

Abstract
Background
Platelet activation and arterial thrombosis on a ruptured atherosclerotic plaque is a major cause of myocardial infarction. Dual antiplatelet therapy (DAPT), the combination of platelet aggregation inhibitors, aspirin and a P2Y12 antagonist, is used to prevent arterial thrombosis. However, many people continue to have arterial thrombosis and myocardial infarction despite DAPT, indicating that additional therapies are required where DAPT is insufficient.
Objectives
To determine whether antagonists of protease‐activated receptors (PARs) can prevent occlusive thrombosis under conditions where DAPT is insufficient.
Methods
We used human whole blood in a microfluidic model of occlusive thrombosis to compare conditions under which DAPT is effective to those under which DAPT was not. Cangrelor (a P2Y12 antagonist) and aspirin were used to mimic DAPT. We then investigated whether the PAR1 antagonist vorapaxar or the PAR4 antagonist BMS 986120, alone or in combination with DAPT, prevented occlusive thrombosis.
Results and Conclusions
A ruptured plaque exposes collagen fibers and is often rich in tissue factor, triggering activation of platelets and coagulation. Occlusive thrombi formed on type I collagen in the presence or absence of tissue factor (TF). However, although DAPT prevented occlusive thrombosis in the absence of TF, DAPT had little effect when TF was also present. Under these conditions, PAR antagonism was also ineffective. However, occlusive thrombosis was prevented by combining DAPT with PAR antagonism. These data demonstrate that PAR antagonists may be a useful addition to DAPT in some patients and further demonstrate the utility of in vitro models of occlusive thrombosis.
Keywords: blood platelets; dual antiplatelet therapy; microfluidics; receptor, proteinase‐activated; thrombosis
Essentials.
Protease‐activated receptors (PARs) on platelets are potential targets for antithrombotic drugs.
We investigated the effect of blocking PARs in an occlusive thrombosis microfluidic model.
PAR antagonists prevented occlusive thrombosis when combined with P2Y12 inhibition and aspirin.
PAR antagonists may be useful antithrombotics when P2Y12 inhibition and aspirin is insufficient.
1. INTRODUCTION
Arterial thrombosis on a ruptured atherosclerotic plaque is a major problem in ischemic heart disease, the leading cause of death worldwide. 1 The ruptured plaque exposes collagen fibers that allow platelet adhesion, activation, and aggregation. 2 The plaque core is also rich in tissue factor (TF), which initiates the coagulation cascade, leading to thrombin generation and fibrin polymerization. Together, the platelet aggregates and polymerized fibrin form a stable thrombus that can occlude the coronary artery, resulting in downstream ischemia and myocardial infarction. Dual antiplatelet therapy (DAPT), the combination of aspirin and a P2Y12 antagonist, is used to prevent arterial thrombosis. 3 , 4 , 5 , 6 However, many people continue to suffer arterial thrombosis and myocardial infarction despite DAPT, indicating that additional therapies are required where DAPT is insufficient.
Protease‐activated receptors (PARs) on platelets are potential antithrombotic targets. 7 PAR1 and PAR4 mediate the platelet response to thrombin, a potent platelet activator. The PAR1 antagonist vorapaxar in addition to DAPT reduces cardiovascular death and ischemic events in at‐risk patients, albeit at the cost of increased major bleeding, including intracranial hemorrhage, which has limited its use. 8 Moreover, although vorapaxar was approved by the US Food and Drug Administration, it has not received approval from the European Medicines Agency. Although no PAR4 antagonist has yet been approved for clinical use, the reversible antagonist BMS 986120 has shown antithrombotic activity in preclinical animal models and in an ex vivo model in a phase 1 trial. 9 , 10 Further development of PAR antagonists would be helped by better understanding the relative contribution of these two receptors to arterial thrombosis and whether PAR antagonism would be beneficial in addition to DAPT or instead of DAPT.
In vivo and in vitro models of thrombosis have made a major contribution to our understanding of thrombosis and the roles of potential anti‐thrombotic drug targets such as PARs. However, in vivo thrombosis models such as the murine carotid artery model have been hampered by interspecies differences between human and murine platelets. 11 , 12 For example, whereas human platelets express PAR1 and PAR4, murine platelets lack PAR1 and instead express PAR3 and PAR4. 13 , 14 This expression difference clouds our understanding of the relative contributions of each PAR to thrombosis. In vitro models of thrombosis, in contrast, often use microfluidic devices to recapitulate arterial blood flow and have the important advantage of being able to use human blood cells. However, a limitation of many in vitro thrombosis models is that they do not result in occlusive thrombosis. To address this, several in vitro occlusive thrombosis models have been described, including by us. 15 , 16 , 17 , 18 Our model uses a bifurcated microfluidic device that allows blood flow to be diverted down one arm while an occlusive thrombus is developing on a patch of prothrombotic trigger in the other. Additionally, coagulation is quenched downstream of the thrombus, such that the time to occlusion is reliably dependent on the thrombus developing on the prothrombotic triggers. Our microfluidic model allows us to investigate the effect of antiplatelet drugs on the time to occlusion in human blood.
In this study, we used our microfluidic model of occlusive thrombosis to compare conditions under which DAPT is effective to those under which DAPT was not. We then investigated whether PAR antagonists, alone or in combination with DAPT, prevented occlusive thrombosis. When DAPT was ineffective, PAR antagonism alone was also ineffective. However, occlusive thrombosis was prevented by combining DAPT with PAR antagonism. These data demonstrate that PAR antagonists may be a useful addition to DAPT in some patients and further demonstrate the utility of in vitro models of occlusive thrombosis.
2. METHODS
2.1. Fabrication of an occlusive thrombosis‐on‐a‐chip microfluidic device
We have previously described the occlusive thrombosis‐on‐a‐chip model in detail. 18 The device contains a symmetric bifurcation with the prothrombotic trigger placed in one arm. Blood is perfused into the device at a constant flow rate. However, as a thrombus grows at the prothrombotic site, blood flow is diverted down the other arm of the bifurcation. This prevents an increase in pressure developing at the thrombus and allows occlusion to occur. Coagulation downstream of the prothrombotic site is quenched by addition of EDTA. The rate of blood flow leaving the device is continuously monitored as the increase in weight of effluent, measured by a sensitive balance. This gives a measure of blood flow rate and is used to determine time to occlusion. For a schematic of the device, see Figure S1.
In brief, devices were fabricated as described previously 18 using polydimethylsiloxane (PDMS) molded to a silicon wafer generated by photolithography. PDMS was vacuum sealed to a glass coverslip such that a 2 μl spot of prothrombotic trigger was positioned in one channel of the device. The prothrombotic trigger was Horm Collagen (0.1 mg ml−1; Takeda Pharmaceutical Company, Tokyo, Japan) ± tissue factor (200 pM; Dade Innovin, Sysmex, UK), as indicated in the main text. All tubing, syringes, and needles were blocked with bovine serum albumin (2%) for 2 hours before use.
2.2. Human blood preparation and drug treatments
Blood was obtained by venipuncture from healthy, drug‐free volunteers who had given written, informed consent. Use of blood from volunteers was approved by the University of Cambridge Human Biology Research Ethics Committee in accordance with the Declaration of Helsinki. Following a discard draw, blood was drawn into Vacuettes containing 3.2% sodium citrate (Greiner‐Bio One, Monroe, NC, USA). Blood was labeled with DiOC6 (1 μM), to label platelets and leukocytes, and Alexa Fluor‐546–conjugated fibrinogen (3.33 μg ml−1; Thermo Fisher Scientific, Waltham, MA, USA). Where indicated, blood was treated with aspirin (500 μM), cangrelor (10 μM), vorapaxar (20 μM), and/or BMS 986120 (0.1 μM) for 30 minutes. The concentrations of vorapaxar and BMS 986120 were established as effective in whole blood in the flow cytometry experiments described in Figure 3.
FIGURE 3.
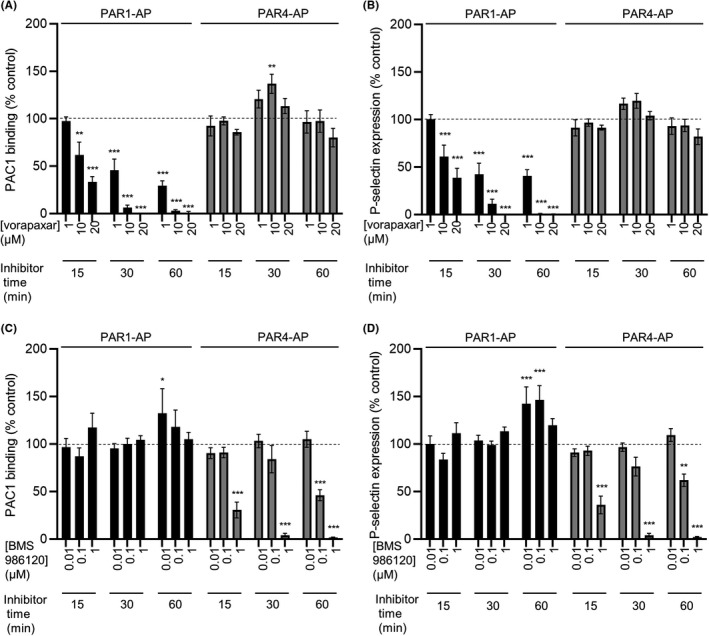
Validation of optimum conditions for PAR1 and PAR4 antagonism. Whole blood was treated with vorapaxar (A, B) or BMS 986120 (C, D) at various concentrations and times as indicated. Blood was then stimulated with PAR1‐AP (10 μM) or PAR4‐AP (100 μM) for 10 minutes, stained with PAC1‐FITC and anti‐P‐selectin‐PE, and analyzed by flow cytometry. Vorapaxar inhibited PAR1‐AP–triggered platelet activation in a concentration‐ and time‐dependent manner, with no inhibition of PAR4‐AP‐triggered platelet activation (A, B). Conversely, BMS 986120 inhibited PAR4‐AP–triggered platelet activation in a concentration‐ and time‐dependent manner, with no inhibition of PAR1‐AP‐triggered platelet activation (C, D). Some weak enhancement of PAR1‐AP‐triggered platelet activation by BMS 986120 was noted. Concentrations and incubation time of antagonists for future experiments was chosen such that the primary target was completely inhibited but the other PAR was not affected. Data are mean ± standard error; n = 4 (*P < .05; **P < .01; ***P < .001 compared to vehicle‐treated control). FITC, fluorescein isothiocyanate; PAR, protease‐activated receptor
2.3. Perfusion of whole human blood
Blood was perfused through the microfluidic device at a constant rate by a syringe pump such the initial shear rate at the prothrombotic trigger was 1000 s−1. Immediately before entering the device, blood was recalcified by addition of coagulation buffer (75 mM CaCl2, 37.5 mM MgCl2, added 1:9) via a Y connector. EDTA was added downstream of the prothrombotic trigger by an additional line delivering 50 mM EDTA (in 4‐[2‐hydroxyethyl]‐1‐piperazineethanesulfonic acid–buffered isotonic saline). The chaotic mixer ensures mixing of blood and EDTA. The total perfusion time was 24 minutes.
2.4. Imaging and analysis
Thrombi in the device were imaged using an LSM150 confocal microscope (10× objective; Zeiss, Oberkochen, Germany) to generate z‐stacks excited at 488 nm (for DiOC6) and 543 nm (for Alexa Fluor‐546–fibrinogen) across the entire channel height (70 μm) every 2 minutes. Data on platelet/leukocyte or fibrin accumulation were analyzed by two‐way repeated measures analysis of variance followed by Tukey’s post hoc multiple comparisons test (Prism version 9; GraphPad Software, San Diego, CA, USA).
2.5. Measurement of blood flow and analysis of occlusion time
Blood flow was measured as the rate of increase in weight of fluid leaving the outlet downstream of the thrombus. Occlusion was defined as a rate of <0.001 mL/min−1 for 3 minutes or longer. Data on occlusion time were analyzed using a one‐way repeated measures nonparametric Friedman’s test, followed by Dunn’s post hoc multiple comparisons test. Channels that did not occlude were assigned a value of 40 minutes for the purposes of statistical testing.
2.6. Whole blood flow cytometry
Flow cytometry analysis was performed on whole blood collected into H‐D‐Phe‐Pro‐Arg‐chloromethylketone hydrochloride (PPACK); to maintain normal divalent cation concentrations but prevent coagulation). Blood was incubated with receptor antagonists at the concentrations and times described in the Results, then stimulated with either the PAR1 agonist PAR1‐AP (10 μM; SFLLRN‐amide), or the PAR4 agonist PAR4‐AP (100 μM; AYPGKF‐amide), for 10 minutes. Blood was stained with phycoerythrin (PE)‐cyanine (Cy) 7–conjugated anti‐CD41 antibody, fluorescein isothiocyanate–conjugated PAC1 antibody (to measure activated integrin αIIbβ3) and PE‐conjugated anti‐P‐selectin (CD62P) antibody (to measure α‐granule secretion) for 15 minutes, diluted in Fix‐Lyse solution (eBioscience, Thermo Fisher Scientific) analyzed by flow cytometry (BD Accuri C6; BD Biosciences, Franklin Lakes, NJ, USA). Compensation beads (eBioscience) were used to account for overlap of fluorescence spectra. PE‐Cy7‐CD41 fluorescence was used to identify platelets.
3. RESULTS
3.1. Efficacy of aspirin plus cangrelor (DAPT) on platelet aggregation and occlusive thrombosis is affected by the presence of tissue factor
We have recently described a novel arterial thrombosis‐on‐a‐chip microfluidic device that allows occlusive thrombus formation under arteriolar shear and measurement of time to occlusion. 18 A key feature of this device is that coagulation downstream of the thrombotic trigger is quenched by EDTA, preventing off‐site coagulation obscuring the measured time to occlusion. For a schematic of the microfluidic device, see Figure S1. We previously demonstrated that this device effectively demonstrated the antithrombotic action of eptifibatide, a glycoprotein IIb/IIIa antagonist. In this study, we first investigated the effects of P2Y12 inhibition and aspirin on occlusive thrombosis.
The microfluidic devices were prepared with either collagen alone (Figure 1) or collagen plus TF (Figure 2) Whole human blood was perfused through the microfluidic device such that flow over the collagen patch was initially at arteriolar shear rate (1000 s−1). To visualize thrombus formation within the devices, platelets (and leukocytes) were labeled with the lipid dye, DiOC6. Fibrin was detected using fibrinogen Alexa Fluor‐546 conjugate. Representative images are shown in Figures 1A and 2A. Blood flow through the device was measured by monitoring the mass of fluid leaving the device as previously described. 18 Platelets and leukocytes built up into three‐dimensional aggregates on the collagen patch in the absence or presence of TF. The integrated fluorescence densities at the end of perfusion are shown in Figures 1B and 2B. Fibrin also accumulated in the absence or presence of TF. Most devices (5/6) occluded within 24 minutes in the absence of TF (Figure 1). In the presence of TF, all devices occluded within 24 minutes (Figure 2C).
FIGURE 1.
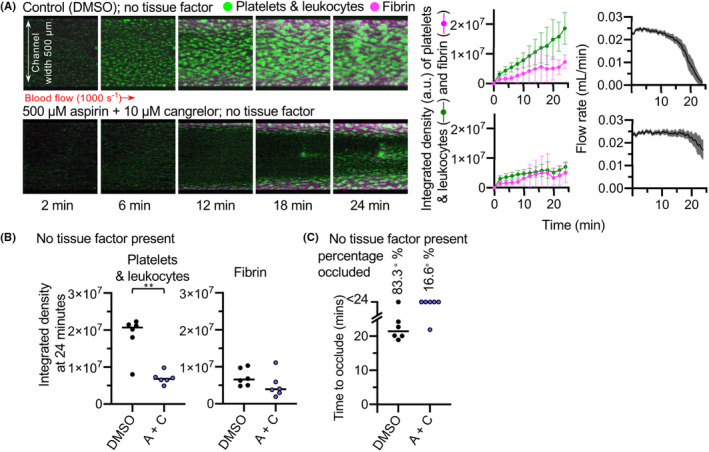
Dual antiplatelet therapy of aspirin + cangrelor effectively prevents thrombosis in the absence of tissue factor. (A) Whole blood was treated with aspirin (500 μM) + cangrelor (10 μM) (A + C) to provide DAPT, or their vehicles as control, before perfusion over collagen in a bifurcated microfluidic model of occlusive thrombosis. Platelets (and leukocytes) were labeled with DiOC6. Fibrin accumulation was monitored using Alexa Fluor‐546–labeled fibrinogen. Platelet (and leukocyte) and fibrin accumulation on collagen were visualized using confocal microscopy over a 24‐minute time course. The images are representative of 6 donors. Mean platelet (green) and fibrin (pink) accumulation ± standard deviation (SD), and mean flow rate are shown in the panels to the right (SD is shaded are for flow rate; n = 6). (B) The platelet and fibrin integrated density values at 24 minutes (**P < .05; no statistically significant difference in fibrin formation was found between control and treated experiments). (C) Time to occlude for each experiment. In control experiments, 1 of 6 channels did not occlude as is marked >24. In A + C‐treated experiments, occlusion was prevented in 5 of 6 channels. DMSO, dimethyl sulfoxide
FIGURE 2.
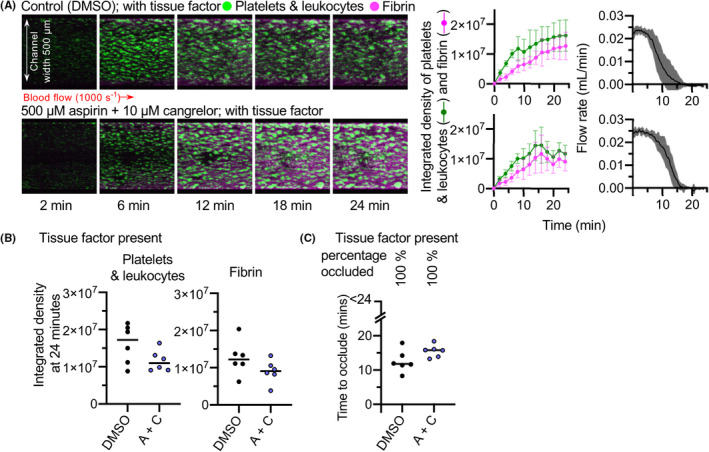
Aspirin + cangrelor does not prevent occlusive thrombosis triggered by both collagen and tissue factor. Whole blood was treated with aspirin + cangrelor (A + C) before perfusion across collagen and tissue factor. Images of platelet (and leukocyte) and fibrin accumulation are shown in (A) and are representative of six independent experiments using blood from different donors. Mean platelet (green) and fibrin (pink) accumulation ± standard deviation (SD), and mean flow rate are shown in the panels to the right (SD is shaded are for flow rate; n = 6). (B) The platelet and fibrin integrated density values at 24 minutes. The differences were not statistically significantly different. (C) Time to occlude for each experiment. All 12 channels occluded within the 24 minutes of each experiment. The time to occlusion was not significantly different following treatment with A + C. DMSO, dimethyl sulfoxide
DAPT, consisting of aspirin plus a P2Y12 inhibitor, is the first‐line therapy for prevention of myocardial infarction. 19 To assess the effects of DAPT, blood was incubated for 30 minutes with 500 μM aspirin and for 5 minutes with 10 μM cangrelor (A + C). In the absence of TF, preincubation of blood with aspirin + cangrelor significantly inhibited the aggregation of platelets within the channel compared to the control. Platelet deposition in the treated experiments mostly built from the edges of the device, and the center of the channels remained mostly clear of platelets over the course of the experiment (Figure 1A,B). Flow remained relatively unobstructed, with flow rate only decreasing toward the ends of some experimental runs and occlusion not occurring within 24 minutes in most (5/6) experiments (Figure 1C). These data indicate that aspirin + cangrelor effectively prevents thrombosis on collagen.
In contrast, the same aspirin + cangrelor had no comparatively little effect on thrombosis on collagen plus TF (Figure 2A,B). Occlusion occurred in all experiments, and there was no significant difference in the time to occlusion (Figure 2C). Moreover, aspirin + cangrelor under these conditions had no effect on platelet and leukocyte accumulation. Together, these data suggest that the antithrombotic efficacy of DAPT is context dependent, with little effect on occlusive thrombosis in the presence of a high level of TF. This suggests that under these conditions there are further drivers of platelet activation and thrombosis.
3.2. Vorapaxar and BMS 986120 do not prevent platelet accumulation and occlusive thrombosis in the presence of TF
One potential explanation for the lack of efficacy of DAPT in the presence of TF in our model is that the TF activates additional platelet activation pathways. TF triggers the coagulation cascade, leading to generation of thrombin, a potent activator of platelets via PAR1 and PAR4. 14 , 20 To investigate this, we used vorapaxar, an antagonist of PAR1, and BMS 986120, an antagonist of PAR4. To validate these antagonists, we monitored platelet activation in whole blood by flow cytometry. Vorapaxar inhibited platelet activation in response to the PAR1‐AP, in a concentration‐ and time‐dependent manner. In contrast, no inhibition of platelet activation in response to PAR4‐AP, was seen with vorapaxar at the concentrations tested. Conversely, BMS 986120 inhibited PAR4‐AP–induced platelet activation in a concentration‐ and time‐dependent manner, with no inhibition of PAR1‐AP–induced platelet activation (Figure 3). These data demonstrate the efficacy and selectivity of these antagonists.
To investigate the role of PAR1 and PAR4 in occlusive thrombosis in the presence of TF, blood was pretreated with vorapaxar, BMS 986120, or both combined, before perfusion through our occlusive thrombosis microfluidic system. However, these antagonists had no effect on thrombosis. There were no significant differences in platelet and leukocyte accumulation or fibrin accumulation (Figure 4). All channels occluded within the time frame of the experiment, and although a small increase in time to occlude was observed when blocking both PARs, this was not statistically significant (Figure 4). These data indicate that inhibition of either PAR1 or PAR4, or both PARs together, is not an effective strategy for the prevention of occlusive thrombi in our model.
FIGURE 4.
Incubation of whole blood with vorapaxar, BMS 986120, or both compounds together, does not inhibit occlusive thrombus formation. Whole blood was treated with vorapaxar (20 μM; V), BMS 986120 (0.1 μM; B), both combined (V + B), or DMSO as vehicle control, before perfusion across collagen and tissue factor at an initial shear rate of 1000 s−1. (A) Representative images from a single experiment (same blood donor, same blood draw) are shown, with mean ± standard deviation shown on the right (n = 8). (B) The platelet and fibrin integrated density values at 24 minutes. No statistically significant differences in platelet or fibrin accumulation were noted in any of the conditions tested. (C) Time to occlude for each experiment. A small apparent increase in time to occlude was noted when blood was pretreated with both vorapaxar and BMS 986120, but this was not statistically significant. All channels occluded within 24 minutes. DMSO, dimethyl sulfoxide
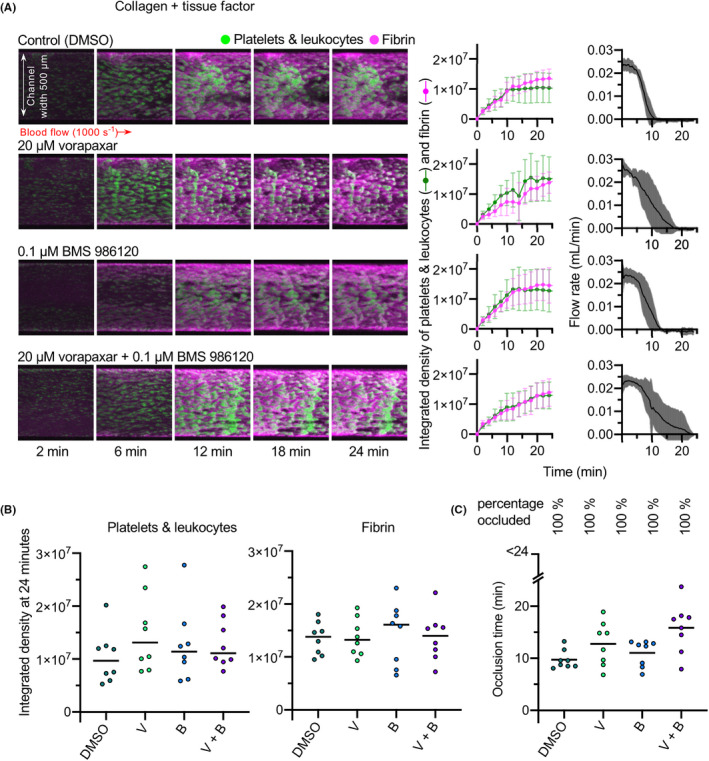
3.3. Vorapaxar and BMS 986120 combined with aspirin + cangrelor effectively prevents occlusive thrombosis
As neither DAPT nor PAR antagonism alone was sufficient to prevent occlusive thrombus formation in collagen + TF‐coated channels, we next investigated whether combining these drug treatments would be effective. Blood was pretreated with aspirin + cangrelor alone or in addition to vorapaxar and/or BS 986120, before perfusion through collagen + TF‐coated channels. As above, aspirin + cangrelor alone did not prevent occlusive thrombosis. Aspirin + cangrelor + vorapaxar inhibited occlusion during the time course of the experiment in some experiments, but with substantial variation between experiments. This is clearly seen in the wide variation around average flow rate under these conditions. Time to occlude was significantly inhibited compared to control (although the difference in time to occlude compared to aspirin + cangrelor was not statistically significantly different). The accumulation of platelets and leukocytes was also significantly inhibited (Figure 5). In contrast, aspirin + cangrelor + BMS 986120 had no effect on occlusion time or the accumulation of platelets and leukocytes or fibrin. The most effective combination was aspirin + cangrelor + vorapaxar + BMS 986120. Under these conditions, occlusion was prevented in every experiment, and accumulation of platelets and leukocytes was significantly inhibited. Despite this, the accumulation of fibrin was not inhibited under these conditions. These data suggest that combination therapy of P2Y12 inhibition, aspirin, vorapaxar, and BMS 986120 might provide effective protection against occlusive thrombosis even in the presence of high levels of TF.
FIGURE 5.
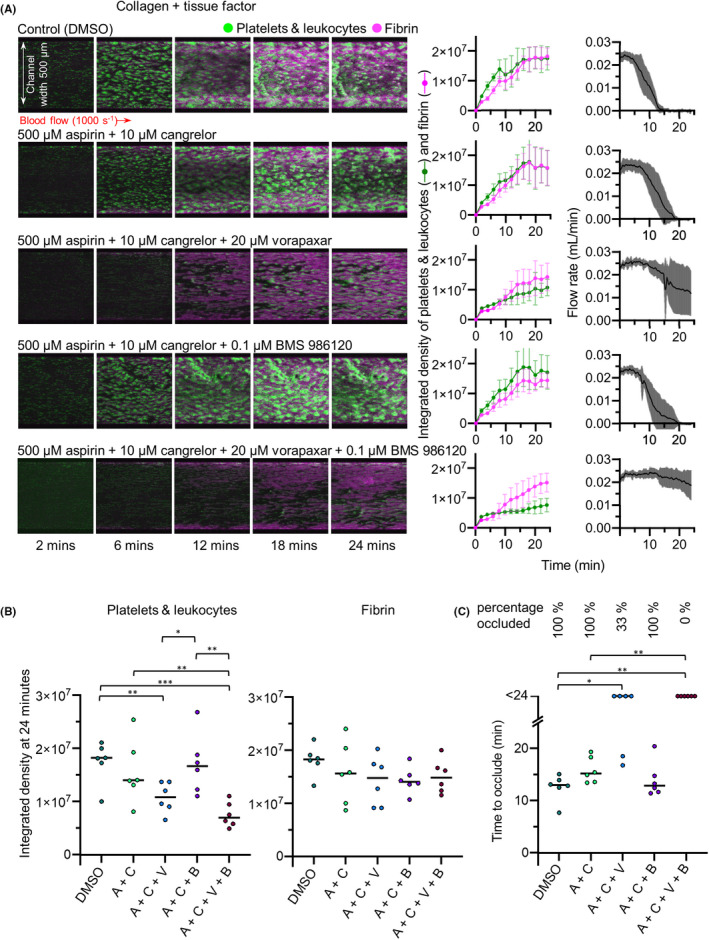
Combined block of P2Y12, COX, PAR1, and PAR4 prevents occlusive thrombus formation. Whole blood was treated with aspirin + cangrelor (A + C; to provide DAPT) either alone or with vorapaxar (A + C + V), BMS 986120 (A + C + B), both combined (A + C + V + B), or DMSO as vehicle control, before perfusion across collagen and tissue factor at an initial shear rate of 1000 s−1. (A) Representative images from a single experiment (same blood donor, same blood draw) are shown, with mean ± standard deviation shown on the right (n = 6). (B) The platelet and fibrin integrated density values at 24 minutes. Fibrin accumulation was not statistically significantly different to control in any treatment condition. (C) Time to occlude for each experiment. Treating with A + C + V significantly increased time to occlude compared to control, with four of six donors failing to occlude within the time frame of the experiment. Incubation with all four inhibitors was the most effective approach, with all six experiments failing to occlude within the time frame of the experiment. For (B, C) *P < .05, **P < .01, ***P < .001, compared to DMSO control. COX, cyclooxygenase; DMSO, dimethyl sulfoxide; PAR, protease‐activated receptor
4. DISCUSSION
Our study suggests that PAR antagonists may be beneficial antithrombotic agents under conditions where DAPT is insufficient. In this study, we used our recently described model of occlusive thrombosis to investigate whether PAR antagonism, either alone or in addition to DAPT, might effectively prevent occlusive thrombosis. We demonstrate that DAPT is ineffective in this model when thrombosis is triggered by collagen and TF, allowing us to investigate what alternative or additional antithrombotic agents may be protective. Under these conditions, addition of PAR antagonists to DAPT prevented occlusive thrombosis.
DAPT is a widely used strategy to prevent coronary thrombosis. Despite its success, however, many patients continue to suffer arterial thrombosis, suggesting that there are circumstances under which DAPT may be insufficient. In our occlusive thrombosis model, DAPT successfully prevented occlusion when the prothrombotic trigger was collagen alone. However, DAPT did not prevent thrombosis in our model when the prothrombotic trigger were collagen and TF together. Although the lack of effect of DAPT may appear surprising, and in contrast to the widely studied role of P2Y12 in thrombus formation and stability, 21 , 22 , 23 , 24 , 25 our data are nonetheless consistent with previous studies using in vitro thrombosis models. Consistent with our data, P2Y12 inhibitors reduce thrombosis in a variety of in vitro thrombosis models when the trigger was collagen alone. 26 , 27 Although many in vitro thrombosis models lack coagulation, P2Y12 inhibitors continue to be effective under coagulating conditions in some studies. 22 , 24 However, when TF or human plaque material containing TF was used, P2Y12 inhibition was less effective 28 or ineffective. 29 In murine in vivo models, the contribution of P2Y12 also depends on the thrombotic conditions. Although occlusion is absent in P2Yr12 −/− mice or clopidogrel‐treated mice in several studies, the contribution of P2Y12 depends on the extent of arterial injury. For example, in an extensive in vivo study using the murine FeCl3‐induced carotid artery thrombosis model, 30 occlusion was prevented in P2ry12 −/− or clopidogrel‐treated mice at the lowest level of injury. In contrast, occlusion was unaffected or slightly delayed at the highest level of injury. Similarly, clopidogrel was 10‐fold less potent at a higher level of injury in another study. 31 Different extents of injury may expose different repertoires of prothrombotic triggers, resulting in varying sensitivity to P2Y12 inhibition. Furthermore, as these arteries were healthy before injury, they may lack some prothrombotic triggers found in atherosclerotic plaques.
Moreover, the blood flow conditions of our microfluidic model differ from many in vitro models. Occlusive thrombi do not form in many published models, as blood flow as a syringe pump delivers blood at constant flow. In contrast, the bifurcated design of our model allows thrombosis to continue until the channel is occluded. 18 Our data are consistent with a study using the Total Thrombus‐Formation System, a commercially available microfluidic model of occlusive thrombosis. In that study, occlusive thrombosis on collagen alone was substantially inhibited by prior ingestion of aspirin and clopidogrel, but these drugs had no effect on occlusive thrombosis on collagen and TF. 32 Similar results were seen in a related study with the P2Y12 antagonist ticagrelor. 16 Together, these studies demonstrate that the efficacy of P2Y12 inhibition depends on the nature of the prothrombotic stimulus and possibly also on blood flow. Indeed, an important advantage of microfluidic thrombosis models is the ability to vary the prothrombotic stimulus, simulating a range of pathological conditions. The extent of lipid and TF accumulation in plaques, and the extent of plaque rupture, may vary between individual thrombotic episodes and between patients. We propose that our models with collagen alone, and with collagen and TF together, represent two positions on the spectrum of plaque rupture events. As the more extreme model (collagen with TF) is not inhibited by DAPT, it provides a means to investigate what other treatments might be effective.
PAR antagonists, either in addition to or instead of DAPT, have been proposed as antithrombotic agents. 7 , 33 , 34 Nonocclusive in vitro thrombosis models have largely supported this, although the results on which PAR is best to target are conflicting. One PAR1 antagonist partially reduced thrombosis when blood was perfused over collagen at 1500 s−1. 35 Similarly, another PAR1 antagonist inhibited nonocclusive thrombosis on collagen at 600 s−1, although not at a higher shear of 1800 s−1. 36 Conversely, thrombosis over collagen was reduced by a PAR4 inhibitor. 35 Similarly, a PAR4‐blocking antibody inhibited platelet procoagulant activity under arterial shear over collagen and reduced fibrin formation. 37 Moreover, prior BMS 986120 ingestion reduced thrombosis in an ex vivo Badimon perfusion chamber, in which blood is perfused over porcine aorta without intima and with the extracellular matrix exposed. 9 However, although collagen I is exposed in this model, TF is not likely to be exposed. 38 Together, previous studies suggest that PAR1 and/or PAR4 antagonists would reduce nonocclusive thrombosis when blood is perfused over collagen alone. However, P2Y12 inhibition also prevents occlusive and nonocclusive thrombosis on collagen alone, suggesting that additional PAR inhibition may not be required under these conditions.
In contrast, under conditions where DAPT is insufficient, our data suggest that PAR antagonism alone, even of PAR1 and PAR4 together, would not prevent occlusive thrombosis; instead, combined DAPT and antagonism of PARs was required. PAR1 antagonism by vorapaxar, combined with DAPT, significantly inhibited platelet and leukocyte accumulation and delayed or prevented occlusion in some experiments. Although PAR4 antagonism by BMS 986120 had little effect on thrombosis, combining vorapaxar with BMS 986120 (with DAPT) further inhibited platelet accumulation and prevented occlusive thrombosis. These data indicate that PAR antagonism will reduce thrombosis in situations where DAPT is insufficient. This conclusion is supported by the extensive in vivo study described above. 30 Following extensive carotid artery injury, arterial occlusion occurred in Par4 −/− mice. However, no occlusion occurred in P2ry12 −/−. Par4 −/− mice. 30 These data demonstrate that combined inhibition of PARs and P2Y12 is needed to prevent occlusive thrombosis following extensive arterial injury. Notably, however, the mouse models cannot be used to compare the benefit of PAR1 and PAR4 antagonists, as mouse platelets lack PAR1. 7 , 14 Human microfluidic models reach the same conclusion without the extensive use of mice and in a manner directly translatable to human antithrombotic therapy.
Microfluidic models of thrombosis have provided many insights into the mechanisms and pharmacology of thrombosis but are not without limitations and could be further refined. Atherosclerotic plaque development is accompanied by progressive stenosis of the coronary artery. Several microfluidic devices have incorporated such stenoses, uncovering the importance of high shear at the apex of stenosis and the gradient of the post‐stenosis deceleration zone as factors that contribute to thrombosis. 39 , 40 , 41 Notably, such factors are often also missing from in vivo injury models. An advantage of microfluidics is the ability to add factors such as a range of stenotic geometries to previously described devices, further refining our understanding of the mechanisms of thrombosis across a spectrum of plaque rupture scenarios.
Bleeding risk is a concern with antiplatelet therapies, particularly as vorapaxar in addition to DAPT increased bleeding complications in clinical trials. 8 Although BMS 986120 may be safer, 9 it also increased bleeding in primates. 10 Combination therapy of P2Y12 inhibition, aspirin, vorapaxar, and BMS 986120 might therefore be expected to further increase bleeding risk. For many patients, this risk may be unacceptably high, especially if DAPT alone is sufficient to reduce their thrombosis risk. For patients with the highest risk of thrombosis, however, the benefits of preventing occlusive thrombosis may potentially outweigh the bleeding risk. Microfluidic models of hemostasis could help assess this bleeding risk. However, bespoke microfluidic models of hemostasis would be best for assessing bleeding risk, rather than extrapolating from thrombosis models, as the biorheology of hemostasis is different from that of occlusive arterial thrombosis, including occurring in different blood vessels with different flow and shear rates, and extraluminal platelet activation during hemostasis compared to intraluminal thrombosis. 2 Several microfluidic hemostasis models have been described 42 , 43 that could be used alongside occlusive thrombosis models as part of a comparative assessment of antithrombotic benefit and bleeding risk.
This study demonstrates that comparing different prothrombotic triggers in microfluidic thrombosis models has the potential to develop a broader understanding of antithrombotic drug efficacy. Combining different triggers with different stenotic geometries could further deepen this understanding. However, translating these insights to patient benefit will require an understanding of the variation in triggers and geometries that are expressed in different patients. Conceivably, a combination of risk factor analysis and advances in imaging could one day be used to indicate which parameters of a microfluidic device best represent thrombotic risk on a patient‐by‐patient basis, opening the way for personalization of antithrombotic therapy.
RELATIONSHIP DISCLOSURE
There are no conflicts of interest to declare.
AUTHOR CONTRIBUTIONS
JB designed, performed, and analyzed experiments, and edited the manuscript. MTH designed experiments and wrote the manuscript. All authors read and approved the manuscript.
Supporting information
Fig S1
Berry J, Harper MT. Protease‐activated receptor antagonists prevent thrombosis when dual antiplatelet therapy is insufficient in an occlusive thrombosis microfluidic model. Res Pract Thromb Haemost. 2022;6:e12703. doi: 10.1002/rth2.12703
Handling Editor: Yotis Senis
Funding information
This work was funded by a National Centre for the Replacement, Refinement and Reduction of Animals in Research/British Heart Foundatio PhD studentship (NC/N002350/1).
REFERENCES
- 1. World Health Organization . World Health Statistics 2021: Monitoring Health for the SDGs, Sustainable Development Goals. World Health Organization; 2021. [Google Scholar]
- 2. Jackson SP. Arterial thrombosis‐insidious, unpredictable and deadly. Nat Med. 2011;17(11):1423‐1436. [DOI] [PubMed] [Google Scholar]
- 3. McFadyen JD, Schaff M, Peter K. Current and future antiplatelet therapies: emphasis on preserving haemostasis. Nat Rev Cardiol. 2018;15(3):181‐191. [DOI] [PubMed] [Google Scholar]
- 4. Cattaneo M, Podda GM. State of the art of new P2Y12 antagonists. Intern Emerg Med. 2010;5(5):385‐391. [DOI] [PubMed] [Google Scholar]
- 5. van der Meijden PEJ, Heemskerk JWM. Platelet biology and functions: new concepts and clinical perspectives. Nat Rev Cardiol. 2019;16(3):166‐179. [DOI] [PubMed] [Google Scholar]
- 6. Wilson SJ, Newby DE, Dawson D, et al. Duration of dual antiplatelet therapy in acute coronary syndrome. Heart. 2017;103(8):573‐580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hamilton JR. Protease‐activated receptors as targets for antiplatelet therapy. Blood Rev. 2009;23(2):61‐65. [DOI] [PubMed] [Google Scholar]
- 8. Morrow DA, Braunwald E, Bonaca MP, et al. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. 2012;366(15):1404‐1413. [DOI] [PubMed] [Google Scholar]
- 9. Wilson SJ, Ismat FA, Wang Z, et al. PAR4 (protease‐activated receptor 4) antagonism with BMS‐986120 inhibits human ex vivo thrombus formation. Arterioscler Thromb Vasc Biol. 2018;38(2):448‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wong PC, Seiffert D, Bird JE, et al. Blockade of protease‐activated receptor‐ 4(PAR4) provides robust antithrombotic activity with low bleeding. Sci Transl Med. 2017;9(371):eaaf5294. [DOI] [PubMed] [Google Scholar]
- 11. Balkenhol J, Kaltdorf KV, Mammadova‐Bach E, et al. Comparison of the central human and mouse platelet signaling cascade by systems biological analysis. BMC Genom. 2020;21(1):1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ware J. Dysfunctional platelet membrane receptors: from humans to mice. Thromb Haemost. 2004;92(3):478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. French SL, Paramitha AC, Moon MJ, et al. Humanizing the protease‐activated receptor (PAR) expression profile in mouse platelets by knocking Par1 into the Par3 locus reveals Par1 expression is not tolerated in mouse platelets. PLoS One. 2016;11(10):e0165565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Coughlin SR. Thrombin signalling and protease‐activated receptors. Nature. 2000;407(6801):258‐264. [DOI] [PubMed] [Google Scholar]
- 15. Colace TV, Muthard RW, Diamond SL. Thrombus growth and embolism on tissue factor‐bearing collagen surfaces under flow: role of thrombin with and without fibrin. Arterioscler Thromb Vasc Biol. 2012;32(6):1466‐1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Al Ghaithi R, Mori J, Nagy Z, et al. Evaluation of the total thrombus‐formation system (T‐TAS): application to human and mouse blood analysis. Platelets. 2018;30(7):893‐900. doi: 10.1080/0953710420181535704 [DOI] [PubMed] [Google Scholar]
- 17. Li M, Hotaling NA, Ku DN, Forest CR. Microfluidic thrombosis under multiple shear rates and antiplatelet therapy doses. PLoS One. 2014;9(1):e82493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Berry J, Peaudecerf FJ, Masters NA, et al. An “occlusive thrombosis‐on‐a‐chip” microfluidic device for investigating the effect of anti‐thrombotic drugs. Lab Chip. 2021;21:4104‐4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jamasbi J, Ayabe K, Goto S, et al. Platelet receptors as therapeutic targets: past, present and future. Thromb Haemost. 2017;117(7):1249‐1257. [DOI] [PubMed] [Google Scholar]
- 20. Tatsumi K, Mackman N. Tissue factor and atherothrombosis. J Atheroscler Thromb. 2015;22:543‐549. [DOI] [PubMed] [Google Scholar]
- 21. Nergiz‐Unal R, Cosemans J, Feijge M, et al. Stabilizing role of platelet P2Y(12) receptors in shear‐dependent thrombus formation on ruptured plaques. PLoS One. 2010;5(4):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maloney SF, Brass LF, Diamond SL. P2Y12 or P2Y1 inhibitors reduce platelet deposition in a microfluidic model of thrombosis while apyrase lacks efficacy under flow conditions. Integr Biol (Camb). 2010;2(4):183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dorsam RT, Kunapuli SP. Central role of the P2Y12 receptor in platelet activation. J Clin Invest. 2004;113(3):340‐345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cosemans JMEM, Munnix ICA, Wetzker R, et al. Continuous signaling via PI3K isoforms β and γ is required for platelet ADP receptor function in dynamic thrombus stabilization. Blood. 2006;108(9):3045‐3052. [DOI] [PubMed] [Google Scholar]
- 25. Baig AA, Haining EJ, Geuss E, et al. TMEM16F‐mediated platelet membrane phospholipid scrambling is critical for hemostasis and thrombosis but not thromboinflammation in mice—brief report. Arterioscler Thromb Vasc Biol. 2016;36(11):2152‐2157. [DOI] [PubMed] [Google Scholar]
- 26. Savage JS, Williams CM, Konopatskaya O, et al. Munc13‐4 is critical for thrombosis through regulating release of ADP from platelets. J Thromb Haemost. 2013;11(4):771‐775. [DOI] [PubMed] [Google Scholar]
- 27. Li R, Grosser T, Diamond SL. Microfluidic whole blood testing of platelet response to pharmacological agents. Platelets. 2017;28(5):457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Penz SM, Reininger AJ, Toth O, et al. Glycoprotein Ibα inhibition and ADP receptor antagonists, but not aspirin, reduce platelet thrombus formation in flowing blood exposed to atherosclerotic plaques. Thromb Haemost. 2007;97:435‐443. [PubMed] [Google Scholar]
- 29. Bossavy J‐P, Thalamas C, Sagnard L, et al. A double‐blind randomized comparison of combined aspirin and ticlopidine therapy versus aspirin or ticlopidine alone on experimental arterial thrombogenesis in humans. Blood. 1998;92(5):1518‐1525. [PubMed] [Google Scholar]
- 30. Cornelissen I, Palmer D, David T, et al. Roles and interactions among protease‐activated receptors and P2ry12 in hemostasis and thrombosis. Proc Natl Acad Sci USA. 2010;107(43):18605‐18610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang X, Xu L. An optimized murine model of ferric chloride‐induced arterial thrombosis for thrombosis research. Thromb Res. 2005;115(1):95‐100. [DOI] [PubMed] [Google Scholar]
- 32. Arima Y, Kaikita K, Ishii M, et al. Assessment of platelet‐derived thrombogenicity with the total thrombus‐formation analysis system in coronary artery disease patients receiving antiplatelet therapy. J Thromb Haemost. 2016;14(4):850‐859. [DOI] [PubMed] [Google Scholar]
- 33. Li S, Tarlac V, Hamilton JR. Using par4 inhibition as an anti‐thrombotic approach: why, how, and when? Int J Mol Sci. 2019;20:5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cunningham M, McIntosh K, Bushell T, et al. Proteinase‐activated receptors (PARs) as targets for antiplatelet therapy. Biochem Soc Trans. 2016;44:606‐612. [DOI] [PubMed] [Google Scholar]
- 35. Lin Y‐C, Ko Y‐C, Hung S‐C, et al. Selective inhibition of PAR4 (protease‐activated receptor 4)‐mediated platelet activation by a synthetic nonanticoagulant heparin analog. Arterioscler Thromb Vasc Biol. 2019;39(4):694‐703. [DOI] [PubMed] [Google Scholar]
- 36. Lee H, Sturgeon SA, Jackson SP, Hamilton JR. The contribution of thrombin‐induced platelet activation to thrombus growth is diminished under pathological blood shear conditions. Thromb Haemost. 2012;107(2):328‐337. [DOI] [PubMed] [Google Scholar]
- 37. French SL, Arthur JF, Lee H, et al. Inhibition of protease‐activated receptor 4 impairs platelet procoagulant activity during thrombus formation in human blood. J Thromb Haemost. 2016;14(8):1642‐1654. [DOI] [PubMed] [Google Scholar]
- 38. Giesen PLA, Rauch U, Bohrmann B, et al. Blood‐borne tissue factor: another view of thrombosis. Proc Natl Acad Sci USA. 1999;96(5):2311‐2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Westein E, van der Meer AD, Kuijpers MJE, et al. Atherosclerotic geometries exacerbate pathological thrombus formation poststenosis in a von Willebrand factor–dependent manner. Proc Natl Acad Sci USA. 2013;110(4):1357‐1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Holme PA, Ørvim U, Hamers MJAG, et al. Shear‐induced platelet activation and platelet microparticle formation at blood flow conditions as in arteries with a severe stenosis. Arterioscler Thromb Vasc Biol. 1997;17(4):646‐653. [DOI] [PubMed] [Google Scholar]
- 41. Receveur N, Nechipurenko D, Knapp Y, et al. Shear rate gradients promote a bi‐phasic thrombus formation on weak adhesive proteins, such as fibrinogen in a VWF‐dependent manner. Haematologica. 2020;105(10):2471‐2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schoeman RM, Rana K, Danes N, et al. A microfluidic model of hemostasis sensitive to platelet function and coagulation. Cell Mol Bioeng. 2017;10(1):3‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sakurai Y, Hardy ET, Ahn B, et al. A microengineered vascularized bleeding model that integrates the principal components of hemostasis. Nat Commun. 2018;9(1):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1