , Sarry, J. E. , Poirot, M. , Silvente‐Poirot, S. , & Récher, C. (2020). Dendrogenin A Enhances Anti‐Leukemic Effect of Anthracycline in Acute Myeloid Leukemia. Cancers (Basel) 12(10), 2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeid, M. , Tesniere, A. , Ghiringhelli, F. , Fimia, G. M. , Apetoh, L. , Perfettini, J.‐L. , Castedo, M. , Mignot, G. , Panaretakis, T. , Casares, N. , Métivier, D. , Larochette, N. , Van Endert, P. , Ciccosanti, F. , Piacentini, M. , Zitvogel, L. , & Kroemer, G. (2007). Calreticulin exposure dictates the immunogenicity of cancer cell death. Nature Medicine 13, 54–61. [DOI] [PubMed] [Google Scholar]
- Ortiz, A. , Gui, J. , Zahedi, F. , Yu, P. , Cho, C. , Bhattacharya, S. , Carbone, C. J. , Yu, Q. , Katlinski, K. V. , Katlinskaya, Y. V. , Handa, S. , Haas, V. , Volk, S. W. , Brice, A. K. , Wals, K. , Matheson, N. J. , Antrobus, R. , Ludwig, S. , Whiteside, T. L. , … Fuchs, S. Y. (2019). An Interferon‐Driven Oxysterol‐Based Defense against Tumor‐Derived Extracellular Vesicles. Cancer Cell 35, 33–45 e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski, M. , Carmo, N. B. , Krumeich, S. , Fanget, I. , Raposo, G. , Savina, A. , Moita, C. F. , Schauer, K. , Hume, A. N. , Freitas, R. P. , Goud, B. , Benaroch, P. , Hacohen, N. , Fukuda, M. , Desnos, C. , Seabra, M. C. , Darchen, F. , Amigorena, S. , Moita, L. F. , Thery, C. , (2010) Rab27a and Rab27b control different steps of the exosome secretion pathway. Nature Cell Biology 12, 11–13. [DOI] [PubMed] [Google Scholar]
- Peinado, H. , Alečković, M. , Lavotshkin, S. , Matei, I. , Costa‐Silva, B. , Moreno‐Bueno, G. , Hergueta‐Redondo, M. , Williams, C. , García‐Santos, G. , Ghajar, C. M. , Nitadori‐Hoshino, A. , Hoffman, C. , Badal, K. , Garcia, B. A. , Callahan, M. K. , Yuan, J. , Martins, V. R. , Skog, J. , Kaplan, R. N. , … Lyden, D. (2012). Melanoma exosomes educate bone marrow progenitor cells toward a pro‐metastatic phenotype through MET. Nature Medicine 18, 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin‐Cocon, L. , Aublin‐Gex, A. , Diaz, O. , Ramière, C. , Peri, F. , André, P. , & Lotteau, V. (2018). Toll‐like receptor 4‐induced glycolytic burst in human monocyte‐derived dendritic cells results from p38‐dependent stabilization of HIF‐1alpha and increased hexokinase II expression. Journal of Immunology 201, 1510–1521. [DOI] [PubMed] [Google Scholar]
- Perrin‐Cocon, L. , Aublin‐Gex, A. , Sestito, S. E. , Shirey, K. A. , Patel, M. C. , André, P. , Blanco, J. C. , Vogel, S. N. , Peri, F. , & Lotteau, V. (2017). TLR4 antagonist FP7 inhibits LPS‐induced cytokine production and glycolytic reprogramming in dendritic cells, and protects mice from lethal influenza infection. Science Reports 7, 40791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt, J. M. , Charrier, M. , Viaud, S. , André, F. , Besse, B. , Chaput, N. , Zitvogel, L. , Dendritic cell‐derived exosomes as immunotherapies in the fight against cancer. Journal of Immunology 193, 1006–1011 (2014). [DOI] [PubMed] [Google Scholar]
- Pontini, L. , & Marinozzi, M. (2021).Shedding light on the roles of liver X receptors in cancer by using chemical probes. British journal of pharmacology 178(16), 3261–3276. 10.1111/bph.15200 [DOI] [PubMed] [Google Scholar]
- Record, M. , Silvente‐Poirot, S. , Poirot, M. , & Wakelam, M. O. (2018). Extracellular vesicles: Lipids as key components of their biogenesis and functions. Journal of Lipid Research 59, 1316–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Record, M. , Subra, C. , Silvente‐Poirot, S. , & Poirot, M. (2011). Exosomes as intercellular signalosomes and pharmacological effectors. Biochemical Pharmacology 81, 1171–1182. [DOI] [PubMed] [Google Scholar]
- Robbins, P. D. , & Morelli, A. E. (2014). Regulation of immune responses by extracellular vesicles. Nature Reviews Immunology 14, 195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer, M. , Schmitz, G. , & Liebisch, G. (2010). Simultaneous quantification of cardiolipin, bis(monoacylglycero)phosphate and their precursors by hydrophilic interaction LC‐MS/MS including correction of isotopic overlap. Analytical Chemistry 82, 8794–8799. [DOI] [PubMed] [Google Scholar]
- Segala, G. , David, M. , de Medina, P. , Poirot, M. C. , Serhan, N. , Vergez, F. , Mougel, A. , Saland, E. , Carayon, K. , Leignadier, J. , Caron, N. , Voisin, M. , Cherier, J. , Ligat, L. , Lopez, F. , Noguer, E. , Rives, A. , Payré, B. , Saati, T. A. l. , … Silvente‐Poirot, S. (2017). Dendrogenin A drives LXR to trigger lethal autophagy in cancers. Nature Communication 8, 1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan, N. , Mouchel, P. L. , Medina, P. , Segala, G. , Mougel, A. , Saland, E. , Rives, A. , Lamaziere, A. , Despres, G. , Sarry, J. E. , Larrue, C. , Vergez, F. , Largeaud, L. , Record, M. , Récher, C. , Silvente‐Poirot, S. , & Poirot, M. (2020). Dendrogenin A synergizes with cytarabine to kill acute myeloid leukemia cells in vitro and in vivo. Cancers (Basel) 12(7), 1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvente‐Poirot, S. , Dalenc, F. , & Poirot, M. (2018). The effects of cholesterol‐derived oncometabolites on nuclear receptor function in cancer. Cancer Research 78, 4803–4808. [DOI] [PubMed] [Google Scholar]
- Subra, C. , Grand, D. , Laulagnier, K. , Stella, A. , Lambeau, G. , Paillasse, M. , de Medina, P. , Monsarrat, B. , Perret, B. , Silvente‐Poirot, S. , Poirot, M. , & Record, M. (2010). Exosomes account for vesicle‐mediated transcellular transport of activatable phospholipases and prostaglandins. Journal of Lipid Research 51, 2105–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkach, M. , Kowal, J. , Zucchetti, A. E. , Enserink, L. , Jouve, M. , Lankar, D. , Saitakis, M. , Martin‐Jaular, L. , & Théry, C. (2017). Qualitative differences in T‐cell activation by dendritic cell‐derived extracellular vesicle subtypes. Embo Journal 36, 3012–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti, R. , Huber, V. , Filipazzi, P. , Pilla, L. , Sovena, G. , Villa, A. , Corbelli, A. , Fais, S. , Parmiani, G. , & Rivoltini, L. (2006). Human tumor‐released microvesicles promote the differentiation of myeloid cells with transforming growth factor‐beta‐mediated suppressive activity on T lymphocytes. Cancer Research 66, 9290–9298. [DOI] [PubMed] [Google Scholar]
- Vidal, M. , Mangeat, P. , & Hoekstra, D. (1997). Aggregation reroutes molecules from a recycling to a vesicle‐mediated secretion pathway during reticulocyte maturation. Journal of Cell Science 110(Pt 16), 1867–1877. [DOI] [PubMed] [Google Scholar]
- Viennois, E. , Esposito, T. , Dufour, J. , Pommier, A. , Fabre, S. , Kemeny, J.‐L. , Guy, L. , Morel, L. , Lobaccaro, J.‐M. , & Baron, S. (2012). Lxralpha regulates the androgen response in prostate epithelium. Endocrinology 153, 3211–3223. [DOI] [PubMed] [Google Scholar]
- Whiteside, T. L. (2016). Exosomes and tumor‐mediated immune suppression. Journal of Clinical Investigation 126, 1216–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfers, J. , Lozier, A. , Raposo, G. , Regnault, A. , Théry, C. , Masurier, C. , Flament, C. , Pouzieux, S. , Faure, F. , Tursz, T. , Angevin, E. , Amigorena, S. , & Zitvogel, L. (2001). Tumor‐derived exosomes are a source of shared tumor rejection antigens for CTL cross‐priming. Nature Medicine 7, 297–303. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, Y. , Brenner, M. , & Hearing, V. J. (2007). The regulation of skin pigmentation. Journal of Biological Chemistry 282, 27557–27561. [DOI] [PubMed] [Google Scholar]
- Zitvogel, L. , Regnault, A. , Lozier, A. , Wolfers, J. , Flament, C. , Tenza, D. , Ricciardi‐Castagnoli, P. , Raposo, G. , & Amigorena, S. (1998). Eradication of established murine tumors using a novel cell‐free vaccine: Dendritic cell‐derived exosomes. Nature Medicine 4, 594–600. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information
Supplementary information
Supplementary information
Supplementary information
Supplementary information
Supplementary information
Supplementary information
Supplementary information
Supplementary information
Supplementary information
Supplementary information
Supplementary information
Supplementary information
Supplementary information
Supplementary information
Supplementary information
Supplementary information
Data Availability Statement
The data for the study are available from the corresponding authors upon request.