

Abstract
Chemotherapy is still regarded as the main modality for cancer treatment. However, it often suppresses host immune system, resulting in limited therapeutic effects. It is desirable to design a novel chemotherapeutic agent to reduce the level of immunosuppression. Herein, we designed bovine serum albumin (BSA)-bioinspired iron oxide nanoparticles (IONPs) as a nanocarrier to load anticancer drug mitoxantrone (MTX) for enhanced chemotherapy of orthotopic breast cancer. The IONPs@BSA-MTX complexes treatment increased much more CD3+CD4+ and CD3+CD8+ T lymphocytes than free MTX. The complexes effectively restored host immune system and produced a better anticancer efficacy than free MTX. It was worth noting that the BSA-inspired IONPs were a satisfactory contrast agent for magnetic resonance imaging of tumors and lymph nodes. Our work provides a novel strategy for enhanced chemotherapy with low levels of immunosuppression in the treatment of breast cancer and other cancers.
Graphical Abstract
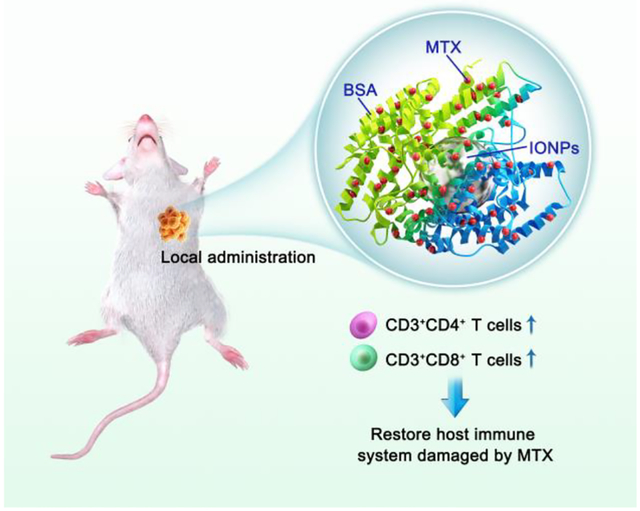
BSA-bioinspired iron oxide nanoparticles as a nanocarrier to load anticancer drug mitoxantrone for enhanced chemotherapy with low levels of immunosuppression.
Introduction
To date, chemotherapy, a systemic treatment method, is still considered as a major modality for cancer treatment for its effectiveness and rapidity in killing cancer cells and controlling tumors. However, most chemotherapeutic drugs face various clinical challenges, such as nonspecificity, poor solubility/stability, rapid release, high-dosage requirement, severe toxic side effects on normal cells, and suppression of host immune system.1–4
To overcome these limitations, various nanoplatforms including proteins,5–8 micelles,9 liposomes,10–12 polymers,13–16 dendrimers,17–21 and mesoporous silica nanoparticles (MSNPs),22–25 have been introduced as powerful vehicles to load chemotherapeutic drugs for cancer treatment. For example, polyethylene glycol-polyethyleneimine (PEG-PEI) copolymer modified with MSNPs can significantly reduce the hydrodynamic size of the particle, and improve the passive delivery of monodispersed.25 After PEG-PEI-MSNPs loading with anticancer drug doxorubicin (DOX), about 12% of the PEG-PEI-MSNPs-DOX complexes passively accumulated in the tumor site, which can significantly reduce the drug dose and systemic side effects. In our previous work, we used targeting agents (e.g., folic acid or hyaluronic acid) to modify PEGylated hyperbranched PEI or dendrimers as a nanoplatform to load DOX for targeted cancer chemotherapy.26–28 These drug delivery systems were water soluble, released DOX in a pH-dependent manner steadily, and displayed specific antitumor efficacy. It is well known that these drug delivery systems through systemic administration, which need to overcome many obstacles such as immune system clearance and tumor matrix barrier in the process of entering the blood and delivering to tumor tissue, result in low drug delivery efficiency.29 In contrast, local administration has a good performance in efficiency promotion and off-target toxicity reduction. Additionally, local administration can break immune tolerance, resulting in antitumor immunity without systemic exposure.30 Local administration was widely used in phototherapy, radiotherapy, immunotherapy, and combination therapy.31–38 The U.S. Food and Drug Administration (FDA) has approved the first intratumoral injection of therapeutic agents to treat non-metastatic mast cell tumors in dogs in 2020 (https://www.fda.gov/news-events).
Recently, IONPs are able to induce pro-inflammatory macrophage polarization, upregulate M1-related markers (e.g., Tumor Necrosis Factor Alpha, TNFα), and downregulate host immunosuppression caused by M2-related markers (e.g., interleukin 10, IL-10).38–41 Bovine serum albumin (BSA) is the main protein species found in bovine plasma, and has been used as an ideal carrier to encapsulate NPs or load drug molecules in the biomedical field because of its good biocompatibility, absorbability, non-antigenicity, and non-immunogenicity.33, 42–44 Here, BSA-coated IONPs were synthesized to improve the biocompatibility of IONPs via electrostatic interaction, which could be as a nanocarrier to load anticancer drug mitoxantrone (MTX) through BSA absorbability for enhanced chemotherapy of orthotopic breast cancer with low levels of immunosuppression. The formed IONPs@BSA-MTX complexes were delivered to the tumor site through intratumoral injection to reduce the dose and off-target toxicity. In addition, owing to IONPs, the level of activated T lymphocytes, usually reduced by free MTX, could be significantly increased by IONPs@BSA-MTX complexes. Therefore, the complexes resulted in a better tumor inhibition and longer animal survival time than free MTX. Besides, the BSA-inspired IONPs can be utilized as a satisfactory magnetic resonance (MR) contrast agent for imaging of tumors and lymph nodes.
Experimental
Synthesis of IONPs@BSA-MTX complexes
BSA protein was used to coat IONPs to form IONPs@BSA through electrostatic interaction. Briefly, amine functionalized IONPs (5 mL, 1 mg/mL Fe) with positive surface charges and BSA (25 mg) with negative surface charges were mixed using water solution (10 mL) under violent stirring overnight. The mixture was then centerfield using 14000 rpm for 10 min and washed by water twice to remove the remaining BSA to form IONPs@BSA. Next, MTX·2HCl solution (2 mL, 0.5 mg/mL MTX in water) was added into IONPs@BSA aqueous solution (10 mL, 0.5 mg/mL Fe) and the mixture was vigorously stirred in dark overnight. Then, the mixture solution was centrifuged at 14000 rpm for 10 min and washed by water twice to get the product of IONPs@BSA-MTX complexes.
Transwell assay
We used a 6 well dual-chamber transwell system (3 μm-microporous membranes for macrophages to pass through) to evaluate cytokine secretion by macrophages and tumor cytotoxicity. RAW264.7 macrophages (1 × 106) and 4T1 breast tumor cells (2 × 105) were plated in each upper and lower chamber, respectively, and co-incubated for 6 hours with IONPs@BSA at a Fe concentration of 1mg/mL. All transwell assays were performed with Dulbecco’s Modified Eagle’s Medium (DMEM), supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Subsequently, the bottom chambers were isolated and cells were stained by Double Stain Apoptosis Detection Kit (calcein-AM/PI) before observation using Leica DM IL LED inverted phase contrast microscope (Wetzlar, Germany).
To assess M1- and M2-related cytokine secretion in vitro, macrophages from the upper chamber (transwell system with 0.4 μm-sized microporous membranes and 12 h incubation time) were harvested, followed by enzyme-linked immunosorbent assay (ELISA) analysis according to the manufacturer’s protocols.38, 45
In vivo immune response and antitumor efficacy
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH) and approved by the University of Oklahoma Health Sciences Center, Institutional Animal Care and Use Committee. Six to eight week-old BALB/C female mice were purchased from Charles River (Wilmington, MA). 4T1 cells (5 × 104 in 100 μL PBS) were subcutaneously injected into the right flank of each mouse. MTX, IONPs@BSA, IONPs@BSA-MTX, or PBS (100 μL, as a control) were injected into tumors when tumor size reached a size of 100–150 mm3. We harvested lymph nodes and spleen on day 3 and 7 after the treatments, respectively. To assess DC maturation, lymphocytes from lymph nodes were stained with anti-mouse CD11c-Alexa Fluor 647 and anti-mouse CD86-PE antibodies, and then were analyzed by flow cytometry according to the manufacturer’s protocols.46 To evaluate T cell activation, splenocytes were stained with APC anti-mouse CD3-Cy7, anti-mouse CD4-PE, and anti-mouse CD8-Alexa Flour 647 antibodies (Biolegend, San Diego, CA), and then were analyzed by flow cytometry according to the manufacturer’s protocols.47
To evaluate the antitumor efficacy of IONPs@BSA-MTX complexes in vivo, we established orthotopic 4T1 breast tumors in mice, with 5 × 104 cells being injected orthotopically into left mammary fat. When tumor volumes reached 100–150 mm3, these tumor-bearing mice were divided at random into 4 groups (n = 6 for each group) and treated with MTX, IONPs@BSA, IONPs@BSA-MTX, and PBS respectively for 3 times (the dosing interval was 3 days). Tumor sizes were measured every two days and the survivals of these mice were carefully monitored until the endpoint of the study.
Results and discussion
Construction and characterization of IONPs@BSA-MTX complexes
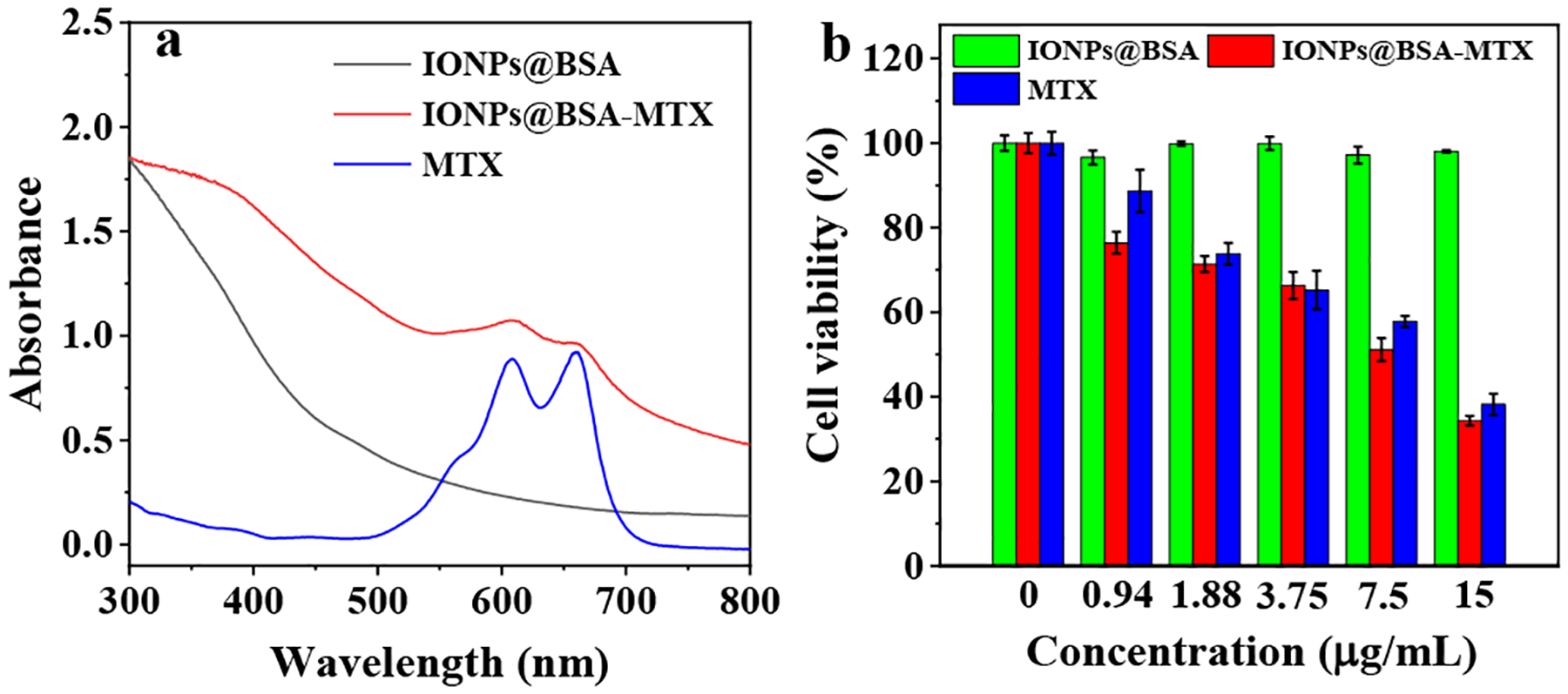
The composition and structure of IONPs@BSA-MTX complexes were characterized by various techniques. Transmission electron microscopy (TEM) images showed that the IONPs displayed uniform morphology (Fig. 1a,b) with a mean diameter of 6.8 nm, and the lattice was clearly visible (Inset of Fig. 1b). Elemental mapping of the IONPs indicated that the particles were mainly comprised of Fe and O elements (Fig. 1 c–f), which was further confirmed by energy-dispersive X-ray spectroscopy (Fig. S1, ESI†). As shown in Fig. 2a, IONPs@BSA-MTX displayed two peaks at 609 nm and 661 nm in the UV-Vis spectroscopy, in accordance with what the peaks of free MTX displayed, indicating the successful loading of MTX to IONPs@BSA. Based on the standard calibration curve of MTX water solution, the MTX encapsulation efficiency (weight of MTX in the IONPs@BSA-MTX/theoretical amount of MTX) and loading capacity (weight of MTX in the IONPs@BSA-MTX/weight of IONPs@BSA-MTX) were 57.3% and 6.0%, respectively (Fig. S2, ESI†). The content of BSA protein (weight of BSA in IONPs@BSA-MTX/weight of IONPs@BSA-MTX) was measured to be 21.4% by Pierce™ bicinchoninic acid (BCA) protein assay (Fig. S3, ESI†).
Fig. 1.

TEM images (a, b) of IONPs (inset in panel b shows the high-resolution TEM image of the particle). Dark-field image of IONPs (c), and the elemental mapping of iron (d), oxygen (e), and merged image (f).
Fig. 2.
(a) UV-vis spectra of IONPs@BSA, IONPs@BSA-MTX, and MTX, respctively. (b) Viability of 4T1 cells treated with IONPs@BSA, IONPs@BSA-MTX, and MTX at different MTX concentrations for 24 h.
It is well known that that the hydrodynamic size is usually larger than TEM size given the same particle.48, 49 As we recorded, the hydrodynamic size of IONPs was 36.6 ± 1.6 nm, and increased to 383.2 ± 55.6 nm after BSA coating, suggesting that the IONPs had been successfully coated with BSA protein (Table S1, ESI†). When MTX was loaded, the hydrodynamic size of IONPs@BSA-MTX was measured to be 376.8 ± 31.1 nm (Table S1, ESI†), which indicated that the load of MTX did not change the hydrodynamic size of IONPs@BSA. The reason may be that TEM only measures a single IONPs core particle, whereas DLS measures clusters of particles that may consist of many single IONPs, in accordance with the literature.48 Because of the functionalization by positive amine groups, the zeta potential of IONPs was 3.2 ± 1.3 mV (Fig. S4, ESI†). However, after IONPs being coated with negative BSA protein, the zeta potential of IONPs@BSA reduced to −13.2 ± 0.2 mV (Fig. S4, ESI†), further confirming the success of BSA in coating onto the surface of IONPs. Notably, we found no obvious change in zeta potential of IONPs@BSA-MTX (−12.8 ± 0.9 mV) in comparison with IONPs@BSA, indicating no obvious impact of MTX on the zeta potential of IONPs@BSA (Fig. S4, ESI†).
For in vitro drug release, as shown in Fig. S5, the IONPs@BSA-MTX complexes can be released both in acid pH (pH = 5.5) and PBS buffer (pH = 7.4) conditions. We found that MTX can be sustainably released from the complexes, and showed a faster release speed under acidic pH conditions than PBS conditions. The MTX release rates were measured to be 52.2% in acid pH condition and 33.3% in PBS condition at 30 h time point, respectively (Fig. S5, ESI†). The faster MTX release rate under acidic pH conditions is likely because a majority of MTX is protonated with high water solubility, and the positively charged MTX may repel each other under acidic pH conditions.
Cell viability assay
CCK-8 assay was utilized to study the cell viability of IONPs@BSA-MTX complexes. For IONPs@BSA, it did not show obvious cytotoxicity at different concentrations, because the cell viability of all given concentrations was higher than 90% (Fig. 2b). This result indicated that IONPs@BSA as a nanocarrier displayed an excellent cytocompatibility. Whereas IONPs@BSA-MTX complexes and free MTX can both induce cell death at different MTX concentrations. The cell viabilities of IONPs@BSA-MTX and free MTX both reduced to approximately 50% at the MTX concentration of 7.5 μg/mL, respectively (Fig. 2b).
We then used cell morphology observation to evaluate their cell viability. 4T1 tumor cells treated with IONPs@BSA at the same equivalent concentrations of IONPs@BSA-MTX ([MTX] = 15 μg/mL) displayed a similar morphology to PBS control (Fig. S6, ESI†). In contrast, cells treated with IONPs@BSA-MTX or free MTX at the MTX concentration of 15 μg/mL had a large portion of round and detached cells, suggesting a significant portion of cell death (Fig. S6, ESI†). These results were consistent with CCK-8 data.
Transwell assay
A 6 well dual chamber transwell system was used to assess cytokine secretion by macrophages and tumor cytotoxicity (Fig. S7, ESI†). Enzyme-linked immunosorbent assay (ELISA) using a dual-chamber transwell system with 0.4 μm microporous membranes (macrophages cannot migrate through the pores) was utilized to analyze cytokine secretion by macrophages. We found that IONPs@BSA-exposed tumor cells and macrophages significantly increased the production of M1-related TNFα (Fig. 3a), however, no obvious production of M2-related IL-10 (Fig. 3b) was observed. It suggested that IONPs@BSA-induced tumor cytotoxicity was related to M1 macrophage polarization. The tumor cytotoxicity induced by IONPs@BSA was further confirmed by live/dead double assay using calcein-AM/PI kit. As shown in Fig. 3c, the group of IONPs@BSA + macrophages + cancer cells displayed obvious PI fluorescence, whereas other groups displayed little, suggesting a good performance of IONPs@BSA in inducing tumor cytotoxicity
Fig. 3.
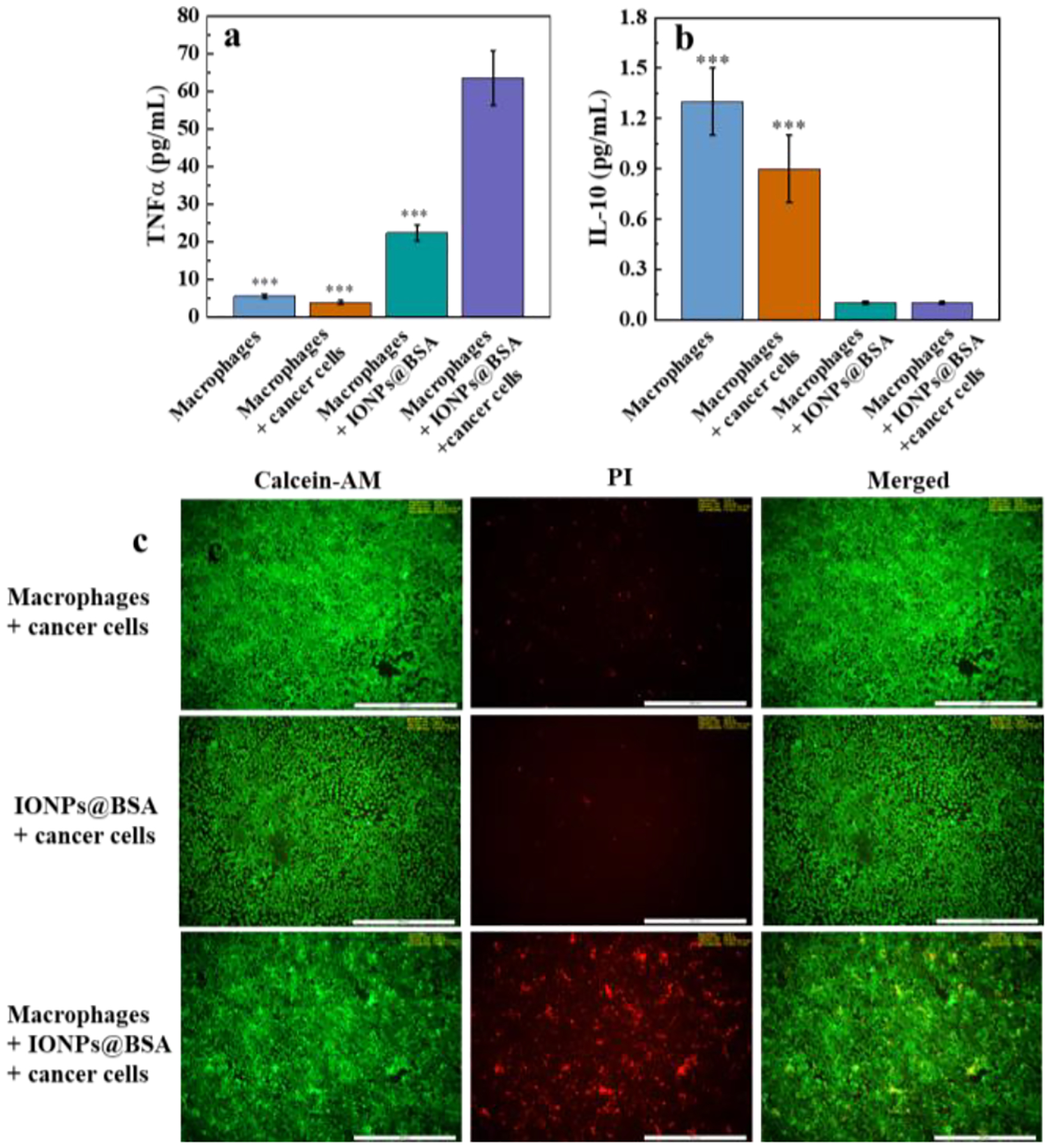
The TNFα (a) and IL-10 (b) secretion of macrophages in cocultures with IONPs@BSA and cancer cells (n = 3, ***p < 0.001, vs. IONPs@BSA + macrophages + cancer cells). (c) Fluorescence images of cancer cells after various treatments. Scare bar = 500 μm.
In vivo immune responses and antitumor efficacy
We first evaluated in vivo immune responses induced by IONPs@BSA-MTX complexes (Fig. 4a). As shown in Fig. 4b,d, mice treated by IONPs@BSA-MTX (25.8% of CD11c+CD86+) displayed a similar level of DC maturation with PBS (25% of CD11c+CD86+) and IONPs@BSA (27.25% of CD11c+CD86+), which was much higher than that with MTX (19.4% of CD11c+CD86+). Importantly, the splenocytes from mice treated with free MTX showed much lower percentages of CD3+CD4+ (34%, Fig. 4c,e) and CD3+CD8+ (13.2%, Fig. 4c,f) T cells than those of other treatments. While, owing to the existence of IONPs, mice treated by IONPs@BSA-MTX displayed similar levels of activated T cells (45.4% of CD3+CD4+ and 18.2% of CD3+CD8+). These results demonstrated that IONPs@BSA-MTX could significantly restore the level of host immune DC and T cell reduced by MTX.
Fig. 4.
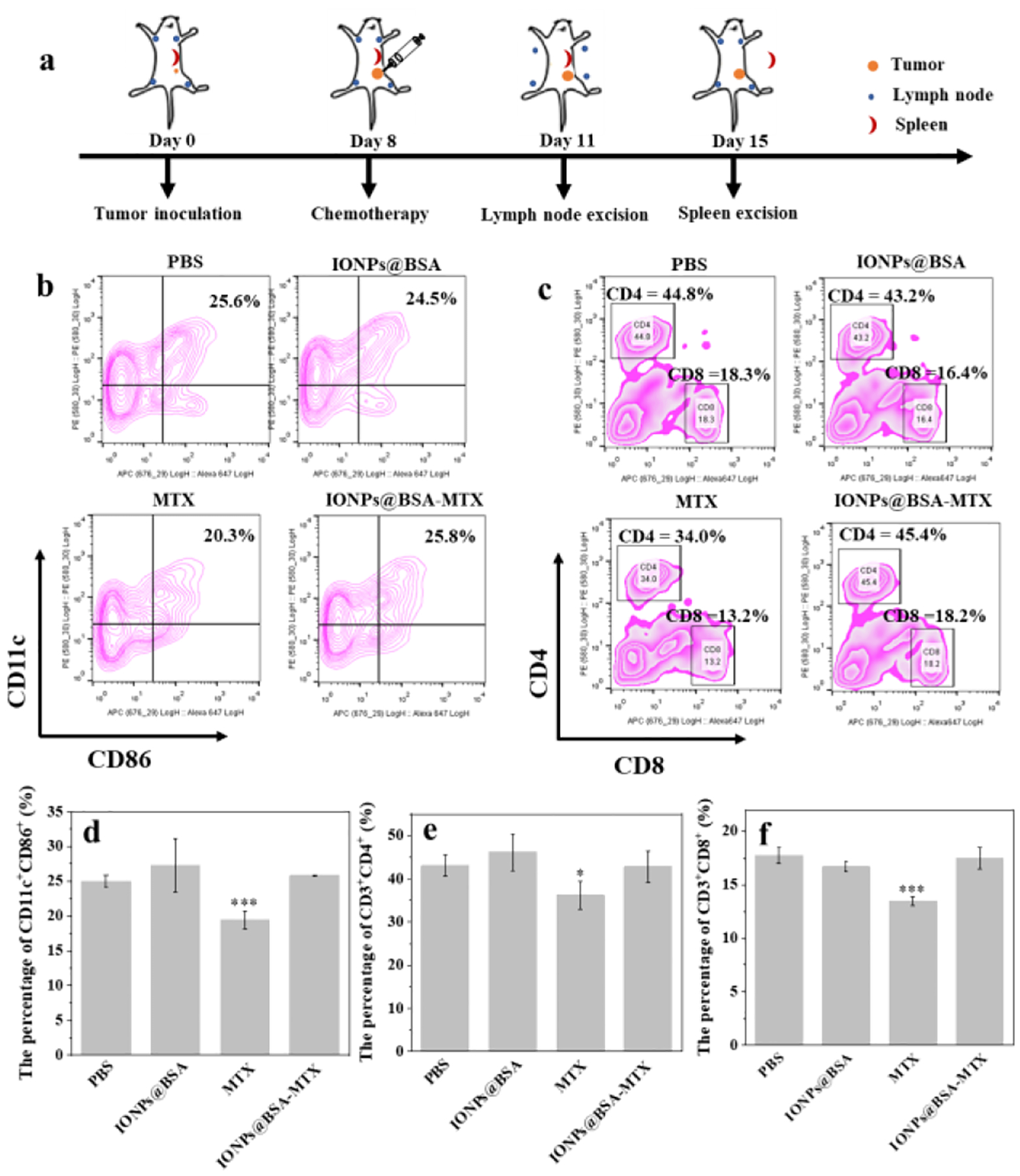
(a) Schematic illustration of experiment design for in vivo immune responses. Representative flow plots (b) and the percentages (c) of DC maturation (CD11c+CD86+) in lymph nodes on day 3 after various treatments (n = 3, ***P < 0.001, vs. PBS group). Representative flow plots (d) and the percentages (e and f) of activated T cells (CD3+CD4+ and CD3+CD8+) in spleen on day 3 after various treatments (n = 3, *P < 0.05, ***P < 0.001, vs. PBS group).
We further evaluated in vivo antitumor efficacy of IONPs@BSA-MTX complexes by an orthotopic 4T1 breast tumor model. The groups of IONPs@BSA-MTX and free MTX both can control the tumor growth more effectively compared with PBS and IONPs@BSA groups (Fig. 5a), suggesting that IONPs alone did not significantly kill tumor cells, although IONPs could reduce host immunosuppression. It should be noted that although free MTX and IONPs@BSA-MTX groups could effectively inhibit tumor growth within 18 days after treatments, it is difficult to completely inhibit tumor growth and metastasis by chemotherapy alone owing to high levels of metastasis of 4T1 tumor cells. Therefore, free MTX and IONPs@BSA-MTX groups cannot cure 4T1 tumors, but could appropriately prolong the survival time of mice. Importantly, intratumoral injection of IONPs@BSA-MTX could more significantly prolong the survival time of mice (Fig. 5b). The last mouse died 52 days after being treated with IONPs@BSA-MTX, which was much longer than that of MTX (36 days), IONPs@BSA (26 days), and PBS (24 days). These results demonstrated a better in vivo antitumor efficacy of IONPs@BSA-MTX than that of free MTX. What caused it may be the existence of IONPs, which can recover the host immunologic function. In addition, the body weight of the tumor-bearing mice treated with IONPs@BSA-MTX had no obvious change when compared to the groups of IONPs@BSA and PBS, while the MTX group had the least body weight at the same time points (Fig. 5c). This implied that free MTX may display toxicity in mice. Importantly, the injection of IONPs@BSA-MTX did not show any significant histological change of all the major organs, compared with the PBS control group, suggesting that the complexes displayed excellent biocompatibility in vivo (Fig. S8, ESI†), while free anticancer drugs (e.g., DOX) would significantly cause organ toxicity in vivo (e.g., heart and lung).26, 50
Fig. 5.
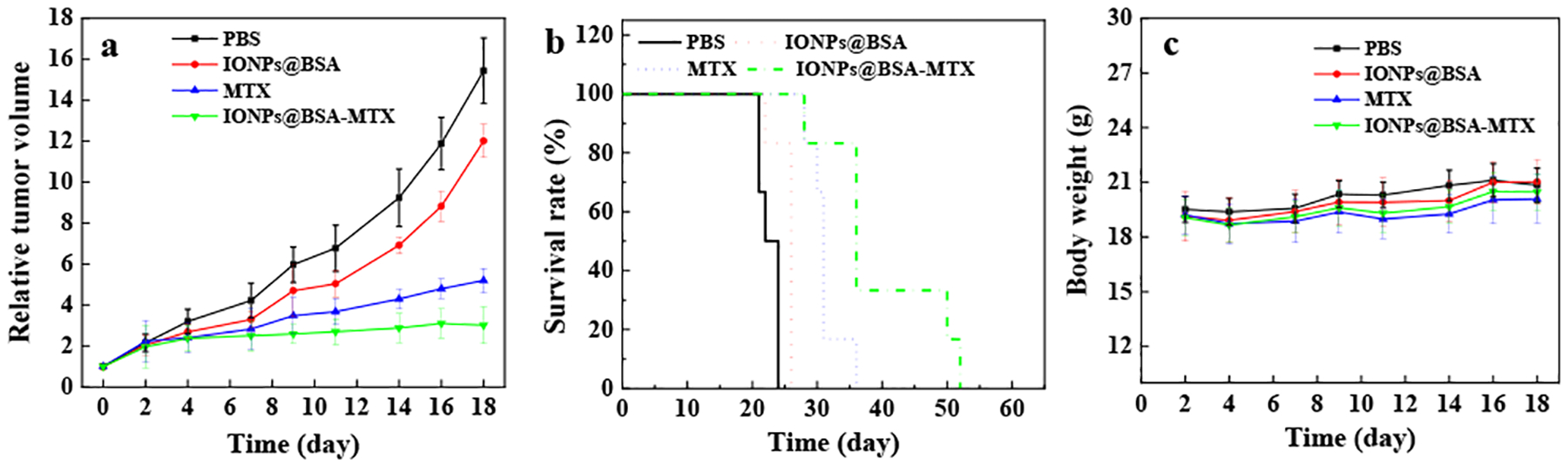
Relative tumor volumes (a), survival rates (b) and body weight (c) of mice after various treatments as a function of time post-tumor inoculation.
In vivo MR imaging
The designed IONPs@BSA not only served as a nanocarrier for drug delivery but also as a satisfactory T2-weighted MR contrast agent for the imaging of orthotopic breast tumors and lymph nodes. After intravenous injection with IONPs@BSA, the tumor region became dark gradually in MR imaging, suggesting that the particles can be delivered into the tumor site (Fig. 6a). It should be noted that when we enlarged tumor region before injection and 3 h postinjection, T2 MR signals reduced distinctly (Fig. 6b). Quantitative analysis of the MR signal/noise ratio (SNR) of tumor sites showed that the SNR of tumor decreased from 3.1 ± 0.2 (before injection) to 1.5 ± 0.1 (3 h postinjection), and then returned to 2.5 ± 0.1 at 24 h postinjection (Fig. 6c), which was consistent with the results shown in Fig. 6a. These results indicated that the particles can be effectively accumulated at tumor sites through enhanced permeability and retention (EPR) effect, and can be gradually metabolized from the tumor region after 24 h postinjection.
Fig. 6.
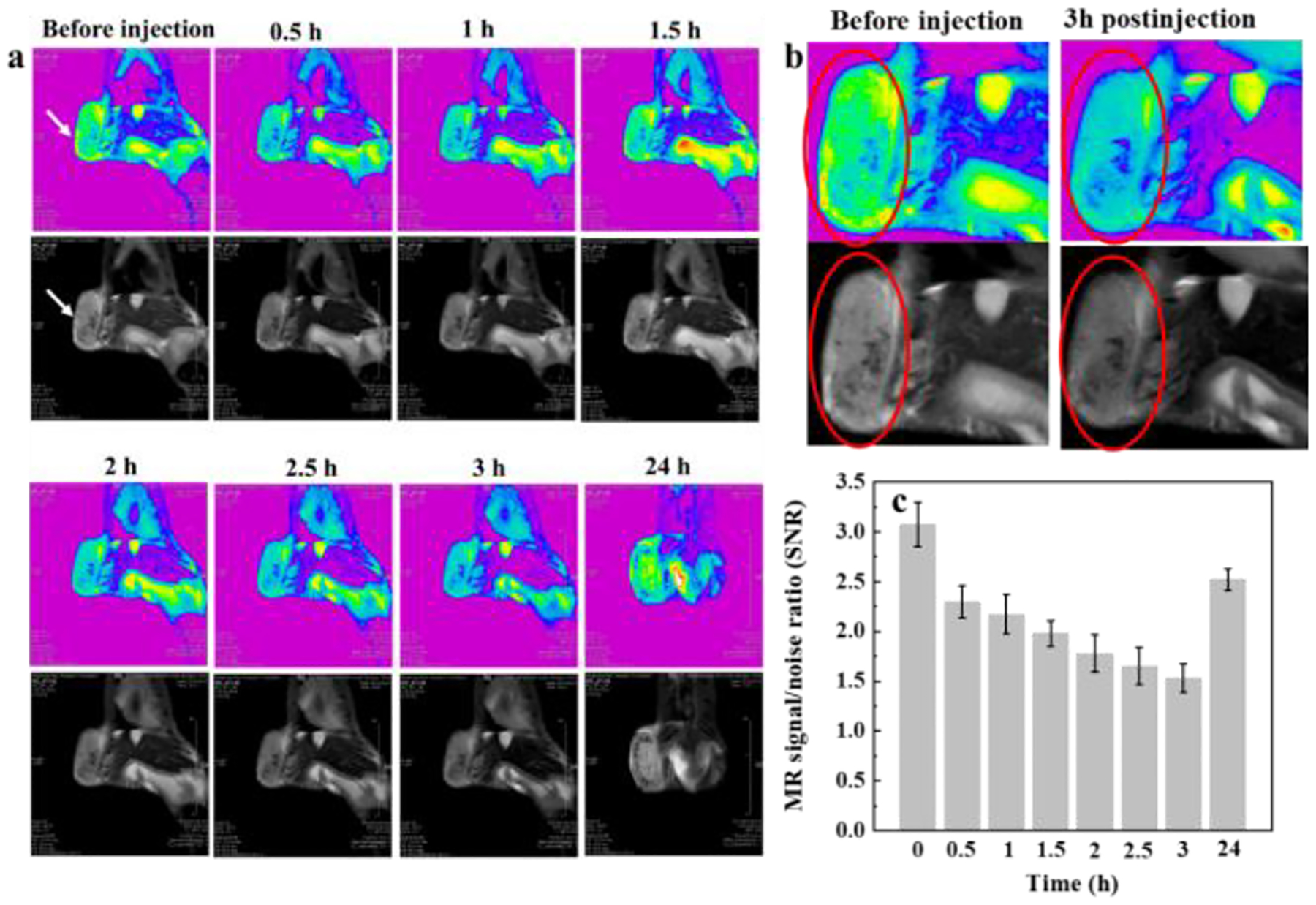
T2-weighted MR images (a) and SNR (c) of orthotopic 4T1 breast tumor in mouse after intravenous injection of IONPs@BSA (white arrows refer to tumors). (b) Local enlargement of the MR images of tumor areas before and 3 h after injection (red circles refer to tumors).
The lymphatic system is a regular route for tumor metastasis.51 Therefore, it is of great significance to develop an MR contrast agent for detecting tumor metastasis via lymph node imaging. In addition, the particles could also be used as a contrast agent for the detection of lymph nodes (Fig. 7). The left and right lymph nodes both became dark after injection with IONPs@BSA (Fig. 7a). When we enlarged them before injection and 2.5 h postinjection, the change of T2-weighted MR signals became obvious (Fig. 7b). Owing to the limitation in the size of mouse lymph nodes, it is difficult to accurately analyze the MR signal variation of lymph nodes quantitatively. It should be noted that the lymph nodes got brightened after 24 h injection, indicating that the particle was gradually metabolized from the lymph nodes (Fig. 7a).
Fig. 7.
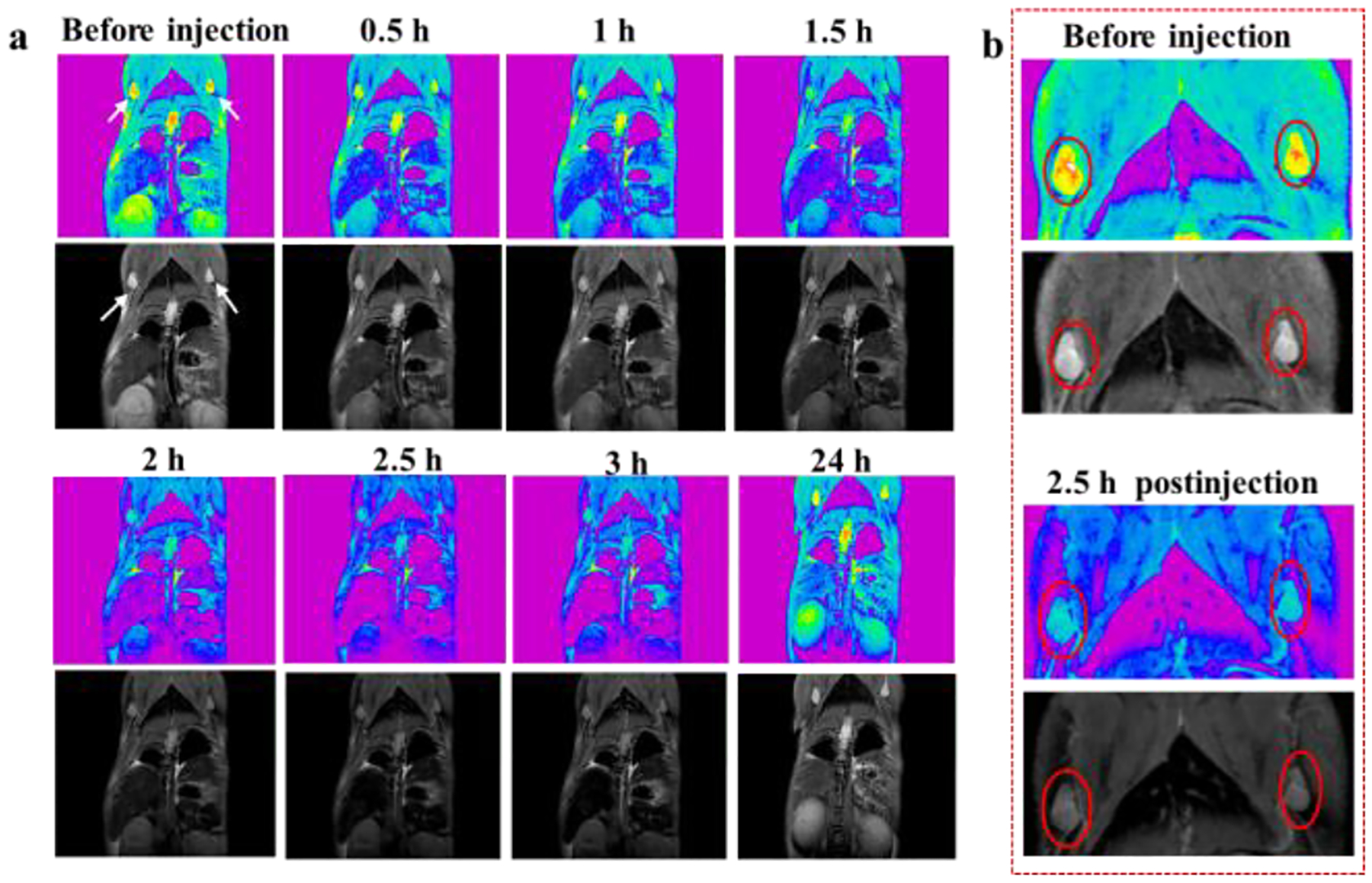
T2-weighted MR images (a) of lymph nodes of orthotopic 4T1 breast tumor-bearing mouse after intravenous injection of IONPs@BSA (white arrows refer to lymph nodes). (b) Local enlargement of the MR images of lymph nodes before and 2.5 h after injection (red circles refer to lymph nodes).
Conclusions
In summary, we designed BSA-inspired IONPs loaded with an anticancer drug for enhanced chemotherapy of breast cancer with low levels of immunosuppression. The IONPs@BSA-MTX complexes can effectively induce tumor cell death in vitro. More importantly, owing to the existence of IONPs, the IONPs@BSA-MTX complexes could significantly increase the level of CD3+CD4+ and CD3+CD8+ T lymphocytes in treated mice, which were usually reduced by free MTX-related chemotherapy. As a result, the complexes had better tumor inhibition and longer animal survival time than free MTX. In addition, the IONPs@BSA was regarded as a satisfactory T2-weighted MR contrast agent for the detection of tumors and lymph nodes. We believe that BSA-inspired IONPs loaded with an anticancer drug showing low levels of immunosuppression hold great potential for the treatment of breast and other cancers.
Supplementary Material
Acknowledgements
This work is supported by the National Natural Science Foundation of China (61805161), the Natural Science Foundation of Guangdong Province (2021A1515010441), and 2020 Li Ka Shing Foundation Cross-Disciplinary Research Grant (2020LKSFG05C). It is also supported by the U.S. National Institutes of Health (R01CA205348), and the Oklahoma Center for the Advancement of Science and Technology (HR16-085 and HF20-019).
Footnotes
Electronic Supplementary Information (ESI) available: Additional experimental results. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Zhou J, Yu GC and Huang FH, Chem. Soc. Rev, 2017, 46, 7021–7053. [DOI] [PubMed] [Google Scholar]
- 2.Moorthi C, Manavalan R and Kathiresan K, J. Pharm. Pharm. Sci, 2011, 14, 67–77. [DOI] [PubMed] [Google Scholar]
- 3.Yang S, Shim MK, Kim WJ, Choi J, Nam GH, Kim J, Kim J, Moon Y, Kim HY, Park J, Park Y, Kim IS, Ryu JH and Kim K, Biomaterials, 2021, 272, 120791. [DOI] [PubMed] [Google Scholar]
- 4.Lan JS, Liu L, Zeng RF, Qin YH, Hou JW, Xie SS, Yue S, Yang J, Ho RJY, Ding Y and Zhang T, Chem. Eng. J, 2021, 407, 127212. [Google Scholar]
- 5.Hawkins MJ, Soon-Shiong P and Desai N, Adv. Drug Delivery Rev, 2008, 60, 876–885. [DOI] [PubMed] [Google Scholar]
- 6.Zhao D, Zhao X, Zu Y, Li J, Zhang Y, Jiang R and Zhang Z, Int. J. Nanomed, 2010, 5, 669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li ZL, Hu Y, Miao ZH, Xu H, Li CX, Zhao Y, Li Z, Chang ML, Ma Z, Sun Y, Besenbacher F, Huang P and Yu M, Nano Lett, 2018, 18, 6778–6788. [DOI] [PubMed] [Google Scholar]
- 8.Kuai R, Yuan W, Son S, Nam J, Xu Y, Fan Y, Schwendeman A and Moon JJ, Sci. Adv, 2018, 4, eaao1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niu D, He J, Qin X, Liu Y, Liu H, Hu P, Li Y and Shi J, Nano Lett, 2021, 21, 9388–9397. [DOI] [PubMed] [Google Scholar]
- 10.Mross K, Niemann B, Massing U, Drevs J, Unger C, Bhamra R and Swenson C, Cancer Chemother. Pharmacol, 2004, 54, 514–524. [DOI] [PubMed] [Google Scholar]
- 11.Barenholz Y, J. Controlled Release, 2012, 160, 117–134. [DOI] [PubMed] [Google Scholar]
- 12.Dong ZL, Feng LZ, Chao Y, Hao Y, Chen MC, Gong F, Han X, Zhang R, Cheng L and Liu Z, Nano Lett., 2019, 19, 805–815. [DOI] [PubMed] [Google Scholar]
- 13.Torchilin VP, J. Controlled Release, 2001, 73, 137–172. [DOI] [PubMed] [Google Scholar]
- 14.Cheng R, Meng F, Deng C, Klok H-A and Zhong Z, Biomaterials, 2013, 34, 3647–3657. [DOI] [PubMed] [Google Scholar]
- 15.Merino S, Martin C, Kostarelos K, Prato M and Vazquez E, ACS Nano, 2015, 9, 4686–4697. [DOI] [PubMed] [Google Scholar]
- 16.Su T, Cheng F, Pu Y, Cao J, Lin S, Zhu G and He B, Chem. Eng. J, 2021, 411, 128561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.D’Emanuele A and Attwood D, Adv. Drug Delivery Rev, 2005, 57, 2147–2162. [DOI] [PubMed] [Google Scholar]
- 18.Li X, Takashima M, Yuba E, Harada A and Kono K, Biomaterials, 2014, 35, 6576–6584. [DOI] [PubMed] [Google Scholar]
- 19.Xiong Z, Wang Y, Zhu W, Ouyang Z, Zhu Y, Shen M, Xia J and Shi X, Small Methods, 2021, 5, 2100204. [DOI] [PubMed] [Google Scholar]
- 20.Li D, Lin LZ, Fan Y, Liu L, Shen MW, Wu R, Du LF and Shi XY, Bioact. Mater, 2021, 6, 729–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Z, Guo Y, Fan Y, Chen J, Wang H, Shen M and Shi X, Adv. Mater, 2021, 2107009. [DOI] [PubMed] [Google Scholar]
- 22.Zhang ZJ, Wang LM, Wang J, Jiang XM, Li XH, Hu ZJ, Ji YH, Wu XC and Chen CY, Adv. Mater, 2012, 24, 1418–1423. [DOI] [PubMed] [Google Scholar]
- 23.Barkat A, Beg S, Panda SK, Alharbi KS, Rahman M and Ahmed FJ, Semin. Cancer Biol, 2021, 69, 365–375. [DOI] [PubMed] [Google Scholar]
- 24.Chen Y, Chen HR and Shi JL, Adv. Mater, 2013, 25, 3144–3176. [DOI] [PubMed] [Google Scholar]
- 25.Meng H, Xue M, Xia T, Ji ZX, Tarn DY, Zink JI and Nel AE, ACS Nano, 2011, 5, 4131–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou B, Zhao L, Shen M, Zhao J and Shi X, J. Mater. Chem . B, 2017, 5, 1542–1550. [DOI] [PubMed] [Google Scholar]
- 27.Chen C, Zhou BQ, Zhu XY, Shen M and Shi XY, RSC Adv, 2016, 6, 9232–9239. [Google Scholar]
- 28.Fu FF, Zhou BQ, Ouyang ZJ, Wu YL, Zhu JY, Shen MW, Xia JD and Shi XY, Chin. J. Polym. Sci, 2019, 37, 129–135. [Google Scholar]
- 29.Ye W, Chen X, Li X, Liu Y, Jia F, Jin Q and Ji J, ACS Appl. Mater. Interfaces, 2020, 12, 22560–22571. [DOI] [PubMed] [Google Scholar]
- 30.Park CG, Hartl CA, Schmid D, Carmona EM, Kim H-J and Goldberg MS, Sci. Transl. Med, 2018, 10, eaar1916. [DOI] [PubMed] [Google Scholar]
- 31.Wang M, Song J, Zhou F, Hoover AR, Murray C, Zhou B, Wang L, Qu J and Chen WR, Adv. Sci, 2019, 6, 1802157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou B, Liu J, Lin M, Zhu J and Chen WR, Coord. Chem. Rev, 2021, 442, 214009. [Google Scholar]
- 33.Zhou B, Song J, Wang M, Wang X, Wang J, Howard EW, Zhou F, Qu J and Chen WR, Nanoscale, 2018, 10, 21640–21647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Y, Hu Y, Sun W, Zhou B, Zhu J, Peng C, Shen M and Shi X, Nanoscale, 2017, 9, 12746–12754. [DOI] [PubMed] [Google Scholar]
- 35.Lu K, He C, Guo N, Chan C, Ni K, Lan G, Tang H, Pelizzari C, Fu Y-X, Spiotto MT, Weichselbaum RR and Lin W, Nat. Biomed. Eng, 2018, 2, 600–610. [DOI] [PubMed] [Google Scholar]
- 36.Ni K, Aung T, Li S, Fatuzzo N, Liang X and Lin W, Chem, 2019, 5, 1892–1913. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Pan J, Wang Y, Zhang C, Wang X, Wang H, Wang J, Yuan Y, Wang X, Zhang X, Yu C, Sun S-K and Yan X-P, Adv. Mater, 2018, 30, 1704408. [Google Scholar]
- 38.Zhou B, Wu Q, Wang M, Hoover A, Wang X, Zhou F, Towner RA, Smith N, Saunders D, Song J, Qu J and Chen WR, Chem. Eng. J, 2020, 396, 125239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gu Z, Liu T, Tang J, Yang Y, Song H, Tuong ZK, Fu J and Yu C, J. Am. Chem. Soc, 2019, 141, 6122–6126. [DOI] [PubMed] [Google Scholar]
- 40.Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, Pajarinen JS, Nejadnik H, Goodman S, Moseley M, Coussens LM and Daldrup-Link HE, Nat. Nanotechnol, 2016, 11, 986–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chiang C-S, Lin Y-J, Lee R, Lai Y-H, Cheng H-W, Hsieh C-H, Shyu W-C and Chen S-Y, Nat. Nanotechnol, 2018, 13, 746–754 [DOI] [PubMed] [Google Scholar]
- 42.Yang W, Guo W, Le W, Lv G, Zhang F, Shi L, Wang X, Wang J, Wang S and Chang J, ACS nano, 2016, 10, 10245–10257. [DOI] [PubMed] [Google Scholar]
- 43.Cobo I, Li M, Sumerlin BS and Perrier S, Nature materials, 2015, 14, 143–159. [DOI] [PubMed] [Google Scholar]
- 44.Chen B, He X-Y, Yi X-Q, Zhuo R-X and Cheng S-X, ACS Appl. Mater. Interfaces, 2015, 7, 15148–15153. [DOI] [PubMed] [Google Scholar]
- 45.Sun H, Yu T, Li X, Lei Y, Li J, Wang X, Peng P, Ni D, Wang X and Luo Y, J. Nanobiotechnol, 2021, 19, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Q, Xu L, Liang C, Wang C, Peng R and Liu Z, Nat. Commun, 2016, 7, 13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Min Y, Roche KC, Tian S, Eblan MJ, McKinnon KP, Caster JM, Chai S, Herring LE, Zhang L, Zhang T, DeSimone JM, Tepper JE, Vincent BG, Serody JS and Wang AZ, Nat. Nanotechnol, 2017, 12, 877–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou B, Wang R, Chen F, Zhao L, Wang P, Li X, Banyai I, Ouyang Q, Shi X and Shen M, ACS Appl. Mater. Interfaces, 2018, 10, 6146–6154. [DOI] [PubMed] [Google Scholar]
- 49.Zhou B, Xiong Z, Wang P, Peng C, Shen M, Mignani S, Majoral J-P and Shi X, Drug Delivery, 2018, 25, 178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhuang Y, Zhou LZ, Zheng LF, Hu Y, Ding L, Li X, Liu CC, Zhao JH, Shi XY and Guo R, ACS Biomater. Sci. Eng, 2017, 3, 431–442. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y, Xiong ZJ, He Y, Zhou BQ, Qu J, Shen MW, Shi XY and Xia JD, Mater. Sci. Eng., C, 2018, 83, 9–16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.