

Abstract
Abacavir (formerly 1592U89) is a carbocyclic nucleoside analog with potent anti-human immunodeficiency virus (anti-HIV) activity when administered alone or in combination with other antiretroviral agents. The population pharmacokinetics and pharmacodynamics of abacavir were investigated in 41 HIV type 1 (HIV-1)-infected, antiretroviral naive adults with baseline CD4+ cell counts of ≥100/mm3 and plasma HIV-1 RNA levels of >30,000 copies/ml. Data for analysis were obtained from patients who received randomized, blinded monotherapy with abacavir at 100, 300, or 600 mg twice-daily (BID) for up to 12 weeks. Plasma abacavir concentrations from sparse sampling were analyzed by standard population pharmacokinetic methods, and the effects of dose, combination therapy, gender, weight, and age on parameter estimates were investigated. Bayesian pharmacokinetic parameter estimates were calculated to determine the peak concentration of abacavir in plasma (Cmax) and the area under the concentration-time curve from time zero to infinity (AUC0–∞) for individual subjects. The pharmacokinetics of abacavir were dose proportional over the 100- to 600-mg dose range and were unaffected by any covariates. No significant correlations were observed between the incidence of the five most common adverse events (headache, nausea, diarrhea, vomiting, and malaise or fatigue) and AUC0–∞. A significant correlation was observed between Cmax and nausea by categorical analysis (P = 0.019), but this was of borderline significance by logistic regression (odds ratio, 1.45; 95% confidence interval, 0.95 to 2.32). The log10 time-averaged AUC0–∞ minus baseline (AAUCMB) values for HIV-1 RNA and CD4+ cell count correlated significantly with Cmax and AUC0–∞, but with better model fits for AUC0–∞. The increase in AAUCMB values for CD4+ cell count plateaued early for drug exposures that were associated with little change in AAUCMB values for plasma HIV-1 RNA. There was less than a 0.4 log10 difference over 12 weeks in the HIV-1 RNA levels with the doubling of the abacavir AUC0–∞ from 300 to 600 mg BID dosing. In conclusion, pharmacodynamic modeling supports the selection of abacavir 300 mg twice-daily dosing.
A continuing need for new antiretroviral agents against human immunodeficiency virus type 1 (HIV-1) exists due to toxicity and development of drug resistance. Abacavir (formerly 1592U89) is a novel purine carbocyclic nucleoside that is phosphorylated by a unique metabolic pathway to carbocyclic guanosine triphosphate, a potent inhibitor of HIV-1 reverse transcriptase (8). Abacavir has only limited cross-resistance with other nucleoside reverse transcriptase inhibitors in vitro (18; J. W. Mellors, K. Hertogs, F. Peeters, R. Lanier, V. Miller, N. Graham, B. Larder, P. Stoffels, and R. Pauwels, Abstracts 5th Conf. Retroviruses Opportunistic Infections, abstr. 687, p. 208, 1998), which suggests that it could be a useful addition to anti-HIV therapy.
Following the administration of single or multiple doses to adults and children, abacavir is rapidly absorbed, with peak concentrations in plasma (Cmax) occurring within 1 to 2 h after dosing, an elimination half-life (t1/2) of 1 to 2 h (11, 12), and penetration into cerebrospinal fluid (13; J. R. Ravitch, S. S. Good, J. E. Humpreys, J. W. Poli, W. H. Robertson, and J. L. Jarrett, Abstr. 5th Conf. Retroviruses Opportunistic Infections, abstr. 636, p. 199, 1998). The pharmacokinetics of abacavir are dose dependent at steady state following the administration of multiple oral doses of >600 mg daily to adults (14).
In order to further define the optimum dose of abacavir and to evaluate the durability of its antiretroviral effects in HIV-1-infected subjects, a phase II clinical trial (CNAB-2002) was conducted to evaluate the antiretroviral activity and safety of three twice-daily (BID) dosage regimens of abacavir monotherapy of up to 24 weeks. The clinical efficacy and safety results of the full trial reported previously have shown that abacavir is a potent nucleoside reverse transcriptase inhibitor, providing a median reduction in plasma HIV-1 RNA level of over 1.5 log10 copies/ml by 4 weeks when administered as monotherapy (16). The sustained reductions in plasma HIV-1 RNA levels for up to 72 weeks achieved by combination therapy with abacavir, lamivudine, and zidovudine in the same study (S. Staszewski, C. Katlama, T. Harrer, P. Massip, P. Yeni, A. Cutrell, and H. M. Steel, Abstr. 12th Int. Conf. AIDS, abstr. 12212, 1998) were associated with the slow development of resistance to abacavir (18). We report here the results of a substudy which was designed to determine the population pharmacokinetics of abacavir with the nonlinear mixed-effects model (NONMEM) (2), the association of pharmacokinetic parameters with demographic or disease-related variables, and the pharmacodynamic modeling of the effects of abacavir exposure on safety and antiretroviral activity over the first 12 weeks of monotherapy.
MATERIALS AND METHODS
Study population and design.
The study design and criteria for study participation have been described in a previous publication (16). Briefly, subjects were eligible for study entry if they were ≥18 years of age with confirmed HIV-1 infection, were antiretroviral therapy naive, and presented at screening (within 14 days prior to study drug administration) with a CD4+ cell count of ≥100/mm3 and a plasma HIV-1 RNA level of more than 30,000 copies/ml. Enrollment was planned for a total of 60 subjects (20 subjects per treatment group) in order to provide an 80% power to detect treatment differences of 0.45 log10 copies/ml in time-averaged HIV-1 RNA profiles. Subject participation in the pharmacokinetic component of the study was optional. Sparse samples for population pharmacokinetic analysis were obtained at the week 12 clinic visit for a subset of the study subjects. In the randomized, double-blind monotherapy phase, subjects were randomly assigned to receive 100, 300, or 600 mg of abacavir orally every 12 h (BID). Subjects could add additional therapy (zidovudine and lamivudine were provided by the study) if they met prespecified criteria (16). These criteria included a plasma HIV-1 RNA load reduction from baseline of <0.7 log10 copies/ml at week 4, a plasma HIV-1 RNA load of >5,000 copies/ml after week 12, a CD4+ cell count that returned to the baseline count, or a new Centers for Disease Control and Prevention AIDS-defining event after 4 weeks.
Blood sampling.
Blood samples (3 ml) for determination of plasma abacavir concentrations were collected by venipuncture and placed into Vacutainer tubes that contained powdered dipotassium EDTA. Blood samples were obtained from each subject in the intervals of 0.5 to 1, 1 to 2, 2 to 3, and 3 to 4 h postdosing, with at least 30 min between the times of sample collection. Blood samples were centrifuged within 1 h of collection and were stored upright in labeled biofreeze tubes at −20°C or lower until shipment to Glaxo Wellcome for assay of the abacavir concentration.
Abacavir assay.
Plasma samples were analyzed for abacavir concentration by a validated reverse-phase high-performance liquid chromatography assay with UV detection over a quantifiable range of 25 to 5,000 ng/ml (12). Briefly, plasma samples (0.2 ml) were mixed with 0.1 ml of 10% trichloroacetic acid, and the mixture was centrifuged at 8,800 × g for 10 min. Supernatant (0.1 ml) was injected onto a Rainin (4.6 by 250 mm) C18 Microsorb MV column. The mobile phase of the column consisted of 40% methanol in 25 mM ammonium phosphate–0.3% triethylamine (pH 7.2) at a flow rate of 1.0 ml/min. Abacavir was detected by measurement of the UV absorbance at 284 nm. The retention time for abacavir was approximately 9 to 10 min under these conditions. The interday variability (coefficient of variation) was <8%, and the bias of the assay was 0%.
Efficacy and safety assessments.
The primary efficacy measures assessed during abacavir monotherapy administration were changes from baseline in the log10 HIV-1 RNA copies per milliliter and changes in the CD4+ lymphocyte cell count. The plasma HIV-1 RNA was measured by the Roche HIV-1 RNA PCR technique (limit of quantification, 400 copies/ml; Amplicor HIV-1 Monitor test; Roche Molecular Systems, Branchburg, N.J.). CD4+ cell counts were assessed by standard flow cytometry methods at each site. Only data collected up to the time of pharmacokinetic evaluation (week 12) were included in related pharmacodynamic analyses. During this interval, assessments were performed at the baseline (day 0) and at weeks 2, 4, 8, and 12.
Adverse events were managed by investigators unaware of the treatment assignments by using predetermined guidelines, and the severity of the adverse events was graded according to criteria developed by the AIDS Clinical Trials Group, Division of AIDS, National Institute for Allergy and Infectious Diseases (4).
Data analysis. (i) Population pharmacokinetic analysis.
Mixed-effect modeling techniques with the software package NONMEM, version 4, level 2 (2), were used to develop a model that describes abacavir population pharmacokinetics after oral administration and to evaluate the influence of specific covariates. The model-building process involved establishment of a base pharmacokinetic compartmental model, selected by graphical observation of the concentration-time data and information from historical pharmacokinetic experience. This model included no covariates. Subsequent NONMEM runs were executed separately for each potential covariate in order to evaluate the effect of inclusion of the covariate on a pharmacokinetic parameter. Following these univariate analyses, fixed effects (e.g., measurable covariates such as age and weight) that were considered potentially significant were combined in a multivariate analysis. Backward elimination of one fixed-effect covariate at a time (each time replacing the other covariates) was then performed to select the final model.
The base model selected to describe abacavir pharmacokinetics was a one-compartment model with first-order absorption and elimination (specified to NONMEM by the routines ADVAN2 and TRANS2). The pharmacokinetic parameters directly estimated by NONMEM with this model specification were apparent oral clearance (CL/F), apparent volume of distribution (V/F), and absorption rate constant (Ka). The elimination rate constant (λz) was determined by the ratio of clearance to volume (CL/V). Ka was constrained to be greater than CL/V. Bayesian pharmacokinetic parameter estimates for individual subjects were obtained by specification of the POST-HOC option to NONMEM. Individual estimates of the steady-state Cmax, time to Cmax (Tmax) at steady state, area under the concentration-time curve from time zero to infinity (AUC0–∞), and t1/2 were then derived from Bayesian estimates of CL/F, V/F, and Ka (10).
The covariate measures considered for evaluation of the effect on abacavir pharmacokinetics included the presence or absence of combination antiretroviral therapy, dose, and demographic traits of age, gender, and body weight. In combination with the statistical NONMEM output, examination of scatter plots of Bayesian parameter estimates for individual subjects versus these fixed effects were used to select meaningful covariates for inclusion in the pharmacokinetic model.
Proportional error models were used throughout the analysis for both interindividual and intraindividual (residual) variability. The covariance step was executed with each NONMEM run to obtain standard errors of the parameter estimates, the variance-covariance matrix, and the correlation matrix. The results were also examined graphically with scatter plots that included predicted versus observed concentrations, weighted residuals versus time, and weighted residuals versus predicted concentrations.
The criteria for acceptance of a NONMEM model estimation included the following: (i) convergence of the objective function (i.e., a “successful termination” statement from the NONMEM program), (ii) attainment of parameter estimates free of boundary conditions, (iii) standard error estimates <30% of the estimate itself, (iv) termination of the covariance step without warnings, and (v) correlations between model parameters of <0.95. A model was declared superior over another one when the value of the objective function was reduced by >7.8 with the inclusion of one additional parameter. A superior model was also expected to reduce the intersubject variance terms and/or the residual error term. Finally, a superior model should improve the random distribution of residuals.
(ii) Pharmacokinetic-pharmacodynamic analyses. (a) Correlation of pharmacokinetics with efficacy.
The efficacy measurements used in this analysis included changes in HIV-1 RNA (in log10 copies per milliliter) and CD4+ cell count (in number of cells per cubic millimeter) from the baseline values. As an initial analysis, nonparametric Spearman's rank-order correlation analysis was performed with SAS software, version 6.12 (SAS Inc., Cary, N.C.), to assess the degree of association between pharmacokinetic parameters and efficacy measures.
To more fully characterize the relationship between antiviral activity and drug exposure, pharmacodynamic modeling was performed. Since changes in CD4+ cell count while on therapy are highly correlated with baseline values (5, 7, 16), log transformation of the CD4+ cell count provided an assessment of the difference from the baseline count, similar to the determination of a percent change from baseline. In this way, the absolute change in the CD4+ cell count would no longer be linked to the patient's baseline value. The HIV-1 RNA load was log transformed given its exponential distribution. After log10 transformation of the HIV-1 RNA load and CD4+ cell count, calculation of the area under the curve, subtraction of the baseline value, and division by the duration of monotherapy (up to 12 weeks) were performed in order to yield a time-weighted average change from the baseline (denoted as the time-weighted area under the curve minus baseline [AAUCMB] value).
Two pharmacodynamic modeling analyses were performed because not all of the subjects in the pharmacokinetic substudy remained on abacavir monotherapy up to the week 12 evaluation. Specifically, of the 41 subjects who participated in the pharmacokinetic substudy, 27 subjects remained on their initial randomized monotherapy regimen through week 12 and 14 subjects switched from monotherapy to combination therapy prior to the week 12 pharmacokinetic sampling. Of the latter group, nine subjects switched from 100-mg-BID or 600-mg-BID abacavir monotherapy to combination antiretroviral therapy with 300 mg of abacavir BID and five subjects switched from monotherapy with abacavir at 300 mg BID to combination antiviral therapy without a change in the abacavir regimen. These subjects switched therapies at or after the week 8 visit. For the first analysis, only those subjects who finished the first 12 weeks of monotherapy without changing their dose of abacavir were analyzed (n = 27). For the second analysis, the dose-adjusted estimates of Cmax and AUC0–∞ were used for all subjects for whom pharmacokinetic data were available at week 12 and who had switched from their initial randomized monotherapy regimen to the combination antiretroviral therapy with 300 mg of abacavir BID prior to the week 12 pharmacokinetic sampling (n = 41). Adjusted Cmax and AUC0–∞ values were determined, assuming dose proportionality, by scaling the original estimates to the monotherapy dose. In this way, pharmacokinetic parameters could be obtained for the same dose for which pharmacodynamic measures were taken. For both pharmacokinetic-efficacy analyses, since the effect of abacavir monotherapy was to be investigated, efficacy data obtained after subjects had switched to open-label combination therapy were excluded.
Pharmacokinetic and pharmacodynamic modeling was conducted with the WinNonlin software package (WinNonlin-Pro version; Scientific Consulting, Inc., Cary, N.C.). The analyses included standard Emax and sigmoid Emax models with uniform weighting. The general form of the model equation used was given by
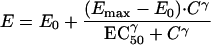 |
1 |
where E is effect, E0 is the baseline effect (possibly fixed equal to zero), Emax is the maximal effect, C denotes the pharmacokinetic variable (e.g., AUC0–∞ or Cmax), EC50 is the value of the pharmacokinetic variable that corresponds to 50% of the maximum effect, and γ denotes the shape parameter that describes the degree of sigmoidicity. For the simple Emax model, γ was assigned a fixed value of unity.
Estimation of E0 was problematic for the CD4 analyses in that final estimates generally converged toward any lower bound placed on the parameter. This convergence was a result of the roughly hyperbolic profile given by the available data. A lower bound of −0.4 was selected for the analysis to be consistent with the data and the maximal possible physiological change in the CD4 count in the absence of therapy (about a 40% decline) (17). Use of other lower bounds such as −0.2 or −0.3 made little difference in estimated EC50 or Emax.
Goodness of fit was assessed from adjusted r2 (Adj r2) and associated P values. Adjusted r2 was calculated by using the following standard formula:
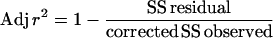 |
2 |
where the residual and corrected sums of squares (SS) were those supplied by the WinNonlin-Pro output. The associated P values were determined from the r value after calculation of the z statistic for a normal distribution from the standard equation
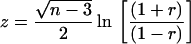 |
3 |
Akaike's information criterion (1) was used to differentiate models by comparison of the same exposure-activity relationships.
(b) Correlation of pharmacokinetics with safety.
Safety data from the first 12 weeks were included in the analyses without regard to combination therapy status as long as the subjects continued to receive abacavir. For this analysis, the abacavir AUC0–∞ and Cmax values used for a given subject were those which corresponded to the abacavir dose received over the majority of the 12-week interval. A subanalysis which excluded any adverse event that occurred after the end of monotherapy was also conducted.
Statistical analyses of the associations between pharmacokinetic measures of drug exposure (AUC0–∞ and Cmax) and measures of safety were determined by both categorical analysis and logistic regression methods. Subject-specific estimates of AUC0–∞ and Cmax were determined from Bayesian pharmacokinetic parameter estimates for individual subjects generated by the final population model. The five most common adverse events (headache, nausea, diarrhea, vomiting, and malaise or fatigue) were selected for analysis on the basis of their incidence (the number of subjects who experienced the adverse event during the first 12 weeks of therapy) and potential for attribution of the adverse event to the study drug.
Categorical analysis was performed by interval analyses in which the range of AUC0–∞ values was divided into three equally populated intervals (tertiles) and the range of Cmax estimates was divided into two equal intervals on the basis of the median value. The frequency of each adverse event was represented as the number of subjects who fell into each of the respective intervals. The association between adverse event incidence and drug exposure was evaluated by Mantel-Haenszel chi-square statistics with modified ridit scores (SAS PROC FREQ).
Logistic regression (SAS PROC LOGISTIC) was conducted to estimate the relationship between the AUC0–∞ or Cmax and each adverse event. Odds ratios were determined along with their 95% confidence intervals, in which the odds ratio denotes the scalar multiplier for the likelihood of a given adverse event associated with a unit change in AUC0–∞ or Cmax.
RESULTS
The demographic and baseline characteristics for the subjects in the pharmacokinetic substudy are summarized in Table 1. All characteristics were similar across the three treatment groups.
TABLE 1.
Demographic and baseline characteristics
| Characteristic | 100 mg BID (n = 8) | 300 mg BID (n = 22) | 600 mg BID (n = 11) | Total (n = 41) |
|---|---|---|---|---|
| No. of men/no. of women | 7/1 | 19/3 | 8/3 | 34/7 |
| Race (no. of subjects) | ||||
| Caucasian | 8 | 21 | 11 | 40 |
| Other | 0 | 1 | 0 | 1 |
| Age (yr [mean ± SD]) | 37 ± 10 | 38 ± 8 | 41 ± 11 | 39 ± 9 |
| Body wt (kg [mean ± SD]) | 70 ± 13 | 77 ± 14 | 71 ± 12 | 74 ± 14 |
| CD4+ count (no. of cells/mm3) | ||||
| Median | 387 | 338 | 404 | 359 |
| Range | 311–720 | 140–641 | 177–648 | 140–720 |
| HIV-1 RNA load (log10 copies/ml) | ||||
| Median | 4.54 | 5.09 | 5.02 | 5.00 |
| Range | 3.77–5.06 | 4.31–6.13 | 4.43–5.44 | 3.77–6.13 |
Population pharmacokinetic analysis.
The population pharmacokinetic parameter estimates (95% confidence intervals of the estimates) associated with the base model were 60.0 liters/h (53.5 to 66.5 liters/h) for CL/F, 67.6 liters (57.6 to 77.6 liters) for V/F, and 1.70 h−1 (1.04 to 2.43 h−1) for Ka. The estimated intersubject coefficient of variation in CL/F was 34% (20% to 43%), and that for Ka was 101% (79% to 120%). Intersubject variability in V/F could not be estimated from the study data. This same result was obtained for model executions with or without inclusion of the intersubject variability parameter for Ka.
Results obtained from the models that incorporated fixed-effect covariates are presented in Table 2. No differences in the CL/F or V/F of abacavir were observed between subjects who received abacavir as monotherapy and subjects who received abacavir in combination with other antiretroviral therapy. Multiplicative models that related CL/F to dose and V/F to dose did not produce a significant reduction in the objective function, indicating dose proportionality over the dose range examined. There were no significant associations of age, gender, and body weight with pharmacokinetic parameters. Evaluation of the effect of age on CL/F resulted in an apparently significant reduction in the objective function (change = −8.55), but the value of the added parameter in this model was not significantly different from its null value (i.e., the 95% confidence interval of the estimate included zero).
TABLE 2.
Summary of NONMEM model executions
| Covariate model (label) | Pharmacokinetic parameter evaluated | NONMEM model parameters | Change in objective functiona |
|---|---|---|---|
| Combination therapy (COMB)b | Clearance | CL = Θ1*(1 − COMB) + Θ2*(COMB) | −3.214 |
| V = Θ3 | |||
| Ka = CL/V + Θ4 | |||
| Combination therapy (COMB)b | Volume of distribution | CL = Θ1 | −0.010 |
| V = Θ2*(1 − COMB) + Θ3*(COMB) | |||
| Ka = CL/V + Θ4 | |||
| Dose (DOSE)c | Clearance | CL = Θ1*(DOSE/300)**Θ2 | −0.510 |
| V = Θ3 | |||
| Ka = CL/V + Θ4 | |||
| Dose (DOSE)c | Volume of distribution | CL = Θ1 | −2.159 |
| V = Θ2*(DOSE/300)**Θ3 | |||
| Ka = CL/V + Θ4 | |||
| Age (AGE)d | Clearance | CL = Θ1*(AGE/39)**Θ2 | −8.555 |
| V = Θ3 | |||
| Ka = CL/V + Θ4 | |||
| Age (AGE)d | Volume of distribution | CL = Θ1 | −3.595 |
| V = Θ2*(AGE/39)**Θ3 | |||
| Ka = CL/V + Θ4 | |||
| Gender (SEX)e | Clearance | CL = Θ1*(1 − SEX) + Θ2*(SEX) | −0.022 |
| V = Θ3 | |||
| Ka = CL/V + Θ4 | |||
| Gender (SEX)e | Volume of distribution | CL = Θ1 | −4.505 |
| V = Θ2*(1 − SEX) + Θ3*(SEX) | |||
| Ka = CL/V + Θ4 | |||
| Weight (WT)f | Clearance | CL = Θ1*(WT/74)**Θ2 | −5.312 |
| V = Θ3 | |||
| Ka = CL/V + Θ4 | |||
| Weight (WT)f | Volume of distribution | CL = Θ1 | −0.856 |
| V = Θ2*(WT/74)**Θ3 | |||
| Ka = CL/V + Θ4 |
Change in value of objective function from that of the base model.
The values of COMB are 0 for monotherapy and 1 for combination antiretroviral therapy.
Subject dose expressed as a fraction of the 300-mg dose.
Subject age expressed as a fraction of the median age for the population included in the pharmacokinetic analysis (39 years).
Values of SEX are 0 for men and 1 for women.
Subject weight expressed as a fraction of the median weight for the population included in the pharmacokinetic analysis (74 kg).
Mean Bayesian pharmacokinetic parameter estimates for each dose group are presented in Table 3. Cmax and AUC0–∞ estimates for the 300- and 600-mg doses were approximately three and seven times greater, respectively, than those for the 100-mg dose. Estimates of Ka, Tmax, and t1/2 were similar among the dose cohorts. CL/F estimates appeared to decrease with increasing dose, although as indicated above, this was not found to be significant.
TABLE 3.
Summary of Bayesian pharmacokinetic parameter estimates for individual subjectsa
| Parameter | 100 mg BID (n = 8) | 300 mg BID (n = 22) | 600 mg BID (n = 11) |
|---|---|---|---|
| Cmax (μg/ml) | 0.64 ± 0.17 (27) | 2.17 ± 0.47 (22) | 4.38 ± 1.19 (27) |
| AUC0–∞ (μg · h/ml) | 1.69 ± 0.85 (51) | 5.47 ± 1.38 (25) | 13.0 ± 3.00 (23) |
| Tmax (h) | 0.97 ± 0.45 (47) | 0.89 ± 0.31 (35) | 1.02 ± 0.30 (30) |
| t1/2 (h) | 0.79 ± 0.40 (51) | 0.85 ± 0.22 (25) | 1.02 ± 0.23 (23) |
| CL/F (liters/h) | 67.5 ± 19.3 (29) | 58.0 ± 13.2 (23) | 48.8 ± 11.8 (24) |
| Ka (1/h) | 1.65 ± 1.38 (83) | 1.79 ± 0.86 (48) | 1.60 ± 0.98 (61) |
Values are means ± standard deviation (percent coefficient of variation).
Pharmacokinetic and pharmacodynamic analyses. (i) Correlation of pharmacokinetics with efficacy.
Spearman's rank correlation analysis showed that AUC0–∞ and Cmax were significantly correlated with a change in HIV-1 RNA from baseline to the end of 12 weeks of monotherapy (P < 0.05; data not shown). In contrast, AUC0–∞ and Cmax were significantly correlated with a change in CD4+ cell count only up to 4 weeks of monotherapy (data not shown). The significant results obtained from this initial correlation analysis led to further characterization of the relationships by pharmacokinetic-pharmacodynamic modeling (see Materials and Methods).
Simple Emax models were found to best describe the data (i.e., inclusion of the exponential shape parameter, γ, was not justified in any of the model fits). The final model parameters for the relationships between AAUCMB values for HIV-1 RNA level or CD4+ cell count over the 12-week evaluation period versus the abacavir AUC0–∞ and Cmax values are summarized for the two data sets in Table 4. Graphical results for the model-fit relationships with abacavir AUC0–∞ are presented in Fig. 1 and 2. AUC0–∞ produced better model fits than Cmax for both data sets, as indicated by the r2 values (Table 4). As predicted by the Emax model, the doubling of the level of abacavir exposure from 300 mg BID to 600 mg BID dosing was associated with a 0.23- to 0.38-log10 difference in the time-averaged change in the log10 HIV-1 RNA change from baseline, depending on the population modeled (Table 5). When data for all subjects (n = 41) up through just 8 weeks of treatment were modeled, the results were similar (data not shown). The increase in drug exposure from 300 to 600 mg BID was also associated with small increases (approximately 12%) in the CD4+-cell count (or an increase of approximately 43 cells/mm3 from the baseline median cell count of 359 cells/mm3) for both sets of analyses (Table 5). The maximum increase in the log10 CD4+ cell count from baseline (Emax) was 0.12 (Table 4), which represents a 31.8% increase in the CD4+ cell count from baseline (or an increase of approximately 114 cells/mm3 from the baseline median cell count of 359 cells/mm3). The increase in the CD4+ cell count also reached a plateau at AUC0–∞ values that were associated with minimal changes in HIV-1 RNA load, regardless of the data set used (Fig. 1 and 2).
TABLE 4.
Pharmacodynamic-pharmacokinetic modeling: model parameter estimates for log10 AAUCMB values of HIV-1 RNA load and CD4+ cell count versus AUC0–∞ and Cmax
| Parameter | 12-wk monotherapy group (n = 27) | All subjects (n = 41) |
|---|---|---|
| Log10 HIV-1 RNA load vs AUC0–∞ | ||
| EC50 | 1.87 | 5.00 |
| Emax | 1.87 | 2.00 |
| Adjusted r2 | 0.45a | 0.30a |
| Log10 HIV-1 RNA load vs Cmax | ||
| EC50 | 0.74 | 1.63 |
| Emax | 1.89 | 1.88 |
| Adjusted r2 | 0.35b | 0.23b |
| Log10 CD4+ count vs AUC0–∞ | ||
| EC50 | 0.49 | 0.43 |
| Emax | 0.12 | 0.11 |
| E0 | −0.40 | −0.38 |
| Adjusted r2 | 0.37b | 0.33a |
| Log10 CD4+ count vs Cmax | ||
| EC50 | 0.30 | 0.24 |
| Emax | 0.12 | 0.11 |
| E0 | −0.22 | −0.24 |
| Adjusted r2 | 0.23c | 0.23b |
P < 0.0001.
P < 0.001.
P < 0.01.
FIG. 1.

Effect of abacavir AUC0–∞ on HIV-1 RNA suppression. Shown here are data for individual subjects, the mean ± 95% confidence intervals for each dose cohort, and the model fit curve. Ordinal values denote the AAUCMB values for the log10 plasma HIV-1 RNA load in plasma. Model parameters are given in Table 4. The mean ± standard deviation HIV-1 RNA AAUCMB values for each dose cohort are given in Table 5. Smooth curves denote the pharmacokinetic- pharmacodynamic model fit from the Emax model. (A) Pharmacokinetic-pharmacodynamic relationship for those subjects who were maintained on abacavir monotherapy for 12 weeks (n = 27). (B) Pharmacokinetic-pharmacodynamic relationship for all subjects while on abacavir monotherapy for up to 12 weeks (n = 41).
FIG. 2.

Effect of abacavir AUC0–∞ on CD4+ cell count. Shown here are data for individual subjects, the mean ± 95% confidence intervals for each dose cohort, and the model fit curve. Ordinal values denote the AAUCMB values for the log10 CD4+ cell count in plasma. Model parameters are given in Table 4. The mean ± standard deviation AAUCMB CD4+ values for each dose cohort are given in Table 5. Smooth curves denote the pharmacokinetic-pharmacodynamic model fit from the Emax model. (A) Pharmacokinetic-pharmacodynamic relationship for those subjects who were maintained on abacavir monotherapy for 12 weeks (n = 27). (B) Pharmacokinetic-pharmacodynamic relationship for all subjects while on abacavir monotherapy for up to 12 weeks (n = 41).
TABLE 5.
Actual and model-fit log10 AAUCMB values for HIV-1 RNA and CD4+-cell count versus abacavir AUC0–∞a
| Group and parameter | 100 mg BID | 300 mg BID | 600 mg BID |
|---|---|---|---|
| 12-wk monotherapy group | |||
| AAUCMB (HIV-1 RNA load)b | 0.86 ± 0.28 | 1.32 ± 0.26 | 1.69 ± 0.28 |
| Model-fit AAUCMB (HIV-1 RNA load)c | 0.89 | 1.41 | 1.64 |
| AAUCMB (CD4+ cell count)b | −0.012 ± 0.075 | 0.097 ± 0.057 | 0.095 ± 0.035 |
| Model-fit AAUCMB (CD4+ cell count)d | −0.11 | 0.02 | 0.07 |
| All subjects | |||
| AAUCMB (HIV-1 RNA load)b | 0.59 ± 0.23 | 1.03 ± 0.33 | 1.39 ± 0.41 |
| Model-fit AAUCMB (HIV-1 RNA load)e | 0.52 | 1.05 | 1.43 |
| AAUCMB (CD4+ cell count)b | 0.006 ± 0.04 | 0.089 ± 0.04 | 0.089 ± 0.031 |
| Model-fit AAUCMB (CD4+ cell count)d | −0.08 | 0.03 | 0.08 |
Mean AAUCMB values were calculated from the simple Emax model by using the mean pharmacokinetic parameter estimates (AUC0–∞ and Cmax) given in Table 3 and the model parameters given in Table 4.
An increase in exposure from 300 mg BID to 600 mg BID is associated with a 0.23 log10 difference in the AAUCMB value for the HIV-1 RNA load.
An increase in exposure from 300 mg BID to 600 mg BID is associated with a 12% (or 100.05) increase in the AAUCMB value for the CD4+ cell count (absolute difference of 0.05 log10).
An increase in exposure from 300 mg BID to 600 mg BID is associated with a 0.38 log10 difference in the AAUCMB value for the HIV-1 RNA load.
(ii) Correlation of pharmacokinetics with safety.
No significant associations were found between the incidence of the adverse events and the abacavir AUC0–∞ from the Mantel-Haenszel analysis (data not shown). A statistically significant association was noted for nausea and Cmax of ≥1.93 μg/ml (P = 0.019). No significant association with the abacavir Cmax was seen for the other adverse events. Logistic regression analyses between the abacavir AUC0–∞ and the incidence of adverse events did not reveal any statistically significant associations. There was a borderline association with increasing Cmax and nausea (odds ratio, 1.45; 95% confidence interval, 0.95 to 2.32).
DISCUSSION
The results of the population analysis for this study indicate that the pharmacokinetics of abacavir are dose proportional over the range of 100 to 600 mg BID. The analysis was unable to detect any pharmacokinetic differences associated with combination antiretroviral therapy, age, gender, or body weight. The power to detect effects due to these factors may have been limited by the relatively small number of subjects in the analysis. The pharmacokinetic-pharmacodynamic relationships for changes from the baseline values in both time-averaged HIV-1 RNA load and CD4+ cell count were strongly associated with abacavir AUC0–∞. The EC50 for the time-averaged change in the HIV-1 RNA load was always appreciably greater than that for the CD4+ cell count, indicating the early saturation of the CD4+ cell count change, with little associated change in HIV-1 RNA load. This finding is consistent with the pharmacokinetic analysis of indinavir monotherapy (7). A modest increase in HIV-1 RNA suppression, but no increase in the CD4+ cell count, was observed at 600 mg BID relative to 300 mg BID as monotherapy. No differences in abacavir pharmacokinetics were observed when the drug was administered as monotherapy or in combination with other antiretroviral medications. This finding is consistent with results from earlier pharmacokinetic drug interaction studies (14, 19).
The present study did not reveal any effects of gender on abacavir pharmacokinetics. A significant gender effect was found in a previous trial that used higher levels of exposure to abacavir (14). In that study, women (n = 11) demonstrated a 54% greater AUC0–∞ and a 30% greater Cmax than men (n = 68). Reasons for the difference between the population pharmacokinetic findings and those from the previous trial are unclear and may be related to the small number of female subjects in both studies. Abacavir has been well tolerated—except for dose-related nausea—at single doses of up to 1,250 mg and multiple doses of up to 1,800 mg/day (600 mg three times daily). Thus, if a gender effect did exist, it is unlikely to be of clinical significance at the dosage regimen intended for further clinical investigation (300 mg BID).
In addition to the present study, pharmacokinetic analysis of abacavir after 12 weeks of dosing with 300 mg BID was previously investigated with nine subjects (14). In the present study, the mean ± standard deviation individual Bayesian estimates were 58.0 ± 13.2 liters/h for CL/F and 67.6 liters for V/F. λz was calculated to be 0.86 ± 0.20 h−1. Estimates obtained in the study by McDowell et al. (14) were 55.0 ± 22.7 liters/h for CL/F, 99.3 ± 20.9 liters for V/F, and 0.54 ± 0.13 h−1 for λz. The estimates for CL/F are in good agreement between the two studies. The reasons for the difference in estimates of the volume of distribution between these two studies are not clear but may be due to the sparse sampling used in the present study and the inability to estimate interindividual variability for V/F.
For pharmacodynamic measures, cumulative assessments of antiviral activity were obtained by calculating AAUCMB for the log10 HIV-1 RNA load and the log10 CD4+ cell count over the first 12 weeks of therapy. The pharmacokinetic-pharmacodynamic relationships evaluated by these measures are less subject to the biological variability associated with evaluation at a single time point and represent the global effect in the HIV-infected population over a meaningful interval for evaluation of clinical activity. The initial pharmacokinetic-pharmacodynamic analysis included subjects who remained on fixed-dose abacavir monotherapy for the 12 weeks up to the time of pharmacokinetic evaluation (n = 27). This analysis thus excluded subjects who switched from abacavir monotherapy because of recognized treatment failure and may therefore be potentially biased toward subjects in whom abacavir had greater or more prolonged antiviral activity. The second pharmacokinetic-pharmacodynamic analysis was conducted with data for all subjects who participated in the pharmacokinetic study (n = 41) and used either all available monotherapy data or a subset of data for up to 8 weeks of therapy. Because 12-week exposure data for 9 of the 41 subjects had to be adjusted for their prior dosage regimens, there is the possibility of error in estimation to the extent that strict dose proportionality may not apply. Any overestimation of exposure at the 100-mg dose as a consequence of this assumption could result in a decrease in the slope of the pharmacokinetic-pharmacodynamic relationships (i.e., a shift to the right) and result in higher EC50s and/or lower Emaxs.
The pharmacodynamic relationships from the analyses in this study indicate a small incremental effect of 600 mg BID versus that of 300 mg BID in HIV-1 RNA load suppression. The pharmacodynamic effects associated with both regimens are on the near-plateau portion of the Emax relationship. Depending on the model, this doubling of the abacavir dose resulted in a 0.23- to 0.38-log10 difference in the time-averaged change in log10 HIV-1 RNA load from baseline. This modest difference would be difficult to detect clinically, especially since abacavir would be used in combination therapy under usual clinical circumstances. The wide range of exposures below those that result in the maximum effect is somewhat different from that observed for other nucleosides; a more pronounced Emax or sigmoid Emax relationship has been described for zidovudine, didanosine, and 3′-deoxy-3′-fluorothymidine (5, 6, 9), although the relationship for zidovudine did not attain significance. Each of these nucleosides has some rate-limiting step in their phosphorylation pathways. By comparison, the pharmacodynamic relationships observed with abacavir in this study are consistent with in vitro data that show a linear relationship between the level of the active triphosphate of abacavir and exposure to abacavir over the range of 0.1 to 100 μM (3). Thus, while it appears that some additional incremental antiviral activity can be obtained with increasing doses of abacavir, it is clear from the Emax relationships that such increases would be progressively smaller with increasing systemic exposure. Results from analyses of the relationship between safety and abacavir exposure in the present study are consistent with those of other studies that have indicated an increase in nausea with increasing doses of abacavir (15, 16).
In conclusion, the pharmacokinetic-pharmacodynamic results of this study support the selection of abacavir at 300 mg BID as the approved dose. (Also, the results from these trials have shown that abacavir in combination with other antiretroviral agents provides potent and durable suppression of HIV-1 RNA [15, 16; M. Fischl, S. Greenberg, N. Clumeck, B. Peters, R. Rubio, B. Pobiner, and L. Verity, 12th World AIDS Conference, abstr. 12230, 1998].)
ACKNOWLEDGMENTS
We gratefully acknowledge the assistance of Bill Mahony with performing the bioanalytical studies, Keith Muir for helpful discussions and review of the manuscript, and Belinda Ha for assistance with manuscript preparation.
REFERENCES
- 1.Akaike A. Posterior probabilities for choosing a regression model. Ann Inst Math Stat. 1978;30:A9–A14. [Google Scholar]
- 2.Beal S L, Sheiner L B. NONMEM user's guide. San Francisco: University of California, San Francisco; 1989. [Google Scholar]
- 3.Daluge S M, Good S S, Faletto M B, Miller W H, St. Clair M H, Boone L R, Tisdale M, Parry N R, Reardon J E, Dornsife R E, Averett D R, Krenitsky T A. 1592U89, a novel carbocyclic nucleoside analog with potent, selective, anti-human immunodeficiency virus activity. Antimicrob Agents Chemother. 1997;41:1082–1093. doi: 10.1128/aac.41.5.1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Division of AIDS, National Institute of Allergy and Infectious Diseases. Division of AIDS table for grading severity of adult adverse experiences. Rockville, Md: National Institute of Allergy and Infectious Diseases; 1996. [Google Scholar]
- 5.Drusano G L, Yuen G J, Lambert J S, Seidlin M, Dolin R, Valentine F T. Relationship between dideoxyinosine exposure, CD4 counts, and p24 antigen levels in human immunodeficiency virus infection. Ann Intern Med. 1992;116:562–566. doi: 10.7326/0003-4819-116-7-562. [DOI] [PubMed] [Google Scholar]
- 6.Drusano G L, Balis F M, Gitterman S R, Pizzo P A. Quantitative relationships between zidovudine exposure and efficacy and toxicity. Antimicrob Agents Chemother. 1994;38:1726–1731. doi: 10.1128/aac.38.8.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drusano G L, Stein D S. Mathematical modeling of the interrelationship of CD4 lymphocyte count and viral load changes induced by the protease inhibitor indinavir. Antimicrob Agents Chemother. 1998;42:358–361. doi: 10.1128/aac.42.2.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faletto M B, Miller W H, Garvey E P, St. Clair M H, Daluge S M, Good S S. Unique intracellular activation of the potent anti-human immunodeficiency virus agent 1592U89. Antimicrob Agents Chemother. 1997;41:1099–1107. doi: 10.1128/aac.41.5.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flexner C, van der Horst C, Jacobson M A, et al. Relationship between plasma concentrations of 3′-deoxy-3′-fluorothymidine (alovudine) and antiretroviral activity in two concentration-controlled trials. J Infect Dis. 1994;170:1394–1403. doi: 10.1093/infdis/170.6.1394. [DOI] [PubMed] [Google Scholar]
- 10.Gibaldi M, Perrier D. Pharmacokinetics. 2nd ed. New York, N.Y: Marcel Dekker, Inc.; 1982. Multiple dosing, first order absorption; pp. 132–142. [Google Scholar]
- 11.Hughes W, McDowell J A, Shenep J, Flynn P, Kline M W, Yogev R, Symonds W, Lou Y, Hetherington S. Safety and single-dose pharmacokinetics of abacavir (1592U89) in human immunodeficiency virus type 1-infected children. Antimicrob Agents Chemother. 1999;43:609–615. doi: 10.1128/aac.43.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar P N, Sweet D E, McDowell J A, Symonds W, Lou Y, Hetherington S, LaFon S. Safety and pharmacokinetics of abacavir (1592U89) following oral administration of escalating single doses in human immunodeficiency virus type 1-infected adults. Antimicrob Agents Chemother. 1999;43:603–608. doi: 10.1128/aac.43.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDowell J A, Chittick G E, Ravitch J R, Polk R E, Kerkering T M, Stein D S. Pharmacokinetics of [14C]abacavir, a human immunodeficiency virus type 1 (HIV-1) reverse transcriptase inhibitor, administered in a single, oral dose to HIV-1-infected adults: a mass balance study. Antimicrob Agents Chemother. 1999;43:2855–2861. doi: 10.1128/aac.43.12.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McDowell J A, Lou Y, Symonds W S, Stein D S, Muir K. Multiple-dose pharmacokinetics and pharmacodynamics of abacavir alone and in combination with zidovudine in human immunodeficiency virus-infected adults. Antimicrob Agents Chemother. 2000;44:2061–2067. doi: 10.1128/aac.44.8.2061-2067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saag M S, Sonnerborg A, Torres R A, Lancaster D, Gazzard B G, Schooley R T, Romero C, Kelleher D, Spreen W, LaFon S the Abacavir Phase 2 Clinical Team. Antiretroviral effect and safety of abacavir and abacavir in combination with zidovudine in HIV-infected adults. AIDS. 1998;12:F203–F209. doi: 10.1097/00002030-199816000-00002. [DOI] [PubMed] [Google Scholar]
- 16.Staszewski S, Katlama C, Harrer T, Massip P, Yeni P, Cutrell A, Tortell S M, Harrigan P R, Steel H, Lanier E R, Pearce G. A dose-ranging study to evaluate the safety and efficacy of three doses of abacavir (1592U89) alone or in combination with zidovudine and lamivudine in antiretroviral treatment naive subjects. AIDS. 1998;12:F197–F202. doi: 10.1097/00002030-199816000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Stein D S, Korvick J A, Vermund S H. CD4+ lymphocyte cell enumeration for prediction of clinical course of human immunodeficiency virus disease: a review. J Infect Dis. 1992;165:352–363. doi: 10.1093/infdis/165.2.352. [DOI] [PubMed] [Google Scholar]
- 18.Tisdale M, Alnadaf T, Cousens D. Combination of mutations in human immunodeficiency virus type 1 reverse transcriptase required for resistance to the carbocyclic nucleoside 1592U89. Antimicrob Agents Chemother. 1997;41:1094–1098. doi: 10.1128/aac.41.5.1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L H, Chittick G E, McDowell J A. Single-dose pharmacokinetics and safety of abacavir (1592U89), zidovudine, and lamivudine administered alone and in combination in adults with HIV infection. Antimicrob Agents Chemother. 1999;43:1708–1715. doi: 10.1128/aac.43.7.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]